

Abstract
Glioblastoma stands as the most frequent primary brain tumor. Despite the multimodal therapy for glioblastoma patients, the survival rate is very low, highlighting the need for novel therapies that improve patient outcomes. Immune checkpoint blockade strategies are achieving promising results in a myriad of tumors and several studies have reported its efficacy in glioblastoma at a preclinical level. ILT2 is a novel immune checkpoint that exerts an inhibitory effect via the interaction with classical and non‐classical HLA class‐I molecules. Herein, we report that ILT2 blockade promotes antitumor responses against glioblastoma. In silico and immunohistochemical analyses revealed that the expression of ILT2 and its ligands HLA‐A, ‐B, ‐C, and ‐E are highly expressed in patients with glioblastoma. Disruption of ILT2 with blocking monoclonal antibodies increased natural killer cell‐mediated IFN‐γ production and cytotoxicity against glioblastoma, partially reverting the immunosuppression linked to this malignancy. In addition, co‐treatment with temozolomide strengthened the antitumor capacity of anti‐ILT2‐treated immune cells. Collectively, our results establish the basis for future studies regarding the clinical potential of ILT2 blockade alone or in combination regimens in glioblastoma.
Keywords: Glioblastoma, ILT2 protein, immune checkpoint proteins, immunotherapy, NK cells
The inhibitory immune checkpoint ILT2 is highly expressed in glioblastoma. Its blockade partially restores antitumor responses in this tumor and combination with temozolomide enhances this anti‐glioblastoma effect.

Abbreviations
- CAR
Chimeric antigen receptor
- GEO
Gene Expression Omnibus
- HLA
Human leukocyte antigen
- ICB
Immune checkpoint blockade
- IFN‐γ
Interferon‐gamma
- mAb
Monoclonal antibody
- MFI
Mean fluorescence intensity
- NK
natural killer
- PBMCs
Peripheral blood mononuclear cells
- SEM
Standard error of the mean
- TCGA
The Cancer Genome Atlas
1. INTRODUCTION
Glioblastoma is a high‐grade glioma (WHO grade IV) characterized by the presence of poorly differentiated and pleomorphic astrocytes. 1 It is the most common primary brain tumor in adults, with higher incidence in individuals ranging from 55 to 60 years old. Due to its poor prognosis, short‐term survival rates remain low, with only 4% of patients surviving at 5 years after diagnosis. 2
Treatment of glioblastoma consists of a multimodal procedure, with surgical resection as the first standard approach, followed by chemoradiation and adjuvant chemotherapy using the alkylating agent temozolomide. 3 Despite numerous studies having demonstrated the clinical benefits of maximal safe resection in glioblastoma, complete surgical elimination of the tumor is not always feasible. 1 , 4 Glioblastoma is a poorly responsive tumor and tumors generally recur within 8 months, demonstrating the need for novel therapies that improve clinical efficacy and prolong patient survival. 5
Patients with glioblastoma exhibit local and systemic immunosuppression. 6 Upregulation of immune checkpoints, including PD‐1 or LAG‐3, coupled to reduced effector functions, are two key features of T and natural killer (NK) cell dysfunction in this malignancy. 7 , 8 , 9 This major immunosuppression associated with glioblastoma has prompted the development of new immunotherapeutic approaches. Despite the disappointing preliminary results in clinical trials, 10 , 11 immune checkpoint blockade (ICB) therapies might prove their efficacy in glioblastoma as neoadjuvants, as demonstrated by pre‐surgical administration of pembrolizumab, 12 or in combination regimens, such as anti‐BTLA plus anti‐PD‐1 treatment. 13 These strategies are typically focused on the activation of T cell responses. However, NK cells from patients with glioblastoma may retain certain antitumor properties in defiance of the immunosuppressive pressure exerted by the tumor. 14 , 15 NK cells are the only known immune cell able to eliminate glioblastoma stem cells without prior stimulation. 8 , 16 This ability turns NK cells into an interesting target for immunotherapeutic approaches in this malignancy. Indeed, chimeric antigen receptor (CAR) NK cell therapies have produced encouraging results in preliminary studies, 17 further supporting the potential of NK cells as a therapeutic tool in glioblastoma.
The inhibitory checkpoint ILT2 (LILRB1/CD85j) is widely known for its role in NK cell function. 18 This receptor exerts its inhibitory effect via recognition and binding of classical and non‐classical human leukocyte antigen (HLA)‐I molecules, albeit exhibiting higher affinity for HLA‐G upon comparison to classical HLA‐I molecules. 19 Disruption of the signaling pathway activated by ILT2 has been thoroughly studied as an alternative to classical ICB therapies in certain types of cancer. For instance, ILT2 blockade rescued NK cell‐mediated responses in chronic lymphocytic leukemia 20 and breast cancer. 21 Combination regimens including ILT2 blockade have also rendered positive results in preclinical studies. 22 , 23 Still, despite the increasing evidence highlighting the potential of ILT2 blockade in a myriad of malignancies, the role of this checkpoint in glioblastoma remains to be elucidated.
Herein, we report that ILT2 and its ligands are highly expressed in glioblastoma. In line with this, ILT2 blockade partially restored antitumor responses against glioblastoma in vitro, an effect strengthened by combination with temozolomide. Collectively, our studies suggest that ILT2 blockade may constitute a novel therapeutic alternative for the management of patients with glioblastoma. Nonetheless, further studies are necessary to determine the clinical potential of this strategy alone or in combination regimens.
2. MATERIALS AND METHODS
2.1. Cell culture
LN‐18, T98G, A172, and U87MG cells (ATCC) were cultured in DMEM (Lonza) supplemented with 10% heat‐inactivated FBS (Sigma‐Aldrich), 1 mM sodium pyruvate, 2 mM L‐glutamine, 100 U/mL penicillin, and 10 μg/mL streptomycin at 37°C and 5% CO2. Buffy‐coats from healthy donors were collected from Centro Comunitario de Sangre y Tejidos de Asturias following the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by ficoll (Biowest) density gradient centrifugation and cultured in supplemented RPMI 1640 (Lonza).
2.2. Evaluation of human leukocyte antigen expression
Surface levels of HLA‐I molecules were determined on glioblastoma cell lines using the following antibodies: anti‐HLA‐(A,B,C)‐PE (clone W6/32), anti‐HLA‐E‐PE (clone 3D12), anti‐HLA‐F‐PE (clone 3D11/HLA‐F), and anti‐HLA‐G‐PE (clone 87G) (all from Biolegend). PE mouse IgG1 (clone MOPC‐21) or PE mouse IgG2a (clone MOPC‐173) antibodies (both from Biolegend) were employed as isotype controls. 7‐AAD (Immunostep) was used to discriminate viable cells. Cells were analyzed in a Cytoflex S flow cytometer with CytExpert 2.3 software (Beckman Coulter). For the analysis of surface expression, raw MFI values were corrected by subtracting isotype values and, where indicated, normalized to the control condition as a fold induction.
2.3. Immunohistochemistry
Tissue specimens from resected glioblastoma tumors were analyzed by immunohistochemistry employing the EnVision FLEX Mini Kit and Dako Autostainer system (Agilent) following the manufacturer’s instructions. In brief, paraffin‐embedded tissues (3 μm) were deparaffinized and rehydrated and epitope retrieval was performed by heat induction (HIER) at 95°C for 20 min and pH = 9 in the PT Link, Pre‐Treatment Module. Endogenous peroxidase activity was blocked with EnVision FLEX Peroxidase‐Blocking Reagent for 5 min. Afterwards, sections were incubated with Protein Block, Serum‐Free buffer for 60 min. Tissue sections were stained with the following polyclonal antibodies: anti‐human HLA‐A (1:400, PA5‐116980), anti‐human HLA‐B (1:200, PA5‐35345), anti‐human HLA‐C (1:2000, PA5‐79367), anti‐human ILT2 (1:1000, PA5‐98738) (all from Invitrogen), or anti‐human HLA‐E (1:200, HPA031454, Merck) polyclonal antibodies and counterstained with hematoxylin. Signals were detected using diaminobenzidine chromogen as substrate in EnVision FLEX HRP (Agilent). Isotype controls were processed by omitting the primary antibody. Each specimen was individually reviewed and expression intensity was scored using a scale from 0 to 3. Sample classification was performed as follows: 0 as negative staining, 1 as dim, and 2–3 as positive.
2.4. Immune cell‐glioblastoma cocultures
PBMCs from healthy donors were cocultured with glioblastoma cell lines at a 5:1 ratio for 24 h. Afterwards, cells were stained with anti‐CD3‐FITC, anti‐CD56‐APC (Cytognos), anti‐CD8‐APC‐C750 (Immunostep), and anti‐ILT2‐PE (clone GHI/75, Biolegend) and analyzed by flow cytometry. PE mouse IgG2b antibodies (clone 27–35, Biolegend) were employed as isotype control.
2.5. Intracellular staining
Production of interferon‐gamma (IFN‐γ) and granzyme B was assessed as described elsewhere. 24 In short, PBMCs from healthy donors were treated with 10 μg/mL anti‐ILT2 blocking monoclonal antibody (mAb) (clone HP‐F1, kindly provided by Miguel López‐Botet, Universitat Pompeu Fabra) or control IgG (Biolegend) for 72 h. Afterwards, PBMCs were cocultured with glioblastoma cell lines in a 5:1 ratio for 4 h. For IFN‐γ detection, cocultures were plated in the presence of 50 nM PMA and 1 μg/mL ionomycin. Following incubation, cells were stained with anti‐CD3‐FITC, anti‐CD56‐APC, and anti‐CD8‐APC‐C750 and anti‐IFN‐γ‐PE (clone 4S.B3; Biolegend) or anti‐granzyme B‐PE (clone QA16A02; Biolegend).
2.6. Cytotoxicity
NK cell‐mediated tumor elimination was studied by calcein‐AM staining as previously described. 25 Briefly, glioblastoma cells were incubated with 10 μM calcein‐AM (Biolegend) for 30 min at 37°C. Following staining, tumor cells were cocultured with PBMCs, treated as designated for each experiment, at 10:1, 25:1 and 50:1 (effector: target) ratio for 4 h. Calcein release was measured on a Synergy H1 microplate reader (Biotek). For the indicated experiments, LN‐18 cells were pretreated with temozolomide (100 μM, Selleckchem) for 72 h.
2.7. Determination of ILT2/HLA‐I binding
First, glioblastoma cells were incubated with anti‐HLA‐(A,B,C) (clone W6/32; Biolegend), anti‐HLA‐E (clone 3D12HLA‐E, ThermoFisher Scientific), or anti‐HLA‐G (clone 87G; Biolegend) blocking antibodies. After washing, cells were incubated with 20 μg/mL human ILT2 protein, Fc tag (AcroBiosystems) and then stained with PE‐conjugated goat anti‐human IgG Fc secondary antibodies (ThermoFisher Scientific). All incubations were performed for 45 min at 4°C. Samples were analyzed in a Cytoflex S flow cytometer with CytExpert 2.3 software (Beckman Coulter).
2.8. In silico data analysis
Expression levels of the genes of interest were analyzed in glioblastoma tumors and normal tissue from The Cancer Genome Atlas (TCGA) database using ClickGene (http://www.clickgenome.org/). 26 Survival of glioblastoma patients stratified by mRNA levels of selected genes was studied via TIMER2.0 platform (http://timer.cistrome.org/). 27 The protein–protein interaction network was analyzed for HLA‐E using STRING (https://string‐db.org/). 28 LILRB1 mRNA analyses were performed employing transcriptome data available on the Gene Expression Omnibus (GEO) repository (dataset GSE4412 29 ) via ShinyGeo (https://gdancik.shinyapps.io/shinyGEO/). 30 Correlations between mRNA expression levels of distinct genes were determined employing the cBioportal platform (http://cbioportal.org/). 31 TISIDB repository (http://cis.hku.hk/TISIDB/) was employed to evaluate the correlation between gene expression and glioblastoma‐infiltrating immune subsets. 32
2.9. Statistics
Sample distribution was determined using the Shapiro–Wilk test. The relationship between continuous and categorical prognostic variables was evaluated by independent samples Student’s t‐test, and paired samples Student’s t‐test was applied for intra‐group comparisons. For mRNA expression comparisons, adjusted p‐values [p(adj)] were calculated applying the false discovery rate (FDR) method. In all cases, differences were considered statistically significant for p‐values: p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Statistical analyses were performed using GraphPad Prism 8 software (GraphPad Software). A heatmap representing Spearman’s correlation coefficients was built with Morpheus software (https://software.broadinstitute.org/morpheus).
3. RESULTS
3.1. Classical and non‐classical HLA‐I molecules are highly expressed in glioblastoma
To evaluate the relevance of classical and non‐classical HLA‐I molecules in glioblastoma, we first interrogated publicly available genomic data from the TCGA database. Greater mRNA expression of HLA‐A, ‐B, and ‐C was detected in glioblastoma samples compared to normal tissue (p(adj) = 6e‐07, 6e‐07 and 8e‐07, respectively) (Figure 1A). HLA‐E and, to a lesser extent, HLA‐F mRNA expression was also significantly higher in glioblastoma (p(adj) = 1.44e‐05 and 9.3e‐05, respectively), 33 , 34 whereas no significant difference in HLA‐G levels (p(adj) = 0.256) was found (Figure 1B). In line with this, strong expression of HLA‐(A,B,C) was detected on glioblastoma cell lines (Figure 1C). These tumor cells also exhibited remarkable levels of HLA‐E (Figure 1D).
FIGURE 1.
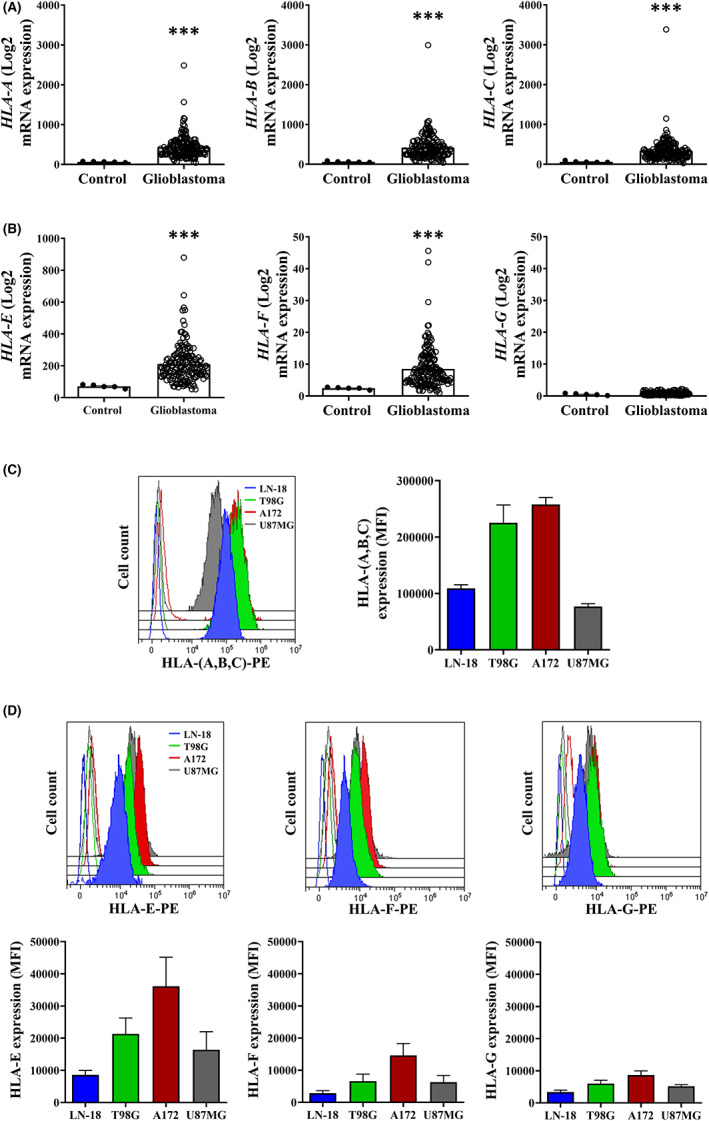
Analysis of expression of classical and non‐classical HLA‐I molecules on glioblastoma. (A,B) mRNA expression levels of the indicated genes on glioblastoma tissue specimens (GBM, n = 168) and normal tissue (control, n = 5) from TCGA database were determined using the ClickGene tool (mean ± standard error of the mean [SEM]). (C,D) Surface expression of classical (C) and non‐classical (D) HLA‐I molecules on glioblastoma cell lines were detected by flow cytometry (n = 4). Histograms depict a representative experiment, and graphs show the mean fluorescence intensity (MFI) ± SEM. Blank histograms correspond to isotype controls. ***p < 0.001
The impact of HLA‐I molecules on the survival of patients with glioblastoma was assessed by a Kaplan–Meier survival analysis via TIMER2.0. As depicted in Figure 2A, mRNA expression of HLA‐I, including classical and non‐classical molecules, did not affect survival in glioblastoma patients. Nonetheless, lower mRNA levels of each of these molecules significantly correlated with improved overall survival in patients with low‐grade glioma (Figure 2B).
FIGURE 2.
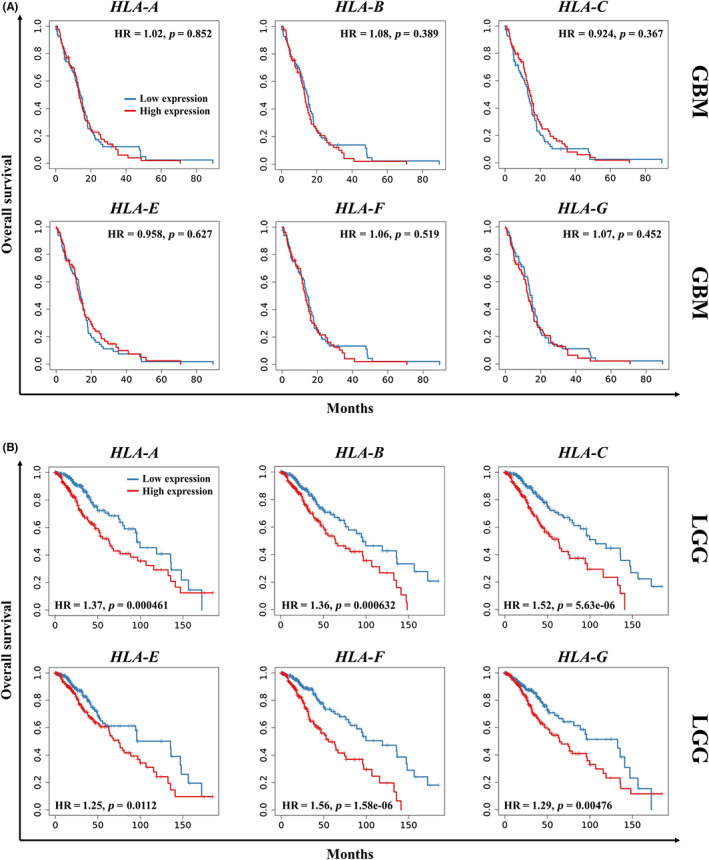
HLA‐I molecules impact survival in low‐grade glioma but not glioblastoma. Kaplan–Meier survival analysis categorized by mRNA levels of the indicated genes in glioblastoma (GBM, n = 153) (A) and low‐grade glioma (LGG, n = 516) (B). HR, hazard ratio
Given the high expression of HLA‐A, ‐B, ‐C, and ‐E observed in glioblastoma, as well as their central role in antitumor immunity, we further studied the expression of these proteins in patients with glioblastoma by immunohistochemistry (n = 40). HLA‐A was vastly expressed on tumor cells, with 36 (90%) patients showing positive staining (Figure 3A). In contrast, strong HLA‐B staining was only detected in 6 (15%) patients, although diffuse expression was found in 12 (30%) patients (Figure 3B). A total of 24 (60%) patients positively stained for HLA‐C, with 10 (25%) of them displaying weak staining (Figure 3C). HLA‐E staining was detected in 29 (72.5%) patients, 22 (55%) of which exhibited diffuse positive expression and 7 (17.5%) showed weak patchy expression on tumor cells (Figure 3D). Since HLA‐E is also expressed on immune cells, we further performed sequential staining of CD45, glial fibrillary acidic protein (GFAP), and HLA‐E on a glioblastoma case. HLA‐E staining was observed on tumor cells (Figure S1A), although it is worth mentioning that certain CD45+ cells within the tissue expressed this protein as well (Figure S1B). Tumor cell localization within tissue samples was evaluated by GFAP staining (Figure S2).
FIGURE 3.
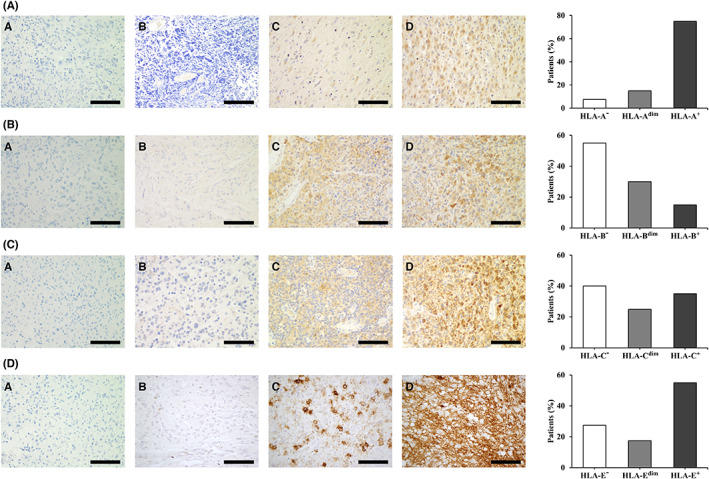
HLA‐I molecules are expressed in human glioblastoma in vivo. Immunohistochemical staining of HLA‐I proteins in tissue sections of different cases of glioblastoma. Graphs depict the percentage of patients with glioblastoma stratified based on immunohistochemical staining (n = 40). Images labeled as (a) correspond to isotype controls for each staining. (A) HLA‐A expression in a negative (b), weakly positive (c), and moderate to intense positivity (d) case. (B) HLA‐B expression in a negative (b), weakly positive in a patchy distribution, (c) and strong positive expression (d) case. (C) HLA‐C expression in a negative (b), weak positive (c), and diffuse strong positive (d) case. (D) HLA‐E expression in a mostly negative (b), patchy positive (c), and diffuse positive (d) case. Scale bars = 100 μm
3.2. ILT2 is highly expressed in glioblastoma and correlates to immune infiltration
HLA‐E, along with classical HLA‐I proteins, is part of a complex network of interacting proteins tightly related to immune modulation and acts as ligand for an array of immune receptors, including the inhibitory checkpoints NKG2A and ILT2 (Figure S3). Thus, we analyzed the mRNA expression of KLRC1 (gene coding for NKG2A) and LILRB1 (gene coding for ILT2) in glioblastoma from the TCGA database. KLRC1 was barely expressed on either condition studied and no different expression was found (p(adj) = 0.761). However, glioblastoma exhibited elevated levels of LILRB1 compared to the control (p(adj) = 0.002) (Figure 4A), suggesting that ILT2 might play a role in this malignancy. ILT2 and, to a lesser degree, NKG2A surface expression was detected on glioblastoma cell lines as well (Figure S4). Interestingly, LILRB2‐4 expression was increased in glioblastoma samples compared to normal tissue (p(adj) = 6e‐05 for LILRB2, 1.44e‐05 for LILRB3, and 1.6e‐05 for LILRB4), whereas no change was detected in LILRB5 levels (p(adj) = 0.758) (Figure 4A).
FIGURE 4.
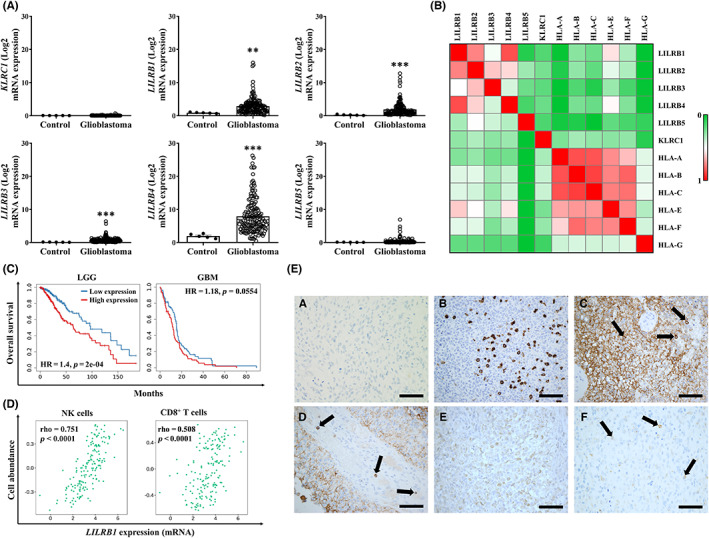
Analysis of expression of inhibitory LILR family receptors in glioblastoma. (A) mRNA expression levels of the indicated genes on glioblastoma tissue specimens (n = 168) and normal tissue (control, n = 5) from TCGA database were determined using ClickGene tool (mean ± SEM). (B) Heatmap representation of Spearman’s correlation values corresponding to the indicated genes in glioblastoma (n = 152) calculated via the cBioportal platform. (C) Kaplan–Meier survival analysis categorized by LILRB1 mRNA expression in low‐grade glioma (LGG, n = 516) and glioblastoma (GBM, n = 153). (D) Correlation between LILRB1 expression and immune infiltration in glioblastoma (n = 166) determined by TISIDB tool. (E) Immunohistochemical staining of CD57 and ILT2 in tissue sections of glioblastoma. (a) Isotype control. (b) CD57+ NK cells in lymph node. (c–d) Scattered CD57+ NK cells in glioblastoma (black arrows). In (d), CD57+ NK cells are mostly intravascular. (e) Mild ILT2 expression on glioblastoma cells. (f) Moderate expression of ILT2 on NK cells (black arrows). Bars = 100 μm. HR, hazard ratio. **p < 0.01; ***p < 0.001
In addition to the correlation between HLA‐I molecules, in silico analysis revealed that LILRB1 correlated to HLA‐E in glioblastoma (Spearman’s coefficient of 0.687) (Figure 4B). Similar to HLA‐I molecules, Kaplan–Meier analysis revealed that LILRB1 expression significantly affected the overall survival of patients with low‐grade glioma (Figure 4C). A similar trend was observed in glioblastoma, although it did not reach significance (p = 0.0554) (Figure 4C). Still, the expression levels of this receptor are increased in grade IV gliomas compared to grade III tumors (p = 0.0002), as shown by in silico analysis of previously published data from the GEO database (dataset GSE4412 29 ) (Figure S5A). Of note, high LILRB2 and LILRB3 levels negatively influence overall survival in patients with glioblastoma compared to other LILRB receptors, although nearly all of them influence survival in low‐grade glioma (Figure S5B).
Overexpression of ILT2 on immune cells is frequently linked to an exhausted phenotype that leads to immune evasion in cancer. Therefore, we next assessed whether ILT2 might be expressed by immune cells in glioblastoma. LILRB1 expression was highly correlated to infiltrating NK cell numbers in glioblastoma, and CD8+ T cells displayed lower correlation (Spearman’s coefficient of 0.751 vs 0.508, respectively) (Figure 4D). To validate these findings, expression of ILT2, as well as CD57 (as a marker for NK cells), was assessed by immunohistochemistry. Although mild expression of ILT2 was observed on tumor cells, immunostaining of this protein was primarily detected on NK cells (Figure 4E), supporting the results obtained from in silico analysis.
3.3. ILT2 is upregulated on immune cells upon coculture with glioblastoma
To gain further insight into the dysregulation of ILT2 on infiltrating immune cells in glioblastoma, PBMCs from healthy donors were cocultured with glioblastoma cell lines, and surface expression of ILT2 was evaluated by flow cytometry (Figure 5A). Coculture with tumor cells triggered an upregulation of ILT2 on NK cells (Figure 5B). CD8+ T cells expressed higher levels of ILT2 upon coculture with LN‐18 and U87MG cells as well (Figure 5B). Noticeably, no changes on ILT2 expression were detected for T98G cells. Glioblastoma‐conditioned media failed to modulate ILT2 surface expression on healthy immune cells (data not shown), suggesting that this effect might be mediated by cell‐to‐cell contact. We next investigated whether glioblastoma cells affected the antitumor function of NK cells. As depicted in Figure 5C, PBMCs previously exposed to LN‐18 cells exhibited reduced cytotoxicity compared to the control, pointing out that glioblastoma cells hinder NK cell antitumor activity.
FIGURE 5.
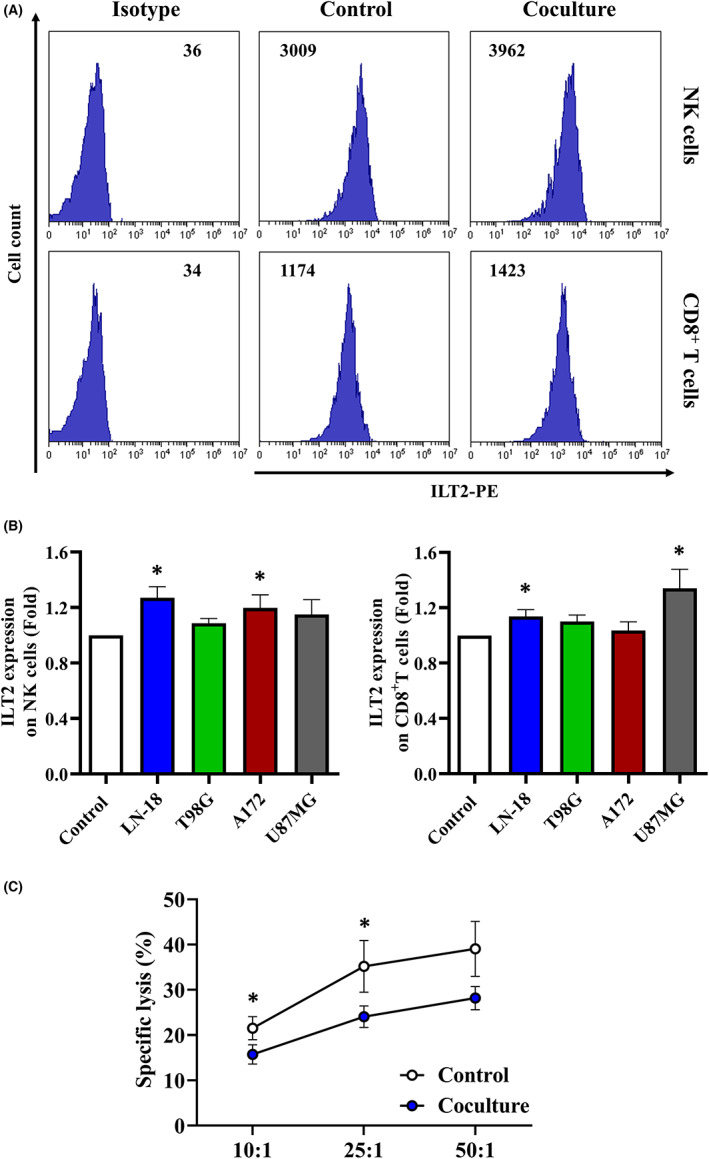
Coculture with glioblastoma cells induces ILT2 expression on immune cells and reduces their cytotoxic activity. PBMCs from healthy donors (n = 10) were cocultured with glioblastoma cell lines at a 5:1 ratio for 24 h. (A,B) ILT2 expression on immune subsets was evaluated by flow cytometry. Histograms depict a representative experiment employing LN‐18 cells. Numbers correspond to MFI values. Bars correspond to ILT2 expression on NK cells and CD8+ T cells represented as normalized MFI (mean ± SEM). (C) PBMCs were then coincubated with fresh LN‐18 cells at three different E:T (effector: target) ratios for 4 h. The cytotoxic activity against tumor cells was evaluated by calcein‐AM staining. Graph depicts the percentage of specific lysis (mean ± SEM). *p < 0.05
3.4. ILT2 blockade partially restores antitumor immune responses against glioblastoma
To assess whether ILT2 takes part in the glioblastoma‐induced suppression of NK cell function, PBMCs from healthy donors were treated with anti‐ILT2 blocking mAb, and cytokine production in response to glioblastoma cells was assessed by flow cytometry (Figure 6A). ILT2 blockade induced IFN‐γ production by NK cells (Figure 6B). Lower, yet significant, IFN‐γ increase was observed for CD8+ T cells (Figure 6C). Granzyme B levels were significantly higher in anti‐ILT2‐treated NK cells exposed to LN‐18 cells (Figure 6D). In contrast, ILT2 blockade did not affect granzyme B production by CD8+ T cells (Figure 6E). Treatment with anti‐ILT2 blocking mAb also boosted NK cell‐mediated lysis of glioblastoma cells (Figure 7A).
FIGURE 6.
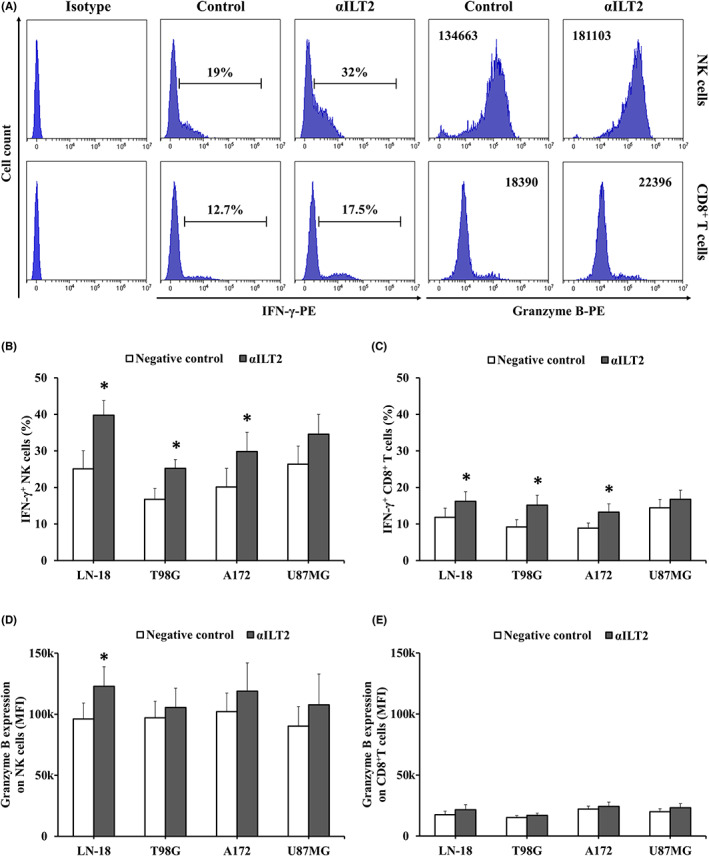
ILT2 blockade enhances IFN‐γ production by immune cells. Peripheral blood mononuclear cells (PBMCs) from healthy donors (n = 10) were treated with anti‐ILT2 blocking antibodies or control IgG (10 μg/mL) for 72 h and then cocultured with glioblastoma cells at a 5:1 ratio for 4 h. IFN‐γ and granzyme B production were assessed by intracellular flow cytometry. (A) Histograms illustrate a representative experiment using LN‐18 cells. Numbers correspond to percentage of positive cells for IFN‐γ and MFI for granzyme B. (B,C) Graphs show the percentage of IFN‐γ+ NK and CD8+ T cells. (D,E) Granzyme B expression on NK and CD8+ T cells represented as MFI (mean ± SEM). *p < 0.05
FIGURE 7.
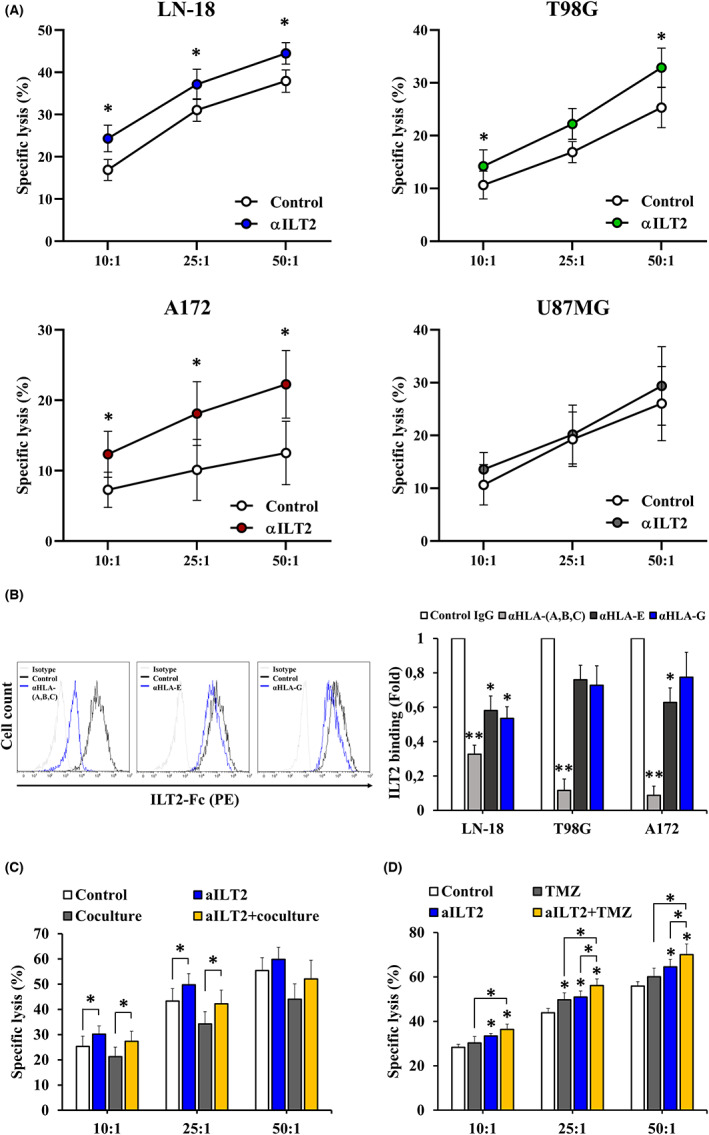
ILT2 blockade promotes natural killer (NK) cell‐mediated cytotoxicity against glioblastoma. The cytotoxic activity of pretreated peripheral blood mononuclear cells (PBMCs) from healthy donors against glioblastoma cells was evaluated at three different E:T (effector:target) ratios by calcein‐AM staining. Graphs depict the percentage of specific lysis (mean ± SEM). (A) PBMCs from healthy donors (n = 10) were treated with anti‐ILT2 blocking antibodies or control IgG (10 μg/mL) for 72 h and then cocultured with glioblastoma cell lines for 4 h. (B) ILT2 binding was evaluated on glioblastoma cells after HLA‐(A,B,C), HLA‐E, or HLA‐G blockade by flow cytometry (n = 3). Histograms illustrate a representative experiment employing LN‐18 cells. Bars correspond to normalized MFI ± SEM. (C) PBMCs from healthy donors (n = 8) were cocultured with LN‐18 cells in the presence of anti‐ILT2 blocking antibodies or control IgG (10 μg/mL) for 24 h, followed by coculture with fresh LN‐18 cells for 4 h. (D) PBMCs from healthy donors (n = 6) were treated with anti‐ILT2 blocking antibodies or control IgG (10 μg/mL) for 72 h. LN‐18 cells were treated with temozolomide (TMZ, 100 μM) for 72 h. Afterwards, cells were cocultured for 4 h. *p < 0.05; **p < 0.01
Next, we sought to elucidate whether ILT2‐mediated increased cytotoxicity is related to ILT2/HLA‐I interactions. In this context, HLA‐E blockade significantly decreased ILT2 binding to glioblastoma cells, in a similar extent to HLA‐G blockade (Figure 7B). siRNA‐mediated HLA‐E knockdown failed to neutralize the increase in cytotoxicity mediated by ILT2 blockade (Figure S6A,B). In the same line, no changes were detected in NK cell‐mediated elimination after blocking HLA‐E on glioblastoma cells (Figure S6C). Of note, NKG2A blockade did not enhance tumor cell elimination (Figure S7), which agrees with the results detected upon HLA‐E blockade, because NKG2A stands as a key receptor for HLA‐E. Despite partially decreasing ILT2 binding (Figure 7B), HLA‐G blockade did not enhance glioblastoma cell killing either (Figure S8A). Notably, ILT2 binding was impaired in a greater fashion upon blockade of classical HLA‐I ligands (Figure 7B), which significantly boosted the cytotoxicity of glioblastoma cells (Figure S8B), suggesting that ILT2 takes part in a complex signaling network. It is worth mentioning that blocking classical HLA‐I molecules did not affect surface expression of their non‐classical counterparts (Figure S9), ensuring the specificity of these results. Additionally, ILT2 blockade partly rescued the cytotoxic capacity of PBMCs previously exposed to LN‐18 cells, thus counteracting, up to a certain point, the immunosuppressive pressure brought about by the tumor (Figure 7C).
Temozolomide stands as the first‐line chemotherapy approved for postoperative treatment in patients with glioblastoma. To evaluate the effect of combining ILT2 blockade with this chemotherapeutic drug on NK cell‐mediated cytotoxicity, LN‐18 cells were exposed to temozolomide and then cocultured with PBMCs from healthy donors previously treated with anti‐ILT2 antibodies. As illustrated in Figure 7D, not only did temozolomide treatment sensitized glioblastoma cells to immune‐mediated elimination, but it significantly increased tumor lysis by anti‐ILT2‐treated immune cells. In agreement with previous studies, discreet MICA upregulation was observed on temozolomide‐treated LN‐18 cells (Figure S10A). Yet, NKG2D blockade did not counteract the increase in tumor cell elimination brought about by this chemotherapeutic agent (data not shown). It is worth mentioning that temozolomide treatment also led to downregulation of non‐classical HLA‐I molecules on glioblastoma cells (Figure S10B). In general, these findings reveal that a combination of ILT2 blockade with clinically available therapies might prove beneficial in reestablishing the immune function in patients with glioblastoma.
4. DISCUSSION
The survival rate of patients with glioblastoma remains low despite the aggressive multimodal therapy approved for their clinical management. Total surgical resection is not always viable, and postoperative chemoradiation generally fails to eliminate the tumor, leaving these patients with adjuvant treatments in an effort to delay cancer progression. Hence, the development of alternative therapeutic approaches is of utmost importance in glioblastoma. The marked exhaustion reported on intratumor T lymphocytes has prompted the study of classical ICB therapies in glioblastoma at a clinical level. 7 Still, PD‐1 blockade has rendered disappointing results in recurrent glioblastoma, with only 8% of patients showing durable responses in a phase III clinical trial, 10 which calls for novel checkpoints that can make a difference in the treatment of glioblastoma.
Various studies have described the dysregulation of key inhibitory checkpoints and their ligands on glioblastoma, including HVEM or TIM‐3, among others. 35 , 36 In line with these results, in silico analysis revealed high expression of classical (HLA‐A, ‐B, and ‐C) and non‐classical (HLA‐E and ‐F) HLA‐I molecules in glioblastoma. Further, an important proportion of glioblastoma patients exhibited detectable levels of HLA‐A (90%) and HLA‐E (72.5%). These data agree with previous studies reporting altered expression of HLA‐E 34 and HLA‐F 37 in human gliomas in vivo. Interestingly, higher expression of these HLA‐I molecules was associated with lower survival in patients with low‐grade glioma but not glioblastoma, which might account for the rapid progression of the latter.
HLA‐E is arising as an interesting target in the field of cancer immunotherapy due to its immunomodulatory role as ligand for immune inhibitory checkpoints, such as NKG2A. 38 , 39 , 40 Nonetheless, our analysis showed that KLRC1, which encodes NKG2A, was not differentially expressed in glioblastoma, and its expression levels were exceptionally low. In contrast, elevated levels of LILRB1, which ILT2, an inhibitory receptor that can bind to HLA‐E 41 and other HLA‐I members, 19 was found in this tumor. Deeper analysis of the LILRB protein family revealed that LILRB2‐4 mRNA expression is elevated in glioblastoma tissue as well. Interestingly, high LILRB receptor levels, except for LILRB2, negatively affected survival in low‐grade glioma patients, whereas only low LILRB2 and LILRB3 expression correlated to improved survival in glioblastoma. LILRB2 expression has been reported in the stem cell compartment at mRNA level, 8 which might explain its connection to patient survival because this cell population typically drives treatment resistance. 42 Interestingly, LILRB2 blockade has proven beneficial in reprogramming tumor‐associated macrophages and promoting antitumor responses, 43 one of the many reasons why the therapeutic potential of this inhibitory checkpoint in glioblastoma deserves future study.
ILT2 is widely expressed by immune cells, including lymphocytes, and its upregulation is frequently linked to immune exhaustion in cancer. 20 , 44 In glioblastoma, LILRB1 expression correlated to NK cell infiltration and, to a small extent, CD8+ T cell abundance. ILT2 staining was primarily detected on NK cells in vivo, although mild expression was observed on glioblastoma cells as well. Consequently, this checkpoint is likely to be mainly expressed on immune cells in this malignancy, and its overexpression might contribute to the pronounced immunosuppression associated with glioblastoma.
Upregulation of inhibitory receptors on effector immune cells is a well‐known immune evasion strategy in cancer. Indeed, tumor‐infiltrating lymphocytes isolated from patients with glioblastoma exhibit higher levels of numerous inhibitory checkpoints, such as PD‐1 and LAG3, compared to its counterparts from peripheral blood. 7 , 35 This evasion mechanism exploited by glioblastoma might be extendable to ILT2 because coculture with glioblastoma cells reduced NK cell‐mediated cytotoxicity and induced ILT2 upregulation on NK cells and CD8+ T cells. Therefore, ILT2 might work as an immune exhaustion marker in glioblastoma, similar to receptors such as PD‐1 or TIGIT. Its upregulation might contribute to the immune suppression exerted by the tumor, as it occurs in other types of cancer. 20 , 44 , 45 In agreement, ILT2 blockade boosted IFN‐γ production and NK cell cytotoxicity against glioblastoma cells and partially restored tumor lysis in immune cells previously cocultured with glioblastoma cells. HLA‐E blockade partially prevented ILT2 binding to glioblastoma cells, supporting the notion that the ILT2/HLA‐E axis might be at work in this tumor. Still, tumor cell killing by ILT2‐treated immune cells was not counteracted upon siRNA‐mediated HLA‐E knockdown, revealing that this ligand might not be responsible for the cytotoxic effect of ILT2 blocking mAb in glioblastoma and pointing out in the direction of alternative HLA‐. Again, neither HLA‐G nor HLA‐E blockade boosted tumor cell killing. Classical HLA‐I molecules stand as ligands for ILT2, and its blockade has been extensively reported to promote NK cell responses. 19 , 23 , 46 , 47 Herein, ILT2 binding was significantly impaired by HLA‐(A,B,C) blockade, which, coupled with the high expression of these molecules in glioblastoma and the significant increase in cytotoxicity upon their blockade, brings forth HLA‐A, ‐B, and ‐C as likely players in the inhibitory role of ILT2 in this malignancy. These data highlight the complexity of the ILT2 equation, which might ultimately account for an integrative network of activating signals that trigger immune cell suppression. Overall, these findings bring to light that disruption of ILT2 signaling promotes, to a certain extent, antitumor immune responses, supporting the therapeutic potential of this strategy in glioblastoma.
Because of the strong immune exhaustion linked to glioblastoma, a combination of ILT2 blockade with other immunotherapeutic approaches might potentiate immune activation. In fact, a phase I/II clinical trial is currently evaluating the efficacy of BND‐22, an anti‐ILT2 blocking mAb, in combination with pembrolizumab in advanced solid tumors (NCT04717375). In glioblastoma, despite the initial results of classical ICB therapies used as adjuvants, recent studies have described the benefits of anti‐PD‐1 treatment prior to surgical resection, 12 , 48 highlighting the advantages of employing ICB treatments as neoadjuvants. Recent works have revealed that co‐blockade of PD‐1 and novel checkpoints, such as BTLA or TIGIT, have therapeutic potential in glioblastoma, 13 , 49 opening the door to the study of ILT2 blockade in combination with other ICBs. The clinical value of certain chemotherapeutic agents relies, in part, on eliciting antitumor immune responses. At this respect, temozolomide treatment slightly increased MICA expression on glioblastoma cells, which agrees with previously reported data, 50 , 51 thus providing rationale for its use in combination with ILT2 blockade to counteract NK cell suppression. Indeed, treatment of LN‐18 glioblastoma cells with temozolomide potentiated tumor lysis by immune cells pretreated with an anti‐ILT2 blocking mAb, suggesting that this combination might counteract, to a certain degree, the immunosuppressive pressure exerted by the tumor. NKG2D blockade failed to counteract the antitumor cytotoxicity triggered by temozolomide. Hence, the added tumor elimination observed with the combination might answer to the modulation of ILT2 ligands detected on glioblastoma cells upon temozolomide exposure, among other mechanisms yet to be revealed.
Altogether, our results provide evidence of the dysregulation of the inhibitory checkpoint ILT2 and its ligands in glioblastoma. Treatment with anti‐ILT2 blocking mAb promoted antitumor immune responses, and its combination with temozolomide led to greater tumor cell elimination, opening a window for the study of ILT2 blockade as an alternative therapeutic approach alone or in combination in glioblastoma.
AUTHOR CONTRIBUTIONS
S. L.‐H. designed and performed experiments, analyzed data and wrote the manuscript. C. S.‐B. performed experiments and analyzed data. A. M.‐P., M. D. C.‐T. and I. F.‐V. performed experiments. M. P. S.‐H. collected and analyzed clinical data. S. G. supervised the research and wrote the manuscript. All the authors reviewed and approved the final version of the manuscript.
FUNDING INFORMATION
This work was supported by a grant from the Spanish Instituto de Salud Carlos III (PI19/01353) and FEDER European Union. C.S‐B holds a Severo Ochoa grant (BP19‐066) by FICYT. S.L‐H is currently supported by Instituto Universitario de Oncología del Principado de Asturias (IUOPA) (SV‐PA‐21‐01).
CONFLICT OF INTEREST
The authors have no conflict of interest. No author works as an Editor or Editorial Board Member of Cancer Science.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank Dr López‐Botet for providing the anti‐ILT2 blocking mAb.
Lorenzo‐Herrero S, Sordo‐Bahamonde C, Martínez‐Pérez A, et al. Immunoglobulin‐like transcript 2 blockade restores antitumor immune responses in glioblastoma. Cancer Sci. 2023;114:48‐62. doi: 10.1111/cas.15575
Contributor Information
Seila Lorenzo‐Herrero, Email: seilalorenzoherrero@gmail.com.
Segundo González, Email: segundog@uniovi.es.
DATA AVAILABILITY STATEMENT
In silico analysis were performed employing publicly available RNAseq data from the TCGA database using ClickGene (http://www.clickgenome.org/), TIMER2.0 (http://timer.cistrome.org/), TISIDB (http://cis.hku.hk/TISIDB/) and cBioportal (http://cbioportal.org/) tools, and dataset GSE4412 from GEO repository via ShinyGeo (https://gdancik.shinyapps.io/shinyGEO/).
REFERENCES
- 1. Young RM, Jamshidi A, Davis G, Sherman JH. Current trends in the surgical management and treatment of adult glioblastoma. Ann Transl Med. 2015;3:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poon MTC, Sudlow CLM, Figueroa JD, Brennan PM. Longer‐term (>/= 2 years) survival in patients with glioblastoma in population‐based studies pre‐ and post‐2005: a systematic review and meta‐analysis. Sci Rep. 2020;10:11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J Clin. 2020;70:299‐312. [DOI] [PubMed] [Google Scholar]
- 4. Bloch O, Han SJ, Cha S, et al. Impact of extent of resection for recurrent glioblastoma on overall survival: clinical article. J Neurosurg. 2012;117:1032‐1038. [DOI] [PubMed] [Google Scholar]
- 5. Chaichana KL, Jusue‐Torres I, Navarro‐Ramirez R, et al. Establishing percent resection and residual volume thresholds affecting survival and recurrence for patients with newly diagnosed intracranial glioblastoma. Neuro Oncol. 2014;16:113‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grabowski MM, Sankey EW, Ryan KJ, et al. Immune suppression in gliomas. J Neurooncol. 2021;151:3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woroniecka K, Chongsathidkiet P, Rhodin K, et al. T‐Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clini Cancer Res. 2018;24:4175‐4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Close HJ, Stead LF, Nsengimana J, et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin Exp Immunol. 2020;200:33‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF‐beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12:7‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1003‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nayak L, Molinaro AM, Peters K, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27:1048‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti‐PD‐1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi J, Medikonda R, Saleh L, et al. Combination checkpoint therapy with anti‐PD‐1 and anti‐BTLA results in a synergistic therapeutic effect against murine glioblastoma. Onco Targets Ther. 2021;10:1956142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kmiecik J, Poli A, Brons NH, et al. Elevated CD3+ and CD8+ tumor‐infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71‐83. [DOI] [PubMed] [Google Scholar]
- 15. Lee SJ, Kang WY, Yoon Y, et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Castriconi R, Daga A, Dondero A, et al. NK cells recognize and kill human glioblastoma cells with stem cell‐like properties. J Immunol. 2009;182:3530‐3539. [DOI] [PubMed] [Google Scholar]
- 17. Burger MC, Zhang C, Harter PN, et al. CAR‐engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front Immunol. 2019;10:2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sordo‐Bahamonde C, Lorenzo‐Herrero S, González‐Rodríguez AP, González S. Checkpoint inhibition in the fight against cancer: NK cells have some to say in it. Successes and challenges of NK. In: Bonavida B, Jewett A, eds. Successes and Challenges of NK Immunotherapy. Academic Press; 2021; 267‐304. [Google Scholar]
- 19. Shiroishi M, Tsumoto K, Amano K, et al. Human inhibitory receptors Ig‐like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA‐G. Proc Natl Acad Sci U S A. 2003;100:8856‐8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Villa‐Alvarez M, Sordo‐Bahamonde C, Lorenzo‐Herrero S, et al. Ig‐like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol. 2018;9:2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberti MP, Julia EP, Rocca YS, et al. Overexpression of CD85j in TNBC patients inhibits cetuximab‐mediated NK‐cell ADCC but can be restored with CD85j functional blockade. Eur J Immunol. 2015;45:1560‐1569. [DOI] [PubMed] [Google Scholar]
- 22. Kim A, Han CJ, Driver I, et al. LILRB1 blockade enhances bispecific T cell engager antibody‐induced tumor cell killing by effector CD8(+) T cells. J Immunol. 2019;203:1076‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Godal R, Bachanova V, Gleason M, et al. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin‐like receptor‐negative natural killer cells after NKG2A and LIR‐1 blockade. Biol Blood Marrow Transplant. 2010;16:612‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sordo‐Bahamonde C, Lorenzo‐Herrero S, Gonzalez S, Lopez‐Soto A. A cytofluorimetric assay to evaluate intracellular cytokine production by NK cells. Methods Enzymol. 2020;631:343‐355. [DOI] [PubMed] [Google Scholar]
- 25. Lorenzo‐Herrero S, Sordo‐Bahamonde C, Gonzalez S, Lopez‐Soto A. Evaluation of NK cell cytotoxic activity against malignant cells by the calcein assay. Methods Enzymol. 2020;631:483‐495. [DOI] [PubMed] [Google Scholar]
- 26. Bi JH, Tong YF, Qiu ZW, et al. ClickGene: an open cloud‐based platform for big pan‐cancer data genome‐wide association study, visualization and exploration. BioData Min. 2019;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li T, Fu J, Zeng Z, et al. TIMER2.0 for analysis of tumor‐infiltrating immune cells. Nucleic Acids Res. 2020;48:W509‐W514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res. 2019;47:D607‐D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Freije WA, Castro‐Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503‐6510. [DOI] [PubMed] [Google Scholar]
- 30. Dumas J, Gargano MA, Dancik GM. shinyGEO: a web‐based application for analyzing gene expression omnibus datasets. Bioinformatics. 2016;32:3679‐3681. [DOI] [PubMed] [Google Scholar]
- 31. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ru B, Wong CN, Tong Y, et al. TISIDB: an integrated repository portal for tumor‐immune system interactions. Bioinformatics. 2019;35:4200‐4202. [DOI] [PubMed] [Google Scholar]
- 33. Chen X, Sun N, Li R, et al. Targeting HLA‐F suppresses the proliferation of glioma cells via a reduction in hexokinase 2‐dependent glycolysis. Int J Biol Sci. 2021;17:1263‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu Z, Liang J, Wang Z, Li A, Fan X, Jiang T. HLA‐E expression in diffuse glioma: relationship with clinicopathological features and patient survival. BMC Neurol. 2020;20:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lombardo SD, Bramanti A, Ciurleo R, et al. Profiling of inhibitory immune checkpoints in glioblastoma: potential pathogenetic players. Oncol Lett. 2020;20:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li G, Wang Z, Zhang C, et al. Molecular and clinical characterization of TIM‐3 in glioma through 1,024 samples. Onco Targets Ther. 2017;6:e1328339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng E, Liang T, Wang X, et al. Correlation of alteration of HLA‐F expression and clinical characterization in 593 brain glioma samples. J Neuroinflammation. 2019;16:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borst L, van der Burg SH, van Hall T. The NKG2A‐HLA‐E axis as a novel checkpoint in the tumor microenvironment. Clin Cancer Res. 2020;26:5549‐5556. [DOI] [PubMed] [Google Scholar]
- 39. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. 2019;129:2094‐2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McWilliams EM, Mele JM, Cheney C, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Onco Targets Ther. 2016;5:e1226720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR‐1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity. 1999;11:603‐613. [DOI] [PubMed] [Google Scholar]
- 42. Prager BC, Bhargava S, Mahadev V, Hubert CG, Rich JN. Glioblastoma stem cells: driving resilience through chaos. Trends Cancer. 2020;6:223‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen HM, van der Touw W, Wang YS, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor‐associated myeloid cells and promotes antitumor immunity. J Clin Invest. 2018;128:5647‐5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dumont C, Jacquier A, Verine J, et al. CD8(+)PD‐1(−)ILT2(+) T cells are an intratumoral cytotoxic population selectively inhibited by the immune‐checkpoint HLA‐G. Cancer Immunol Res. 2019;7:1619‐1632. [DOI] [PubMed] [Google Scholar]
- 45. Villa‐Alvarez M, Lorenzo‐Herrero S, Gonzalez‐Rodriguez AP, et al. Ig‐like transcript 2 (ILT2) suppresses T cell function in chronic lymphocytic leukemia. Onco Targets Ther. 2017;6:e1353856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bernson E, Christenson K, Pesce S, et al. Downregulation of HLA class I renders inflammatory neutrophils more susceptible to NK cell‐induced apoptosis. Front Immunol. 2019;10:2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koh CY, Blazar BR, George T, et al. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood. 2001;97:3132‐3137. [DOI] [PubMed] [Google Scholar]
- 48. Schalper KA, Rodriguez‐Ruiz ME, Diez‐Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470‐476. [DOI] [PubMed] [Google Scholar]
- 49. Hung AL, Maxwell R, Theodros D, et al. TIGIT and PD‐1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Onco Targets Ther. 2018;7:e1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fluh C, Chitadze G, Adamski V, et al. NKG2D ligands in glioma stem‐like cells: expression in situ and in vitro. Histochem Cell Biol. 2018;149:219‐233. [DOI] [PubMed] [Google Scholar]
- 51. Weiss T, Schneider H, Silginer M, et al. NKG2D‐dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin Cancer Res. 2018;24:882‐895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
In silico analysis were performed employing publicly available RNAseq data from the TCGA database using ClickGene (http://www.clickgenome.org/), TIMER2.0 (http://timer.cistrome.org/), TISIDB (http://cis.hku.hk/TISIDB/) and cBioportal (http://cbioportal.org/) tools, and dataset GSE4412 from GEO repository via ShinyGeo (https://gdancik.shinyapps.io/shinyGEO/).