

Abstract
Retinoic acid receptor–related orphan receptor α (RORα) is a transcription factor involved in nuclear gene expression and a known tumor suppressor. RORα was the first identified substrate of lysine methylation–dependent degradation. However, the mechanisms of other post‐translational modifications (PTMs) that occur in RORα remain largely unknown, especially in liver cancer. Arginine methylation is a common PTM in arginine residues of nonhistone and histone proteins and affects substrate protein function and fate. We found an analogous amino acid disposition containing R37 at the ROR N‐terminus compared to histone H3 residue, which is arginine methylated. Here, we provide evidence that R37 methylation–dependent degradation is carried out by protein arginine methyltransferase 5 (PRMT5). Further, we discovered that PRMT5 regulated the interaction between the E3 ubiquitin ligase ITCH and RORα through RORα arginine methylation. Arginine methylation–dependent ubiquitination‐mediated RORα degradation reduced downstream target gene activation. H2O2‐induced reactive oxygen species (ROS) decreased PRMT5 protein levels, consequently increasing RORα protein levels in HepG2 liver cancer cells. In addition, ROS inhibited liver cancer progression by inducing apoptosis via PRMT5‐mediated RORα methylation and the ITCH axis. Our results potentiate PRMT5 as an elimination target in cancer therapy, and this additional regulatory level within ROS signaling may help identify new targets for therapeutic intervention in liver cancer.
Keywords: arginine methylation, liver cancer, methyl‐degron, RORα‐PRMT5‐ITCH, ROS
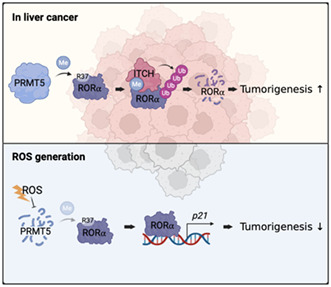
The schematic diagram of the mechanism of arginine methylation‐mediated RORα degradation by PRMT5 and ITCH. In liver cancer, PRMT5‐catalyzed arginine methylation of RORα induces RORα ubiquitination by facilitating RORα and E3 ligase ITCH interactions. When the cells were exposed to excessive ROS, the cells inhibited PRMT5 to dissociate ITCH from RORα, resulting in RORα stabilization.
Abbreviations
- CHX
cycloheximide
- DEN
diethylnitrosamine
- DMSO
dimethyl sulfoxide
- H2O2
hydrogen peroxide
- ITCH
itchy E3 ubiquitin protein ligase
- LKO
liver‐specific knockout
- mRNA
messenger ribonucleic acid
- p21
cyclin‐dependent kinase inhibitor 1
- PRMT
protein arginine methyltransferase
- PTMs
post‐translational modifications
- qRT‐PCR
quantitative reverse‐transcription PCR
- RORE
ROR‐responsive element
- RORα
retinoic acid–related orphan nuclear receptor α
- ROS
reactive oxygen species
1. INTRODUCTION
Liver cancer is the sixth most common cancer worldwide and has the second‐highest mortality rate. 1 Patients are primarily diagnosed at advanced stages, and if not detected and treated early, cancer survival rates can continue to decline and contribute to poor prognosis. Liver cancer incidence is increasing but effective treatment options and an understanding of cancer‐related processes are still limited. Therefore, finding a critical factor that fuels and sustains cancers will help us precisely comprehend tumorigenesis and provide the rationale for liver cancer treatment options.
Retinoic acid–related orphan nuclear receptor α (RORα) is a member of the orphan nuclear receptor family and functions as a transcription factor. 2 , 3 RORα recruits to a specific DNA sequence called the ROR‐responsive element (RORE) and, together with various coregulators, dynamically regulates target gene expression. Alternative splicing produces four human RORα isoforms, referred to as ROR α1‐α4. RORα consists of two conserved regions, a DNA‐binding domain (DBD), a ligand‐binding domain (LBD), and a hinge domain that links them. The N‐terminal domain (NTD) is a region with specificity among the RORα isoforms and performs distinct functions in a context‐dependent manner. 4 RORα is widely involved in pathophysiological processes, such as the circadian rhythm, lipid metabolism, immune infection, and tumorigenesis. 5 , 6 , 7 In particular, accumulating evidence has demonstrated that RORα is downregulated in various cancers and suppresses many malignancies. Under DNA damage conditions, p53 induces RORα, which positively regulates p53 stability, thereby increasing p53‐mediated apoptosis in colon cancer cells. 7 Wnt5a‐dependent phosphorylation of RORα attenuates the canonical Wnt/β‐catenin signaling pathway and reduces RORα phosphorylation compared with normal counterparts in colorectal tumor tissues. 8 Indeed, the NTD of RORα also suppresses the proliferation and metastatic potential of prostate cancer cells through Wnt target gene expression downregulation. 9 Moreover, RORα inhibits breast tumor growth, migration, and invasion through semaphorin‐3F (SEMA3F) transcriptional activation. 10 These findings provide evidence that RORα is commonly considered a tumor suppressor in many cancers and that reduced RORα expression is necessary for cancer‐related processes.
Post‐translational modification (PTM) is the enzymatic mechanism after protein synthesis that confers diverse roles by providing functional groups to the amino acids of proteins. 11 We previously reported that enhancer of zeste homolog 2 (EZH2), a methyltransferase of histone H3 lysine 27 (H3K27), imparts a methyl group to lysine 38 of the RORα protein. 12 A specific adapter, DCAF1, recognizes methylated RORα, recruits the Cullin 4 (CUL4) E3‐ligase complex, and degrades it accordingly. An inverse correlation between EZH2 and RORα in breast tumor patient samples compared with their normal counterparts reflects this methylation‐dependent RORα degradation. Indeed, RORα restoration by EZH2 and DCAF1 ablation led to a significant reduction in colony number, suggesting that RORα degradation is critical in tumorigenesis progression.
Arginine methylation, another PTM, is mediated by protein arginine methyltransferase (PRMT) enzymes that catalyze methylarginine. 13 PRMTs transfer methyl groups from S‐adenosyl methionine (SAM) to the guanidinium group of the arginine residue. PRMT5 catalyzes monomethylarginine and symmetric dimethylarginine and is crucial for various cellular processes, such as development, differentiation, and cancer. 13 , 14 Accumulating evidence suggests that PRMT5 has oncogenic activities, and its expression is correlated with poor prognosis. 15 In recent years, some studies revealed that PRMT5 can methylate motifs containing GRG, RGG, or RG sequences. 16 , 17 , 18 However, motifs that do not have the sequences, such as H3R2, H3R8, p53 R337, and BCL6 R305, are also methylated by PRMT5. 19 , 20 , 21
Here, we found a PRMT5‐mediated arginine methylation site of RORα. Although the site does not include the RG sequence, it is similar to the close vicinity of H3R8. Furthermore, our results provide another PTM to modulate RORα protein stability regulated by PRMT5 and E3 ligase ITCH interplay. PRMT5 induced methylation at arginine 37 residue of RORα, and subsequently, arginine methylation acted as a degradation signal for ITCH. Our study revealed that reactive oxygen species (ROS) generation induced dynamic changes in RORα and PRMT5 expression in liver cancer cells. Restoring RORα expression and activation suppressed tumor cell proliferation and transformation activities. Therefore, reinstituting RORα expression via ROS generation presents a new liver cancer treatment strategy.
2. MATERIALS AND METHODS
2.1. Cell culture
HepG2 cells were maintained in RPMI 1640 medium (WELGENE; LM 011–01) supplemented with 10% FBS (GenDEPOT; F0900‐050) and 1% PS (WELGENE; LS202‐02‐AC). HEK293T cells were maintained in Dulbecco's modified Eagle's medium (WELGENE; LM 001–05) supplemented with 10% FBS and 1% PS at 37°C in a humidified atmosphere of 5% CO2.
2.2. Supplemental experimental procedures
Other detailed experimental procedures are described in Supplemental information.Docx.
3. RESULTS
3.1. RORα inhibits oncogenic effects in liver cancer cells
RORα functions as a tumor suppressor in various cancers, such as prostate, colon, and breast cancers. 8 , 10 , 22 We analyzed the change in oncogenic effects in HepG2 cells following RORα introduction or RORα agonist treatment to define the function of RORα in liver cancer cell tumorigenesis. RORα overexpression significantly reduced invasion activity compared with control cells (Figure 1A). Treatment with the RORα agonist SR1078 also showed the same tendency to suppress metastatic potential in HepG2 cells (Figure 1B).
FIGURE 1.

RORα functions as a tumor suppressor in liver cancer cells. A, HepG2 cells transfected with FLAG‐mock or RORα were seeded per upper chambers for the invasion assays. After 48 h, invaded cells were stained and counted. B, For the invasion assays, HepG2 cells were seeded per upper chambers with DMSO or the SR1078 treatment. After 48 h, invaded cells were stained and counted. C, Cell proliferation assay using a manual cell count. HepG2 cells were plated into 12‐well plates, and treated with DMSO or 10 μM SR1078 at the indicated concentration for 3 d. DMSO was used as a drug control (CTL). D, Cell proliferation assay with Ki67 and quantitative data of Ki67‐positive cells. HepG2 cells treated with DMSO or 10 μM SR1078 at the indicated concentration for 48 h. Green, Ki‐67; blue, nuclear DNA (DAPI). Scale bar = 125 μm. E, Cell proliferation assay using a manual cell count. HepG2 cells were plated in 12‐well plates, and after 15 h, cells were transfected with si‐CTL or si‐RORα. Cells were measured for 2 d. si‐CTL was used as a siRNA control (CTL). F, HepG2 cells were plated in 96‐well plates, and after 1 d, cells were treated with SR1078 or SR3335 at the indicated concentrations for 5 d. Cell viability was measured via CCK8 assay. G, c‐Myc protein and mRNA levels in HepG2 cells after transfection with FLAG‐mock or RORα. β‐actin was used as the loading control. The mRNA levels were normalized to r18S expression. Statistical analysis was performed using Student's unpaired t test for comparisons between two groups and two‐way ANOVA with Šidák's post hoc tests for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are expressed as mean ± SEM
Next, we performed cell‐counting assays and Ki67 staining to examine whether RORα inhibited cancer cell growth. Consistently, we observed a delay in cell growth (Figure 1C) and a reduction in the proliferation marker Ki67 (Figure 1D) in SR1078‐treated HepG2 cells. On the contrary, RORα depletion using siRNA promoted cell proliferation and increased invasion ability (Figures 1E and S1A‐C). In addition, we investigated whether treatment with the RORα antagonist SR3335 or agonist SR1078 affected HepG2 cell viability. SR1078 treatment significantly decreased cell viability in a dose‐dependent manner. However, SR3335 did not alter cell growth activity (Figure 1F). These results showed that RORα reduced the proliferation and metastatic potential of liver cancer cells. As RORα suppresses cancer progression via canonical Wnt/β‐catenin signaling inhibition in colon cancer, 8 we examined whether β‐catenin activity mediated tumor‐suppressive function of RORα in liver cancer cells. Quantitative reverse transcription PCR (qRT‐PCR) and Western blotting analysis revealed that c‐Myc mRNA and protein levels were downregulated in RORα‐transfected HepG2 cells compared with those in control cells (Figure 1G). Collectively, our results demonstrate that RORα plays a crucial role as a tumor suppressor in liver cancer cells.
3.2. Oxidative damage–induced RORα stabilization is functional in liver cancer
We generated mice with a specific RORα allele deletion in the hepatocytes (RORαf/f; Alb‐Cre, RORα liver‐specific KO [LKO]) to confirm tumor‐suppressive function of RORα in liver cancer. WT (RORαf/f) and RORαLKO mice were intraperitoneally injected with diethylnitrosamine (DEN) at 2 weeks of age and subjected to tumorigenesis analysis (Figure 2A). The number of tumor formations and the largest tumor size among tumors in liver tissues were comparable between 8‐month‐old WT and RORαLKO mice (Figure 2B,C). Moreover, increased liver cancer proliferation was confirmed by the substantial increase in levels of PCNA in RORαLKO mice (Figure 2D). These results demonstrate that a reduction in RORα expression can critically contribute to promoting liver cancer progression, consistent with the finding that enhanced RORα activities regressed liver cancer cell proliferation (Figure 1C,D,F).
FIGURE 2.
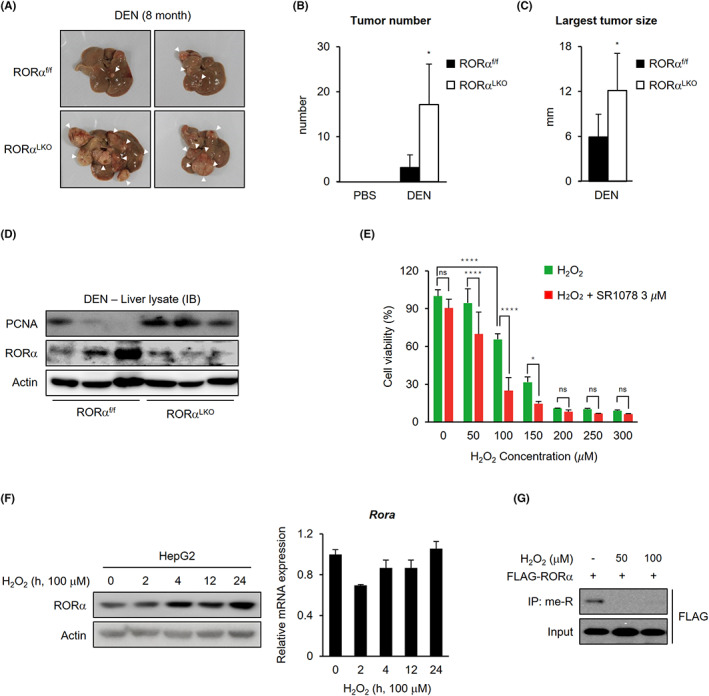
Enhanced tumor formation in liver‐specific RORα null mice treated with damage signals. A, Representative images of tumor‐bearing livers from 8‐m‐old WT and RORα LKO mice treated with DEN. B, C, Bar graph for tumor number (B) and largest tumor size (C) in 8‐m‐old DEN‐treated WT and RORα LKO mice liver tissues. D, Western blot images of tumor‐bearing livers from 8‐m‐old WT and RORα LKO mice treated with DEN. E, HepG2 cells were plated into 96‐well plates, and after 1 d, cells were treated with H2O2 only or H2O2 plus 3 μM SR1078 at the indicated concentrations for 5 d. Cell viability was measured by CCK8 assay. F, RORα protein and mRNA levels in HepG2 cells after 100 μM H2O2 treatment for the indicated lengths of time. The protein and mRNA levels were normalized to β‐actin expression. G, Arginine methylated FLAG‐RORα levels in 293 T cells after 0, 50, and 100 μM H2O2 treatment for 6 h. Statistical analysis was performed using Student's unpaired t test for comparisons between two groups and two‐way ANOVA with Šidák's post hoc tests for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are expressed as mean ± SEM
Given that RORα protein levels are responsible for damage‐induced liver cancer progression, it is reasonable to expect that physiologically relevant functions underlie the correlation between damage and RORα protein levels. In liver cancer, ROS‐induced oxidative damage may act as an antiproliferative and proapoptotic signal 23 and therefore may be used as an anticancer reagent. 24 , 25 We performed a cell viability analysis after treatment with a RORα agonist (Figure 2E) to determine whether RORα activity alters ROS‐triggered reduction in cell proliferation. The result supported that changed activities of RORα are significant downstream of the action of ROS in liver cancer. Furthermore, H2O2 treatment of HepG2 liver cancer cells stimulated the stabilization of RORα at the protein level (Figure 2F). Under these conditions, however, the mRNA levels of RORα were not affected, suggesting that ROS signaling influences protein stability of RORα. Therefore, we aimed to find out the molecular mechanism of RORα stabilization induced by oxidative stress. H2O2 treatment inhibited arginine methylation of RORα (Figure 2G). In addition, although RORα1 and RORα4 are coexpressed in HepG2 cells (Figure S2B), 26 specific arginine methylation of RORα1 was confirmed and diminished in response to ROS (Figures 2G and S2C). These results might describe that the difference in the NTD between RORα1 and RORα4 caused RORα1‐selective methylation (Figure S2A). Together, these data indicated that controlling RORα protein levels via ROS‐mediated oxidative damage signals is critical in liver cancer cells.
3.3. RORα is arginine methylated at R37 residue by PRMT5, and PRMT5 attenuates RORα activity via destabilization
Next, we examined the functional consequences of changes in RORα at the post‐translational level and the subsequent control of its protein expression. Protein arginine and lysine residues are commonly methylated in eukaryotic cells. 27 Arginine methylation is an influential PTM that occurs on nonhistone and histone proteins, affecting their interactions, such as protein‐protein and protein–nucleic acid interactions. 28 In mammals, nine enzymes, PRMTs 1‐9, promote arginine methylation. 29 RORα lysine methylation is known for the methylation‐dependent ubiquitination machinery of “methyl degron.” EZH2‐mediated RORα K38 methylation facilitates polyubiquitination by the E3 ubiquitin ligase complex DCAF1/DDB1/CUL4. 12 However, the functions and mechanisms of arginine methylation in RORα remain unclear. Interestingly, conserved arginine and surrounding residues comparable to those of histone H3 regulated by PRMTs were present in NTD of RORα (Figure 3A). PRMT5 mediates H3R8 methylation, leading to transcriptional repression, and PRMT4 (known as CARM1) mediates H3R17/26 methylation, thereby promoting gene expression. 30 We hypothesized that PRMT5 and PRMT4 might methylate R37 and R46/55 of RORα, respectively, because the arginine residues were arranged in a similar sequence to R8 and R17/26 of H3, respectively. We primarily investigated which PRMTs methylate RORα. Our data revealed that PRMT5 overexpression elevated arginine methylation of RORα, but other PRMTs, such as PRMT1‐4, did not (Figure 3B). Therefore, we focused on the relationship between PRMT5 and RORα. PRMT5 interacts with RORα and subsequently methylates it. In contrast, PRMT5 E444Q, an enzymatically dead mutant, 31 was bound to RORα but did not methylate it (Figures 3C and S3A). In addition, the aberrant methylation of RORα induced by PRMT5 overexpression occurred only in a specific isoform RORα1 (Figure S3B). Next, we analyzed the methylation of RORα using the PRMT5 inhibitor GSK3326595. When we treated GSK3326595, RORα methylation level was significantly reduced compared with the control (Figures 3D and S3C). Next, we constructed RORα arginine‐to‐alanine (R‐to‐A) or arginine‐to‐lysine (R‐to‐K) substitution mutants. Wild‐type RORα was arginine methylated, but R37A and R37K mutants were not (Figure 3E), and RORα R37K or R37A overexpression significantly increased RORα target gene p21 reporter activity (Figures 3F and S3D). 32
FIGURE 3.
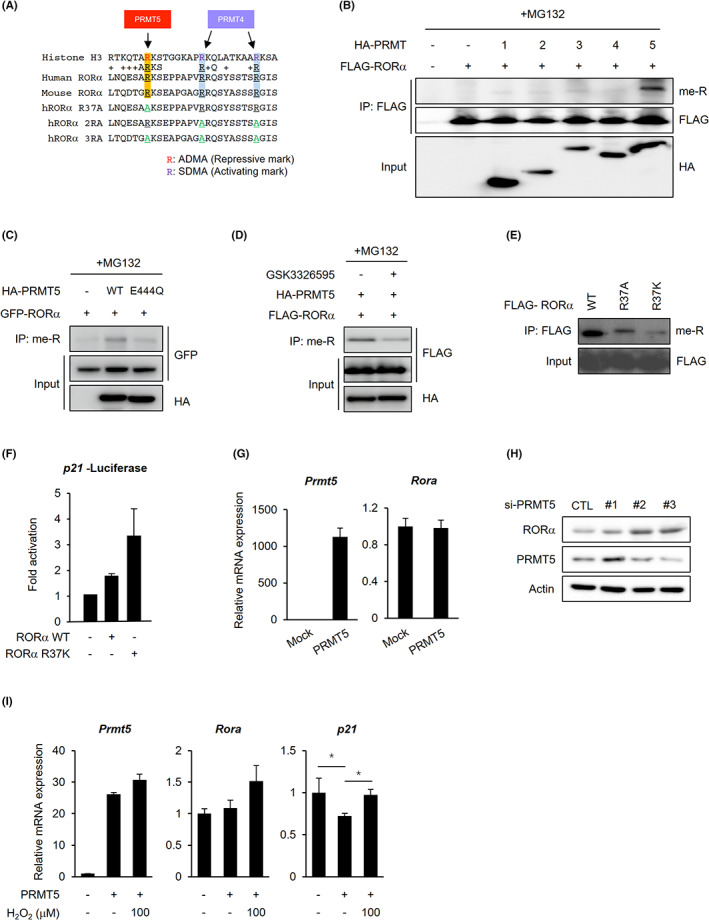
PRMT5 induces RORα degradation via methylating the R37 residue of RORα. A, Comparison between predicted RORα arginine methylation sites and PRMTs‐mediated methylation sites of histone H3. Arginine (R) to alanine (A) substitutions are highlighted in green. B, Arginine methylated FLAG‐RORα levels in 293 T cells after cotransfection with HA‐mock or PRMT1‐5. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 4 h. C, Arginine methylated GFP‐RORα levels in 293 T cells after cotransfection with HA‐mock, PRMT5 WT, or E444Q. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 4 h. D, Arginine methylated FLAG‐RORα levels in 293 T cells after cotransfection with HA‐PRMT5 in the absence or presence of GSK3326595 (5 μM, 24 h). Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 6 h. E, Arginine methylated FLAG‐RORα levels in 293 T cells after transfection with RORα WT, R37A, or R37K. F, Luciferase assay using a p21 promoter in 293 T cells transfected with RORα WT or R37K. G, qRT‐PCR analysis for relative Rora mRNA levels in 293 T cells transfected with HA‐mock or HA‐PRMT5. The mRNA levels were normalized to β‐actin expression. H, RORα and PRMT5 protein levels in HepG2 cells after transfection with control siRNA (si‐CTL), si‐PRMT5. β‐actin was used as a loading control. I, qRT‐PCR analysis for relative Rora and p21 mRNA levels in HepG2 cells transfected with HA‐mock or HA‐PRMT5. The cells were incubated with or without 100 μM H2O2 treatment for 6 h. The mRNA levels were normalized to β‐actin expression. Statistical analysis was performed using two‐way ANOVA with Tukey's post hoc tests. *p < 0.05. Data are expressed as mean ± SEM
We tested the methylation levels of RORα mutants (R37A, 2RA, and 3RA) after PRMT4 or PRMT5 overexpression to confirm whether PRMT4 methylated RORα R46/55 residues. Although the co‐immunoprecipitation (co‐IP) assay revealed that PRMT4 interacted with RORα (Figure S4A), PRMT4 overexpression did not induce RORα WT, 2RA (R46/55A), or 3RA (R37/46/55A) methylation (Figures 3B and S4B). These results suggest that enhanced binding to RORα via induced expression of PRMT4 could not trigger direct arginine methylation by PRMT4. PRMT5 catalyzed the WT or 2RA methylation, but the 3RA methylation did not rise, even after PRMT5 overexpression. As arginine methylation disappeared when R37 was altered to alanine, R37 might be a crucial residue for arginine methylation. PRMT4 did not seem to methylate RORα directly but might indirectly interact with RORα in a CREB‐binding protein (CBP)‐dependent manner (Figure S4C). CBP is a PRMT4 coactivator and can be activated as a histone acetyltransferase (HAT) by PRMT4‐mediated arginine methylation. 33 , 34 CBP and PRMT4 coexpression synergistically increased RORα‐dependent transcriptional activation of the RORE‐luciferase reporter and RORα target gene transcript levels (Figure S4D,E).
Protein methyltransferases are responsible for numerous regulatory pathways, such as cancer development, progression, and therapeutic response. 19 In recent years, accumulating evidence suggested that PRMT5, as an oncogene, is overexpressed and promotes tumor cell proliferation, invasion, and migration in several cancers, including hepatocellular carcinoma. 35 , 36 PRMT5 regulates gene expression via histone and transcription factor methylation. PRMT5‐mediated arginine methylation affects transcription factor activity, recruitment, and stability. 19 Therefore, we checked how PRMT5 regulates transcription factor RORα through arginine methylation. PRMT5 overexpression did not regulate RORα expression at the mRNA level (Figure 3G), but PRMT5 knockdown increased RORα protein levels (Figure 3H). p21 mRNA levels were downregulated in PRMT5‐overexpressed HepG2 cells (Figure 3I). However, H2O2 treatment restored the p21 mRNA expression levels despite PRMT5 overexpression. These data suggest that PRMT5 might destabilize RORα protein by methylation of R37 residue. Arginine methylation by the PRMT family can regulate the ubiquitination of substrates by determining the interaction between the E3 ligase and substrates. 37 , 38 , 39 In particular, PRMT5 enhances dual specificity phosphatase 14 (DUSP14) and CRAF degradation 40 , 41 or, conversely, attenuates CFLARL and Krüppel‐like factor 4 (KLF4) dilapidation by regulating their interactions with the E3 ligase. 38 , 42 Previous studies have shown that PRMT5‐catalyzed arginine methylation is highly associated with the ubiquitination of substrates and is important for development and tumorigenicity. 43 Thus, we hypothesized that PRMT5 methylates RORα and induces its ubiquitination, resulting in tumorigenesis by reducing tumor suppressor RORα.
3.4. E3 ligase ITCH polyubiquitinates RORα via the K441 linkage
We attempted to identify a candidate E3 ligase responsible for degrading RORα in response to arginine methylation by PRMT5. We performed E3 ligase screening to find the candidate (Figure 4A). We detected green fluorescent signals when the N‐terminal RORα amino acid sequence, including R37 and biotin, bound to the E3 ligase. In particular, the fluorescence intensity was the highest when the sequence interacted with ITCH or CA150 WW2. Therefore, we analyzed the potential E3 ligase roles of ITCH and CA150 in RORα ubiquitination. A co‐IP assay confirmed that ITCH and CA150 interacted with RORα (Figure 4B). However, the RORE‐luciferase reporter assay indicated that ITCH WT decreased RORα‐dependent RORE activity, and CA150 synergistically upregulated RORE activity together with RORα overexpression (Figure 4C). In contrast, ITCH WT, ITCH mutant (MT), and CA150 alone did not affect transcriptional activity without RORα overexpression. If ITCH or CA150 are E3 ligases that ubiquitinate RORα to degrade, they repress RORα target gene transcription. Furthermore, although the CA150 WW2 domain interacted with the RORα N‐terminal sequence during screening (Figure 4A), CA150 is a known transcription elongation factor rather than an E3 ligase. 44 WW domains are important modules that lead to protein‐protein binding. 45 The C2 and WW domains of Nedd4 family E3 ligases are crucial in interacting with adaptors and recognizing substrates. Therefore, the CA150 WW2 domain and RORα binding might only represent the significance of the WW domain in interaction with RORα as a substrate.
FIGURE 4.
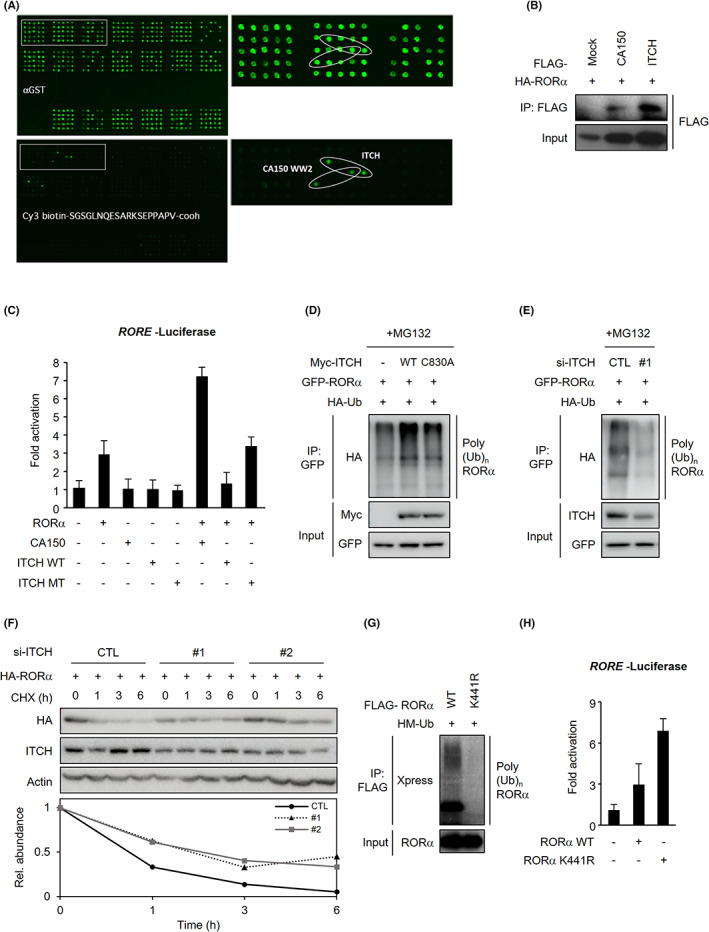
E3 ligase ITCH polyubiquitinates RORα. A, Putative E3 ligases of the RORα N‐terminal sequence (27‐45 aa) were screened by the E3 ligases array. B, Interactions of HA‐RORα with FLAG‐CA150 or ITCH in 293 T cells after cotransfection with the above vectors. C, Luciferase assay using RORE promoters in 293 T cells transfected with a vector, RORα, CA150, ITCH WT, or MT. D, Ubiquitination assay of GFP‐RORα in 293 T cells after cotransfection with HA‐Ub and Myc‐mock, ITCH WT, or C830A. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 4 h. E, Ubiquitination assay of GFP‐RORα in 293 T cells after cotransfection with HA‐Ub and si‐CTL, or si‐ITCH. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 4 h. F, Protein levels of ITCH and HA‐RORα in HepG2 cells after cotransfection with si‐CTL, si‐ITCH. Cells were treated with 20 μg/ml protein synthesis inhibitor CHX for 0, 1, 3, and 6 h. β‐actin was used as a loading control. G, Ubiquitination assay of FLAG‐RORα in 293 T cells after cotransfection with RORα WT or K441R and HM‐Ub. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 4 h. H, Luciferase assay using RORE promoters in 293 T cells transfected with mock, RORα WT, or K441R
We observed that ITCH WT polyubiquitinated RORα (Figure 4D). However, the catalytically dead mutant ITCH C830A failed to promote RORα polyubiquitination. Similarly, silencing ITCH via siRNA markedly reduced RORα polyubiquitination (Figure 4E) and facilitated RORα stabilization when measuring protein stability after treatment with the protein synthesis inhibitor cycloheximide (CHX) (Figure 4F). Thus, RORα ubiquitination requires ITCH. We conducted a ubiquitination assay after substituting putative ubiquitination sites to examine which RORα residue is ubiquitinated by ITCH. Notably, replacing the RORα lysine 441 residue with arginine almost completely abolished RORα polyubiquitination (Figure 4G). Also, the K441‐to‐R substitution mutant enhanced RORα transcriptional activity (Figure 4H). Collectively, our findings demonstrate that ITCH‐mediated RORα ubiquitination triggers its degradation in a K441‐dependent manner.
3.5. PRMT5 is involved in RORα degradation by regulating ITCH binding
We investigated whether PRMT5 affects RORα and ITCH interactions by methylating RORα. PRMT5‐mediated arginine methylation upregulated the RORα and ITCH interaction (Figure 5A). Conversely, PRMT5 E444Q overexpression attenuated this interaction. Additionally, PRMT5 WT overexpression, not E444Q, increased RORα ubiquitination and decreased RORα stability (Figure 5B,C). Moreover, the methylation‐deficient RORα R37A mutation significantly prevented its own ubiquitination (Figure 5D) and stabilized itself at the protein level (Figure 5E). Our results suggested that PRMT5 facilitates RORα ubiquitination by methylating the R37 residue. The results were reconfirmed by treating with PRMT5 inhibitor GSK3326595 that augmented the RORα stability (Figure 5F). These observations revealed crosstalk between PRMT5‐specific methylation and ubiquitination to regulate RORα stability.
FIGURE 5.

PRMT5 Is involved in RORα degradation by regulating ITCH binding. A, Interactions of GFP‐RORα with FLAG‐ITCH in 293 T cells after cotransfection with HA‐PRMT5 WT or E444Q. B, Protein levels of FLAG‐RORα in HepG2 cells after cotransfection with HA‐mock, PRMT5 WT, or E444Q. Cells were treated with 20 μg/ml CHX for 0, 1, 3, and 6 h. β‐actin was used as a loading control. C, Ubiquitination assay of GFP‐RORα in 293 T cells after cotransfection with HM‐Ub and HA‐mock, PRMT5 WT, or E444Q. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 6 h. D, Ubiquitination assay of FLAG‐RORα in 293 T cells after cotransfection with HA‐Ub and RORα WT or R37A. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 6 h. E, Protein levels of FLAG‐RORα in HepG2 cells transfected with RORα WT or R37A. Cells were treated with 20 μg/ml CHX for 0, 3, 6, and 12 h. β‐actin was used as a loading control. F, Protein levels of FLAG‐RORα in HepG2 cells transfected in the absence or presence of GSK3326595 (5 μM, 24 h). Cells were treated with 20 μg/ml CHX for 0, 1, 3, and 6 h. β‐actin was used as a loading control. G, Schematic of ITCH WT and deletion mutants. H, Interaction of GFP‐RORα with FLAG‐ITCH truncations in 293 T cells after cotransfection with the above vectors. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 6 h. I, Interaction of GFP‐RORα WT or R37A with FLAG‐ITCH WT or WW1‐4 in 293 T cells transfected with the above vectors. Cells were treated with 5 μg/ml proteasomal inhibitor MG132 for 6 h
Next, we determined which ITCH domains were responsible for binding with RORα. The E3 ligase ITCH contains a C2 domain, four WW domains, and a HECT domain. Nedd4 family E3s, including ITCH, bind to E2 and transfer ubiquitin from E2 to substrates through the HECT domain. The N‐terminal C2 and four WW domains are involved in subcellular localization and substrate recognition. 46 We generated isolated ITCH domains tagged with a FLAG epitope (Figure 5G). Intriguingly, the co‐IP assay showed that RORα strongly interacted with the C2 or WW1‐4 domains compared with full‐length ITCH (Figure 5H). However, the HECT domain did not bind to RORα. Next, we examined RORα methylation–dependent recognition by ITCH within the WW domain. WW domains are known as substrate‐binding domains, and the association between the WW 1‐4 domains of ITCH and RORα appeared to be methylation‐dependent, as only ROR WT was able to interact with ITCH, while RORα R37A exhibited a significantly reduced interaction (Figure 5I). Together, these data suggest that PRMT5‐dependent RORα arginine methylation is crucial for its direct association with the E3 ligase ITCH.
3.6. ROS facilitates RORα stabilization through PRMT5 inhibition in liver cancer cells
Our results showed that H2O2 treatment elevated protein levels of RORα. However, there were no significant changes in Rora mRNA levels (Figure 2F). We explored the molecular mechanism of H2O2‐induced RORα regulation. We performed Western blotting to detect RORα, PRMT5, and ITCH in HepG2 cells treated with H2O2 to evaluate the influence of H2O2 stimulation on expression of PRMT5 and ITCH accelerating RORα degradation. Notably, RORα was negatively correlated with PRMT5 and ITCH levels after exposure to oxidative stress. RORα protein levels increased in response to H2O2 treatment (Figure 6A), and p21 expression also increased (Figure 6B). H2O2 treatment remarkably downregulated PRMT5 expression, but ITCH slowly reduced in a time‐dependent manner. H2O2 might control PRMT5 and ITCH at the protein level because these mRNA levels did not change (Figure 6A,B). As PRMT5 is responsible for RORα degradation, it is assumed that there must be an upstream modulation regulating PRMT5 enzymatic activity. As Figure 6C,D shows, ROS signaling dramatically increased PRMT5 polyubiquitination and abolished the ability of PRMT5 to methylate RORα. These observations suggested that oxidative damage reduced PRMT5 protein levels and activity. Finally, we verified the effect of ITCH and H2O2‐induced oxidative damage on RORα target genes (Figure 6E). While ITCH overexpression decreased RORα target gene p21, ROS generation by H2O2 enhanced p21 mRNA levels even in ITCH‐overexpressing HepG2 cells. This might imply that ROS interrupts ITCH‐involved degradation of RORα and upregulates RORα target genes. Together, these data strongly support that the inverse correlation between PRMT5 and RORα protein levels under ROS generation is conferred by methylation‐dependent ubiquitination in liver cancer (Figure 6F).
FIGURE 6.
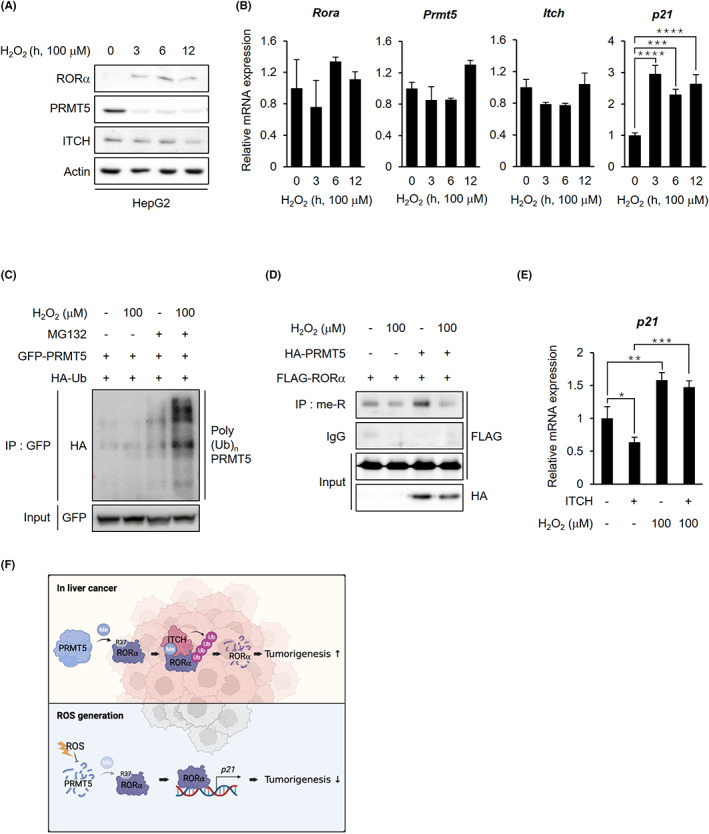
ROS facilitates RORα stabilization through PRMT5 inhibition in liver cancer cells. A, Endogenous protein levels of RORα, PRMT5, and ITCH in HepG2 cells after H2O2 treatment. Cells were treated with 100 μM H2O2 for the indicated lengths of time. β‐actin was used as a loading control. B, qRT‐PCR analysis for relative Rora, Prmt5, Itch, and p21 mRNA levels in HepG2 cells treated with 100 μM H2O2 for 0, 3, 6, and 12 h. The mRNA levels were normalized to GAPDH expression. C, Ubiquitination assay of GFP‐PRMT5 in 293 T cells cotransfected with HA‐Ub after H2O2 treatment. Cells were incubated with or without 100 μM H2O2 and 5 μg/ml proteasomal inhibitor MG132 for 6 h. D, Arginine methylated FLAG‐RORα levels in 293 T cells after cotransfection with HA‐mock or PRMT5 WT. Cells were incubated with or without 100 μM H2O2 for 6 h. E, qRT‐PCR analysis for relative p21 mRNA levels in HepG2 cells after transfection with FLAG‐mock or ITCH. Cells were treated with or without 100 μM H2O2 for 6 h. The mRNA levels were normalized to HPRT expression. F, The schematic diagram of the mechanism of arginine methylation‐mediated RORα degradation. In liver cancer, PRMT5‐catalyzed arginine methylation of RORα induces RORα ubiquitination by facilitating RORα and E3 ligase ITCH interactions. When the cells were exposed to excessive ROS, the cells inhibited PRMT5 from dissociating ITCH from RORα, resulting in RORα stabilization. Statistical analysis was performed using one‐way ANOVA and two‐way ANOVA with Tukey's post hoc tests. *p < 0.05. Data are expressed as mean ± SEM
4. DISCUSSION
Previous reports have frequently linked RORα to anti‐tumorigenesis, and it is generally considered a tumor suppressor, with only a few exceptions. 8 , 10 , 22 , 47 In this study, we determined that RORα functions as a tumor suppressor under ROS signaling in liver cancer. Interestingly, this is the first report to describe the critical role of RORα arginine methylation by PRMT5 in liver cancer and the series of molecular events involved in this process. We identified PRMT5 as a direct RORα arginine methyltransferase that dimethylates R37 residue. It destabilizes RORα by enhancing the binding of RORα with ITCH, leading to ubiquitination at K441 residue. This, in turn, promotes liver cancer cell tumorigenesis. Therefore, we have unraveled a critical novel function of the PRMT5‐ITCH‐RORα axis in liver tumorigenesis.
PRMTs are consistently upregulated in various cancers. 48 However, the downstream PRMT5 methylation events in cancer progression, especially via nonhistone substrates, remain poorly understood. We confirmed that RORα is a direct nonhistone substrate of PRMT5 and identified R37 as an arginine methylation site. Notably, recent studies indicate that the function of PRMT5 is complicated and context‐dependent in cancer progression, as it operates as both a tumor suppressor and oncogene. 19 Several studies have shown the oncogenic activities of PRMT5 in liver cancer. 36 , 49 , 50 However, the molecular basis of its activity as a methyltransferase remains largely unexplored. Our report demonstrates that ROS signaling downregulates PRMT5 protein levels and is negatively correlated with protein levels of the tumor suppressor RORα, suggesting an oncogenic role of PRMT5 in liver cancer.
Protein ubiquitination is a highly controlled process, 51 and we have provided novel evidence that RORα arginine methylation induces its ubiquitination. Additionally, the molecular level of arginine‐methylated RORα potentiates its association with the E3 ligase ITCH, leading to RORα polyubiquitination. Previous studies revealed that ITCH is crucial in tumor progression by destabilizing several target substrates, such as large tumor suppressor 1 (LATS1), p63, and p73. 52 , 53 , 54 In line with these findings, our study illustrated that inducing the association of ITCH with its substrate via other PTMs allows arginine methylation to stimulate liver cancer cell carcinogenesis.
Recently, regulating redox homeostasis by controlling ROS generation in anticancer therapies has received significant attention. 55 , 56 , 57 The acceleration of accumulated ROS disturbs redox homeostasis, resulting in severe damage to cancer cells. 58 Increasing ROS under H2O2 treatment reduces liver cancer cell proliferation depending on the functions of RORα, suggesting that regulating RORα protein levels is the underlying crucial molecular basis of ROS signaling effects.
In conclusion, we have demonstrated a series of molecular events in which PRMT5 dimethylated and degraded RORα by promoting ITCH recruitment by providing a direct link between arginine methylation and polyubiquitination, which led to the progression of liver cancer cells. Collectively, involvement of the PRMT5‐ITCH‐RORα axis under ROS signaling in liver cancer cell carcinogenesis and stimulation of this axis by inhibitors weakened HepG2 cell migration and invasion abilities. This molecular basis may provide fundamental knowledge to develop a potential therapeutic strategy for liver cancer intervention by controlling the members of this axis according to the pro‐oxidants for the ROS‐inducing approach.
ACKNOWLEDGEMENT
CMV10‐RORα and p21 luciferase constructs were kindly gifted by Sung Hee Baek from Seoul National University.
FUNDING INFORMATION
This work was supported by the Basic Science Research Program (NRF‐2021R1C1C1008780 to J.M.L. and NRF‐2020R1C1C1010489 to H.K.) from the National Research Foundation (NRF) grant funded by the Korea government.
DISCLOSURE
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: N/A.
Informed Consent: N/A.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: N/A.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Supporting information S1.
Im H, Baek H‐j, Yang E, et al. ROS inhibits RORα degradation by decreasing its arginine methylation in liver cancer. Cancer Sci. 2023;114:187‐200. doi: 10.1111/cas.15595
Hyuntae Im and Hee‐ji Baek contributed equally to this work.
[Correction added on 09 November 2022, after first online publication: The text “Hyuntae Im and Hee‐ji Baek contributed equally to this work” has been added in this version.]
Contributor Information
Hyunkyung Kim, Email: hyunkkim@korea.ac.kr.
Ji Min Lee, Email: jimin.lee@kaist.ac.kr.
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 2. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841‐850. [DOI] [PubMed] [Google Scholar]
- 3. Giguere V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20:689‐725. [DOI] [PubMed] [Google Scholar]
- 4. Park SC, Park IG, Kim H, Lee JM. N‐terminal domain mediated regulation of RORalpha1 inhibits invasive growth in prostate cancer. Int J Mol Sci. 2019;20:1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lau P, Fitzsimmons RL, Raichur S, Wang SC, Lechtken A, Muscat GE. The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: staggerer (SG/SG) mice are resistant to diet‐induced obesity. J Biol Chem. 2008;283:18411‐18421. [DOI] [PubMed] [Google Scholar]
- 6. Oh SK, Kim D, Kim K, et al. RORalpha is crucial for attenuated inflammatory response to maintain intestinal homeostasis. Proc Natl Acad Sci U S A. 2019;116:21140‐21149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim H, Lee JM, Lee G, et al. DNA damage‐induced RORalpha is crucial for p53 stabilization and increased apoptosis. Mol Cell. 2011;44:797‐810. [DOI] [PubMed] [Google Scholar]
- 8. Lee JM, Kim IS, Kim H, et al. RORalpha attenuates Wnt/beta‐catenin signaling by PKCalpha‐dependent phosphorylation in colon cancer. Mol Cell. 2010;37:183‐195. [DOI] [PubMed] [Google Scholar]
- 9. Moretti RM, Montagnani Marelli M, Motta M, Limonta P. Role of the orphan nuclear receptor ROR alpha in the control of the metastatic behavior of androgen‐independent prostate cancer cells. Oncol Rep. 2002;9:1139‐1143. [PubMed] [Google Scholar]
- 10. Xiong G, Wang C, Evers BM, Zhou BP, Xu R. RORalpha suppresses breast tumor invasion by inducing SEMA3F expression. Cancer Res. 2012;72:1728‐1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Platero JS, Hartnett T, Eissenberg JC. Functional analysis of the chromo domain of HP1. EMBO J. 1995;14:3977‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee JM, Lee JS, Kim H, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell. 2012;48:572‐586. [DOI] [PubMed] [Google Scholar]
- 13. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37‐50. [DOI] [PubMed] [Google Scholar]
- 14. Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263‐272. [DOI] [PubMed] [Google Scholar]
- 15. Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72:2041‐2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Musiani D, Bok J, Massignani E, et al. Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci Signal. 2019;12:eaat8388. [DOI] [PubMed] [Google Scholar]
- 17. Fong JY, Pignata L, Goy PA, et al. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell. 2019;36:194.e9‐209.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Branscombe TL, Frankel A, Lee JH, et al. PRMT5 (Janus kinase‐binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem. 2001;276:32971‐32976. [DOI] [PubMed] [Google Scholar]
- 19. Kim H, Ronai ZA. PRMT5 function and targeting in cancer. Cell Stress. 2020;4:199‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jansson M, Durant ST, Cho EC, et al. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431‐1439. [DOI] [PubMed] [Google Scholar]
- 21. Lu X, Fernando TM, Lossos C, et al. PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood. 2018;132:2026‐2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moretti RM, Montagnani Marelli M, Sala A, Motta M, Limonta P. Activation of the orphan nuclear receptor RORalpha counteracts the proliferative effect of fatty acids on prostate cancer cells: crucial role of 5‐lipoxygenase. Int J Cancer. 2004;112:87‐93. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Q, Cui C, Chen C‐Q, et al. Anti‐proliferative and pro‐apoptotic activities of Alpinia oxyphylla on HepG2 cells through ROS‐mediated signaling pathway. J Ethnopharmacol. 2015;169:99‐108. [DOI] [PubMed] [Google Scholar]
- 24. Dewaele M, Maes H, Agostinis P. ROS‐mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 2010;6:838‐854. [DOI] [PubMed] [Google Scholar]
- 25. Regmi S, Fung TS, Lim S, Luo KQ. Fluidic shear stress increases the anti‐cancer effects of ROS‐generating drugs in circulating tumor cells. Breast Cancer Res Treat. 2018;172:297‐312. [DOI] [PubMed] [Google Scholar]
- 26. Chauvet C, Bois‐Joyeux B, Danan JL. Retinoic acid receptor‐related orphan receptor (ROR) alpha4 is the predominant isoform of the nuclear receptor RORalpha in the liver and is up‐regulated by hypoxia in HepG2 human hepatoma cells. Biochem J. 2002;364:449‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emery‐Corbin SJ, Hamey JJ, Ansell BRE, et al. Eukaryote‐conserved methylarginine is absent in diplomonads and functionally compensated in giardia. Mol Biol Evol. 2020;37:3525‐3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu J, Richard S. Cellular pathways influenced by protein arginine methylation: implications for cancer. Mol Cell. 2021;81:4357‐4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qin J, Xu J. Arginine methylation in the epithelial‐to‐mesenchymal transition. FEBS J. 2021. 10.1111/febs.16152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fulton MD, Cao M, Ho MC, Zhao X, Zheng YG. The macromolecular complexes of histones affect protein arginine methyltransferase activities. J Biol Chem. 2021;297:101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma D, Yang M, Wang Q, et al. Arginine methyltransferase PRMT5 negatively regulates cGAS‐mediated antiviral immune response. Sci Adv. 2021;7:eabc1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schrader M, Danielsson C, Wiesenberg I, Carlberg C. Identification of natural monomeric response elements of the nuclear receptor RZR/ROR. They also bind COUP‐TF homodimers. J Biol Chem. 1996;271:19732‐19736. [DOI] [PubMed] [Google Scholar]
- 33. Streubel G, Bouchard C, Berberich H, et al. PRMT4 is a novel coactivator of c‐Myb‐dependent transcription in haematopoietic cell lines. PLoS Genet. 2013;9:e1003343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ceschin DG, Walia M, Wenk SS, et al. Methylation specifies distinct estrogen‐induced binding site repertoires of CBP to chromatin. Genes Dev. 2011;25:1132‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao W, Chen X, Liu L, Shu Y, Zhang M, Zhong Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed Pharmacother. 2019;114:108790. [DOI] [PubMed] [Google Scholar]
- 36. Zhang B, Dong S, Li Z, et al. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta‐catenin. J Transl Med. 2015;13:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Tran NT, Su H, et al. Cross‐talk between PRMT1‐mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife. 2015;4:e07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li M, An W, Xu L, Lin Y, Su L, Liu X. The arginine methyltransferase PRMT5 and PRMT1 distinctly regulate the degradation of anti‐apoptotic protein CFLARL in human lung cancer cells. J Exp Clin Cancer Res. 2019;38:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi S, Jung CR, Kim JY, Im DS. PRMT3 inhibits ubiquitination of ribosomal protein S2 and together forms an active enzyme complex. Biochim Biophys Acta. 2008;1780:1062‐1069. [DOI] [PubMed] [Google Scholar]
- 40. Yang CY, Chiu LL, Chang CC, Chuang HC, Tan TH. Induction of DUSP14 ubiquitination by PRMT5‐mediated arginine methylation. FASEB J. 2018;32:fj201800244RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andreu‐Perez P, Esteve‐Puig R, de Torre‐Minguela C, et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci Signal. 2011;4:ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu D, Gur M, Zhou Z, et al. Interplay between arginine methylation and ubiquitylation regulates KLF4‐mediated genome stability and carcinogenesis. Nat Commun. 2015;6:8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sengupta S, West KO, Sanghvi S, et al. PRMT5 promotes symmetric Dimethylation of RNA processing proteins and modulates activated T cell alternative splicing and ca(2+)/NFAT signaling. Immunohorizons. 2021;5:884‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goldstrohm AC, Albrecht TR, Sune C, Bedford MT, Garcia‐Blanco MA. The transcription elongation factor CA150 interacts with RNA polymerase II and the pre‐mRNA splicing factor SF1. Mol Cell Biol. 2001;21:7617‐7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang SS, Hsu LJ, Chang NS. Functional role of WW domain‐containing proteins in tumor biology and diseases: insight into the role in ubiquitin‐proteasome system. FASEB Bioadv. 2020;2:234‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao W, Shan Z, Gu A, Fu M, Shi Z, Wen W. WW domain‐mediated regulation and activation of E3 ubiquitin ligase suppressor of Deltex. J Biol Chem. 2018;293:16697‐16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song H, Chu JW, Park SC, et al. Isoform‐specific lysine methylation of RORalpha2 by SETD7 is required for association of the TIP60 coactivator complex in prostate cancer progression. Int J Mol Sci. 2020;21:1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jarrold J, Davies CC. PRMTs and arginine methylation: Cancer's best‐kept secret? Trends Mol Med. 2019;25:993‐1009. [DOI] [PubMed] [Google Scholar]
- 49. Jiang H, Zhu Y, Zhou Z, et al. PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 2018;7:869‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jeon JY, Lee JS, Park ER, et al. Protein arginine methyltransferase 5 is implicated in the aggressiveness of human hepatocellular carcinoma and controls the invasive activity of cancer cells. Oncol Rep. 2018;40:536‐544. [DOI] [PubMed] [Google Scholar]
- 51. Guo HJ, Tadi P. Biochemistry, Ubiquitination. StatPearls; 2021. [PubMed] [Google Scholar]
- 52. Ho KC, Zhou Z, She YM, Chun A, Cyr TD, Yang X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability. Proc Natl Acad Sci U S A. 2011;108:4870‐4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rossi M, De Laurenzi V, Munarriz E, et al. The ubiquitin‐protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rossi M, Aqeilan RI, Neale M, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci U S A. 2006;103:12753‐12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang H, Wang H, De Ridder M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018;438:154‐164. [DOI] [PubMed] [Google Scholar]
- 56. Dong L, Gopalan V, Holland O, Neuzil J. Mitocans revisited: mitochondrial targeting as efficient anti‐cancer therapy. Int J Mol Sci. 2020;21:7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weng Q, Sun H, Fang C, et al. Catalytic activity tunable ceria nanoparticles prevent chemotherapy‐induced acute kidney injury without interference with chemotherapeutics. Nat Commun. 2021;12:1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim SJ, Kim HS, Seo YR. Understanding of ROS‐inducing strategy in anticancer therapy. Oxid Med Cell Longev. 2019;2019:5381692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Supporting information S1.