

Abstract
The ability of a patient tumor to engraft an immunodeficient mouse is the strongest known independent indicator of poor prognosis in early-stage non–small cell lung cancer (NSCLC). Analysis of primary NSCLC proteomes revealed low-level expression of mitochondrial aconitase (ACO2) in the more aggressive, engrafting tumors. Knockdown of ACO2 protein expression transformed immortalized lung epithelial cells, whereas upregulation of ACO2 in transformed NSCLC cells inhibited cell proliferation in vitro and tumor growth in vivo. High level ACO2 increased iron response element binding protein 1 (IRP1) and the intracellular labile iron pool. Impaired cellular proliferation associated with high level ACO2 was reversed by treatment of cells with an iron chelator, whereas increased cell proliferation associated with low level ACO2 was suppressed by treatment of cells with iron. Expression of CDGSH iron-sulfur (FeS) domain-containing protein 1 [CISD1; also known as mitoNEET (mNT)] was modulated by ACO2 expression level and inhibition of mNT by RNA interference or by treatment of cells with pioglitazone also increased iron and cell death. Hence, ACO2 is identified as a regulator of iron homeostasis and mNT is implicated as a target in aggressive NSCLC.
Implications:
FeS cluster–associated proteins including ACO2, mNT (encoded by CISD1), and IRP1 (encoded by ACO1) are part of an “ACO2–Iron Axis” that regulates iron homeostasis and is a determinant of a particularly aggressive subset of NSCLC.
Introduction
Lung cancer is the number one cause of cancer related death in both men and women worldwide (1). Nearly 85% of lung cancers are non–small cell lung cancer (NSCLC), the 5-year survival rate of which is only 21% (1). With the rapid adoption of next generation sequencing technologies, in-depth large-scale genomic and transcriptomic studies on tumors and model systems have been carried out to comprehensively characterize NSCLC to discover drivers that may be clinically actionable (2–6). However, these genomics analyses reveal only a minor fraction of NSCLC as candidates for targeted therapies aimed at molecularly defined targets including for example mutationally activated protein tyrosine kinases such as EGFR, ROS1, and ALK (7, 8). Proteins are the effectors of genomes and transcriptomes and determinants of normal and disease phenotypes. However, the role of proteome signatures in tumor aggressiveness and patient outcomes is only just emerging (9–12).
Patient-derived xenograft (PDX) tumors are powerful preclinical models for NSCLC (13, 14) and other cancers (15, 16). In NSCLC ∼30% of primary tumors can engraft, and tumor engraftment is the strongest known prognostic indicator of poor outcome (13). Therefore, tumor engraftment and PDX establishment are features of more aggressive primary tumors. Consistent with this, studies show that PDX formation is a prognostic indicator of worse clinical outcomes across multiple types of cancers in addition to NSCLC (13, 17) including acute myeloid leukemia (18), colorectal cancer (19), uveal melanoma (20), and cancers of kidney (21), breast (22), pancreas (23), bladder (24), and ovary (15, 16).
The significant correlation between engraftability and poor prognosis extends to patients with NSCLC with tumors lacking mutations in known cancer drivers such as EGFR, KRAS, PIK3CA, and BRAF (13). This suggests the existence of unknown NSCLC cancer drivers involved in PDX formation and tumor aggressiveness. However, knowledge of molecular features that distinguish between engrafting (XG) and non-engrafting (Non-XG) tumors is limited. Multi-omic analyses of NSCLC have revealed that proteins involved in metabolism (i.e., the metabolism proteome) are particularly highly conserved between primary and cognate PDX tumors (25), and that proteome signatures conserved between primary and PDX tumors have prognostic impact (25, 26). More metabolically active NSCLC tumors, defined by fluorodeoxyglucose uptake, were better able to engraft (27). These observations suggest that metabolism proteome remodeling plays a key role in the elaboration of metabolic states that drive aggressive cancer phenotypes. Indeed, changes in cellular energy metabolism are crucial for neoplastic transformation and a hallmark of cancer (28). However, the role of metabolism proteome signatures and metabolic states in tumor engraftment and patient prognosis are not fully understood. Given the prognostic impact of lung tumor engraftment and metabolism proteome signatures, we posited the existence of metabolism protein determinants of aggressive cancer phenotypes.
In this study, significant proteome remodeling was associated with NSCLC PDX formation, including a metabolism protein signature that predicts outcomes in NSCLC. Low-level protein expression of mitochondrial aconitase (ACO2), an iron-sulfur (FeS) cluster containing enzyme that catalyzes interconversion of citrate and iso-citrate as part of the tricarboxylic acid (TCA) cycle, is identified as most highly associated with cases of aggressive, engrafting NSCLC having worse outcome. Downregulation of ACO2 transforms normal lung epithelial cells in vitro, whereas upregulation of ACO2 inhibits proliferation of transformed NSCLC cells in vitro and tumor growth in vivo. ACO2 was found to regulate cellular iron levels. High-level expression of ACO2 increased the translation factors iron response element binding proteins 1 and 2 (IRP1 and IRP2), and the intracellular labile iron pool (LIP). Conversely, low level ACO2 decreased intracellular iron. Impaired cell proliferation associated with high level ACO2 was reversed by treatment of cells with an iron chelator, whereas increased proliferation associated with low level ACO2 was suppressed by treatment of cells with iron. Modulation of ACO2 protein levels altered the levels of mitoNEET (mNT, encoded by CISD1). mNT delivers mitochondrial synthesized FeS clusters to IRP1 (encoded by ACO1; refs. 29–32), which inhibits its translation factor activity and activates its enzymatic activity as a nonessential cytosolic aconitase (33). Knockdown (KD) mNT expression or treatment of cells with the mNT inhibitor pioglitazone also caused increased labile iron like that seen with upregulated ACO2 expression. These data provide mechanistic insight into a previously unrecognized ACO2–Iron axis and a role for ACO2 in the regulation of iron homeostasis and cell proliferation and as a determinant of the aggressive cancer phenotype embodied by tumor engraftment.
Materials and Methods
Material availability
This study did not generate new unique reagents. PDX models generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Human subjects
Snap-frozen samples of 52 patients with previously untreated NSCLC were acquired from the UHN Biobank. This cohort of patients was a subset of patients analyzed in a previous study (13). The University Health Network Human Research Ethics Board approved the protocol. Patient tumors with any mutation in EGFR, KRAS, PIK3CA, or BRAF were excluded (13). All samples were pathologically examined. Details of the patients and their tumors are provided in Supplementary Data 1. Unique identifiers used for these patients are 5-digit numbers. A fragment of tumor tissues was embedded in optimal cutting temperature (OCT) compound.
Clinical data annotation
The demographics, histopathologic, stage and relevant clinical information is summarized in Supplementary Data S1.
Preparation of patient tissues
Patient tissue samples were prepared as previously described (34). In brief, snap-frozen NSCLC patient tumor fragments were embedded in OCT compound (Sakura), cut into 8-μmol/L serial sections at −21°C by using a cryostat (Leica CM1950) and serially mounted on glass slides (Thermo Fisher Scientific). To enrich for tumor cells, a frozen patient tissue section was stained with hematoxylin and eosin (H&E) to discriminate non-tumor area and tumor area. Then this first H&E staining section was used as a guide for scraping away the surrounding non-tumor area in the next 4 consecutive unstained slices. The tumor-enriched materials were stored at −80°C until use. Additional 70% ethanol and water washing steps were used to remove OCT compound from patient slices prior to protein extraction by using urea buffer (8 mol/L urea in 0.1 mol/L Tris/HCl pH 7.6).
Preparation of super-SILAC standard
Three common NSCLC cultured cell lines derived from respectively lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), and lung large cell/neuroendocrine carcinoma origins were labeled by metabolic incorporation of lysine and arginine containing stable isotopes 13C and 15N (Fig. 1A). The isotopically labelled “heavy” proteomes were combined to generate a super-SILAC standard mixture (35). This mixture was combined with tumor proteome samples and served as an internal reference, allowing sample proteomes to be normalized and quantitatively measured by comparison with the internal reference heavy proteome. NSCLC cell lines (HCC827, H520, and H460) that differ in histology were grown in RPMI1640 Minus L-Lysine and L-Arginine media supplemented with 13C615N2-Lysine (Lys-8), 13C615N4-Arginine (Arg-10), and 10% dialyzed FBS (all from Cambridge Isotope Laboratories, Inc.) and 1% penicillin–streptomycin (Sigma-Aldrich) until their proteomes were fully labeled by the heavy lysing and arginine. Proteins were extracted in urea buffer (8 mol/L urea in 0.1 mol/L Tris/HCl pH 7.6) and combined in a ratio of 1:1:1 (protein amount) to generate the super-SILAC standard.
Figure 1.

Low level ACO2 protein is most strongly associated with engraftment and worse survival. A, Super-SILAC quantitative proteome analysis of 52 NSCLC patient tumors. B, Spearman correlation hierarchal clustering of 52 NSCLC proteomes and including CES. C, Statistical analysis performed to identify protein expression predictive of engraftment. D, Venn-diagram of proteins selected in (C). Twelve proteins highlighted in purple predict XG from Non-XG in both LUAD and LUSC and do not interact with histology. E, Leave-one-out cross validation analysis showed that 45 of 52 samples were classified correctly based on risk scores. F, The 12 metabolism signature proteins that are predictive of engraftment. G, Volcano plot showing the significantly differential proteins between XG and Non-XG (Welch t test permutation-based FDR < 0.05). H, Enriched pathways by 194 significantly differential proteins between XG and Non-XG using IPA. Pathways shown in boldface/purple bars include ACO2. H, OS of low versus high ACO2 expressing tumors in an independent LUAD patient cohort based on TMA. I, DFS of low versus high ACO2 expressing tumors in an independent LUAD patient cohort based on TMA. J, OS of low vs. medium versus high ACO2 expressing tumors in an independent LUSC patient cohort based on TMA (see also Supplementary Fig. S1A–C). K and L, DFS of low versus medium versus high ACO2 expressing tumors in an independent LUSC patient cohort based on TMA (see also Supplementary Fig. S1A–S1C).
Sample preparation for mass spectrometry analysis for primary tumors
Proteins extracted from the patient specimens were mixed with equal amounts of the super-SILAC standard and reduced with 4.5 mmol/L DTT at 60°C for 20 minutes and then alkylated with 10 mmol/L iodoacetamide for 30 minutes in dark. After overnight digestion by trypsin, peptides were desalted and purified by using Pierce C18 Spin Tips (Thermo Fisher Scientific).
LC/MS-MS analysis
Purified peptides were separated by a ultra-high performance liquid chromatography system (Easy- nLC1000, Thermo Fisher Scientific) coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) through a nano-electrospray ion source (EASY-SPRAY, Thermo Fisher Scientific). Peptides resuspended in 0.1% formic acid were loaded onto a 50-cm column (75 μmol/L inner diameter) packed with PepMapRSLC C18 resin and heated at 60°C with a flow rate of 250 nL/min from 5% to 30% acetonitrile in 0.1% formic acid over 224 minutes, followed by 30% to 90% acetonitrile in 0.1% formic acid in 2 minutes and then 90% acetonitrile in 0.1% formic acid for 12 minutes to clean up the column.
Mass spectra were obtained by using one full mass spectrometry (MS) scan (400–1,200 m/z) in the Orbitrap mass analyzer followed by 10 data-dependent MS2 scans. For MS scans, automatic gain control target was set at 1,000,000 with a maximum ion injection time of 200 ms, and resolution was 240,000 (full-width half-maximum) at m/z 400 in the Orbitrap. For MS2 scans, up to 10 most intensive ions were selected and fragmented by collision-induced dissociation (CID) with normalized collision energies at 30 in the ion trap. Automatic gain control target for MS2 was 10,000 with a maximum injection time of 50 ms. Dynamic exclusion duration was set at 50 s. Maximum exclusion list size was 500.
MS data analysis
Raw MS files were analyzed by MaxQuant (refs. 36, 37; version 1.5.2.8). Peaks were searched against the UniProt human database (released Feb. 2015; http://www.uniprot.org) using Andromeda search engine (38) implemented in MaxQuant. Two mis-cleavages were allowed and a minimum of 7 amino acids per identified peptide was required. FDR for peptides and proteins was set at 1%. “Match between runs” option was turned on with an alignment time window of 20 minutes and a matching time window of 2 minutes. A minimum ratio count of 1 was chosen for quantification of SILAC pairs. “Re-quantify” option was selected.
Selection of metabolism protein signatures
Group LASSO (least absolute shrinkage and selection operator) model was used to select metabolism protein signature to predict NSCLC tumor engraftability. 359 metabolism proteins that were quantified in all 52 tumors were analyzed. To determine which subset of proteins was included in the group LASSO model, three analyses were conducted (Fig. 1C). This preselection of a smaller subset of proteins was for obtaining a more stable group LASSO result. Wilcoxon–Mann–Whitney tests were used to establish which proteins were associated with xenograft formation. This test was conducted separately on antibody–drug conjugate (ADC) and non-ADC samples. Proteins that affected formation of xenografts significantly (i.e., Wilcoxon P < 0.1 in either ADC or non-ADC subcohort analysis) were included in the following group LASSO analysis. Another logistic regression model was used to determine if the effect of a protein on xenograft formation varied between histologic subtypes (i.e., LUAD vs. non-LUAD). Thus, this model contained the protein, histology type and the interaction between protein and histology and was fit for each protein. Proteins that interacted significantly with histology (i.e., P value of the interaction between protein and histology < 0.1) were included in the following group LASSO analysis. Group LASSO does variable selection on defined groups of variables (i.e., proteins and/or their interactions with histology in this analysis) rather than single variables and is an extension of the ordinary LASSO. A group LASSO model (gglasso package in R) was fit with the 106 significant proteins and histology type. Each protein expression values (log2 transformed) were standardized across samples. If a protein had a significant interaction with histology (i.e., P value of interaction < 0.1 in the logistic regression analysis), the interaction term was also kept in the group LASSO model, and the protein and its interaction term were considered a group. To ensure histology type was included in the model, it was not penalized. Histology equals 1 if a sample is LUAD and 0 otherwise. A risk score was calculated on the basis of the proteins and interactions selected by the group LASSO and their coefficients, which had the formula as: Risk score = βhistology × Histology (Histology = 1 if a sample is ADC, otherwise 0) + β1 × Protein1 + (β2 × Protein2 + β2 × Protein2 × Histology) + …, where β is the coefficient value for histology or selected protein, Proteinn is the standardized protein n expression value, and as an example shown in this formula, protein2 has a significant interaction with histology, thus there is an additional item (i.e., β2 × Protein2 × Histology) including the Histology information. The risk score was dichotomized using a cut-off percentile describing the proportion of Non-XG samples in the full cohort. Leave-one-out cross validation (LOOCV) was used to evaluate the performance of group LASSO model.
For histology subgroup analysis, Elastic Net procedure was used (glmnet package in R) to construct a multivariable model. 400 metabolism proteins quantified in all 24 NSCLC LUAD tumors and 389 in 25 LUSC tumors were included in LUAD and LUSC subgroup analysis respectively. Protein expression values (log2 transformed) were standardized across samples in preparation for using the Elastic Net analysis. The outcome is binary (XG vs. Non-XG). The shrinkage parameter, λ was obtained based on 5-fold cross-validation. The elastic-net mixing parameter, α, controls how much LASSO and how much ridge penalization is imposed. For example, if α = 1 the LASSO penalty is used, and for α = 0, the ridge penalty is used. Alpha was set to 0.1, 0.3, 0.5, 0.7, and 1 and was decided using leave-one-out cross validation. For each α setting, a risk score was calculated on the basis of the selected proteins and their coefficients obtained from the Elastic Net procedure. The risk score was then dichotomized using a cut-off percentile that describes the proportion of Non-XG in the analyzed cohort. The strategy to choose λ was the same for the leave-one-out cross validation and each sample had a predicted risk score based on the coefficients obtained in the subcohort when that case was left out. The cut-off point for the LOOCV predicted risk scores was the one calculated by using the original cohort. The proportion of agreement between the observed XG vs. Non-XG dichotomy and the ones defined based on leave-one-out cross validation was calculated for each of the α levels. Alpha with the largest agreement proportion was chosen. If the proportion agreement was the same for several levels of α the one with the best sensitivity (i.e., the number of cases that our model predicts as XG divided by the number of cases that are actually XG) and specificity (the number of cases that our model predicts as Non-XG divided by the number of cases that are actually Non-XG) was chosen. In the case that the sensitivity and specificity were also the same, the α which corresponded to the fewest number of proteins selected by the Elastic Net procedure was chosen. If there were poor agreement at all levels of α tested, no α was chosen and Elastic Net results were not reported.
Tandem-Mass-Tag-based MS quantitative measurement of H226 xenografts and BEAS2B proteome
Protein extraction: lung xenograft tumors were sliced and per 100 μg of tumor was lysed in 100 μL of lysis buffer composed of 0.5 mol/L Tris pH 8.0, 2% SDS, 1% NP40, 1% Trition-X100, 5 mmol/L EDTA, 50 mmol/L NaCl, 10 mmol/L Tris-2-Catboxyethyl phosphine hydrochloride and 40 mmol/L Chloroacetamide. Samples were then sonicated for 15 to 20 s at 4 watts. The samples were then heated in the Thermomixer at 95°C for 20 minutes at 1,000 rpm. Samples were then clarified by centrifugation (20,000 g for 5 minutes) and supernatants were then cleaned by using SP3 beads (GE Healthcare, catalog no. 45152105050350) and then digested with LysC/Trypsin (Promega, catalog no. V507A) in 50 μL of 50 mmol/L triethylammonium bicarbonate buffer (14 hours, 37°C) with shaking at 1,000 rpm. For tandem mass tag (TMT) labeling, 20 μL of anhydrous acetonitrile was added to the 0.2 mg of TMT labels (Thermo Fisher: 909309). Then 25 μg of peptide was added to each 20 μL reconstituted TMT label reagent. The reaction proceded at room temperature for 1 hour and then quenched by adding 1 μL of 5% hydroxylamine and incubation at room temperature for 15 minutes. The samples were then mixed at a 1:1:1:1:1:1:1:1:1:1 ratio. The mixture was then fractionated to 8 fractions by using a Pierce high pH reversed-phase peptide fractionation kit (Thermo Fisher, 84868). The 8 fractions were then concatenated to 4 and dried by centrifugation under vacuum. The dried samples were diluted in 14 μL 0.1% formic acid, and 7 μL was used for the MS run. Each fraction was then run for 3 hours. Tandem MS data were collected by using an Orbitrap Fusion Lumos instrument configured with an EasySpray NanoSource (Thermo Scientific). Survey scans were performed in the Orbitrap mass analyzer (resolution 120,000), and data-dependent MS2 scans performed in the linear ion trap using CID (collision energy 35) following isolation in the quadrupole. Reporter ion detection was performed in the Orbitrap mass analyzer (resolution 50,000) using MS3 scans following synchronous precursor isolation of the top 10 ions in the linear ion trap, and higher-energy collisional dissociation in the ion-routing multipole (normalized collision energy 32).
Metabolite extraction from cells and media
Dox was added from day 0. A full 6-well plate was used for each condition and an additional plate with medium only was included as the background group and processed in parallel. On day 2, media was changed to 30-mL fresh media prepared with 10% dialyzed FBS. On day 3 (roughly 16 h post media change from day 3), metabolites in media were first extracted: 250 μL growth media was taken out and centrifuged at 20,000 × g for 2 minutes to remove cell debris, then 200 μL supernatant was transferred to a precooled (on dry ice) tube containing 800 μL methanol and stored at −80°C. Then culture media was changed to fresh media prepared with dialyzed FBS. Three hours later, metabolites in cells were extracted: 80/20 (volume/volume) mixture of methanol/high performance liquid chromatography (HPLC)-grade water was used as the extraction buffer and precooled and kept on dry ice, media was aspirated, and 1 mL extraction buffer was added to each well immediately, and the plate was stored overnight at −80°C. Only one plate was processed at a time. On day 4, material in each well was scraped and collected and an additional 400 μL of extraction buffer was added and residual material was collected. The plate and tubes were kept on dry ice and samples were stored at −80°C. Samples were centrifuged at 14,000 rpm and supernatant was transferred to a new tube. Residual pellet was dissolved in 250 μL buffer (20 mmol/L Tris-HCl pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 1 mmol/L sodium orthovanadate) to extract and measure total RNA in each replicate of metabolite extractions from cells by using Quant-iT RiboGreen RNA Reagent and Kit (Invitrogen) as indicators of cell amounts. Extraction solvent in the supernatant was evaporated from metabolite extracts using N2 gas. Dried extracts were reconstituted in HPLC-grade water by vortexing for 1 minutes. One of four of dried metabolite extracts from each sample were reconstituted in 15-μL HPLC-grade water and combined with an equal volume of 13C, 15N isotopically labeled metabolites extracted from S. cerevisiae to provide an internal reference for quantifying biologically derived compounds. The yeast is a mixture of exponentially grown, heat stressed, oxidatively stressed, and stationary phase cells. Remaining insoluble material was vacuum filtered out by an AcroPrep polypropylene membrane (PALL part number 8084) using vacuum filtration. Filtered samples were transferred into polypropylene vials (Agilent).
LC/MS of metabolites
LC/MS analysis for organic acids was performed using an Agilent Technologies 6540 Q-TOF equipped with a Jet Spray ESI source operated in negative ionization mode at a 3 Hz scan rate in high resolution mode across mass range 50 to 1,700 m/z. The source parameters were 150°C drying 76 gas, 12 L/min drying gas, nebulizer pressure of 45 psi, sheath gas temperature 325°C, sheath gas flow 12 L/min, capillary at 2,000 V, nozzle at 0 V, fragmentor 90 V, skimmer 35 V, octopole RF 750 V. Five microliter of each sample was injected using an HTC autosampler into a 2 μL loop supplied by an Agilent 1260 binary pump. The column was a 2.1 mm × 100 mm Waters HSS-T3 C18 column with 1.8 μm packing. Buffer A was 20 mmol/L ammonium formate pH 2.9; buffer B was acetonitrile. Gradient separation began with 0.5% buffer B at 0.4 mL/min, held until 2.5 minutes, then buffer B was increased to 99.5% at 4 minutes. The stream was switched from the LC/MS to waste, and for regeneration, flow was increased to 0.5 mL/min at 5 minutes and 1 mL/min at 5.5 minutes, held until 8.5 minutes, and then decreased to 0.5 mL/min at 9 minutes. The column was re-equilibrated by a shift to 0.5% buffer B at 10.5 minutes, flow as decreased to 0.4 mL/min at 13 minutes, and held until 14 minutes. Other compounds were analyzed by ion-paired reverse phase analysis in negative mode on a 6550 Q-TOF.
Metabolite data analysis
For raw data analysis, metabolite levels were extracted from raw data using MassHunter software packages (Agilent) and R. Neat standards were used to annotate the retention time and exact masses for metabolites of interest. Metabolite levels were determined by integrating the area under each chromatographic peak and correcting to the co-eluting isotopically labeled reference.
NSCLC tissue analysis and IHC
Tissue microarray (TMA) blocks of 51 NSCLC SCC and 216 NSCLC ADC cases were stained with anti-ACO2 antibody (Sigma-Aldrich, HPA001097) at The Applied Molecular Profiling Laboratory, University Health Network. Pathologists reviewed slides and scored ACO2 staining intensities. ACO2 staining intensity in tumor cells was graded with barely visible at objective lens of 4× as 1+, visible at objective lens of 4× as 2+, and very strong at objective lens of 4× as 3+. Percentage of stained tumor cells was also recorded. For each case, 1 to 3 tumor cores were scored separately. H-score for each core was calculated by using the formula: H-score = (1 × % of staining at 1+) + (2 × % of staining at 2+) + (3 × % staining at 3+). Average of H-scores from duplicate cores was used as the final H-score for each patient. Data and association with survival were further assessed by using R as described in survival analysis section.
Cell cultures
Unless otherwise indicated, cell lines were obtained from ATCC without additional authentication. Cells were maintained under sub-confluent conditions and used for experiments within 4 months of resuscitation and tested as Mycoplasma-free by the SickKids Microbiology Laboratory within 4 months of experimentation. NSCLC cell lines H226 and MGH7 (of SCC histology) were grown in RPMI1640 medium (R8758, Sigma-Aldrich) supplemented with 10% FBS (HyClone) and 1% penicillin–streptomycin (Sigma-Aldrich). For selection in NSCLC cell lines, hygromycin (50 mg/mL stock) was used at 100 μg/mL and blasticidin (10 mg/mL) was used at 2 μg/mL. Doxycycline (50 mg/mL stock) was used at 1:50,000 dilution.
Generation of NSCLC cell lines with inducible ACO2 overexpressing lentiviral constructs
Vectors from Tet-on Advanced pLenti CMV tight series (Addgene) were used to make lentivirus and generate inducible ACO2-overexpression cell lines. A repressor vector of pLenti CMV rtTA3 Blast (Addgene) and a destination vector of pLenti CMVtight Hygro DEST (Addgene) were used. ACO2 sequence was obtained from Human Ultimate Open Reading Frame collection, which matched Homo sapiens aconitase 2, mitochondrial, mRNA (cDNA clone MGC:33908 IMAGE:5264179), was shuttled into the pLenti CMVtight Hygro DEST vector (SPARC BioCentre, Toronto). Lentiviral particles containing pLenti CMV rtTA3 Blast and pLenti CMVtight Hygro DEST-ACO2 were produced respectively and used to infect NSCLC cell lines.
T-REx 293 inducible overexpressing ACO2 cell lines
To make inducible ACO2 expressing cell lines in T-REx 293cells, ACO2 sequence obtained from Human Ultimate Open Reading Frame collection (cDNA clone MGC:33908 IMAGE:5264179) in pLenti vectors were cloned to pcDNA5.1 by adding NotI and HindIII restriction sites and FLAG sequence at the 3′. Forward and reverse primers used to do this were:
Forward: 5′ AA AAGCTT ACCA ATG GCGCCCTACAGCCTAC 3′
Reverse: 5′ CAGAATGAAGGAACTGCAACAG GATTACAAGGATGACGACGATAAGTGAGCGGCCGC AA 3′
shCISD1 cell lines: pGIPZ clones targeting CISD1 V2LHS-276497 with sequence TTATCATAGCTTTATTTCG and V3LHS-358841 with the sequence TTAACATAAAATCTTTTGT
shIDH2 cell lines: pGIPZ clones targeting IDH2 V2LHS-233824 with the sequence TAGTAGCTAAATATGGCAC and V3LHS-412536 with the sequence ATGCTTTTAAAAACATCTG were obtained from Dharmacon and were used to make viruses to transfect HEK293 T-REx inducible overexpressing ACO2 cells.
Normal Lung epithelial cells BEAS-2B (CRL-9609) from Cedarlane was used to produce ACO2-KD with Non-Targeting Inducible Vector control for SMART vector system provided by Dharmacon horizon discovery (Catalogue #VSC6572)
1-V3SH11252–224977242 TGGGACTTGCACTTGAGGC (targets ORF)
2-V3SH11252–226388817 GGCTGTGACAATCTCTGGG (targets ORF)
3-V3SH11252–228633609 GCCAGGACAATGCCACCCA (targets ORF)
All three constructs worked as expected, number 3 is used for Further experiments.
10,000 shACO2-BEAS2B cells were counted and seeded in 12 well plates in BEGM media and supplements (Lonza, Catalog No. CC-3170) and tetracycline was added to induce ACO2-KD at final concentration of 1 μg/mL. Cells were trypsinized for 3 minutes at 37°C and counted with Vi-CELL TM Beckman Coulter cell counter after 5 days.
Western blotting
Cell pellets or xenograft tissues were washed in PBS twice and lysed in NP40 lysis buffer (50 mmol/L Tris/HCl pH7.5, 100 mmol/L NaCl, 1% NP40, 0.1% sodium deoxycholate, 10% glycerol, 1 mmol/L EDTA, 10 mmol/L NaF, protease inhibitor tablets-EDTA-free (Thermo Fisher Scientific)) with gentle agitation for 30 minutes at 4°C. Insoluble cell debris was removed by centrifuging at 12,000 × g for 10 minutes at 4°C and protein concentrations were measured by using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). ACO2 antibody (A22) was from Santa Cruz Biotechnology (sc-130677) and used at 1:500 dilution. β-Actin antibody was from Cell Signaling Technology (#4967) and used at 1:1,000 dilution.
Cell proliferation measurement
20,000 cells were counted using Vi-CELL cell counter (Beckman Coulter) and seeded in 24-well plates and tetracycline (Bioshop TET 70125) was added to induce protein expression at final working concentration of 1 μg/mL. After 3 days, cells were lifted off using 100 μL trypsin and 37°C incubation for 3 minutes, after which 400 μL media was added to lift cells, which were then counted (Vi-CELL TM Cell counter, Beckman Coulter).
Tumorgenicity assay
H226 cells (2 ×106) were counted by using a Vi-CELL counter (Beckman Coulter) and then injected subcutaneously into flanks of 4 to 6 weeks old female NOD/SCID mice. In the ACO2 overexpression group, doxycycline (2 mg/mL with 2% sucrose) was added into mice drinking water 3 days before tumor cell injection and changed twice a week. Tumor volume was measured by calipering in two dimensions and calculated as 0.5 × length × width2. To generate growth curves of xenografts, mean ± SD of the tumor volume was presented. Growth rates of ACO2 overexpressing and control xenografts were compared using a mixed effect model to account for correlations within a mouse. Xenograft volume was considered the outcome, and mouse was included as a random effect. An auto-regressive correlation structure was assumed.
Aconitase activity measurement
HEK293 T-Rex ACO2 WT or HEK293 T-Rex ACO2 triple mutant cells (0.6×106) were seeded in 6-well plates and incubated overnight in a 37°C incubator so that they reached 70% to 80% confluence the next day. At 24, 20, 16, 12, 8, and 4 hours prior to assay, tetracycline (Bioshop TET 70125) was added to a final concentration of 1 μg/mL to induce protein expression. At time zero, cells were counted and aliquoted at 106 cells per tube. Aconitase activity was measured by using Biovision Aconitase Activity Colorimetric Assay Kit (K716–100) according to the manufacturer's instructions.
LIP measurement
Cells were washed three times with PBS. 2×106 cells were incubated with 250 nmol/L Calcein at 37°C for 30 minutes and then collected by centrifugation. Cells were resuspended in 300 μL PBS and placed in 96-well plates. 100 μmol/L PIH was added, and fluorescent signals measured at time 0 and 10 minutes at excitation 488 nm and emission 517 nm as described previously (39). The increase in fluorescent signal indicates the LIP level.
Soft agar colony formation assay
Cells were pretreated with tetracycline and cultured in soft agar medium for 21 to 28 days. Following this incubation period, formed colonies were analyzed by quantifying the number of colonies formed per well. To prepare the base agar layer 1% Nobel Agar (Sigma, A5431) was melted in a microwave oven and then cooled to 40°C and mixed with an equal volume of 2x-RPMI with 20% FBS and antibiotics and then plated at 1 mL per well in 12-well plates, and then incubated at room temperature (approx. 21°C) for 5 minutes to allow the agar to set. Therefore, final concentrations of the base agar were 0.5% Agar, 1X RPMI, 10% FBS. Top agarose comprised 0.7% melted agarose at 40°C mixed with an equal volume of 2X RPMI and 20% FBS at the same temperature. Cells (5,000/mL) were added and plated as 1 mL per well, and then incubated for 15 minutes at room temperature to allow the agar to set. Top media was BEBG media with or without 1 μg/mL Tetracycline. Plates were stained with Tetrazolium Blue Chloride (Sigma, T4375) for 1 hour at 37°C.
Pioglitazone administration
20,000 HEK293 WT ACO2 cells were seeded in 24-well plates and Tetracycline (Bioshop TET 70125) was added to induce protein expression at final working concentration of 1 μg/mL. Pioglitazone hydrochloride (PZH; Sigma Aldrich E6910) was added at the indicated final concentration. After 3 days, cells were lifted off by using 100-μL trypsin and 37°C incubation for 3 minutes, after which 400-μL media was added to lift cells, which were counted (Vi-CELL TM Cell counter, Beckman Coulter).
Pyridoxal isonicotinoyl hydrazone (PIH) administration
HEK293 WT ACO2 cells (20, 000) were seeded in 24-well plates and Tetracycline (Bioshop TET 70125) was added to induce protein expression at final working concentration of 1 μg/mL. Pyridoxal isonicotinoyl hydrazone (Santa Cruz-737–86–0) was added at a final concentration of 10 μmol/L/mL, 30 μmol/L/mL, and 50 μmol/L/mL. After 3 days, cells were lifted by using 100-μL trypsin and 37°C incubation for 3 minutes, after which 400-μL media was added.
Quantification and statistical analyses
Disease-free survival (DFS) up to 5 years was used as the endpoint of interest. DFS was calculated as the time difference (in years) between the date of surgery and the date of recurrence, death, or last follow-up. Patients for whom death or recurrence was not observed were censored. Patients with more than 5 years follow-up were censored at 5 years as relapse or deaths occurring after 5 years were not likely to be the first lung cancer-related. The Kaplan–Meier method was used to estimate DFS probabilities. The log-rank test was used to compare DFS between groups. A two-sided P value of 0.05 was considered statistically significant. HRs and 95% confidence intervals (CI) were calculated using a Cox proportional hazards model. All analyses were performed using the survival package in the open-source software R.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (40) with the dataset identifier PXD028556.
Results
Quantitative proteome profiling of 52 NSCLC patient tumors
To investigate NSCLC proteome characteristics associated with xenograft formation, we analyzed a cohort of 52 NSCLC primary tumors freshly frozen and embedded in OCT compound. All materials and methods are described in Supplementary Methods. Some 29 of 52 tumors were previously shown to XG, whereas 23 were Non-XG (13). All 52 specimens were collected from patients with previously non-treated NSCLC lacking known mutations in EGFR, KRAS, PIK3CA, or BRAF (13). Clinical and pathologic features that associated with higher engraftment rate in this cohort include gender (male), squamous cell carcinoma histology, and grading (poor differentiation; Supplementary Table S1), which was consistent with previous reports (13, 17, 41).
To minimize effects of experimental and technical variance and facilitate accurate quantification of protein expression levels, super-SILAC methodology was used, wherein a metabolically labeled cultured cell extract is mixed with tissue samples as an internal reference and control (Fig. 1A; refs. 42, 43). Using this methodology, a total of 4,920 proteins were quantified (Supplementary Data 1).
Proteome features associated with engraftment
To test the hypothesis that XG and Non-XG tumor proteomes are different, we clustered all 52 samples by a subset of proteins that were identified in all samples by using unsupervised Spearman hierarchical clustering (Fig. 1B). This resulted in two major clusters. One cluster (group 1) contained 23 samples comprising mostly (18/23) Non-XG tumors and the other group (group 2) comprised of 29 samples of which most (24/29) were XG tumors (Fig. 1B). As visible by cluster enrichment score (CES; Fig. 1B), separation of the two clusters was most strongly associated with engraftment (Fisher Exact t test P < 0.0001) and to a lesser degree histology (P = 0.0245), inflammation (P = 0.0354), and grade (P = 0.0495; Fig. 1B). These data indicate that the proteomes of XG and Non-XG tumors are distinctly different.
ACO2 expression is the strongest negative predictor of engraftment
The expression of metabolism proteins is highly conserved between patient-matched primary and PDX lung tumors (25). Therefore, we reasoned that the ability of primary NSCLC tumors to engraft might be predictable based on metabolism protein expression. To derive a metabolism proteome signature for engraftment prediction, we focused on metabolic enzymes and small molecule transporters. To select for a subset of metabolism proteins for engraftability prediction, we employed a group least absolute shrinkage and selection operator (LASSO) regression model. Some 38 LUAD proteins and 74 LUSC proteins that differ significantly between XG and Non-XG were identified and included in the LASSO analysis. In addition, 35 proteins that interacted significantly with histology were included in the group LASSO analysis (Fig. 1C). The model selected a subset of 12 metabolism proteins significantly associated with and able to predict engraftment (Fig. 1D). The 12-protein metabolism-related signature was tested by leave-one-out cross validation analysis, which showed that 45 of 52 samples were classified correctly based on their calculated risk scores (Fig. 1E). The sensitivity was 90% (26/29) and the specificity was 83% (19/23). ACO2 had the largest absolute value in risk score (1.56), indicating a strong correlation between low level ACO2 protein expression and engraftment (Fig. 1F). The range of relative ACO2 protein expression was 11.6-fold.
To identify a subset of proteins that significantly discriminated XG from Non-XG tumors in a supervised manner, differential analysis was performed with all 4609 proteins comparing XG and Non-XG groups. This identified 194 protein groups as significantly differentially expressed in XG and Non-XG primary tumors (Permutation-based adjusted Welch t test P value < 0.05). Within this set of 194 protein groups, 80 were significantly upregulated in XG and 114 were upregulated in Non-XG (Fig. 1G; Supplementary Data 2). To further investigate biological processes and pathways enriched in the set of 194 proteins, we conducted Ingenuity Pathway Analysis (IPA; Fig. 1H; Supplementary Data 2). Pathways significantly enriched (P value < 0.01) and containing at least 2 proteins are shown. Biological pathways that differed between XG and Non-XG and included ACO2 were TCA cycle and iron homeostasis (Fig. 1H).
Validation of low ACO2 association with worse survival in independent NSCLC cohorts
To validate the prognostic impact of ACO2, independent cohorts of LUAD and LUSC patient tumor samples were assessed for ACO2 protein expression by using TMA analysis. In the LUAD cohort, consistent with our initial observations, low ACO2 expressing tumors had a significantly worse overall survival (OS) and DFS compared with high expressing ACO2 tumors (Fig. 1I and J). In the LUSC cohort patients were grouped into low, medium, and high expressing ACO2 tumors, and a significant difference in OS was observed between low and high ACO2 expressing tumors (Fig. 1K), but not when they were grouped into only low and high ACO2 groups (Supplementary Fig. S1A–S1C). DFS trended towards worse in low expressing ACO2 tumors in LUSC, however this did not reach statistical significance (Fig. 1L).
Elevated ACO2 changed proteome landscapes and suppressed cell proliferation in vitro and in vivo
Since ACO2 expression level is higher in less aggressive (Non-XG) tumors and in normal lung tissue, we tested if elevation of ACO2 levels in a NSCLC cancer cell line would reduce cell proliferation. ACO2 was ectopically expressed in the LUSC-derived cell line H226, which has a relatively low level of endogenous ACO2 protein expression (Supplementary Fig. S1D). Quantitative MS analysis revealed a 7-fold increase in ACO2 protein because of ectopic expression (Fig. 2A). Increased ACO2 expression was associated with a significant suppression of H226 cell proliferation in vitro (Fig. 2B). Similarly, growth of NSCLC-derived MGH7 cells and HEK293 cells was also inhibited by ectopic expression of ACO2 (Supplementary Fig. S1E). These results suggested that elevated ACO2 at the protein level suppresses cell proliferation. When endogenous ACO2 expression was reduced in H226 cells by using an inducible short hairpin RNA targeting ACO2, no significant changes in proliferation were detected in vitro or in vivo (Supplementary Fig. S1F–S1H). This is perhaps due to the already low levels of ACO2 in H226 cell line in comparison with normal tissue and cell lines (Supplementary Fig. S1D).
Figure 2.

Elevated ACO2 decreases proliferation in H226 cell line in vitro and in vivo. A, Top, relative ACO2 levels in H226 cell lines between control and elevated ACO2 (mean±SD). Bottom, Western blot showing ACO2 levels in H226 cell line between control and elevated ACO2. B, Proliferation rate determined by cell number of inducible H226 NSCLC cell line between control and elevated ACO2 (mean±SD). C, Spearman hierarchal correlation of 3 control and 3 elevated ACO2 replicates of H226. D, Volcano plot showing 183 significantly differential proteins between control and elevated ACO2 (Student t test permutation-based FDR < 0.05). E, Significantly enriched pathways by IPA matched with XG versus Non-XG; ACO2-containing pathways are shown in boldface/purple bars. F, ACO2 expression reduces tumor growth in H226 NSCLC xenograft in NOD-SCID mice (mean±SD). G, Western blot showing resected H226 xenograft tumors ACO2 levels of control versus elevated ACO2. Resected H226 xenograft control versus elevated ACO2 tumors for: G, ACO2 levels via Western blotting; H, H&E staining; I, ACO2 levels via IHC; J, Ki67 as a marker of proliferation via IHC. K, CASP3 as a marker of apoptosis via IHC. L, Quantification of Ki67 positive cells show a significant decrease in number of proliferative cells (mean±SD). M, Quantification of CASP3 positive cells show no difference in number of cells undergoing apoptosis (mean±SD). N, Percentage composition of Human, Mouse, and ambiguous Human/Mouse proteins in total proteomics of H226 xenograft tumors. O, ACO2 levels of control versus elevated ACO2 H226 xenografts quantitated by MS (mean±SD). P, Spearman's correlation hierarchal clustering of 3 control and 3 elevated ACO2 H226 xenografts proteome (Q) Spearman's correlation of each protein with ACO2 are ranked based on P value with P value < 0.05 shown in red that was used for IPA. R, Significantly enriched pathways by IPA. Two-sided Student t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.00005.
The proteomes of H226 cells with and without a 3-day induction of ectopic ACO2 expression were measured. In total approximately 3,500 proteins were characterized (Supplementary Data 1). Spearman's correlation hierarchal clustering of these samples produced two main clusters with the 3 control replicates forming one group and elevated ACO2 replicates forming the other (Fig. 2C). This result suggested that elevated ACO2 expression significantly altered the proteome. Differential analysis revealed 183 proteins with significantly altered expression associated with elevated ACO2 (Student t test permutation-based FDR < 0.05; Supplementary Data 2). Some 92 proteins were upregulated and 91 downregulated in response to increased ACO2 protein expression (Fig. 2D). Significantly enriched pathways containing more than one protein in their gene set affected by altering ACO2 are reported in Supplementary Data 2. The topmost significantly enriched pathways that contained ACO2 in their gene sets were again TCA cycle and iron homeostasis (Fig. 2E).
To determine if elevated ACO2 levels affect tumor formation and growth, the engineered H226-ACO2 cell line was implanted into NOD-SCID mice with (ACO2) and without (control) doxycycline in their water to induce ectopic ACO2 expression. Control (i.e., non-induced) H226-ACO2 cells formed tumors, whereas mice with induced ectopic expression of ACO2 showed slower tumor growth (Fig. 2F). Resected tumors from these mice were assessed for expression of ACO2 by Western blot and immunocytochemistry, which confirmed ACO2 expression was elevated in the doxycycline treated group (Fig. 2G and I). Resected H226 xenograft tissue sections were visualized by H&E staining (Fig. 2H), where infiltrated stroma appears to have increased in the more slowly growing tumors with elevated ACO2 (Fig. 2H). A significant decrease in Ki67-positivity and no significant change in CASP3 positivity was seen in tumors with elevated ACO2 expression suggesting the decreased tumor growth associated with elevated ACO2 is due to decreased proliferation and not increased apoptosis (Fig. 2J–M).
Total proteomes of 3 control and 3 elevated ACO2 xenograft tumors were measured. This identified ∼6,300 proteins, of which 37% were human, 32% mouse and 31% ambiguous with no species-unique peptides (Fig. 2N) (Supplementary Data 1). The MS results showed that induced ACO2 levels were increased 3-, 5-, and 11-fold compared with controls (Fig. 2O). Clustering analysis indicated significant proteome remodeling (Fig. 2P) including 1,227 proteins that significantly changed in abundance with ACO2 expression (Fig. 2Q; Supplementary Data 2). Significantly enriched pathways associated with these proteins included iron homeostasis, which has ACO2 as part of its gene set (Fig. 2R; Supplementary Data 2).
Suppression of ACO2 in lung epithelial cells alters the proteome, increases cell proliferation, and stimulates colony formation
Since lower ACO2 levels were seen in the more aggressive XG tumors, we tested if lowering ACO2 protein expression would drive the transformation of non-transformed lung epithelial cells. Inducible KD of ACO2 (shACO2) and a scrambled shRNA control (shScr) were tested in immortalized normal lung epithelial BEAS-2B cells. KD of ACO2 led to increased proliferation while the shScr control did not (Fig. 3A and B; Supplementary Fig. S2A and S2B). ACO2 KD in BEAS-2B stimulated growth and colony formation in soft agar approximately 4-fold compared with the control (Fig. 3C and D).
Figure 3.

Decreasing ACO2 levels in normal epithelial BEAS-2B cells increases proliferation rate in vitro and increases colony formation and ACO2’s role in iron regulation. A, Number of BEAS-2B cells in vitro with and without ACO2 KD at day 5 (mean±SD). B, Western blot showing the KD of ACO2 at day 5 with dox. C, Colony formation in agar of BEAS-2B cells with and without dox in control shScr and shACO2. D, Quantification for colony formation in agar of BEAS2B cells with and without dox in control shScr and shACO2 (mean±SD). E, ACO2 levels quantified in MS experiment with and without dox (mean±SD). F, Spearman's rho correlation-based hierarchical clustering of ∼6,000 proteins separate control cells from ACO2 KD. G, Volcano analysis show the significantly differential proteins (Student t test permutation-based FDR < 0.05). H, Top ten most significantly enriched pathways of the differentially altered proteome in (G). I, Comparative pathway analysis showing top 7 pathways that reverse in activity between ACO2-up and ACO2-down systems. J, Essentiality Score across 1,070 DepMAP cell lines indicates sensitivity to loss of ACO2 correlates with sensitivity to loss of TFRC, which may reflect their shared roles in iron regulation. K, Western blot showing IRP2 levels in control versus elevated ACO2 in H226 cells. L, Western blot showing IRP2 levels in control versus ACO2 KD in BEAS-2B cells. M, LIP levels increase with elevated ACO2 in H226 cells (mean±SD). N, LIP levels decrease with ACO2 KD in BEAS-2B cells (mean±SD). O, Iron chelator PIH rescues proliferation defect caused by elevated ACO2 in H226 cells at 30–50 μmol/L (mean±SD). P, Proliferation advantage seen with ACO2 KD is reversed by 5 to 30 μmol/L FeCl3 (mean±SD). Two-sided Student t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.00005.
The proteomes of the BEAS-2B cell types with modulated ACO2 levels were characterized by MS. Approximately 6,000 proteins were quantified (Supplementary Data 1). ACO2 expression was 3-times less in ACO2 KD cells (Fig. 3E) and KD of ACO2 led to significant proteome remodeling (Fig. 3F). Approximately 2,800 proteins were significantly differentially expressed between shACO2 and control cells (Student t test permutation-based FDR < 0.05; Fig. 3G). IPA was applied to investigate biological processes and pathways enriched within the set of proteins whose expression was affected by ACO2 KD (Fig. 3H) (Supplementary Data 2). The ten most significantly enriched pathways associated with ACO2 KD (Fig. 3H) included activation of mTOR signaling and remodeling of epithelial adherens junctions.
Increased ACO2 protein expression causes elevated cellular iron levels
Biological pathways in the ACO2 KD and elevated systems were compared with an emphasis on pathways that reverse in activity between the KD and elevated ACO2 systems. Sixty pathways were identified (Supplementary Data 2) and the seven most active pathways in each system (i.e., ACO2 suppression or elevation) are shown in Fig. 3I. Markers of ferroptosis, i.e., cell death resulting from high iron levels (44), decreased with ACO2 KD and increased significantly with ACO2 elevation (Fig. 3I). In ACO2 overexpressing cells markers indicative of ferroptosis included increased transferrin receptor (TFRC) and BID, and lower expression of GPX4 and GSS. The trends in these markers were reversed under conditions of ACO2 KD (Fig. 3I; Supplementary Fig. S2C). Furthermore, iron homeostasis which included ACO2 in the gene set was enriched in the sub-proteome that differed between XG and Non-XG primary tumors, and in the control vs. high/low ACO2 model systems (Figs. 1H, 2E and R, and 3H).
ACO2 translation is regulated by virtue of an iron response element in its transcript that interacts with IRP1 and IRP2 during iron deficiency (33, 45). Our in vitro and in vivo data suggests that ACO2 protein levels modulate the expression of key iron homeostasis regulatory proteins (Figs. 2E and R, and 3H). Analysis of Cancer Dependency Map (DepMap) data showed that cells sensitive to losing ACO2 are also most highly sensitive to losing TFRC, a major iron transporter (Fig. 3J), further linking ACO2 to iron homeostasis. A reliable proxy of the intracellular iron state is IRP2 level as it is ubiquitinated and degraded in iron-replete cells (46). IRP2 was significantly downregulated in H226 cells when ACO2 was increased in vitro suggesting that iron levels were increased (Fig. 3K; Supplementary Fig. S2D). Conversely, KD of ACO2 in BEAS-2B cells led to increased IRP2 indicating that decreased ACO2 caused decreased cellular iron levels (Fig. 3L). These observations suggest that ACO2 is not only a downstream effector of iron homeostasis but might also have a regulatory role.
To further examine the consequences of ACO2 expression on iron homeostasis cellular labile iron levels were measured as a function of ACO2 protein expression. Increased ACO2 led to concordant increases in cellular iron (Fig. 3M) and ACO2 KD led to concordant decrease in cellular iron (Fig. 3N). To assess whether the anti-proliferative role of ACO2 was due to increased cellular iron, the iron chelator pyridoxal isonicotinoyl hydrazine (PIH) was administered to see if it would alleviate the ACO2 effect. In a dose dependent manner PIH rescued the proliferation impairment caused by ACO2 in H226 (Fig. 3O) and HEK293 cells (Fig. S2E). Conversely, the proliferation advantage associated with ACO2 KD was eliminated by administration of iron in the form of iron chloride (Fig. 3P) or ferric ammonium citrate (Supplementary Fig. S2F), which did not affect control cells. These observations suggest that modulation of cell proliferation by ACO2 protein level is a consequence of its effects on the LIP.
ACO2 modulates mNT and the iron starvation response
Since ACO2 contains a 4Fe-4S iron-sulphur cluster, we wondered whether its influence on iron homeostasis involved dysregulation of other FeS proteins. To investigate this, a list of 70 proteins that require FeS for function or are involved in the biogenesis or transport of FeS clusters was curated (Supplementary Data 2). Comparing the FeS proteins in BEAS-2B with KD of ACO2 versus control showed mNT, as the most increased FeS protein (Fig. 4A). Conversely, in H226 xenograft tumors, increased ACO2 was associated with decreased mNT (Fig. 4B). The effect of ACO2 on FeS-associated proteins in the four systems studied herein (i.e., NSCLC primary tumors, H226 with elevated ACO2 in vitro and in vivo, and BEAS-2B with ACO2 KD) was compared. Venn analysis revealed mNT as the only FeS protein significantly altered across all 4 systems as a function of ACO2 modulation (Fig. 4C).
Figure 4.

ACO2 decreases FeS transport by inhibiting CISD1 leading to an iron starvation response. A, Control versus shACO2 in BEAS-2B shows CISD1 highest increased in shACO2 cells among FeS-ome. B, CISD1 decreases with increase of ACO2 in H226 xenograft tumors with spearman rho of −1. C, Overlap of significantly altered FeS proteins show CISD1 as the only significantly altered protein across all 4 different model systems used in this study. D, The proposed model. E, Change in TCA cycle metabolites levels with increase in ACO2 in H226 (mean±SD). F, Change in NADPH levels with increased ACO2 in T-REx 293 cells (mean±SD). G, Proliferation effect of ACO2 in IDH2 KD background in T-REx cells. H, Viability assay in T-REx 293 with ACO2 and PZH at 50 μmol/L (mean±SD). I, Viability assay in T-REx 293 cells with and without ACO2 and shCISD1 (mean±SD). J, Change in LIP levels across days of ACO2 expression in T-REx 293 cells (mean±SD). K, Change in cytosolic aconitase activity levels in 4-hour intervals within 24 hours of ACO2 expression (mean±SD). L, MS-quantified expression of IRP1 and its targets in BEAS-2B without (control) and with KD of ACO2 (shACO2) (mean±SD). M, MS-quantified expression of IRP1 targets in H226 xenograft control versus elevated ACO2 tumors. N, Viability assay for BEAS-2B cells control and shACO2 with administration of PZH at 10 pmol/L-10 μmol/L (mean±SD). Two-sided Student t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.00005.
mNT is a mitochondrial outer membrane (2Fe-2S) protein that delivers FeS clusters synthesized in the mitochondrion to specific cytosolic acceptor proteins. Among the proteins provided an FeS by mNT is IRP1 (encoded by ACO1), which contains an FeS binding site that becomes occupied under conditions of replete iron and in that holo-IRP1 state functions as a nonessential FeS-dependent cytosolic aconitase (29, 47). mNT is inhibited by NADPH, the downstream product of sequential aconitase-isocitrate dehydrogenase (IDH) reactions of the TCA cycle (31). We posited a model (Fig. 4D) in which NADPH produced by coupled ACO2-IDH2 activity leads to inhibition of mNT and consequently decreased cytosolic aconitase activity as it becomes depleted of FeS and converted into IRP1. IRP1 (like IRP2) functions as a translation factor and binds to stem-loop IREs located in iron-regulated transcripts and controls their translation or stability (48). Under conditions of iron deficiency IRP1 binds to the IRE in heavy- and light-ferritin mRNAs and blocks their translation, whereas IRP1 binding to IREs in TFRC 1 mRNA protects it from degradation. The combination of decreased ferritin and increased TFRC results in increased iron uptake and mobilization within cells (Fig. 4D). Consistent with the model, we observed that among TCA cycle metabolites, decreased citrate and increased α-ketoglutarate, the product of ACO2 plus IDH2, were significantly changed with elevated ACO2 (Fig. 4E). In addition, we observed a significant increase in cellular NADPH with elevated ACO2 in T-REx 293 cells (Fig. 4F). The antiproliferative effect of ACO2 is dependent on IDH2 because with IDH2 KD in T-REx 293 cells, elevated ACO2 had no effect on cell proliferation (Fig. 4G).
To address whether the anti-proliferative activity of ACO2 involves inhibition of mNT, we compared the effects of inhibiting mNT with and without elevated ACO2. mNT was inhibited by using the drug PZH, which has been shown to bind mNT and stabilize its bound FeS cluster and thereby prevent FeS transport to cytosolic acceptor proteins including ACO1/IRP1 (49, 50). PZH was administered in the absence or presence of induced ectopic ACO2 expression in the T-REx 293 system. PZH significantly impaired cell proliferation to an extent similar to, but not additive with that caused by ectopic ACO2 (Fig. 4H). In addition, mNT protein expression was suppressed by using shRNA (Supplementary Fig. S2G), which also inhibited cell proliferation in a manner that was not additive with that caused by ACO2 overexpression in T-REx 293 system (Fig. 4I). These results suggest that ACO2 and mNT modulate cell proliferation through a similar target or pathway.
Our model predicts that elevated ACO2 leads to inhibition of mNT leading to conversion of ACO1 into IRP1, and consequently a loss of cytosolic aconitase activity. To test this, ACO2 expression was increased over time and cytosolic aconitase activity and labile iron levels were measured in T-Rex 293 cells. In agreement with the model, increased ACO2 was accompanied by a concomitant decrease in cytosolic aconitase activity as a surrogate for conversion of ACO1 into IRP1, and increased iron levels (Fig. 4J and K). Increased ACO2 expression in the H226 system was also associated with a modest decrease (0.43-fold, FDR 0.025; Supp.Data 2) in ACO1/IRP1 at the protein level, which might reflect its documented vulnerability to proteolysis in its apo form (51).
The results suggest that increased ACO2 drives ACO1 into its apo form, IRP1. Shotgun MS quantification of BEAS-2B cells consistently showed that decreased ACO2 in BEAS-2B cells led to increased ACO1/IRP1 protein, decreased TFRC, and decreased FTL and FTH1 (Fig. 4L). In H226 XG tumors proteome analysis showed that increased ACO2 was associated with a significant decreased ferritin subunits FTL and FTH1 and increased TFRC (Fig. 4M). These results suggest that increased ACO2 causes conversion of ACO1 into IRP1 that caused increased TFRC and decreased ferritin. To test the hypothesis that the proliferation stimulation associated with low ACO2 was dependent on the activity of mNT, we examined the consequences of PZH treatment on proliferation stimulated by ACO2 KD (shACO2). We observed that BEAS-2B cells with low ACO2 were sensitive to PZH (10 nmol/L-10 μmol/L), whereas control cells with normal levels of ACO2 were not affected (Fig. 4N).
Discussion
The molecular determinants of engraftment may overlap with those responsible for more aggressive cancer phenotypes. By comparing the proteomes of XG and Non-XG NSCLC, we identified low level ACO2 as the protein feature most statistically significantly associated with engraftment. In cancers where lower ACO2 levels or activity have been reported, speculation was that low ACO2 may shift metabolism towards glycolysis and citrate-dependent lipogenesis (52, 53). However, our investigation revealed ACO2 as a regulator of cellular iron levels as a mechanism linked to cell proliferation and transformation.
The mitochondrion is a focal point for iron utilization including heme and FeS cluster biogenesis (54). It follows that mitochondria may communicate iron requisites and control cellular iron homeostasis. However, no mechanism has been elucidated as to how this occurs (55). Our findings suggest ACO2 levels communicate mitochondrial iron demands by modulating IRP1 activity, and consequently cellular iron uptake and availability. Indeed, elevated ACO2 led to increased NADPH, IRP1 and cellular iron. Our data support a model (Fig. 4D) in which mNT is inhibited downstream of ACO2+IDH-generated NAPDH. NADPH induces release of FeS from mNT (56). Elevated ACO2 was also accompanied by decreased levels of mNT, which may reflect decreased expression and/or proteolytic destruction of apo-mNT downstream of ACO2. This is consistent with observations by Crooks and colleagues (57) that lack of FeS cluster binding may be associated with decreased apo-protein abundance. Our findings suggest that mitochondrial ACO2 modulates the ACO1⇌IRP1 equilibrium and consequently cellular iron uptake and storage.
We showed that ACO2-mediated increased iron inhibits cell proliferation in vitro, and we suggest a model wherein ACO2 modulation of proliferation is a component of the aggressive phenotype required for tumorigenesis and the engraftment phenomenon. This in turns suggests that increased iron levels associated with high level ACO2 expression decreases engraftability. Consistent with our findings, positive selection for mechanisms that decreased cellular iron levels was observed in lung cancer (58), while overexpression of IRP1 in H1299 human lung cancer cells increased cellular iron and suppressed xenograft growth (59). These findings suggest that treatments that increase iron uptake be investigated as a therapeutic modality in aggressive lung cancers.
Inhibition of mNT by KD or PZH or possibly ACO2-IDH2 coupled NADPH production led to increased cellular iron and impaired cell proliferation. Interestingly, diabetes patients treated with thiozolidinediones including PZH are at significantly lower risks for lung cancer (60). ACO2 KD in normal cells stimulated cell proliferation that was inhibited by PZH. This suggests that mNT inhibition may have therapeutic potential specifically in aggressive NSCLC associated with low ACO2.
Supplementary Material
Statistical association of clinical and pathologic characteristics with engraftment
S1. The associations of ACO2 expression with NSCLC patient outcomes, and with cell line proliferation in vitro and in vivo.
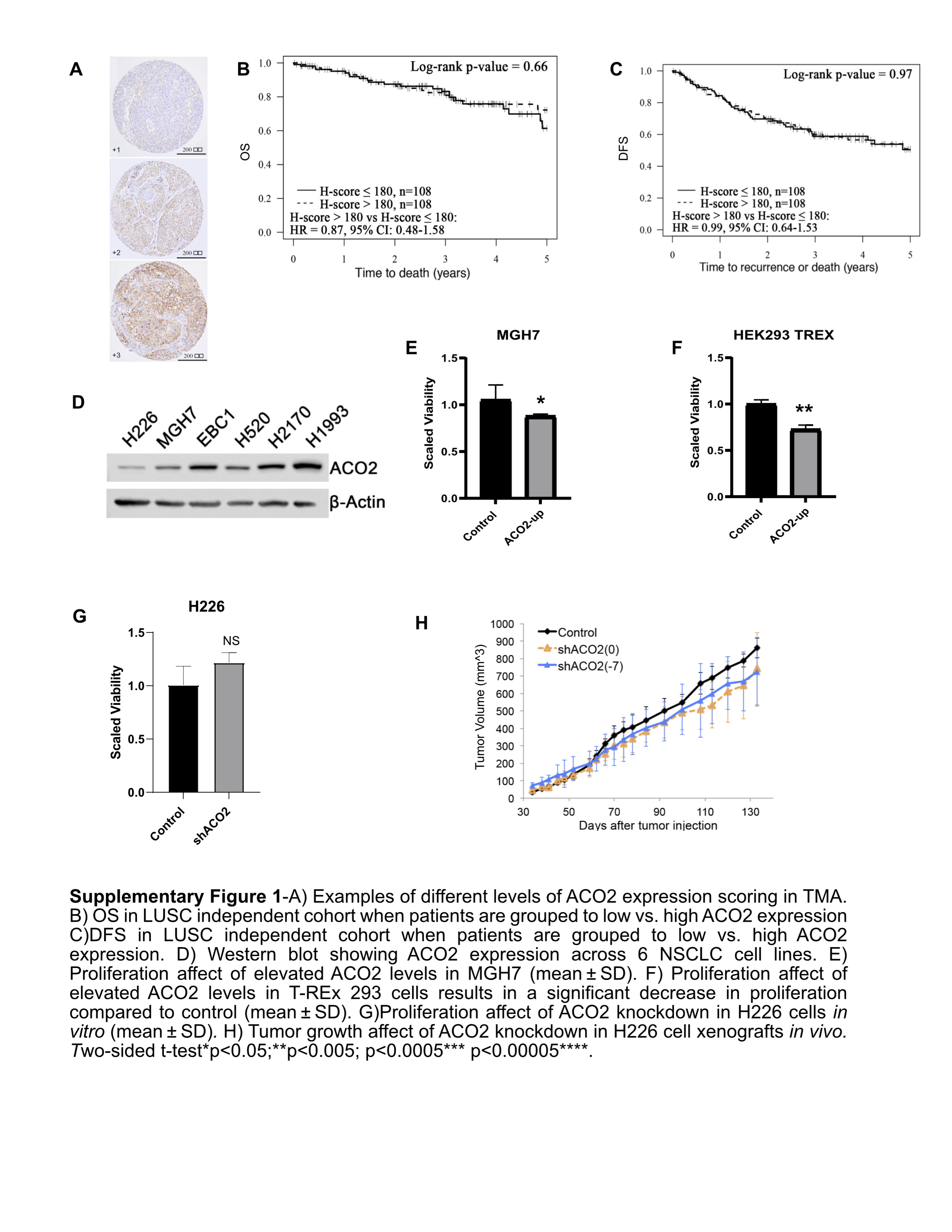{kind=link}
S2. The associations of ACO2 expression and iron on cell proliferation and expression of proteins involved in iron regulation and ferroptosis.
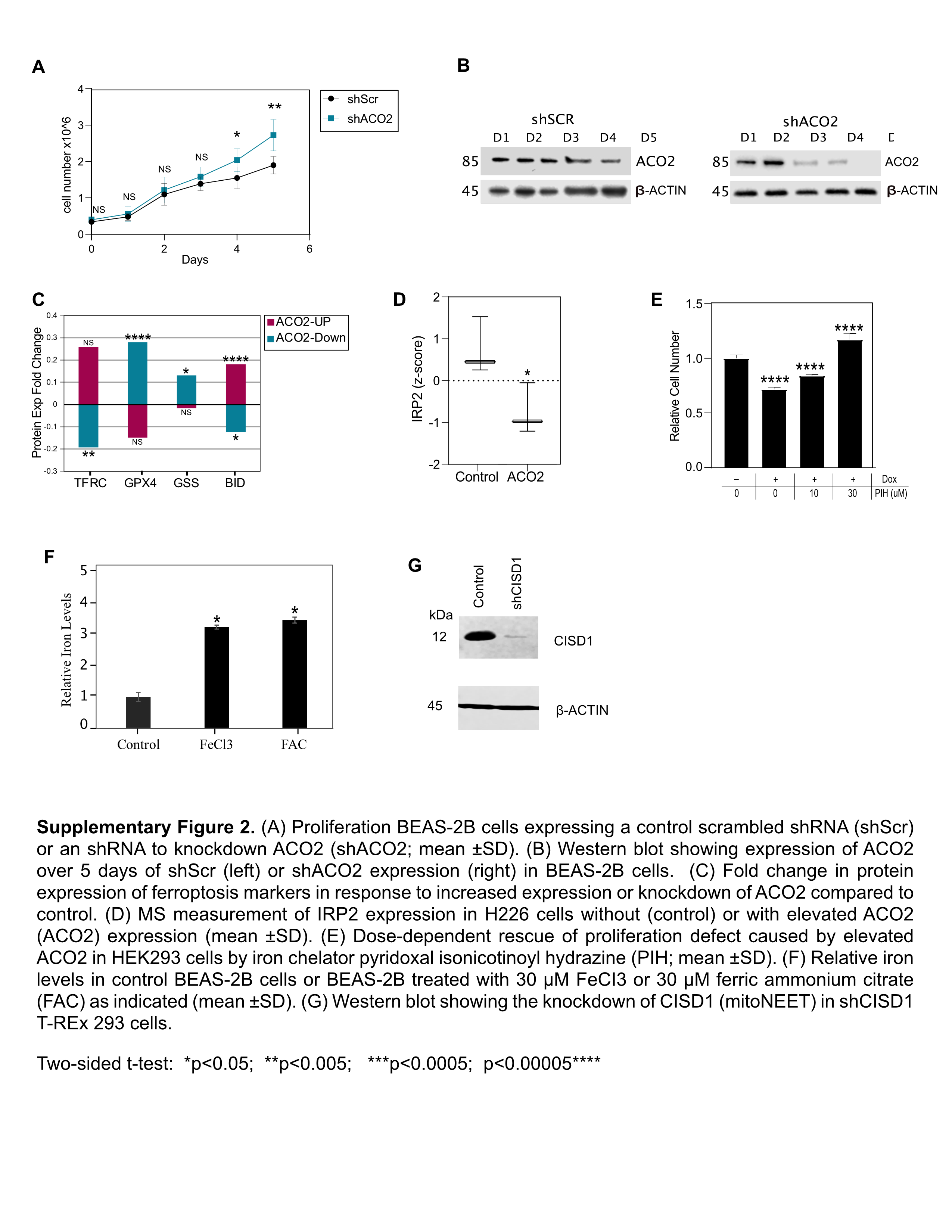{kind=link}
Clinical information and proteome quantification
Differentially expressed proteins and pathways analyses
Acknowledgments
Funds in support of this research were provided by the Canadian Cancer Society and Canadian Institutes of Health Research (to M.F. Moran and M.S. Tsao) and Canada Foundation for Innovation (CFI), Natural Sciences and Engineering Research Council of Canada, and Canada Research Chairs (to M.F. Moran). MS analyses at SPARC BioCentre were supported by CFI (#36294) and the Pan-Canadian Proteomics Centre Genomic Technology Platform Program (264PRO) of Genome Canada and Genome British Columbia.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors' Disclosures
B. Pitcher reports other support from F. Hoffmann-La Roche Ltd outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
S. Mirhadi: Conceptualization, formal analysis, investigation, writing–original draft. W. Zhang: Formal analysis, investigation, methodology, writing–original draft. N.-A. Pham: Formal analysis, investigation, project administration, writing–review and editing. F. Karimzadeh: Investigation, methodology. M. Pintilie: Formal analysis, methodology. J. Tong: Investigation, methodology. P. Taylor: Formal analysis, investigation. J. Krieger: Data curation, investigation. B. Pitcher: Data curation, formal analysis. J. Sykes: Formal analysis. L. Wybenga-Groot: Data curation, methodology, writing–review and editing. C. Fladd: Resources. J. Xu: Investigation. T. Wang: Investigation. M. Cabanero: Investigation, methodology. M. Li: Investigation, methodology. J. Weiss: Data curation, formal analysis. S. Sakashita: Investigation, methodology. O. Zaslaver: Formal analysis, investigation. M. Yu: Investigation. A.A. Caudy: Investigation. J. St-Pierre: Formal analysis, investigation. C. Hawkins: Formal analysis, investigation, methodology. T. Kislinger: Conceptualization, data curation, formal analysis, supervision, investigation, writing–review and editing. G. Liu: Conceptualization, formal analysis, writing–review and editing. F.A. Shepherd: Formal analysis, supervision, funding acquisition, writing–review and editing. M.S. Tsao: Resources, formal analysis, supervision, funding acquisition, writing–review and editing. M.F. Moran: Conceptualization, formal analysis, supervision, funding acquisition, writing–review and editing.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR, et al. Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin Cancer Res 2010;16:4864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayes DN, Monti S, Parmigiani G, Gilks CB, Naoki K, Bhattacharjee A, et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol 2006;24:5079–90. [DOI] [PubMed] [Google Scholar]
- 4. Chen F, Zhang Y, Parra E, Rodriguez J, Behrens C, Akbani R, et al. Multiplatform-based molecular subtypes of non–small cell lung cancer. Oncogene 2017;36:1384–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma: The cancer genome atlas research network. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hammerman PS, Voet D, Lawrence MS, Voet D, Jing R, Cibulskis K, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferrara R, Mezquita L, Besse B. Progress in the management of advanced thoracic malignancies in 2017. J Thorac Oncol 2018;13:301–22. [DOI] [PubMed] [Google Scholar]
- 8. Lewis DR, Check DP, Caporaso NE, Travis WD, Devesa SS. US lung cancer trends by histologic type. Cancer 2014;120:2883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu JY, Zhang C, Wang X, Zhai L, Ma Y, Mao Y, et al. Integrative proteomic characterization of human lung adenocarcinoma. Cell 2020;182:245–61. [DOI] [PubMed] [Google Scholar]
- 10. Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 2020;182:200–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y-J, Roumeliotis TI, Chang Y-H, Chen H-Y, Yang P-C, Correspondence Y-JC, et al. Proteogenomics of nonsmoking lung cancer in east Asia delineates molecular signatures of pathogenesis and progression ll proteogenomics of non-smoking lung cancer in east Asia delineates molecular signatures of pathogenesis and progression. Cell 2020;182:226–44. [DOI] [PubMed] [Google Scholar]
- 12. Stewart PA, Welsh EA, Slebos RJC, Fang B, Izumi V, Chambers M, et al. Proteogenomic landscape of squamous cell lung cancer. Nat Commun 2019; 10:3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. John T, Kohler D, Pintilie M, Yanagawa N, Pham N-A, Li M, et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non–small cell lung cancer. Clin Cancer Res 2011;17:134–41. [DOI] [PubMed] [Google Scholar]
- 14. Moro M, Bertolini G, Tortoreto M, Pastorino U, Sozzi G, Roz L. Patient-derived xenografts of non–small cell lung cancer: Resurgence of an old model for investigation of modern concepts of tailored therapy and cancer stem cells. J Biomed Biotechnol 2012;2012:568567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 2014;4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumor xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9:338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ilie M, Nunes M, Blot L, Hofman V, Long-Mira E, Butori C, et al. Setting up a wide panel of patient-derived tumor xenografts of non–small cell lung cancer by improving the preanalytical steps. Cancer Med 2015;4:201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood 2006;107:1166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oh BY, Lee WY, Jung S, Hong HK, Nam D-H, Park YA, et al. Correlation between tumor engraftment in patient-derived xenograft models and clinical outcomes in colorectal cancer patients. Oncotarget 2015;6:16059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garrido-Laguna I, Uson M, Rajeshkumar NV, Tan AC, de Oliveira E, Karikari C, et al. Tumor engraftment in nude mice and enrichment in stroma-related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin Cancer Res 2011;17:5793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med 2012;4:137ra75–ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H, et al. Ability to generate patient-derived breast cancer xenografts is enhanced in chemoresistant disease and predicts poor patient outcomes. PLoS One 2015;10:e0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pergolini I, Morales-Oyarvide V, Mino-Kenudson M, Honselmann KC, Rosenbaum MW, Nahar S, et al. Tumor engraftment in patient-derived xenografts of pancreatic ductal adenocarcinoma is associated with adverse clinicopathological features and poor survival. PLoS One 2017;12:e0182855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skowron KB, Pitroda SP, Namm JP, Balogun O, Beckett MA, Zenner ML, et al. Basal tumor cell isolation and patient-derived xenograft engraftment identify high-risk clinical bladder cancers. Sci Rep 2016;6:35854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li L, Wei Y, To C, Zhu CQ, Tong J, Pham NA, et al. Integrated Omic analysis of lung cancer reveals metabolism proteome signatures with prognostic impact. Nat Commun 2014;5:5469. [DOI] [PubMed] [Google Scholar]
- 26. Mirhadi S, Tam S, Tong J, Li Q, Moghal N, Pham N, et al. Integrative analysis of non–small cell lung cancer patient-derived xenografts identifies unique proteotypes associated with patient outcomes. Nat Commun 2022;13:1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moro M, Bertolini G, Caserini R, Borzi C, Boeri M, Fabbri A, et al. Establishment of patient-derived xenografts as functional testing of lung cancer aggressiveness. Sci Rep 2017;7:6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ferecatu I, Gonçalves S, Golinelli-Cohen M-P, Clémancey M, Martelli A, Riquier S, et al. The diabetes drug target MitoNEET governs a novel trafficking pathway to rebuild an Fe-S cluster into cytosolic aconitase/iron regulatory protein 1. J Biol Chem 2014;289:28070–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golinelli-Cohen MP, Lescop E, Mons C, Gonçalves S, Clémancey M, Santolini J, et al. Redox control of the human iron-sulfur repair protein MitoNEET activity via its iron-sulfur cluster. J Biol Chem 2016;291:7583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuris JA, Ali SS, Yeh H, Nguyen TA, Nechushtai R, Paddock ML, et al. NADPH inhibits (2Fe-2S) cluster protein transfer from diabetes drug target MitoNEET to an apo-acceptor protein. J Biol Chem 2012;287:11649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huynh N, Ou Q, Cox P, Lill R, King-Jones K. Glycogen branching enzyme controls cellular iron homeostasis via iron regulatory protein 1 and mitoNEET. Nat Commun 2019;10:5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katsarou A, Pantopoulos K. Basics and principles of cellular and systemic iron homeostasis. Mol Aspects Med 2020;75:100866. [DOI] [PubMed] [Google Scholar]
- 34. Zhang W, Wei Y, Ignatchenko V, Li L, Sakashita S, Pham NA, et al. Proteomic profiles of human lung adeno and squamous cell carcinoma using super-SILAC and label-free quantification approaches. Proteomics 2014;14:795–803. [DOI] [PubMed] [Google Scholar]
- 35. Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y, et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc 2011;6:147–57. [DOI] [PubMed] [Google Scholar]
- 36. Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc 2009;4:698–705. [DOI] [PubMed] [Google Scholar]
- 37. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72. [DOI] [PubMed] [Google Scholar]
- 38. Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011;10:1794–805. [DOI] [PubMed] [Google Scholar]
- 39. Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik I. Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem 1997;248:31–40. [DOI] [PubMed] [Google Scholar]
- 40. Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019;47:D442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang XC, Zhang J, Li M, Huang XS, Yang XN, Zhong WZ, et al. Establishment of patient-derived non–small cell lung cancer xenograft models with genetic aberrations within EGFR, KRAS and FGFR1: useful tools for preclinical studies of targeted therapies. J Transl Med 2013;11:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shenoy A, Geiger T. Super-SILAC: Current trends and future perspectives. Expert Review of Proteomics 2014;12:13–9. [DOI] [PubMed] [Google Scholar]
- 43. Geiger T, Cox J, Ostasiewicz P, Wisniewski JR, Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat Methods 2010;7:383–5. [DOI] [PubMed] [Google Scholar]
- 44. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology, and role in disease. Nat Rev Mol Cell Biol 2021;22:266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schalinske KL, Chen OS, Eisenstein RS. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. Implications for iron regulatory proteins as regulators of mitochondrial citrate utilization. J Biol Chem 1998;273:3740–6. [DOI] [PubMed] [Google Scholar]
- 46. Takahashi-Makise N, Ward DMV, Kaplan J. On the mechanism of iron sensing by IRP2: new players, new paradigms. Nat Chem Biol 2009;5:874–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tan G, Liu D, Pan F, Zhao J, Li T, Ma Y, et al. His-87 ligand in mitoNEET is crucial for the transfer of iron sulfur clusters from mitochondria to cytosolic aconitase. Biochem Biophys Res Commun 2016;470:226–32. [DOI] [PubMed] [Google Scholar]
- 48. Wilkinson N, Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol 2014;5:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab 2004;286:E252-E60. [DOI] [PubMed] [Google Scholar]
- 50. Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, et al. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci 2007;104:14342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J, Fillebeen C, Chen G, Biederbick A, Lill R, Pantopoulos K. Iron-dependent degradation of apo-IRP1 by the ubiquitin-proteasome pathway. Mol Cell Biol 2007;27:2423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang P, Mai C, Wei YL, Zhao JJ, Hu YM, Zeng ZL, et al. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med Oncol 2013;30:552. [DOI] [PubMed] [Google Scholar]
- 53. You X, Tian J, Zhang H, Guo Y, Yang J, Zhu C, et al. Loss of mitochondrial aconitase promotes colorectal cancer progression via SCD1-mediated lipid remodeling. Mol Metab 2021;48:101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem 2020;89:471–99. [DOI] [PubMed] [Google Scholar]
- 55. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci 2016;41:274–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou T, Lin J, Feng Y, Wang J. Binding of reduced nicotinamide adenine dinucleotide phosphate destabilizes the iron−sulfur clusters of human mitoNEET. Biochemistry 2010;49:9604–12. [DOI] [PubMed] [Google Scholar]
- 57. Crooks DR, Maio N, Lane AN, Jarnik M, Higashi RM, Haller RG, et al. Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J Biol Chem 2018;293:8297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumors and protects cells from ferroptosis. Nature 2017;551:639–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun 2016;478:838–44. [DOI] [PubMed] [Google Scholar]
- 60. Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, et al. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol 2007;25:1476–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical association of clinical and pathologic characteristics with engraftment
S1. The associations of ACO2 expression with NSCLC patient outcomes, and with cell line proliferation in vitro and in vivo.
S2. The associations of ACO2 expression and iron on cell proliferation and expression of proteins involved in iron regulation and ferroptosis.
Clinical information and proteome quantification
Differentially expressed proteins and pathways analyses
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (40) with the dataset identifier PXD028556.