

Abstract
Background
Human coenzyme Q4 (COQ4) is essential for coenzyme Q (CoQ10) biosynthesis. Pathogenic variants in COQ4 cause childhood-onset neurodegeneration. We aimed to delineate the clinical spectrum and the cellular consequences of COQ4 deficiency.
Methods
Clinical course and neuroradiological findings in a large cohort of paediatric patients with COQ4 deficiency were analysed. Functional studies in patient-derived cell lines were performed.
Results
We characterised 44 individuals from 36 families with COQ4 deficiency (16 newly described). A total of 23 different variants were identified, including four novel variants in COQ4. Correlation analyses of clinical and neuroimaging findings revealed three disease patterns: type 1: early-onset phenotype with neonatal brain anomalies and epileptic encephalopathy; type 2: intermediate phenotype with distinct stroke-like lesions; and type 3: moderate phenotype with non-specific brain pathology and a stable disease course. The functional relevance of COQ4 variants was supported by in vitro studies using patient-derived fibroblast lines. Experiments revealed significantly decreased COQ4 protein levels, reduced levels of cellular CoQ10 and elevated levels of the metabolic intermediate 6-demethoxyubiquinone.
Conclusion
Our study describes the heterogeneous clinical presentation of COQ4 deficiency and identifies phenotypic subtypes. Cell-based studies support the pathogenic characteristics of COQ4 variants. Due to the insufficient clinical response to oral CoQ10 supplementation, alternative treatment strategies are warranted.
INTRODUCTION
Coenzyme Q10 (CoQ10 or ubiquinone) is a lipid-soluble cofactor that is an essential component of the mitochondrial oxidative phosphorylation (OXPHOS) system, shuttling electrons from NADH: coenzyme Q oxidoreductase (complex I) and succinate: coenzyme Q oxidoreductase (complex II) to coenzyme Q: cytochrome c oxidoreductase (complex III).1 In addition, CoQ10 is one of the main cellular antioxidants that protects cell membranes and plasma lipoproteins.2
The CoQ10 molecule is composed of a benzoquinone ring attached to a species-specific poly-isoprenoid side chain. Most CoQ10 required by human cells originates from endogenous de novo synthesis through an intricate biosynthetic pathway that requires the interplay of at least 10 different proteins.3
Primary CoQ10 deficiencies are caused by pathogenic variants in genes encoding proteins directly involved in the synthesis of CoQ10, while secondary CoQ10 deficiencies can be caused by defects in other components of the mitochondrial OXPHOS system. Recessive disorders leading to primary CoQ10 deficiency present with a broad clinical spectrum that overlaps with other mitochondrial diseases. Causative biallelic variants in genes relevant for CoQ10 synthesis have been identified in PDSS1 (OMIM *607429), PDSS2 (OMIM *610564), COQ2 (OMIM *609825), COQ4 (OMIM *612898), COQ5 (OMIM *616359), COQ6 (OMIM *614647), COQ7 (OMIM *601683), COQ8A/ADCK3 (OMIM *606980), COQ8B/ADCK4 (OMIM *615573) and COQ9 (OMIM *612837).4
COQ4 encodes a ubiquitously expressed protein associated with the mitochondrial inner membrane.5 Although the precise function of COQ4 remains elusive, it appears to play a crucial role in stabilising the CoQ multienzyme complex.6 Biallelic, pathogenic variants in the COQ4 gene underlie a severe paediatric disorder. A total of 29 affected individuals with biallelic variants in COQ4 have been reported to date.7–13 The phenotypic spectrum includes neonatal-onset encephalopathy,7,8 infantile developmental delay and epilepsy with or without hypertrophic cardiomyopathy,9,12 as well as childhood-onset ataxia with stroke-like episodes.10,11 In view of the clinical heterogeneity of COQ4 deficiency, investigations of larger patient cohorts are urgently needed to guide the clinical management and counselling of families. In addition, treatment with CoQ10 has been suggested for COQ4 deficiency, but its efficacy remains unclear.14
In this study, we systematically analysed a cohort of 44 patients from 36 families with 23 rare biallelic variants in COQ4, among which we identified four novel missense variants (c.437T>G, c.458C>T, c.469C>Aand c.718C>T) by exome sequencing.
MATERIALS AND METHODS
Clinical assessment
The 16 previously unreported patients were systematically assessed according to standardised criteria (online supplemental table S1). Previously published data from patients were reassessed using the same standards, including genetic and deep phenotyping, neuroradiological reports and MRI data (DICOM format). The dosage of CoQ10 treatment was recorded, and the effect was approximately rated according to descriptions in medical records as no effect, stabilisation or clinical improvement (online supplemental table S1). Informed consent was obtained from the 16 unpublished individuals or their legal representatives according to local regulations (study number #5238). Siblings (Family ID 16b and 26b) who presented with clinical symptoms of COQ4 deficiency but lacked genetic confirmation were not included in the statistical analyses.
Genetic sequencing
COQ4 variants were identified by Sanger, exome or genome sequencing. Detected variants were annotated in the Ensemble Variant Effect Predictor15 and the megSAP pipeline (https://github.com/imgag/megSAP) (online supplemental file 2, Genetic data). Variant confirmation and carrier testing were conducted by Sanger sequencing.
MRI analysis
Twenty sets of MRI results were available for analysis, 19 from newly diagnosed patients, seven of which were follow-up MRI results over a maximal time course of 4.4 years, and one already published MRI result.11 Digital brain MRI results, including T1-weighted, T2-weighted, diffusion-weighted sequences and fluid attenuated inversion recovery T2 (FLAIR) images, were assessed by two independent physicians. Comments and statistical analyses were performed on the MRI data available at the latest time-point available during follow-up. Cerebral atrophy was assessed according to Gburek-Augustat et al.16 Cerebellar atrophy was defined as rarefication of foliae with enlarged sulci and cerebellar hypoplasia as short foliae with poor branching. Brain MRI findings reported in the literature were systematically reviewed (online supplemental table S1). After the visual inspection, a pattern recognition approach was used to group the findings into three subtypes. In 24 cases, MRI data with nine additional follow-up studies were reported in the literature or clinical reports. Pathological MRI features that were not listed in the already published MRI reports were counted as non-existent.
MRI volumetry
The volumetric analysis was performed using available MRI datasets (n=18, including four follow-up studies) and DICOM images as previously described.17 Findings were compared with healthy age-matched and sex-matched controls, as previously described.17
Cell culture
Fibroblast cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (all from Life Technologies) at 37°C in a humidified atmosphere of 5% CO2. For CoQ10 supplementation experiments, fibroblasts were treated with 5μM CoQ10 (SanoMitQ10; mse-pharma) for 1week. The use of patient-derived cell lines was approved by the local ethics committee (study number #5238).
Measurements of CoQ10 and 6-DMQ levels
Measurements of CoQ10 and 6-DMQ levels were performed as described previously18 using ultra-performance liquid chromatography-electrospray tandem mass spectrometry (UPLC-ESI-MS/MS).
Dried blood spot (DBS) and cerebrospinal fluid (CSF) sample extraction
An 8mm disc was punched from the centre of the dried blood spot (DBS) sample and transferred to a 2mL Eppendorf Safe-Lock tube. A total of 500μL of extraction solvent (methanol/dichloromethane; 1:1) including 5ng of internal standard (d9-CoQ10) was added, and the tube was shaken at 300rpm for 30min at 37°C. The extract was then centrifuged at 14000rpm for 10 min, and the supernatant was evaporated to dryness under a nitrogen stream. The dried samples were reconstituted in 50 μL of methanol/hexane (95/5, v/v).
Two hundred microlitres of the cerebrospinal fluid (CSF) sample were mixed with 300μL isopropanol containing 5ng of internal standard (d9-CoQ10). After vortexing and centrifugation at 14000rpm for 10min, the organic phase was separated. One millilitre of hexane was added to the organic supernatant. After gentle shaking for 5min at room temperature, the extract was stained for 15min at 4°C. The hexane phase was then evaporated to dryness. The dried samples were reconstituted in 50 μL of methanol/hexane (95/5, v/v).
Measurements of CoQ10 levels in DBS and CSF
DBS and CSF samples were analysed using UPLC-MS/MS. The system consisted of an Acquity UPLC-I Class (Waters, UK) coupled to a Waters Xevo TQ-S tandem mass spectrometer (Waters, UK) equipped with an ESI source operating in the positive ion mode. Quantitative data were collected in multiple reaction monitoring mode. Chromatographic separation was performed using a Waters UPLC BEH Shield RP18 column (100 mm length, 2.1 mm inner diameter, 1.7 μm particle size; Waters) using methanol and water (98/2 including 0.05% formic acid; v/v) as the mobile phases. The run time and flow rate of this analysis were 5 min and 0.4 mL/min, respectively. For quantitative analysis, the following mass transitions were used: 863.7>197 (CoQ10) and 872.7>206 (d9-CoQ10 as internal standard). The chemicals CoQ10 and d9-CoQ10 were purchased from Sigma (Germany) and IsoSciences (USA).
COQ4 immunoblotting
Western blotting of cell lysates was performed as described previously.19 Primary antibodies against COQ4 (rabbit polyclonal antibody; 1:500; Proteintech), COQ7 (rabbit polyclonal antibody; 1:1000; Proteintech), COQ9 (rabbit polyclonal antibody; 1:1000; Proteintech) or SDHA (anti-mouse, 1:1000; Abcam, ab14715) were used.
Cell proliferation
Cell proliferation was determined using the crystal violet assay as described previously.19
Live/dead assay
Cell viability was measured using the Live/Dead Assay (Invitrogen) according to the manufacturer’s protocol.
Statistical analyses
Graph Pad Prism V.9 software (Graph Pad Software, La Jolla, California, USA) was used for statistical analyses. Student’s t-test was used to compare the mean values of repeated CoQ10 and 6-DMQ measurements. Kaplan-Meier analysis was used to evaluate survival and age at first presentation.
RESULTS
Genetic variant annotation
We analysed 44 patients from 36 families, including 16 previously unreported individuals. Patients originated from 17 different countries. The male-to-female ratio was 7:15, and consanguinity was reported for one-third of patients (online supplemental file 2). Nineteen biallelic variants in the COQ4 gene (ENST00000300452.8) have already been described, including three truncating variants (one frameshift, one stop-gain and one splice donor), 15 missense variants and one in-frame deletion (n=1; figure 1A and online supplemental table S2). We did not identify biallelic loss-of-function (LoF) variants. We identified four novel missense variants (figure 1A, labelled in red). Twenty-one of 23 reported variants were classified as disease-causing based on the American College of Medical Genetics and Genomics criteria. The c.311G>T and c.356C>T variants were classified as variants of unknown significance, as both variants were located on the same allele in a compound heterozygous state with a probable disease-causing frameshift variant (c.23_33del).9 Although the variants are spread throughout the entire COQ4 gene, exon 4 and exon five harbour most of the reported variants. Based on reported data, the c.370G>A variant can be identified as a Chinese founder variant.8,13 Prediction scores and allele frequency support the pathogenicity of the novel variants (see online supplemental table S2)
Figure 1.
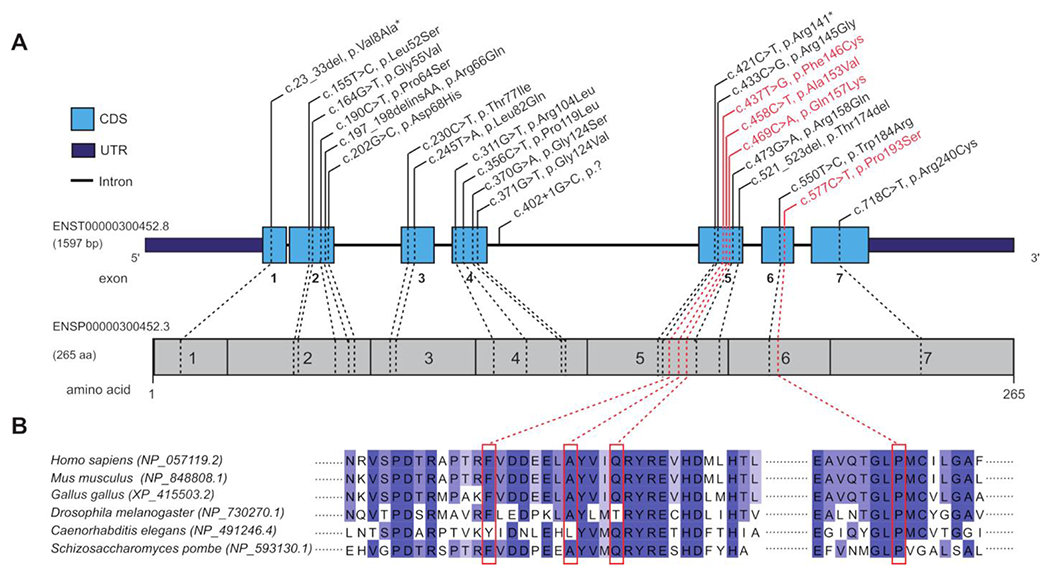
(A) COQ4 gene structure. Schematic overview of all variants identified in this study in relation to COQ4 protein structure. COQ4 variants were identified in all exons. Already published variants are marked in black; novel variants are in red. (B) Conservation of novel variants based on Jalview software.
Clinical characterization
Half of the patients exhibited clinical features in the first week of life, but the age at first presentation ranged from the prenatal period to a maximum of 9 years (median 0.82 months) (figure 2A). The median age at last examination was 2.25 years (range: first day of life to 27 years). Seventeen out of 44 affected individuals had died at the time of the study at a median age of 2.3 months (range: first day of life to 6 years, figure 2B). The major cause of death was cardiorespiratory failure (53%). Abnormalities during pregnancy were reported for nine patients, including intrauterine growth restriction (4), cerebral malformation (3), suspected cardiomyopathy (2) and decreased fetal movements (1) (online supplemental figure S1A). The majority of patients were born at term; only 5 of 37 patients were born prematurely (gestational age ranged from 32 to 36 weeks; online supplemental table S1). Respiratory distress (18/43) and/or seizures were the most frequent initial disease features (17/43) manifesting in the neonatal period (figure 2D and S1B).
Figure 2.
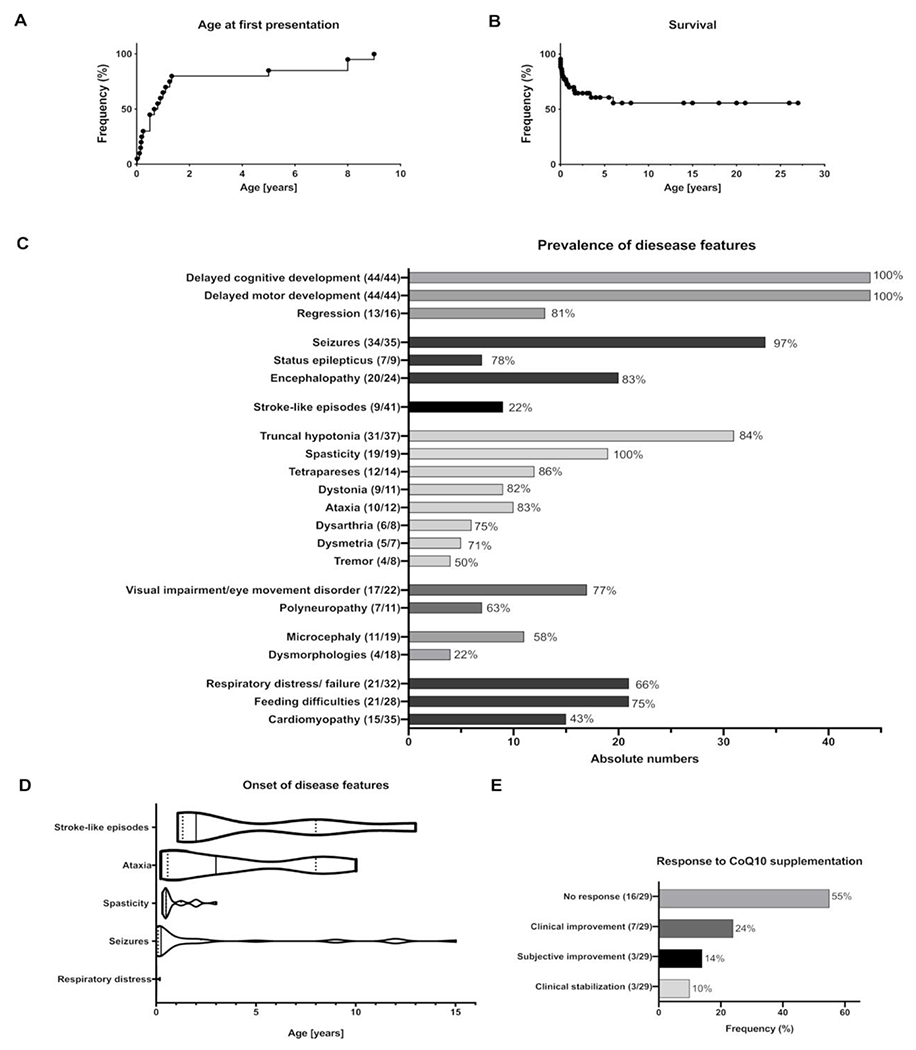
Phenotypic spectrum of COQ4 deficiency. (A) Age at first presentation: Kaplan-Meier analysis of disease onset. Censored subjects: 0, events: 44. Median onset 2.3 months. (B) Survival. Kaplan-Meier analysis of survival. Due to the limited number of subjects aged >7 years, estimates above age 7 years should be interpreted with caution. Censored subjects: 27, subjects deceased: 17. Median survival was undefined, as more than 50% of patients were alive at the time of last exam. (C) Prevalence of clinical features in patients with COQ4 deficiency (n=44). Numerators in brackets indicate the absolute numbers of affected patients versus the number of patients who were evaluated for the clinical feature. Due to the death of 17 patients and the young median age of the probands, clinical signs that manifest later in the disease course were evaluated only in a very limited number of surviving individuals. (D) Age of onset of disease features. Numbers of affected patients with available information regarding the age of onset for stroke-like episodes n=7, ataxia n=9, spasticity n=19, seizures n=33, respiratory distress n=19. Violin plots represent medians (lines), quartiles (dots) and minimum to maximum intervals. Age of onset classified as infantile was calculated as 0.5 years; hence, estimates should be interpreted with caution. (E) Effect of CoQ10 treatment. The numerator and denominator in brackets indicate the number of patients with a feature and the number of patients assessed for this feature, respectively. Treatment response based on clinical reports suggests no response to CoQ10 supplementation in 55% of patients. COQ4, coenzyme Q4; CoQ10, coenzyme Q10.
The central nervous system (CNS) was the most frequently affected organ system. Global developmental delay was observed in all patients (100%, 44/44) (figure 2C). The degree of cognitive impairment varied between moderate and profound. In 13 patients, developmental regression was observed, and in 20 patients, development was described as stagnant (figure 2C). The second most predominant clinical sign was seizures, which were present in 34 of 35 patients. The median onset was at 2 months of age (range: 0-12 years, figure 2D). In seven affected patients, repetitive status epilepticus was reported. Epileptic encephalopathy was diagnosed in 20 out of 24 patients. Nine patients suffered from clinically variable stroke-like episodes (online supplemental table S1). During the disease course, 19 patients developed spasticity. In 12 out of 14 patients, spastic tetraparesis or paraparesis was reported. Ataxia was reported in 10 of 12 patients with a median disease onset at 3 years (range 3 months to 10 years) (figure 2D). Other movement disorders reported included dystonia (9/11), dysmetria (5/7), dysarthria (6/8) and tremor (4/8). Nerve conduction studies were performed in 11 patients, and seven individuals were diagnosed with sensory polyneuropathy (figure 2C). Additional frequent disease features were visual impairment or eye movement disorders, observed in 17 out of 22 cases. Secondary findings included respiratory distress in 24 of 34 patients and feeding difficulties in 20 of 28 patients. Hypertrophic cardiomyopathy was reported in 15 of 35 patients. Dysmorphic facial features were only observed in four patients (figure 2C).
Laboratory and biochemical investigations
Hyperlactaemia was reported in 22 patients (of 31 with reported data). In 13 patients, abnormalities in the metabolic profile were reported, including elevated glutaric acid levels (5/24), elevated pyruvate levels (2/24), hyperammonaemia (3/24) and/or hyperalaninaemia (4/24) (online supplemental figure S1C). Biochemical analyses were performed using fibroblasts, muscle, CSF and blood from patients. CoQ10 levels in fibroblasts were reduced in 12 samples (out of 13 measured samples from different patients) and in muscle tissue in three samples (out of four) (online supplemental figure S1C). Respiratory chain complex I and III deficiency was prevalent in 8 of 10 fibroblast samples and in four of nine muscle samples.
Response to CoQ10 supplementation
Administration of CoQ10 was initiated in 29 cases (dosage range: 15mg/kg body weight/day to 60mg/kg body weight/day; online supplemental table S1). No response to CoQ10 supplementation was reported in 16 cases (figure 2E), and limited clinical improvement or stabilisation was noted in 12 cases (figure 2E). An association between dosage and response was not evident; however, treatment efficacy was not analysed quantitatively. For most individuals, there was no detailed information regarding the form of CoQ10 administered (ubiquinone vs ubiquinol). However, in the cases with detailed information, we found no differences regarding the neurological outcome between ubiquinol (eg, ID 06: no response, ID 11: no response, ID10: no response), ubiquinone (ID 05: no response) and idebenone (ID 07: no neurological improvement, possible stabilisation of cardiomyopathy).
In two individuals (ID 06 and ID 10), CoQ10 levels were measured in DBSs using UPLC-ESI-MS/MS analysis to determine CoQ10 uptake during substitution therapy (ID 06 received 34 mg/kg body weight/day and ID 10 received 30mg/kg body weight/day). CoQ10 levels were markedly increased in both children (ID 6: 35.7μM; ID 10: 34.2μM; control range: 1-4μM). In addition, in individual ID 10, CSF levels of CoQ10 were measured and elevated as well (28.2nM, control range 5-10nM).
Neuroimaging findings
Prenatal imaging data (ultrasound and MRI) were available for nine patients, and three of these patients were diagnosed with cerebellar hypoplasia based on a prenatal ultrasound (online supplemental table S1 and figure S1A). One prenatal MRI was rated as unremarkable. For 36 patients, postnatal brain MRI was reported, including follow-up studies in 16 cases. After reviewing all patients, the median age at the time of last reported cerebral imaging was found to be 16 months (range first day of life to 27 years).
The most prevalent finding was mild to profound cerebral atrophy, seen in 18 patients (figure 3A and B). Cerebellar atrophy was described in 15 out of 36 patients (plus in two cases based on ultrasound data) and/or cerebellar hypoplasia in 10 cases (plus three additional cases based on postnatal ultrasound and/or postmortem pathology findings, figure 3A and B). Three patients showed cystic degeneration of the cerebellar hemispheres (figure 3C). In eight patients, stroke-like lesions with occipito-parietal localisation were described (figure 3D). Bilateral basal ganglia and thalamic lesions were noted less frequently (7/36) (figure 3E). Delayed myelination was reported in six out of 36 cases. Magnetic resonance spectrometry was performed in 13 patients and revealed lactate peaks (11/13) mostly in the basal ganglia and/or, less frequently, a reduced N-acetylaspartate signal (3/13) (figure 3A and B).
Figure 3.
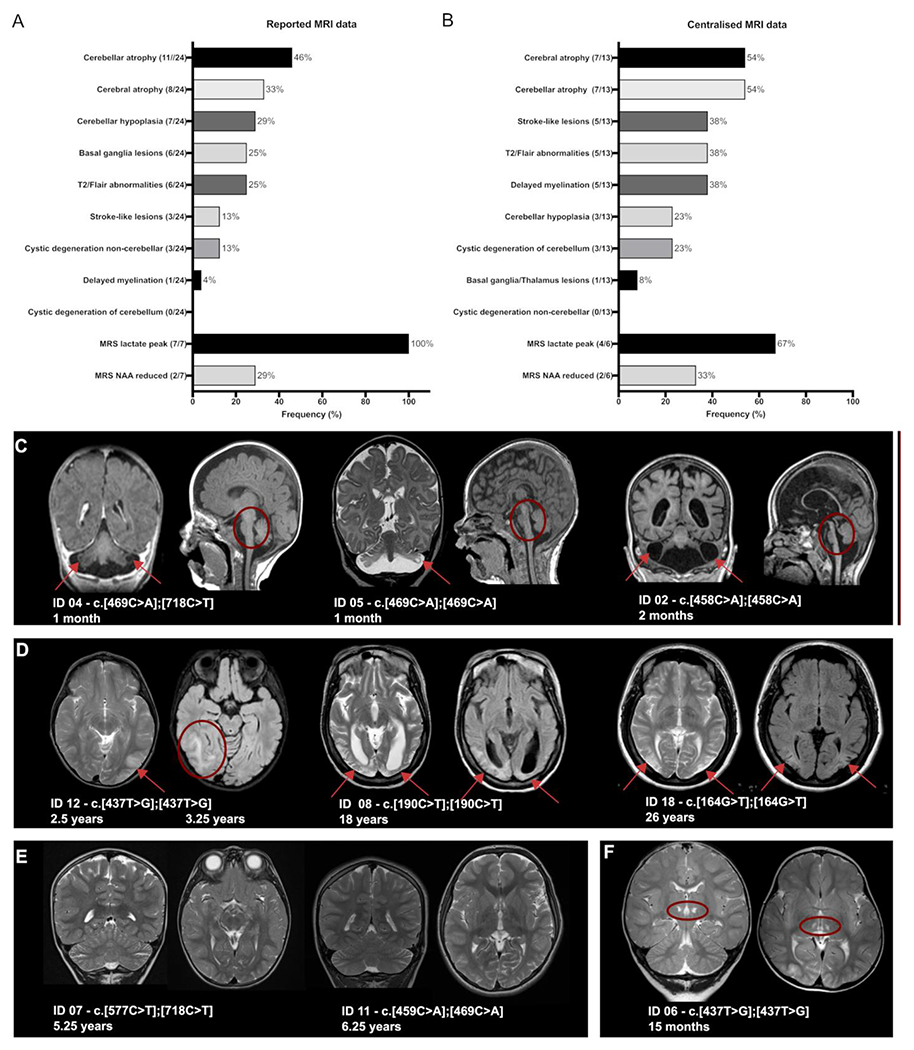
MRI features of COQ4 deficiency. (A) Reported MRI findings. The numerator and denominator in brackets indicate the number of patients with a feature and the number of patients with reported MRI data, respectively. (B) Centralised analysis of original MRI images from 13 patients and six follow-up studies by two independent physicians. (C) Representative cMRI images of type 1 highlighting cystic cerebellar degeneration and cerebellar atrophy (ID 04, ID 05 and ID 01). Coronal and sagittal sections of ID 04 (T1 weighted) and ID 05 (coronal T2-weighted and sagittal FLAIR sequence) show bilateral cystic changes in the cerebellum (red arrow) and hypoplasia of the brainstem and pons (red circle). ID 01 displays an enlargement of ventricles indicating severe cerebral and cerebellar atrophy. (D) Representative cMRI images of type 2 show infarcts at different stages in the parasagittal and parieto-occipital areas (red arrows). ID 12 displays acute strokes in the right and left parieto-occipital areas (axial FLAIR sequences) at different ages. Images of ID 8 and ID 18 reveal bilateral chronic, postinfarct changes. (E) Representative cMRI images of type 3 reveal slightly delayed myelination (ID 07, coronal and sagittal section, T2 weighted). Interestingly, ID 11 had no pathological MRI findings. (F) Bilateral thalamic lesions (red arrow) on ID 06 (coronal and sagittal section, T2 weighted) without pattern-specific changes. COQ4, coenzyme Q4; FLAIR, fluid attenuated inversion recovery.
After the visual inspection and recording of pathological findings referring to the MRI data available for centralised reanalyzes, a pattern recognition approach was used to group the findings: type 1 patients presenting with predominant cerebral atrophy and a mixed picture of cerebellar atrophy and hypoplasia (figure 3C, online supplemental figure S2), type 2 patients presenting with distinct stroke-like lesions (figure 3D) and mild global atrophy and type 3 patients displaying nonspecific changes with mild, generalised atrophy, slightly delayed myelination or even normal MRI scans (figure 3E).
Type 1 was seen in 4 out of 13 patients and was related to a severe clinical course with primary and severe developmental delay (ID 01, ID 04, ID 05 and ID 16). Brain volumetry revealed a reduction in the volume of the cerebellum, brainstem and pons even within the first months of life (online supplemental figure S2, ID 01, ID 04 and ID 05).
Type 2 was identified in 5 of 13 patients characterised by a later disease onset and less rapid disease progression. Volumetric quantification showed comparably normal volumes. However, general atrophy on visual inspection manifested over the disease course secondary to stroke-like lesions (online supplemental figure S2, ID 08, ID 09, ID 10, ID 12 and ID 018).
Type 3 was seen in 3 out of 13 patients with a rather non-specific disease course and moderate developmental delay. All measured brain areas showed normal volumes (online supplemental figure S2, ID 03, ID 07 and ID 11).
MRI results of ID 06 showed bilateral thalamic lesions and an otherwise unremarkable scan. As a result, we did not assign ID 06 to any of the suggested patterns (figure 3F).
Biochemical analysis in fibroblast lines
Investigation of patient-derived fibroblasts revealed a variable reduction in COQ4 protein levels in all cell lines (figure 4A). In addition, for several cell lines, we observed a variable reduction in the levels of the CoQ10 biosynthesis enzymes COQ7 and COQ9 (ID 01, ID 05 and ID 06).
Figure 4.
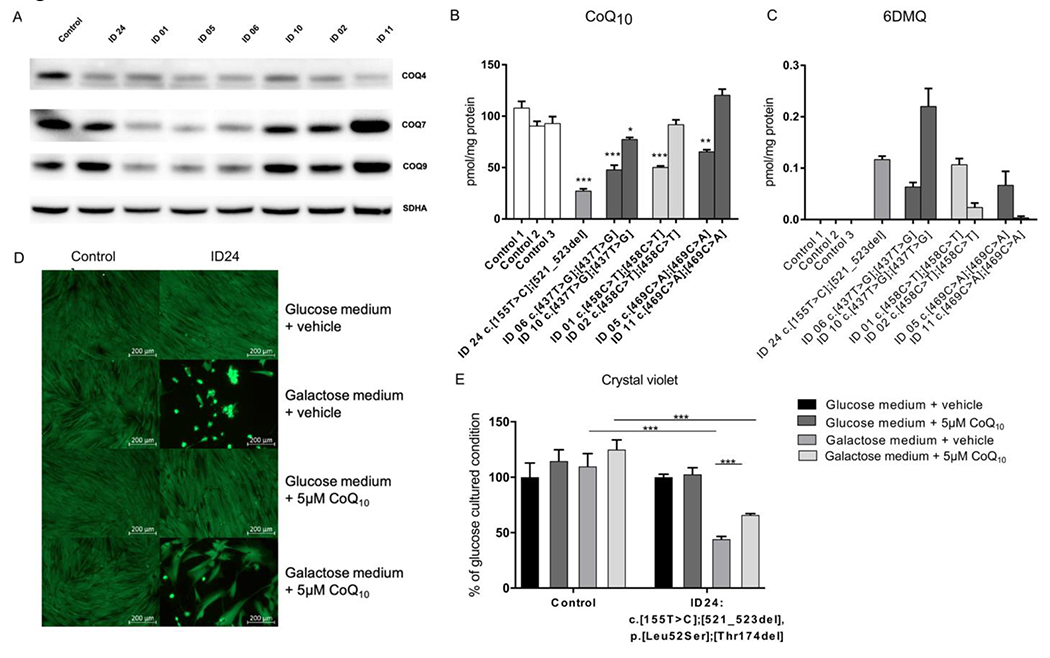
In vitro analyses (A) representative immunoblot analysis of the CoQ10-biosynthesis proteins COQ4, COQ7 and COQ9 in control and patient-derived fibroblast lines (ID 01, ID 02, ID 05, ID 06, ID 11 and ID 24). Succinate dehydrogenase complex flavoprotein subunit A (SDHA) was used as a loading control. Experiments demonstrate reduced COQ4 protein levels in all patient-derived cell lines. Moreover, a variable reduction in COQ7 and COQ9 protein levels was observed. (B) Quantification of cellular CoQ10 levels using UPLC-ESI-MS/MS analysis. Most patient-derived fibroblast lines show a significant reduction in CoQ10 levels (average values are presented as the mean±SEM; *p<0.05, **p<0.01 and ***p<0.001 significantly different from pooled control data based on Student’s t-test). (C) Determination of the metabolic intermediate 6-demethoxyubiquinone (6-DMQ) in control and patient-derived fibroblast lines. 6-DMQ was undetectable in three control cell lines, but levels were variably increased in all cell lines with COQ4 deficiency. (D) Quantification of cell proliferation using the crystal violet assay. The results are expressed relative to values obtained with the vehicle-treated condition cultured in glucose medium, which was set at 100% for each individual condition. The results indicate a severe growth defect in the COQ4-deficient cell line in galactose medium. This growth defect was partially improved with CoQ10 treatment. (E) Live/dead assay of control and COQ4-deficient fibroblasts (derived from individual ID 24) in glucose or galactose medium containing either 5 μM CoQ10 or similar amounts of ethanol (vehicle). CoQ10 treatment partially rescues the cell viability of galactose-cultured COQ4-deficient cells. COQ4, coenzyme Q4; CoQ10, coenzyme Q10; UPLC-ESI-MS/MS, ultra-performance liquid chromatography-electrospray tandem mass spectrometry.
To gain further insights into the reasons for reduced COQ4 protein amounts, we performed RT-qPCR in three selected cell lines (ID 1, ID 6 and ID 24). We observed no differences in COQ4 expression compared with controls (see online supplemental figure 3). This suggests a problem with protein stability or protein turnover resulting in reduced COQ4 protein levels. However, also impaired antibody binding due to the genetic variants cannot be fully excluded.
At the functional level, we observed significantly reduced cellular CoQ10 levels in most of the patient-derived fibroblast cell lines (figure 4B). Only two cell lines (ID 02 and ID 11) displayed normal CoQ10 levels. In addition, we detected increased levels of the metabolic intermediate 6-DMQ (figure 4C), which is known to accumulate in patients with CoQ10 biosynthesis defects that affect the stability of the CoQ synthome.19
Of note, when comparing data from immunoblotting with the results of biochemical CoQ10 and 6-DMQ measurements, certain discrepancies became apparent. Several patient cell lines with identical COQ4 variants showed divergent results. Most strikingly, ID 11 showed a drastically reduced COQ4 protein signal on immunoblotting but CoQ10 measurements were normal and 6-DMQ was only marginally elevated. In contrast, ID 05 showed similar immunoblotting results as ID 11, but CoQ10 levels were significantly reduced and 6-DMQ was clearly elevated. A comparable pattern was observed for ID 01 and ID 02 or ID 06 and ID 10. It is difficult to explain these discrepancies but one interesting observation might be that ID 02, ID 10 and ID 11 show normal or even increased levels of the CoQ10 biosynthesis enzymes COQ7 and COQ9 on immunoblotting, whereas ID 01, ID 05 and ID 06 show clearly decreased levels of COQ7 and COQ9. These results suggest a different downstream effect on CoQ10 biosynthesis in the cell lines (eg, in ID 02, ID 10 and ID 11: it is upregulated/in ID 01, ID 05 and ID 06: it is downregulated). Based on our data, it is unclear why the cell lines show different compensation strategies. When looking at the clinical phenotypes, no clear correlation between CoQ10 levels and clinical outcome can be established.
Besides the experiments for validation of genetic variants, we investigated the effect of CoQ10 supplementation therapy in vitro (figure 4D and E). To this end, control and patient-derived fibroblasts were cultured in glucose or galactose medium containing either 5μM CoQ10 or similar amounts of ethanol (vehicle). We specifically used fibroblasts from ID 24 for these experiments because this cell line has very low CoQ10 levels and is unable to survive in galactose medium, which forces the cells to use the respiratory chain for energy production. Cell survival experiments indicated a benefit of the COQ4-deficient cell line, with a partial recovery of cell viability upon CoQ10 supplementation. Quantification of cell proliferation confirmed this finding. Of note, apart from CoQ10, we also investigated the effects of the drugs idebenone (100 nM and 200 nM) and MitoQ (mitochondria-targeted ubiquinone; 10nM and 100nM) in this model system. However, no benefit on cell survival and proliferation was observed (data not shown).
DISCUSSION
The heterogeneity of the clinical manifestations of primary CoQ10 deficiencies is challenging for clinicians and geneticists.4–20 For family counselling and based on the recommended treatment option with CoQ10 supplementation, early diagnosis is very important. Nevertheless, many uncertainties exist regarding the clinical management and prognosis of affected individuals.14
Disease course of human COQ4 deficiency
A reanalysis of reported cases7 8 12 13 and characterisation of 16 unreported individuals revealed that COQ4 deficiency manifests as an early-onset neurodegenerative disorder. All individuals presented with a variable but mostly profound developmental disability. In most patients, clinical course was complicated by seizures. Severely affected individuals suffered from recurrent status epilepticus. Over time, patients developed variable movement disorders, including ataxia, spasticity and/or dystonia, leading to progressive immobility. In nine patients, recurrent stroke-like episodes complicated the disease course. The clinical presentation during these stroke-like episodes was variable, ranging from motor impairment to worsening of epilepsy. Only few patients experienced a nearly complete recovery, while others exhibited long-term sequelae and gliosis in the occipito-parietal region (figure 3D). The majority of patients did not benefit from CoQ10 supplementation and repetitive strokes occurred despite therapy. Only in few patients a subjective stabilisation was reported (online supplemental table S1). In addition to CNS and skeletal muscle involvement, hypertrophic cardiomyopathy emerged as an important organ manifestation. The overall survival was unfavourable, as only 5 of 44 patients reached adulthood. The cause of death was mainly cardiorespiratory failure, which most likely resulted from progressive CNS damage and secondary complications.
Analysis of clinical and neuroimaging data
By performing a systematic analysis of clinical and neuroimaging data, we searched for specific disease patterns within the patient cohort. Our results distinguished three conditions.
Type 1: Patients with this phenotype exhibit a severe cerebellar disorder with hypoplasia in combination with cerebellar atrophy and/or distinct cystic degeneration (figure 3C). Imaging studies indicated that this neurodegenerative disorder is already observed during prenatal development. This phenomenon has also been reported for other severe mitochondrial diseases.21 22 However, the cystic degeneration of cerebellar hemispheres detected in several patients appeared unusual and has, to our knowledge, not been described in other mitochondrial disorders. The clinical course of type 1 patients is severe and characterised by a profound developmental disability and poor prognosis (the oldest patient was in middle childhood). In addition to cerebellar hypoplasia, bilateral basal ganglia lesions were reported in the literature in six patients with a type 1 phenotype. However, basal ganglia lesions were not observed in the cohort available for the repeated MRI analysis.
Type 2: The neuroimaging pattern of this phenotype is characterised by stroke-like episodes. When reanalysing the nine patients with stroke-like lesions on MRI,7,10,11 we noted that the localisation of these infarcts was always in the parieto-occipital region, which appears to be specific to COQ4 deficiency. Patients exhibit an intermediate clinical phenotype with a slightly later onset of seizures, movement disorder (ataxia, dystonia) and improved long-term survival (the oldest patient was in late 20s).
Type 3: his phenotype accounts for an as yet unspecified clinical pattern. Neuroimaging suggested mild generalised atrophy or normal brain volumes. Consistent with the mild neuroradiological pathology, patients present with moderate developmental delay and a rather stable disease course. Only one patient with a stable disease course exhibited isolated bilateral thalamic hyperintensities (ID 06).
Interestingly, delayed myelination was detected in approximately half of all MRI studies that were evaluated using centralised standards. These neuroimaging features were not discussed in previous reports on COQ4 deficiency.
Genotype–phenotype correlation
Although genotype–phenotype correlations are difficult to establish, some clinicogenetic patterns were identified. One important finding appeared to be that truncating variants in a compound heterozygous state with missense variants led to a type 1 phenotype. Consistent with this finding, the residual amounts of COQ4 protein and the levels of CoQ10 in fibroblasts were lower in these patients than in individuals carrying biallelic missense variants (figure 4A and B). LoF variants were not detected in individuals with a type 2 or 3 phenotype. Interestingly, biallelic LoF variants were not identified, suggesting that they result in a non-viable phenotype.
The phenotypic spectrum and disease course were consistent among affected siblings within the same family, underlining the effects of epigenetic factors. Regarding the comparison of individuals from different families, the phenotypic spectrum was more variable. For example, ID 10 and ID 12 clinically and radiologically exhibited repetitive strokes and harboured the same homozygous missense variants as ID 03 and ID 06. However, ID 03 and ID 06 presented with very different clinical features. ID 03 suffered from severe epileptic encephalopathy, and brain MRI has not shown stroke-like lesions to date. ID 06 presented with a moderate developmental delay and late-onset seizures (2.5 years). Brain MRI showed bilateral thalamic lesions and no signs of metabolic strokes.
The heterogeneity of the biochemical phenotype due to biallelic missense variants regarding variable COQ4 protein and functional CoQ10 levels mirrors the broad clinical spectrum and suggests the hypothesis of alternative, compensatory pathways of CoQ10 synthesis.
More consistently, the functional impact of LoF variants in compound heterozygous state with any missense variant becomes clinically and biochemically evident.
Taken together, despite some interesting observations, further reliable statistical analyses of genotype–phenotype correlations are not yet applicable.
Laboratory findings in COQ4 deficiency
Similar to many other mitochondrial disorders, no specific parameters on clinical chemistry or metabolic screening that clearly indicate a COQ4 deficiency are available. Metabolic findings such as hyperlactaemia are highly variable depending on the clinical condition of the patient. As depicted in supplementary figure 1C, approximately 25% of patients did not show increased lactate levels in blood. Investigation of CSF appeared to be more reliable, although values were reported only for a small subset of patients. Comparably, metabolic work-up in tissue biopsies showed results that were more consistent, although some degree of variability was observed. Of note, biochemical tissue analysis was performed in different laboratory facilities, which might add to this variability. Due to the lack of specific disease markers, we suggest that the diagnostic assessments should be performed according to recommendations for other mitochondrial diseases.23
Biochemical consequences of COQ4 deficiency
Consistent with the predicted pathogenicity of COQ4 variants, we observed a reduction in COQ4 protein levels in all cell lines investigated. Moreover, cellular CoQ10 levels were significantly lowered in most fibroblast lines. Nevertheless, two patient-derived cell lines showed normal CoQ10 levels, consistent with other publications on CoQ10 deficiency disorders, indicating that24 biochemical defects are not always detectable in fibroblasts.
Interestingly, there was no correlation between the genetic and biochemical phenotypes (eg, fibroblasts of individuals ID 01 and ID 02, ID 05 and ID 11, ID 06 and ID 10). These individuals have similar COQ4 variants but display clearly different results regarding COQ4 levels (figure 54). This finding suggests that additional regulatory processes modulate CoQ10 biosynthesis. In this context, the upregulation or downregulation of other CoQ10 biosynthesis enzymes might play a role. As detailed in the results section, we observed reduced expression levels of the proteins COQ7 and COQ9 in cell lines with low CoQ10 levels and normal or even increased expression COQ7 and COQ9 levels in cell lines with normal CoQ10 levels. This phenomenon might point to different cellular adaptation strategies, which need to be addressed in follow-up research studies.
Measurements of the metabolic intermediate 6-DMQ revealed elevated levels in all patient-derived cell lines. Of note, this metabolite is processed by the enzyme COQ7 and the lipid-binding protein COQ9.25 Elevated 6-DMQ levels have been suggested as an indicator of a disturbance/instability in the CoQ synthome, and the metabolite is absent in healthy controls.18 Based on our findings, elevated 6-DMQ levels are a consistent marker of COQ4-deficient cell lines. However, 6-DMQ is not specific to COQ4 deficiency since it can also be elevated in other CoQ10 biosynthesis defects (eg, COQ7 and COQ9). Unfortunately, 6-DMQ was not detected in the DBS analysis of affected individuals, indicating that this metabolite might not be suitable as a screening parameter.
In addition to the aforementioned experiments, we also investigated the effects of CoQ10 treatment on patient-derived fibroblasts. We observed a severe growth defect in fibroblasts from individual ID 24 after culture in galactose-containing medium, which was partially restored on CoQ10 supplementation. However, we did not observe a full recovery, despite the administration of a relatively high dose of CoQ10, suggesting that exogenous CoQ10 supplementation might be insufficient to compensate for the biosynthesis defect.
Clinical response to CoQ10 treatment
The treatment response of individuals with COQ4 deficiency to CoQ10 supplementation was mostly unsatisfactory (figure 2E). In particular, children with early-onset encephalopathy showed a rapid clinical decline. No objective measure of the treatment effect was available in these patients (eg, no improvement in epilepsy, etc). The efficacy of CoQ10 supplementation in patients with a slower disease progression was not studied in a systematic manner. Notably, patients continued to suffer from stroke-like episodes, despite long-term supplementation with CoQ10,1110.
As explained in the results section, information on the use of ubiquinone versus ubiquinol was limited underlying the need of controlled studies. The reasons for CoQ10 treatment failure remain unclear. One factor might be the poor intestinal bioavailability of CoQ10.18 However, we detected highly elevated levels of CoQ10 in DBS from ID 06 and ID 10 during supplementation therapy, indicating sufficient intestinal uptake. Another problem with CoQ10 treatment might be limited transport across the blood–brain barrier. However, we detected elevated CoQ10 levels in the CSF of ID 10.
Although measurements of CoQ10 levels in DBS and CSF were obtained from single samples, our findings suggest that other factors influence the poor treatment response. In this context, the intracellular transport of CoQ10 might be important. Endogenously synthesised CoQ10 requires the shuttle proteins COQ10A and COQ10B to reach the respiratory chain. Researchers have not clearly determined if exogenous CoQ10 effectively enters this pathway. This idea is consistent with the results obtained after CoQ10 treatment in vitro, which revealed only a partial improvement in cell growth. Accordingly, medical approaches to reactivate endogenous CoQ10 biosynthesis might constitute a promising alternative. However, for patients with COQ4 deficiency, no precursor compounds for such metabolic bypass therapies have been identified to date.18 Currently, we must be aware of the limited treatment options that are available for patients with COQ4 deficiency.
CONCLUSIONS
The data presented here provide detailed insights into the clinical presentation of COQ4 deficiency and highlight the fact that distinct subtypes of disease severity can be distinguished.
This is important for adequate counselling of affected families regarding prognosis and potential complications during the disease course. A detailed description of distinct neuroimaging findings seen in COQ4 deficiency will help in patient phenotyping and interpretation of genetic findings. Moreover, brain regions that are specifically vulnerable to COQ4 deficiency are identified in our study, which might have implications for future studies addressing the tissue-specific role of COQ4. Our in vitro experiments provide a set of key parameters (eg, protein expression, endogenous CoQ10 production and 6-DMQ levels) for functional validation of COQ4 variants. In our view, this strategy proved to be very helpful to complement genetic analysis. Regarding treatment of COQ4-deficiency, it is important to note that oral CoQ10 supplementation showed only limited benefits in vitro and in vivo. The reasons for this unfavourable treatment response is unclear. Analysis of blood and CSF in selected cases suggest that this phenomenon cannot only be explained by problems with oral bioavailability or inadequate uptake across the blood brain barrier. Accordingly further research on treatment options for human COQ4 deficiency is urgently required.
Supplementary Material
Acknowledgements
All authors thank the families for participating in the study.
Funding
SG and IK-M are members of the European Reference Network for Rare Neurological Diseases - Project ID No 739510.The study was supported by a grant of the German Research Foundation/Deutsche Forschungsgemeinschaft (DI 1731/2-2 to FD) and by a grant from the “Elterninitiative Kinderkrebsklinik e.V.” (Düsseldorf; #701900167). PEB is supported by NIH NINDS RO1 NS08372
Footnotes
Competing interests None declared.
Patient consent for publication Not applicable.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement
Data are available in a public, open access repository. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
REFERENCES
- 1.Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis 2015;38:145–56. [DOI] [PubMed] [Google Scholar]
- 2.López-Lluch G, Rodríguez-Aguilera JC, Santos-Ocaña C, Navas P. Is coenzyme Q a key factor in aging? Mech Ageing Dev 2010;131:225–35. [DOI] [PubMed] [Google Scholar]
- 3.Bentinger M, Tekle M, Dallner G. Coenzyme Q--biosynthesis and functions. Biochem Biophys Res Commun 2010;396:74–9. [DOI] [PubMed] [Google Scholar]
- 4.Herebian D, López LC, Distelmaier F. Bypassing human CoQ 10 deficiency. Mol Genet Metab 2018;123:S1096-7192(17)30704-7:289–91. [DOI] [PubMed] [Google Scholar]
- 5.Casarin A, Jimenez-Ortega JC, Trevisson E, Pertegato V, Doimo M, Ferrero-Gomez ML, Abbadi S, Artuch R, Quinzii C, Hirano M, Basso G, Ocaña CS, Navas P, Salviati L. Functional characterization of human COQ4, a gene required for coenzyme Q10 biosynthesis. Biochem Biophys Res Commun 2008;372:35–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marbois B, Gin P, Gulmezian M, Clarke CF. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim Biophys Acta 2009;1791:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brea-Calvo G, Haack TB, Karall D, Ohtake A, Invernizzi F, Carrozzo R, Kremer L, Dusi S, Fauth C, Scholl-Bürgi S, Graf E, Ahting U, Resta N, Laforgia N, Verrigni D, Okazaki Y, Kohda M, Martinelli D, Freisinger P, Strom TM, Meitinger T, Lamperti C, Lacson A, Navas P, Mayr JA, Bertini E, Murayama K, Zeviani M, Prokisch H, Ghezzi D. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet 2015;96:S0002-9297(15)00002-6:309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung WK, Martin K, Jalas C, Braddock SR, Juusola J, Monaghan KG, Warner B, Franks S, Yudkoff M, Lulis L, Rhodes RH, Prasad V, Torti E, Cho MT, Shinawi M. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet 2015;52:627–35. [DOI] [PubMed] [Google Scholar]
- 9.Sondheimer N, Hewson S, Cameron JM, Somers GR, Broadbent JD, Ziosi M, Quinzii CM, Naini AB. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ10 deficiency. Mol Genet Metab Rep 2017;12:23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosch AM, Kamsteeg E-J, Rodenburg RJ, van Deutekom AW, Buis DR, Engelen M, Cobben J-M. Coenzyme Q10 deficiency due to a Coq4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol Genet Metab Rep 2018;17:19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caglayan AO, Gumus H, Sandford E, Kubisiak TL, Ma Q, Ozel AB, Per H, Li JZ, Shakkottai VG, Burmeister M. Coq4 mutation leads to childhood-onset ataxia improved by COQ10 administration. Cerebellum 2019;18:665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu M, Zhou Y, Wang Z, Xia Z, Ren J, Guo Q. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J Hum Genet 2019;64:297–304. [DOI] [PubMed] [Google Scholar]
- 13.Yu MH-C, Tsang MH-Y, Lai S, Ho MS-P, Tse DML, Willis B, Kwong AK-Y, Chou Y-Y, Lin S-P, Quinzii CM, Hwu W-L, Chien Y-H, Kuo P-L, Chan VC-M, Tsoi C, Chong S-C, Rodenburg RJT, Smeitink J, Mak CC-Y, Yeung K-S, Fung JL-F, Lam W, Hui J, Lee N-C, Fung C-W, Chung BH-Y. Primary coenzyme Q10 deficiency-7: expanded phenotypic spectrum and a founder mutation in southern Chinese. NPJ Genom Med 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salviati L, Trevisson E, Doimo M, Navas P. Primary coenzyme Q10 deficiency. In: Adam MP, Ardinger HH, Pagon RA, eds. Gene reviews. Seattle, WA: University of Washington, 1993: 1993–2018. [PubMed] [Google Scholar]
- 15.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gburek-Augustat J, Groeschel S, Kern J, Beck-Woedl S, Just J, Harzer K, Stampfer M, Kraegeloh-Mann I. Comparative analysis of cerebral magnetic resonance imaging changes in nontreated infantile, juvenile and adult patients with niemann-pick disease type C. Neuropediatrics 2020;51:037–44. [DOI] [PubMed] [Google Scholar]
- 17.Ekert K, Groeschel S, Sánchez-Albisua I, Frölich S, Dieckmann A, Engel C, Krägeloh-Mann I. Brain morphometry in pontocerebellar hypoplasia type 2. Orphanet J Rare Dis 2016;11:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herebian D, Seibt A, Smits SHJ, Bünning G, Freyer C, Prokisch H, Karall D, Wredenberg A, Wedell A, López LC, Mayatepek E, Distelmaier F. Detection of 6-demethoxyubiquinone in CoQ10 deficiency disorders: Insights into enzyme interactions and identification of potential therapeutics. Mol Genet Metab 2017;121:S1096-7192(17)30100-2:216–23. [DOI] [PubMed] [Google Scholar]
- 19.Herebian D, Seibt A, Smits SHJ, Rodenburg RJ, Mayatepek E, Distelmaier F. 4-Hydroxybenzoic acid restores CoQ10 biosynthesis in human COQ2 deficiency. Ann Clin Transl Neurol 2017;4:902–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alcázar-Fabra M, Trevisson E, Brea-Calvo G. Clinical syndromes associated with coenzyme Q10 deficiency. Essays Biochem 2018;62:377–98. [DOI] [PubMed] [Google Scholar]
- 21.Baertling F, Klee D, Haack TB, Prokisch H, Meitinger T, Mayatepek E, Schaper J, Distelmaier F. The many faces of paediatric mitochondrial disease on neuroimaging. Childs Nerv Syst 2016;32:2077–83. [DOI] [PubMed] [Google Scholar]
- 22.Baertling F, Alhaddad B, Seibt A, Budaeus S, Meitinger T, Strom TM, Mayatepek E, Schaper J, Prokisch H, Haack TB, Distelmaier F. Neonatal encephalocardiomyopathy caused by mutations in VARS2. Metab Brain Dis 2017;32:267–70. [DOI] [PubMed] [Google Scholar]
- 23.Wortmann SB, Mayr JA, Nuoffer JM, Prokisch H, Sperl W. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics 2017;48:309–14. [DOI] [PubMed] [Google Scholar]
- 24.Barca E, Musumeci O, Montagnese F, Marino S, Granata F, Nunnari D, Peverelli L, DiMauro S, Quinzii CM, Toscano A. Cerebellar ataxia and severe muscle CoQ10 deficiency in a patient with a novel mutation in ADCK3. Clin Genet 2016;90:156–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohman DC, Aydin D, Von Bank HC, Smith RW, Linke V, Weisenhorn E, McDevitt MT, Hutchins P, Wilkerson EM, Wancewicz B, Russell J, Stefely MS, Beebe ET, Jochem A, Coon JJ, Bingman CA, Dal Peraro M, Pagliarini DJ. An isoprene lipid-binding protein promotes eukaryotic coenzyme Q biosynthesis. Mol Cell 2019;73:e10:763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in a public, open access repository. Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.