

Abstract
EP4, a prostaglandin E2 receptor, has shown an immunosuppressive activity on cancer cells. This first‐in‐human study evaluated ONO‐4578, a highly selective EP4 antagonist, as monotherapy and in combination with nivolumab in patients with advanced or metastatic solid tumors. A daily dose ranging from 30 mg to 100 mg of ONO‐4578 monotherapy and that ranging from 2 mg to 60 mg of ONO‐4578 with biweekly nivolumab 240 mg were administered. A total of 31 patients were enrolled, 10 receiving monotherapy and 21 receiving combination therapy. Overall, 26 patients experienced treatment‐related adverse events. Dose‐limiting toxicities were observed in three patients; one of six patients receiving 100 mg monotherapy developed grade 3 duodenal ulcer and two of six patients receiving 60 mg combination therapy developed either grade 3 erythema multiforme or grade 3 increased amylase and grade 4 increased lipase. One patient with small‐cell lung cancer who received 40 mg combination therapy had a partial response, and three patients with monotherapy and six patients with combination therapy had stable disease. Pharmacodynamics analyses showed that ONO‐4578 had EP4 antagonistic activity at doses as low as 2 mg. In conclusion, the maximum tolerated dose of ONO‐4578 alone or in combination with nivolumab was not reached. ONO‐4578 was well tolerated at the tested doses and showed signs of antitumor activity. Considering safety, efficacy, and pharmacokinetics/pharmacodynamics results, ONO‐4578 40 mg daily with nivolumab 240 mg biweekly was selected as the recommended dose for future clinical trials. (Registration: JapicCTI‐173,496 and NCT03155061).
Keywords: maximum tolerated dose, nivolumab, phase I, prostaglandin E2 receptor EP4, solid tumor
ONO‐4578 is a selective antagonist of the EP4 receptor for prostaglandin E2. ONO‐4578 alone or in combination with nivolumab was well tolerated and showed signs of anti‐tumor activity in patients with advanced or metastatic solid tumors. Considering safety, efficacy, and pharmacokinetics/pharmacodynamics results, ONO‐4578 40 mg daily with nivolumab 240 mg biweekly was selected as the recommended dose for future clinical trials.
Abbreviations
- AE
adverse event
- AUC
area under the concentration‐time curve
- C max
maximum plasma concentration
- DLT
dose‐limiting toxicity
- G‐CSF
granulocyte colony stimulating factor
- HNSTD
highest nonseverely toxic dose
- ICI
immune checkpoint inhibitor
- LPS
lipopolysaccharide
- MTD
maximum tolerated dose
- NSAID
nonsteroidal anti‐inflammatory drug
- PD
progressive disease
- PD‐1
programmed death‐1
- PGE2
prostaglandin E2
- PGEM
prostaglandin E2 metabolite
- PPI
proton pump inhibitor
- PR
partial response
- SD
stable disease
- TNF‐α
tumor necrosis factor‐α
- TRAE
treatment‐related adverse event
1. INTRODUCTION
Immunotherapy, which activates patients’ inherent immune surveillance system, is a recent breakthrough in cancer treatment. Immune checkpoint inhibitors, such as the anti‐PD‐1 Abs nivolumab and pembrolizumab, have established favorable efficacy and safety in various cancer types 1 , 2 , 3 , 4 , 5 and have been approved in over 65 countries. 6 However, it has also become evident that only a limited number of patients have responded to currently approved ICIs and that certain cancer types do not respond to current ICIs. 5 , 6 , 7 , 8 Tumors in nonresponders to current ICIs could evade the immune surveillance system through mechanisms other than immune checkpoint pathways. 9
The signaling pathway of PGE2 and the PGE2 receptor EP4 subtype would be a promising therapeutic target for cancer immunotherapy with a different mode of action than ICIs. Prostaglandin E2 is widely produced in the body and is an important mediator of fever, pain, and inflammation. 10 , 11 EP4 is a G‐protein‐coupled receptor for PGE2 that is found in the cell membrane of gastrointestinal epithelial cells, vascular smooth muscle cells, and other cells. 12 , 13 Prostaglandin E2 and its primary receptor EP4 have shown an elevated expression in cancer patients and to have immunosuppressive activity by inducing the differentiation of immune‐suppressive cells and by blocking T cell activation. 12 , 14 , 15 , 16
ONO‐4578 is a highly selective small‐molecule EP4 antagonist. A preclinical study in tumor‐bearing mice has reported that ONO‐4578 reduces infiltration of M2 macrophages in tumors when compared with control mice and had potent antitumor activity. When ONO‐4578 and anti‐mouse PD‐1 Abs were given concomitantly, tumor‐bearing mice had a higher complete response rate and longer survival than those receiving either ONO‐4578 or anti‐mouse PD‐1 Abs alone. The AUC for 24 h of ONO‐4578 at the effective dose of 3 mg/kg twice a day in tumor‐bearing mice was estimated to be 10.1 μg·h/ml. Toxicity tests on rats and monkeys showed a manageable safety profile.
In this first‐in‐human phase I study (ONO‐4578‐01 study), we evaluated ONO‐4578 as a monotherapy and in combination with nivolumab in patients with advanced or metastatic solid tumors. Here we report the results of the dose‐escalation parts of the ONO‐4578‐01 study, including DLTs, the MTD, safety, efficacy, pharmacokinetics, and pharmacodynamics.
2. MATERIALS AND METHODS
2.1. Study design
The dose‐escalation parts of the open‐label ONO‐4578‐01 study were undertaken at a single institute (National Cancer Center Hospital, Tokyo, Japan). In part A, patients were given 30, 60, or 100 mg ONO‐4578 orally every day in 28‐day cycles (ONO‐4578 monotherapy cohorts). The initial dose of ONO‐4578 at 30 mg was set considering the findings of preclinical pharmacology and toxicology of ONO‐4578; estimated pharmacokinetics in humans predicted that a dose of 30 mg was roughly equal to the human equivalent dose of the effective dose in mice and less than one‐sixth of the HNSTD. Part B evaluated a combination therapy of ONO‐4578 and nivolumab. The dose‐escalation design of part B was amended to take into account the pharmacokinetic results of ONO‐4578 in humans in part A, and patients were treated with 2, 5, 10, 20, 40, or 60 mg ONO‐4578 orally every day and 240 mg nivolumab intravenously every 2 weeks. The initial dose of ONO‐4578 in part B was set at 2 mg on the basis of the pharmacokinetics of ONO‐4578 observed in part A as well as the results of preclinical pharmacology and toxicology studies of ONO‐4578 with anti‐PD‐1 Ab. This dose was deemed greater than the modified human equivalent dose of the effective dose in mice, and the calculated AUC for 24 h of ONO‐4578 at 2 mg was sufficiently less than the AUC at the HNSTD. Considering the results in preclinical toxicity studies and in part A, a dose of 60 mg was selected as the maximum dose tested in part B. The minimum number of patients in each monotherapy and combination therapy cohort was one and three, respectively; the dose for the next cohort was determined using the continual reassessment method. The treatment was continued until showing PD according to RECIST 1.1, 17 the experience of unacceptable toxicities, or withdrawal of informed consent. Because gastrointestinal disorders such as erosions and ulcers were observed in preclinical studies of ONO‐4578, and EP4 antagonists may disrupt the secretion of mucin and HCO3 −, which protect epithelial cells and mucosa from acid peptic injury, 18 PPIs were allowed to be used in the treatment and prophylaxis of gastrointestinal disorders.
2.2. Patients
Patients with histologically or cytologically confirmed advanced or metastatic solid tumors were eligible in this study. Eligible patients were aged 20 years or older, had one or more measurable lesions according to RECIST 1.1 (only for the combination therapy cohorts), were refractory or intolerant to standard therapy or had no standard therapy, other than nivolumab in case of patients in the combination therapy cohorts, and had an ECOG performance status score of 0 or 1, a life expectancy of at least 3 months, and adequate organ function. Patients with asymptomatic brain metastases were eligible if no treatment was required.
2.3. Assessments
UGT1A1 is a uridine diphosphate glucuronosyltransferase that was considered the major metabolic enzyme for ONO‐4578. UGT1A1 polymorphisms UGT1A1*6 and UGT1A1*28 19 were assessed using serum samples collected before the first treatment with the study drugs.
Adverse events that appeared by 28 days after discontinuation of the study drugs were assessed and graded using the NCI's Common Terminology Criteria for Adverse Events, version 4.0. 20 Possible links between AEs and the study treatment were suggested by investigators. Prespecified DLTs that occurred during the first 28‐day cycle were grade 3 thrombocytopenia requiring platelet transfusion, grade 4 thrombocytopenia, grade ≥3 febrile neutropenia without supportive care with G‐CSF preparation, grade 4 neutropenia lasting ≥8 days without supportive care with G‐CSF preparation, grade ≥2 central nervous system symptoms, grade ≥2 uveitis requiring systemic therapy, and grade ≥3 nonhematologic toxicity.
Computed tomography and MRI of the chest, abdomen, and pelvis were carried out before the enrollment and every 4 weeks by 8 weeks, every 8 weeks by 56 weeks, and every 12 weeks thereafter. Measurable lesions were assessed according to RECIST 1.1, and the best overall response was graded as a complete response, PR, SD, or PD.
For pharmacokinetic analyses of ONO‐4578, blood samples were collected before and 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after treatment on days 1 and 28 of cycle 1. Plasma concentrations of ONO‐4578 were determined in a central laboratory by liquid chromatography and tandem mass spectrometry. The pharmacokinetic parameters were determined with the Phoenix WinNonlin software version 7.0 (Certara).
2.4. Pharmacodynamic evaluation
Lipopolysaccharide‐stimulated production of TNF‐α was inhibited by PGE2, 21 , 22 and EP4 was found to be involved in this inhibitory pathway. 23 Thus, EP4 antagonism counteracts this inhibitory pathway, increasing TNF‐α production. The ex vivo TNF‐α release assay 24 was chosen to assess the target engagement for EP4 antagonism. Whole blood samples were collected before ONO‐4578 treatment (baseline) and 4 h after treatment on day 1, and before treatment on days 2 and 28. Diclofenac was added at 10 μM to the whole blood samples. The blood samples were stimulated by 1 ng/ml LPS, and PGE2 was added to a final concentration of 10 nM. The samples were incubated at 37°C for 4 h followed by centrifugation to collect the plasma. The TNF‐α concentrations were determined using Quantikine ELISA Human TNF‐α Immunoassay (R&D Systems) according to the manufacturer's protocol. The ex vivo TNF‐α release assay was done in triplicate, and the mean value was calculated.
To evaluate PGEM in plasma, whole blood was collected before ONO‐4578 treatment on days 1 and 28, mixed with EDTA‐2 K anticoagulant, and kept on ice. Indomethacin, a COX inhibitor, was immediately added to the blood sample at a final concentration of 18 μg/ml to prevent further production of PGE2 and PGEM during sample preparation. Plasma samples were prepared by centrifugation at 4°C before being applied to an Oasis MAX cartridge (Waters). The cartridges were washed with ammonium acetate solutions, methanol, ethyl acetate, and hexane/ethyl acetate solutions, and analytes were eluted with ethyl acetate/formic acid solutions. The concentration of the PGEM, 13, 14‐dihydro‐15‐keto prostaglandin E2, was measured using a triple‐quadrupole linear ion trap mass spectrometer (QTRAP6500; AB Sciex) equipped with HPLC (Nexera UHPLC; Shimadzu).
To assess PGE2 metabolites (tetranor‐PGEM) in urine, urine was collected continuously for 24 h before ONO‐4578 treatment on day 1 and day 28, and stored in a polypropylene bottle at 2°C–8°C. The urine volume was calculated using the total urine weight and the urine‐specific gravity. Urine samples (150 μl) were applied to Oasis HLB μElution plates, the plates were washed with 0.1% acetic acid, and the analytes were eluted with methanol and 0.1% acetic acid. The urinary concentration of tetranor‐PGEM was measured by a triple‐quadrupole mass spectrometer (API5000; AB Sciex) equipped with HPLC (NANOSPACE system; Shiseido). The amount of urine tetranor‐PGEM was calculated using the concentration and the total urine volume.
2.5. Statistics
Adverse events were assessed in all patients who received at least one dose of the study drugs. Efficacy, pharmacokinetics, and pharmacodynamics were evaluated in patients with available data as of September 27, 2020.
3. RESULTS
3.1. Patient demographics and disposition
Between April 18, 2017, and January 8, 2020, 31 patients were enrolled in three monotherapy (N = 10) and six combination therapy cohorts (N = 21). In the overall population, the most common cancer types were pancreatic cancer (19%), bile duct cancer (16%), and colorectal cancer (16%); 22 patients (71%) had received three or more prior regimens (Table 1). At the date of data cut‐off (April 26, 2021), all patients discontinued the study drug, primarily due to disease progression (28 patients, 90%).
TABLE 1.
Baseline demographics and disease characteristics of patients with solid tumors treated with ONO‐4578
|
Monotherapy N = 10 |
Combination therapy N = 21 |
|
|---|---|---|
| Median age, years (range) | 57 (41–73) | 59 (33–73) |
| Male, n (%) | 5 (50) | 13 (62) |
| ECOG PS, n (%) | ||
| 0 | 3 (30) | 12 (57) |
| 1 | 7 (70) | 9 (43) |
| Cancer type, n (%) | ||
| Bile duct | 1 (10) | 4 (19) |
| Breast | 0 (0) | 2 (10) |
| Colorectal | 2 (20) | 3 (14) |
| Ovarian | 1 (10) | 0 (0) |
| Pancreatic | 1 (10) | 5 (24) |
| Prostate | 1 (10) | 0 (0) |
| Small cell lung | 0 (0) | 1 (5) |
| Other solid tumors | 4 (40) a | 6 (29) b |
| Number of prior regimens, n (%) | ||
| 0 | 0 (0) | 1 (5) c |
| 1 | 0 (0) | 1 (5) |
| 2 | 2 (20) | 5 (24) |
| 3 | 3 (30) | 3 (14) |
| ≥4 | 5 (50) | 11 (52) |
| Prior radiotherapy | 4 (40) | 9 (43) |
Abbreviation: PS, performance status.
Included alveolar soft part sarcoma, cervical cancer, medulloblastoma, and urothelial cancer.
Included ameloblastoma, apocrine adenocarcinoma, cervical cancer, gallbladder cancer, thymic cancer, and uterine body cancer.
A patient with ameloblastoma had not received a prior therapy due to the lack of standard therapy.
3.2. Safety
The duration of treatment is summarized in Table S1. No DLTs were observed in the 30 mg or 60 mg monotherapy cohorts, whereas one DLT (grade 3 duodenal ulcer) was observed in one of six patients receiving 100 mg monotherapy (Table 2). Two of the six patients in the 60 mg combination therapy cohort experienced DLTs (one patient with grade 3 erythema multiforme and the other patient with grade 3 increased amylase and grade 4 increased lipase). Although MTD was not reached in either monotherapy (30–100 mg) or combination therapy (2–60 mg), the combination of ONO‐4578 40 mg daily with nivolumab 240 mg biweekly was chosen as the recommended dose for the next trials, in part because the 60 mg combination therapy caused DLTs, and the 60 mg monotherapy developed grade 2 duodenal ulcer (Table 2).
TABLE 2.
Adverse events (AEs) in patients with solid tumors treated with ONO‐4578 alone or in combination with nivolumab
| ONO‐4578 dose | Monotherapy | Combination therapy | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
30 mg n = 1 |
60 mg n = 3 |
100 mg n = 6 |
2 mg n = 3 |
5 mg n = 3 |
10 mg n = 3 |
20 mg n = 3 |
40 mg n = 3 |
60 mg n = 6 |
|
| Any AEs | 1 (100) | 2 (67) | 6 (100) | 3 (100) | 3 (100) | 2 (67) | 2 (67) | 3 (100) | 6 (100) |
| Grade 3–4 | 0 (0) | 1 (33) | 3 (50) | 1 (33) | 0 (0) | 1 (33) | 0 (0) | 3 (100) | 3 (50) |
| Any TRAEs | 1 (100) | 2 (67) | 6 (100) | 2 (67) | 2 (67) | 2 (67) | 2 (67) | 3 (100) | 6 (100) |
| Grade 3–4 | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 2 (33) |
| Action taken because of TRAEs | |||||||||
| Discontinuation | 0 (0) | 1 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) |
| Grade 3–4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) |
| Dose delay | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) |
| Grade 3–4 | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) |
| TRAEs in ≥2 patients in any cohorts and any grade 3–4 TRAEs | |||||||||
| Anemia | 0 (0) | 0 (0) | 2 (33) | 1 (33) | 0 (0) | 1 (33) | 2 (67) | 1 (33) | 1 (17) |
| Grade 3–4 | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Rash | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 1 (33) | 0 (0) | 0 (0) | 2 (67) | 3 (50) |
| Erythema multiforme | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) |
| Grade 3–4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) a |
| Amylase increased | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) |
| Grade 3–4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) a |
| ALT increased | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) |
| AST increased | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (50) |
| Lipase increased | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) |
| Grade 3–4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (17) a |
| Lymphocyte count decreased | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 1 (33) | 0 (0) |
| Grade 3–4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) |
| White blood cell count decreased | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 1 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Duodenal ulcer | 0 (0) | 2 (67) | 1 (17) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 0 (0) | 0 (0) |
| Grade 3–4 | 0 (0) | 0 (0) | 1 (17) a | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Note: Data are shown as n (%).
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; TRAE, treatment‐related AE.
Dose‐limiting toxicities.
In the overall cohort, 26 (84%) patients experienced TRAEs (Table 2). The most frequent TRAEs were anemia (26%) and rash (23%). In the monotherapy cohorts, one patient on 60 mg discontinued the study drug due to grade 2 gastrointestinal disorders (duodenal ulcer, duodenitis, and gastritis), and two patients on 100 mg had dose delay due to grade 3 anemia and grade 3 duodenal ulcer. In the combination therapy cohorts, two patients on 60 mg discontinued due to grade 2 gastritis and grade 3 erythema multiforme, and two patients on 60 mg had dose delays due to either grade 2 gastric ulcer or grade 4 increased lipase and grade 3 increased amylase. No AEs led to death; 23 patients (74%) died by the date of data cut‐off, primarily (n = 22) due to tumor progression.
The study drug was continued in all three patients with grade 2 duodenal ulcer along with PPIs. The patient with a grade 3 duodenal ulcer was cured of the DLT by dose delay of the study drug and treatment with PPIs. One of the 11 patients who received prophylactic PPIs during the study drug administration developed grade 2 gastric ulcer, while six of the 20 patients who did not receive prophylactic PPIs developed gastritis (3 patients), duodenitis (1), gastric ulcer (1), and/or duodenal ulcer (4); except for two patients with gastritis, five patients underwent endoscopy at the first onset of gastric or duodenal inflammation.
3.3. Efficacy
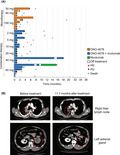
Responses were assessed in a total of 27 patients. Across all cohorts, one patient with small‐cell lung cancer who received the 40 mg combination therapy had the best overall response of PR; the time to and duration of the response was 0.92 and 17.15 months, respectively (Figure 1A). Figure 1B shows computed tomography images of the patient with PR. One patient with pancreatic cancer who received the 2 mg combination therapy experienced a 34% reduction in the target tumors before treatment discontinuation due to newly developed brain metastases (Figure S1). Stable disease occurred in nine patients (29%; one patient each in the 30, 60, and 100 mg monotherapy cohorts, and two, one, and three patients in the 2, 5, and 60 mg combination therapy cohorts, respectively).
FIGURE 1.
(A) Swimmer plot showing the duration of treatment with ONO‐4578 alone or in combination with patients with solid tumors treated with nivolumab in patients with solid tumors. (B) In a patient with small‐cell lung cancer, computed tomography highlighted metastatic tumors at the right hilar lymph node and the left adrenal gland before and 11.7 months after treatment with 40 mg combination therapy. PD, progressive disease; PR, partial response
3.4. Pharmacokinetics
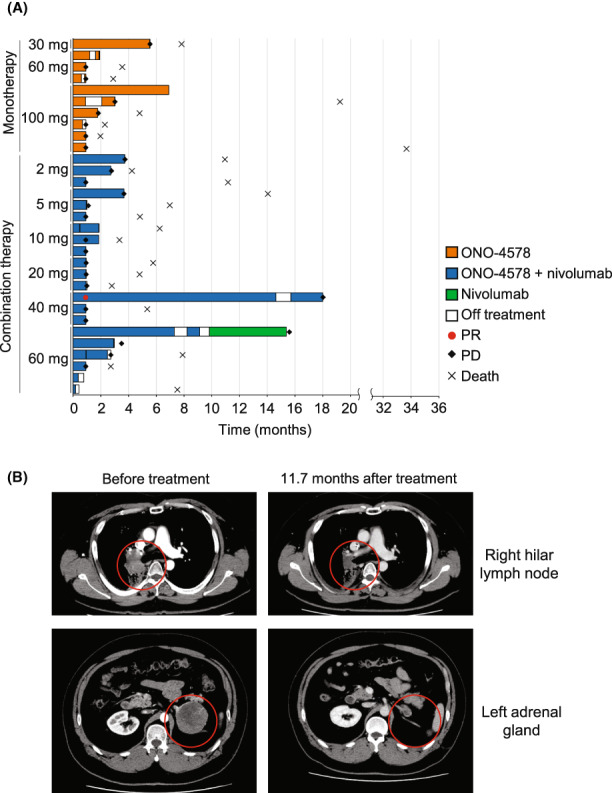
On day 1 of cycle 1, the plasma concentration of ONO‐4578 peaked at 4 h, and then decreased with a mean terminal half‐life of 22–53 h (Table S2). Repeated treatment accumulated ONO‐4578, and on day 28 of cycle 1, C max and the AUC for 24 h in the 100 mg monotherapy cohort reached a mean of 14.5 μg/mL and 292 μg·h/mL, respectively. (Figure 2, Table S2). The C max and AUC for 24 h increased in proportion to dose (Figure S2), and the mean time to C max and the mean terminal half‐life were comparable across all cohorts.
FIGURE 2.
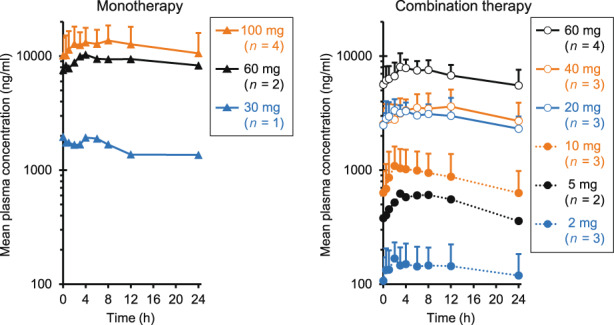
Plasma concentration of ONO‐4578 at day 28 of cycle 1 in patients with solid tumors treated with ONO‐4578 monotherapy or in combination with nivolumab. Mean value of each patient's data is shown. Error bars represent standard deviation. In the 60 and 100 mg monotherapy cohorts and the 5 and 60 mg combination therapy cohorts, some patients' data were missing
An exploratory subgroup analysis showed that the Cmax and AUC for 24 h were comparable between patients who received PPIs concurrently and those who did not (Figure S3). Although quantitative comparisons were limited due to data limitation, a trend of higher Cmax and AUC in heterozygous and homozygous carriers of a UGT1A1 polymorphism, UGT1A1*6 and/or UGT1A1*28, was observed.
3.5. Pharmacodynamics
Tumor necrosis factor‐α production stimulated by LPS/PGE2 was increased in whole blood isolated after ONO‐4578 treatment, but no dose dependency was observed (Figure 3A). The plasma concentration of PGEM and the excretion of tetranor‐PGEM in urine increased in 24/27 (89%) and 27/29 (93%) patients, respectively, after repeated treatment with ONO‐4578; however, a dose–response relationship was not clearly observed (Figure 3B,C).
FIGURE 3.
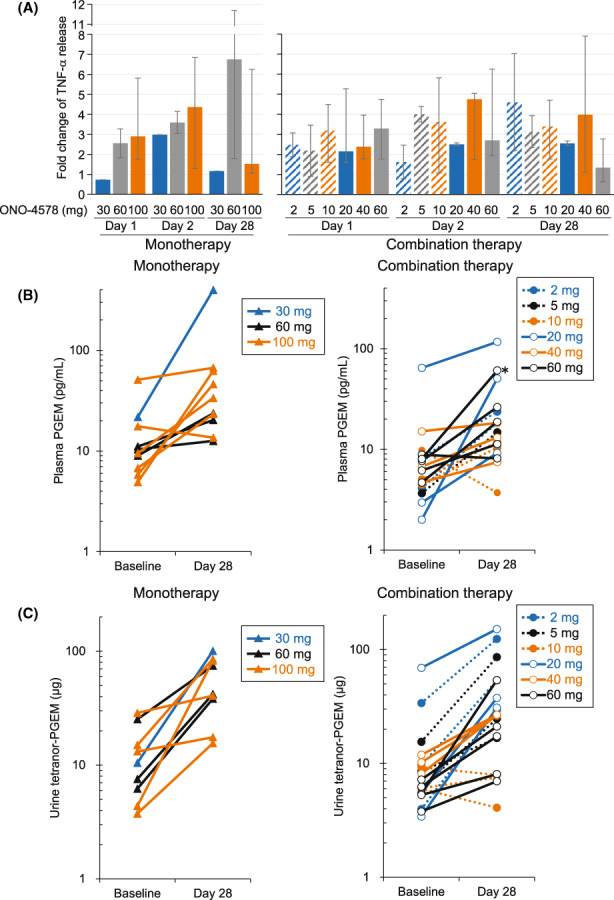
Pharmacodynamic parameters in patients with solid tumors treated with ONO‐4578 monotherapy or in combination with nivolumab. (A) Tumor necrosis factor‐α (TNF‐α) release in whole blood samples collected on days 1, 2, and 28 were divided by baseline. Change in the median fold is shown. Error bars represent minimum and maximum values. (B, C) Plasma prostaglandin E2 metabolites (PGEM) (B) and tetranor‐PGEM in urine (C) of individual patients are shown. Data were missing in some patients due to discontinuation of study treatment before the completion of the 28‐day cycle or technical difficulties in the analysis
4. DISCUSSION
This first‐in‐human study evaluated ONO‐4578, a selective EP4 antagonist, as monotherapy and in combination with nivolumab in patients with advanced or metastatic solid tumors. The MTD was not reached in either monotherapy (30–100 mg) or combination therapy (260 mg). In the monotherapy cohorts, one of six patients receiving 100 mg had a DLT. When three patients receiving 100 mg have completed the tolerability assessment, 200 mg is the recommended dose for the next cohort, according to the continual reassessment method. However, given the occurrence of AEs including DLT, and the fact that the exposure of ONO‐4578 after repeated doses of 100 mg (AUC for 24 h, 84.6 μg·h/ml) was sufficiently high compared to the steady‐state exposure of ONO‐4578 at the effective dose in tumor‐bearing mice (AUC for 24 h, 10.1 μg·h/ml), the dose escalation to 200 mg was deemed unnecessary, and three more patients were added at 100 mg to complete the dose escalation. In the combination therapy cohorts, two of six patients receiving 60 mg ONO‐4578 had DLTs despite having comparable pharmacokinetic and pharmacodynamic parameters to those in the other patients. Although the dose of 60 mg was deemed tolerable, the dose was not increased to more than 60 mg per the predetermined dose‐escalation plan.
Gastrointestinal TRAEs such as duodenal ulcer, duodenitis, gastric ulcer, and gastritis were common. Prostaglandin E2 and EP4 stimulated the secretion of gastrointestinal mucin, which is a high‐molecular‐weight glycoprotein that protects epithelial cells. 12 , 18 , 25 HCO3 − secretion has also been shown to protect the duodenum mucosa from acid peptic injury and to be inhibited by an EP4 antagonist. 12 , 18 , 26 ONO‐4578 likely inhibited these protective roles of PGE2 and EP4, similarly to NSAIDs, which can cause gastrointestinal TRAEs. Anemia observed in this study could be caused by peptic ulcers, as gastrointestinal injuries caused by NSAID treatments have been linked to occult blood loss and anemia. 27 Nonetheless, ONO‐4578‐related gastrointestinal TRAEs, like NSAID‐induced gastrointestinal disorders, were manageable with PPI treatment. Notably, concomitant administration of PPIs did not affect the pharmacokinetics of ONO‐4578, which could be attributed to the increased solubility of ONO‐4578 in higher pH solutions. As the incidence of gastric or duodenal inflammation was 30% in patients who did not receive prophylactic PPIs, compared to 9% in patients who did receive prophylactic PPIs, prophylactic administration of PPIs could be considered in ONO‐4578 therapy, which should be confirmed in future studies.
Tumor necrosis factor‐α release in LPS‐stimulated whole blood and urinary tetranor‐PGEM alteration with ONO‐4578 treatment were evaluated to assess an on‐target pharmacodynamic modulation by ONO‐4578. Increase in the ex vivo TNF‐α release was observed in almost all patients, suggesting that ONO‐4578 showed EP4 antagonistic activity at a dose as low as 2 mg. Although the plasma concentration of ONO‐4578 appeared to increase as the dose increased, no clear dose dependency was observed in the pharmacodynamic analysis. Because the efficiency of ex vivo TNF‐α release observed in this study was comparable to that observed with other EP4 inhibitors, such as LY3127760 and CJ‐042794, 24 , 28 the lowest dose of ONO‐4578 tested in this study might be high enough to induce the ex vivo TNF‐α release. Alternatively, the sensitivity of this assay could be insufficient to detect the dose‐dependency of ONO‐4578. In urine, tetranor‐PGEM is the major metabolite of PGE2. Some clinical studies have found a link between urinary PGEM levels and cancer progression. 29 , 30 A recent study evaluated the pharmacological profiles of a selective EP4 antagonist, LY3127760, and a COX‐2 inhibitor, celecoxib, in healthy people. 24 LY3127760 increased the excretion of PGEM while celecoxib inhibited urinary PGEM excretion. Similarly, ONO‐4578 treatment enhanced the excretion of urinary tetranor‐PGEM in cancer patients. This observation suggests that ONO‐4578 target modulation possibly leads to a feedback mechanism that promotes the production of PGE2 and/or the metabolism of the accumulated free PGE2 as a result of the EP4 antagonistic activity of ONO‐4578, but the detailed mechanism needs to be further elucidated. In summary, we demonstrated on‐target pharmacodynamic modulation of ONO‐4578 in cancer patients, thereby supporting the recommended dose selection of 40 mg ONO‐4578 in future clinical trials.
Although their best overall responses were SD, two patients in monotherapy cohorts experienced tumor shrinkage, suggesting a promising efficacy of ONO‐4578 monotherapy. One patient with small‐cell lung cancer who received 40 mg ONO‐4578 in combination with nivolumab showed PR and remained on the study drug for more than 18 months. One unconfirmed PR was also observed in a patient with pancreatic cancer who received 2 mg ONO‐4578 with nivolumab. In recent phase III trials, nivolumab has failed to demonstrate survival benefits in patients with small‐cell lung cancer, 31 , 32 and pancreatic cancer is thought to be more resistant to ICIs. 33 , 34 Although we cannot rule out the possibility that these observed responses are solely due to nivolumab, ONO‐4578 could have an additive effect on improving the immunosurveillance tumor microenvironment. As a result, similar to a combination therapy of nivolumab with ipilimumab that showed promising efficacy for multiple advanced malignant tumors including melanoma, non‐small‐cell lung cancer, and malignant pleural mesothelioma, 35 , 36 , 37 nivolumab plus ONO‐4578 would warrant further investigation in future clinical trials.
In conclusion, treatment with ONO‐4578 was well tolerated in patients with advanced or metastatic solid tumors. Given the occurrence of AEs, including DLTs, as well as the results of efficacy, pharmacokinetics, and pharmacodynamics, a combination of ONO‐4578 40 mg daily and nivolumab 240 mg biweekly was selected as the recommended dose and will be evaluated in future clinical trials.
AUTHOR CONTRIBUTIONS
SI, TK, MN, SK, KS, KY, TY, KT, TS, YF, SK, AS, and JS: Investigation, writing review and editing. FY, HI: Formal analysis, methodology, writing original draft, and editing. MK: Conceptualization, methodology, project administration, writing review, and editing. NY: Conceptualization, investigation, methodology, project administration, writing review, and editing.
FUNDING INFORMATION
Ono Pharmaceutical, Co., Ltd.
DISCLOSURE
SI received research grants from Bristol Myers Squibb, Daiichi Sankyo, Eisai, Ono, Pfizer, and Taiho; and honoraria from Bristol Myers Squibb and Ono. TK received research grants from Daiichi Sankyo, Eli Lilly, Novartis, Ono, and PACT Pharma; and honoraria from Chugai and Sysmex. MN received research grants from Ono; and honoraria from AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Chugai, Eli Lilly, MSD, Novartis, Pfizer, Ono, and Taiho. SK received research grants from Ono. KS received research grants from AstraZeneca, Daiichi Sankyo, NanoCarrier, and Ono; and honoraria from AstraZeneca, Eisai, and Pfizer. KY received research grants from AstraZeneca, Boehringer Ingelheim, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, Genmab, Haihe, Kyowa Hakko Kirin, MSD, Nihon Kayaku, Novartis, Ono, Pfizer, Sanofi, Seagen, Seattle Genetics, Taiho, and Takeda; consulting fees from AstraZeneca, Chugai, Eisai, Genmab, Novartis, OncXerna, and Takeda; and honoraria from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Chugai, Eisai, Eli Lilly, Fujifilm, MSD, Ono, Pfizer, and Takeda. TY received research grants from AbbVie, Amgen, AstraZeneca, Blueprint, Bristol Myers Squibb, Chugai, Daiichi Sankyo, Merck, MSD, Novartis, Ono, and Takeda; honoraria from AstraZeneca, ArcherDX, Boehringer Ingelheim, Bristol Myers Squibb, Chugai, Invitae, Eli Lilly, MSD, Novartis, Ono, Roche, Taiho, and Takeda; and fees for participation on a Data Safety Monitoring Board or Advisory Board from AstraZeneca, Amgen, Chugai, MSD, and Novartis. KT is an Associate Editor of Cancer Science; and received research grants from Ono. TS received research grants from 3D Medicines, AbbVie, Astellas, AstraZeneca, Bristol Myers Squibb, Chordia Therapeutics, Daiichi Sankyo, Eisai, Eli Lilly, Five Prime, Incyte, Loxo Oncology, Novartis, Ono, Pfizer, PharmaMar, SymBio, and Takeda; honoraria from Boehringer Ingelheim, Chugai, Eli Lilly, MSD, and Taiho; support for attending meetings and/or travel from AbbVie, Chugai, Daiichi Sankyo, Eisai, MSD, Taiho, and Takeda Oncology; and fees for participation on a Data Safety Monitoring Board or Advisory Board from AbbVie, Chordia Therapeutics, Daiichi Sankyo, and Takeda Oncology. YF received research grants from Amgen, AnHeart Therapeutics, Bristol Myers Squibb, Chugai, Eli Lilly, and Ono; consulting fees from AstraZeneca, Chiome Bioscience, Daiichi Sankyo, and Otsuka; and honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, Chugai, Daiichi Sankyo, Eli Lilly, Micron, MSD, Novartis, Ono, Pfizer, Taiho, Takeda, and Yakult. SK received research grants from Astellas, AstraZeneca, Boehringer Ingelheim, Chugai, Daiichi Sankyo, Eisai, Gilead, Japan Agency for Medical Research and Development, Japan Society for the Promotion of Science, MSD, Ono, PACT Pharma, Pfizer, Regeneron, and Takara Bio; honoraria from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Chugai, Daiichi Sankyo, Eisai, GlaxoSmithKline, MSD, Novartis, Ono, Pfizer, Pharmaceuticals and Medical Devices Agency, Regeneron, and Taiho; and fees for participation on an Advisory Board from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Chugai, Eisai, GlaxoSmithKline, ImmuniT Research, MSD, Novartis, Ono, Pfizer, Rakuten Medical, Regeneron, and Sumitomo Dainippon. AS received research grants from AstraZeneca, Chugai, Daiichi Sankyo, Eisai, Mochida, and Taiho; and honoraria from AstraZeneca, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, Kyowa Kirin, MSD, Novartis, Ono, Pfizer, and Takeda. FY, HI, and MK are employees of Ono. NY received research grants from AbbVie, Astellas, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Carna Biosciences, Chiome Bioscience, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, Genmab, GlaxoSmithKline, Janssen, Kyowa Hakko Kirin, Merck, MSD, Novartis, Ono, Otsuka, Pfizer, Quintiles, Shionogi, Sumitomo Dainippon, Taiho, and Takeda; honoraria from AstraZeneca, Bristol Myers Squibb, Chugai, Eisai, Lilly, Ono, Pfizer, and Sysmex; and consulting fees from Boehringer Ingelheim, Cimic, Eisai, Otsuka, and Takeda.
ETHICS STATEMENT
Approval of the research protocol: The study protocol and all amendments were approved by the institutional review board.
Informed consent: This study was conducted following the protocol, Good Clinical Practice guidelines of the International Council for Harmonization, and the Declaration of Helsinki. All of the patients signed informed consent.
Registry and registration no. of the study/trial: JapicCTI‐173,496 at the Japan Pharmaceutical Information Center; NCT03155061 at https://ClinicalTrials.gov.
Animal studies: N/A.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank the patients and their supportive families for participating in this phase I study. We would like to thank the staff at the National Cancer Center Hospital (Tokyo, Japan) for making this trial possible. Masatoshi Esaki, Ph.D., of Ono Pharmaceutical Co., Ltd, assisted with the medical writing. This research was supported by funding from Ono Pharmaceutical, Co., Ltd.
Iwasa S, Koyama T, Nishino M, et al. First‐in‐human study of ONO‐4578, an antagonist of prostaglandin E2 receptor 4, alone and with nivolumab in solid tumors. Cancer Sci. 2023;114:211‐220. doi: 10.1111/cas.15574
DATA AVAILABILITY STATEMENT
Qualified researchers may request Ono Pharmaceutical Co., Ltd. to disclose individual patient‐level data from clinical studies through the following website: https://www.clinicalstudydatarequest.com/. For more information on the policy of Ono Pharmaceutical Co., Ltd. for the Disclosure of Clinical Study Data, please visit https://www.ono.co.jp/eng/rd/policy.html.
REFERENCES
- 1. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti–PD‐1) in melanoma. N Engl J Med. 2013;369(2):134‐144. doi: 10.1056/NEJMoa1305133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;373(2):123‐135. doi: 10.1056/NEJMoa1504627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med. 2015;372(21):2018‐2028. doi: 10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 4. Kang Y‐K, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390:2461‐2471. doi: 10.1016/S0140-6736(17)31827-5 [DOI] [PubMed] [Google Scholar]
- 5. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320‐330. doi: 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 6. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021;184(21):5309‐5337. doi: 10.1016/j.cell.2021.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med. 2015;373(19):1803‐1813. doi: 10.1056/NEJMoa1510665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moskovitz J, Moy J, Ferris RL. Immunotherapy for head and neck squamous cell carcinoma. Curr Oncol Rep. 2018;20:22. doi: 10.1007/s11912-018-0654-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14(8):561‐584. doi: 10.1038/nrd4591 [DOI] [PubMed] [Google Scholar]
- 10. Lin CR, Amaya F, Barrett L, et al. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther. 2006;319(3):1096‐1103. doi: 10.1124/jpet.106.105569 [DOI] [PubMed] [Google Scholar]
- 11. McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110(5):651‐658. doi: 10.1172/JCI15528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010‐1052. doi: 10.1124/pr.112.007195 [DOI] [PubMed] [Google Scholar]
- 13. Konya V, Marsche G, Schuligoi R, Heinemann A. E‐type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther. 2013;138:485‐502. doi: 10.1016/j.pharmthera.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Obermajer N, Kalinski P. Generation of myeloid‐derived suppressor cells using prostaglandin E2 . Transplant Res. 2012;1:15. doi: 10.1186/2047-1440-1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ylöstalo JH, Bartosh TJ, Coble K, Prockop DJ. Human mesenchymal stem/stromal cells cultured as spheroids are self‐activated to produce prostaglandin E2 (PGE2) that directs stimulated macrophages into an anti‐inflammatory phenotype. Stem Cells. 2012;30:2283‐2296. doi: 10.1002/stem.1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okano M, Sugata Y, Fujiwara T, et al. E prostanoid 2 (EP2)/EP4‐mediated suppression of antigen‐specific human T‐cell responses by prostaglandin E2 . Immunology. 2006;118:343‐352. doi: 10.1111/j.1365-2567.2006.02376.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. doi: 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 18. Takeuchi K, Amagase K. Roles of cyclooxygenase, prostaglandin E2 and EP receptors in mucosal protection and ulcer healing in the gastrointestinal tract. Curr Pharm Des. 2018;24(18):2002‐2011. doi: 10.2174/1381612824666180629111227 [DOI] [PubMed] [Google Scholar]
- 19. Stingl JC, Bartels H, Viviani R, Lehmann ML, Brockmöller J. Relevance of UDP‐glucuronosyltransferase polymorphisms for drug dosing: A quantitative systematic review. Pharmacol Ther. 2014;141:92‐116. doi: 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 20. National Cancer Institute . Common Terminology Criteria for Adverse Events (CTCAE). Accessed May 9, 2022. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm
- 21. Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D. Prostaglandin E2 regulates macrophage‐derived tumor necrosis factor gene expression. J Biol Chem. 1988;263(11):5380‐5384. doi: 10.1016/S0021-9258(18)60727-6 [DOI] [PubMed] [Google Scholar]
- 22. Strassmann G, Patil‐Koota V, Finkelman F, Fong M, Kambayashi T. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2 . J Exp Med. 1994;180(6):2365‐2370. doi: 10.1084/jem.180.6.2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamane H, Sugimoto Y, Tanaka S, Ichikawa A. Prostaglandin E2 receptors, EP2 and EP4, differentially modulate TNF‐α and IL‐6 production induced by lipopolysaccharide in mouse peritoneal neutrophils. Biochem Biophys Res Commun. 2000;278:224‐228. doi: 10.1006/bbrc.2000.3779 [DOI] [PubMed] [Google Scholar]
- 24. Jin Y, Smith C, Hu L, et al. LY3127760, a selective prostaglandin E4 (EP4) receptor antagonist, and celecoxib: a comparison of pharmacological profiles. Clin Transl Sci. 2018;11:46‐53. doi: 10.1111/cts.1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kunikata T, Tanaka A, Miyazawa T, Kato S, Takeuchi K. 16,16‐Dimethyl prostaglandin E2 inhibits indomethacin‐induced small intestinal lesions through EP3 and EP4 receptors. Dig Dis Sci. 2002;47(4):894‐904. doi: 10.1023/a:1014725024519 [DOI] [PubMed] [Google Scholar]
- 26. Aoi M, Aihara E, Nakashima M, Takeuchi K. Participation of prostaglandin E receptor EP4 subtype in duodenal bicarbonate secretion in rats. Am J Physiol Gastrointest Liver Physiol. 2004;287:G96‐G103. doi: 10.1152/ajpgi.00038.2004 [DOI] [PubMed] [Google Scholar]
- 27. Tai FWD, McAlindon ME. Non‐steroidal anti‐inflammatory drugs and the gastrointestinal tract. Clin Med. 2021;21(2):131‐134. doi: 10.7861/clinmed.2021-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murase A, Taniguchi Y, Tonai‐Kachi H, Nakao K, Takada J. In vitro pharmacological characterization of CJ‐042794, a novel, potent, and selective prostaglandin EP4 receptor antagonist. Life Sci. 2008;82:226‐232. doi: 10.1016/j.lfs.2007.11.002 [DOI] [PubMed] [Google Scholar]
- 29. Kawamoto H, Hara H, Araya J, et al. Prostaglandin E‐major urinary metabolite (PGE‐MUM) as a tumor marker for lung adenocarcinoma. Cancer. 2019;11:768. doi: 10.3390/cancers11060768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Csiki I, Morrow JD, Sandler A, et al. Targeting cyclooxygenase‐2 in recurrent non‐small cell lung cancer: A phase II trial of celecoxib and docetaxel. Clin Cancer Res. 2005;11(18):6634‐6640. doi: 10.1158/1078-0432.CCR-05-0436 [DOI] [PubMed] [Google Scholar]
- 31. Owonikoko TK, Park K, Govindan R, et al. Nivolumab and ipilimumab as maintenance therapy in extensive‐disease small‐cell lung cancer: CheckMate 451. J Clin Oncol. 2021;39(12):1349‐1359. doi: 10.1200/JCO.20.02212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spigel DR, Vicente D, Ciuleanu TE, et al. Second‐line nivolumab in relapsed small‐cell lung cancer: CheckMate 331. Ann Oncol. 2021;32(5):631‐641. doi: 10.1016/j.annonc.2021.01.071 [DOI] [PubMed] [Google Scholar]
- 33. Wandmacher AM, Letsch A, Sebens S. Challenges and future perspectives of immunotherapy in pancreatic cancer. Cancer. 2021;13(16):4235. doi: 10.3390/cancers13164235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bian J, Almhanna K. Pancreatic cancer and immune checkpoint inhibitors—still a long way to go. Transl Gastroenterol Hepatol. 2021;6:6. doi: 10.21037/tgh.2020.04.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hellmann MD, Paz‐Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small‐cell lung cancer. N Engl J Med. 2019;381(21):2020‐2031. doi: 10.1056/NEJMoa1910231 [DOI] [PubMed] [Google Scholar]
- 36. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122‐133. doi: 10.1056/NEJMoa1302369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baas P, Scherpereel A, Nowak AK, et al. First‐line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open‐label, phase 3 trial. Lancet. 2021;397:375‐386. doi: 10.1016/S0140-6736(20)32714-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Qualified researchers may request Ono Pharmaceutical Co., Ltd. to disclose individual patient‐level data from clinical studies through the following website: https://www.clinicalstudydatarequest.com/. For more information on the policy of Ono Pharmaceutical Co., Ltd. for the Disclosure of Clinical Study Data, please visit https://www.ono.co.jp/eng/rd/policy.html.