

Abstract
With its nuclear dualism, the ciliate Paramecium constitutes a unique model to study how host genomes cope with transposable elements (TEs). P. tetraurelia harbors two germline micronuclei (MICs) and a polyploid somatic macronucleus (MAC) that develops from one MIC at each sexual cycle. Throughout evolution, the MIC genome has been continuously colonized by TEs and related sequences that are removed from the somatic genome during MAC development. Whereas TE elimination is generally imprecise, excision of approximately 45,000 TE-derived internal eliminated sequences (IESs) is precise, allowing for functional gene assembly. Programmed DNA elimination is concomitant with genome amplification. It is guided by noncoding RNAs and repressive chromatin marks. A subset of IESs is excised independently of this epigenetic control, raising the question of how IESs are targeted for elimination. To gain insight into the determinants of IES excision, we established the developmental timing of DNA elimination genome-wide by combining fluorescence-assisted nuclear sorting with high-throughput sequencing. Essentially all IESs are excised within only one endoreplication round (32C to 64C), whereas TEs are eliminated at a later stage. We show that DNA elimination proceeds independently of replication. We defined four IES classes according to excision timing. The earliest excised IESs tend to be independent of epigenetic factors, display strong sequence signals at their ends, and originate from the most ancient integration events. We conclude that old IESs have been optimized during evolution for early and accurate excision by acquiring stronger sequence determinants and escaping epigenetic control.
Transposable elements (TEs) have colonized the genomes of most living species and constitute a significant fraction of extant genomes, from a few percentages in yeast (Bleykasten-Grosshans and Neuvéglise 2011) to ∼85% in some plant genomes (Bennetzen and Park 2018). TEs are often considered as genomic parasites threatening host genome integrity, even though they can be a source of genetic innovation (Cosby et al. 2019; Capy 2021). Host defense pathways counteract the potentially detrimental effects of transposon invasion. In eukaryotes, small RNA (sRNA)-dependent post-transcriptional and transcriptional silencing mechanisms inactivate TE expression and transposition, both in germline and somatic cells (Ketting et al. 1999; Tabara et al. 1999; Zilberman et al. 2003; Brennecke et al. 2007). TE transcriptional inactivation is associated with heterochromatin formation, through DNA methylation and histone H3 methylation on lysine 9 (Deniz et al. 2019; Choi and Lee 2020). Another epigenetic mark, H3K27me3, also contributes to TE silencing in several species (Déléris et al. 2021).
Because of their germline–soma nuclear dualism (Prescott 1994; Cheng et al. 2020), ciliates are unique unicellular eukaryotic models to study the dynamics of TEs within genomes, at both the developmental and evolutionary timescales (Arnaiz et al. 2012; Hamilton et al. 2016; Kapusta et al. 2017; Sellis et al. 2021). Paramecium species harbor one to four transcriptionally silent diploid germline micronuclei (MICs) (Görtz 1988) that coexist with a polyploid somatic macronucleus (MAC) responsible for gene expression. During sexual processes (conjugation of compatible reactive partners or a self-fertilization process called autogamy), the MICs undergo meiosis and transmit the germline genome to the diploid zygotic nucleus through fertilization and karyogamy (Bétermier and Duharcourt 2014). In the meantime, the old MAC splits into about 30 fragments that continue to ensure gene expression, whereas new MICs and MACs differentiate from division products of the zygotic nucleus. The formation of a functional new MAC is essential to take over gene expression once old MAC fragments have disappeared from the cell. New MAC development covers two cell cycles after the zygotic nucleus is formed. During this period, massive genome amplification takes place within each developing MAC (also called anlagen) to reach the final endoduplication level of mature MACs (about 800C to 1600C in Paramecium tetraurelia) (Preer 1976). Concomitantly with genome amplification, programmed DNA elimination (PDE) removes ∼30% of germline DNA from the new MAC genome, going from a 98- to 151-Mbp haploid genome size in the MIC to 72–75 Mbp in the mature MAC (Aury et al. 2006; Guérin et al. 2017; Sellis et al. 2021). Because eliminated DNA includes TEs and related sequences (Arnaiz et al. 2012; Guérin et al. 2017; Sellis et al. 2021), PDE in Paramecium, as in other ciliates, can be viewed as an extreme mechanism to inactivate TEs in the somatic genome.
Two types of germline sequences, referred to as “MIC-limited” DNA, are removed during PDE in Paramecium (Bétermier and Duharcourt 2014). At least 25% of the MIC genome, including DNA repeats (TEs, minisatellites), is eliminated imprecisely, alternatively leading to chromosome fragmentation (with de novo telomere addition to heterogeneous new MAC chromosome ends) or to intrachromosomal deletions between variable boundaries (Baroin et al. 1987; Le Mouël et al. 2003; Guérin et al. 2017). In contrast, approximately 45,000 internal eliminated sequences (IESs) scattered throughout the germline genome (including inside coding sequences [CDSs]) are excised precisely, allowing assembly of functional open reading frames (Arnaiz et al. 2012). Paramecium IESs are mostly short (93% < 150 bp) noncoding sequences, with a damped sinusoidal size distribution extending from 25 bp to a few kilobase pairs. They are consistently flanked by two TA dinucleotides, one on each side, and leave a single TA on MAC chromosomes upon excision. Two independent studies—the first relying on the analysis of paralogous gene quartets originating from successive whole-genome duplications in a single species, P. tetraurelia (Arnaiz et al. 2012), the other on phylogenetic analyses across nine Paramecium species (Sellis et al. 2021)—have made it possible to date ∼40% of P. tetraurelia IES insertions and define groups of old, intermediate, and young IESs according to their evolutionary age. The oldest IESs, thought to have colonized the germline genome before divergence of Paramecium caudatum and the Paramecium aurelia clade, tend to be very short (26–30 bp) (Sellis et al. 2021). Several families of larger and younger IESs, some sharing homology with known Paramecium TEs, appear to have been mobile recently at the timescale of Paramecium evolution: intermediate IESs were acquired after the divergence of P. caudatum; young IESs were gained after the burst of P. aurelia speciation. This is consistent with IESs being relics of ancestral TEs that have decayed during evolution through the reduction in size and loss of coding capacity, while remaining under selection for precise excision from the MAC (Klobutcher and Herrick 1997; Dubois et al. 2012).
IES excision occurs through a “cut-and-repair” mechanism involving double-strand DNA cleavage around each flanking TA (Gratias and Bétermier 2003), followed by excision site closure through precise nonhomologous end joining (NHEJ) (Kapusta et al. 2011; Bétermier et al. 2014). Several components of the core IES excision machinery are known. The PiggyMac (Pgm) endonuclease, a catalytically active domesticated transposase (Baudry et al. 2009; Dubois et al. 2012), and its five Pgm-like partners, PgmL1 to PgmL5 (Bischerour et al. 2018), are essential for the introduction of DNA double-strand breaks at IES ends. In the absence of Pgm, all IESs are retained in the anlagen, and most imprecise DNA elimination is also impaired, except for ∼3 Mbp of germline sequences, the elimination of which seems to be Pgm-independent (Guérin et al. 2017). A specialized NHEJ factor, the Ku70/Ku80c (Ku) heterodimer, also appears to be an essential component of the core endonuclease machinery: Ku is able to interact with Pgm, tethers it in the anlagen, and licenses DNA cleavage at IES ends (Marmignon et al. 2014; Abello et al. 2020; Bétermier et al. 2020).
Paramecium IES ends display a weak consensus (5′ TAYAGTNR 3′), which includes the palindromic flanking TA dinucleotide conserved at each boundary (Arnaiz et al. 2012). This consensus defines an internal inverted repeat at IES ends but is too poorly conserved to serve as a specific recognition sequence for the endonuclease. Additional epigenetic factors, including noncoding RNAs and histone modifications, control the recognition of eliminated DNA by the core machinery (Chalker et al. 2013; Bétermier and Duharcourt 2014; Allen and Nowacki 2020). According to the “scanning” model, sRNAs processed from meiotic MIC transcripts by the Dicer-like proteins Dcl2 and Dcl3 (called “scnRNAS”) are subtracted against old MAC sequences, resulting in the selection of a subpopulation of scnRNAs covering the MIC-limited fraction of the germline genome (Lepère et al. 2008, 2009). MIC-limited scnRNAs are thought to target elimination of their homologous sequences by pairing with TFIIS4-dependent noncoding nascent transcripts in the anlagen (Maliszewska-Olejniczak et al. 2015), thereby triggering H3K9 and K27 trimethylation by the PRC2 complex containing the histone methyltransferase Ezl1 (Lhuillier-Akakpo et al. 2014; Frapporti et al. 2019; Miró-Pina et al. 2022; Wang et al. 2022). The H3K9me3 and H3K27me3 heterochromatin marks are required for the elimination of TEs and ∼70% of IESs (Lhuillier-Akakpo et al. 2014; Guérin et al. 2017). A second population of sRNAs (called iesRNAs), produced by the Dcl5 protein from excised IES transcripts, was proposed to further assist IES excision (Sandoval et al. 2014; Allen et al. 2017). Both types of sRNAs appear to act synergistically. Indeed, although DCL2/3 or DCL5 knockdowns (KDs) each impair excision of only a small fraction of IESs (∼7% in a DCL2/3 KD, ∼5% in a DCL5 KD) (Lhuillier-Akakpo et al. 2014; Sandoval et al. 2014), a triple DCL2/3/5 KD inhibits excision of ∼50% of IESs that coincide with the set of TFIIS4-dependent IESs (Swart et al. 2017).
Although our knowledge of the molecular mechanisms involved in the epigenetic control and catalysis of PDE in P. tetraurelia has increased over the past decade, little is known about the relative timing of DNA replication and PDE during MAC development. Molecular data obtained for a handful of IESs suggested that excision starts following several endoreplication rounds in the anlagen (Bétermier et al. 2000). In the present study, we have investigated at the genome-wide level the elimination timing of all 45,000 IESs and other MIC-limited sequences, including TEs. To follow the progression of IES excision during MAC development, we monitored for each IES the fraction of excised molecules that were present in purified anlagen at each developmental stage, which we have referred to as the excision score (ES). This allowed us to establish the timing of PDE during MAC development, address whether a mechanistic link exists between IES excision and DNA replication, and examine whether the temporal and epigenetic control of PDE may be related to the evolutionary age of eliminated DNA.
Results
A fluorescence-activated nuclear sorting strategy to purify new MACs
Because old MAC fragments containing the rearranged genome are present in Paramecium cells throughout the sexual cycle, we set up a protocol to selectively purify developing new MACs during an autogamy time course (tc). We adapted a published flow cytometry procedure that was initially designed to sort anlagen from old MAC fragments at a late developmental stage, when the two types of nuclei can clearly be distinguished based on their size and DNA content (Guérin et al. 2017). Because at early stages (DEV1 and DEV2; see Methods), anlagen and old MAC fragments have similar sizes (Fig. 1A), we selectively labeled the new MACs using a specific α-PgmL1 antibody raised against a component of the IES excision machinery (Bischerour et al. 2018). We first confirmed that immunofluorescence staining of whole cells yielded a strong and specific signal in the anlagen throughout DEV1 to DEV4 (Fig. 1A; Supplemental Fig. S1A–C), which corresponds to the time-window (T3 to T30) when programmed double-strand breaks are detected at IES boundaries (Gratias et al. 2008; Baudry et al. 2009). Using the α-PgmL1 antibody to label unfixed nuclei harvested at DEV1 and DEV2 during an autogamy time course of wild-type (wt) cells, we confirmed that PgmL1 labeling can be used to separate anlagen from old MAC fragments using flow cytometry (Fig. 1B; Supplemental Fig. S1D,E).
Figure 1.
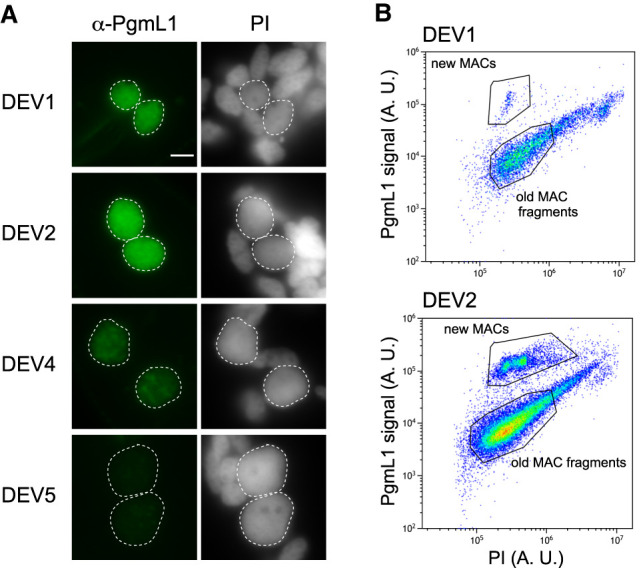
PgmL1 immunostaining during autogamy. (A) Whole-cell immunostaining at different stages of autogamy time course 1 (tc1). New MACs and fragments are counterstained with propidium iodide (PI). Developing MACs are surrounded by a white dotted line. Scale bar is 5 µm. Developmental stages (DEV1 to DEV5) are defined in the Methods. (B) Flow cytometry analysis of immunostained nuclei at the DEV1 and DEV2 stages of autogamy time course 2 (tc2). Following gating of total nuclei (see Supplemental Fig. S1D), the population of new MACs was separated based on their PgmL1 signal. The PI axis is indicative of DNA content. (A. U.) Arbitrary units in log scale.
Most IES excision takes place within one round of replication
We used autogamy to purify new MACs by fluorescence-activated nuclear sorting (FANS) at different DEV stages despite the asynchrony of this sexual process (Berger 1986), because it allows us to collect large amounts of material. The distribution of propidium iodide (PI) fluorescence intensities revealed a series of three discrete peaks from DEV1 to DEV3 for PgmL1-labeled new MACs (Fig. 2A). This is indicative of the presence of nuclear populations with a defined DNA content. Previously published work suggested that at least four discontinuous peaks of DNA synthesis, corresponding to around five doublings of DNA content, take place in anlagen before the first cell fission in P. tetraurelia, whereas 4.5 additional doublings occur with a more continuous pattern during the second cell cycle (Berger 1973). We therefore made the reasonable assumption that each peak observed in flow cytometry corresponds to one whole-genome doubling following a pulse of genome replication, and focused on these populations to draw the sorting gates for further purification. We calculated the DNA content for each peak (C-value in Mbp; see Methods) (Supplemental Fig. S2; Supplemental Table S1) and further defined the corresponding amplification level of the genome (C-level), using an approximate 1C-value of 100 Mbp for the unrearranged P. tetraurelia MIC genome (Guérin et al. 2017; Sellis et al. 2021). We attributed the closest power of two to each resulting amplification level and defined an estimated C-level of approximately 32C (DEV1/DEV2), 64C (DEV2/DEV3), and 128C (DEV3) for each population (tc4 in Supplemental Table S1). At DEV4, which is the final stage at which PgmL1 staining can be detected, we observed an enlargement of the 128C peak, indicative of a mixed population with a more variable amount of DNA (see Discussion).
Figure 2.

IES excision kinetics and endoreplication. (A) Flow cytometry sorting of nuclei during the different stages of an autogamy time course (tc4). (Upper panels) Plots of PgmL1 fluorescence intensity (y-axis) versus PI fluorescence intensity (x-axis) for nuclei collected at different developmental stages. (Lower panels) Histograms of PI-stained nuclei gated in the upper panel. Sorted new MAC peaks are indicated by light green shading. The estimated C-level for each sorted peak is indicated above. For DEV4 nuclei, the whole PgmL1-labeled population was sorted (light green), but the major peak was used for calculation of the C-level. As a control, old MAC fragments were sorted from the DEV1 stage. (B) Distribution of IES excision scores (ESs) in the different sorted new MAC populations. Samples are named according to the developmental stage (DEV1 to DEV4 from tc4) and the C-level of the sorted population. A schematic representation of the IES+ and IES− Illumina sequencing reads that were counted to calculate the ES is presented on the left. An ES of zero or one corresponds to no or complete IES excision, respectively. The black dot is the median, and the vertical black line delimitates the second and third quartiles. (C) Flow cytometry sorting of nuclei following aphidicolin treatment. PI histograms of PgmL1-labeled nuclei are presented for each stage or condition (DEV1, DEV3 DMSO, and DEV3 Aphi). The C-level for the indicated peaks was estimated as described in Supplemental Table S1. For each stage, all PgmL1-labeled nuclei were sorted. Old MAC fragments were sorted as a control from the DEV3 DMSO nuclear preparation. The dotted line is indicative of a PI value of 103. (D) ES distribution in the sorted new MAC populations in the aphidicolin time course. Sample names correspond to the sorted samples shown in C.
We further sorted the populations of nuclei issued from each peak (Fig. 2A) and extracted their DNA for deep sequencing (tc4 in Supplemental Table S2). Thanks to the absence of old MAC contamination (see Methods) (Supplemental Fig. S3A–C), molecules lacking an inserted IES (designated IES−) only correspond to de novo excision junctions. Therefore, the power of the FANS procedure allows us for the first time to calculate a real ES, which varies from zero (no excision) to one (complete excision), for each of the 45,000 IESs (Fig. 2B). At DEV1 ∼ 32C, few IESs have been excised, with a median ES value of 0.15. The median ES rises to one at DEV3 ∼ 64C, indicating that nearly all IESs are excised within one round of replication. To investigate whether the fifth endoreplication round itself is mandatory for DNA elimination, we performed a replicate time course experiment in which we treated autogamous cells with aphidicolin, a specific inhibitor of eukaryotic replicative DNA polymerases (Byrnes 1984; Cheng and Kuchta 1993), after they reached DEV1 ∼32C (Fig. 2C). Comparison of the flow cytometry profiles confirmed that the new MACs of control cells (+DMSO) have undergone their fifth replication round at DEV3, whereas those of aphidicolin-treated cells are blocked at ∼32C. We further sorted anlagen from the DEV1, DEV3 DMSO, and DEV3 Aphi samples for DNA sequencing (Supplemental Table S2). For the control replicate, we confirmed that most IES excision is completed within one round of replication, between ∼32C and ∼64C (median ES at DEV3 ∼ 64C is 0.99) (Fig. 2D). For aphidicolin-treated anlagen, the median ES is 0.98, indicating that inhibiting the fifth endoreplication round does not impair IES excision.
Imprecise elimination is delayed relative to IES excision
To strengthen our analysis of the timing of DNA elimination, we included sorted samples from four additional replicate time course experiments (Supplemental Fig. S4A–C; Supplemental Tables S1, S2). The resulting ES distributions confirm our conclusion that IES excision takes place between DEV1 ∼ 32C and DEV3 ∼ 64C (Fig. 3A). We used the same sequencing data (Supplemental Table S2) to study the timing of imprecise DNA elimination during MAC development. Because this process yields heterogeneous MAC junctions, preventing us from calculating an ES, we analyzed sequencing data by read coverage (Fig. 3B). Using this procedure, we confirmed that the sequencing coverage drops between DEV1 ∼ 32C and DEV3 ∼ 64C for IESs, consistent with the excision profile obtained by ES calculation (Fig. 3A). After this valdation, we analyzed TE coverage as a proxy for imprecise DNA elimination. We observed a delayed decrease relative to IES coverage, with a drop starting at DEV3 ∼ 64C. Analysis of the percentage of coverage of the whole MIC genome revealed a similar decrease (from 97% to 90%) between DEV3 ∼ 64C and DEV3 ∼ 128C, which likely corresponds to the elimination of MIC-specific DNA. During DNA elimination, however, TE sequences may be found in two forms: nonexcised intrachromosomal molecules and not yet degraded extrachromosomal elimination products. Because sequence coverage analysis cannot discriminate between these two forms, TE elimination may have started before the drop of sequence coverage. We therefore used another marker of imprecise elimination: the formation of de novo telomeric ends that accompanies the removal of TE-containing MIC-specific sequences (Le Mouël et al. 2003). We observed that telomeric reads only increase at DEV3 ∼ 64C (Fig. 3C), supporting the idea that imprecise elimination does not begin before DEV3. Of note, the majority of telomere addition sites are localized at a >100-nt distance from IES boundaries, confirming that they are not related to precise IES elimination. We also noticed that the whole MIC genome coverage at DEV4 ∼ 128C is still higher than the genome coverage of fragments (which harbor a fully rearranged genome), indicating that imprecise elimination is not totally completed in the new MAC at this stage.
Figure 3.

Kinetics of precise IES excision and imprecise DNA elimination. (A) Distribution of ESs in all samples. Samples are named and ordered according to developmental stage (DEV1 to DEV4), time course (tc2, tc3, tc4, tc5, tc6), and C-level (indicated above the plot). Hierarchical clustering of ESs confirmed that samples from the same developmental stage (DEV1 to DEV4) group together (Supplemental Fig. S4C). Inside each developmental stage, for a given C-level, we have ordered the samples using their median ES score. For each time course, old MAC fragments (FRAG) were also sorted as controls. The black dot is the median, and the vertical black line spans the second and third quartiles. (B) TE and IES coverage during autogamy. The mean depth coverage distribution is represented as a boxplot. For each data set described in A, the gray boxplot shows TE coverage; the white, IES coverage. The percentage of the MIC genome covered by the sequencing reads is indicated above each pair of boxplots. (C) Abundance of telomere addition sites during autogamy. The schema above the bars illustrates the method for detection of telomere addition sites using the sequencing data. For each data set, the bar shows the normalized number (per million mapped reads [RPM]) of detected telomere addition sites localized at <10 nt (black), between 10 and 100 nt (dark gray), and >100 nt (light gray) from an IES. (D) Quantification of IES–IES junctions. All putative molecules resulting from ligation of excised IES ends (see Supplemental Fig. S5A,B) are counted and normalized using sequencing depth. The percentage of IESs involved in at least one IES–IES junction is indicated above the barplot.
Genome-wide detection of transient IES–IES junctions
We took advantage of the purity of FANS-sorted anlagen to increase our ability to detect transient DNA molecules produced during IES excision. Based on a few Southern blot experiments, IESs were proposed to be excised as linear molecules, and subsequent formation of closed DNA circles was documented for a few long IESs (Gratias and Bétermier 2001). More recently, excised IESs were proposed to concatenate through the NHEJ pathway into end-to-end joined circular molecules that are used as substrates for transcription and Dcl5-dependent production of iesRNAs, before being eventually degraded (Allen et al. 2017). The existence of multi-IES concatemers, however, was only supported by the sequencing of reverse-transcribed RNA molecules, and direct evidence for concatemerized DNA molecules was still lacking. We therefore developed a new bioinformatic method to quantify IES excision products from high-throughput DNA sequencing data.
Among the three expected types of excision products (linear molecules, single-IES circles, and multi-IES concatemers), only IES–IES junctions from circles or concatemers were analyzed. Indeed, the sequencing reads that map within IESs cannot be used to unambiguously count linear excised molecules, because they do not discriminate between intrachromosomal (not excised) or extrachromosomal (excised) IES forms. We confirmed that new MACs indeed contain DNA molecules corresponding to single-IES circles and multi-IES concatemers (Fig. 3D; Supplemental Fig. S5A). Because the normalized count of IES–IES junctions is maximal at DEV2 ∼ 32C (Fig. 3D), the stage at which the ES increases (Fig. 3A), we infer that IES–IES junctions may be formed concomitantly with MAC junctions but are still detected at DEV3 ∼64C, when IES excision is completed. This confirms, at the genome-wide level, that excised IES products are not degraded immediately and persist in the new MACs (Bétermier et al. 2000). Our data also reveal that the vast majority of IESs (97.2% considering all data sets and 86% at DEV2 ∼ 32C) are involved in the formation of IES–IES junctions (Fig. 3D). Based on read counts, single-IES circles represent <2% of excised IES junctions (Supplemental Fig. S5B), indicating that concatemers are the major products of IES-end joining following excision. This can be explained by the size distribution of IESs, 93% being <150 bp (Arnaiz et al. 2012), a size corresponding to the persistence length of double-stranded DNA, below which self-circularization is inefficient (Schleif 1992; Bates et al. 2013). Consistently, we find that the size distribution of single-IES circles is centered around 200 bp with a sharp drop for IESs <150 bp (Supplemental Fig. S5C). Our sequencing data do not allow us to determine the size range of IES concatemers but previous experimental observations have indicated that concatemeric and single-IES circles have the same size range (>200 bp) (Gratias and Bétermier 2001; Allen et al. 2017), and support our conclusion that only the longest IESs can self-circularize.
Sequential timing of excision is associated with specific IES features
Previously published molecular data suggested that not all IESs are excised at exactly the same time (Gratias and Bétermier 2001). To gain deeper insight into the differential timing of IES excision, we used the ES values obtained for the 45,000 IESs across all samples to group IESs into four clusters according to their excision timing (“very early,” “early,” “intermediate,” and “late”) (Fig. 4A; Supplemental Fig. S6A; Supplemental Table S3). Very early IESs are almost all excised at DEV2 ∼ 32C, whereas excision of most late IESs takes place between DEV2 ∼ 64C and DEV3 ∼ 64C. Detection of IES–IES junctions follows the same excision timing: IESs from the very early and early clusters contribute to the majority of junctions detected at the earliest developmental stages, whereas IESs from the intermediate and late clusters become dominant at late developmental stages (Supplemental Fig. S6B). It has been previously observed that the excision machinery sometimes generates different types of errors caused by the use of misplaced alternative TA boundaries (Supplemental Fig. S7A; Duret et al. 2008; Bischerour et al. 2018). At the stage when all IESs are completely excised (DEV4 ∼ 128C), we observe sevenfold fewer excision errors for very early relative to late excised IESs (Fig. 4B), indicating that very early IESs are much less error-prone. Of note, the maximum of excision errors during the excision time course never exceeds the error level observed in old MAC fragments (Supplemental Fig. S7B,C).
Figure 4.

Excision timing defines IES classes with different characteristics. (A) Heatmap of ESs for all IESs. IESs are sorted by hierarchical clustering, with each row corresponding to one IES. The ES is encoded from zero (dark blue; no excision) to one (white; complete excision). IESs are separated in four classes according to their excision profile by k-means clustering of their ES (very early: N = 10,994; early: N = 14,490; intermediate: N = 12,353; late: N = 6548). (B) Abundance of IES excision errors in the four excision profile groups counted in the DEV4 128C (tc4) sample. In this analysis, we focused on error types that would be the least impacted by IES length (external, overlap, and partial external) (see Supplemental Fig. S7A). The number of IESs in each excision profile group is indicated above the bars. (C) IES fraction for IES length categories in the four excision profile groups compared with all IESs. (D) Sequence logos of the eight bases at IES ends for all IESs and IESs belonging to the very early and late clusters. IESs are grouped in three length categories as described in C. (E) Venn diagram showing how the 44,385 reference IESs are distributed according to their sensitivity to EZL1, TFIIS4, DCL2/3, and DCL5 RNAi with regard to excision. The group “excision complex only” represents IESs that do not depend on any of these factors but do depend upon Pgm. The Venn diagram has been simplified to display only overlaps representing >1% of the total number of IESs. (F) IES proportions in the four groups of excision profiles for the data sets defined in E. The numbers above the barplots indicate the number of IESs in each data set. “All” is the random expectation for all IESs. (G) IES proportions in the four groups of excision profiles relative to the age of IES insertion during evolution of the Paramecium lineage. (Old) Insertion predating the divergence between P. caudatum and the P. aurelia clade; (pre-aurelia) insertion before the radiation of the P. aurelia complex; (post-aurelia) insertion after the radiation of the P. aurelia complex (Sellis et al. 2021).
With regard to genomic location, we found that late IESs are underrepresented in genes, particularly in CDSs versus introns, whereas the inverse trend is observed for very early and early IESs (Supplemental Fig. S8A). We also observed a strong enrichment of late excised IESs and a depletion of very early and early IESs at the extremities of MAC scaffolds, which is consistent with these regions being gene-poor (Supplemental Fig. S8B). Underrepresentation of late excised IESs within genes might be explained by a selective pressure for accurate excision to avoid the formation of nonfunctional open reading frames.
As for IES intrinsic properties, we detected an impressive size bias between IESs from the different clusters, with very early excised IESs tending to be much shorter than expected from the global IES size distribution. In contrast, short IESs are underrepresented among late IESs (Fig. 4C; Supplemental Fig. S9). We then examined whether IESs have different sequence properties at their ends depending on the cluster to which they belong. Because the consensus of IES ends varies at positions 3, 4, and 5 (position 1 being the T from the TA boundary) as a function of IES length (Swart et al. 2014), we compared the sequence logos of IES ends in the different clusters according to the IES size category (Fig. 4D; Supplemental Fig. S10A,B). For 25- to 33-bp IESs, we found an overrepresentation of the TATAG boundary among very early IESs compared with late IESs, with a significant increase of G frequency at the fifth base position (62% vs. 35% for very early compared to late IESs). For 42- to 140-bp IESs, we observed an even stronger sequence bias with an overrepresentation of the TACAG boundary among very early IESs, with the increase of the C frequency at the third position being highly significant (77% vs. 30% for very early vs. late IESs, respectively). We conclude that very early IESs <140 bp tend to show a stronger nucleotide sequence signal at their ends than late excised IESs. No significant sequence difference between very early and late IESs was observed for longer IESs (>140 bp).
We further studied the link between excision timing and dependence upon known factors involved in the epigenetic control of IES excision (Fig. 4E,F; Supplemental Fig. S11). We found an underrepresentation of the very early excised cluster among the subset of IESs whose excision depends on the deposition of H3K9me3 and H3K27me3 marks (i.e., IESs retained in an EZL1 KD) (Lhuillier-Akakpo et al. 2014). A similar bias was observed among IESs depending on the production of TFIIS4-dependent transcripts from the anlagen (Maliszewska-Olejniczak et al. 2015) and was exacerbated for sRNA-dependent IESs (retained in DCL2/3 or DCL5 KDs) (Lhuillier-Akakpo et al. 2014; Sandoval et al. 2014). In the DCL RNAi data sets, IESs from the very early cluster are totally absent, whereas IESs from the intermediate and the late clusters are overrepresented. In contrast, very early excised IESs are strongly enriched (∼60%) among the 12,414 IESs that are excised independently of the above factors (“excision complex only”). Considering the overlap between IES dependencies (Fig. 4E), our data indicate that IESs depending on known heterochromatin-targeting factors tend to take longer to be excised during MAC development. We observed that, consistent with late IESs being error-prone (Fig. 4B), there were more errors for Dcl2/3- or Ezl1-dependent IESs than for IESs depending on the “excision complex only” (Supplemental Fig. S7D).
Finally, we examined the relationship between IES evolutionary age (Sellis et al. 2021) and excision timing (Fig. 4G). Our data indicate that old IESs that invaded the Paramecium genome before the divergence of the P. caudatum and P. aurelia lineages tend to be precociously excised. Reciprocally, we observed that the younger the IESs, the later their excision during MAC development.
Identification of new IESs in MIC-limited regions
The presence of IESs nested in MIC-limited regions was reported previously, but only a few examples have been described (Duharcourt et al. 1998; Mayer et al. 1998; Mayer and Forney 1999; Le Mouël et al. 2003). We took advantage of the sequencing data we obtained during the course of MAC development to pinpoint precise excision events within late eliminated regions, therefore identifying new bona fide IESs (see Supplemental Methods). Their excision could be transiently observed before complete elimination of the surrounding DNA. We could identify a set of 167 “buried” IESs localized in imprecisely eliminated regions and 226 “internal” IESs located inside IESs from the reference set (Supplemental Fig. S12A,B; Supplemental Tables S4, S5). We found that buried IESs are strongly biased toward short sequences, whereas internal IESs present no major difference in size compared to the reference IESs (Supplemental Fig. S12C).
To assess whether these newly identified IESs depend on heterochromatin marks for excision, we analyzed their retention in Ezl1-depleted cells (Supplemental Fig. S12D; Supplemental Table S6). As previously published (Denby Wilkes et al. 2016), we calculated their retention scores (IES retention score [IRS]), varying from zero for no retention to one for full retention. We found two contrasting situations for internal IESs: 26% are not affected in Ezl1-depleted relative to control cells (IRS ∼ 0) whereas 20% are strongly retained (IRS ∼ 1). We also noticed that retained IESs are much longer than the unaffected ones (Supplemental Fig. S12D, boxplot). A similar size bias was reported for the Ezl1-dependent IESs from the reference set (Lhuillier-Akakpo et al. 2014). For their related encompassing IESs (n = 223), we observed that 94% are significantly retained in Ezl1-depleted cells, consistent with their late excision timing. Our results suggest that internal IESs show similar characteristics to those of the reference IES set in terms of length and epigenetic control and, therefore, might share the same evolutionary history. With regard to the excision mechanism, molecular data indicate that IESs can be excised while retaining a nested IES (Bétermier et al. 2000; Gratias and Bétermier 2001). This suggests that excision of internal IESs is not a systematic prerequisite for the elimination of their encompassing IESs, reminiscent of the sequential splicing of introns inserted within introns (Hafez and Hausner 2015). Thus, the existence of internal IESs adds to the list of features shared by IESs and introns (Arnaiz et al. 2012; Sellis et al. 2021).
In contrast to internal IESs, we found that most buried IESs are independent of Ezl1-mediated heterochromatin marks for their excision (Supplemental Fig. S12D). Moreover, we noticed that the most independent are the shortest, with a breakpoint size of 33 nt (Supplemental Fig. S12E). The finding of buried IESs confirms that IESs are scattered all along MIC chromosomes, including MIC-limited regions, as previously hypothesized (Sellis et al. 2021). The properties of buried IESs, however, raise the question of their origin. Most IESs are derived from TEs, but a previous report showed that genomic fragments can be co-opted to become IESs (Singh et al. 2014). Even though buried IESs have no stronger sequence end logos than the set of all IESs (Supplemental Fig. S12F), we speculate that buried IESs are excision-prone genomic fragments recognized by the excision machinery independently of histone mark deposition. Why these genomic fragments are excised as IESs remains an open question.
Discussion
Developmental timing of sequential DNA elimination
The present study aimed at unravelling the links between two intertwined DNA-driven mechanisms underlying somatic nuclear differentiation in Paramecium: genome amplification and PDE. Setting up the FANS procedure allowed us to show that genome amplification during MAC development is an endocycling process, defined as alternating S and G phases without mitosis (Lilly and Duronio 2005). In the time-window during which PDE takes place, we identified three peaks representing discrete new MAC populations differing in their DNA content. The estimated C-levels of the first two peaks (32C, 64C) are consistent with their resulting from successive whole-genome doublings. The range of C-levels obtained for the third peak fits less well with 128C, which can be explained by ongoing massive DNA elimination between 64C and 128C causing variability in the actual 1C-value of the anlagen.
We determined at an unprecedented resolution the timing of IES excision and imprecise DNA elimination genome-wide, across successive endoreplication cycles (Fig. 5A). Our data show that DNA elimination is an ordered process. We found that most IESs are excised between DEV1 ∼ 32C and DEV3 ∼ 64C under standard conditions (see Methods), whereas imprecise elimination only starts at DEV3 ∼ 64C. We established four classes of IESs according to their excision timing (Fig. 4A). We also showed that the progression of IES elimination, once it has started, is independent of replication, suggesting that the excision machinery is recruited to its chromatin targets independently of replication fork passage.
Figure 5.

Schematic view of DNA elimination timing in Paramecium and a model for IES evolution. (A) Relative timing of DNA amplification and PDE during new MAC development. The wavy gray lines stand for imprecisely eliminated sequences. The endoreplication level (C-level) is indicated as a green bar on the left. The black double arrowheads schematize the telomeric ends of MAC chromosomes. At each step of PDE, only one representative copy of the new MAC genome is drawn. (B) Model for evolutionary optimization of IESs. Old IESs have become independent of sRNAs and histone mark deposition for their excision. They have acquired strong sequence information at their ends (red arrowheads), promoting their efficient excision.
We observed that little IES excision has already taken place at the earliest stage of our study (median ES = 0.15 at DEV1 ∼32C), suggesting that the onset of PDE is controlled during MAC development. Given that Paramecium IESs are mostly intragenic (Arnaiz et al. 2012), starting their excision after a few endoreplication rounds have taken place may have been advantageous to limit the detrimental effects of excision errors on functional gene assembly. The temporal control of PDE may be explained by the expression profile of genes encoding components of the core excision machinery, many of which (e.g., PGM, PGMLs, KU80C) are not expressed during early autogamy stages and reach their maximum transcription level at DEV2 (Arnaiz et al. 2017). In addition, as previously suggested (Bétermier et al. 2000), the three to four endoreplication rounds preceding IES excision might also contribute to chromatin remodeling to provide a suitable substrate for the excision machinery. In support of the latter hypothesis, several chromatin remodelers and histone chaperones are known to control PDE in P. tetraurelia (Ignarski et al. 2014; de Vanssay et al. 2020; Singh et al. 2022), but the temporal and mechanistic details of their action remain to be precisely understood.
The existence of a sequential DNA elimination program may provide Paramecium with a peculiar mechanism to fine-tune zygotic gene expression during MAC development. Developmental regulation was previously proposed for genes located inside IESs or embedded in imprecisely eliminated MIC-limited regions (Sellis et al. 2021). Such germline-specific genes may be expressed when zygotic transcription starts in the new MAC, until their encompassing DNA is removed from the genome. PDE-mediated regulation of germline-limited genes was shown in the ciliate Euplotes crassus for a development-specific de novo telomerase gene (Karamysheva et al. 2003) and in Tetrahymena thermophila for the gene encoding Tpb6p, a protein involved in excision of intragenic IESs (Feng et al. 2017). It has also been reported in other organisms that eliminate germline-specific genes during development (Smith et al. 2012; Wang et al. 2012; Chen et al. 2014; Kinsella et al. 2019; Torgasheva et al. 2019). In Paramecium, IESs could block zygotic gene expression as long as they are present within CDSs or in gene regulatory regions, as suggested for the PTIWI10 gene (Furrer et al. 2017). An even more sophisticated regulatory scheme could be proposed with a first IES excision event turning on an anlagen-specific gene that would subsequently be turned off by a second DNA elimination event. In the future, monitoring the timing of PDE in other Paramecium species and annotating the sequential versions of the rearranged genome should allow us to assess whether temporal control of IES excision has been conserved during evolution and to what extent it may contribute to gene regulation.
DNA elimination timing reveals evolutionary optimization of TE-derived sequences for efficient excision
DNA and RNA transposons that have colonized the Paramecium germline genome during evolution are eliminated in an imprecise manner from the new MAC during PDE (Arnaiz et al. 2012; Guérin et al. 2017). We report here that imprecise elimination of TEs and other MIC-limited regions occurs at a late stage during MAC development (DEV3 to DEV4). TE elimination was previously shown to depend upon scnRNA-driven deposition of H3K9me3 and H3K27me3 histone marks, which are enriched on TEs at a developmental stage corresponding to DEV2 and accumulate in the anlagen up to DEV3 (Lhuillier-Akakpo et al. 2014; Frapporti et al. 2019). The deposition of heterochromatin marks thus appears to be a late process, which might explain why TEs and other MIC-limited sequences are eliminated at a late developmental stage.
The Paramecium MIC genome harbors TEs belonging to different families, most of which are eliminated imprecisely during MAC development (Guérin et al. 2017). Paramecium IESs have been proposed to originate from Tc1/mariner TEs, a particular family of transposons that duplicate their TA target site upon integration (Arnaiz et al. 2012). Target site duplication generates potential Pgm cleavage sites at the boundaries of newly inserted Tc1/mariner copies, which has provided these TA-flanked TEs with the ability to be excised precisely and behave as IESs right after their integration. We propose that, due to this ability, only Tc1/mariner TEs could be maintained within genes in the germline and were allowed to decay, giving rise to extant IESs. This evolutionary scenario was enriched by the finding that a handful of multicopy noncoding IESs was recently mobilized in trans by transposases expressed from active TEs (Sellis et al. 2021). These newly inserted IES copies are thought to evolve under the same constraints as all other IESs with regard to their precise somatic excision.
In P. tetraurelia, IESs have shortened down to a minimal size range of 25 to 33 bp, representing ∼30% of all IESs. A phylogenetic analysis of IESs across the P. aurelia species showed that shortening has been accompanied by a switch in their excision mechanism. Indeed, the most recently inserted IESs (i.e., the youngest) were shown to depend on scnRNAs and heterochromatin marks, whereas old IESs have become independent of these epigenetic factors (Sellis et al. 2021). Here we show that late excised IESs tend to be the youngest and that, similar to their TE ancestors, their elimination depends on scnRNAs and histone marks. In addition, excision of late IESs tends to depend on the presence of iesRNAs, which have been proposed to boost excision through a positive-feedback loop (Sandoval et al. 2014). The stimulatory contribution of iesRNAs might explain why excision of late IESs precedes imprecise elimination of TEs and other MIC-limited sequences during MAC development. We also report that early excised IESs tend to be the oldest and are enriched for smaller sizes (54.5% belong to the 25- to 33-bp peak). They are also mostly independent of sRNAs and heterochromatin marks and tend to be the least error-prone. Our data therefore provide experimental support to the proposed evolutionary scenario of Paramecium IESs, showing that their excision timing reflects their evolutionary age (Fig. 5B).
We provide evidence that IESs have evolved through optimization for efficient excision, combining an early and accurate excision process. Closer analysis of the intrinsic properties of very early excised IESs furthermore revealed a strong nucleotide sequence signal at their ends, which varies according to IES size (TATAG for 25- to 33-bp IESs, TACAG for 42- to 140-bp IESs). In contrast, late excised IESs only show a conserved TA dinucleotide at their ends. These observations suggest that acquisition of a stronger sequence motif has allowed “optimized” IESs to loosen their requirement for sRNAs for excision. Sequence-dependent determination of efficient excision would explain the previous observation that 25- to 33-bp MAC genome segments flanked by terminal TATAG inverted repeats are underrepresented in the somatic MAC genome (Swart et al. 2014), possibly because such sequences are highly excision-prone. MAC genome segments of any size flanked by terminal TACAG inverted repeats are overall poorly represented as well in the Paramecium genome, thus precluding their harmful excision (Swart et al. 2014). The present study therefore points to the joint contribution of IES nucleotide sequence and size as intrinsic determinants of efficient IES excision. Several hypotheses might explain how these determinants could work. By facilitating the formation of particular DNA structures, they could help to target specific sequences for elimination, either through a passive mechanism involving nucleosome exclusion to increase their accessibility or by actively promoting the assembly of the Pgm-endonuclease complex. The IES size-dependent consensus sequences might also be related to a distinct spatial organization of IES ends within the excision complex formed for very short versus long IESs (Arnaiz et al. 2012). Another nonexclusive hypothesis might be that conserved sequence motifs help to position the Pgm catalytic domain on its cleavage sites. Why two different sequence logos have evolved for different sizes of early excised IES remains to be investigated. It could be linked to preferential recognition by different subunits of the excision complex (e.g., PgmLs), which are all coexpressed with Pgm (Bischerour et al. 2018).
Studying the Paramecium model, with its nuclear dimorphism and ability to precisely excise TE-related IESs even when inserted inside CDSs, provides a unique opportunity to monitor how TEs have degenerated within their host genomes. Further work on Paramecium PDE will make it possible to decipher the evolutionary and mechanistic switch from sRNA- and heterochromatin-mediated TE silencing to efficient elimination of TE-related sequences from the genome. The characterization of a set of efficiently excised IESs, whose excision has become independent of sRNAs and the heterochromatin pathway, paves the way to future biochemical studies that will address the longstanding question of how domesticated PiggyBac transposases are recruited to specific DNA cleavage sites to carry out precise DNA excision.
Methods
Cell growth and autogamy time courses
Culture of P. tetraurelia wt 51 new (Gratias and Bétermier 2003) or its mutant derivative 51 nd7-1 (Dubois et al. 2017) was performed using standard conditions. Briefly, cells were grown in medium made of wheat grass infusion (WGP) inoculated with Klebsiella pneumoniae and supplemented with β-sitosterol (0.8 µg/mL) before use (Beisson et al. 2010). For autogamy time courses, cells (around 20–30 vegetative fissions) were seeded at a final concentration of 250 cells/mL in inoculated WGP medium with an OD600 nm adjusted to 0.1. Autogamy was triggered by starvation the next day. We performed six independent autogamy time courses (tc1 to tc6). For each time course, the T0 time point was defined as the time (in hours) when 50% of the cells in the population have a fragmented MAC. We further defined five developmental stages: DEV1 (T2.5-T3), DEV2 (T7-T12), DEV3 (T20-T24), DEV4 (T30), and DEV5 (T48), with time points following T0 as previously described (Arnaiz et al. 2017). At each selected time point, 0.7–2 L of culture (at a concentration of 1500–3500 cells/mL) was processed for nuclear preparation or 30 mL for whole-cell immunofluorescence. To inhibit DNA replication during autogamy, aphidicolin (Sigma-Aldrich A0781) was added at T2.5 at a final concentration of 15 μM, and the same volume was added a second time at T10. Cells were harvested, and nuclei were isolated at T20. For all time courses, the survival of postautogamous progeny was tested as described before (Dubois et al. 2017).
Immunofluorescence analysis
A peptide corresponding to PgmL1 amino acid sequence 1 to 266 and carrying a C-terminal His tag was used for guinea pig immunization (Proteogenix). Sera were purified by antigen affinity purification to obtain highly specific α−PgmL1-GP antibodies (0.8 mg/mL). RNAi targeting the PGML1 gene during autogamy, immunofluorescence labeling of whole cells, and quantification of PgmL1 signal were performed as described previously (Bischerour et al. 2018). Cells were extracted with ice-cold PHEM (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2 at pH 6.9) + 1% Triton X-100 before fixation and immunostaining with α-PgmL1-GP (1:2000).
Isolation of nuclei and immunostaining
Nuclear preparations enriched in developing MACs were obtained as previously described (Arnaiz et al. 2012) with few modifications: The cell pellet was resuspended in 6–10 volumes of lysis buffer (0.25 M sucrose, 10 mM MgCl2, 10 mM Tris at pH 6.8, 0.2% Nonidet P-40) supplemented with 2× protease inhibitor cocktail set I (PICS; Cabiochem 539131), kept on ice for 15 min, and disrupted with a Potter–Elvehjem homogenizer (100 to 400 strokes). Lysis efficiency was monitored with a Zeiss Lumar V12 fluorescence stereo-microscope, following the addition of 66 µg/mL DAPI. Nuclei were collected through centrifugation at 1000g for 2 min and washed four times with 10 volumes of washing buffer (0.25 M sucrose, 10 mM MgCl2, 10 mM Tris at pH 7.4). The nuclear pellet either was diluted twofold in washing buffer containing glycerol (13% final concentration) and frozen as aliquots at −80°C or was diluted twofold in washing buffer supplemented with 2× PICS and loaded on top of a 3-mL sucrose (2.1 M) layer before ultra-centrifugation in a swinging rotor for 1 h at 210,000g. After gentle washes, the pellet was resuspended in 1 volume of washing buffer containing glycerol (13% final concentration) and frozen as aliquots at −80°C. New MAC labeling was adapted from a published method (Sardo et al. 2017). Nuclear preparations were immunostained on ice for 1 h in TBS (10 mM Tris at pH 7.4, 0.15 M NaCl) + 3% BSA containing α-PgmL1-GP (1:1000). Nuclei were washed twice in TBS + 3% BSA and stained for 45 min with Alexa Fluor (AF) 488-conjugated goat anti-guinea pig IgG (1:500; Thermo Fisher Scientific A11073). Nuclei were finally washed twice in TBS + 3% BSA and resuspended in PI/RNase staining buffer (BD Pharmingen 550825). All centrifugation steps were performed at 500g for 1 min at 4°C. Samples were kept in the dark at 4°C until processing.
Flow cytometry
Stained nuclei were filtered through sterile 30-µm cell strainers (Sysmex filters, CellTrics 04-004-2326) and processed for flow cytometry. Immunostained nuclei were analyzed on a CytoFlex S cytometer (Beckman Coulter) with a 488-nm laser for scatter measurements (forward scatter [FSC], side scatter [SSC]) and AF488 excitation, and a 561-nm laser for PI excitation. AF488 and PI staining signals were, respectively, collected using a 525/40-nm band pass filter and a 610/20-nm band pass filter. Immunostained nuclei were sorted on a Moflow Astrios EQ cell sorter (Beckman Coulter) with a 488-nm laser for scatter measurements (FCS and SSC) and AF488 excitation, and a 561-nm laser for PI excitation. AF488 and PI staining signals were, respectively, collected using a 526/52-nm band pass filter and a 614/20-nm band pass filter. Phosphate buffered saline-like (puraflow sheath fluid, Beckman Coulter) was used as sheath and run at a constant pressure of 10 or 25 PSI. Frequency of drop formation was 26 or 43 kHz. Purify mode was used for sorting in order to reach a maximum rate of purity (>95%). The instrument used a 100-μm nozzle. A threshold on the PI signal was optimized to increase collecting speed (about 1000 events per second). Data were collected using Summit software (Beckman Coulter). Nuclei were first gated based on their side scatter (SSC-area) and high PI signal (PI-area) and sorted according to their AF488 signal. AF488-positive events were backgated onto SSC versus PI to optimize the gating. Doublets were discarded using PI-area and PI-height signals. Nuclei (fewer than 30,000) were collected into 100 μL of buffer AL (QIAamp DNA micro kit, Qiagen) and immediately lysed by pulse-vortexing. The final volume was adjusted to 200 μL with PI/RNase staining buffer. We confirmed that the FANS procedure yields pure anlagen in an experiment (tc3) in which old MAC fragments contained a marker transgene absent from the anlagen (Supplemental Fig. S3A–C; Supplemental Methods).
Estimation of new MAC DNA content by flow cytometry
Estimation of the absolute DNA content (C-value, in Mbp) in the new MAC populations was based on a previously described method (Bourge et al. 2018). The DNA content was calculated using the linear relationship between the fluorescent signal from the new MAC peaks and a known internal standard (tomato nuclei, Solanum lycopersicum L. cv. Montfavet 63-5, 2C = 1946 Mbp). Briefly, leaves were chopped with a razor blade in a Petri dish with PI/RNase staining buffer, filtered through 30-µm cell strainers, and added in a constant ratio to an aliquot of stained and filtered Paramecium nuclei. The C-value (Cnew MACs) for each new MAC subpopulation was calculated using its PI mean fluorescence intensity (MFInew MACs), the PI mean fluorescence intensity of the 2C tomato standard (MFIstandard), and the 2C-value of the tomato standard (2Cstandard) (Supplemental Fig. S2):
The endoreplication level for each new MAC population (C-level) was further estimated by dividing the C-value for each new MAC population by the DNA content of the unrearranged P. tetraurelia MIC genome (1C = 100 Mbp) (Guérin et al. 2017; Sellis et al. 2021): C-level = Cnew MACs/1Cmic.
Genomic DNA extraction and high-throughput sequencing
DNA extraction was performed using the QIAamp DNA micro kit (Qiagen) as recommended by the manufacturer, with minor modifications. Following a 10-min incubation with Proteinase K (2 mg/mL) in buffer AL, the nuclear lysate was directly loaded onto the purification column. Elution was performed with 20–50 μL buffer AE in DNA LoBind Eppendorf tubes. DNA concentration was determined using the QBit high-sensitivity kit (Invitrogen) before storage at −20°C. Sequencing libraries were prepared using 1.5 to 8.5 ng of DNA with the TruSeq NGS library prep kit from Westburg (WB9024) following the manufacturer's instructions. Alternatively, genomic DNA was fragmented with the S220 focused-ultrasonicator (Covaris). Fragments were processed with NEBNext Ultra II end prep reagents (NEB E7546), and TruSeq adapters were ligated using the NEBNext quick ligation kit (NEB E6056). Libraries were amplified by PCR using Kapa HiFi DNA polymerase (10–14 cycles). Library quality was checked with an Agilent Bioanalyzer instrument (Agilent high-sensitivity DNA kit). Sequencing was performed on 75- to 75-bp paired-end runs, with an Illumina NextSeq 500/550 instrument, using the NextSeq 500/550 MID output cycle kit. Demultiplexing was performed with bcl2fastq2-2.18.12 (https://emea.support.illumina.com/), and adapters were removed with cutadapt 1.15 (Martin 2011); only reads >10 bp were retained.
Software and R packages
Sequencing reads were mapped on genome references using Bowtie 2 (v2.2.9 ‐‐local ‐‐X 500) (Langmead and Salzberg 2012). The resulting alignments were analyzed using SAMtools (v1.9) (Li et al. 2009), ParTIES (v1.05) (Denby Wilkes et al. 2016; https://github.com/oarnaiz/ParTIES), and BEDTools (v2.26) (Quinlan and Hall 2010). R (v4) packages were used to generate images (ggplot2 v3.3.5; ComplexHeatmap v2.6.2; GenomicRanges v1.42) (Lawrence et al. 2013; Gu et al. 2016; Wickham 2016; R Core Team 2021). The IES sequence end logos were generated using WebLogo (v3.6.0 ‐‐composition 0.28 ‐‐units bits) (Crooks et al. 2004).
Reference genomes and data sets
Paired-end sequencing data were mapped on P. tetraurelia strain 51 MAC (ptetraurelia_mac_51.fa), MAC + IES (ptetraurelia_mac_51_ with_ies.fa), or MIC (ptetraurelia_mic2.fa) reference genomes (Arnaiz et al. 2012; Guérin et al. 2017). Gene annotation v2.0 (ptetraurelia_mac_51_annotation_v2.0.gff3), IES annotation v1 (internal_eliminated_sequence_PGM_ParTIES.pt_51.gff3), and TE annotation v1.0 (ptetraurelia_mic2_TE_annotation_v1.0.gff3) were used in this study (Arnaiz et al. 2012, 2017; Guérin et al. 2017). All files are available from the ParameciumDB download section (Arnaiz et al. 2020; https://paramecium.i2bc.paris-saclay.fr/download/Paramecium/tetraurelia/51/). DNA sequencing data of Paramecium cells depleted of Ezl1, TFIIS4, Dcl2/3, or Dcl5 were previously published (Lhuillier-Akakpo et al. 2014; Sandoval et al. 2014; Maliszewska-Olejniczak et al. 2015). ParTIES (MIRET module) was used to determine the IESs that were significantly retained compared to the control (Denby Wilkes et al. 2016). An IES is considered to be dependent on the depleted factor for its excision (31,505, 20,524, 3439, and 2475 IESs sensitive to EZL1, TFIIS4, DCL2/3, and DCL5 RNAi, respectively), if at least one IES boundary in at least one replicate shows significant retention.
ES calculation and IES classification
Mapping of sequencing reads on the MAC and the MAC + IES references was used to calculate an IES ES (ES = IES−/(IES+ + IES−) using ParTIES (MIRET default parameters). An ES of zero means no excision, and an ES of one means complete IES excision. The violin plots show the distribution of the mean ES score for the two IES boundaries of all IESs. Excision profile classification was performed on the 44,928 annotated IESs, after removing 543 IESs with ES < 0.8, in at least one FRAG sample, which indicates imperfect excision in the old MAC (group defined as “none”). K-Means clustering (iter.max = 100, k-means R function from “stats” package) was used to define four groups based on the ES in all conditions.
TE and genome coverage
The mean sequencing depth (SAMtools depth -q 30 -Q 30), normalized by the number of reads mapped on the MIC reference genome, was calculated on TE copies (500 nt minimum length and localized on MIC contigs > 2 kb) and IESs. Only fully mapped reads overlapping at least 4 nt of the annotated feature were considered. As previously described (Guérin et al. 2017), the same window coverage approach was used to estimate genome coverage at each time point. The coverage (multicov -q 30) was calculated for nonoverlapping 1-kb windows and then normalized by the total number of mapped reads (RPM). An empirical cutoff of 2.5 RPM was used to decide if the window is covered or not.
Detection of de novo telomere addition sites
De novo telomere addition sites were identified on the MIC genome, with the requirement of at least three consecutive repeats of either G4T2 or G3T3 on mapped reads. A telomere addition site was identified if the read alignment stops at the exact position at which the telomeric repeat starts. The number of telomere addition sites was normalized by the number of reads mapped on the MIC genome.
IES–IES junctions
The ParTIES concatemer module, developed for this study, was used with default parameters to identify concatemers of excised IESs. Reads were recursively mapped to the IES sequences, as shown in Supplemental Figure S5A. At each round, reads are mapped to IES sequences and selected if the alignment begins or ends at an IES extremity. If the read is partially aligned, then the unmapped part of the read is reinjected into the mapping and the selection procedure continues until the entire read has been mapped.
IES excision errors
The ParTIES MILORD module was used with the MAC + IES reference genome to identify IES excision errors. Only error types described in Supplemental Figure S7A were considered. The number of nonredundant errors was normalized by the number of mapped reads. PCR duplicates were removed using SAMtools rmdup.
Data access
Genes used in this study are accessible in ParameciumDB (https://paramecium.i2bc.paris-saclay.fr/) as follows: PGM (PTET.51.1.G0490162), PGML1 (PTET.51.1.G0110267), EZL1 (PTET.51.1.G1740049), TFIIS4 (PTET.51.1.G0900102), DCL2 (PTET.51.1.G0210241), DCL3 (PTET.51.1.G0990073), DCL5 (PTET.51.1.G0070121), and ND7 (PTET.51.1.G0050374) (Arnaiz et al. 2020). The sequencing data generated for this study have been submitted to the ENA database (https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB49315. The statistical data, scripts (Supplemental Codes), and raw images have been deposited at Zenodo (https://doi.org/10.5281/zenodo.6534539). The cytometry data generated in this study have been submitted to the FlowRepository database (http://flowrepository.org/id/RvFrl4FUJTnaAIDEsEqK3MzxKwQZpkfp7yqzGGSco3tuuLfuAHKrPI2fP65KehpH).
Supplementary Material
Acknowledgments
We thank Cindy Mathon and Pascaline Tirand for their technical assistance and all members of the Bétermier laboratory for stimulating and fruitful discussions. We give special thanks to Joël Acker, Julien Bischerour, and Linda Sperling for critical reading of the manuscript. This study was supported by intramural funding from the Centre National de la Recherche (CNRS) and by grants from the Agence Nationale de la Recherche (LaMarque ANR-18-CE12-0005-02 and CURE ANR-21-CE12-0019-01 to M. Bétermier, POLYCHROME ANR-19-CE12-0015 to O.A.) and the Fondation pour la Recherche Médicale (FRM EQU202103012766 to M. Bétermier). The present work has benefited from the expertise of the Imagerie-Gif core facility, supported by the Agence Nationale de la Recherche (ANR-11-EQPX-0029/Morphoscope, ANR-10-INBS-04/FranceBioImaging, ANR-11-IDEX-0003-02/ Saclay Plant Sciences). We acknowledge the sequencing and bioinformatics expertise of the I2BC High-throughput sequencing facility, supported by France Génomique (funded by the French National Program “Investissement d'Avenir” ANR-10-INBS-09).
Author contributions: C.Z., M. Bourge, M. Bétermier, O.A., and V.R. designed research and analyzed data. C.Z., K.G., L.E., M. Bourge, N.M., O.A., V.R., and Y.J. performed research. O.A. developed bioinformatic pipelines and conducted bioinformatic data analysis. C.Z., K.G., M. Bourge, M. Bétermier, O.A., and V.R. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277027.122.
Competing interest statement
The authors declare no competing interests.
References
- Abello A, Régnier V, Arnaiz O, Le Bars R, Bétermier M, Bischerour J. 2020. Functional diversification of Paramecium Ku80 paralogs safeguards genome integrity during precise programmed DNA elimination. PLoS Genet 16: e1008723. 10.1371/journal.pgen.1008723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SE, Nowacki M. 2020. Roles of noncoding RNAs in ciliate genome architecture. J Mol Biol 432: 4186–4198. 10.1016/j.jmb.2019.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SE, Hug I, Pabian S, Rzeszutek I, Hoehener C, Nowacki M. 2017. Circular concatemers of ultra-short DNA segments produce regulatory RNAs. Cell 168: 990–999. 10.1016/j.cell.2017.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Mathy N, Baudry C, Malinsky S, Aury JM, Denby Wilkes C, Garnier O, Labadie K, Lauderdale BE, Le Mouël A, et al. 2012. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet 8: e1002984. 10.1371/journal.pgen.1002984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Van Dijk E, Bétermier M, Lhuillier-Akakpo M, de Vanssay A, Duharcourt S, Sallet E, Gouzy J, Sperling L. 2017. Improved methods and resources for Paramecium genomics: transcription units, gene annotation and gene expression. BMC Genomics 18: 483. 10.1186/s12864-017-3887-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Meyer E, Sperling L. 2020. ParameciumDB 2019: integrating genomic data across the genus for functional and evolutionary biology. Nucleic Acids Res 48: D599–D605. 10.1093/nar/gkz948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, et al. 2006. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444: 171–178. 10.1038/nature05230 [DOI] [PubMed] [Google Scholar]
- Baroin A, Prat A, Caron F. 1987. Telomeric site position heterogeneity in macronuclear DNA of Paramecium primaurelia. Nucleic Acids Res 15: 1717–1728. 10.1093/nar/15.4.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates AD, Noy A, Piperakis MM, Harris SA, Maxwell A. 2013. Small DNA circles as probes of DNA topology. Biochem Soc Trans 41: 565–570. 10.1042/BST20120320 [DOI] [PubMed] [Google Scholar]
- Baudry C, Malinsky S, Restituito M, Kapusta A, Rosa S, Meyer E, Bétermier M. 2009. PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia. Genes Dev 23: 2478–2483. 10.1101/gad.547309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisson J, Bétermier M, Bré M-H, Cohen J, Duharcourt S, Duret L, Kung C, Malinsky S, Meyer E, Preer JR, et al. 2010. Paramecium tetraurelia: the renaissance of an early unicellular model. Cold Spring Harb Protoc 2010: pdb.emo140. 10.1101/pdb.emo140 [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Park M. 2018. Distinguishing friends, foes, and freeloaders in giant genomes. Curr Opin Genet Dev 49: 49–55. 10.1016/j.gde.2018.02.013 [DOI] [PubMed] [Google Scholar]
- Berger JD. 1973. Nuclear differentiation and nucleic acid synthesis in well-fed exconjugants of Paramecium aurelia. Chromosoma 42: 247–268. 10.1007/BF00284774 [DOI] [PubMed] [Google Scholar]
- Berger JD. 1986. Autogamy in Paramecium. Cell cycle stage-specific commitment to meiosis. Exp Cell Res 166: 475–485. 10.1016/0014-4827(86)90492-1 [DOI] [PubMed] [Google Scholar]
- Bétermier M, Duharcourt S. 2014. Programmed rearrangement in ciliates: Paramecium. Microbiol Spectr 2: MDNA3-0035-2014. 10.1128/microbiolspec.MDNA3-0035-2014 [DOI] [PubMed] [Google Scholar]
- Bétermier M, Duharcourt S, Seitz H, Meyer E. 2000. Timing of developmentally programmed excision and circularization of Paramecium internal eliminated sequences. Mol Cell Biol 20: 1553–1561. 10.1128/MCB.20.5.1553-1561.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétermier M, Bertrand P, Lopez BS. 2014. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 10: e1004086. 10.1371/journal.pgen.1004086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétermier M, Borde V, de Villartay J-P. 2020. Coupling DNA damage and repair: an essential safeguard during programmed DNA double-strand breaks? Trends Cell Biol 30: 87–96. 10.1016/j.tcb.2019.11.005 [DOI] [PubMed] [Google Scholar]
- Bischerour J, Bhullar S, Denby Wilkes C, Régnier V, Mathy N, Dubois E, Singh A, Swart E, Arnaiz O, Sperling L, et al. 2018. Six domesticated PiggyBac transposases together carry out programmed DNA elimination in Paramecium. eLife 7: e37927. 10.7554/eLife.37927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleykasten-Grosshans C, Neuvéglise C. 2011. Transposable elements in yeasts. C R Biol 334: 679–686. 10.1016/j.crvi.2011.05.017 [DOI] [PubMed] [Google Scholar]
- Bourge M, Brown SC, Siljak-Yakovlev S. 2018. Flow cytometry as tool in plant sciences, with emphasis on genome size and ploidy level assessment. GenApp 2: 1–12. 10.31383/ga.vol2iss2pp1-12 [DOI] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. 10.1016/j.cell.2007.01.043 [DOI] [PubMed] [Google Scholar]
- Byrnes JJ. 1984. Structural and functional properties of DNA polymerase Δ from rabbit bone marrow. Mol Cell Biochem 62: 13–24. 10.1007/BF00230073 [DOI] [PubMed] [Google Scholar]
- Capy P. 2021. Taming, domestication and exaptation: trajectories of transposable elements in genomes. Cells 10: 3590. 10.3390/cells10123590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Meyer E, Mochizuki K. 2013. Epigenetics of ciliates. Cold Spring Harb Perspect Biol 5: a017764. 10.1101/cshperspect.a017764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bracht JR, Goldman AD, Dolzhenko E, Clay DM, Swart EC, Perlman DH, Doak TG, Stuart A, Amemiya CT, et al. 2014. The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell 158: 1187–1198. 10.1016/j.cell.2014.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CH, Kuchta RD. 1993. DNA polymerase ε: aphidicolin inhibition and the relationship between polymerase and exonuclease activity. Biochemistry 32: 8568–8574. 10.1021/bi00084a025 [DOI] [PubMed] [Google Scholar]
- Cheng CY, Orias E, Leu JY, Turkewitz AP. 2020. The evolution of germ–soma nuclear differentiation in eukaryotic unicells. Curr Biol 30: R502–R510. 10.1016/j.cub.2020.02.026 [DOI] [PubMed] [Google Scholar]
- Choi JY, Lee YCG. 2020. Double-edged sword: the evolutionary consequences of the epigenetic silencing of transposable elements. PLoS Genet 16: e1008872. 10.1371/journal.pgen.1008872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosby RL, Chang NC, Feschotte C. 2019. Host–transposon interactions: conflict, cooperation, and cooption. Genes Dev 33: 1098–1116. 10.1101/gad.327312.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déléris A, Berger F, Duharcourt S. 2021. Role of Polycomb in the control of transposable elements. Trends Genet 37: 882–889. 10.1016/j.tig.2021.06.003 [DOI] [PubMed] [Google Scholar]
- Denby Wilkes C, Arnaiz O, Sperling L. 2016. ParTIES: a toolbox for Paramecium interspersed DNA elimination studies. Bioinformatics 32: 599–601. 10.1093/bioinformatics/btv691 [DOI] [PubMed] [Google Scholar]
- Deniz O, Frost JM, Branco MR. 2019. Regulation of transposable elements by DNA modifications. Nat Rev Genet 20: 417–431. 10.1038/s41576-019-0106-6 [DOI] [PubMed] [Google Scholar]
- de Vanssay A, Touzeau A, Arnaiz O, Frapporti A, Phipps J, Duharcourt S. 2020. The Paramecium histone chaperone Spt16-1 is required for Pgm endonuclease function in programmed genome rearrangements. PLoS Genet 16: e1008949. 10.1371/journal.pgen.1008949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois E, Bischerour J, Marmignon A, Mathy N, Régnier V, Bétermier M. 2012. Transposon invasion of the Paramecium germline genome countered by a domesticated PiggyBac transposase and the NHEJ pathway. Int J Evol Biol 2012: 436196. 10.1155/2012/436196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois E, Mathy N, Regnier V, Bischerour J, Baudry C, Trouslard R, Betermier M. 2017. Multimerization properties of PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements. Nucleic Acids Res 45: 3204–3216. 10.1093/nar/gkx571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duharcourt S, Keller A-M, Meyer E. 1998. Homology-dependent maternal inhibition of developmental excision of internal eliminated sequences in Paramecium tetraurelia. Mol Cell Biol 18: 7075–7085. 10.1128/MCB.18.12.7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Cohen J, Jubin C, Dessen P, Goût J-F, Mousset S, Aury J-M, Jaillon O, Noël B, Arnaiz O, et al. 2008. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Res 18: 585–596. 10.1101/gr.074534.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang G, Hamilton EP, Xiong J, Yan G, Chen K, Chen X, Dui W, Plemens A, Khadr L, et al. 2017. A germline-limited piggyBac transposase gene is required for precise excision in Tetrahymena genome rearrangement. Nucleic Acids Res 45: 9481–9502. 10.1093/nar/gkx652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frapporti A, Miró Pina C, Arnaiz O, Holoch D, Kawaguchi T, Humbert A, Eleftheriou E, Lombard B, Loew D, Sperling L, et al. 2019. The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat Commun 10: 2710. 10.1038/s41467-019-10648-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer DI, Swart EC, Kraft MF, Sandoval PY, Nowacki M. 2017. Two sets of Piwi proteins are involved in distinct sRNA pathways leading to elimination of germline-specific DNA. Cell Rep 20: 505–520. 10.1016/j.celrep.2017.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görtz HD. 1988. Paramecium. Springer-Verlag, Berlin. [Google Scholar]
- Gratias A, Bétermier M. 2001. Developmentally programmed excision of internal DNA sequences in Paramecium aurelia. Biochimie 83: 1009–1022. 10.1016/S0300-9084(01)01349-9 [DOI] [PubMed] [Google Scholar]
- Gratias A, Bétermier M. 2003. Processing of double-strand breaks is involved in the precise excision of Paramecium internal eliminated sequences. Mol Cell Biol 23: 7152–7162. 10.1128/MCB.23.20.7152-7162.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratias A, Lèpere G, Garnier O, Rosa S, Duharcourt S, Malinsky S, Meyer E, Bétermier M. 2008. Developmentally programmed DNA splicing in Paramecium reveals short-distance crosstalk between DNA cleavage sites. Nucleic Acids Res 36: 3244–3251. 10.1093/nar/gkn154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M. 2016. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849. 10.1093/bioinformatics/btw313 [DOI] [PubMed] [Google Scholar]
- Guérin F, Arnaiz O, Boggetto N, Denby Wilkes C, Meyer E, Sperling L, Duharcourt S. 2017. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics 18: 327. 10.1186/s12864-017-3713-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafez M, Hausner G. 2015. Convergent evolution of twintron-like configurations: One is never enough. RNA Biol 12: 1275–1288. 10.1080/15476286.2015.1103427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton EP, Kapusta A, Huvos PE, Bidwell SL, Zafar N, Tang H, Hadjithomas M, Krishnakumar V, Badger JH, Caler EV, et al. 2016. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. eLife 5: e19090. 10.7554/eLife.19090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarski M, Singh A, Swart EC, Arambasic M, Sandoval PY, Nowacki M. 2014. Paramecium tetraurelia chromatin assembly factor-1-like protein PtCAF-1 is involved in RNA-mediated control of DNA elimination. Nucleic Acids Res 42: 11952–11964. 10.1093/nar/gku874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Matsuda A, Marmignon A, Ku M, Silve A, Meyer E, Forney JD, Malinsky S, Bétermier M. 2011. Highly precise and developmentally programmed genome assembly in Paramecium requires ligase IV–dependent end joining. PLoS Genet 7: e1002049. 10.1371/journal.pgen.1002049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Suh A, Feschotte C. 2017. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci 114: E1460–E1469. 10.1073/pnas.1616702114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamysheva Z, Wang L, Shrode T, Bednenko J, Hurley LA, Shippen DE. 2003. Developmentally programmed gene elimination in Euplotes crassus facilitates a switch in the telomerase catalytic subunit. Cell 113: 565–576. 10.1016/S0092-8674(03)00363-5 [DOI] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp TH, van Luenen HG, Plasterk RH. 1999. mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell 99: 133–141. 10.1016/S0092-8674(00)81645-1 [DOI] [PubMed] [Google Scholar]
- Kinsella CM, Ruiz-Ruano FJ, Dion-Côté A-M, Charles AJ, Gossmann TI, Cabrero J, Kappei D, Hemmings N, Simons MJP, Camacho JPM, et al. 2019. Programmed DNA elimination of germline development genes in songbirds. Nat Commun 10: 5468. 10.1038/s41467-019-13427-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klobutcher LA, Herrick G. 1997. Developmental genome reorganization in ciliated protozoa: the transposon link. Prog Nucleic Acid Res Mol Biol 56: 1–62. 10.1016/S0079-6603(08)61001-6 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Mouël A, Butler A, Caron F, Meyer E. 2003. Developmentally regulated chromosome fragmentation linked to imprecise elimination of repeated sequences in paramecia. Eukaryot Cell 2: 1076–1090. 10.1128/EC.2.5.1076-1090.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepère G, Bétermier M, Meyer E, Duharcourt S. 2008. Maternal noncoding transcripts antagonize the targeting of DNA elimination by scanRNAs in Paramecium tetraurelia. Genes Dev 22: 1501–1512. 10.1101/gad.473008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepère G, Nowacki M, Serrano V, Gout JF, Guglielmi G, Duharcourt S, Meyer E. 2009. Silencing-associated and meiosis-specific small RNA pathways in Paramecium tetraurelia. Nucleic Acids Res 37: 903–915. 10.1093/nar/gkn1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier-Akakpo M, Frapporti A, Denby Wilkes C, Matelot M, Vervoort M, Sperling L, Duharcourt S. 2014. Local effect of enhancer of zeste-like reveals cooperation of epigenetic and cis-acting determinants for zygotic genome rearrangements. PLoS Genet 10: e1004665. 10.1371/journal.pgen.1004665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly MA, Duronio RJ. 2005. New insights into cell cycle control from the Drosophila endocycle. Oncogene 24: 2765–2775. 10.1038/sj.onc.1208610 [DOI] [PubMed] [Google Scholar]
- Maliszewska-Olejniczak K, Gruchota J, Gromadka R, Denby Wilkes C, Arnaiz O, Mathy N, Duharcourt S, Bétermier M, Nowak JK. 2015. TFIIS-dependent non-coding transcription regulates developmental genome rearrangements. PLoS Genet 11: e1005383. 10.1371/journal.pgen.1005383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmignon A, Bischerour J, Silve A, Fojcik C, Dubois E, Arnaiz O, Kapusta A, Malinsky S, Bétermier M. 2014. Ku-mediated coupling of DNA cleavage and repair during programmed genome rearrangements in the ciliate Paramecium tetraurelia. PLoS Genet 10: e1004552. 10.1371/journal.pgen.1004552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Mayer KM, Forney JD. 1999. A mutation in the flanking 5′-TA-3′ dinucleotide prevents excision of an internal eliminated sequence from the Paramecium tetraurelia genome. Genetics 151: 597–604. 10.1093/genetics/151.2.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer KM, Mikami K, Forney JD. 1998. A mutation in Paramecium tetraurelia reveals functional and structural features of developmentally excised DNA elements. Genetics 148: 139–149. 10.1093/genetics/148.1.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miró-Pina C, Charmant O, Kawaguchi T, Holoch D, Michaud A, Cohen I, Humbert A, Jaszczyszyn Y, Chevreux G, Del Maestro L, et al. 2022. Paramecium Polycomb repressive complex 2 physically interacts with the small RNA-binding PIWI protein to repress transposable elements. Dev Cell 57: 1037–1052.e8. 10.1016/j.devcel.2022.03.014 [DOI] [PubMed] [Google Scholar]
- Preer JR. 1976. Quantitative predictions of random segregation models of the ciliate macronucleus. Genet Res 27: 227–238. 10.1017/S0016672300016426 [DOI] [PubMed] [Google Scholar]
- Prescott DM. 1994. The DNA of ciliated protozoa. Microbiol Rev 58: 233–267. 10.1128/mr.58.2.233-267.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2021. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/. [Google Scholar]
- Sandoval PY, Swart EC, Arambasic M, Nowacki M. 2014. Functional diversification of Dicer-like proteins and small RNAs required for genome sculpting. Dev Cell 28: 174–188. 10.1016/j.devcel.2013.12.010 [DOI] [PubMed] [Google Scholar]
- Sardo L, Lin A, Khakhina S, Beckman L, Ricon L, Elbezanti W, Jaison T, Vishwasrao H, Shroff H, Janetopoulos C, et al. 2017. Real-time visualization of chromatin modification in isolated nuclei. J Cell Sci 130: 2926–2940. 10.1242/jcs.205823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleif R. 1992. DNA looping. Annu Rev Biochem 61: 199–223. 10.1146/annurev.bi.61.070192.001215 [DOI] [PubMed] [Google Scholar]
- Sellis D, Guérin F, Arnaiz O, Pett W, Lerat E, Boggetto N, Krenek S, Berendonk T, Couloux A, Aury JM, et al. 2021. Massive colonization of protein-coding exons by selfish genetic elements in Paramecium germline genomes. PLoS Biol 19: e3001309. 10.1371/journal.pbio.3001309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DP, Saudemont B, Guglielmi G, Arnaiz O, Goût J-F, Prajer M, Potekhin A, Przybòs E, Aubusson-Fleury A, Bhullar S, et al. 2014. Genome-defence small RNAs exapted for epigenetic mating-type inheritance. Nature 509: 447–452. 10.1038/nature13318 [DOI] [PubMed] [Google Scholar]
- Singh A, Maurer-Alcalá XX, Solberg T, Häußermann L, Gisler S, Ignarski M, Swart EC, Nowacki M. 2022. Chromatin remodeling is required for sRNA-guided DNA elimination in Paramecium. EMBO J 41: e111839. 10.15252/embj.2022111839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Baker C, Eichler EE, Amemiya CT. 2012. Genetic consequences of programmed genome rearrangement. Curr Biol 22: 1524–1529. 10.1016/j.cub.2012.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, Wilkes CD, Sandoval PY, Arambasic M, Sperling L, Nowacki M. 2014. Genome-wide analysis of genetic and epigenetic control of programmed DNA deletion. Nucleic Acids Res 42: 8970–8983. 10.1093/nar/gku619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart EC, Denby Wilkes C, Sandoval PY, Hoehener C, Singh A, Furrer DI, Arambasic M, Ignarski M, Nowacki M. 2017. Identification and analysis of functional associations among natural eukaryotic genome editing components. F1000Res 6: 1374. 10.12688/f1000research.12121.1 [DOI] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. 1999. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 99: 123–132. 10.1016/S0092-8674(00)81644-X [DOI] [PubMed] [Google Scholar]
- Torgasheva AA, Malinovskaya LP, Zadesenets KS, Karamysheva TV, Kizilova EA, Akberdina EA, Pristyazhnyuk IE, Shnaider EP, Volodkina VA, Saifitdinova AF, et al. 2019. Germline-restricted chromosome (GRC) is widespread among songbirds. Proc Natl Acad Sci 116: 11845–11850. 10.1073/pnas.1817373116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mitreva M, Berriman M, Thorne A, Magrini V, Koutsovoulos G, Kumar S, Blaxter ML, Davis RE. 2012. Silencing of germline-expressed genes by DNA elimination in somatic cells. Dev Cell 23: 1072–1080. 10.1016/j.devcel.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Solberg T, Maurer-Alcalá XX, Swart EC, Gao F, Nowacki M. 2022. A small RNA-guided PRC2 complex eliminates DNA as an extreme form of transposon silencing. Cell Rep 40: 111263. 10.1016/j.celrep.2022.111263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York. https://ggplot2.tidyverse.org [Google Scholar]
- Zilberman D, Cao X, Jacobsen SE. 2003. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299: 716–719. 10.1126/science.1079695 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.