

Abstract
Background
We analyzed the FGF/FGFR and co-alteration cancer landscape, hypothesizing that combination therapy might be useful in the presence of co-drivers.
Materials and methods
We describe FGF/FGFR-altered pathways, prognosis, and co-alterations [cBioPortal (N = 7574)] and therapeutic outcomes [University of California San Diego Molecular Tumor Board (MTB) (N = 16)].
Results
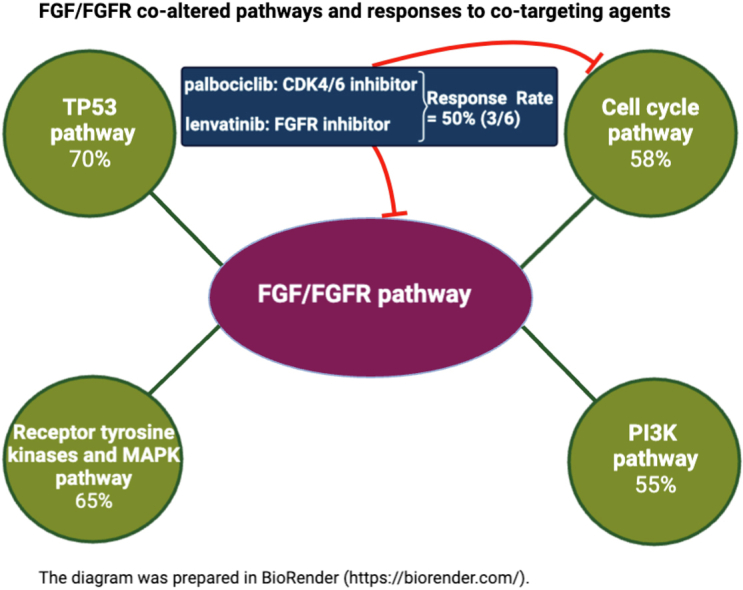
Patients whose cancers harbored FGF/FGFR alterations (N = 1074) versus those without them (N = 6500) had shorter overall survival (OS) (median: 23.1 versus 26.4 months, P = 0.038) (cBioPortal). Only 6.1% (65/1074 patients) had no pathogenic co-alterations accompanying FGF/FGFR axis abnormalities. The most frequently co-altered pathways/genes involved: TP53 (70%); cell cycle (58%); PI3K (55%); and receptor tyrosine kinases and mitogen-activated protein kinase (MAPK) (65%). Harboring alterations in both FGF/FGFR and in the TP53 pathway or in the cell cycle pathway correlated with shorter OS (versus FGF/FGFR-altered without those co-altered signals) (P = 0.0001 and 0.0065). Four of 16 fibroblast growth factor receptor (FGFR) inhibitor-treated patients presented at MTB attained durable partial responses (PRs) (9, 12, 22+, and 52+ months); an additional two, stable disease (SD) of ≥6 months (13+ and 15 months) [clinical benefit rate (SD ≥ 6 months/PR) = 38%]. Importantly, six patients with cyclin pathway co-alterations received the CDK4/6 inhibitor palbociclib (75 mg p.o. 3 weeks on, 1 week off) and the multikinase FGFR inhibitor lenvatinib (10 mg p.o. daily); three (50%) achieved a PR [9 (ovarian), 12 (biliary), and 52+ months (osteosarcoma)]. Palbociclib and lenvatinib were tolerated well.
Conclusions
FGF/FGFR alterations portend a poor prognosis and are frequently accompanied by pathogenic co-aberrations. Malignancies harboring co-alterations that activate both cyclin and FGFR pathways can be co-targeted by CDK4/6 and FGFR inhibitors.
Key words: precision cancer medicine, drug resistance, tyrosine kinase inhibitor, matched therapy, combination therapy, FGFR
Graphical abstract
Highlights
-
•
Overall, 93.9% of cancers bearing FGF/FGFR axis genomic alterations also harbored deleterious co-alterations.
-
•
Co-alterations occurred in critical pathways: TP53, cell cycle, PI3K, tyrosine kinase, and MAPK.
-
•
TP53 and cell cycle co-alterations correlated with shorter survival versus FGF/FGFR-altered without those co-aberrations.
-
•
Patients (3/6) with FGF/FGFR and cyclin alterations responded to lenvatinib and palbociclib (FGFR and CDK4/6 inhibitor).
Introduction
Fibroblast growth factor receptors (FGFRs) are highly conserved transmembrane tyrosine kinase receptors, which regulate basic biologic process such as development, differentiation, cell survival, migration, angiogenesis, and carcinogenesis.1 FGFR forms a family of four tyrosine kinase receptors (FGFR1-4), and one that lacks an intracellular tyrosine kinase domain (FGFR5).2 In humans, >20 unique ligands are identified, which are known as fibroblast growth factors (FGFs).3 Ligand binding to FGFR leads to intracellular phosphorylation of receptor kinase domains, a cascade of intracellular signaling, and gene transcription.4 Downstream signaling is triggered by intracellular receptor substrates FGFR substrate 2 (FRS2) and phospholipase Cg (PLC-g), leading to subsequent up-regulation of mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), and signal transducer and activator of transcription (STAT) signaling pathways.5
Aberrant FGFR signaling as a result of gene amplification, point mutations, and gene fusions has been observed in different tumor types; these alterations are promising targets for cancer therapeutics.6 An aberrantly activated FGF/FGFR signaling axis is likely to contribute to tumor growth and proliferation, promote neo-angiogenesis, and participate in acquired resistance to anticancer therapies. Using next-generation sequencing (NGS) with a review of nearly 5000 cancer patients, FGFR aberrations were found in 7.1% of malignancies.7
There are several non-selective multikinase inhibitors that target FGF/FGFR pathways. Food and Drug Administration (FDA)-approved agents include: ponatinib, regorafenib, pazopanib, and lenvatinib. FGFR alteration status is not required to use those medications. Recently, selective FGFR inhibitors have been investigated specifically for FGFR-altered solid tumors.8 To date, the FDA has approved erdafitinib for urothelial cancer with FGFR2 or FGFR3 alterations, as well as pemigatinib and infigratinib for cholangiocarcinoma with FGFR2 fusions or other rearrangements.9, 10, 11
Although multiple studies have found that the aberrant FGFR signaling pathway is an attractive therapeutic target, FGFR inhibitor-based therapies do not always benefit patients, even if one selects for FGFR-altered cancers.6 The potential reasons for the variable efficacy of FGFR targeting may be related to multiple factors, but we hypothesized that, in some patients, concomitant oncogenic alterations that appear along with FGFR abnormalities could be associated with primary and/or secondary resistance.7,12,13 Targeting one specific signal in a complex network of genomic drivers may not be sufficient to control cancer progression.
We have previously shown that dual inhibition of MAPK and cell cycle pathways, when both were co-altered, could be effective, even if single-agent targeting was mostly inactive.14 This has been suggested to be akin to ‘whack a mole’.15 Furthermore, recent tumor agnostic studies demonstrate that the greater the proportion of genomic alterations targeted, the better the outcome, and studies targeting single alterations may not always show salutary effects.16, 17, 18, 19, 20, 21 Combination approaches may conceivably overcome the limitation of single-agent FGFR suppression. However, there are limited data reflecting actual clinical practice regarding matched therapy targeting both the FGFR pathway and the co-alterations.
Herein, we provide evidence that FGFR pathway alterations are associated with a poor prognosis. Even so, co-targeting FGF/FGFR axis alterations and concomitant deleterious alterations showed activity in advanced refractory malignancies.
Materials and methods
Analysis of 1074 patients from cBioPortal
A total of 1074 patients with FGF/FGFR alterations of FGF3/4/19 and FGFR1/2/3/4, and 6500 patients without those FGF/FGFR alterations were analyzed from the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) Clinical Sequencing Cohort from cBioPortal (cBio cancer genomics portal) for cancer genomics to evaluate correlations between FGF/FGFR alterations and other pathway perturbations at the genomic level, as well as outcome (Supplementary Figure S1, available at https://doi.org/10.1016/j.esmoop.2022.100647).22
Study population receiving FGFR-targeted treatment after MTB presentation
A total of 715 patients with malignancies and available molecular diagnostics were discussed at the (face-to-face) University of California San Diego (UCSD) Molecular Tumor Board (MTB) (Supplementary Figure S2, available at https://doi.org/10.1016/j.esmoop.2022.100647).20 Among these patients, 16 patients who received an FGFR inhibitor to target alterations in FGF3/4/6/14/19/23, FGFR1/2/3/4, and FRS2 were identified [note: fibroblast growth factor receptor substrate 2 (FRS2) is an adaptor protein that plays a critical role in FGFR signaling].23
Somatic alteration identification and annotation
Somatic alterations were identified in tumor tissues by hybrid capture-based targeted DNA sequencing using FoundationOne CDx.24 Somatic alterations in blood-derived cell-free DNA (cfDNA) were detected by Guardant360.25 Both assays apply NGS of cancer-related genes. The figure of the landscape of somatic mutations was created using the visualized data feature by the OncoPrinter tool in cBioPortal.26,27 Somatic mutation annotation of the biological and oncogenic effects was extracted from the OncoKB knowledge base.28
Molecular matching
The MTB made recommendations to optimize molecular matching for each patient, trying to cover as many aberrant genomic alterations by cognate therapies as possible.16,18,20,21 Physicians, however, ultimately chose the patient’s therapy and they could decide to follow MTB recommendations or not.
Statistical analysis
Summary statistics were used to describe patient characteristics. We evaluated progression-free survival (PFS), which was defined as the time between the start of the treatment and disease progression confirmed by imaging or clinical findings. Overall survival (OS) was measured as the time between the onset of therapy until the last follow-up. Patients without progression at the last follow-up date were censored for PFS at that date. Patients alive at last follow-up were censored for OS at that point. PFS and OS data were represented by Kaplan–Meier estimation and the survival endpoints were compared using log-rank tests. OS data from cBioPortal represents the time to metastatic/advanced disease to death or censoring as above. Hazard ratio (HR) was computed by log-rank test and Cox regression analysis. All P values ≤ 0.05 were considered significant. All statistical analyses were carried out using R version 4.1.1.
Ethics statement
Our study was carried out according to the guidelines of the UCSD Institutional Review Board [Profile Related Evidence Determining Individualized Cancer Therapy (PREDICT), NCT02478931] and I-PREDICT (NCT02534675) and for any investigational interventions for which the patients consented.
Data for our study will be made available by the corresponding author upon reasonable request.
Results
Patients with FGF/FGFR-altered cancers have a worse prognosis than patients with wild-type FGF/FGFR
When compared to patients with FGF/FGFR-unaltered cancers (N = 6500), patients who harbored cancers with FGF/FGFR gene alterations (N = 1074) had significantly shorter OS [median OS: 26.4 months versus 23.1 months, HR: 0.89, 95% confidence interval (CI): 0.79-0.99, P = 0.038; Figure 1].
Figure 1.

Kaplan–Meier survival curves for overall survival stratified by FGF/FGFR pathway alterations (N = 7574, data from cBio cancer genomics portal). Data derived from cBio cancer genomics portal, MSK-IMPACT Clinical Sequencing Cohort http://www.cbioportal.org/). Tick marks represent censored time points for patients still alive at last follow-up. Overall survival from time of metastatic/advanced disease comparing patients with FGF/FGFR pathway-unaltered and FGF/FGFR pathway-altered cancers. FGF/FGFR pathway alterations included FGF3/4/19, FGFR1/2/3/4, and FGF6/23; FRS2 genes that were found in the UCSD cohort were not listed since they were not assessed in the MSK-IMPACT Clinical Sequencing Cohort. Overall survival analysis was based on FGF/FGFR alteration status. When compared to FGF/FGFR pathway-unaltered cases (N = 6500), FGF/FGFR pathway-altered cases (N = 1074) had worse overall survival with a HR of 0.89 (95% CI 0.79-0.99, P = 0.038). CI, confidence interval; HR, hazard ratio; MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; UCSD, University of California San Diego.
FGF/FGFR pathway alterations were frequently accompanied by alterations in other signaling pathways
When interrogating the cBioPortal cohort (N = 1074), the most frequently co-altered pathways/genes were: TP53 [70% (753/1074)]; cell cycle [58% (624/1074)]; PI3K pathway [55% (588/1074)]; receptor tyrosine kinases and MAPK pathway [65% (694/1074)]; and other genomic alterations [78% (834/1074) patients] (Figure 2A). Only 65 patients of 1074 (6.1%) had no co-alterations accompanying their FGF/FGFR axis genomic abnormalities.
Figure 2.


Summary of selected co-altered oncogenic genes along withFGF/FGFRpathway alterations. (A) Integrated view of oncogenic genes co-occurring with FGF/FGFR alterations from cBio cancer genomics portal (N = 1074, MSK-IMPACT Clinical Sequencing Cohort, data from cBio cancer genomics portal, http://www.cbioportal.org/). Each column denotes an individual patient, and each row displays a gene. Genetic alterations are color coded by the type of alterations. Co-alterations in oncogenic pathways were observed in TP53 pathway genes (70%), cell cycle–associated genes (58%), receptor tyrosine kinases and MAPK pathway-associated genes (65%), PI3K signaling-associated genes (55%), and other genomic alteration genes (78%). FGF6, FGF23, FRS2, TERC, ZNF703, CHD4, and PRKCL genes that were found in the UCSD cohort were not listed since they were not assessed in the MSK-IMPACT Clinical Sequencing Cohort. Selected genes of relevant pathways were listed. The diagram was prepared in OncoPrinter tool using cBio cancer genomics portal (http://www.cbioportal.org/). (B) Integrated view of co-occurring somatic oncogenic alterations in 16 cases with FGF/FGFR pathway alterations who received FGFR inhibitors (N = 16, UCSD patients). Each column denotes an individual patient, and each row displays a gene. Genetic alterations are color coded by the type of alterations. Co-alterations in oncogenic pathways were observed in TP53 pathway genes (71%), cell cycle–associated genes (53%), receptor tyrosine kinases and MAPK pathway-associated genes (35%), PI3K signaling-associated genes (41%), and other genomic alteration genes (76%). The diagram was prepared in OncoPrinter tool using cBio cancer genomics portal (http://www.cbioportal.org/). MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; UCSD, University of California San Diego.
The landscape of FGF/FGFR pathway and co-occurring alternations in the UCSD cohort of FGFR inhibitor-treated patients (N = 16) is shown in Figure 2B and in Tables 1 and 2 and is similar to those derived from the larger cBioPortal cohort with regard to the commonly co-altered genes/pathways accompanying FGF/FGFR axis aberrations. The median number of alterations in the FGF/FGFR genes was 1 (range, 1-4). Alterations in FGF3, FGF4, FGF19, and CCND1 often co-occurred, probably because they reside on the same amplicon on 11q13. Alterations in MDM2 and FRS2 were mostly concomitant, probably because they reside on the same amplicon on 12q15. Among UCSD patients with FGF/FGFR pathway alterations, a wide variety of gene co-alterations were seen (Figure 2B). The most frequently co-altered pathways/genes were: TP53 [69% (11/16)]; cell cycle [50% (8/16)]; PI3K pathway [38% (6/16)]; receptor tyrosine kinases and MAPK pathway [31% (5/16)]; and other genomic alterations [75% (12/16)] patients.
Table 1.
Characteristics of patients with FGF/FGFR pathway gene alterations who received FGFR inhibitor-based therapies (N = 16; UCSD cohort)
| Patient characteristics | N = 16 |
|---|---|
| Median agea (range), year | 61 (21-82) |
| Sex, n (%) | |
| Men | 6 (37.5) |
| Women | 10 (62.5) |
| Type of cancer, n (%) | |
| Gastroesophageal | 4 (25.0) |
| Biliary | 3 (18.8) |
| Bladder | 1 (6.3) |
| Urothelial | 1 (6.3) |
| Ovarian | 1 (6.3) |
| Endometrial | 1 (6.3) |
| Glioneuronal | 1 (6.3) |
| Osteosarcoma | 1 (6.3) |
| Gastrointestinal stromal | 1 (6.3) |
| Adenoid cystic carcinoma | 1 (6.3) |
| Undifferentiated sarcoma | 1 (6.3) |
| FGF/FGFR alterations,bn (%) | |
| Amplification | 12 (75.0) |
| Single-nucleotide variant | 3 (18.9) |
| Fusion | 2 (12.5) |
| Type of FGFR inhibitor, n (%)c | |
| Lenvatinib | 12 (75.0) |
| Ponatinib | 2 (12.5) |
| Pazopanib | 1 (6.3) |
| Infigratinib | 1 (6.3) |
| Number of molecularly matched drugs, n (%) | |
| Monotherapy | 5 (31.3) |
| ≥Two matched therapies | 11 (68.8) |
| Best response, n (%) | |
| Progressive disease or stable disease <6 months | 10 (62.5) |
| Stable disease ≥6 months | 3 (18.8) |
| Partial response | 3 (18.8) |
| Median number of lines of prior therapies (range) | 2 (1-12) |
| Median number of deleterious co-alterations (range)d | 5 (0-10) |
| Median progression-free survival, month (range) | 4.6 (2.8-51.7) |
| Median overall survival, month (range) | 13.5 (1.2-51.7) |
FGFR, fibroblast growth factor receptor; UCSD, University of California San Diego.
Age at the time of metastatic/locally advanced disease.
One patient had both amplification and a single-nucleotide variant.
See Supplementary Table S1, available at https://doi.org/10.1016/j.esmoop.2022.100647 for 50% inhibitory concentrations (IC50) of these drugs.
Variants of unknown significance excluded.
Table 2.
Clinical and molecular characteristics as well as clinical outcomes of patients treated with FGFR inhibitor-based therapies (N = 16)
| ID | Age, years | Sex | Diagnosis | Molecular characteristics (laboratory vendor, source) | PD-L1 CPSa | TMB (muts/Mb) | Treatment regimen | Number of prior line of therapies | PFS (month) | OS (month) | Best response |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 37 | F | Gastroesophageal cancer | (FM, tissue) FGFR2 amplification CDKN2A loss MYC amplification APC I1307K ARID1A P2139fs∗62 TP53 F113C |
Unknown | Unknown | Ponatinib | 6 | 2.3 | 2.3 | PD |
| 2 | 78 | M | Bladder cancer | (FM, tissue) FGFR1 amplification NF1 Q1218∗ TP53 R267G ERBB2 I767M MLL2 P3668fs∗5, splice site 177-1G>T ZNF703 amplification |
Unknown | Unknown | Pazopanib | 3 | 1.2 | 1.2 | PD |
| 3 | 72 | M | Urothelial cancer | (FM, tissue) FRS2 amplification AKT2 amplification BRIP1 truncation PIK3CA H450_V461>GS RAF1 amplification MDM2 amplification MYC amplification RNF43 S262∗ ARID2 S889∗ (GH, blood) FGFR2 V516L, G131R |
Unknown | Unknown | Lenvatinib, olaparib | 2 | 5.8 | 10.2 | SD <6 months |
| 4 | 33 | F | Biliary cancer |
(FM, tissue) FGFR2-BICC1 fusion |
Unknown | Unknown | Infigratinib | 2 | 21.6+ | 21.6+ | PR |
| 5 | 47 | F | Biliary cancer | (FM, tissue) FGFR2-BICC2 fusion POLE R446Q (GH, blood) PIK3CA amplification |
Unknown | 2.4 | Lenvatinib, everolimus | 2 | 3.7 | 3.7 | SD <6 months |
| 6 | 81 | M | Biliary cancer |
(FM, tissue) FRS2 amplification MDM2 amplification CDKN2A p16INK4a R80∗, p14ARF P94L CEBPA G103_G104del |
Unknown | Unknown | Lenvatinib, palbociclib | 1 | 11.8 | 12.0 | PR |
| 7 | 30 | F | Osteosarcoma |
(FM, tissue) FGF23 amplification FGF6 amplification FRS2 amplification CDK4 amplification CCND2 amplification MDM2 amplification |
Unknown | 3 | Lenvatinib, palbociclib | 4 | 51.7+ | 51.7+ |
PR (per PERCIST) Note: At 32 months, the patient had a new lesion that was resected; she has continued on therapy, doing well at 51.7+ months |
| 8 | 60 | M | Gastroesophageal cancer | (FM, tissue) FGF19 amplification FGF3 amplification FGF4 amplification CCND1 amplification CDK6 amplification MET amplification ARID1A R1276∗ TERC amplification TP53 P278L |
10 | 8 | Lenvatinib, palbociclib, nivolumab | 2 | 0.9 | 2.5+ | PD |
| 9 | 21 | F | Glioneuronal tumor |
(FM, tissue) FGFR1 K656E |
Unknown | 2 | Lenvatinib | 2 | 15.1 | 30.6+ | SD ≥6 months |
| 10 | 73 | F | Endometrial cancer | (FM, tissue) FGFR2 N549K PIK3CA G1049R PTEN K125N ARID1A Q2115fs∗33 CHD4 R975H CTCF S282fs∗21 MLL3 S2123∗ TP53 Y163C |
2 | 1 | Lenvatinib, everolimus | 1 | 3.2 | 22.1 | PD |
| 11 | 54 | M | Gastroesophageal cancer | (FM, tissue) FGF19 amplification FGF3 amplification FGF4 amplification CCND1 amplification CDK6 amplification CDKN2A/B loss PIK3CA amplification SOX2 amplification PIK3CG amplification PRKCI amplification TERC amplification TP53 G245D |
1 | 7 | Lenvatinib, palbociclib, nivolumab | 1 | 2.7 | 2.7 | PD |
| 12 | 82 | M | Gastroesophageal cancer | (FM, tissue) FGFR2 amplification TP53 A159V |
Unknown | 4 | Lenvatinib | 1 | 4.7 | 6.5 | SD <6 months |
| 13 | 63 | F | Ovarian cancer |
(FM, tissue) FGFR4 amplification FLT4 amplification PDGFRB amplification CDK6 amplification TP53 K132R (GH, blood) TP53 K132, K120M PIK3CA E545K MET amplification PDGFRA amplification KIT amplification |
Unknown | 6 | Lenvatinib, palbociclib | 12 | 8.9 | 17.6+ | PR |
| 14 | 72 | F | Gastrointestinal stromal tumor |
(FM, tissue) KIT K558_E562del, N822K, V654A ARID1A truncation NOTCH2 P6fs∗27 (GH, blood) FGFR1 amplification MYC amplification ERBB2 amplification |
Unknown | 7 | Lenvatinib, pembrolizumab | 4 | 13.1+ | 13.1+ | SD ≥6 months |
| 15 | 61 | F | Adenoid cystic carcinoma | (FM, tissue) FGF19 amplification FGF3 amplification FGF4 amplification CCND1 amplification FANCA F1263del (GH, blood) FGFR1 amplification |
Unknown | Unknown | Lenvatinib, palbocicib | 4 | 2.8 | 2.8+ | PD |
| 16 | 47 | F | Undifferentiated sarcoma | (FM, tissue) FGFR3 amplification AKT2 amplification BRCA2 R1190W CCNE1 amplification CDK4 amplification MDM2 amplification AR amplification |
Unknown | Unknown | Pazopanib, everolimus | 1 | 4.4 | 15.2 | SD <6 months |
CPS, combined positive score; FM, Foundation Medicine; GH, Guardant Health; muts/Mb, mutations per megabase; PD progressive disease; PD-L1, programmed death ligand-1; PR, partial response; SD, stable disease; TMB, tumor mutational burden.
Cases with SD >6 months/PR were highlighted in bold.
PD-L1 was assessed with the 22C3 antibody.
Patients harboring cancers with FGF/FGFR pathway gene alterations accompanied by TP53 pathway or cell cycle pathway alterations had worse overall survival compared to patients with FGF/FGFR-altered cancers without those co-alterations
Interrogating all 1074 individuals with FGF/FGFR-altered tumors, those harboring alterations in both FGF/FGFR and in the TP53 pathway (HR: 0.61, 95% CI 0.48-0.79, P = 0.0001; Figure 3A) or in the cell cycle pathway (along with the FGF/FGFR genes) (HR: 0.74, 95% CI 0.59-0.92, P = 0.0065; Figure 3B) had significantly shorter OS when compared to patients with FGF/FGFR-altered cancers without those co-altered anomalies. However, patients who had both FGF/FGFR abnormalities and co-alterations in receptor tyrosine kinases or MAPK or PI3K pathway showed no significant difference in OS when compared to patients with cancers harboring FGF/FGFR pathway alterations without those co-altered pathways (Figure 3C and D).
Figure 3.


Overallsurvival among patients with FGF/FGFR pathway alterations stratified by co-occurring alterations in TP53 pathway (TP53, MDM2), cell cycle pathway (CDKN2A, CDKN2B, CCND1, CCND2, CDK4, CDK6), receptor tyrosine kinases and MAPK pathway (EGFR, ERBB2, ERBB3, ERBB4, MET, PDGFRA, KIT, IGF1R, RET, ROS1, ALK, FLT3, NTRK1, NTRK2, NTRK3, NF1, KRAS, HRAS, NRAS, ARAF, BRAF, RAF1, MAP2K1, MAP2K2, MAPK1), and PI3K pathway (PIK3CA, AKT2, PTEN, PIK3CG (N = 1074, data from cBio cancer genomics portal). Data derived from cBioPortal cancer genomics, MSK-IMPACT Clinical Sequencing Cohort. (A) Overall survival comparing the impact of FGF/FGFR and TP53 pathway alterations. When compared to FGF/FGFR pathway-altered/TP53 pathway-unaltered cases (N = 321), FGF/FGFR pathway-altered/TP53 pathway-altered cases (N = 753) had significantly worse overall survival (HR of 0.61, 95% CI 0.48-0.79, P < 0.001). (B) Overall survival comparing the impact of FGF/FGFR and cell cycle pathway alterations. When compared to FGF/FGFR pathway-altered/cell cycle pathway-unaltered cases (N = 450), FGF/FGFR pathway-altered/cell cycle pathway-altered cases (N = 624) had significantly worse overall survival (HR of 0.74, 95% CI 0.59-0.92, P = 0.007). (C) Overall survival comparing the impact of FGF/FGFR and receptor tyrosine kinases and MAPK pathway alterations. When compared to FGF/FGFR pathway-altered/receptor tyrosine kinases and MAPK pathway-unaltered cases (N = 380), FGF/FGFR pathway-altered/receptor tyrosine kinases and MAPK pathway-altered cases (N = 694) did not demonstrate a significant difference in overall survival (HR of 0.88, 95% CI 0.71-1.10, P = 0.27). (D) Overall survival comparing the impact of FGF/FGFR and PI3K pathway alterations. When compared to FGF/FGFR pathway-altered/PI3K pathway-unaltered cases (N = 486), FGF/FGFR pathway-altered/PI3K pathway-altered cases (N = 588) did not demonstrate a significant difference in overall survival (HR of 0.99, 95% CI 0.80-1.23, P = 0.96). See Figure 2A and B for the list of selected genes associated with the pathway alterations. CI, confidence interval; HR, hazard ratio; NA, not available.
Characteristics of patients with FGF/FGFR-altered cancers who received FGFR inhibitor therapy
Sixteen patients with FGF/FGFR pathway alterations received FGFR inhibitors (Supplementary Figure S2, available at https://doi.org/10.1016/j.esmoop.2022.100647) at UCSD. Demographics and patient characteristics are shown in Table 1. The median age was 61 years. Six patients (37.5%) were men. The most common diagnosis was gastroesophageal cancer [25.0% (4/16)], followed by biliary cancer [18.8% (3/16)]. FGF/FGFR amplification occurred in 12 patients, single-nucleotide variant (SNV) in three patients, and fusions in two patients (Table 1). One patient had both amplification and SNV. Five patients (31%) received FGFR inhibitors as monotherapy; 11 patients (69%) received a customized combination. The median number of prior therapies was 2 (range, 1-12). The median number of co-alterations, excluding FGF/FGFR alterations, was 5 (range, 0-10). The median PFS and OS were 4.6 months and 13.5 months, respectively.
FGFR inhibitors have activity in the UCSD cohort of patients with FGF/FGFR axis alterations, especially in combination with cyclin inhibitors when cyclin-activating genes are co-altered
Overall, 16 patients with FGF/FGFR alterations were treated with drugs with potent FGFR inhibitory activity (5 with monotherapy and 11 with an FGFR inhibitor combined with one or more other drugs that matched co-activated signals) (Supplementary Tables S1 and S2, available at https://doi.org/10.1016/j.esmoop.2022.100647). Partial responses (PRs) were seen in four patients (9, 12, and 22+, and 52+ months) and an additional two patients achieved stable disease (SD) ≥6 months (13+ and 15 months). Therefore, 6 of 16 patients attained clinical benefit (SD ≥ 6 months/PR) [clinical benefit rate (CBR): 38%] (Table 2).
Five patients received monotherapy with an FGFR inhibitor; two achieved SD ≥ 6 months/PR—both with single alterations on NGS [FGFR2 fusion (biliary tract cancer treated with infigratinib) and FGFR K656E mutation (glioneuronal tumor treated with lenvatinib)]; the other patients, who did not respond, had pathogenic genomic co-alterations in their cancers.
Four of 11 patients (36%) who received customized combination therapy achieved SD ≥ 6 months/PR; all of these patients had tumors harboring pathogenic co-alterations in addition to their FGF/FGFR genomic anomalies. These four patients had malignancies as follows (Table 2 and Supplementary Table S2, available at https://doi.org/10.1016/j.esmoop.2022.100647): (i) a biliary cancer with an FRS2 amplification and CDKN2A alteration treated with a combination of the FGFR inhibitor lenvatinib and the CDK4/6 inhibitor palbociclib (achieving a PR for ∼12 months); (ii) a patient with osteosarcoma whose tumor harbored amplifications in FGF23, FGF6, FRS2 as well as CDK4 and CCND2 amplifications [treated with lenvatinib and palbociclib with an ongoing PERCIST 1.0 (PET) response at 52+ months29]; (iii) an ovarian cancer harboring FGFR4 and CDK6 amplification (also treated with lenvatinib and palbociclib with a PR that lasted ∼9 months); and (iv) a gastrointestinal stromal tumor (status after four prior systemic therapies) with FGFR1 amplification as well as MYC and ERBB2 amplification, and KIT, ARID1A, and NOTCH2 mutations treated with lenvatinib and pembrolizumab (ARID1A alterations may induce sensitivity to immune checkpoint blockade30) (achieving SD ongoing at 13.1+ months).
Importantly, six patients received a combination of drugs that included the CDK4/6 inhibitor palbociclib (administered because of the presence of alterations in the cyclin pathway) and the multi-tyrosine kinase inhibitor (including FGFR inhibitor) lenvatinib. Three of the six patients (50%) achieved a PR (lasting 9, 12, and 52+ months) (Table 2). The doses used were palbociclib 75 mg p.o. 3 weeks on/1 week off (approved dose = 125 mg p.o. 3 weeks on and 1 week off) and lenvatinib 10 mg p.o. daily (approved dose = 24 mg p.o. daily). In each case, the lower modified dose was the starting dose, and because tolerability was good and efficacy was seen, the doses were not further titrated upwards. The most common side effects in these patients were grade 1-2 rash and fatigue, which were manageable without dose adjustments. There was one patient who experienced grade 3 neutropenia and required dose modification of palbociclib (dose decreased to 75 mg p.o. 1 week on and 1 week off).
Discussion
Herein we report prognostic and predictive observations related to the presence of FGF/FGFR genomic alterations in a group of pan-cancer patients. Our results indicate that FGF/FGFR axis alterations portend a poor prognosis, as reflected by the observation that patients whose malignancies harbor FGF/FGFR genomic alterations have significantly shorter survival than those with malignancies that are wild type for FGF/FGFR. These results are consistent with prior observations of worse prognosis in patients whose tumors harbor FGFR amplification.31,32
Importantly, 94% of 1074 cancers harbored genomic co-alterations in addition to the FGF/FGFR genomic abnormalities. The most common co-altered pathways/genes were in the TP53 axis (TP53 or MDM2 genes) (70% of cancers); cell cycle (58%); PI3K pathway (55%); and receptor tyrosine kinases and MAPK pathway (65%). Additional genomic alterations were observed in 78% of patients (Figure 2A). The presence of FGF/FGFR axis genomic alterations along with TP53/MDM2 or cell cycle pathway alterations correlated with worse OS compared to individuals with FGF/FGFR-altered cancers without those co-alterations. Co-alterations in receptor tyrosine kinases or MAPK or PI3K pathway did not show prognostic significance.
Sixteen patients with diverse malignancies presented at the UCSD face-to-face MTB20 had FGF/FGFR pathway genomic alterations and were treated with cognate FGFR inhibitors, either alone or together with agents that targeted pathogenic co-alterations. Targeting FGFR signaling alone or together with co-alterations achieved a clinical benefit rate (SD ≥ 6 months/PR/CR) of 38% (6/16) and a median PFS of 14 months among those six patients. Most of the tumors [88% (14/16)] had genomic co-alterations along with the FGF/FGFR pathway abnormalities. These co-alterations could conceivably be associated with primary resistance to FGFR inhibition. Indeed, it has been recognized that targeting FGF/FGFR pathway alterations is complicated.13 Some challenges include differences in antitumor activity among different cancer diagnoses, and variable responses depending on the type of FGFR alterations [fusions and mutations have been reported to achieve higher clinical benefit from FGFR-tyrosine kinase inhibitors (TKIs) when compared to amplification].8,33 Moreover, multiple different mechanisms for primary and acquired resistance to FGFR-targeted therapies have been revealed.34, 35, 36
We hypothesized that resistance to FGFR inhibitors in FGF/FGFR-altered cancers could be due, in some cases, to genomic complexity with co-alterations that differ from patient to patient. In our cohort, in line with our hypothesis, 14 of our 16 patients had ≥1 potentially important molecular co-alterations in their cancer. A similar phenomenon was observed in the MSK-IMPACT Clinical Sequencing Cohort. The median number of deleterious co-alterations, excluding FGF/FGFR alterations, in the UCSD cohort was 5 (range, 0-10).
With regard to FGFR inhibitor therapy, altogether 6 out of 16 of our patients derived clinical benefit [SD ≥ 6 months (N = 2)/PR (N = 4)]. The two patients with FGFR alterations (a fusion and a mutation) and no co-alterations responded to single-agent FGFR inhibitors. The other four responders received a combination regimen that targeted FGFR and one or more of the co-alterations.
The most common combination utilized included palbociclib (CDK4/6 inhibitor) (given due to cyclin pathway genomic abnormalities) and the potent multi-tyrosine kinase FGFR inhibitor lenvatinib (given to six patients). Three of the six patients (50%) attained a PR (duration 9, 12, and 52+ months). The doses used were palbociclib 75 mg p.o. for 3 weeks on and 1 week off (approved dose = 125 mg p.o. 3 weeks on and 1 week off) and lenvatinib 10 mg p.o. daily (approved dose = 24 mg p.o. daily). In each case, there were no serious adverse events and efficacy was observed in half of the patients, despite the fact that doses were substantially lower than those approved by the FDA for the individual drugs.
It has been reported that tumors with FGFR fusions appear to respond to FGFR inhibition.13,37,38 In our cohort, there were two patients with an FGFR fusion. One patient attained a durable PR, and the other patient did not respond; both patients had biliary cancers. There were several potential reasons for different clinical outcomes. First, the patient who achieved a PR had no co-alterations, while the other patient had concomitant molecular alterations, including in the PI3K pathway, a signal transduction pathway downstream of FGFR pathway.39 Co-occurrence of FGF/FGFR and PI3K pathway alterations could imply cooperation as potential dual oncogenic drivers.40, 41, 42 Therefore, an FGFR inhibitor as a monotherapy would not disrupt oncogenic signaling driven by PI3K pathway alterations. Preclinical studies have reported a high synergistic activity between PI3K and FGFR inhibitors.43, 44, 45 However, the early phase combination trial was challenging because of difficulty with tolerability with the high rates of treatment interruptions and dose modification.46 Another factor may be that, while lenvatinib is a potent FGFR inhibitor, infigratinib (which was the drug administered to the patient who responded) has an even lower IC50 for FGFR (Supplementary Table S1, available at https://doi.org/10.1016/j.esmoop.2022.100647). Finally, the fusion FGFR2-BICC1 in the case of the patient who attained a PR is known to be a typical oncogenic driver, while FGFR2-BICC2 in the other patient may not be as clear-cut.8
There are several limitations to the current report. Firstly, the therapeutic arm of the study has a small sample size and a variety of histologies (though the latter may also suggest generalizability of results). Secondly, molecular characteristics of tumors could evolve, particularly with therapeutic pressure, and serial molecular follow-up was not available. Future investigations may require molecular sequential analysis, such as monitoring by circulating tumor cell-free DNA analysis. Thirdly, some FGF/FGFR pathway alterations might not be pivotal for cancer cells, and targeting other accompanied signaling pathways might have a greater effect. Finally, most of the FGFR inhibitors used in this study were not selective to the FGF/FGFR pathway. However, despite these limitations, the current research targeting FGF/FGFR signaling in real-world practice provides evidence for the importance of impacting the products of co-occurring alterations, especially in the cyclin pathway.
In conclusion, we demonstrate that FGF/FGFR-altered tumors have a poor prognosis and frequent co-alterations in several important pathways. Co-targeting of cyclin and FGF/FGFR alterations with the CDK4/6 inhibitor palbociclib and the FGFR inhibitor lenvatinib can be carried out safely, with responses seen in three of six patients, including ongoing benefit in a patient with refractory osteosarcoma who continues to do well at 52+ months. Further prospective evaluation of this strategy is warranted in patients whose tumors harbor FGF/FGFR pathway alterations.
Funding
None declared.
Disclosure
SI received speaker’s fee from Roche, Chugai pharmaceutical Co., Merck, AstraZeneca, MSD, Taho pharmaceutical Co., Novartis, Boehringer Ingelheim, Act Med, Bayer, Takeda, Guardant Health, IQVIA, and Cannon Medical. He received research funding from ACT Genomics, Cannon Medical, and Hitachi Systems. SK serves as a consultant for Medpace, Foundation Medicine, NeoGenomics, and CureMatch. He receives speaker’s fees from Roche and Bayer, and advisory board for Pfizer. He has research funding from ACT Genomics, Sysmex, Konica Minolta, OmniSeq, and Personalis. JKS receives research funding from Novartis Pharmaceuticals, Amgen Pharmaceuticals, and Foundation Medicine; consultant fees from Grand Rounds, Loxo, and Deciphera; and speaker’s fees from Roche and Deciphera. He also owns stocks in Personalis. RK has received research funding from Biological Dynamics, Boehringer Ingelheim, Debiopharm, Foundation Medicine, Genentech, Grifols, Guardant, Incyte, Konica Minolta, Medimmune, Merck Serono, Omniseq, Pfizer, Sequenom, Takeda, and TopAlliance; as well as consultant and/or speaker fees and/or advisory board for Actuate Therapeutics, AstraZeneca, Bicara Therapeutics, Biological Dynamics, Daiichi, EISAI, EOM Pharmaceuticals, Iylon, Merck, NeoGenomics, Neomed, Pfizer, Prosperdtx, Roche, TD2/Volastra, Turning Point Therapeutics, X-Biotech; has an equity interest in CureMatch Inc., CureMetrix, and IDbyDNA; serves on the Board of CureMatch and CureMetrix, and is a co-founder of CureMatch. All other authors have declared no conflicts of interest.
Supplementary data
References
- 1.Helsten T., Schwaederle M., Kurzrock R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: biologic and clinical implications. Cancer Metastasis Rev. 2015;34(3):479–496. doi: 10.1007/s10555-015-9579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sleeman M., Fraser J., McDonald M., et al. Identification of a new fibroblast growth factor receptor. FGFR5. Gene. 2001;271(2):171–182. doi: 10.1016/s0378-1119(01)00518-2. [DOI] [PubMed] [Google Scholar]
- 3.Mossahebi-Mohammadi M., Quan M., Zhang J.S., Li X. FGF signaling pathway: a key regulator of stem cell pluripotency. Front Cell Dev Biol. 2020;8:79. doi: 10.3389/fcell.2020.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105–122. doi: 10.1038/s41571-018-0115-y. [DOI] [PubMed] [Google Scholar]
- 5.Touat M., Ileana E., Postel-Vinay S., André F., Soria J.C. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21(12):2684–2694. doi: 10.1158/1078-0432.CCR-14-2329. [DOI] [PubMed] [Google Scholar]
- 6.Babina I.S., Turner N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318–332. doi: 10.1038/nrc.2017.8. [DOI] [PubMed] [Google Scholar]
- 7.Helsten T., Elkin S., Arthur E., Tomson B.N., Carter J., Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–267. doi: 10.1158/1078-0432.CCR-14-3212. [DOI] [PubMed] [Google Scholar]
- 8.Facchinetti F., Hollebecque A., Bahleda R., et al. Facts and new hopes on selective FGFR inhibitors in solid tumors. Clin Cancer Res. 2020;26(4):764–774. doi: 10.1158/1078-0432.CCR-19-2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Javle M., Roychowdhury S., Kelley R.K., et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021;6(10):803–815. doi: 10.1016/S2468-1253(21)00196-5. [DOI] [PubMed] [Google Scholar]
- 10.Abou-Alfa G.K., Sahai V., Hollebecque A., et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–684. doi: 10.1016/S1470-2045(20)30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loriot Y., Necchi A., Park S.H., et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338–348. doi: 10.1056/NEJMoa1817323. [DOI] [PubMed] [Google Scholar]
- 12.Zuo W., He Y., Li W., et al. Landscape of FGF/FGFR alterations in 12,372 Chinese cancer patients. J Cancer. 2020;11(22):6695–6699. doi: 10.7150/jca.49269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chae Y.K., Hong F., Vaklavas C., et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH trial (EAY131) Subprotocol W. J Clin Oncol. 2020;38(21):2407–2417. doi: 10.1200/JCO.19.02630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato S., Adashek J.J., Shaya J., et al. Concomitant MEK and cyclin gene alterations: implications for response to targeted therapeutics. Clin Cancer Res. 2021;27(10):2792–2797. doi: 10.1158/1078-0432.CCR-20-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groisberg R., Subbiah V. Combination therapies for precision oncology: the ultimate whack-a-mole game. Clin Cancer Res. 2021;27(10):2672–2674. doi: 10.1158/1078-0432.CCR-21-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sicklick J.K., Kato S., Okamura R., et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–750. doi: 10.1038/s41591-019-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tourneau C.L., Delord J.P., Gonçalves A., et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 18.Schwaederle M., Parker B.A., Schwab R.B., et al. Precision oncology: the UC San Diego Moores Cancer Center PREDICT experience. Mol Cancer Ther. 2016;15(4):743–752. doi: 10.1158/1535-7163.MCT-15-0795. [DOI] [PubMed] [Google Scholar]
- 19.Rodon J., Soria J.C., Berger R., et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25(5):751–758. doi: 10.1038/s41591-019-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato S., Kim K.H., Lim H.J., et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun. 2020;11(1):4965. doi: 10.1038/s41467-020-18613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sicklick J.K., Kato S., Okamura R., et al. Molecular profiling of advanced malignancies guides first-line N-of-1 treatments in the I-PREDICT treatment-naïve study. Genome Med. 2021;13(1):155. doi: 10.1186/s13073-021-00969-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zehir A., Benayed R., Shah R.H., et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang K., Chu K., Wu X., et al. Amplification of FRS2 and activation of FGFR/FRS2 signaling pathway in high-grade liposarcoma. Cancer Res. 2013;73(4):1298–1307. doi: 10.1158/0008-5472.CAN-12-2086. [DOI] [PubMed] [Google Scholar]
- 24.Frampton G.M., Fichtenholtz A., Otto G.A., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanman R.B., Mortimer S.A., Zill O.A., et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10(10) doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerami E., Gao J., Dogrusoz U., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao J., Aksoy B.A., Dogrusoz U., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakravarty D., Gao J., Phillips S.M., et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017 doi: 10.1200/PO.17.00011. PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tirkes T., Hollar M.A., Tann M., Kohli M.D., Akisik F., Sandrasegaran K. Response criteria in oncologic imaging: review of traditional and new criteria. Radiogr. 2013;33(5):1323–1341. doi: 10.1148/rg.335125214. [DOI] [PubMed] [Google Scholar]
- 30.Okamura R., Kato S., Lee S., Jimenez R.E., Sicklick J.K., Kurzrock R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J Immunother Cancer. 2020;8(1) doi: 10.1136/jitc-2019-000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim E.K., Cho Y.A., Koh Y.W., Shin H.A., Cho B.C., Yoon S.O. Prognostic implications of Fibroblast growth factor receptor 1 (FGFR1) gene amplification and protein overexpression in hypopharyngeal and laryngeal squamous cell carcinoma. BMC Cancer. 2020;20(1):348. doi: 10.1186/s12885-020-06792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang J., Liu X., Wang S., et al. Prognostic value of FGFR gene amplification in patients with different types of cancer: a systematic review and meta-analysis. PloS One. 2014;9(8) doi: 10.1371/journal.pone.0105524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikanjam M., Okamura R., Barkauskas D.A., Kurzrock R. Targeting fusions for improved outcomes in oncology treatment. Cancer. 2020;126(6):1315–1321. doi: 10.1002/cncr.32649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byron S.A., Chen H., Wortmann A., et al. The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia. 2013;15(8):975–988. doi: 10.1593/neo.121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., Mikse O., Liao R.G., et al. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene. 2015;34(17):2167–2177. doi: 10.1038/onc.2014.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herrera-Abreu M.T., Pearson A., Campbell J., et al. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013;3(9):1058–1071. doi: 10.1158/2159-8290.CD-12-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nogova L., Sequist L.V., Perez Garcia J.M., et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol. 2017;35(2):157–165. doi: 10.1200/JCO.2016.67.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Luca A., Esposito Abate R., Rachiglio A.M., et al. FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention. Int J Mol Sci. 2020;21(18):6856. doi: 10.3390/ijms21186856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner N., Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 40.Hortobagyi G.N., Chen D., Piccart M., et al. Correlative analysis of genetic ;alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2015;34(5):419–426. doi: 10.1200/JCO.2014.60.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Packer L.M., Geng X., Bonazzi V.F., et al. PI3K inhibitors synergize with FGFR inhibitors to enhance antitumor responses in FGFR2mutant endometrial cancers. Mol Cancer Ther. 2017;16(4):637–648. doi: 10.1158/1535-7163.MCT-16-0415. [DOI] [PubMed] [Google Scholar]
- 42.Markman B., Tao J.J., Scaltriti M. PI3K pathway inhibitors: better not left alone. Curr Pharm Des. 2013;19(5):895–906. [PubMed] [Google Scholar]
- 43.Holzhauser S., Lukoseviciute M., Andonova T., et al. Targeting fibroblast growth factor receptor (FGFR) and phosphoinositide 3-kinase (PI3K) signaling pathways in medulloblastoma cell lines. Anticancer Res. 2020;40(1):53–66. doi: 10.21873/anticanres.13925. [DOI] [PubMed] [Google Scholar]
- 44.Dieci M.V., Arnedos M., Andre F., Soria J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3(3):264–279. doi: 10.1158/2159-8290.CD-12-0362. [DOI] [PubMed] [Google Scholar]
- 45.Holzhauser S., Kostopoulou O.N., Ohmayer A., et al. In vitro antitumor effects of FGFR and PI3K inhibitors on human papillomavirus positive and negative tonsillar and base of tongue cancer cell lines. Oncol Lett. 2019;18(6):6249–6260. doi: 10.3892/ol.2019.10973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyman D.M., Tran B., Paz-Ares L., et al. Combined PIK3CA and FGFR inhibition with alpelisib and infigratinib in patients with PIK3CA-mutant solid tumors, with or without FGFR alterations. JCO Precis Oncol. 2019;3:1–13. doi: 10.1200/PO.19.00221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.