

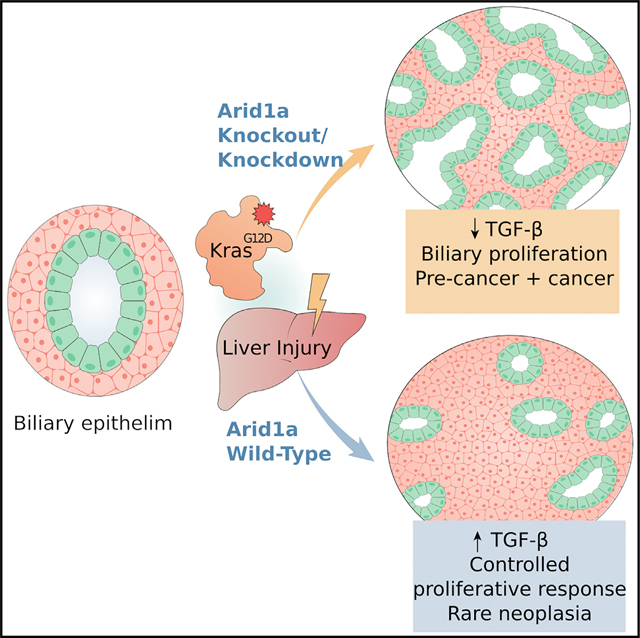
SUMMARY
Activating KRAS mutations and functional loss of members of the SWI/SNF complex, including ARID1A, are found together in the primary liver tumor cholangiocarcinoma (CC). How these mutations cooperate to promote CC has not been established. Using murine models of hepatocyte and biliary-specific lineage tracing, we show that Kras and Arid1a mutations drive the formation of CC and tumor precursors from the biliary compartment, which are accelerated by liver inflammation. Using cultured cells, we find that Arid1a loss causes cellular proliferation, escape from cell-cycle control, senescence, and widespread changes in chromatin structure. Notably, we show that the biliary proliferative response elicited by Kras/Arid1a cooperation and tissue injury in CC is caused by failed engagement of the TGF-β-Smad4 tumor suppressor pathway. We thus identify an ARID1A-TGF-β-Smad4 axis as essential in limiting the biliary epithelial response to oncogenic insults, while its loss leads to biliary pre-neoplasia and CC.
In brief
Guo et al. show that loss of the ARID1A subunit of the SWI/SNF complex suppresses TGF-β signaling activation in biliary epithelium, synergizes with oncogenic Kras mutation in inducing proliferative phenotypes with widespread transcriptomic and chromatin accessibility changes, and drives the development of cholangiocarcinoma from a biliary cell of origin.
Graphical Abstract
INTRODUCTION
Cholangiocarcinoma (CC) is the second most common primary liver tumor, with a rising incidence and mortality and poor prognosis (Banales et al., 2020). DNA sequencing (DNA-seq) demonstrates that genes encoding chromatin remodeling complexes are frequently deleteriously mutated in CC, including ARID1A (AT-rich interactive domain-containing protein 1A) in 7%–36% CC (Chan-On et al., 2013; Jiao et al., 2013; Zou et al., 2014). ARID1A, a DNA binding subunit and the most commonly mutated gene in CC within the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex, broadly governs chromatin structure and accessibility, and has been shown to have differing cancer-related activities depending on the tissue types studied, the genetic context, and even the timing of mutation (Mathur et al., 2017; Sun et al., 2016; Wang et al., 2019a, 2019b). Among other frequently altered genes in human CC, activating KRAS mutations regularly co-occur with ARID1A mutations (Cerami et al., 2012; Farshidfar et al., 2017; Lowery et al., 2018; Wardell et al., 2018) and are commonly found together in other gastrointestinal epithelial malignancies, including esophagogastric, colon, and pancreatic tumors (Cerami et al., 2012; Wang et al., 2019b).
In the liver, ARID1A has been found to regulate liver cell proliferation, migration, angiogenesis, inflammation, and hepatocyte differentiation (Chan-On et al., 2013; Fang et al., 2015; Hu et al., 2018; Sun et al., 2016). In the context of active oncogenic signaling, the role of ARID1A becomes nuanced, effectively enabling early transformation events while inhibiting activities such as migration and metastasis of advanced cancers (Sun et al., 2017). Although several studies have shown the important role of ARID1A in hepatocellular carcinoma (HCC) (Fang et al., 2015; Hu et al., 2018; Sun et al., 2017) and SWI/SNF complex in regulating key CC-related pathways in other contexts (Banales et al., 2020; Li et al., 2019; Ringel et al., 2020; Suryo Rahmanto et al., 2020; Xi et al., 2008), the specific role of ARID1A mutation in CC tumorigenesis is less clear. How Kras and Arid1a mutations cooperate to drive biliary carcinogenesis, what compartments (hepatocyte versus biliary cell) these genes cooperate to transform, and the molecular pathways they cooperatively act upon are incompletely understood.
KrasG12D mutations have been used to model murine CC formation in the context of Trp53 heterozygous deletion (O’Dell et al., 2012), IDH mutations (Saha et al., 2014), and Pten deletion (Ikenoue et al., 2016), revealing perturbed pathways and processes in each specific context. We have shown that, depending on oncogenic context and environmental factors, KrasG12D can drive CC from either mature hepatocytes or cholangiocytes (Hill et al., 2018), demonstrating that CC may derive from either of these cell types. How incipient Arid1a mutation affects the cell of origin in the pathogenesis of CC is unknown. To address this, we studied the effects of coincident Kras activation and Arid1a deletion (KA) in murine hepatic epithelial compartments to evaluate the cooperative role in liver tumorigenesis and in mouse embryonic fibroblasts (MEFs) to determine the overall impact on gene expression and chromatin architecture. Using these systems, we show that Kras and Arid1a mutations drive the formation of CC and tumor precursors from the biliary compartment when incited by inflammation, and that Arid1a loss causes proliferation, escape from cell-cycle control, widespread changes in chromatin structure, and failed engagement of the transforming growth factor β (TGF-β)-Smad4 tumor suppressor pathway. We thus identify an ARID1A-TGF-β-Smad4 axis as essential to limiting biliary epithelial response to oncogenic insults, while its loss leads to biliary pre-neoplasia and CC.
RESULTS
KrasG12D and Arid1a−/− mutations cooperate to cause CC
Given the coincidence of KRAS and ARID1A mutations in human CC (Farshidfar et al., 2017; Lowery et al., 2018; Wardell et al., 2018), we targeted the mutations to the liver epithelium using the Alb-Cre allele, which allows late embryogenic CRE recombinase expression in both the hepatocyte and cholangiocyte compartments (Figure 1A) (Postic et al., 1999). To induce liver inflammation, mice of different genotypes, including Alb-Cre (Alb), Alb-Cre;Arid1aL/L (AA), Alb-Cre;KrasLSL-G12D (AK), and Alb-Cre; KrasLSL-G12D;Arid1aL/L (AKA), were fed 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet (Figure 1B), a non-carcinogenic liver injury model that causes ductular reaction, fibrosis, and inflammation, and has been used to accelerate CC formation in other models (Banales et al., 2020; Chan-On et al., 2013; Hill et al., 2018; Jiao et al., 2013; Zou et al., 2014). Survival was diminished in the AKA cohorts (restricted mean survival AKA 61.1 ± 8.1, AK 92.4 ± 11.9, AA 97.8 ± 11.0, and Alb 119.7.9 ± 5.2 weeks) with lethargy and abdominal distention developing starting at 33 weeks of age (Figures 1C and 1D; Table S1). Gross and pathologic analysis revealed that varying proportions of AKA, AK, and AA animals developed primary hepatic carcinomas, including CC and HCC. The burden of HCC was comparable between AKA and AK cohorts (11/12 versus 7/8), with an increase in HCC formation of AA cohort (4/11) over the Alb-Cre (Alb, 0/15) controls (Table S1), as previously reported (Fang et al., 2015). The burden of biliary neoplastic pathology, however, was significantly increased in AKA mice, including intrahepatic CC (ICC), mixed and undifferentiated malignancies, as well as ICC precursors, including biliary hamartomas (also known as Von Meyenburg complexes [VMCs]), intraductal papillary neoplasm of the bile ducts (IPNB), biliary in traepithelial neoplasia (BilIN), and proliferative biliary lesions with varying degrees of atypia including atypical biliary proliferations (ABPs) (Figure 1E; Table S1). In contrast, these biliary pathologies were rarely observed in AK, AA, and Alb cohorts. The finding of multifocal pre-malignant and malignant biliary pathologies was significantly increased in the AKA cohort (10/12, 83% penetrance) as compared with either single mutant group or controls (0%, 25%, 0% penetrance for AA, AK, and Alb, p < 0.001, p = 0.019, p < 0.001, respectively) (Figure 1E). Non-DDC-treated cohorts were also followed longitudinally and were normal and healthy up to an age of over 40 weeks (Figures S1A–S1D), when livers were evaluated pathologically following necropsy. Consistent with the cooperation of Kras activation and Arid1a loss in driving CC in the setting of liver injury, histological analysis revealed CC and precursor lesions in the non-DDC-treated AKA group (3/10 and 1/10, respectively), while 0 of 13 AK mice developed CC or multifocal biliary precursors (p = 0.024; Figures S1A and S1B; Table S1). Thus, Kras activation and Arid1a loss cooperate in vivo to promote neoplastic precursors and advanced biliary cancer formation, which is significantly exacerbated by liver injury and inflammation.
Figure 1. Arid1a inactivation cooperates with oncogenic Kras mutation in liver tumorigenesis.
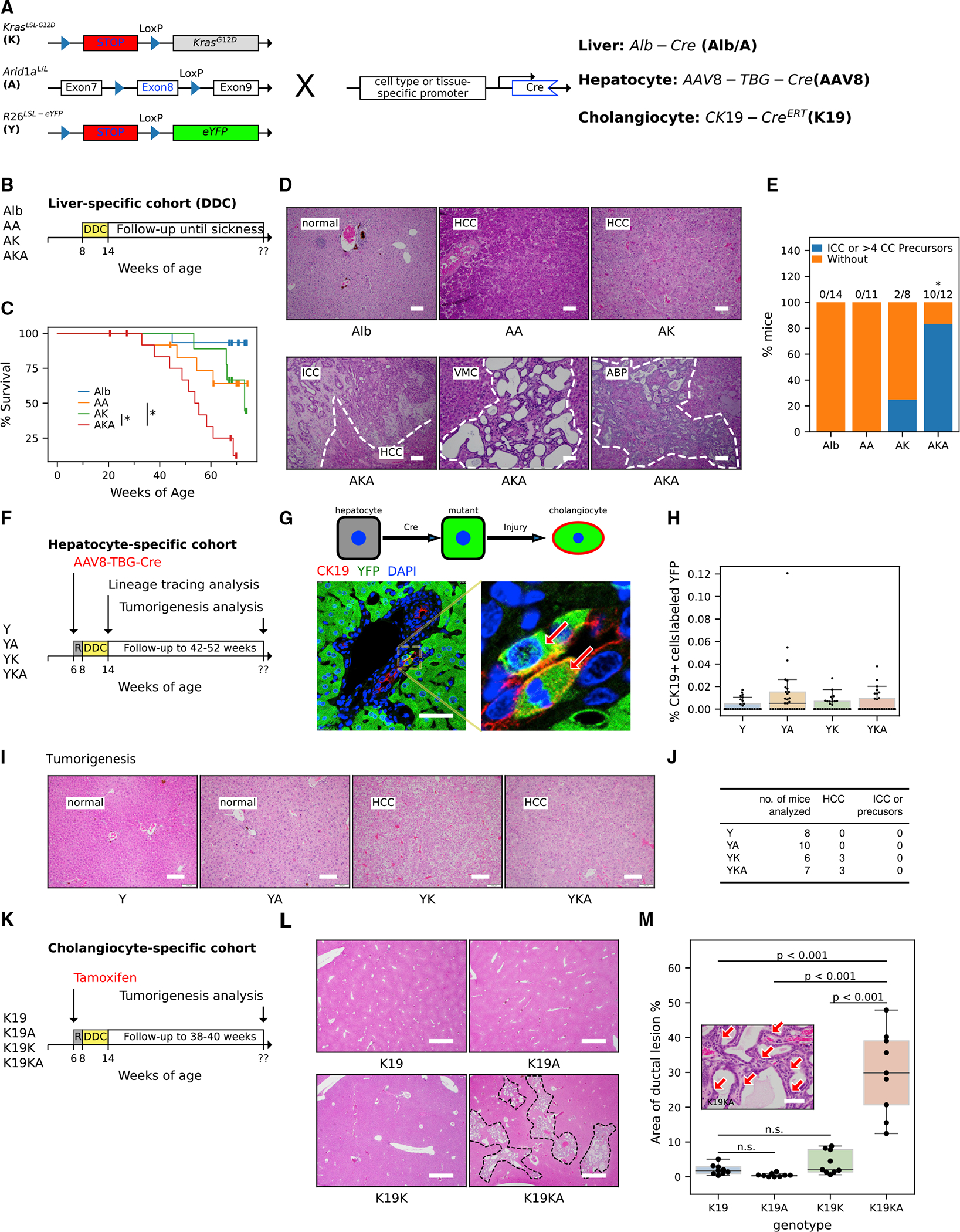
(A) Schematic for the generation of (1) liver epithelium-specific mutants, Alb-Cre; KrasLSL-G12D; Arid1aL/L (AKA) and control mice, Alb-Cre; KrasLSL-G12D (AK) and Alb-Cre; Arid1a L/L (AA), using Alb-Cre allele (Alb), (2) hepatocyte-specific mutants by using AAV8-TBG-Cre adenoviruses (AAV8-Cre) to recombine double (YKA) and single KrasLSL-G12D (YK) and/or Arid1aL/L (YA) alleles along with the Rosa26LSL-eYFP reporter allele (Y) in hepatocytes, and (3) biliary epithelium-specific mutants by targeting double (KA) and single KrasLSL-G12D (K) and/or Arid1aL/L (A) alleles to cholangiocytes using the tamoxifen-inducible CK19-CreERT allele (K19).
(B) Experimental protocol for the Alb-Cre DDC cohorts. Mice of indicated genotypes and age were fed with DDC for 6 weeks and followed up until illness.
(C) Kaplan-Meier survival curve of DDC-fed AKA mice (n = 14) compared to DDC-fed AK (n = 13), AA (n = 13), and Alb (n = 15) mice. The asterisks indicate that the survival differences of the compared pairs of groups were statistically significant (log rank test, p values are 0.03 for AKA versus AA and 0.01 for AKA versus AK).
(D) Representative H&E images of DDC-injured livers from AKA, AK, and AA mice that were followed up until sickness. (HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma; VMC, Von Meyenburg complexes; ABP, atypical biliary proliferation). Scale bar, 100 μm.
(E) Frequency of mice harboring ICC tumors or ICC precursors in AKA (n = 12), AK (n = 8), AA (n = 11), and Alb (n = 14) mice from the Alb-Cre DDC cohorts. Frequency in the AKA cohort is different from the other 3 genotypes (Fisher’s exact test, p < 0.05). Details are available in Table S1.
(F) Experimental protocol of AAV8-TBG-Cre DDC cohorts for reprogramming (harvested when 6-week DDC feeding was complete) and tumorigenesis (followed up to 52 weeks) analyses. “R” represents a 2-week waiting period to allow mice to recover after injection of the virus.
(G) Illustration of lineage tracing analysis. AAV8-TBG-Cre recombines the R26LSL-eYFP allele and allows cells of hepatocytic origin to express EYFP (green). Gaining additional CK19 (red) expression (EYFP+/CK19+ cells, arrows) after DDC-induced injury indicates transdifferentiation to cholangiocytes. See Figures S1E and S1F for information on Cre-recombination efficiency. Scale bar, 50 μm.
(H) Quantification of transdifferentiation in YKA, YK, YA, and Y livers harvested immediately after 6 weeks of DDC diet. n = 3 mice per genotype and 10 portal fields per mouse from different lobes of the liver were analyzed; each field was represented by a point in the graph. The percentage of co-labeled cells (ratio of K19+/YFP+ cell count to K19+ cell count) was used to indicate transdifferentiation. Nested ANOVA did not reject the null hypothesis (there was no difference among genotypes, p = 0.06). See Figures S1G and S1H for detailed cell counting.
(I) Representative H&E images of YKA, YK, YA, and Y livers after extended observation for ~40 weeks after AAV8 virus infection. See detailed analysis in Table S1. Scale bar, 100 μm.
(J) Frequency of tumor/precursor types in YKA and control mice from the AAV8-TBG-Cre DDC tumorigenesis cohort in (F).
(K) Experimental protocol for the CK19-CreERT DDC cohorts. The DDC-injured mice were followed up for ~30 weeks after intraperitoneal tamoxifen injection. “R” represents a 2-week waiting period to allow mice to recover after injection of tamoxifen.
(L) H&E staining of livers from the CK19-CreERT DDC cohorts. The areas circled by a dotted line indicate the extensive ductal lesions in the liver. Scale bar, 1,000 μm.
(M) Quantification of ductal lesions (arrows in the inset image) in livers (n = 3 mice per genotype and 3 fields per mouse analyzed) from the CK19-CreERT DDC cohorts. Nested ANOVA suggests ductal lesion areas were significantly different among genotypes (p < 0.001). p values of pairwise comparison using Tukey’s honestly significant difference (HSD) tests were indicated in the graph. Scale bar, 50 μm.
Kras activation and Arid1a loss drive neoplastic biliary progression and do not affect hepatocyte to cholangiocyte reprogramming
CC may originate from either of the major hepatic epithelial cell compartments, biliary or hepatocyte. We have previously shown that KrasG12D, in the context of concurrent Trp53 mutation and liver injury, may induce CC when targeted to either mature biliary cells or hepatocytes (Hill et al., 2018). Essentially, both biliary and hepatocyte compartments have the potential to serve as the cellular origin of CC and that the genetic events and environmental context can affect this. Given the known role of ARID1A in controlling cellular differentiation in other tissue compartments (Mathur et al., 2017; Wang et al., 2019a; Wilson et al., 2019) and outstanding questions about how Arid1a mutation may affect CC tumor progression and the cellular origins of CC, we chose to evaluate the cooperation of KrasG12D and Arid1a−/− in both compartments of the injured adult liver, hepatocyte, and biliary.
To specifically target hepatocytes, AAV8-TBG-Cre viruses were injected into compound mutant mice harboring a Rosa26LSL-eYFP reporter allele (Y) along with either KrasLSL-G12D (YK) or Arid1aL/L alleles (YA) or both (YKA) (Figures 1A and 1F). The recombination efficiency was confirmed by RT-qPCR of Arid1a and immunofluorescence of the reporter protein enhanced yellow fluorescent protein (EYFP) (Figures S1E and S1F). We have found previously that Trp53 mutations enable CC induction from hepatocytes, and that Trp53 mutation causes increased transdifferentiation of hepatocytes, taking on cellular features of cholangiocytes or biliary cells, proceeding the development of CC. To evaluate this in the above models, liver damage was induced by the DDC diet for 6 weeks before histologic and lineage tracing analyses (Figures 1F and 1G). The ratio of CK19+/YFP+ cells to CK19+ cells was used to indicate transdifferentiation from hepatocytes to cholangiocytes (Figure 1G) as previously reported (Hill et al., 2018; Yanger et al., 2013). The percentages of co-labeled cells were similar among Y, YK, and YKA cohorts. The YA cohort demonstrated a higher degree of variability of co-labeled cells among regions studied and broader distribution of data, raising the possibility that with larger scale experiments, a small difference could be found, although no significant difference between among YKA, YK, YA, and Y genotypes was observed (Figures 1H, S1G, and S1H). We simultaneously followed up separate cohorts of DDC-fed Y, YA, YK, and YKA (n = 8, 10, 6, and 7, respectively) to 40 weeks of age in the event that cell fate switches occurred with a longer latency over the course of carcinogenesis. We found that no CC or biliary precursor lesions developed in any of the four genotypes, while 3/7 YKA and 3/6 YK animals developed HCC (Figures 1I and 1J; Table S1). Thus, Kras mutations drive the mature hepatocyte lineage to hepatocellular cancer. Concurrent hepatocyte-directed Arid1a mutation does not cause observable transdifferentiation of hepatocytes toward a biliary fate or the development of CC.
To target KrasG12D and Arid1a−/− to cholangiocytes, we used the tamoxifen-inducible CK19-CreERT models followed by 6 weeks of DDC diet or standard diet (Figure 1K) (Means et al., 2008). Upon the observation of the cohort for signs of illness, a necropsy was necessitated (by 30 weeks after tamoxifen treatment) due to cancer development in other CK19-expressing organs, including the oral cavity, pharynx, and stomach. Livers from all cohorts, CK19-CreERT (K19), CK19-CreERT;KrasLSL-G12D (K19K), CK19-CreERT;Arid1aL/L (K19A), CK19-CreERT;KrasLSL-G12D;Arid1aL/L (K19KA) with regular diets (no hepatic injury) were grossly and histologically normal (Figure S1I), as were DDC-fed (hepatic injury-induced) K19, K19K, and K19A cohorts (Figure S1J). The DDC-treated K19KA cohort developed widespread proliferative biliary lesions characterized as biliary hamartomas, or Von Meyenburg complexes (Figures 1L and 1 M), which are established CC precursors and observed in the AKA cohorts, as described above (Figure 1D). These experiments demonstrated ARID1A loss cooperates with KrasG12D to drive the formation of CC precursor lesions and the malignant conversion of the biliary epithelial compartment. Unlike Trp53, mutations in Arid1a targeted to KrasG12D mutant hepatocytes, even in the setting of hepatic injury, do not enable either transitions of cell fate or the formation of CC.
Arid1a loss drives KrasG12D MEF proliferation
MEFs provide a tractable system to understand the basis of cooperative mutations in cancer (Sun and Taneja, 2007). Given the robustness of MEF systems, we infected day 13.5 MEFs from wild-type (WT), Arid1aL/L (A), KrasLSL-G12D (K), KrasLSL-G12D; Arid1aL/L (KA) embryos with adenoviruses expressing CRE recombinase (Ad5-CMV-Cre) at passage 3–4 (Figure 2A). Recombination efficiency was >90% by RT-qPCR and confirmed by PCR on genomic DNA and immunoblot (Figures S2A–S2C). KA cells had a proliferative advantage evidenced by population doubling (Figures 2B and S2D), and 5-ethynyl-2′-deoxyuridine (EdU) labeling (mean percentage of EdU+ nuclei is 52% in KA versus 28%, 23%, and 20% in A, K, and WT controls, respectively) (Figures 2C and S2E) as compared with single-mutant populations, which behaved as previously published (Tuveson et al., 2004). Morphologically, KA MEFs are smaller, form colonies when plated at a low density (Figures 2D and S2F), and have diminished senescence (Figures 2E, 2F, S2G, and S2H) and blunted (adipocyte) differentiation capacity (Figures S2I and S2J). These data show that KrasG12D and Arid1a−/− cooperate in MEFs to confer several cancer-associated phenotypes, including rapid proliferation, senescence escape, colony formation, and diminished differentiation potential.
Figure 2. KrasG12D and Arid1a−/− cooperate in driving mouse embryonic fibroblast proliferation with increased chromatin accessibility in promoters and decreased chromatin accessibility in distal genomic regions.
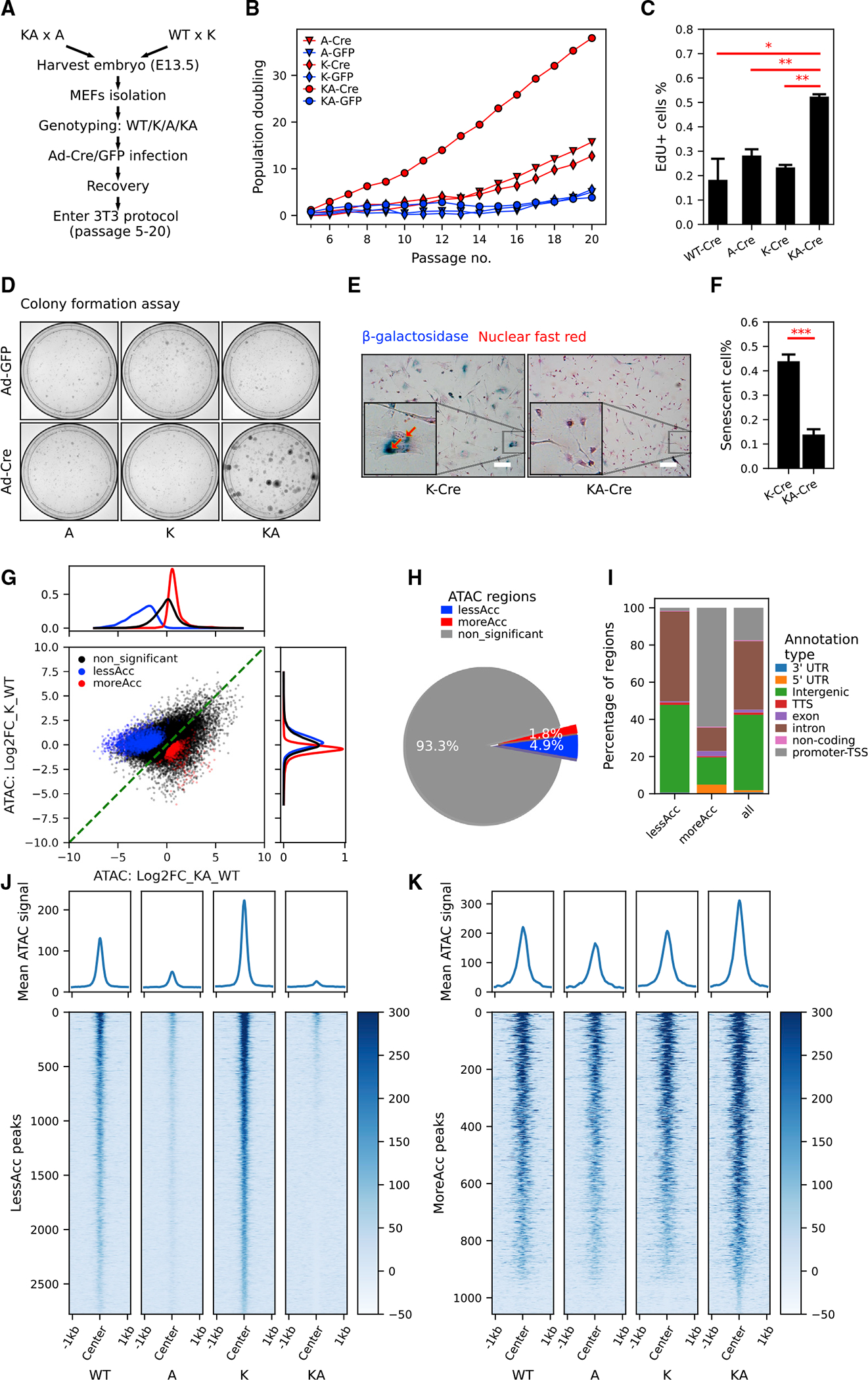
(A) The workflow of generating MEFs with KrasG12D and/or Arid1a−/− for in vitro study.
(B) Population doubling curve of MEFs with no, single, or combined KrasG12D and Arid1a−/− mutations (n = 2 cell lines per genotype. Cre: recombined. GFP: unrecombined). Also see Figure S2D.
(C) Quantification of EdU incorporation of WT-Cre, A-Cre, K-Cre, and KA-Cre MEFs (n = 2 repeats from the same round of infection, Student’s t tests, *p < 0.05, **p < 0.01). See representative images in Figure S2E.
(D) Representative images of colony formation assay of MEFs with indicated genotypes (n = 3).
(E and F) Representative images (E) and quantification (F) of senescence-associated b-galactosidase staining in K-Cre and KA-Cre MEFs (n = 4, Student’s t test, ***p < 0.001). Also, see Figures S2G and S2H for b-galactosidase staining in K-Cre versus K-GFP MEFs. Scale bar, 50 μm.
(G and H) Scatterplot (G) and pie chart (H) characterizing differentially accessible regions between KA and K MEFs. Density plots of log2 fold change in chromatin accessibility in either all ATAC peaks (black), less accessible (lessAcc, blue), or more accessible (moreAcc, red) peaks are provided for the KA versus WT comparison (at top in G) and the K versus WT comparison (at right in G).
(I) Different percentages of ATAC peak annotation types between lessAcc and moreAcc peaks.
(J and K) Heatmaps showing chromatin accessibility in lessAcc (J) and moreAcc (K) regions across genotypes. For the heatmaps, ATAC counts of all 3 samples per genotype were merged. Plots on the top show the average ATAC-signal across regions.
Arid1a inactivation leads to loss of intergenic chromatin accessibility and cooperates with KrasG12D to increase accessibility at promoters
Assay for transposase-accessible chromatin using sequencing (ATAC-seq) of MEFs with either or both mutations differentiated KA and A cells from WT and K cells by principal-component analysis (PCA) across all ATAC regions (Figure S3A) and hierarchical clustering focusing on differentially accessible regions (Figure S3B). Among all ATAC regions, 4.9% were less accessible (lessAcc) and 1.8% more accessible (moreAcc) in KA versus K MEFs (Figures 2G and 2H). Annotation of these regions using Homer (Heinz et al., 2010) suggested a predominant pattern of moreAcc regions located to promoters, whereas the lessAcc regions are intergenic or intronic (Figure 2I), consistent with others’ findings that SWI/SNF perturbation induces both loss of chromatin accessibility in enhancers and gain in promoter regions in an in vitro model (Schick et al., 2021). Heatmaps of aligned lessAcc regions showed reduced accessibility in the A group, with accentuated loss in the KA MEFs; in moreAcc regions, KA cells exhibited increased accessibility compared with all of the other genotypes (Figures 2J and 2K). These data demonstrated that Arid1a loss decreases chromatin accessibility at distal and intronic regions. Conversely, increased chromatin accessibility in cells with cooperating Kras and Arid1a mutations occurred primarily at promoters.
Motifs enriched in moreAcc promoter regions included the transcription factor SP1/5, STAT members, and the RNA polymerase II-interacting CCAAT motifs (Figure S3C). LessAcc regions are enriched with AP-1 motifs, which are involved in enhancer selection (Vierbuchen et al., 2017) and EBF1, NANOG, and SMAD2/3 sites (Ringel et al., 2020; Suryo Rahmanto et al., 2020; Xi et al., 2008) (Figure S3C). Overlay of publicly available chromatin immunoprecipitation sequencing (ChIP-seq) data (Vierbuchen et al., 2017) with our ATAC-seq data showed that lessAcc regions share enrichment of AP-1 and SMARCA4 (an ATPase subunit of the SWI/SNF complex) binding, and are flanked by the active enhancer marker H3K27ac (Figures S3D–S3F). These data support the loss of AP1 binding in Arid1a-null cells and an essential role of AP-1 regulation in ARID1A function as previously reported (Mathur et al., 2017).
Transcriptomic analysis reveals dysregulation in cell-cycle control and TGF-β pathways in Arid1a null KrasG12D MEFs
To explore transcriptomic mechanisms underlying the above cooperation, we performed RNA-seq analysis on 5 of each genotypic conditions (WT, A, K, and KA) that showed intra-group clustering on PCA (Figure 3A), and 10 mock- or Ad5-CMV-EGFP-infected cell lines (Figure S4A). Compared to WT controls, KA, K, and A genotypes had 4,208, 1,073, and 1,117 differentially expressed genes (DEGs), and the majority of these DEGs occurs only in KA but not in K or A MEFs (Figures 3B–3D). To understand the global impact of DNA accessibility on gene transcription, we evaluated log2 fold changes in RNA-seq profiling among the genes nearest to significantly moreAcc or lessAcc regions, as defined above. We found that expression of these genes was largely preserved in either single Kras or Arid1a mutants; however, Kras/Arid1a co-mutant (KA) lines had significant changes in transcript levels, with the nearest genes to the lessAcc regions having lower expression in KA compared to K or A MEFs, and those genes nearest to the moreAcc regions having higher levels in KA MEFs (Figure 3E). Representative individual genes associated with lessAcc and moreAcc also demonstrated corresponding changes in transcription (Figures S3E and S3F). Gene set enrichment analysis (GSEA) of KA versus K upregulated genes revealed HALLMARK E2F_TARGETS, G2M_CHECKPOINT, and MYC_v1/2 as the most enriched pathways (Figures 3F and 3G), consistent with the accelerated cell-cycle phenotype. Given the proliferative phenotype of KA cells and the enrichment of E2F pathway in this transcriptional analysis (Figures 2B–2D and 3F; Table S2), we evaluated moreAcc regions for E2F motifs, which we found to be highly enriched (Figure 3H). Consistent with this and established data, the cell-cycle regulators CYCLIN A2 and CDK4 were suppressed in KA cells compared with K cells (Figures S4C and S4D) (Zhang et al., 2000).
Figure 3. RNA-seq reveals specific gene expression changes in response to oncogenic Kras activation and Arid1a deletion.
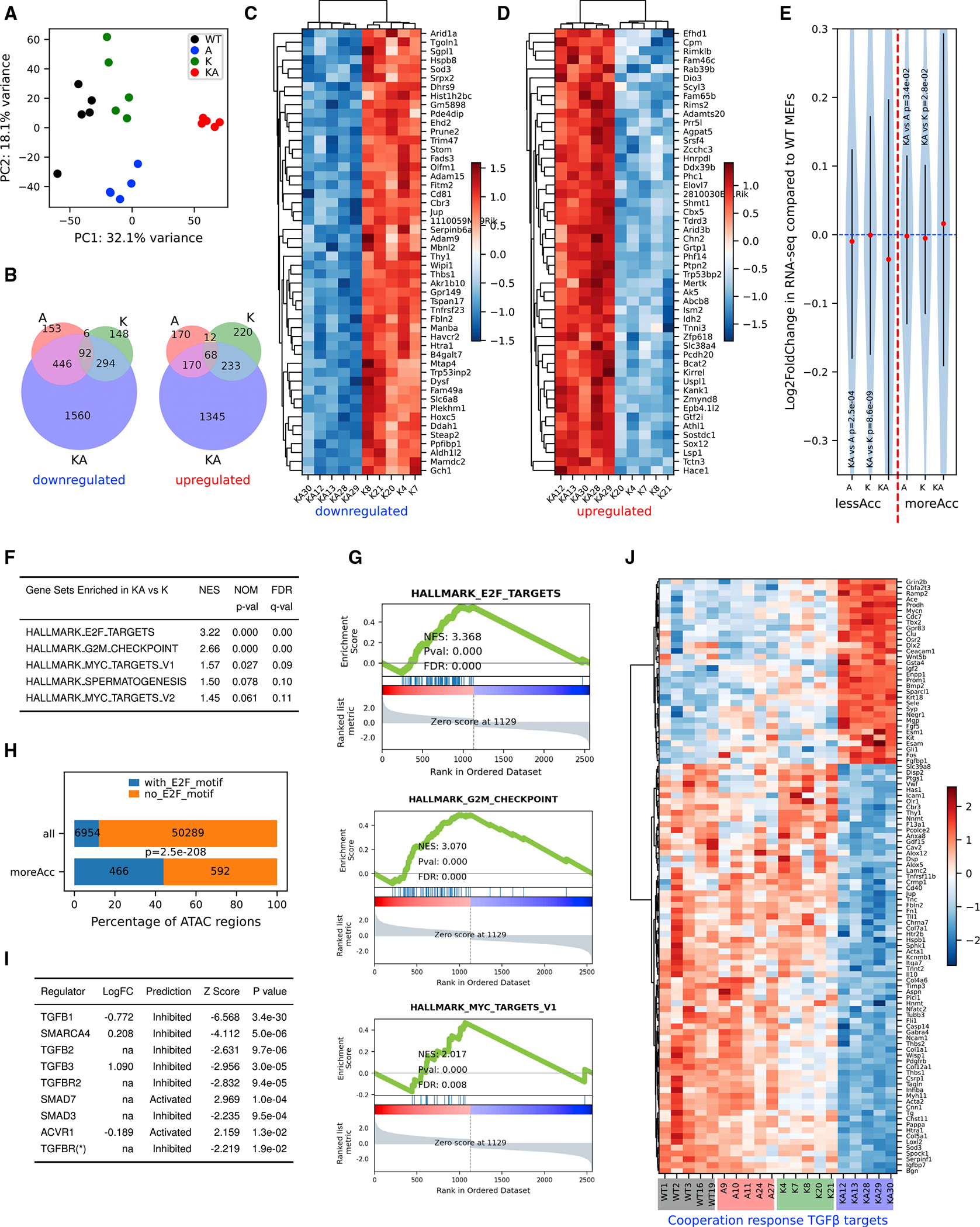
(A) Principal-component analysis using log2 transformed RNA-seq normalized count matrix of Ad5-CMV-Cre recombined MEFs.
(B) Venn diagram of significantly up- (left) and down (right)-regulated genes when MEFs of 3 genotypes (A, K, and KA) are compared to WT MEFs. The significantly differentially expressed genes (DEGs) are defined using criteria of adjusted p < 0.05 and log2 fold change >0.5 or <−0.5.
(C and D) Heatmap of top 50 most significantly downregulated (C) and upregulated (D) genes in KA versus K. Row-wise Z scores of RNA-seq DESeq2-normalized counts in the log scale were clustered and plotted in the heatmap.
(E) Violin plot showing average log2 fold change of RNA-seq signal for nearest genes of less (left of the red dashed line) and more (right of the red dashed line) accessible regions. Student’s t tests, p values are indicated in the plot.
(F) Top gene sets enriched in KA versus K DEGs pre-ranked by log2 fold change.
(G) GSEA plots for the selected gene sets as indicated in the titles. Normalized enrichment score (NES), p value (Pval), and false discovery rate (FDR) values were indicated within the plots.
(H) Homer analysis of E2F motifs in all ATAC regions versus moreACC regions. E2F motif-containing rate is significantly higher in moreAcc peaks than all ATAC peaks (chi-square test p < 0.001).
(I) Ingenuity Pathway Analysis (IPA) upstream regulator analysis of DEGs between KA versus K MEFs predicts inhibited TGF-β pathways and SWI/SNF components.
(J) Heatmap of RNA-seq signal of 103 TGFB1 targets revealing a pattern consistent with cooperation response genes (CRGs). CRGs were identified using CRG score of <0.9 (McMurray et al., 2008).
Ingenuity Pathway Analysis (IPA; Krämer et al., 2014) was used to predict inhibited or activated upstream regulators (Table S2) and identified TGFB1 (Z score = −6.568) as the most significantly inhibited regulator. Other TGF-β pathway components, including TGFB2, TGFB3, TGFBR2, and SMAD3 (R-SMAD), were also identified as inhibited, and SMAD7 (I-SMAD) activated (Figure 3I), suggestive of pervasively inhibited TGF-β-Smad pathways in KA cells. SMARCA4 was also predicted to be inhibited, consistent with the loss of function of SWI/SNF due to Arid1a deletion (Figure 3I). Transcript levels of the majority (65.1%) of TGF-β targets (defined by IPA database) compared between KA and K MEFs were changed in a direction that was consistent with TGFB1 inhibition (Figure S4B). The cooperative impact of concurrent Kras and Arid1a mutations on the TGF-β-SMAD pathway is further supported by the identification of 103 TGFB1 targets from a total of 558 genes, in which expression changes were cooperative in KA versus K and A MEFs (defined previously as more than additive and frequently critical to the maintenance of the cancer phenotype) (McMurray et al., 2008) (Figure 3J). In summary, our MEF model demonstrated that Kras and Arid1a co-mutation leads to both the activation of pro-proliferative transcriptional programs at promoter regions and the loss of chromatin accessibility and diminished gene dosage at AP1-rich intergenic or distal regions. This results in a broad acquisition of cancer phenotypes and dysregulation of pathways governing cell cycling and differentiation pathways, including AP-1, E2F, and predominantly TGF-β-SMAD.
TGF-β-Smad4 signaling is critical to limiting the biliary proliferative response to injury
The identification of mutant Kras and Arid1a cooperation leading to the loss of cell-cycle control and TGF-β-SMAD deregulation in MEFs prompted us to evaluate this in vivo in the biliary compartment. Using the previously described tamoxifen-inducible CK19-CreERT models of pre-malignant biliary disease (Figures 1K–1M), we confirmed active cell proliferation and expansion through immunofluorescence of the cholangiocyte marker CK19 and proliferation marker Ki67 comparing between CK19-CreERT (K19), CK19-CreERT;KrasLSL-G12D (K19K), CK19-CreERT;Arid1aL/L (K19A), and CK19-CreERT;KrasLSL-G12D;Arid1aL/L (K19KA) cohorts (Figures 4A–4C). Given the diminished TGF-β-Smad pathway signaling observed in the KA MEFs, we then evaluated SMAD2/3 phosphorylation (P-SMAD2/3) as a downstream indicator of TGF-β-SMAD signaling within the biliary compartment across these same groups. K19KA biliary cells demonstrated diminished P-SMAD2/3 immunofluorescence as compared with all of the other genotypes (Figures 4D and 4E). To test the primary importance of TGF-β-SMAD4 signaling in controlling proliferative responses of injured biliary epithelium, we conditionally mutated the central transcription factor of the canonical TGF-β pathway, SMAD4, using the CK19-CreERT, Rosa26LSL-eYFP alleles and the conditional knockout allele Smad4L/L (Figure 4F). After tamoxifen injection followed by a 2-week DDC diet to induce biliary inflammation (Figure 4G), the Smad4 knockout mice (K19YS, n = 4) demonstrated a 4-fold greater expansion of cholangiocyte-derived YFP+ cells than the control mice (K19Y, n = 6, p = 0.03; Figures 4H and 4I). These data show that the loss of SMAD4, which is essential to TGF-β-Smad pathway function, leads to an unrestrained cholangiocyte proliferative response to liver injury.
Figure 4. TGF-β/Smad4 signaling is critical to limiting the biliary proliferative response to injury and tumorigenesis.
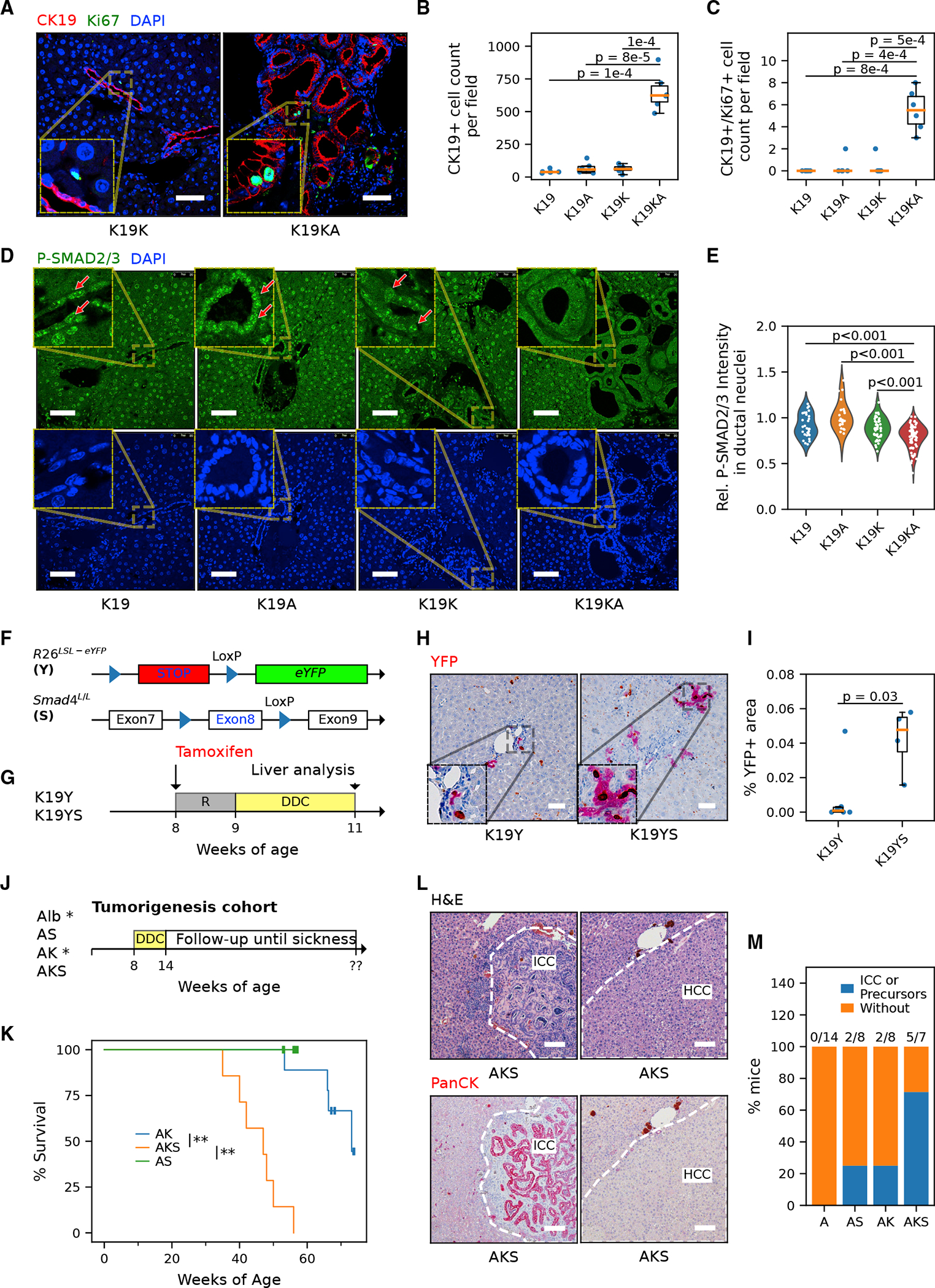
(A) Representative immunostaining images of cholangiocyte marker CK19 and proliferation marker Ki-67 in the liver of the CK19-CreER cohorts. Scale bar, 50 μm.
(B and C) Quantification of cells positive for CK19 (B) and actively proliferating CK19/Ki-67 co-positive cholangiocytes (C) (n = 2 mice for each genotype, 3 fields per mouse analyzed.Student’s t tests, p values are indicated in the plot).
(D) Representative immunostaining image of phospho-SMAD2/3 in the liver of the CK19-CreERT cohort. Red arrows indicate cholangiocytes with positive phospho-SMAD2/3. Scale bar, 50 μm.
(E) Quantification of relative ductal (versus hepatocytic) fluorescence intensity of phospho-SMAD2/3. n = 3 mice per genotype and ≥25 fields per mouse were analyzed. Each point represents the mean P-SMAD2/3 immunofluorescence (IF) signal within nuclei in a ductal region relative to the mean of those in the corresponding non-ductal region in the same field. The difference among genotypes was significant (nested ANOVA p = 0.031). P-SMAD2/3 intensity is significantly lower in K19KA ductal cells compared with K19K, K19A, and K19 (Tukey’s HSD tests, all p < 0.001).
(F) Schematic of generating inducible cholangiocyte-specific Smad4 knockout model with lineage-tracing reporter allele R26LSL-eYFP(K19YS) and its control (K19Y).
(G) Experimental protocol of the K19YS model. Mice were subjected to tamoxifen injection to induce recombination and DDC diet for liver injury for 2 weeks. R, recovery.
(H) Immunohistochemistry for EYFP staining suggests expanded ductal lesions in K19YS versus K19Y livers. Scale bar, 50 μm.
(I) Quantification of the area of ductal reactions in K19YS versus K19Y mice (n = 4 and 6 mice, respectively, Student’s t test, p = 0.03).
(J) Experimental protocol of the AKS DDC model. Alb-Cre; KrasLSL-G12D;Smad4 L/L (AKS) and Alb-Cre;Smad4 L/L (AS) were fed with DDC for 6 weeks and followed up. Historical cohorts of Alb-Cre (Alb*) and Alb-Cre;Kras LSL-G12D (AK*) from a comparable design (Figure 1B) were included for analysis in (K) and (M).
(K) The survival curve of DDC-injured AKS mice (restricted mean ± standard deviation, 45.2 ± 2.4 weeks, n = 7) compared to AK (n = 8) and AS (125 ± 0, n = 8). The asterisks indicate that the survival differences of the compared pairs of groups are statistically significant (log rank test, **p are both <0.01 for AKS versus AK and AKS versus AS).
(L) Representative H&E images and PanCK IHC staining for AKS livers. Scale bar, 100 μm.
(M) Frequency of mice that developed either CC or multiple occurrences of biliary precursor lesions. The absolute counts of CC-prone mice, as well as the total number of mice per genotype, were shown above the bars.
To determine whether Smad4 mutation has an impact similar to that of the Arid1a mutation on mutant KrasG12D-driven liver carcinogenesis, an Alb-Cre;KrasLSL-G12D;Smad4L/L cohort was similarly fed a DDC diet to induce liver injury (Figure 4J), as described earlier (Figure 1B). Compared with a control Alb-Cre; Smad4L/L and the historical control groups (Alb-Cre, Alb-Cre; KrasLSL-G12D, as in Figure 1C), survival was diminished in the Alb-Cre;KrasLSL-G12D;Smad4L/L cohort (45.2 ± 2.4 weeks; Figure 4K) and comparable with the survival of Alb-Cre; KrasLSL-G12D;Arid1aL/L mice (61.1 ± 8.1 weeks). Lethargy and abdominal distention developed starting at ~38 weeks of age in Alb-Cre; KrasLSL-G12D;Smad4L/L mice, with gross and pathologic analysis revealing primary hepatic carcinomas including CC and HCC in all of the animals (Figure 4L; Table S1). The temporal onset, burden of liver cancer, and distribution of pathologies was comparable between Alb-Cre; KrasLSL-G12D;Arid1aL/L and Alb-Cre;KrasLSL-G12D;Smad4L/L cohorts. Mirroring the pathologic analysis of the Kras/Arid1a mutant cohort, the Kras/Smad4 mutants had extensive biliary neoplastic pathology (Figure 4M), including ICC and precursors including biliary hamartomas, ABP, and BilIN (10/12 versus 5/7), as well as a similar incidence of HCC (11/12 versus 7/7). These results demonstrate that the loss of either SMAD4 or ARID1A will accelerate the development of Kras-driven biliary cancers and hepatic epithelial neoplasia, with mirroring tempos and pathologies, including the early stages of CC.
ARID1A loss impairs TGF-β-SMAD pathway activation
To understand the basis of diminished TGF-β pathway activation in the KrasG12D/Arid1a−/− co-mutant setting, we compared the transcription of TGF-β-Smad4 pathway components across WT, K, A, and KA MEFs (Figure 5A). We found altered expression of many genes in this pathway in KA MEFs, including thrombospondin-1 (Thbs1), which operates at the cell surface in the extracellular space to convert TGF-β from its typically translated and cellularly secreted latent form to a smaller proteolytically cleaved active form (Schultz-Cherry and Murphy-Ullrich, 1993). To determine whether suppressed TGF-β-Smad4 signaling in Arid1a-deficient cells could be the consequence of a paucity of activated ligand, we treated KA MEFs with exogenous active TGF-β ligand. Active TGF-β treatment inhibited the colony formation and proliferative index, and increased the phosphorylation of SMAD2/3, demonstrating ongoing sensitivity of Kras/Arid1a co-mutant cells to upstream pathway activation in the presence of the active ligand (Figures 5B–5E and S5A). To directly evaluate the role of ARID1A in governing the TGF-β-Smad4 signaling axis in the biliary lineage, we knocked down ARID1A using lentivirus-mediated small hairpin RNA (shRNA) in the immortalized human cholangiocyte cell line MMNK1 (Maruyama et al., 2004). Knockdown (KD) efficiency was confirmed by both RT-qPCR and western blot (Figures 5F and 5G). Diminished THBS1 protein levels and TGF-β-Smad4 signaling activity as measured by P-SMAD2/3 signaling were confirmed by western blot (Figure 5G). ARID1A KD conferred a growth advantage as determined by increased cell proliferation (Figure 5H) and increased colony formation (Figure 5I). TGF-β-Smad pathway activation is known to direct cholangiocyte lineage differentiation (Schaub et al., 2018). To determine whether ARID1A KD inhibits differentiation, we evaluated tubule formation and branching morphogenesis in MNNK1 cells with or without ARID1A KD. Among the KD cells, we found reduced tubule formation, with the more efficient KD construct revealing the more prominent deficit (shARID1A #89; Figure 5J). RNA-seq analysis of MNNK1 cells with ARID1A KD compared with control revealed similar changes in the expression of upstream components of the TGF-β-Smad pathway (Gene Ontology [GO] :0007179), including THBS1 (Figure 5K), as well as changes in the expression of established TGF-β-Smad targets consistent with TGF-β pathway suppression (Figure 5M). Consistent with a de-differentiated phenotype in ARID1A-depleted biliary cells, we found a diminished expression of genes associated with cholangiocyte development and maturation (Ober and Lemaigre, 2018), including WT1, AQP1, SOX4, HES1, and KRT19 as well as genes in the Notch pathway in a dose-dependent manner (Figures 5L, S5B, and S5C). Arid1a-depleted MMNK1 cells remained sensitive to TGF-β-Smad4 pathway stimulation as exogenous active TGF-β ligand led to the increase in SMAD2/3 phosphorylation (Figures 5G and S5D). TGF-β active ligand treatment inhibited colony formation in Arid1a-depleted cells and led to the restoration of tubule formation and branching morphogenesis consistent with the promotion of cellular differentiation behaviors (Figures 5I and 5J). Taken together, these results demonstrate that ARID1A loss in the biliary compartment leads to suppressed TGF-β-Smad signaling axis, causing unchecked proliferation and growth. Exogenous active TGF-β ligand, leading to pathway activation, is able to rescue many of the associated phenotypes caused by the loss of ARID1A.
Figure 5. ARID1A is critical in upstream TGF-β pathway activation and cholangiocyte homeostasis.
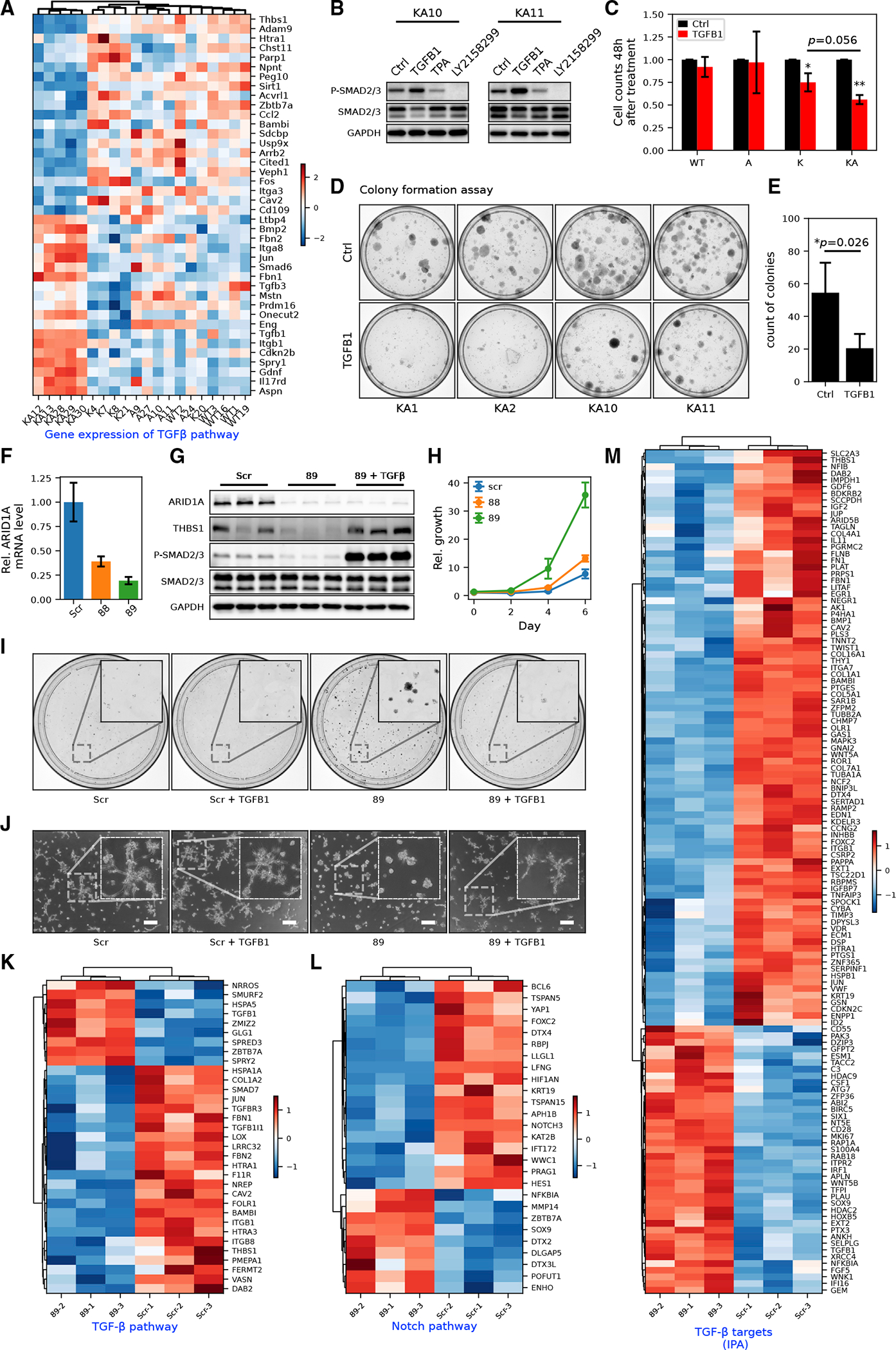
(A) MEF RNA-seq heatmap of genes in the canonical TGF-β pathway (GO: 0007179). Row-wise Z scores of RNA-seq DESeq2-normalized counts in the log scale were clustered and plotted in the heatmap.
(B) Western blot of SMAD2/3 phosphorylation in KA MEFs with or without exogenous TGFB1 (10 ng/mL) or TGFB receptor inhibitor LY-2157299 (10 μM). See Figure S5A for detailed quantification.
(C) Cell counts of MEFs of different genotypes after 48 h of TGFB1 treatment (Student’s t tests, *p < 0.05, ** p < 0.01).
(D and E) Colony formation assay (D) and its quantification (E) in KA MEFs (n = 4 cell lines) with or without exogenous TGFB1 treatment (Student’s t tests, *p < 0.05).
(F) RT-qPCR showing the knockdown (KD) efficiency of shARID1A constructs 88 (Student’s t test, p < 0.01) and 89 (p < 0.01) in MMNK1 cells.
(G) Western blot showing decreased expression of THBS1 and phosphorylation of SMAD2/3 in MMNK1 cells with ARID1A KD and rescue with active TGFB1 treatment. Quantification of protein bands was shown in Figure S5D.
(H) Growth curve of MMNK1 cells with or without ARID1A KD measured by crystal violet absorbance (n = 3).
(I) Representative images from colony formation assay of MMNK1 control and ARID1A KD cells with or without TGFB1 treatment (n = 3).
(J) Representative images from tubule formation assay of MMNK1 control and ARID1A KD cells with or without TGFB1 treatment (n = 3). Scale bar, 200 μm.
(K) RNA-seq heatmap for DEGs in the TGF-β receptor signaling pathway (GO: 0007179) in MMNK1 control versus ARID1A KD cells.
(L) RNA-seq heatmap for DEGs in Notch signaling pathways (GO: 0007219) in MMNK1 control versus ARID1A KD cells.
(M) RNA-seq heatmap for TGF-β targets (from IPA) in MMNK1 control cells versus ARID1A KD cells.
DISCUSSION
ARID1A is one of the most frequently mutated genes in human CC. In this study, we targeted KrasG12D or Arid1a−/−, or both to liver tissue-specific and compartment-specific mouse models, MEFs and human biliary cells. Liver-specific Kras and Arid1a mutations drive the formation of CC and tumor precursors from the biliary compartment that is accelerated by inflammation and liver damage. We see that hepatocyte reprogramming in this genetic context, unlike others, is not a significant factor in Kras and Arid1a-driven biliary carcinogenesis. We found that cells harboring both Kras and Arid1a mutations cause proliferation and circumvention of cell-cycle controls and senescence compared to WT MEFs and MEFs with a single mutation in either gene. Finally, our data support a cooperative role of Arid1a inactivation and Kras activation in driving biliary tumorigenesis through the attenuation of TGF-β-SMAD pathway signaling and that this can be overcome through upstream pathway activation. Changes in DNA accessibility among KrasG12D and Arid1a co-mutant cells are different from those found in either single mutant cell line. As others have shown, we found that Arid1a loss closes chromatin at enhancer regions (Kelso et al., 2017; Mathur et al., 2017). Our studies suggest the synergy of the two mutations on both chromatin accessibility and the transcriptome. In comparison to previous studies (Kelso et al., 2017; Mathur et al., 2017), we have found effects of ARID1A loss on chromatin accessibility at the promoters compared to enhancers consistent with a recent study on immediate accessibility changes after SWI/SNF perturbation (Schick et al., 2021). Increased accessibility at locations enriched with E2F motifs, associated with an increased expression of associated genes are in accordance with the pro-proliferative phenotype in MEFs, as well as work describing the regulatory role of ARID1A in regulating transcription machinery (Trizzino et al., 2018). The underlying cooperative proliferation program driven by these mutations is consistent with effects observed in the pancreatic epithelium in response to the same mutations (Livshits et al., 2018; Wang et al., 2019a, 2019b).
The identification of ARID1A as critical to the function of the TGF-β-Smad pathway in biliary cells and that ARID1A suppresses CC offers insight into the epidemiologic finding that mutations in this pathway are enriched in CCs arising in the settings of primary biliary injury when TGF-β-Smad is likely to function to restrain biliary carcinogenesis (Chan-On et al., 2013; Mu et al., 2016). The dysregulation of the TGF-β axis in the setting of SWI/SNF perturbations has been reported in other systems, including lung, endometrium, intestine, and others (Li et al., 2019; Ringel et al., 2020; Suryo Rahmanto et al., 2020; Xi et al., 2008). While in other systems, the loss of the SWI/SNF complex function led to cellular resistance to exogenous TGF-β treatment, we found that KrasG12D and Arid1a-null MEFs and Arid1a-depleted cholangiocytes maintained their sensitivity to active TGF-β, leading to the reversal of the pro-proliferative and dedifferentiated phenotypes. It will be important to understand whether manipulation of the TGF-β axis could offer a therapeutic strategy in advanced human CCs that harbor ARID1A mutations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aram F. Hezel (Aram_Hezel@URMC.Rochester.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data availability: Sequencing data has been deposited at GEO and is publicly available as of the publication date. Please refer to the “Deposited data” subsection in the key resources table for the accession number.
Custom code availability: This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-ARIDI A (clone D2A8U) | Cell Signaling Technology | Cat# 12354; RRID:AB_2637010 |
| Rabbit monoclonal anti-Smad2/3 (clone D7G7) | Cell Signaling Technology | Cat# 8685; RRID:AB_10889933 |
| Rabbit monoclonal anti-Phospho-Smad2(Ser465/ 467)/Smad3(Ser423/425) (clone D27F4) | Cell Signaling Technology | Cat# 8828; RRID:AB_2631089 |
| Rabbit monoclonal anti-GAPDH (clone 14C10) | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Rabbit monoclonal anti-GFP (clone D5.1) | Cell Signaling Technology | Cat# 2956; RRID:AB_1196615 |
| Mouse monoclonal anti-THBS1 (clone A6.1) | ThermoFisher | Cat# MA5-13398; RRID:AB_10984611 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab66155; RRID:AB_1140752 |
| Rabbit anti-p15(INK4b) | ThermoFisher | Cat# PA5-49749; RRID:AB_2635202 |
| Rabbit anti-CDK4 | ThermoFisher | Cat# MA5-41183; RRID:AB_2898937 |
| Rabbit anti-Cyclin A2 | ThermoFisher | Cat# MA5-32353; RRID:AB_2809634 |
| Rat anti-CK19 | Developmental Studies Hybridoma Bank Univ Iowa | Cat# TROMA-III; RRID:AB_2133570 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV8.TBG.PI.Cre.rBG | Penn Vector Core | Cat# CS1380 |
| Ad5-CMV-Cre | Baylor Gene Vector Core | Cat# 013018 |
| Ad5-CMV-eGFP | Baylor Gene Vector Core | Cat# 050917 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| hTGF-β1 | Cell Signaling Technology | Cat# 8915 |
| mTGF-β 1 | Cell Signaling Technology | Cat# 5231 |
| LY2157299 | MedChemExpress | Cat# HY-13226 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Senescence b-Galactosidase Staining Kit | Cell Signaling Technology | Cat# 9860 |
| RNeasy kit | Qiagen | Cat# 74104 |
| Click-IT EdU | Invitrogen | C10337 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data (SuperSeries) | This paper | [GEO]: GSE207895 |
| MEF RNA-seq (SubSeries) | This paper | [GEO]: GSE207891 |
| MEF ATAC-seq (SubSeries) | This paper | [GEO]: GSE207888 |
| MMNK1 RNA-seq (SubSeries) | This paper | [GEO]: GSE207892 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: primary liver tumor cell lines | This paper | N/A |
| Mouse: MEF cells | This paper | N/A |
| Human: MMNK-1 cells | Maruyama et al., 2004 | Cat# JCRB1554, RRID:CVCL_M266 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Tg(Alb-cre)21Mgn | Gift from Dr. Magnuson | RRID:IMSR_JAX:003574 |
| Mouse: Krastm4Tyj | Gift from Dr. Jacks | RRID:IMSR_JAX:008179 |
| Mouse: Trp53tm1Brn | Gift from Dr. Berns | RRID:IMSR_JAX:008462 |
| Mouse: Arid1atm1.1Zhwa | Gift from Dr. Wang | RRID:IMSR_JAX:027717 |
| Mouse: Krt19tm1(cre/ERT)Ggu | Gift from Dr. Gu | MGI:J:136694JAX:026925 |
| Mouse: Gt(ROSA)26Sortm1(EYFP)Cos/J | Jackson Laboratory | RRID:IMSR_JAX:006148 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See oligonucleotide list in Table S3 | N/A | |
|
| ||
| Recombinant DNA | ||
|
| ||
| Human ARID1A: pLKO shRNA Arid1a | Sigma | TRCN0000059088 |
| Human ARID1A: pLKO shRNA Arid1a | Sigma | TRCN0000059089 |
|
| ||
| Software and algorithms | ||
|
| ||
| bcl2fastq version 2.19.1 | illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| FastP version 0.20.0 | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| STAR 2.7.0f | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Salmon 0.13.1 | Patro et al., 2017 | https://github.com/COMBINE-lab/salmon |
| DESeq2 1.22.1 | Love et al., 2014 | https://doi.org/10.18129/B9.bioc.DESeq2 |
| Trimmomatic | Bolger et al., 2014 | https://github.com/usadellab/Trimmomatic |
| Bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
|
| ||
| Other | ||
|
| ||
| DDC diet | Custom Animal Diets | Cat# AD5001 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
All animal studies were conducted in accordance with ARRIVE guidelines and the AAALAC accredited University Committee on Animal Resources (UCAR). All mouse strains (Alb-Cre, CK19-CreERT, KrasLSL-G12D, Arid1aL/L Rosa26LSL-eYFP, and Smad4L/L) (Bardeesy et al., 2006; Jackson et al., 2001; Means et al., 2008; Postic et al., 1999; Srinivas et al., 2001) have been previously reported. Mice lineages were maintained on a mixed background and bred to obtain desired genotypes as indicated. See detailed genotyping information listed Table S3.
Primary cell cultures
MEFs were derived utilizing the protocol outlined in Method details/Cell culture. To induce recombination of floxed alleles in MEFs, cells at passage 3 were plated at a density of 106/100cm2 and infected with recombinase expressing Ad5-CMV-Cre or Ad5-CMV-eGFP (Vector Development Lab, Baylor) viral particles (MOI = 15,000) overnight in the presence of 1μg/mL polybrene. Cells at passage 5 were used for further experiments.
METHOD DETAILS
Histology and immunostaining
Fresh tissue was fixed in neutralized formalin and processed for paraffin sectioning. 5 μm sections were deparaffinized in xylene and rehydrated in 100%, 95%, 75% ethanol and water. Hematoxylin and Eosin (H&E) staining was conducted according to standard procedures. For immunostaining, slides were immersed in citrate buffer (pH = 6) and heated in a pressure cooker for 30 min for antigen retrieval, and blocked for 30 min (Background Sniper, Biocare Medical, BS966). Primary antibodies were incubated sequentially at 4°C overnight, washed in TBST, and incubated with secondary conjugated antibodies for 2 h at room temperature. Slides were then washed in TBST, stained with Hoechst 33342 at 1:1000 for 45 min (Invitrogen, H3570 10 mg/mL), and mounted with (Prolong Gold Antifade Mountant, Life Technologies, P36930). Images were taken on a confocal microscope (Leica TCS SP5 II) with fixed parameters. Quantification of H&E staining (for pathologic lesion) and immunostaining (for double- or single-positive cells) were conducted by two independent observers blind to genotype. Antibodies are listed in the Key Resources Table.
Cell culture
MMNK-1 cells are an immortalized human cholangiocyte cell line established and characterized by (Maruyama et al., 2004).
Mouse embryonic fibroblast (MEFs) were derived following Jozefczuk’s protocol (Jozefczuk et al., 2012). To generate MEFs of desired genotypes, we set two types of breeders, Arid1a L/L (A) with Arid1aL/L;KrasLSL-G12D (KA), and WT with KrasLSL-G12D (K) so that all generated genotypes (WT, A, K, and, KA) are relevant. MEFs and their embryo tissue were genotyped via PCR of genomic DNA using the same primers mentioned above. MEFs from passage 3 were infected with Adenovirus (Ad5-CMV-Cre or Ad5-CMV-eGFP) with 1 μg/mL polybrene overnight. Cells were expanded for 2 passages. The infected cells at passage 5 and 80% confluency were harvested for RNA-seq and ATAC-seq analyses. Other MEF experiments were also performed with the cells at early passages after infection.
Population doubling
Population doubling was done following the 3T3 protocol described in (Sun and Taneja, 2007). Briefly, we passed the cells every 3 days, plated cells of each genotype at the same density (1 million cells in a 10 cm dish). Absolute cell counts from each dish were measured manually using a hemocytometer. Doubling was calculated as log2 Fold change of harvested vs plated cell counts. The accumulative doubling over passages presented in the population doubling curve.
Adipocyte differentiation and Oil Red O quantification
MEF differentiation and quantification was adopted from (Kraus et al., 2016; Matsumoto et al., 2016). 5000 MEFs were plated in 24-well dish in normal growth media (defined above) for 24 h, and then washed and transferred to adipocyte differentiation media, which was the growth media supplemented with 5 μg/mL insulin (Sigma, I6634), 0.5 mM 3-isobutyl-1-methylxanthine IBMX (Sigma, I5879), 1 μM dexamethasone (Sigma, D1756), and 0.5 μM rosiglitazone (Sigma, R2408) for 6 days, with fresh differentiation media replaced every other day. After 6 days in the differentiation media the cells were washed with PBS, fixed with 4% formaldehyde in PBS at room temperature. Formaldehyde was removed and cells in each well were incubated with 271 μL of a filtered 0.2% Oil Red O solution in 40% isopropanol in ddH20 was for staining. After staining, cells were washed with water 5 times and allowed to air dry. Pictures were taken with an inverted microscope. Then, 542 μL of 100% isopropanol was added to the cells and they were placed on a shaker for 10 min in order to elute Oil Red O. Two 200 μL aliquots were measured from each well by absorption at 510 nm.
EdU incorporation assay
100,000 MEFs were plated on coverslips and processed with (Click-IT EdU, Invitrogen, C10337). At least 10 random fields per sample were imaged and quantified for DAPI positive cells and DAPI/EdU double-positive cells.
β-Gal staining
50,000 MEFs were plated collagen-coated coverslips. 48 h later cells were fixed, stained overnight with Senescence β-Galactosidase Staining Kit (Cell Signaling, 9860), counterstained with Nuclear Fast Red (Vector, H-3403), and mounted with 70% glycerol. 10 fields per sample were analyzed for β-Gal positivity.
Colony formation
For colony formation assays, cells were plated in 10 cm dishes at very low density. Media was changed every other day for 10–14 days and cells were fixed with cold 100% methanol stained with crystal violet.
Crystal violet staining
Cells were fixed with cold 100% methanol, dried, stained with 0.5% crystal violet for 1 h, washed with ddH2O, and dried. Fixed cells were imaged and extracted with 10% acetic acid. Absorbance at 590 nm was measured using a Nanodrop OneC Spectrophotometer.
Lentiviral packaging and shRNA mediated knockdown
A second-generation lentiviral packaging system was used. HEK 293 T cells were transfected with PAX2, VSV-G, and ARID1A shRNA expression vector (TRCN0000059088 or TRCN0000059089) using Lipofectamine™ 2000 Transfection Reagent (Invitrogen, 11668–019). Target cells were subject to infection followed by 48-h recovery and antibiotic selection.
Western blot
Protein was isolated from cells or tissue with RIPA buffer (Cell Signaling, 9806) supplemented with protease inhibitor (Sigma, P8340) and phosphatase inhibitors (Sigma, P5726 & P0044) on ice. Protein samples were denatured in loading buffer, separated on SDS-PAGE gels, and transferred to a PVDF membrane. Blots were blocked with 5% NFDM in TBST and washed with TBST in between incubations. Images were captured at variable exposures on a ChemiDoc MP imaging system (BIO-RAD) following incubation with SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher, 34579). Antibody information is listed in the Key Resources Table.
RT-qPCR
Total RNA was extracted and purified from cells or tissue using the RNeasy kit (Qiagen, cat. no. 74104) with on-column DNase I treatment (Qiagen, 79254). Two-step RT-qPCR was carried out, cDNA was created with the Reverse Transcription Kit (Applied Biosystems, 4374967). cDNA and primers were combined with iTaq Universal SYBR Green Supermix (BIO-RAD, 1725124) and Cq values were obtained using the QuantStudio™ 5 Real-Time PCR System (Thermofisher, A28570). Relative mRNA levels to RhoA were normalized to the control sample using the 2−ΔΔCt method. Primer sequences are provided in Table S3.
mRNA Library Prep
Total RNA concentration and quality was determined using NanopDrop 1000 spectrophotometer and Agilent Bioanalyzer 2100 methods, respectively. mRNA sequencing libraries were constructed using the Illumina TruSeq Stranded mRNA Library Prep kit with 200 ng total RNA following the manufacturer’s specifications. Library quantity and quality were measured using a Qubit fluorometer (ThermoFisher) and Fragment Analyzer 5300 (Agilent), respectively.
RNA-seq
Libraries were sequenced on a Illumina NovaSeq 6000 SP flowcell, generating single-end reads of 100 nucleotides (nt) with at least 25 million raw reads per sample.Raw reads generated from the Illumina basecalls were demultiplexed using bcl2fastq version 2.19.1. Quality filtering and adapter removal were performed using FastP version 0.20.0 with the following parameters: “–length_required 35 –cut_front_window_size 1 –cut_front_mean_quality 13 –cut_front –cut_tail_window_size 1 –cut_tail_mean_quality 13 –cut_tail -y -r”. Processed/cleaned reads were then mapped to the mus musculus reference genome (GRCm37 + Gencode-31 Annotation) using STAR 2.7.0f with the following parameters: “–twopass Mode Basic –runMode alignReads –outSAMtype BAM SortedByCoordinate – outSAMstrandField intronMotif –outFilterIntronMotifs RemoveNoncanonical –outReads UnmappedFastx”. Gene-level read quantification was derived using the subread 1.6.4 package (featureCounts) with a GTF annotation file (Gencode 31) and the following parameters: “-s 2 -t exon -g gene_name”. Transcript-level reads were quantified with Salmon 0.13.1 using default parameters. Differential expression analysis was performed using DESeq2 1.22.1 with an adjusted p value threshold of 0.05 within R version 3.5.1. The differentially expressed genes with an absolute log2FoldChange>0.5 between KA and K MEFs were used as input for IPA upstream regulator analysis. For GSEA analysis, these genes were further ranked by the log2FoldChange and analyzed using the GSEA Preranked algorithm. For cooperation response gene (CRG) analysis, the mean expression of each gene for A, K and KA MEFs (denoted as a, b and d) were calculated as the Fold Change against the WT MEFs. The CRG scores were then calculated as a/d+b/d for genes upregulated in KA vs WT MEFs and (d/a + d/b) for genes downregulated. Genes with a CRG score <0.9 were identified as CRGs (McMurray et al., 2008).
ATAC-seq
Libraries were prepared utilizing methods outlined previously. (Myers et al., 2020) ATAC-seq Raw reads were processed using Trimmomatic (SLIDINGWINDOW:4:20 TRAILING:13 LEADING:13 MINLEN:15). Quality reads (FastQC) were aligned to mm9 using Bowtie2. Alignment quality was evaluated by BamQC and low-quality alignments were filtered using samtools based on MAPQ <10. Significant regions (peaks) were identified using MACS2 based on quality filtered PE alignments (-f BAMPE –nomodel –shift −100 –extsize 200). Peaks called from each biological replicate were concatenated and merged if the inter-interval distance is less than 300 base pair. Within-peak read counts were analyzed for differential chromatin accessibility via DESeq2 1.22.1. Homer annotatepeaks were used to annotate MACS2 enrichments based on their genomic locations. Bedgraph files were converted to bigwig format using a UCSC bedGraphToBigWig for visualization in IGV. BAM files used for peak calling were RPKM normalized using deeptools bamCoverage and a binSize of 10. For heatmap visualization, RPKM normalized bigwig files from biological replicates of each genotype were merged using bigWigMerge which was then used as input for deeptools computeMatrix to obtain ATAC-seq signal that is lined up at the center of peaks. The ATAC-seq results were compared with publicly available ChIP-seq data from GEO series GSE83295. Selected samples include Fos (GSM2198470-GSM2198471), Fosl2 (GSM2198478-SM2198479), H3K27Ac (GSM2198495-GSM2198497), JunD (GSM2198504-GSM2198505) and Smarca4 (GSM2905664-GSM2905666).
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments were repeated at least three times independently unless otherwise indicated. Data were expressed in the form of mean ± sd. For continuous data, Student’s t test was used; for discrete data, the Chi-Square or Fisher’s exact test was used. For multiple-group experiments, nested one-way anova analysis was performed to test if there is difference among groups and a post-hoc Tukey Honestly Significant Difference (HSD) test was used to compare groups pair wisely. For multiple hypothesis testing for RNA-seq and ATAC-seq data, adjusted p values were provided from the DESeq2 package. Survival time was expressed in the form of restricted mean ± se (restricted mean), which uses R package survival and assumes the upper limit of survival to be 125 weeks for all cohorts; Log Rank test was used to compare different survival curves. p value < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Kras/Arid1a mutations drive cholangiocarcinoma synergistically from a biliary origin
Changes in transcriptome and DNA accessibility underlie proliferative phenotypes
TGF-β/Smad4 signaling is a main pathway in regulating biliary tumorigenesis
ARID1A loss impairs upstream activation of the TGF-β-SMAD pathway
ACKNOWLEDGMENTS
This work was supported by 1R01CA172302 (to A.F.H.), 1R35CA197562 (to H.K.L.), and the Wilmot Cancer Research Fellowship (to B.G.). The authors would like to acknowledge the work of Mary Georger at the Wilmot Cancer Institute Histopathology Core as well as the UR Genomics Research Center. The authors thank the Institute of Genome Sciences at the University of Maryland School of Medicine for supporting B.G.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111253.
REFERENCES
- Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, Cardinale V, Carpino G, Andersen JB, Braconi C, et al. (2020). Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 17, 557–588. 10.1038/s41575-020-0310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, and DePinho RA (2006). Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 20, 3130–3146. 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-On W, Nairismägi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, et al. (2013). Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 45, 1474–1478. 10.1038/ng.2806. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JZ, Li C, Liu XY, Hu TT, Fan ZS, and Han ZG (2015). Hepatocyte-specific Arid1a deficiency initiates mouse steatohepatitis and hepatocellular carcinoma. PLoS One 10, e0143042. 10.1371/journal.pone.0143042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, Hinoue T, Hoadley KA, Gibb EA, Roszik J, et al. (2017). Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 18, 2780–2794. 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Alexander WB, Guo B, Kato Y, Patra K, O’Dell MR, McCall MN, Whitney-Miller CL, Bardeesy N, and Hezel AF (2018). Kras and Tp53 mutations cause cholangiocyte- and hepatocyte-derived cholangiocarcinoma. Cancer Res. 78, 4445–4451. 10.1158/0008-5472.CAN-17-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Li W, Tian F, Jiang K, Liu X, Cen J, He Q, Qiu Z, Kienast Y, Wang Z, et al. (2018). Arid1a regulates response to anti-angiogenic therapy in advanced hepatocellular carcinoma. J. Hepatol. 68, 465–475. 10.1016/j.jhep.2017.10.028. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Terakado Y, Nakagawa H, Hikiba Y, Fujii T, Matsubara D, Noguchi R, Zhu C, Yamamoto K, Kudo Y, et al. (2016). A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 6, 23899. 10.1038/srep23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248. 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. (2013). Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 45, 1470–1473. 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozefczuk J, Drews K, and Adjaye J (2012). Preparation of mouse embryonic fibroblast cells suitable for culturing human embryonic and induced pluripotent stem cells. JoVE e3854, 3854. 10.3791/3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, and Hargreaves DC (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife 6. 10.7554/eLife.30506.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus NA, Ehebauer F, Zapp B, Rudolphi B, Kraus BJ, and Kraus D (2016). Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte 5, 351–358. 10.1080/21623945.2016.1240137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Xia J, Fang M, and Xu Y (2019). Epigenetic regulation of lung cancer cell proliferation and migration by the chromatin remodeling protein BRG1. Oncogenesis 8, 66. 10.1038/s41389-019-0174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livshits G, Alonso-Curbelo D, Morris JP, Koche R, Saborowski M, Wilkinson JE, and Lowe SW (2018). Arid1a restrains Kras-dependent changes in acinar cell identity. Elife 7, e35216. 10.7554/eLife.35216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery MA, Ptashkin R, Jordan E, Berger MF, Zehir A, Capanu M, Kemeny NE, O’Reilly EM, El-Dika I, Jarnagin WR, et al. (2018). Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin. Cancer Res. 24, 4154–4161. 10.1158/1078-0432.CCR-18-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama M, Kobayashi N, Westerman KA, Sakaguchi M, Allain JE, Totsugawa T, Okitsu T, Fukazawa T, Weber A, Stolz DB, et al. (2004). Establishment of a highly differentiated immortalized human cholangiocyte cell line with SV40T and hTERT. Transplantation 77, 446–451. 10.1097/01.TP.0000110292.73873.25. [DOI] [PubMed] [Google Scholar]
- Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, and Roberts CWM (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 49, 296–302. 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Kondo K, Shiraki T, Brydun A, Funayama R, Nakayama K, Yaegashi N, Katagiri H, and Igarashi K (2016). Genomewide approaches for BACH1 target genes in mouse embryonic fibroblasts showed BACH1-Pparg pathway in adipogenesis. Gene Cell. 21, 553–567. 10.1111/gtc.12365. [DOI] [PubMed] [Google Scholar]
- McMurray HR, Sampson ER, Compitello G, Kinsey C, Newman L, Smith B, Chen SR, Klebanov L, Salzman P, Yakovlev A, and Land H (2008). Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature 453, 1112–1116. 10.1038/nature06973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means AL, Xu Y, Zhao A, Ray KC, and Gu G (2008). A CK19CreERT knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis. 10.1002/dvg.20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Pradere JP, Affò S, Dapito DH, Friedman R, Lefkovitch JH, and Schwabe RF (2016). Epithelial transforming growth factor-β signaling does not contribute to liver fibrosis but protects mice from cholangiocarcinoma. Gastroenterology 150, 720–733. 10.1053/j.gastro.2015.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JA, Couch T, Murphy Z, Malik J, Getman M, and Steiner LA (2020). The histone methyltransferase Setd8 alters the chromatin landscape and regulates the expression of key transcription factors during erythroid differentiation. Epigenet. Chromatin 13, 16. 10.1186/s13072-020-00337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober EA, and Lemaigre FP (2018). Development of the liver: insights into organ and tissue morphogenesis. J. Hepatol. 68, 1049–1062. 10.1016/j.jhep.2018.01.005. [DOI] [PubMed] [Google Scholar]
- O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, and Hezel AF (2012). Kras G12Dand p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 72, 1557–1567. 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, and Magnuson MA (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315. 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Ringel T, Frey N, Ringnalda F, Janjuha S, Cherkaoui S, Butz S, Srivatsa S, Pirkl M, Russo G, Villiger L, et al. (2020). Genome-scale CRISPR screening in human intestinal organoids identifies drivers of TGF-β resistance. Cell Stem Cell 26, 431–440.e8. 10.1016/j.stem.2020.02.007. [DOI] [PubMed] [Google Scholar]
- Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, et al. (2014). Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114. 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, Donnelly B, Karns RA, Chen F, Rezvani M, Luu HY, et al. (2018). De novo formation of the biliary system by TGFb-mediated hepatocyte transdifferentiation. Nature 557, 247–251. 10.1038/s41586-018-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick S, Grosche S, Kohl KE, Drpic D, Jaeger MG, Marella NC, Imrichova H, Lin J-MG, Hofstätter G, Schuster M, et al. (2021). Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat. Genet. 53, 269–278. 10.1038/s41588-021-00777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Cherry S, and Murphy-Ullrich JE (1993). Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J. Cell Biol. 122, 923–932. 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4. 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, and Taneja R (2007). Analysis of transformation and tumorigenicity using mouse embryonic fibroblast cells. Methods Mol. Biol. 383, 303–310. 10.1007/978-1-59745-335-6_19. [DOI] [PubMed] [Google Scholar]
- Sun X, Chuang JC, Kanchwala M, Wu L, Celen C, Li L, Liang H, Zhang S, Maples T, Nguyen LH, et al. (2016). Suppression of the SWI/SNF component Arid1a promotes mammalian regeneration. Cell Stem Cell 18, 456–466. 10.1016/j.stem.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, Gopal P, Zhu M, Nassour I, Chuang JC, et al. (2017). Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell 32, 574–589.e6. 10.1016/j.ccell.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryo Rahmanto Y, Shen W, Shi X, Chen X, Yu Y, Yu ZC, Miyamoto T, Lee MH, Singh V, Asaka R, et al. (2020). Inactivation of Arid1a in the endometrium is associated with endometrioid tumorigenesis through transcriptional reprogramming. Nat. Commun. 11, 2717–2814. 10.1038/s41467-020-16416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trizzino M, Barbieri E, Petracovici A, Wu S, Welsh SA, Owens TA, Licciulli S, Zhang R, and Gardini A (2018). The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 23, 3933–3945. 10.1016/j.celrep.2018.05.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. (2004). Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5, 375–387. 10.1016/S1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 68, 1067–1082.e12. 10.1016/j.molcel.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Nassour I, Xiao S, Zhang S, Luo X, Lee J, Li L, Sun X, Nguyen LH, Chuang JC, et al. (2019a). SWI/SNF component ARID1A restrains pancreatic neoplasia formation. Gut 68, 1259–1270. 10.1136/gutjnl-2017-315490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Friedland SC, Guo B, O’Dell MR, Alexander WB, Whitney-Miller CL, Agostini-Vulaj D, Huber AR, Myers JR, Ashton JM, et al. (2019b). ARID1A, a SWI/SNF subunit, is critical to acinar cell homeostasis and regeneration and is a barrier to transformation and epithelial-mesenchymal transition in the pancreas. Gut 68, 1245–1258. 10.1136/gutjnl-2017-315541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, Polak P, Kim J, Hatanaka Y, Maejima K, et al. (2018). Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 68, 959–969. 10.1016/j.jhep.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, Adams M, Johnson B, Su R-W, Joshi NR, et al. (2019). ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 10, 3554. 10.1038/s41467-019-11403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, He W, Zhang XH-F, Le H-V, and Massagué J (2008). Genomewide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor β transcriptional program. J. Biol. Chem. 283, 1146–1155. 10.1074/jbc.M707479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, Wells RG, Greenbaum LE, and Stanger BZ (2013). Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 27, 719–724. 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, and Dean DC (2000). Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101, 79–89. 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JSC, Zhao X, Li Y, Li Q, Wang H, et al. (2014). Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 5, 5696. 10.1038/ncomms6696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data availability: Sequencing data has been deposited at GEO and is publicly available as of the publication date. Please refer to the “Deposited data” subsection in the key resources table for the accession number.
Custom code availability: This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-ARIDI A (clone D2A8U) | Cell Signaling Technology | Cat# 12354; RRID:AB_2637010 |
| Rabbit monoclonal anti-Smad2/3 (clone D7G7) | Cell Signaling Technology | Cat# 8685; RRID:AB_10889933 |
| Rabbit monoclonal anti-Phospho-Smad2(Ser465/ 467)/Smad3(Ser423/425) (clone D27F4) | Cell Signaling Technology | Cat# 8828; RRID:AB_2631089 |
| Rabbit monoclonal anti-GAPDH (clone 14C10) | Cell Signaling Technology | Cat# 2118; RRID:AB_561053 |
| Rabbit monoclonal anti-GFP (clone D5.1) | Cell Signaling Technology | Cat# 2956; RRID:AB_1196615 |
| Mouse monoclonal anti-THBS1 (clone A6.1) | ThermoFisher | Cat# MA5-13398; RRID:AB_10984611 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab66155; RRID:AB_1140752 |
| Rabbit anti-p15(INK4b) | ThermoFisher | Cat# PA5-49749; RRID:AB_2635202 |
| Rabbit anti-CDK4 | ThermoFisher | Cat# MA5-41183; RRID:AB_2898937 |
| Rabbit anti-Cyclin A2 | ThermoFisher | Cat# MA5-32353; RRID:AB_2809634 |
| Rat anti-CK19 | Developmental Studies Hybridoma Bank Univ Iowa | Cat# TROMA-III; RRID:AB_2133570 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV8.TBG.PI.Cre.rBG | Penn Vector Core | Cat# CS1380 |
| Ad5-CMV-Cre | Baylor Gene Vector Core | Cat# 013018 |
| Ad5-CMV-eGFP | Baylor Gene Vector Core | Cat# 050917 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| hTGF-β1 | Cell Signaling Technology | Cat# 8915 |
| mTGF-β 1 | Cell Signaling Technology | Cat# 5231 |
| LY2157299 | MedChemExpress | Cat# HY-13226 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Senescence b-Galactosidase Staining Kit | Cell Signaling Technology | Cat# 9860 |
| RNeasy kit | Qiagen | Cat# 74104 |
| Click-IT EdU | Invitrogen | C10337 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data (SuperSeries) | This paper | [GEO]: GSE207895 |
| MEF RNA-seq (SubSeries) | This paper | [GEO]: GSE207891 |
| MEF ATAC-seq (SubSeries) | This paper | [GEO]: GSE207888 |
| MMNK1 RNA-seq (SubSeries) | This paper | [GEO]: GSE207892 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: primary liver tumor cell lines | This paper | N/A |
| Mouse: MEF cells | This paper | N/A |
| Human: MMNK-1 cells | Maruyama et al., 2004 | Cat# JCRB1554, RRID:CVCL_M266 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Tg(Alb-cre)21Mgn | Gift from Dr. Magnuson | RRID:IMSR_JAX:003574 |
| Mouse: Krastm4Tyj | Gift from Dr. Jacks | RRID:IMSR_JAX:008179 |
| Mouse: Trp53tm1Brn | Gift from Dr. Berns | RRID:IMSR_JAX:008462 |
| Mouse: Arid1atm1.1Zhwa | Gift from Dr. Wang | RRID:IMSR_JAX:027717 |
| Mouse: Krt19tm1(cre/ERT)Ggu | Gift from Dr. Gu | MGI:J:136694JAX:026925 |
| Mouse: Gt(ROSA)26Sortm1(EYFP)Cos/J | Jackson Laboratory | RRID:IMSR_JAX:006148 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See oligonucleotide list in Table S3 | N/A | |
|
| ||
| Recombinant DNA | ||
|
| ||
| Human ARID1A: pLKO shRNA Arid1a | Sigma | TRCN0000059088 |
| Human ARID1A: pLKO shRNA Arid1a | Sigma | TRCN0000059089 |
|
| ||
| Software and algorithms | ||
|
| ||
| bcl2fastq version 2.19.1 | illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| FastP version 0.20.0 | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| STAR 2.7.0f | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Salmon 0.13.1 | Patro et al., 2017 | https://github.com/COMBINE-lab/salmon |
| DESeq2 1.22.1 | Love et al., 2014 | https://doi.org/10.18129/B9.bioc.DESeq2 |
| Trimmomatic | Bolger et al., 2014 | https://github.com/usadellab/Trimmomatic |
| Bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
|
| ||
| Other | ||
|
| ||
| DDC diet | Custom Animal Diets | Cat# AD5001 |