

Abstract
Epilepsy is an umbrella term used to define a wide variety of seizure disorders and sudden unexpected death in epilepsy (SUDEP) is the leading cause of death in epilepsy. Although some SUDEP risk factors have been identified, it remains largely unpredictable, and underlying mechanisms remain poorly understood. Most seizures start in the cortex, but the high mortality rate associated with certain types of epilepsy indicates brainstem involvement. Therefore, to help understand SUDEP we discuss mechanisms by which seizure activity propagates to the brainstem. Specifically, we highlight clinical and pre-clinical evidence suggesting how seizure activation of: (i) descending inhibitory drive or (ii) spreading depolarization might contribute to brainstem dysfunction. Furthermore, since epilepsy is a highly heterogenous disorder, we also considered factors expected to favor or oppose mechanisms of seizure propagation. We also consider whether epilepsy-associated genetic variants directly impact brainstem function. Because respiratory failure is a leading cause of SUDEP, our discussion of brainstem dysfunction focuses on respiratory control.
Keywords: cortical-brainstem connectivity, seizure propagation, spreading depolarization (SD), SUDEP (sudden unexpected death in epilepsy), apnea
Introduction
Epilepsy is a chronic disease associated with uncontrolled brain activity that results in recurrent seizures. Approximately 50 million people globally have epilepsy and people with this condition have a two-three-fold higher mortality rate than the general public. Sudden unexpected death in epilepsy (SUDEP)- defined as death in people with epilepsy that are not caused by injury, drowning, or other known reasons- is a leading cause of death in epilepsy patients (Pathak et al., 2022) and is second only to stroke in years of potential life lost to neurological disease, thus making SUDEP a significant public health problem (Thurman et al., 2014). Despite their potential lethality, most seizures are not fatal, and so a frequent question posed by family members of SUDEP victims is “what was it about that [final] seizure that resulted in death”1? Considering seizures typically originate in the cortex and lethality involves disruption of autonomic (Thijs et al., 2021) and respiratory (Teran et al., 2022) function at the level of the brainstem, to address this question, we discuss two likely mechanisms by which cortical seizure activity propagates to the brainstem. We also consider whether the expression of epilepsy-associated genes in the brainstem contributes to epilepsy-associated cardiorespiratory dysfunction (Figure 1). It is our contention that SUDEP is a heterogenous process involving different mechanisms depending on the underlying cause of seizure activity. Highlighted here are what we consider the most likely mechanisms by which cortical seizure activity might propagate to the brainstem; however, it is also important to recognize that other regions and polysynaptic pathways may contribute to descending seizure propagation.
Figure 1.

Putative basis of brainstem dysfunction in epilepsy. (A) Cortical-brainstem connectivity provides an anatomical basis for seizure propagation to the brainstem. Likewise, brainstem-cortical projections have been shown to synchronize cortical network activity and so may influence seizure propensity. (B) Spreading depolarization can propagate through contiguous tissue and if it reaches the brainstem will likely result in respiratory failure. (C) certain epilepsy-associated ion channel mutations are expressed in the brainstem where they directly impair respiratory function. Lower insets, cartoon coronal sections show the relative location of suprapontine regions that project to brainstem respiratory centers implicated in SUDEP (Harper et al., 1998; McKay et al., 2003; Rosin et al., 2006; Yang et al., 2020; Trevizan-Baú et al., 2021; Feinstein et al., 2022; Leitner et al., 2022). IC, insular cortex; LHA, lateral hypothalamus; HYP, hypothalamus; CeA, central amygdala nucleus; SC, superior colliculus; RN, red nucleus; KF, kölliker-fuse; PBN, parabrachial nuclei; raphe, medullary raphe nucleus; RTN, retrotrapezoid nucleus; NTS, nucleus of the solitary tract; pre-bötC, pre-bötzinger complex. Note, that the medullary raphe is part of the reticular activating system.
I. Descending Seizure Propagation Through Synaptic Connectivity
Essential components of the respiratory circuit are located in the brainstem and include inspiratory rhythmogenic neurons in the pre-bötzinger complex (pre-bötC; Smith et al., 1991), neurons in the retrotrapezoid nucleus (RTN; Mulkey et al., 2004) and medullary raphe (Richerson, 2004; Ray et al., 2011) that regulate breathing in response to changes in CO2/H+ (i.e., function as respiratory chemoreceptors), parabrachial neurons that modulate inspiratory-expiratory phase transitions and integrate chemoreceptor, visceral and arousal information between the forebrain and brainstem (Kaur and Saper, 2019), and respiratory motor neurons that serve as the final common output for the respiratory system (Fogarty et al., 2018). These respiratory centers also receive input from suprapontine regions including the insular cortex (McKay et al., 2003), hippocampus (Harper et al., 1998), and amygdala (Feinstein et al., 2022); in humans, stimulation of these regions results in cessation of breathing (Ochoa-Urrea et al., 2020), presumably to allow for voluntary and emotional control of ventilation (Bondarenko et al., 2014; Ashhad et al., 2022). Evidence also suggests communication between brainstem respiratory centers and suprapontine structures is bidirectional; ascending respiratory activity entrains cortical and limbic network oscillations that are thought to be important for emotion and memory consolidation (Herrero et al., 2018; Karalis and Sirota, 2022). Furthermore, brainstem projections from the reticular formation to the thalamus and cortex via the reticular activating system modulate sleep-wake transitions and arousal (Kovalzon, 2016). In the context of epilepsy, cortical-brainstem connections provide an anatomical substrate for cortical seizure propagation to the brainstem, and as such, have long been implicated in seizure-induced cardiorespiratory dysfunction (Frysinger and Harper, 1990).
The amygdala stands out as a hub of the so-called brainstem-homeostatic forebrain connectome (Edlow et al., 2016). This region is located in the temporal lobe and sends extensive inhibitory projections to brainstem respiratory centers where it is thought to regulate fear-related respiratory responses (Nardi et al., 2009; Feinstein et al., 2022), particularly to external perceived threats but not necessarily interoceptive threats (Feinstein et al., 2013; for review see Feinstein et al., 2022). The amygdala is also highly susceptible to seizure activity (Aroniadou-Anderjaska et al., 2008), and animal models (Totola et al., 2019), as well as clinical work from pediatric (Rhone et al., 2020) and adult (Dlouhy et al., 2015; Lacuey et al., 2017; Nobis et al., 2018) epilepsy patients, showed that stimulation or seizure activation could elicit apnea. Consistent with its lack of involvement in interoceptive threats (Feinstein et al., 2022), amygdala-evoked apneas did not occur in conjunction with dyspnea (Dlouhy et al., 2015). Curiously, amygdala-evoked apneas are dependent on attention and nasal breathing (Nobis et al., 2018). This is interesting because cortical respiratory rhythms also depend on nasal breathing (Zelano et al., 2016), suggesting there is a hierarchical organization to cortical-brainstem communication where conscious effort through attention or elicited by mouth breathing can override coordinated activity between regions. This also suggests interventions that facilitate mouth breathing might limit seizure-induced apnea.
It should be noted that the amygdala is composed of multiple sub-nuclei and only a subset of which contribute to the respiratory activity. For example, stimulation of the basolateral, basomedial, and central regions consistently resulted in apnea, whereas stimulation of the more lateral amygdala failed to affect breathing (Rhone et al., 2020). Therefore, not all amygdala seizures result in apnea (Park et al., 2020). It is also worth mentioning that amygdala stimulation elicited apnea even during sleep (Nobis et al., 2018) when SUDEP occurs most frequently (Nobili et al., 2011). By contrast, another putative SUDEP mechanism, namely spreading depolarization (SD), is less likely to be favored during sleep (see next section below). In any case, these mechanisms are not mutually exclusive, but rather may occur simultaneously and in a synergistic manner. For example, SD will result in high extracellular potassium ([K+]o) and this has been shown to facilitate excitatory more so than inhibitory synaptic transmission (Rasmussen et al., 2020), thus promoting synaptic seizure propagation. Likewise, excitatory synaptic transmission has been shown to facilitate SD in a seizure-related mouse model of familial hemiplegic migraine type-1 (Tottene et al., 2009). Furthermore, SUDEP-prone mouse lines showed cortical epileptic activity that correlated with abnormal brainstem electroencephalographic (EEG) oscillations and suppression of brainstem activity (Gu et al., 2022), suggesting cortical-brainstem connectivity facilitated SD propagation to the brainstem.
In addition to individual ictal events causing cardiorespiratory arrest as described above, it is also possible that repeated cortical seizures cause maladaptive changes to the forebrain and brainstem respiratory circuity that render the respiratory system vulnerable to failure. In this case, epilepsy patients are expected to exhibit background cardiorespiratory abnormalities. Indeed, interictal cardiorespiratory problems are common in epilepsy (Barot and Nei, 2019). Typical background autonomic and respiratory symptoms exhibited by Dravet syndrome patients and partly recapitulated in animal models of this disease include diminished heart rate variability, bradycardia, and hypoventilation with increased apnea (Delogu et al., 2011; Kim et al., 2018). The possibility the brainstem is disrupted in epilepsy is also supported by anatomical evidence showing patients with focal epilepsy have diminished brainstem volume (Mueller et al., 2018) including loss of both neurons (Patodia et al., 2021) and astrocytes (Patodia et al., 2019) in respiratory control centers. Evidence also suggests that seizure activity can alter synaptic connectivity between cortical and brainstem structures, and such circuit level changes may alter brainstem function and perpetuate excitotoxicity-related damage (Armada-Moreira et al., 2020). For example, amygdala neurons located ipsilateral to the temporal lobe seizure foci exhibited a stronger convergence of cardiorespiratory information compared to neurons on the contralateral side (Frysinger and Harper, 1990), thus indicating seizure activity enhanced excitatory coupling between cortical and brainstem respiratory areas. Nevertheless, the possibility that altered cortex-brainstem connectivity contributes to brainstem dysfunction or cell death remains unknown. From a bottom-up perspective, it is conceivable that enhanced respiratory-driven oscillations in cortical regions might increase neural network synchronization and seizure propensity.
It has also been speculated that seizure-induced amygdala-dependent apnea is independent of the underlying cause or type of epilepsy (Rhone et al., 2020). However, considering the central nucleus of the amygdala provides a primary output of this region to the brainstem (Feinstein et al., 2022) and since neurons in this region are mostly inhibitory and suppress breathing presumably by synaptic inhibition, it seems unlikely such a mechanism would be effective in forms of epilepsy associated with loss of inhibitory tone. Consistent with this, amygdala neurons that make monosynaptic projections to the parabrachial pneumotaxic center are hypoexcitable in a mouse model of Dravet syndrome (Yan et al., 2021). Also, inhibitory neurons in the amygdala appear prone to seizure-induced damage (Tuunanen et al., 1996), which conceivably will favor epileptogenesis in the amygdala and nearby hippocampus but is less likely to increase inhibitory bombardment of brainstem respiratory centers.
In sum, seizure activation of cortical to brainstem inhibitory projections can disrupt cardiorespiratory function and contribute to SUDEP (Figure 2). Repeated activation of cortical-brainstem circuits may also alter network connectivity in maladaptive ways that compromise cardiorespiratory control, favor seizure propagation to the brainstem or increase synchronized cortical activity to increase seizure propensity.
Figure 2.
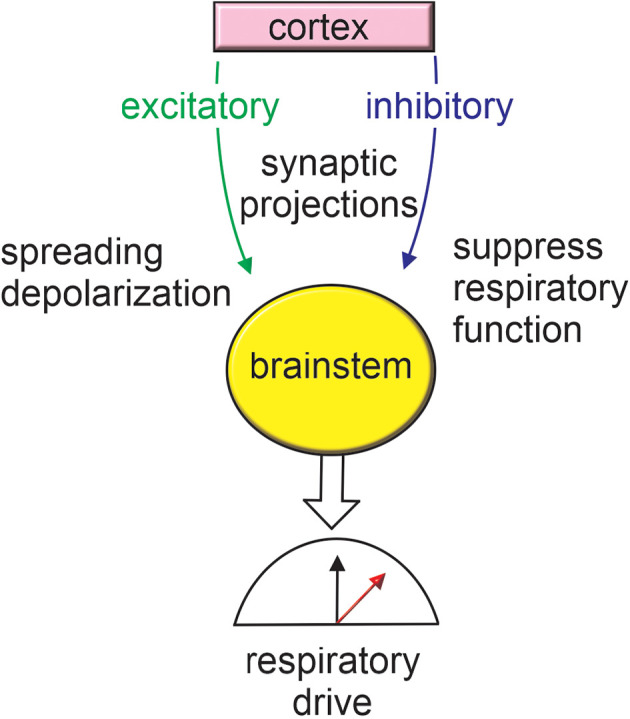
Cortex-brainstem connections provide a framework for seizure-induced respiratory failure. Seizure activation of inhibitory cortical projections to brainstem respiratory centers has been shown to result in respiratory arrest. Seizure activity may also stimulate excitatory brainstem projections thereby resulting in metabolic stress and favoring the propagation of spreading depolarization. For more information on anatomical regions refer to Figure 1.
Ii. Spreading Depolarization
SD is a pathological event associated with migraine headache, ischemic, or traumatic brain injury and epilepsy (for reviews see Pietrobon and Moskowitz, 2014; Cozzolino et al., 2018). It is thought to be triggered by a severe depolarization that leads to large-scale loss of ion and transmitter homeostasis. In particular, a pronounced increase in [K+]o and glutamate can initiate a self-propagating wave of depolarization and cytotoxic edema (Hinzman et al., 2016; Hubel et al., 2017). The ability of such a wave to propagate into and through contiguous tissue is strongly influenced by ongoing neural activity and metabolic status (Aiba and Noebels, 2015). SD is followed shortly thereafter by a wave of neural inactivation (presumably caused by depolarizing block) that results in depression of EEG activity as frequently observed following generalized tonic-clonic seizures (GTCS; Surges et al., 2011). In the cortex, this so-called spreading depression may serve a protective role by limiting further seizure activity (Tamim et al., 2021). However, if such an event were to occur in the brainstem it is expected to have a negative impact on cardiorespiratory control. Consistent with this, GTCS are the most common type of seizure associated with SUDEP (Ryvlin et al., 2019), and mechanisms underlying GTCS are thought to involve dysregulation of the ascending reticular activating system and descending reticulospinal projections to result in characteristic features of GTCS including loss of consciousness and muscle convulsions (Sedigh-Sarvestani et al., 2015).
In epilepsy patients it is unclear whether post-ictal EEG suppression is an independent risk factor SUDEP (Ryvlin et al., 2019); however, pre-clinical experiments using monogenic SUDEP models showed that brainstem SD correlated with cardiorespiratory failure and mortality. For example, cortical-evoked seizures in two SUDEP mouse models (Kv1.1 null and Scn1aR1407X/+ loss of function) resulted in brainstem SD and cardiorespiratory arrest (Aiba and Noebels, 2015). Similar results were also observed in mice expressing a Cav2.1 gain of function mutation associated with familial hemiplegic migraine type 1 (Cacna1aS218L/+; Jansen et al., 2019) and mice expressing a ryanodine receptor-2 gain of function mutation associated with catecholaminergic polymorphic ventricular tachycardia (Aiba et al., 2016). Also, in Cacna1aS218L/+ mice the superior colliculus, a midbrain structure that receives input from both the cortex and brainstem, was particularly effective at propagating seizure-induced SD to the brainstem (Cain et al., 2022), suggesting anatomical connectivity may facilitate SD.
Interestingly, expression of certain epilepsy-associated channel variants in the cortex but not the brainstem can cause cortical seizures and SD but in the absence of increased mortality. For example, Kcnq2 channels produce a subthreshold K+ conductance (Abbott, 2020) and loss of Kcnq2 function is associated with neonatal epileptic encephalopathy (Orhan et al., 2014; Kim H. J. et al., 2021). Conditional deletion of Kcnq2 from forebrain excitatory neurons (Emx1Cre/+::Kcnq2f/f; Kcnq2 cKO) resulted in cortical seizures and SD. Despite this, only a small subset of these animals died prematurely (Aiba and Noebels, 2021). However, a caveat to these experiments is that Emx1cre/+ is not restricted to cortical excitatory neurons but rather is also expressed by peripheral autonomic ganglia that provide modulatory feedback to cardiorespiratory centers (Ning et al., 2022). Excluding potential confounding effects of peripheral nerves, these results suggest compromised brainstem function is required for cortical seizure- or SD-induced respiratory arrest and premature death (see Section “III. Direct effect of epilepsy associated mutations on brainstem function” below for more detail).
The initiation phase of SD is dependent on the concentration of ions and transmitters in the extracellular space which are themselves inversely related to extracellular volume (ECV). Also, ECV increases during sleep (Ding et al., 2016), therefore, we speculate the threshold for SD induction will be higher during sleep when SUDEP is thought to occur most frequently (Nobili et al., 2011). Consistent with this, spontaneous cortical SD in Kcnq2 cKO mice (Aiba and Noebels, 2021), as well as seizure-induced death in Kv1.1 null mice (Moore et al., 2014) and Scn1aR1407X/+ mice (Teran et al., 2019), occurred primarily during the dark/active state when ECV is expected to be minimal and the impact of [K+]o on neural activity is most favored (Ding et al., 2016). Therefore, these results are consistent with the involvement of high [K+]o as a key determinant of SD threshold and propagation. However, these results are not consistent with clinical evidence suggesting SUDEP occurs primarily during sleep (Buchanan et al., 2021). Note that once SD has been initiated the corresponding cellular edema is expected to negate this issue; thus, SD propagation is not expected to be sleep-wake state dependent.
Seizure events can deplete energy availability, thus limiting ion and transmitter buffer capacity and lowering SD induction threshold (Major et al., 2020). Consistent with this, Kv1.1 null and Scn1aR1407X/+ models showed a low threshold for SD elicited by metabolic stress. Furthermore, repeated seizures may facilitate pathological remodeling that can lead to progressively more severe outcomes. For example, mice that express the Scn1a loss of function variant R1648H exhibit a mild/asymptomatic phenotype under resting conditions that can be transformed into a severe phenotype by subjecting the mice to heat- or chemoconvulsant-induced seizures (Dutton et al., 2017; Salgueiro-Pereira et al., 2019). This study also showed that wild type mice subjected to the same seizure induction protocol did not develop a severe seizure phenotype. These results suggest loss of Scn1a function lowered the seizure threshold, as expected, and is required for remodeling following repeated seizures that can lead to severe phenotypes and SUDEP. However, contrary to this, early work with chemoconvulsant models of epilepsy suggests frequent seizures can confer resistance to SD. For example, a pentylenetetrazol rat model of epilepsy showed that frequent seizures increased the SD threshold (Koroleva et al., 1993). Furthermore, patients with chronic epilepsy and pilocarpine-treated rats exhibited a similar high [K+]o threshold for SD (Maslarova et al., 2011). Based on this, it was speculated that chronic seizures promote a compensatory increase in [K+]o buffering capacity.
A critical function of astrocytes is to regulate extracellular ion and transmitter homeostasis and as such are important determinants of [K+]o buffering. The dynamics of [K+]o are complex and depend on several factors. Here, we focus on inward rectifying Kir4.1 channels because these are the main determinant of astrocyte resting membrane potential (by K+ efflux) and can serve as a conduit for K+ uptake when the reversal potential for K+ is depolarized to resting membrane potential as can occur during increased neural activity. Glutamate uptake by astrocytes is also an electrogenic process favored at more negative membrane potentials (Grewer and Rauen, 2005). Astrocytes are also highly sensitive to seizure activity and in chronic epilepsy these cells are known to proliferate (gliosis) and transition into a pro-inflammatory state (so-called reactive astrocyte) characterized by the release of cytokines and growth factors that can increase seizure propensity or promote tissue repair (for review see Wetherington et al., 2008; Verhoog et al., 2020). Although there is some evidence suggesting Kir4.1 expression increased in a pilocarpine model of temporal lobe epilepsy (Nagao et al., 2013) and a mouse model of Dravet syndrome (Miljanovic et al., 2021), most studies suggest the opposite, that astrocyte Kir4.1 channel expression is diminished in epilepsy (Kinboshi et al., 2020; Ohno et al., 2021). Indeed, loss of function variations (missense and nonsense mutations) in KCNJ10 (the gene encoding Kir4.1) causes an epileptic disorder known as EAST/SeSAME syndrome (Bockenhauer et al., 2009) characterized by early onset tonic-clonic seizures, sensorineural deafness, ataxia, intellectual disability, and electrolyte imbalance. Also, astrocyte Kir4.1 expression or function has been shown to be reduced in humans (Heuser et al., 2012; Steinhauser et al., 2012; Kitaura et al., 2018) and various animal models of epilepsy (Harada et al., 2013) including DBA/2 model of audiogenic seizures (Inyushin et al., 2010). Note that Kir4.1 channels also contribute to K+ buffering by oligodendrocytes, and loss of oligodendrocyte Kir4.1 channels also increases seizure activity (Larson et al., 2018). Also, loss of serotonergic signaling by raphe neurons contributed to seizure propensity and respiratory arrest in DBA/2 mice (Cervo et al., 2005). This is interesting because Kir4.1 channels can heteromerize with Kir5.1 to form a CO2/H+ sensitive conductance (Xu et al., 2000), and recent work showed that both Kir4.1 and Kir5.1 transcript are expressed by medullary serotonergic raphe neurons and so may contribute to CO2/H+ detection by these putative chemoreceptors (Puissant et al., 2017). Moreover, loss of Kir5.1 (Kcnj16 gene) resulted in an audiogenic seizure phenotype with increased mortality in a rat model of salt-sensitive hypertension and chronic kidney disease (Manis et al., 2021), probably by a mechanism involving disruption of heteromeric Kir4.1/5.1 channels since Kir5.1 does not form functional homomeric channels (Pessia et al., 1996). Therefore, disruption of homo or heteromeric Kir4.1 channels may be a common substrate for breathing problems and seizure propensity.
As expected, astrocyte-specific deletion of Kcnj10 also disrupted K+ and glutamate uptake and resulted in increased seizure activity and premature death (Djukic et al., 2007). Kir4.1 channels also colocalize with aquaporin-4 (Aqp4) water channels (Nagelhus et al., 2004) and Aqp4 knockout mice exhibited slowed [K+]o clearance (Amiry-Moghaddam et al., 2003) and longer duration seizures following neural stimulation (Binder et al., 2006). Therefore, disruption of Kir4.1 may impact Aqp4 function and consequently regulation of cell size and ECV. This may be important because the ability of astrocytes to influence neural activity by paracrine signaling or regulation of extracellular ions and transmitters is proximity-dependent and inversely related to ECV (Murphy et al., 2017).
It is also possible that the loss of Kir4.1 containing channels will facilitate the release of various neuroactive signaling molecules from astrocytes. In particular, inhibition of Kir4.1 chanenls in cultured astrocytes increased expression of brain-derived neurotrophic factor (BDNF; Kinboshi et al., 2017), a growth factor that signals through TrkB receptors to regulate neural growth, differentiation, and synaptic plasticity (Cowley et al., 1994; Meakin et al., 1999; Huang and Reichardt, 2003). This is of interest because BDNF is a potent modulator of epileptogenesis; BDNF expression reportedly increased in the brains of epileptic patients and animal models of epilepsy (Jankowsky and Patterson, 2001), whereas disruption of BDNF/TrkB signaling suppressed seizure activity in epileptic mouse models (Kokaia et al., 1995; Hagihara et al., 2005; Liu et al., 2013). Although the link between the loss of Kir4.1 and increased BDNF expression remains murky, pharmacological evidence implicates activation of the MAPK/ERK pathway (Kinboshi et al., 2017), possibly in response to the depolarization-induced increase in intracellular Ca2+. Together, these results suggest seizure-induced changes in astrocyte Kir4.1 expression is maladaptive and likely to contribute to epileptogenesis.
It is also important to recognize that increased Kir4.1 expression will not necessarily diminish seizure propensity. For example, KCNJ10 gain of function variants that result in increased channel expression (p.R18Q), diminish proton-dependent inhibition (p.R348H) or increased channel conductance (p.V84M) are also associated with seizure-like behavior (Sicca et al., 2011, 2016). Mechanistically, it is hard to imagine how increased Kir4.1 channel function in astrocytes might promote seizures. One possibility is that increased Kir4.1 channel activity may increase [K+]o buffering kinetics, thereby limiting [K+]o build-up during increased activity. This mechanism may minimize depolarization-induced Na+ channel inactivation and allow neurons to fire at higher frequencies for longer periods of time (Niday and Tzingounis, 2018). Note that increased Kir4.1 expression is not expected to substantially decrease [K+]o because a prerequisite for K+ uptake by astrocytes is high [K+]o and a depolarized K+ reversal potential relative to resting membrane potential. As such, decreasing [K+]o will favor K+ efflux.
Another interesting mechanism by which increased Kir4.1 might favor seizure activity involves dysregulation of brain pH. Astrocytes express high levels of the electrogenic sodium bicarbonate cotransporter (NBC; Turovsky et al., 2016). The most common NBC isoform expressed by astrocytes has 1 Na+: 2 stoichiometry and a predicted reversal potential of around −100 mV (Mulkey and Wenker, 2011). This value is negative to astrocyte resting membrane potential, thus under normal conditions flux through the NBC is directed inward (Mulkey and Wenker, 2011). If this is the case, then increased expression of Kir4.1 is expected to hyperpolarize astrocyte membrane potential and decrease electrogenic transport, thereby resulting in extracellular alkalosis. This is significant because just 0.2 pH unit increase in extracellular pH can cause seizures (Schuchmann et al., 2006).
In sum, there is no doubt that SD, once initiated, can have profound effects on neural activity, and preclinical studies clearly implicate SD as a cause of seizure-induced mortality. However, the correlation between postictal generalized EEG suppression (PGES), which presumably also reflects SD, and SUDEP is a matter of debate in the literature. Some studies suggest there is a correlation between PGES and SUDEP (Lhatoo et al., 2010; Moseley and DeGiorgio, 2015), whereas other studies found PGES duration is not a risk factor for SUDEP (Surges et al., 2011; Lamberts et al., 2013). Also, sleep-wake changes in ECV are not expected to favor the initiation of SD during sleep when SUDEP usually occurs. Furthermore, although chronic seizure activity may result in compensatory cellular responses to limit SD, such adaptations are not likely to involve increased astrocyte Kir4.1 expression since most evidence indicates loss of this channel in epilepsy. For this same reason, astrocyte Kir4.1 channels may have some therapeutic potential in treating epilepsy, possibly by limiting SD. Consistent with this, certain antiepileptic drugs have been shown to stimulate Kir4.1 expression (Mukai et al., 2018).
Iii. Direct Effect of Epilepsy Associated Mutations on Brainstem Function
In addition to promoting seizure activity in the cortex, epilepsy-associated genetic mutations may also be expressed in the brainstem (Kuo et al., 2019) where they increase SUDEP risk in seizure-dependent and -independent manners (Figure 3). For example, as noted above Emx1cre/+::Kcnq2f/f mice (Kcnq2 cKO) showed spontaneous cortical seizures with SD but they did not die prematurely (Aiba and Noebels, 2021), suggesting the brainstem was protected from SD infiltration. Another study used a similar approach (Emx1Cre/+) to express a dominant negative Kcnq2 variant (M547V) in forebrain pyramidal neurons (but with some off-target expression in astrocytes) of Kcnq2 heterozygous knockout mice (C57BL/6 background); unlike Kcnq2 cKO animals, these mice showed a severe phenotype including seizures and premature death (Kim E. C. et al., 2021). In this case, global Kcnq2 haploinsufficiency appears sufficient to allow cortical seizures to disrupt brainstem function and result in mortality.
Figure 3.
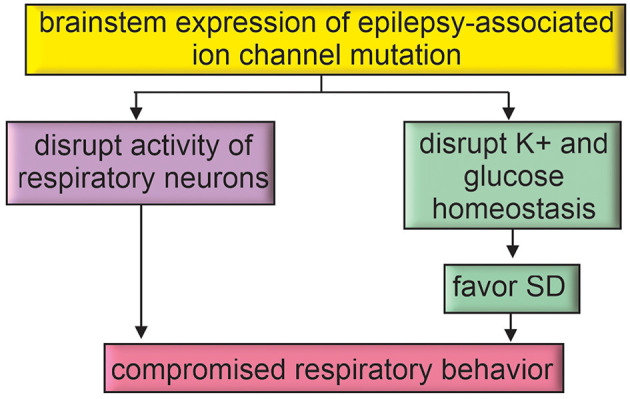
Epilepsy-associated genes may directly disrupt brainstem function. Brainstem expression of epilepsy-associated ion channel mutations may directly impact respiratory function or favor mechanisms for SD propagation to the brainstem.
The possibility that brainstem expression of epilepsy-associated mutations increases the risk of mortality is supported by evidence that SUDEP can occur without overt seizure activity (Lhatoo et al., 2016). Pre-clinical animal experiments also support this possibility. For example, polymorphisms associated with DBA/2 mice (a common model of audiogenic seizures; De Sarro et al., 2017) appear to disrupt brainstem serotonergic signaling and contribute to seizure-induced cardiorespiratory failure. Specifically, DBA/2 mice express a single amino acid substitution in the gene encoding tryptophan hydroxylase-2 that results in limited serotonin production (Cervo et al., 2005), and this likely contributes to seizure-induced respiratory arrest since the systemic application of serotonin reuptake inhibitors improved seizure activity and related apneic events in DBA/2 mice (Faingold et al., 2014). DBA/2 mice also express a Kcnj10 loss of function mutation that has been shown to disrupt Kir4.1-dependent maintenance of extracellular K+ and glutamate (Ferraro et al., 2004; Inyushin et al., 2010) and thus lower seizure threshold (Figure 2). It is also worth noting that Kir4.1 channels together with Kir5.1 may contribute to CO2/H+ chemosensation by serotonergic neurons (Puissant et al., 2017), thus loss of Kir4.1 could further compromise raphe chemoreception and worsen seizure-induced respiratory problems.
The retrotrapezoid nucleus (RTN) is another important respiratory chemoreceptor region implicated in SUDEP (Patodia et al., 2018). For example, in the context of Dravet syndrome (caused by loss of function mutations in SCN1A), we showed that Scn1a transcript is expressed by inhibitory parafacial neurons in the region of the RTN (Kuo et al., 2019). We also showed that inhibitory somatostatin (SST)-expressing neurons in the region of the RTN are inhibited by CO2/H+ and contribute to RTN chemoreception by disinhibition of CO2/H+-activated glutamatergic neurons (i.e., RTN chemoreceptors; Cleary et al., 2021). Therefore, in addition to causing cortical seizure activity, Dravet syndrome-associated Scn1a mutations may disrupt the inhibitory modulation of RTN chemoreception. Consistent with this, inhibitory neurons in the region of the RTN in slices from mice that express a loss of function Scn1a mutation (A1783V) conditionally in inhibitory neurons under the vesicular GABA transporter promoter (Slc32a1cre/+::Scn1aA1783V fl/+) showed lower basal activity compared to control cells and fired fewer action potentials in response to depolarizing current steps (Kuo et al., 2019). Consistent with a disinhibitory mechanism, chemosensitive RTN neurons in slices from Slc32a1cre/+::Scn1aA1783V fl/+ mice showed increased baseline activity and enhanced output in response to increases in CO2 (Kuo et al., 2019). However, at the whole animal level, VgatA1783V/+ mice showed reduced respiratory activity in room air and a blunted ventilatory response to CO2 (Kuo et al., 2019). This outcome is not entirely unexpected because inhibitory signaling in the RTN (Cregg et al., 2017) and at other levels of the respiratory circuit (Baertsch et al., 2018) can facilitate respiratory output. Also, Slc32a1cre/+::Scn1aA1783V fl/+ mice have spontaneous seizures which, for reasons mentioned above, may propagate to the brainstem and disrupt respiratory control in a seizure-dependent manner. This later possibility is an important consideration since deletion of Scn1a only from forebrain inhibitory neurons also resulted in seizures and premature death (Cheah et al., 2012), suggesting in this mouse model that cortical seizure activity can cause brainstem dysfunction and SUDEP. This contrasts with evidence from Kcnq2 cKO that as noted above exhibits cortical seizure activity that did not correlate with premature death (Aiba and Noebels, 2021). Both mouse models are maintained on a similar C56BL/6 background so the reason(s) for these divergent results remains unclear. That said, it is worth mentioning that disruption of Scn1a globally or conditionally only in forebrain inhibitory neurons caused sleep fragmentation with less non-rapid eye movement (NREM) sleep and more frequent waking episodes (Kalume et al., 2015). Although the relationship between sleep, sleep problems, and epilepsy have long been appreciated (Diaz-Negrillo, 2013; Wang et al., 2018), the basis for these associations is not clear. Based on evidence that regulation of the ECV is coupled to sleep-wake status (Ding et al., 2016) and decreased ECV positively correlates with neural activity (Walch et al., 2022), we speculate that disruption of sleep (as seen in Dravet syndrome; Kalume et al., 2015) will decrease ECV, lower seizure threshold and favor propagation of SD (Figure 2).
In sum, epilepsy-associated genes may be expressed by neurons or astrocytes in brainstem respiratory centers and so may contribute to background breathing problems that render the system vulnerable to failure. Altered neural activity or compromised astrocyte regulation of the extracellular milieu may also favor the propagation of SD into the brainstem.
Author Contributions
DM drafted manuscript, drafted figures, and approved the final version. BM edited manuscript and figures, and approved the final version. All authors contributed to the article and approved the submitted version.
Footnotes
1Partners Against Mortality in Epilepsy (PAME) hosts an annual meeting that brings together health care providers, basic scientists and families touched by epilepsy with the goal of improving our understanding of SUDEP. During previous PAME meetings we had the opportunity to talk with family members that lost a loved one to SUDEP. These interactions made a lasting impact on our perception of this disease and continue to motivate our work in this field.
Funding
This work was supported by the following National Institutes of Health Grants: HL104101 (DM), HL137094 (DM), and F31 NS120467 (BM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Abbott G. W. (2020). KCNQs: ligand- and voltage-gated potassium channels. Front. Physiol. 11:583. 10.3389/fphys.2020.00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba I., Noebels J. L. (2015). Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci. Transl. Med. 7:282ra246. 10.1126/scitranslmed.aaa4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba I., Noebels J. L. (2021). Kcnq2/Kv7.2 controls the threshold and bi-hemispheric symmetry of cortical spreading depolarization. Brain 144, 2863–2878. 10.1093/brain/awab141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba I., Wehrens X. H., Noebels J. L. (2016). Leaky RyR2 channels unleash a brainstem spreading depolarization mechanism of sudden cardiac death. Proc. Natl. Acad. Sci. U S A 113, E4895–4903. 10.1073/pnas.1605216113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M., Williamson A., Palomba M., Eid T., de Lanerolle N. C., Nagelhus E. A., et al. (2003). Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc. Natl. Acad. Sci. U S A 100, 13615–13620. 10.1073/pnas.2336064100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armada-Moreira A., Gomes J. I., Pina C. C., Savchak O. K., Goncalves-Ribeiro J., Rei N., et al. (2020). Going the extra (Synaptic) mile: excitotoxicity as the road toward neurodegenerative diseases. Front. Cell. Neurosci. 14:90. 10.3389/fncel.2020.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V., Fritsch B., Qashu F., Braga M. F. (2008). Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 78, 102–116. 10.1016/j.eplepsyres.2007.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashhad S., Kam K., Del Negro C. A., Feldman J. L. (2022). Breathing rhythm and pattern and their influence on emotion. Annu. Rev. Neurosci. 45, 223–247 10.1146/annurev-neuro-090121-014424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertsch N. A., Baertsch H. C., Ramirez J. M. (2018). The interdependence of excitation and inhibition for the control of dynamic breathing rhythms. Nat. Commun. 9:843. 10.1038/s41467-018-03223-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barot N., Nei M. (2019). Autonomic aspects of sudden unexpected death in epilepsy (SUDEP). Clin. Auton. Res. 29, 151–160. 10.1007/s10286-018-0576-1 [DOI] [PubMed] [Google Scholar]
- Binder D. K., Yao X., Zador Z., Sick T. J., Verkman A. S., Manley G. T., et al. (2006). Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53, 631–636. 10.1002/glia.20318 [DOI] [PubMed] [Google Scholar]
- Bockenhauer D., Feather S., Stanescu H. C., Bandulik S., Zdebik A. A., Reichold M., et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970. 10.1056/NEJMoa0810276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko E., Hodgson D. M., Nalivaiko E. (2014). Amygdala mediates respiratory responses to sudden arousing stimuli and to restraint stress in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 306, R951–R959. 10.1152/ajpregu.00528.2013 [DOI] [PubMed] [Google Scholar]
- Buchanan G. F., Gluckman B. J., Kalume F. K., Lhatoo S., Maganti R. K., Noebels J. L., et al. (2021). Proceedings of the sleep and epilepsy workshop: section 3 mortality: sleep night and SUDEP. Epilepsy Curr. 21:15357597211004556. 10.1177/15357597211004556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain S. M., Bernier L. P., Zhang Y., Yung A. C., Kass J., Bohnet B., et al. (2022). Hyperexcitable superior colliculus and fatal brainstem spreading depolarization in a model of sudden unexpected death in epilepsy. Brain Commun. 4:fcac006. 10.1093/braincomms/fcac006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervo L., Canetta A., Calcagno E., Burbassi S., Sacchetti G., Caccia S., et al. (2005). Genotype-dependent activity of tryptophan hydroxylase-2 determines the response to citalopram in a mouse model of depression. J. Neurosci. 25, 8165–8172. 10.1523/JNEUROSCI.1816-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah C. S., Yu F. H., Westenbroek R. E., Kalume F. K., Oakley J. C., Potter G. B., et al. (2012). Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U S A 109, 14646–14651. 10.1073/pnas.1211591109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary C. M., Milla B. M., Kuo F. S., James S., Flynn W. F., Robson P., et al. (2021). Somatostatin-expressing parafacial neurons are CO2/H+ sensitive and regulate baseline breathing. eLife 10:e60317. 10.7554/eLife.60317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley S., Paterson H., Kemp P., Marshall C. J. (1994). Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77, 841–852. 10.1016/0092-8674(94)90133-3 [DOI] [PubMed] [Google Scholar]
- Cozzolino O., Marchese M., Trovato F., Pracucci E., Ratto G. M., Buzzi M. G., et al. (2018). Understanding spreading depression from headache to sudden unexpected death. Front. Neurol. 9:19. 10.3389/fneur.2018.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg J. M., Chu K. A., Dick T. E., Landmesser L. T., Silver J. (2017). Phasic inhibition as a mechanism for generation of rapid respiratory rhythms. Proc. Natl. Acad. Sci. U S A 114, 12815–12820. 10.1073/pnas.1711536114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sarro G., Russo E., Citraro R., Meldrum B. S. (2017). Genetically epilepsy-prone rats (GEPRs) and DBA/2 mice: Two animal models of audiogenic reflex epilepsy for the evaluation of new generation AEDs. Epilepsy Behav. 71, 165–173. 10.1016/j.yebeh.2015.06.030 [DOI] [PubMed] [Google Scholar]
- Delogu A. B., Spinelli A., Battaglia D., Dravet C., De Nisco A., Saracino A., et al. (2011). Electrical and autonomic cardiac function in patients with dravet syndrome. Epilepsia 52, 55–58. 10.1111/j.1528-1167.2011.03003.x [DOI] [PubMed] [Google Scholar]
- Diaz-Negrillo A. (2013). Influence of sleep and sleep deprivation on ictal and interictal epileptiform activity. Epilepsy Res. Treat. 2013:492524. 10.1155/2013/492524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F., O’Donnell J., Xu Q., Kang N., Goldman N., Nedergaard M., et al. (2016). Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352, 550–555. 10.1126/science.aad4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B., Casper K. B., Philpot B. D., Chin L. S., McCarthy K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. 10.1523/JNEUROSCI.0723-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlouhy B. J., Gehlbach B. K., Kreple C. J., Kawasaki H., Oya H., Buzza C., et al. (2015). Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J. Neurosci. 35, 10281–10289. 10.1523/JNEUROSCI.0888-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton S. B. B., Dutt K., Papale L. A., Helmers S., Goldin A. L., Escayg A., et al. (2017). Early-life febrile seizures worsen adult phenotypes in Scn1a mutants. Exp. Neurol. 293, 159–171. 10.1016/j.expneurol.2017.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlow B. L., McNab J. A., Witzel T., Kinney H. C. (2016). The structural connectome of the human central homeostatic network. Brain Connect. 6, 187–200. 10.1089/brain.2015.0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold C. L., Kommajosyula S. P., Long X., Plath K., Randall M. (2014). Serotonin and sudden death: differential effects of serotonergic drugs on seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav. 37, 198–203. 10.1016/j.yebeh.2014.06.028 [DOI] [PubMed] [Google Scholar]
- Feinstein J. S., Buzza C., Hurlemann R., Follmer R. L., Dahdaleh N. S., Coryell W. H., et al. (2013). Fear and panic in humans with bilateral amygdala damage. Nat. Neurosci. 16, 270–272. 10.1038/nn.3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein J. S., Gould D., Khalsa S. S. (2022). Amygdala-driven apnea and the chemoreceptive origin of anxiety. Biol. Psychol. 170:108305. 10.1016/j.biopsycho.2022.108305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro T. N., Golden G. T., Smith G. G., Martin J. F., Lohoff F. W., Gieringer T. A., et al. (2004). Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm. Genome 15, 239–251. 10.1007/s00335-003-2270-3 [DOI] [PubMed] [Google Scholar]
- Fogarty M. J., Mantilla C. B., Sieck G. C. (2018). Breathing: motor control of diaphragm muscle. Physiology (Bethesda) 33, 113–126. 10.1152/physiol.00002.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frysinger R. C., Harper R. M. (1990). Cardiac and respiratory correlations with unit discharge in epileptic human temporal lobe. Epilepsia 31, 162–171. 10.1111/j.1528-1167.1990.tb06301.x [DOI] [PubMed] [Google Scholar]
- Grewer C., Rauen T. (2005). Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics and their impact on synaptic signaling. J. Membr. Biol. 203, 1–20. 10.1007/s00232-004-0731-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B., Levine N. G., Xu W., Lynch R. M., Pardo-Manuel de Villena F., Philpot B. D., et al. (2022). Ictal neural oscillatory alterations precede sudden unexpected death in epilepsy. Brain Commun. 4:fcac073. 10.1093/braincomms/fcac073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagihara H., Hara M., Tsunekawa K., Nakagawa Y., Sawada M., Nakano K., et al. (2005). Tonic-clonic seizures induce division of neuronal progenitor cells with concomitant changes in expression of neurotrophic factors in the brain of pilocarpine-treated mice. Brain Res. Mol. Brain Res. 139, 258–266. 10.1016/j.molbrainres.2005.05.031 [DOI] [PubMed] [Google Scholar]
- Harada Y., Nagao Y., Shimizu S., Serikawa T., Terada R., Fujimoto M., et al. (2013). Expressional analysis of inwardly rectifying Kir4.1 channels in Noda epileptic rat (NER). Brain Res. 1517, 141–149. 10.1016/j.brainres.2013.04.009 [DOI] [PubMed] [Google Scholar]
- Harper R. M., Poe G. R., Rector D. M., Kristensen M. P. (1998). Relationships between hippocampal activity and breathing patterns. Neurosci. Biobehav. Rev. 22, 233–236. 10.1016/s0149-7634(97)00010-9 [DOI] [PubMed] [Google Scholar]
- Herrero J. L., Khuvis S., Yeagle E., Cerf M., Mehta A. D. (2018). Breathing above the brain stem: volitional control and attentional modulation in humans. J. Neurophysiol. 119, 145–159. 10.1152/jn.00551.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser K., Eid T., Lauritzen F., Thoren A. E., Vindedal G. F., Tauboll E., et al. (2012). Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 71, 814–825. 10.1097/NEN.0b013e318267b5af [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman J. M., Wilson J. A., Mazzeo A. T., Bullock M. R., Hartings J. A. (2016). Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J. Neurotrauma 33, 1775–1783. 10.1089/neu.2015.4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E. J., Reichardt L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642. 10.1146/annurev.biochem.72.121801.161629 [DOI] [PubMed] [Google Scholar]
- Hubel N., Hosseini-Zare M. S., Ziburkus J., Ullah G. (2017). The role of glutamate in neuronal ion homeostasis: a case study of spreading depolarization. PLoS Comput. Biol. 13:e1005804. 10.1371/journal.pcbi.1005804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inyushin M., Kucheryavykh L. Y., Kucheryavykh Y. V., Nichols C. G., Buono R. J., Ferraro T. N., et al. (2010). Potassium channel activity and glutamate uptake are impaired in astrocytes of seizure-susceptible DBA/2 mice. Epilepsia 51, 1707–1713. 10.1111/j.1528-1167.2010.02592.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky J. L., Patterson P. H. (2001). The role of cytokines and growth factors in seizures and their sequelae. Prog. Neurobiol. 63, 125–149. 10.1016/s0301-0082(00)00022-8 [DOI] [PubMed] [Google Scholar]
- Jansen N. A., Schenke M., Voskuyl R. A., Thijs R. D., van den Maagdenberg A., Tolner E. A., et al. (2019). Apnea associated with brainstem seizures in Cacna1a (S218L) mice is caused by medullary spreading depolarization. J. Neurosci. 39, 9633–9644. 10.1523/JNEUROSCI.1713-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F., Oakley J. C., Westenbroek R. E., Gile J., de la Iglesia H. O., Scheuer T., et al. (2015). Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol. Dis. 77, 141–154. 10.1016/j.nbd.2015.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalis N., Sirota A. (2022). Breathing coordinates cortico-hippocampal dynamics in mice during offline states. Nat. Commun. 13:467. 10.1038/s41467-022-28090-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S., Saper C. B. (2019). Neural circuitry underlying waking up to hypercapnia. Front. Neurosci. 13:401. 10.3389/fnins.2019.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Bravo E., Thirnbeck C. K., Smith-Mellecker L. A., Kim S. H., Gehlbach B. K., et al. (2018). Severe peri-ictal respiratory dysfunction is common in dravet syndrome. J. Clin. Invest. 128, 1141–1153. 10.1172/JCI94999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J., Yang D., Kim S. H., Won D., Kim H. D., Lee J. S., et al. (2021). Clinical characteristics of KCNQ2 encephalopathy. Brain Dev. 43, 244–250. 10.1016/j.braindev.2020.08.015 [DOI] [PubMed] [Google Scholar]
- Kim E. C., Zhang J., Tang A. Y., Bolton E. C., Rhodes J. S., Christian-Hinman C. A., et al. (2021). Spontaneous seizure and memory loss in mice expressing an epileptic encephalopathy variant in the calmodulin-binding domain of Kv7.2. Proc. Natl. Acad. Sci. U S A 118:e2021265118. 10.1073/pnas.2021265118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinboshi M., Ikeda A., Ohno Y. (2020). Role of astrocytic inwardly rectifying potassium (Kir) 4.1 channels in epileptogenesis. Front. Neurol. 11:626658. 10.3389/fneur.2020.626658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinboshi M., Mukai T., Nagao Y., Matsuba Y., Tsuji Y., Tanaka S., et al. (2017). Inhibition of inwardly rectifying potassium (Kir) 4.1 channels facilitates brain-derived neurotrophic factor (BDNF) expression in astrocytes. Front. Mol. Neurosci. 10:408. 10.3389/fnmol.2017.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura H., Shirozu H., Masuda H., Fukuda M., Fujii Y., Kakita A., et al. (2018). Pathophysiological characteristics associated with epileptogenesis in human hippocampal sclerosis. EBioMedicine 29, 38–46. 10.1016/j.ebiom.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M., Ernfors P., Kokaia Z., Elmer E., Jaenisch R., Lindvall O., et al. (1995). Suppressed epileptogenesis in BDNF mutant mice. Exp. Neurol. 133, 215–224. 10.1006/exnr.1995.1024 [DOI] [PubMed] [Google Scholar]
- Koroleva V. I., Vinogradova L. V., Bures J. (1993). Reduced incidence of cortical spreading depression in the course of pentylenetetrazol kindling in rats. Brain Res. 608, 107–114. 10.1016/0006-8993(93)90780-q [DOI] [PubMed] [Google Scholar]
- Kovalzon V. M. (2016). Ascending reticular activating system of the brain. Transl. Neurosci. Clin. 2, 275–285. 10.18679/CN11-6030_R.2016.034 [DOI] [Google Scholar]
- Kuo F. S., Cleary C. M., LoTurco J. J., Chen X., Mulkey D. K. (2019). Disordered breathing in a mouse model of Dravet syndrome. eLife 8:e43387. 10.7554/eLife.43387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacuey N., Zonjy B., Londono L., Lhatoo S. D. (2017). Amygdala and hippocampus are symptomatogenic zones for central apneic seizures. Neurology 88, 701–705. 10.1212/WNL.0000000000003613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberts R. J., Gaitatzis A., Sander J. W., Elger C. E., Surges R., Thijs R. D., et al. (2013). Postictal generalized EEG suppression: an inconsistent finding in people with multiple seizures. Neurology 81, 1252–1256. 10.1212/WNL.0b013e3182a6cbeb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson V. A., Mironova Y., Vanderpool K. G., Waisman A., Rash J. E., Agarwal A., et al. (2018). Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. eLife 7:e34829. 10.7554/eLife.34829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner D. F., Kanshin E., Askenazi M., Faustin A., Friedman D., Devore S., et al. (2022). Raphe and ventrolateral medulla proteomics in epilepsy and sudden unexpected death in epilepsy. Brain Commun. 4:fcac186. 10.1093/braincomms/fcac186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhatoo S. D., Faulkner H. J., Dembny K., Trippick K., Johnson C., Bird J. M., et al. (2010). An electroclinical case-control study of sudden unexpected death in epilepsy. Ann. Neurol. 68, 787–796. 10.1002/ana.22101 [DOI] [PubMed] [Google Scholar]
- Lhatoo S. D., Nei M., Raghavan M., Sperling M., Zonjy B., Lacuey N., et al. (2016). Nonseizure SUDEP: sudden unexpected death in epilepsy without preceding epileptic seizures. Epilepsia 57, 1161–1168. 10.1111/epi.13419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Gu B., He X. P., Joshi R. B., Wackerle H. D., Rodriguiz R. M., et al. (2013). Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron 79, 31–38. 10.1016/j.neuron.2013.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major S., Huo S., Lemale C. L., Siebert E., Milakara D., Woitzik J., et al. (2020). Direct electrophysiological evidence that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura and a review of the spreading depolarization continuum of acute neuronal mass injury. Geroscience 42, 57–80. 10.1007/s11357-019-00142-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis A. D., Palygin O., Isaeva E., Levchenko V., LaViolette P. S., Pavlov T. S., et al. (2021). Kcnj16 knockout produces audiogenic seizures in the Dahl salt-sensitive rat. JCI Insight 6:e143251. 10.1172/jci.insight.143251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslarova A., Alam M., Reiffurth C., Lapilover E., Gorji A., Dreier J. P., et al. (2011). Chronically epileptic human and rat neocortex display a similar resistance against spreading depolarization in vitro. Stroke 42, 2917–2922. 10.1161/STROKEAHA.111.621581 [DOI] [PubMed] [Google Scholar]
- McKay L. C., Evans K. C., Frackowiak R. S., Corfield D. R. (2003). Neural correlates of voluntary breathing in humans. J. Appl. Physiol. (1985) 95, 1170–1178. 10.1152/japplphysiol.00641.2002 [DOI] [PubMed] [Google Scholar]
- Meakin S. O., MacDonald J. I., Gryz E. A., Kubu C. J., Verdi J. M. (1999). The signaling adapter FRS-2 competes with Shc for binding to the nerve growth factor receptor TrkA. A model for discriminating proliferation and differentiation. J. Biol. Chem. 274, 9861–9870. 10.1074/jbc.274.14.9861 [DOI] [PubMed] [Google Scholar]
- Miljanovic N., Hauck S. M., van Dijk R. M., Di Liberto V., Rezaei A., Potschka H., et al. (2021). Proteomic signature of the Dravet syndrome in the genetic Scn1a-A1783V mouse model. Neurobiol. Dis. 157:105423. 10.1016/j.nbd.2021.105423 [DOI] [PubMed] [Google Scholar]
- Moore B. M., Jerry Jou C., Tatalovic M., Kaufman E. S., Kline D. D., Kunze D. L., et al. (2014). The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP). Epilepsia 55, 1808–1816. 10.1111/epi.12793 [DOI] [PubMed] [Google Scholar]
- Moseley B. D., DeGiorgio C. M. (2015). The sudep risk inventory: association with postictal generalized eeg suppression. Epilepsy Res. 117, 82–84. 10.1016/j.eplepsyres.2015.09.006 [DOI] [PubMed] [Google Scholar]
- Mueller S. G., Nei M., Bateman L. M., Knowlton R., Laxer K. D., Friedman D., et al. (2018). Brainstem network disruption: a pathway to sudden unexplained death in epilepsy? Hum. Brain Mapp. 39, 4820–4830. 10.1002/hbm.24325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T., Kinboshi M., Nagao Y., Shimizu S., Ono A., Sakagami Y., et al. (2018). Antiepileptic drugs elevate astrocytic Kir4.1 expression in the rat limbic region. Front. Pharmacol. 9:845. 10.3389/fphar.2018.00845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey D. K., Stornetta R. L., Weston M. C., Simmons J. R., Parker A., Bayliss D. A., et al. (2004). Respiratory control by ventral surface chemoreceptor neurons in rats. Nat. Neurosci. 7, 1360–1369. 10.1038/nn1357 [DOI] [PubMed] [Google Scholar]
- Mulkey D. K., Wenker I. C. (2011). Astrocyte chemoreceptors: mechanisms of H+ sensing by astrocytes in the retrotrapezoid nucleus and their possible contribution to respiratory drive. Exp. Physiol. 96, 400–406. 10.1113/expphysiol.2010.053140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T. R., Binder D. K., Fiacco T. A. (2017). Turning down the volume: astrocyte volume change in the generation and termination of epileptic seizures. Neurobiol. Dis. 104, 24–32. 10.1016/j.nbd.2017.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao Y., Harada Y., Mukai T., Shimizu S., Okuda A., Fujimoto M., et al. (2013). Expressional analysis of the astrocytic Kir4.1 channel in a pilocarpine-induced temporal lobe epilepsy model. Front. Cell. Neurosci. 7:104. 10.3389/fncel.2013.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus E. A., Mathiisen T. M., Ottersen O. P. (2004). Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR41. Neuroscience 129, 905–913. 10.1016/j.neuroscience.2004.08.053 [DOI] [PubMed] [Google Scholar]
- Nardi A. E., Freire R. C., Zin W. A. (2009). Panic disorder and control of breathing. Respir. Physiol. Neurobiol. 167, 133–143. 10.1016/j.resp.2008.07.011 [DOI] [PubMed] [Google Scholar]
- Niday Z., Tzingounis A. V. (2018). Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist 24, 368–380. 10.1177/1073858418763752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Y., Noebels J. L., Aiba I. (2022). Emx1-Cre is expressed in peripheral autonomic ganglia that regulate central cardiorespiratory functions. eNeuro 9:ENEURO.0093-22.2022. 10.1523/ENEURO.0093-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobili L., Proserpio P., Rubboli G., Montano N., Didato G., Tassinari C. A., et al. (2011). Sudden unexpected death in epilepsy (SUDEP) and sleep. Sleep Med. Rev. 15, 237–246. 10.1016/j.smrv.2010.07.006 [DOI] [PubMed] [Google Scholar]
- Nobis W. P., Schuele S., Templer J. W., Zhou G., Lane G., Rosenow J. M., et al. (2018). Amygdala-stimulation-induced apnea is attention and nasal-breathing dependent. Ann. Neurol. 83, 460–471. 10.1002/ana.25178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa-Urrea M., Dayyani M., Sadeghirad B., Tandon N., Lacuey N., Lhatoo S. D., et al. (2020). Electrical stimulation-induced seizures and breathing dysfunction: a systematic review of new insights into the epileptogenic and symptomatogenic zones. Front. Hum. Neurosci. 14:617061. 10.3389/fnhum.2020.617061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y., Kunisawa N., Shimizu S. (2021). Emerging roles of astrocyte Kir4.1 channels in the pathogenesis and treatment of brain diseases. Int. J. Mol. Sci. 22:10236. 10.3390/ijms221910236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orhan G., Bock M., Schepers D., Ilina E. I., Reichel S. N., Loffler H., et al. (2014). Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394. 10.1002/ana.24080 [DOI] [PubMed] [Google Scholar]
- Park K., Kanth K., Bajwa S., Girgis F., Shahlaie K., Seyal M., et al. (2020). Seizure-related apneas have an inconsistent linkage to amygdala seizure spread. Epilepsia 61, 1253–1260. 10.1111/epi.16518 [DOI] [PubMed] [Google Scholar]
- Pathak S. J., Yousaf M. I. K., Shah V. B. (2022). Sudden Unexpected Death in Epilepsy. Treasure Island, FL: StatPearls. [PubMed] [Google Scholar]
- Patodia S., Paradiso B., Ellis M., Somani A., Sisodiya S. M., Devinsky O., et al. (2019). Characterisation of medullary astrocytic populations in respiratory nuclei and alterations in sudden unexpected death in epilepsy. Epilepsy Res. 157:106213. 10.1016/j.eplepsyres.2019.106213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patodia S., Somani A., O’Hare M., Venkateswaran R., Liu J., Michalak Z., et al. (2018). The ventrolateral medulla and medullary raphe in sudden unexpected death in epilepsy. Brain 141, 1719–1733. 10.1093/brain/awy078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patodia S., Somani A., Thom M. (2021). Review: neuropathology findings in autonomic brain regions in SUDEP and future research directions. Auton. Neurosci. 235:102862. 10.1016/j.autneu.2021.102862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia M., Tucker S. J., Lee K., Bond C. T., Adelman J. P. (1996). Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 15, 2980–2987. [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D., Moskowitz M. A. (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 15, 379–393. 10.1038/nrn3770 [DOI] [PubMed] [Google Scholar]
- Puissant M. M., Mouradian G. C., Jr., Liu P., Hodges M. R. (2017). Identifying candidate genes that underlie cellular pH sensitivity in serotonin neurons using transcriptomics: a potential role for Kir5.1 channels. Front. Cell. Neurosci. 11:34. 10.3389/fncel.2017.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen R., O’Donnell J., Ding F., Nedergaard M. (2020). Interstitial ions: a key regulator of state-dependent neural activity? Prog. Neurobiol. 193:101802. 10.1016/j.pneurobio.2020.101802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R. S., Corcoran A. E., Brust R. D., Kim J. C., Richerson G. B., Nattie E., et al. (2011). Impaired respiratory and body temperature control upon acute serotonergic neuron inhibition. Science 333, 637–642. 10.1126/science.1205295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhone A. E., Kovach C. K., Harmata G. I., Sullivan A. W., Tranel D., Ciliberto M. A., et al. (2020). A human amygdala site that inhibits respiration and elicits apnea in pediatric epilepsy. JCI Insight 5:e134852. 10.1172/jci.insight.134852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson G. B. (2004). Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat. Rev. Neurosci. 5, 449–461. 10.1038/nrn1409 [DOI] [PubMed] [Google Scholar]
- Rosin D. L., Chang D. A., Guyenet P. G. (2006). Afferent and efferent connections of the rat retrotrapezoid nucleus. J. Compar. Neurol. 499, 64–89 10.1002/cne.21105 [DOI] [PubMed] [Google Scholar]
- Ryvlin P., Rheims S., Lhatoo S. D. (2019). Risks and predictive biomarkers of sudden unexpected death in epilepsy patient. Curr. Opin. Neurol. 32, 205–212. 10.1097/WCO.0000000000000668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgueiro-Pereira A. R., Duprat F., Pousinha P. A., Loucif A., Douchamps V., Regondi C., et al. (2019). A two-hit story: Seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol. Dis. 125, 31–44. 10.1016/j.nbd.2019.01.006 [DOI] [PubMed] [Google Scholar]
- Schuchmann S., Schmitz D., Rivera C., Vanhatalo S., Salmen B., Mackie K., et al. (2006). Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nat. Med. 12, 817–823. 10.1038/nm1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedigh-Sarvestani M., Blumenfeld H., Loddenkemper T., Bateman L. M. (2015). Seizures and brain regulatory systems: consciousness, sleep and autonomic systems. J. Clin. Neurophys. 32, 188–193. 10.1097/WNP.0000000000000133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F., Ambrosini E., Marchese M., Sforna L., Servettini I., Valvo G., et al. (2016). Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci. Rep. 6:34325. 10.1038/srep34325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F., Imbrici P., D’Adamo M. C., Moro F., Bonatti F., Brovedani P., et al. (2011). Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol. Dis. 43, 239–247. 10.1016/j.nbd.2011.03.016 [DOI] [PubMed] [Google Scholar]
- Smith J. C., Ellenberger H. H., Ballanyi K., Richter D. W., Feldman J. L. (1991). Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science 254, 726–729. 10.1126/science.1683005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser C., Seifert G., Bedner P. (2012). Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60, 1192–1202. 10.1002/glia.22313 [DOI] [PubMed] [Google Scholar]
- Surges R., Strzelczyk A., Scott C. A., Walker M. C., Sander J. W. (2011). Postictal generalized electroencephalographic suppression is associated with generalized seizures. Epilepsy Behav. 21, 271–274. 10.1016/j.yebeh.2011.04.008 [DOI] [PubMed] [Google Scholar]
- Tamim I., Chung D. Y., de Morais A. L., Loonen I. C. M., Qin T., Misra A., et al. (2021). Spreading depression as an innate antiseizure mechanism. Nat. Commun. 12:2206. 10.1038/s41467-021-22464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teran F. A., Bravo E., Richerson G. B. (2022). Sudden unexpected death in epilepsy: respiratory mechanisms. Handb. Clin. Neurol. 189, 153–176. 10.1016/B978-0-323-91532-8.00012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teran F. A., Kim Y., Crotts M. S., Bravo E., Emaus K. J., Richerson G. B., et al. (2019). Time of day and a ketogenic diet influence susceptibility to SUDEP in Scn1aR1407X/+ mice. Front. Neurol. 10:278. 10.3389/fneur.2019.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs R. D., Ryvlin P., Surges R. (2021). Autonomic manifestations of epilepsy: emerging pathways to sudden death? Nat. Rev. 17, 774–788. 10.1038/s41582-021-00574-w [DOI] [PubMed] [Google Scholar]
- Thurman D. J., Hesdorffer D. C., French J. A. (2014). Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia 55, 1479–1485. 10.1111/epi.12666 [DOI] [PubMed] [Google Scholar]
- Totola L. T., Malheiros-Lima M. R., Delfino-Pereira P., Del Vecchio F., Souza F. C., Takakura A. C., et al. (2019). Amygdala rapid kindling impairs breathing in response to chemoreflex activation. Brain Res. 1718, 159–168. 10.1016/j.brainres.2019.05.015 [DOI] [PubMed] [Google Scholar]
- Tottene A., Conti R., Fabbro A., Vecchia D., Shapovalova M., Santello M., et al. (2009). Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 61, 762–773. 10.1016/j.neuron.2009.01.027 [DOI] [PubMed] [Google Scholar]
- Trevizan-Baú P., Dhingra R. R., Furuya W. I., Stanić D., Mazzone S. B., Dutschmann M., et al. (2021). Forebrain projection neurons target functionally diverse respiratory control areas in the midbrain, pons and medulla oblongata. J. Compar. Neurol. 529, 2243–2264. 10.1002/cne.25091 [DOI] [PubMed] [Google Scholar]
- Turovsky E., Theparambil S. M., Kasymov V., Deitmer J. W., Del Arroyo A. G., Ackland G. L., et al. (2016). Mechanisms of CO2/H+ sensitivity of astrocytes. J. Neurosci. 36, 10750–10758. 10.1523/JNEUROSCI.1281-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuunanen J., Halonen T., Pitkanen A. (1996). Status epilepticus causes selective regional damage and loss of GABAergic neurons in the rat amygdaloid complex. Eur. J. Neurosci. 8, 2711–2725. 10.1111/j.1460-9568.1996.tb01566.x [DOI] [PubMed] [Google Scholar]
- Verhoog Q. P., Holtman L., Aronica E., van Vliet E. A. (2020). Astrocytes as guardians of neuronal excitability: mechanisms underlying epileptogenesis. Front. Neurol. 11:591690. 10.3389/fneur.2020.591690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walch E., Bilas A., Bebawy V., Lam A., Murphy T. R., Sriram S., et al. (2022). Contributions of astrocyte and neuronal volume to CA1 neuron excitability changes in elevated extracellular potassium. Front. Cell. Neurosci. 16:930384. 10.3389/fncel.2022.930384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Q., Zhang M. Q., Li R., Qu W. M., Huang Z. L. (2018). The mutual interaction between sleep and epilepsy on the neurobiological basis and therapy. Curr. Neuropharmacol. 16, 5–16. 10.2174/1570159X15666170509101237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherington J., Serrano G., Dingledine R. (2008). Astrocytes in the epileptic brain. Neuron 58, 168–178. 10.1016/j.neuron.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Cui N., Yang Z., Qu Z., Jiang C. (2000). Modulation of kir4.1 and kir5.1 by hypercapnia and intracellular acidosis. J. Physiol. 524, 725–735. 10.1111/j.1469-7793.2000.00725.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W. W., Xia M., Chiang J., Levitt A., Hawkins N., Kearney J., et al. (2021). Enhanced synaptic transmission in the extended amygdala and altered excitability in an extended amygdala to brainstem circuit in a dravet syndrome mouse model. eNeuro 8:ENEURO.0306-20.2021. 10.1523/ENEURO.0306-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. F., Kim E. J., Callaway E. M., Feldman J. L. (2020). Monosynaptic projections to excitatory and inhibitory prebötzinger complex neurons. Front. Neuroanat. 14:58. 10.3389/fnana.2020.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelano C., Jiang H., Zhou G., Arora N., Schuele S., Rosenow J., et al. (2016). Nasal respiration entrains human limbic oscillations and modulates cognitive function. J. Neurosci. 36, 12448–12467. 10.1523/JNEUROSCI.2586-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]