

Abstract
Background
Accumulation of myofibroblasts is critical to fibrogenesis in idiopathic pulmonary fibrosis (IPF). Senescence and insufficient mitophagy in fibroblasts contribute to their differentiation into myofibroblasts, thereby promoting the development of lung fibrosis. Bone morphogenetic protein 4 (BMP4), a multifunctional growth factor, is essential for the early stage of lung development; however, the role of BMP4 in modulating lung fibrosis remains unknown.
Methods
The aim of this study was to evaluate the role of BMP4 in lung fibrosis using BMP4-haplodeleted mice, BMP4-overexpressed mice, primary lung fibroblasts and lung samples from patients with IPF.
Results
BMP4 expression was downregulated in IPF lungs and fibroblasts compared to control individuals, negatively correlated with fibrotic genes, and BMP4 decreased with transforming growth factor (TGF)-β1 stimulation in lung fibroblasts in a time- and dose-dependent manner. In mice challenged with bleomycin, BMP4 haploinsufficiency perpetuated activation of lung myofibroblasts and caused accelerated lung function decline, severe fibrosis and mortality. BMP4 overexpression using adeno-associated virus 9 vectors showed preventative and therapeutic efficacy against lung fibrosis. In vitro, BMP4 attenuated TGF-β1-induced fibroblast-to-myofibroblast differentiation and extracellular matrix (ECM) production by reducing impaired mitophagy and cellular senescence in lung fibroblasts. Pink1 silencing by short-hairpin RNA transfection abolished the ability of BMP4 to reverse the TGF-β1-induced myofibroblast differentiation and ECM production, indicating dependence on Pink1-mediated mitophagy. Moreover, the inhibitory effect of BMP4 on fibroblast activation and differentiation was accompanied with an activation of Smad1/5/9 signalling and suppression of TGF-β1-mediated Smad2/3 signalling in vivo and in vitro.
Conclusion
Strategies for enhancing BMP4 signalling may represent an effective treatment for pulmonary fibrosis.
Short abstract
BMP4 deficiency results in cellular senescence and defective mitophagy of fibroblasts and promotes fibrosis in the lung. This study provides a proof-of-principle that BMP4 activation may offer a promising therapeutic approach for pulmonary fibrosis. https://bit.ly/3HWIKWS
Introduction
Idiopathic pulmonary fibrosis (IPF), the most common and severe form of idiopathic interstitial pneumonia, is a chronic, aggressive and devastating age-related disease of unknown cause. IPF is characterised by an excessive accumulation of myofibroblasts and extracellular matrix (ECM) that destroys the physiological architecture and function in the lung [1]. IPF affects ~120 000 people in the United States with a steady increase in both morbidity and mortality [2]. There is interindividual variability in the disease trajectory, but ultimately prognosis is worse than some cancers, with a median survival of 3–5 years [3, 4]. The current antifibrotic drugs, pirfenidone and nintedanib (BIBF 1120), slow disease progression, but do not provide a cure for IPF [5]; thus, new therapeutic approaches are needed for this disease.
IPF is the result of repetitive cycles of epithelial cell injury and death that provoke the proliferation, activation and myofibroblast differentiation of fibroblasts [6]. The activated fibroblasts, also called myofibroblasts, are the critical effector cells in a variety of fibrotic diseases including IPF, in which they have stronger contractile activity and account for the enhanced release of ECM molecules with the subsequent scaring and destruction of the tissue architecture [7, 8]. During this process, transforming growth factor (TGF)-β1, considered as the principal profibrotic cytokine, plays a key role in the persistent fibroblast activation and myofibroblast differentiation in fibrotic diseases [9]. TGF-β1 signalling occurs only temporarily in wound healing, but remains active in fibrotic diseases. Fibroblasts isolated from the lungs of patients with IPF show a TGF-β1-biased gene expression signature [10]. Moreover, long-lasting activation of TGF-β1 signalling induces persistent fibroblast activation in lung fibrosis [11]. Hence, therapies blocking TGF-β1 signalling activation have been under clinical trials for IPF [12]. Regardless of the initial trigger, unremitting fibroblast activation and differentiation with subsequent excessive accumulation of ECM play a central role in fibrosis formation and is observed in all types of lung fibrosis.
Autophagy is a conserved catabolic process that mediates the degradation of cytosolic organelles and proteins via lysosome-mediated degradation. Adequate turnover of damaged organelles is cytoprotective during the integrated stress response [13, 14]. Previous reports have highlighted the potential involvement of insufficient autophagy and mitophagy (mitochondria-selective autophagy) in the pathogenesis of IPF through the regulation of fibroblast senescence and myofibroblast differentiation [13, 15]. Cellular senescence, an evolutionarily conserved state of stable replicative arrest, is also considered as a marker for the pathological condition of IPF, and contributes to myofibroblast differentiation [16]. Senescence biomarkers, including p16, p21, p53 and senescence-associated β-galactosidase (SA-β-gal) activity, are abundant within lung fibroblasts of human IPF and experimental pulmonary fibrosis [17, 18]. When these senescent cells are not effectively cleared from the tissues and the regeneration is not fully available, a senescence state occurs, which causes lung fibrosis and organ dysfunction [19]. Thus, identification of genes indicative of mitophagy and cellular senescence might offer potential targeted therapies for IPF.
Bone morphogenetic protein 4 (BMP4), a multifunctional secreted growth factor that belongs to the TGF-β superfamily, can regulate cell proliferation and differentiation during embryonic development [20]. It has been reported that the BMP antagonists Gremlin and Fstl1 are overexpressed and implicated in fibrotic disorders of various organs, including the lungs [21–24]. However, the role of BMP4 in lung fibrosis has not been investigated. In this study, we first investigated whether BMP4 downregulation in human lung is a robust feature of IPF and whether BMP4 expression is associated with IPF fibroblast pathobiology or clinical manifestations of IPF severity. We then determined whether BMP4 signalling suppresses or promotes pulmonary fibrosis through its activity in lung fibroblasts. We identified that BMP4 mediated mitophagy and cellular senescence of fibroblasts during IPF pathogenesis. We found that BMP4 inhibited TGF-β1-induced Smad2/3 phosphorylation by activating Smad1/5/9, thereby preventing fibroblast-to-myofibroblast differentiation and ECM production. Our data provide a proof of principle that BMP4 signalling activation may represent a novel and effective therapeutic approach for pulmonary fibrosis.
Methods
Methods for detailed methods including animal protocols and experimental procedures, see the supplementary material.
Ethics statement
All animal experiments described here complied with the guidelines of the Committee on the Use and Care of Animals of Guangzhou Medical University (Guangzhou, China), and were approved by the Animal Subjects Committee of Guangzhou Medical University. All human studies have been approved by the institutional review board at the First Affiliated Hospital of Guangzhou Medical University, and were performed in compliance with the relevant ethical regulations.
Statistical analysis
All data are presented as mean±sem. Statistical analyses were applied using t-tests and one-way ANOVA to determine statistical significance. Statistical analyses were carried out using the GraphPad Prism software (version 6.07). A two-sided p-value of <0.05 is considered as statistically significant.
Results
BMP4 is downregulated in lungs and fibroblasts from IPF patients and bleomycin-injured mice
To investigate the potential role of BMP4 in the pathogenesis of IPF, we first analysed the expression levels of BMP4 in the lung tissues of IPF patients and control subjects. Western blotting showed that the protein levels of BMP4 were significantly decreased in patients with IPF, accompanied with an increase in myofibroblast biomarker α-smooth muscle actin (SMA) and ECM protein molecules such as Col1 and Col3 (figure 1a and b, and supplementary figure S1). Lungs from human subjects with IPF are characterised by ongoing tissue remodelling with accumulation of α-SMA-expressing myofibroblasts within fibroblastic foci. Within these regions of active fibrosis, a significant reduction in BMP4 expression was observed (figure 1e). Although α-SMA expressing myofibroblasts were largely deficient in BMP4 expression, alveolar epithelial cells represented a relatively high level of BMP4 in both control and IPF lung tissues (figure 1e). This fits well with previously published data that healthy epithelial cells can produce BMP4, which functions to prevent fibroblast activation, and it is loss of this production that releases the brakes on fibroblast activation [25]. Next, we determined the expression of BMP4 in isolated lung fibroblasts from patients with IPF and control subjects, and found that BMP4 levels were decreased in IPF fibroblasts (figure 1c and d). We also tested the expression of BMP4 in the bleomycin (BLM) mouse model of lung fibrosis, a useful tool to investigate treatment options for IPF [26]. As shown in figure 1f–j, BMP4 expression was also decreased in the lungs and freshly isolated lung fibroblasts from BLM-treated mice versus controls. Collectively, these findings indicate that decreased BMP4 signalling in IPF is associated with an enhanced fibrotic response, which contributes to the pathogenesis of lung fibrosis.
FIGURE 1.

Bone morphogenetic protein (BMP)4 is downregulated in lungs and lung fibroblasts from idiopathic pulmonary fibrosis (IPF) patients and bleomycin (BLM)-injured mice. a) Representative Western blot results of BMP4, α-smooth muscle actin (SMA), and Col1 protein expressions in lungs of patients with IPF and control subjects (n=30 per group). b) Densitometric analysis of BMP4, α-SMA, and Col1 in the immunoblots using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the internal reference. c) Representative Western blot results of BMP4, α-SMA and Col1 protein expressions in isolated lung fibroblasts from patients with IPF and control subjects (n=4 per group). d) Densitometric analysis of BMP4, α-SMA and Col1 in the immunoblots using GAPDH as the internal reference. e) Representative images show BMP4 (red), E-cadherin (green), α-SMA (green) and nuclei (blue) in lung sections of control and subjects with IPF. Right-hand panels display magnified areas from images indicated by dashed boxes. f, g) Western blot analysis of BMP4, α-SMA, fibronectin (FN) and Col1 protein expression in lung tissues of C57BL/6 mice at day 21 post-BLM or -saline administration (n=3∼10 per group). β-actin was used as a loading control. h) Western blot analysis of BMP4 expression in the lung fibroblasts isolated from C57BL/6 mice at day 21 post-BLM or -saline administration (n=4 per group). β-actin was used as a loading control. i) Quantitative real-time PCR analysis of BMP4 mRNA level in the lung fibroblasts isolated from C57BL/6 mice at day 21 post-BLM or -saline administration (n=5 per group). j) Representative images show BMP4 (red), E-cadherin (green), α-SMA (green) and nuclei (blue) in lung sections of C57BL/6 mice at day 21 post-BLM or -saline administration. Right-hand panels display magnified areas from images indicated by dashed boxes. Scale bars=50 µm. Data are presented as mean±sem. DAPI: 4′,6-diamidino-2-phenylindole. **: p<0.01; ***: p<0.001; ****: p<0.0001.
BMP4+/− mice are more susceptible to developing BLM-induced lung fibrosis
To determine the role of BMP4 deficiency in mediating fibrogenesis, we determined the fibrotic response to a lower dose of BLM-induced (2.0 mg·kg−1) lung injury in BMP4-haploinsufficient mice. Because homozygous mutation of BMP4 (BMP4−/−) is lethal to the embryo [27], heterozygous BMP4+/− mice were used in this study. BMP4 protein levels in the lung tissue were significantly reduced in BMP4+/− mice, and further decreased by BLM (figure 2h, supplementary figure S2a). BMP4+/− mice were more susceptible to developing BLM-induced lung injury and showed a decrease in survival (figure 2a) and body weight (figure 2b) compared to the wild-type (WT) littermates. Importantly, BMP4+/− mice exhibited enhanced BLM-induced lung fibrosis (more fibrotic phenotype), as illustrated by micro-computed tomography (CT) (figure 2c) and pulmonary function testing (figure 2d). Collagen accumulation was markedly increased in the lungs of BMP4+/− mice, as examined by Masson's trichrome staining (figure 2e) and hydroxyproline content (a biochemical index for collagen deposition; figure 2f). More severe fibrosis in BMP4+/− mice was further supported by increased mRNA and protein levels for fibronectin (FN), TGF-β1, Col1 and Col3 (figure 2g and h, supplementary figure S2a). These in vivo data indicate that BMP4 downregulation is induced in response to lung injury and causally drives pulmonary fibrogenesis.
FIGURE 2.

Bone morphogenetic protein (BMP)4+/− mice demonstrate higher mortality and more severe fibrosis after bleomycin (BLM)-induced lung injury. a) Percentages of surviving BMP4+/− and wild type (WT) (BMP4+/+) mice were plotted over a 21-day period post-BLM (2.0 mg·kg−1) or -saline administration. BMP4+/++saline n=12, BMP4+/−+saline n=10, BMP4+/++BLM n=25, BMP4+/−+BLM n=37. b) The corresponding body weight of mice in each group was shown over a 21-day observation period. c) Representative axial (top row) and their corresponding coronal (bottom row) images from different groups determined by micro-computed tomography showing radiological features (n=4 per group). Healthy lungs are black, and diseased lungs were increasingly white (elevated density). d) Lung function parameters including respiratory resistance and dynamic compliance (Cdyn) among different groups were compared at day 21 post-BLM or -saline administration (n=5∼6 per group). e) Masson trichrome staining of left lungs from BMP4+/+ and BMP4+/− mice at day 21 post-BLM or -saline administration. Images in the lower panels were magnified from the photomicrographs in the upper panels (n=6 per group). f) Hydroxyproline content of lungs from BMP4+/+ and BMP4+/− mice after BLM injury (n=5 per group). g) Quantitative real-time PCR analysis of Col1, Col3, fibronectin (FN) and α-smooth muscle actin (SMA) mRNA levels in lung homogenates of BLM-challenged BMP4+/+ and BMP4+/− mice. h) Representative Western blot results of α-SMA, Col1, Col3, FN, BMP4 and TGF-β1 and i) expression of phosphorylated and total Smad1/5/9, Smad2 and Smad3 in lung homogenates of BLM-challenged BMP4+/+ and BMP4+/− mice (n=6 per group). β-actin was used as a loading control. j) Immunofluorescence analysis of myofibroblasts in lung sections (nucleus, 4′,6-diamidino-2-phenylindole (DAPI); n=3 per group). Representative images of the staining are shown. Scale bars=50 µm. Arrows indicate myofibroblasts with α-SMA- and vimentin-positive staining. Data are presented as mean±sem. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
Since myofibroblasts have been identified as key players in this disease, we investigated whether the more fibrotic phenotype in BMP4+/− mice correlates with the accumulation of myofibroblasts by immunofluorescence staining on lung sections with anti-α-SMA antibodies, a reliable marker of activated fibroblasts/myofibroblasts. As shown in supplementary figure S3, BLM-increased myofibroblast accumulation was more severe in BMP4+/− mice, as compared to WT mice. Additionally, real-time quantitative (q)PCR and Western blot analyses of lung tissues showed that BLM-induced expression of α-SMA in BMP4+/− mice was dramatically increased (figure 2g and h, supplementary figure S2a), implicating the increased myofibroblast accumulation after BLM treatment in BMP4+/− mice. Moreover, dual immunostaining for α-SMA and vimentin (a fibroblast marker) showed similar results with more co-staining myofibroblasts in BMP4+/− mice (figure 2j), suggesting that BMP4 deficiency aggravates the differentiation capacity of lung fibroblasts into myofibroblasts.
To further investigate the effects of the haplodeletion of BMP4 on downstream signalling pathways in pulmonary fibrosis, we performed Western blot of phosphorylated Smad1/5/9 (p-Smad1/5/9), p-Smad2 and p-Smad3 using lung homogenates from BMP4+/− and WT mice 21 days post-BLM administration. Haplodeletion of BMP4 significantly reduced the expression levels of p-Smad1/5/9, but elevated the levels of p-Smad2 and p-Smad3 (figure 2i, supplementary figure S2b), suggesting the inactivation of Smad1/5/9 signalling and the activation of Smad2/3 signalling pathways by BMP4 deficiency. Altogether, BMP4+/− mice are more susceptible to developing BLM-induced lung fibrosis.
Adeno-associated virus 9-mediated overexpression of BMP4 prevents and reverses pulmonary fibrosis
We next investigated whether overexpression of BMP4 in the lung could prevent and reverse experimental pulmonary fibrosis. Adeno-associated virus (AAV), an ideal nonintegrative therapeutic vector, is the leading platform for gene delivery for the therapy of miscellaneous human diseases. Two AAV-based therapeutics have gained regulatory approval recently [28]. Therefore, we selected the AAV9 serotype due to its high viral transduction of the lungs and its low immunogenicity [29, 30], to determine the effect of BMP4 gene therapy in the BLM mouse model of pulmonary fibrosis. To examine the transduction efficiency of the lungs in our present study, we intratracheally injected the C57BL/6 mice with BMP4-expressing AAV9 (AAV9-BMP4; 1.0×1010 viral genomes in 60 μL saline) and observed BMP4 expression in the lungs 3 weeks later, a time point recommended by Povedano et al. [31]. We found that AAV9-BMP4 transduced lungs showed a significant increase in BMP4 protein expression comparing with AAV9-green fluorescent protein (GFP) transduced lungs (supplementary figure S4). Hence, we continue to use this dose of virus (1.0×1010 viral genomes in 60 μL saline per mouse) for the subsequent in vivo preventive or therapeutic experiments.
We first investigated the potential of BMP4 to prevent pulmonary fibrosis. 3 weeks after viral intervention, these AAV9-GFP or AAV9-BMP4 transduced mice were injected intratracheally with a single higher dose of BLM (2.5 mg·kg−1) to induce a relatively severe lung fibrosis (supplementary figure S5a). 3 weeks after BLM administration, micro-CT assay showed that BMP4 treatment efficiently protected the lung structure and alleviated the volume reduction and fibrotic alterations induced by BLM treatment (supplementary figure S5b). Masson's trichrome staining (supplementary figure S5c) and hydroxyproline content (supplementary figure S5d) further showed that overexpression of BMP4 remarkably reduced the extent of pulmonary fibrosis following BLM administration. In agreement with lower collagen, AAV9-BMP4 treated lungs also showed significantly less myofibroblast biomarker α-SMA and ECM components compared to AAV9-GFP-treated mice (supplementary figure S5e and f). These results suggest that BMP4 overexpression prevents the development of BLM-induced fibroblast activation and lung fibrosis.
The “switch” between inflammation and fibrosis appears to occur around day 9 after BLM [26]; we examined whether overexpression of BMP4 protein could reverse the established lung fibrosis by treating mice from day 10 to day 31 after BLM (figure 3). In the context of later intervention, AAV9-BMP4 treatment significantly attenuated pulmonary fibrosis as indicated by a good performance (supplementary videos), micro-CT (figure 3b), lung function (figure 3c), Masson's trichrome staining (figure 3d) and hydroxyproline content (figure 3e). In addition, AAV9-BMP4 treated mice showed a remarkable reduction in α-SMA-positive myofibroblast counts and TGF-β1 expression, as well as the levels of ECM proteins in the lungs at week three after treatment with the viral vectors (figure 3f–j, supplementary figure S6a). Moreover, the BLM-induced increase of vimentin+α-SMA+ co-staining myofibroblasts was significantly downregulated by BMP4 overexpression (figure 3h), further demonstrating the role of BMP4 in the differentiation capacity of lung fibroblasts into myofibroblasts. In addition, we observed that overexpression of BMP4 could reverse the late stage of lung fibrosis by treating mice from day 21 after BLM (supplementary figure S7). These data indicate that BMP4 gene therapy promotes resolution of lung fibrosis in mice.
FIGURE 3.
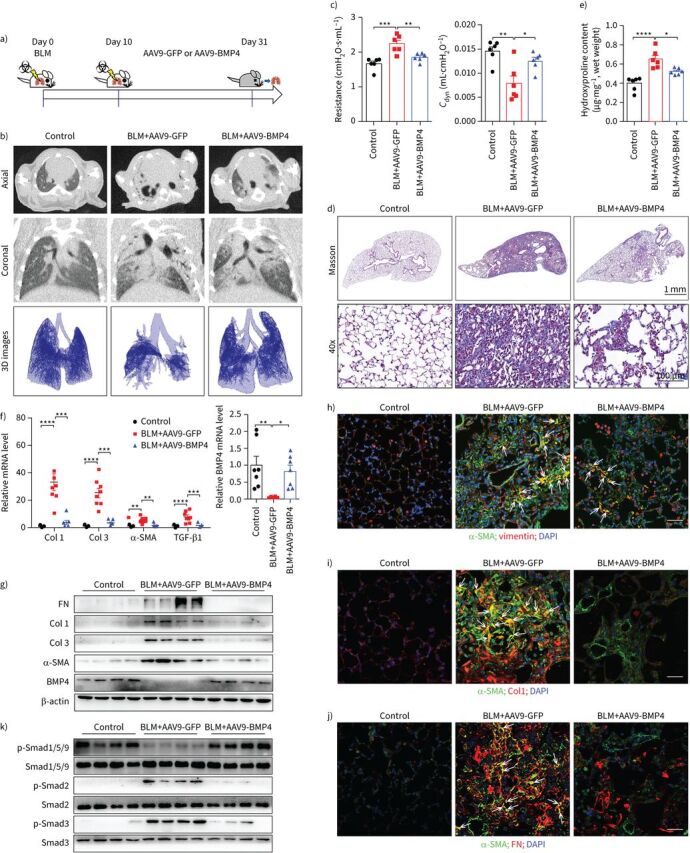
Adeno-associated virus (AAV)9-bone morphogenetic protein (BMP)4 treatment reverses bleomycin (BLM)-induced pulmonary fibrosis. a) Schematic showing the overexpression of BMP4 in the therapeutic BLM mouse lung fibrosis model. Mice were injected intratracheally with AAV9-BMP4 (1.0×1010 viral genomes in 60 μL saline per mouse) starting at day 10, and lungs were assessed on day 31 after BLM (2.5 mg·kg−1) administration. b) Axial and their corresponding coronal micro-computed tomography (CT) images were acquired after BLM administration. The lower panels show the representative three-dimensional (3D) images drawn out from micro-CT images, based on different tissues with varying density (n=4 per group). c) Lung function parameters including respiratory resistance and dynamic compliance (Cdyn) among different groups were compared 21 days after virus instillation (n=6 per group). d) Masson's trichrome staining of lung sections from AAV9-BMP4- or AAV9-green fluorescent protein (GFP)-treated mice in the therapeutic mouse lung fibrosis model (n=6 per group). Scale bars=1 mm. Images in the lower panels were magnified from the photomicrographs in the upper panels. Scale bars=100 µm. e) Hydroxyproline contents in the lungs of AAV9-BMP4- or AAV9-GFP-treated mice in the therapeutic lung fibrosis model (n=6). f) Quantitative real-time PCR analysis of Col1, Col3, α-smooth muscle actin (SMA), transforming growth factor (TGF)-β1 and BMP4 mRNA expressions in the lungs of AAV9-BMP4- or AAV9-GFP-treated mice in the therapeutic lung fibrosis model (n=5∼8 per group). g) Representative Western blot analysis of α-SMA, Col1, Col3, fibronectin (FN) and BMP4 and k) expression of phosphorylated (p) and total Smad1/5/9, Smad2 and Smad3 in lung homogenates of AAV9-BMP4- or AAV9-GFP-treated mice in the therapeutic lung fibrosis model (n=4 per group). β-actin was used as a loading control. h–j) Immunofluorescence analysis of α-SMA, vimentin, FN and Col1 expressions in lung sections (nucleus, 4′,6-diamidino-2-phenylindole (DAPI); n=3 per group). Representative images of the staining are shown. Scale bars=50 µm. Yellow colouration indicates representative double-positive cells. Data are presented as mean±sem. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
To further determine the underlying mechanisms responsible for the BMP4-induced attenuation of lung fibrosis, we also profiled the activation status of Smad1/5/9 and Smad2/3 signalling pathways by Western blot of lung homogenates from AAV9-BMP4- or AAV9-GFP-treated mice after either preventive (supplementary figure S5g) or the therapeutic (figure 3k, supplementary figure S6b) interventions. BMP4 expression using AAV9 vectors clearly increased the activation of Smad1/5/9 signalling and reduced the BLM-induced activation of Smad2/3 signalling pathways in vivo.
BMP4 suppresses TGF-β1-induced fibroblast activation
Activation of TGF-β1 signalling is a common characteristic of fibrotic conditions. Therefore, we investigated the potential effects of TGF-β1 on BMP4 expression in lung fibroblasts. We found that the mRNA (figure 4a and b) and protein (figure 4c and d) levels of BMP4 were dose- and time-dependently decreased in human primary lung fibroblasts incubated with TGF-β1, along with upregulation of fibrosis-related genes (supplementary figure S8) and proteins (figure 4c and d). Similarly, stimulation of mouse primary lung fibroblasts with TGF-β1 also decreased the protein expression levels of BMP4 in a time-dependent manner (data not shown). This suggests a negative correlation between BMP4 content and the development of fibrotic lung disease.
FIGURE 4.

Bone morphogenetic protein (BMP)4 inhibits transforming growth factor (TGF)-β1-induced lung fibroblast activation, differentiation and extracellular matrix (ECM) production. a) Real-time quantitative (q)-PCR analysis of BMP4 mRNA expression in human primary lung fibroblasts stimulated with different concentrations of TGF-β1 for 48 h (n=3). b) Real-time qPCR analysis of BMP4 mRNA expression in human primary lung fibroblasts stimulated with TGF-β1 (10 ng·mL−1) at different time points (n=3). c) Western blot analysis of BMP4, Col1, fibronectin (FN) and α-smooth muscle actin (SMA) protein levels in human primary lung fibroblasts stimulated with different concentrations of TGF-β1 for 48 h (n=3). d) Western blot analysis of BMP4, Col1, FN and α-SMA protein levels in human primary lung fibroblasts stimulated with TGF-β1 (10 ng·mL−1) at different time points (n=3). e) Western blot analysis of α-SMA, FN and Col1 protein expressions in total cell lysates of human idiopathic pulmonary fibrosis (IPF) lung fibroblasts treated with TGF-β1 (10 ng·mL−1, 48 h) in the presence of BMP4 (20 μM) or vehicle (n=6). β-actin was used as a loading control. f) Representative images of Col3 immunostaining in human primary lung fibroblasts treated with TGF-β1 (10 ng·mL−1, 48 h) in the presence of BMP4 (20 μM) or vehicle controls (n=3). g) Western blot analysis of α-SMA, Col1, FN and BMP4 and h) expression of phosphorylated and total Smad1/5/9, Smad2 and Smad3 in total cell lysates of primary lung fibroblasts from BMP4+/+ and BMP4+/− mice treated with TGF-β1 (10 ng·mL−1, 48 h) (n=4). β-actin was used as a loading control. i) Western blot analysis of α-SMA, FN and Col1 protein expressions in total cell lysates of wild type (WT) mouse primary lung fibroblasts treated with TGF-β1 (10 ng·mL−1, 48 h) and/or BMP4 (20 μM; n=6). β-actin was used as a loading control. j) Western blot analysis of phosphorylated and total expression of Smad1/5/9, Smad2 and Smad3 in total cell lysates of NIH/3T3 fibroblasts treated with TGF-β1 (10 ng·mL−1) and/or BMP4 (20 μM; n=4). β-actin was used as a loading control. k) Immunofluorescent staining of myofibroblasts in mouse primary lung fibroblasts treated with TGF-β1 (10 ng·mL−1, 48 h) and/or BMP4 (20 μM). Representative images of the staining are shown (n=3). Arrows indicate myofibroblasts with α-SMA- and vimentin-positive staining. Nuclear, 4′,6-diamidino-2-phenylindole (DAPI). l, m) Representative images of Col1 and Col3 immunostaining of TGF-β1-differentiated mouse lung fibroblasts treated with BMP4 or vehicle (n=3). Data are presented as mean±sem. Scale bars=100 µm. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
In injured lung tissues, fibroblasts are activated and differentiate into myofibroblasts, which are the main effector cells accountable to the secretion of huge amounts of ECM molecules in the lung [7]. To test the functional role of decreased BMP4 levels, mouse lung fibroblasts from BMP4+/− and WT mice was incubated with recombinant TGF-β1. As shown in figure 4g, BMP4 haploinsufficiency significantly enhanced TGF-β1-induced myofibroblast differentiation and ECM production as indicated by Western blot of α-SMA, Col1 and FN. In contrast, administration of exogenous recombinant BMP4 observably attenuated TGF-β1-induced myofibroblast differentiation with reduced protein levels of α-SMA as monitored by high-content imaging of vimentin+α-SMA+ double-positive cells and by Western blot-based quantification of α-SMA in mouse lung fibroblasts (figure 4i and k). The induction of Col1, Col3 and FN protein levels by TGF-β1 was also reduced by addition of BMP4 in these mouse lung fibroblasts (figure 4i, l and m).
The suppression of profibrotic phenotypes by BMP4 was subsequently confirmed in TGF-β1-treated primary lung fibroblasts from patients with IPF. Immunofluorescence and Western blot analysis demonstrated that BMP4 treatment dramatically inhibited TGF-β1-induced fibroblast-to-myofibroblast transition and ECM production (figure 4e and f). Taken together, these data collectively suggest that BMP4 can inhibit TGF-β1-induced lung fibroblast activation, differentiation and ECM production.
To investigate the signalling pathways involved in mediating the inhibitory effect of BMP4 on TGF-β1-induced myofibroblast transition and ECM production in vitro, we evaluated the changes in the phosphorylated Smad1/5/9 and Smad2/3 during 48 h following treatment. Treatment of fibroblasts with BMP4 alone induced a sustained upregulation of p-Smad1/5/9 in a concentration-dependent manner. Co-treatment of cells with BMP4 and TGF-β1 exhibited a significant activation of p-Smad1/5/9 signalling at 48 h, which was significantly higher than that with TGF-β1 alone at this same time point (figure 4j). In addition, treatment with BMP4 significantly reduced TGF-β1-induced activation of Smad2/3 signalling in fibroblasts (figure 4j). In line with these findings, our results showed that haplodeletion of BMP4 enhanced the reduction of p-Smad1/5/9 and the increase of p-Smad2/3 in mouse lung fibroblasts (figure 4h), suggesting that the protection of fibroblast activation occurs downstream of TGF-β1 signalling at the post-transcriptional level.
BMP4 modulates TGF-β1-induced senescence in fibroblasts
An increasing body of evidence implicates accelerated cellular senescence in IPF pathogenesis [32, 33]. Senescence-associated biomarkers, including p16, p21, p53 and senescence-associated SA-β-gal activity, are higher in fibroblasts of IPF lung tissue, which promote the fibroblast-to-myofibroblast transformation [17]. Therefore, we tested the effects of BMP4 on cellular senescence. TGF-β1 stimulation significantly increased SA-β-gal activity (figure 5a) and the levels of prosenescent proteins (i.e. p21 and p53) (figure 5b) in lung fibroblasts as demonstrated by β-gal staining and Western blot assays. These effects were further augmented by the knockdown of BMP4 expression, suggesting that reduced BMP4 expression amplifies TGF-β1-induced senescence. Senescent cells are metabolically active and secrete a great deal of cytokines, chemokines, matrix metalloproteinases and growth factors, which is called senescence-associated secretory phenotype (SASP) [34]. Accordingly, fibroblasts from BMP4+/− mice exhibited a higher SASP compared to WT fibroblasts upon TGF-β1 stimulation (figure 5d–g). Conversely, the TGF-β1-induced cellular senescence was significantly restrained by exogenous BMP4 (figure 5c). These findings indicate that BMP4 can prevent fibroblasts from entering senescence.
FIGURE 5.

Bone morphogenetic protein (BMP)4 inhibits transforming growth factor (TGF)-β1-induced cellular senescence in primary lung fibroblasts. a) Representative images of senescence-associated β-galactosidase (SA-β-gal) staining in primary lung fibroblasts from BMP4+/+ and BMP4+/− mice treated with TGF-β1 (10 ng·mL−1) for 48 h (n=4). Scale bars=200 µm. b) Western blot analysis of p53 and p21 protein expressions in total cell lysates of primary lung fibroblasts from BMP4+/+ and BMP4+/− mice treated with TGF-β1 (10 ng·mL−1, 48 h) (n=4). β-actin was used as a loading control. c) Western blot analysis of p53 and p21 in total cell lysates of mouse wild-type (WT) primary lung fibroblasts treated with TGF-β1 (10 ng·mL−1, 48 h) and/or BMP4 (20 μM) (n=4). d–g) Quantitative real-time PCR analysis of interleukin (IL)-1β, monocyte chemoattractant protein (MCP)-1, matrix metalloproteinase (MMP)-9 and TGF-β1 mRNA levels in primary lung fibroblasts from BMP4+/+ and BMP4+/− mice treated with TGF-β1 (10 ng·mL−1, 48 h) (n=4). Representative microphotographs showing h) intracellular reactive oxygen species (ROS) and i) mitochondrial ROS generation (n=4). Scale bars=100 µm. Data are presented as mean±sem. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
Reactive oxygen species (ROS) can mediate the deleterious effects of cellular damage and contribute to the activation of senescence pathways in cells [35], and we found that treatment with ROS scavenger N-acetylcysteine significantly abolished the TGF-β1-induced cell senescence in lung fibroblasts (data not shown). By determining the levels of cytosolic (figure 5h) and mitochondrial ROS (figure 5i), we found that the TGF-β1-induced production of ROS was significantly enhanced by BMP4 knockdown. Together, these findings suggest that BMP4 may decrease oxidative damage in lung fibroblasts, thereby preventing the activation of specific mechanisms of senescence.
BMP4 promotes mitophagy and restores mitochondrial dynamics in TGF-β1-stimulated lung fibroblasts
Autophagy is a critical stress-mediator mechanism related to senescence. When senescent cells are not effectively cleared from the tissues by autophagy and mitophagy, a senescent state occurs, which causes superfluous ROS for myofibroblast differentiation and organ fibrosis [13, 18]. Thus, we further evaluated the effect of BMP4 on autophagy and mitophagy in lung fibroblasts in response to TGF-β1 stimulation. Western blot assay indicated reduced expression of Pink1 in BMP4+/− fibroblasts when compared to WT fibroblasts after TGF-β1 stimulation (figure 6a, supplementary figure S9a). Changes in autophagic flux, as indicated by altered autophagy proteins LC3B-II, Beclin-1 and p62 protein levels, were also examined. Western blot showed that the TGF-β1-induced downregulation of Beclin-1 and LC3B-II, as well as the upregulation of p62, was significantly enhanced by BMP4 haploinsufficiency (figure 6a, supplementary figure S9a). Mitophagy was also determined by means of co-localisation analysis of confocal microscopy images of MitoTracker (a mitochondrial marker) and LC3B (an autophagosome marker) double fluorescence staining in BMP4+/− and WT lung fibroblasts. We found that TGF-β1-induced impaired mitophagy shown by co-localisation of MitoTracker and LC3B was significantly enhanced by BMP4 haploinsufficiency (figure 6e).
FIGURE 6.

Bone morphogenetic protein (BMP)4 promotes mitophagy and restores mitochondrial dynamics in transforming growth factor (TGF)-β1-stimulated lung fibroblasts. a) Representative Western blot results of Beclin-1, p62, LC3B and Pink1 protein expression in total cell lysates of primary lung fibroblasts from BMP4+/+ and BMP4+/− mice treated with TGF-β1 (10 ng·mL−1, 48 h) (n=4). β-actin was used as a loading control. b) Representative Western blot results of mitofusin (MFN)1, MFN2, dynamin-related protein (DRP)1 and mitochondrial fission 1 protein (FIS1) expressions in total cell lysates of primary lung fibroblasts from BMP4+/+ and BMP4+/− mice treated with TGF-β1 (10 ng·mL−1, 48 h) (n=4). β-actin was used as a loading control. c) Representative Western blot results of MFN1, MFN2, DRP1 and FIS1 expressions in total cell lysates of wild-type (WT) mouse primary lung fibroblasts treated with TGF-β1 (10 ng·mL−1, 48 h) and/or BMP4 (20 μM) (n=4). β-actin was used as a loading control. d) Mitochondrial fission was visualised by MitoTracker Red staining. Scale bars=25 µm. e) Immunofluorescence analysis of LC3B (red) and mitochondria (MitoTracker, green) in primary lung fibroblasts (n=4). Scale bars=25 µm. After Pink1 short-hairpin (sh)RNA (shPink1) or control shRNA (shNC) was transfected into NIH/3T3 fibroblasts, Pink1 protein expression was measured using f) Western blotting and g) immunofluorescence assays (n=4). Scale bars=100 µm. After shPink1 or shNC was transfected into NIH/3T3 fibroblasts for 24 h, cells were treated with TGF-β1 (10 ng·mL−1) and/or BMP4 (20 μM) for 48 h. h) Col1 and α-smooth muscle actin (SMA) protein expressions were examined with Western blot (n=4). i–k) The expression of α-SMA, fibronectin (FN) and Col1 was visualised using confocal microscopy (n=4). Scale bars=100 µm. GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Next, we determined the role of PINK1 in modulating the inhibitory effect of BMP4 on myofibroblast differentiation. When Pink1 was silenced by Pink1-specific shRNA (figure 6f and g), the inhibitory effect of BMP4 on myofibroblast differentiation were absent (figure 6h and i). In addition, we tested the effect of BMP4 on ECM production in lung fibroblasts with TGF-β1 stimulation and Pink1 silencing. When Pink1 was knocked down, the expressions of ECM proteins (Col1 and FN) (figure 6h, j and k) were no longer able to be decreased by BMP4. All of these results further suggest that antifibrotic effects of BMP4 are dependent on Pink1-mediated mitophagy.
Mitophagy is orchestrated by mitochondrial dynamics [36]. Therefore, we measured the expression of mitochondrial fusion and fission proteins, which regulate the mitochondrial dynamics. The results showed that TGF-β1 stimulation significantly reduced the expression of mitochondrial fusion proteins mitofusin (MFN)1 and MFN2, and increased the expression of mitochondrial fission proteins dynamin-related protein (DRP)1 and mitochondrial fission 1 protein (FIS1) in lung fibroblasts, which is consistent with a previous report [37]. Fibroblasts from BMP4+/− mice exhibited a more imbalanced mitochondrial dynamics compared to WT fibroblasts upon TGF-β1 stimulation (figure 6b, supplementary figure S9b). Conversely, treatment with BMP4 significantly reversed the TGF-β1-induced the imbalance of mitochondrial fusion and fission (figure 6c, supplementary figure S9c). Mitochondrial morphology was then visualised using MitoTracker Red staining. Exposure of lung fibroblasts to TGF-β1 resulted in increasing fragmented mitochondria (figure 6d), further indicating TGF-β1-induced mitochondrial fission in lung fibroblasts, whereas BMP4 haploinsufficiency caused more fragmented mitochondria, suggesting reduced BMP4 expression exacerbated TGF-β1-induced mitochondrial fission (figure 6d). These results suggest that BMP4 protects against TGF-β1-induced mitophagy by regulating mitochondrial dynamics.
BMP4 modulates cellular senescence and mitophagy in BLM-injured mice
We further assessed the role of BMP4 in cellular senescence and mitophagy during BLM-induced pulmonary fibrosis in mice. To confirm cellular senescence, real-time qPCR and Western blotting of p53, p21 and p16 were performed in both WT and BMP4+/− mice after BLM stimulation. Real-time qPCR and Western blotting using lung homogenates confirmed significant increases in p53, p21 and p16 mRNA and protein levels in BLM-injured BMP4+/− mouse lungs as compared to injured WT mice (figure 7a–c). Whereas the BLM-induced cellular senescence was attenuated by AAV9-mediated overexpression of BMP4 in the therapeutic lung fibrosis model, as demonstrated by real-time qPCR (figure 7g) and Western blot (figure 7f). In addition, immunofluorescence assay showed that pulmonary fibroblasts in BMP4-treated mice demonstrated less positive staining in both p21 and p16 (figure 7h). We also analysed cytokine secretion of the SASP including tumour necrosis factor (TNF)-α and interleukin (IL)-1β levels in lung homogenates. Consistent with the degree of cellular senescence, TNF-α mRNA levels were significantly increased in BLM-treated BMP4+/− mice as compared with their corresponding WT controls (figure 7d). A mild but significant increase in IL-1β was also observed in BLM-treated BMP4+/− mice (figure 7d).
FIGURE 7.

Bone morphogenetic protein (BMP)4 attenuates bleomycin (BLM)-induced cellular senescence, impaired mitophagy and oxidative stress in mouse lungs. a, b) Western blot analysis of p53, p21 and p16 in lung homogenates of BLM-challenged BMP4+/+ and BMP4+/− mice (n=6 per group). β-actin was used as a loading control. c, d) Real-time quantitative (q)PCR analysis of p53, p21, tumour necrosis factor (TNF)-α and interleukin (IL)-1β mRNA levels in the lungs of BLM-challenged BMP4+/+ and BMP4+/− mice (n=5 per group). e) Western blot analysis of Pink1, NADPH oxidase (NOX)4 and superoxide dismutase (SOD)2 in lung homogenates of BLM-challenged BMP4+/+ and BMP4+/− mice (n=6 per group). β-actin was used as a loading control. f) Western blot analysis of Pink1 and p21 protein expressions in lung homogenates of adeno-associated virus (AAV)9-BMP4- or AAV9-green fluorescent protein (GFP)-treated mice in the therapeutic lung fibrosis model (n=4 per group). β-actin was used as a loading control. g) Real-time qPCR analysis of p53 and p21 mRNA expressions in lung homogenates of BLM-challenged BMP4+/+ and BMP4+/− mice (n=7∼8 per group). h) Immunofluorescence analysis of α-smooth muscle actin (SMA) (green) and p21 (red), as well as α-SMA (green) and p16 (red) expressions in lung sections (nucleus, 4′,6-diamidino-2-phenylindole (DAPI); n=3 per group). Representative images of the staining are shown. Scale bars=50 µm. Data are presented as mean±sem. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
In accordance with the increase in cellular senescence in BLM-treated BMP4+/− mouse lungs, we further investigated the changes in oxidative stress, which is associated with degree of ageing. NADPH oxidase (NOX)4 is a main source of oxidative stress by enhancing ROS generation and superoxide dismutase (SOD)2 is one of the most important enzymes to reduce oxidative stress. Western blot showed higher NOX4 expression and lower SOD2 level in BMP4+/− lungs compared to WT lungs after BLM treatment, indicating increased lung oxidative stress in BMP4-deficient mice (figure 7e).
We investigated if mitophagy was recapitulated in vivo. Western blot analysis showed that Pink1 protein level was apparently decreased in the lungs within BMP4+/− mice compared to WT lungs after BLM exposure (figure 7e), whereas AAV9-mediated overexpression of BMP4 in the therapeutic lung fibrosis model showed an obvious increase of Pink1 protein level in comparing with the BLM-challenged mice as demonstrated by Western blot (figure 7f). These results suggest that Pink1 levels may also reflect accumulation of damaged mitochondrial due to insufficient mitophagy.
Correspondingly, the BLM-induced defective mitophagy and cellular senescence in vivo were also attenuated by AAV9-mediated overexpression of BMP4 in the prophylactic lung fibrosis mouse model (supplementary figure S10). All these findings support the notion that BMP4 promotes mitophagy and suppresses cellular senescence in BLM-treated lungs.
Discussion
IPF is a fatal ageing-related lung disease, closely related to the pathological accumulation of myofibroblasts that secrete ECM, resulting in scarring and destruction of lung architecture, respiration failure, and, ultimately, death [38]. Here, we demonstrate that BMP4 was significantly downregulated in the lungs of IPF patients and BLM-injured mice, and negatively correlated with fibrogenic genes. Furthermore, BMP4 was substantially repressed in primary cultures of human and mouse lung fibroblasts incubated with TGF-β1. BMP4 suppressed TGF-β1-induced fibroblast activation across species by activating Smad1/5/9 signalling, as well as inactivating Smad2/3 signalling. These beneficial effects were associated with the attenuation of insufficient mitophagy and cellular senescence-mediated myofibroblast differentiation, which is dependent on the Pink1 pathway. In addition, we studied the role of BMP4 signalling in experimental pulmonary fibrosis in mice. Haplodeletion of BMP4 enhanced BLM-induced lung fibrosis, whereas AAV9 transfer of BMP4 to mouse lung not only inhibited the development of lung fibrosis, but also blunted established pulmonary fibrosis. Therefore, BMP4 gene therapy may represent an effective treatment for pulmonary fibrosis.
BMP4, a ligand belonging to the TGF-β superfamily, is highly expressed in many tissues, and plays a crucial role in embryogenesis, embryonic development and homeostasis of the organs [20]. The previously published literature shows that BMP4 can cause damage, and promote inflammation and fibrosis across various organs. For instance, BMP4 increased in AngII-induced cardiomyocytes, and thus aggravated cardiomyocyte hypertrophy, apoptosis and cardiac fibrosis [39]. BMP4 expression was significantly increased in the liver of bile duct ligation rats and promoted hepatic stellate cell activation [40]. In asymptomatic smokers and COPD smokers, exaggerated BMP4 signalling promoted human airway basal progenitor cell differentiation to cigarette smoking-related phenotypes [41]. Conversely, Li et al. [27] showed that BMP4 inhibited liposaccharide-induced inflammation in the airway. Mizutani et al. [42] demonstrated that BMP4 mRNA levels continued to decrease at 12–24 h after incubation with TGF-β1 in human peritoneal mesothelial cells from the peritoneal fibrosis patients with high peritoneal transport, and its level was inversely correlated with dialysate-to-plasma ratio for creatinine, which suggests a protective effect of BMP4 for both peritoneal fibrosis and membrane function. BMP4 was also reported to reduce TGF-β2-induced epithelial–mesenchymal transition (EMT) and ECM production in optic nerve head cells, retinal pigment epithelial cells and lens epithelial cells [43–45]. Pegorier et al. [46] showed that BMP4 constrained TGF-β1-induced cellular proliferation in normal human lung fibroblasts, although its association with fibrosis was not investigated. Tan et al. [25] showed that BMP signalling partially mediates the epithelial antifibrotic effect. Probably as a result of this contradictory literature, there is no available evidence in support of the therapeutic effectiveness of BMP4 in IPF or lung fibrosis.
Our study provides several lines of evidence to demonstrate the potential of BMP4 gene therapy as an antifibrotic therapy. This agrees with recent findings which show the profibrotic role of BMPs antagonists such as follistatin-like 1 and Gremlin in pulmonary fibrogenesis [22, 23]. TGF-β1 is the best characterised and most powerful cellular transformer of fibroblasts, and a variety of pathways can regulate its function [47]. We show that BMP4 haploinsufficiency aggravated the TGF-β1-induced myofibroblast differentiation and ECM production in adult IPF and mouse lung fibroblasts, whereas administration of exogenous recombinant BMP4 attenuated these processes. This appears to contradict the effect of BMP4 identified in a previous study showing that BMP4 suppressed proliferation and promoted myocyte differentiation of human fetal lung fibroblasts through Smad1 and JNK pathways [48]. These contradictory results may be explained by the fact that BMP4 can affect myofibroblast differentiation differently according to cell type, age and stress levels, which indicates that metabolic capacity determines the cellular vulnerability to BMP4 deficiency in cells. However, our findings are consistent with recent results that show that BMP4 inhibited TGF-β1-induced vascular smooth muscle cell differentiation and phenotypic changes. Moreover, myofibroblasts are not terminally differentiated cells and myofibroblast dedifferentiation may be critical for normal regenerative versus fibrotic responses to tissue injury [8, 49]. In addition to inducing myofibroblast differentiation [17, 50, 51], senescence also promotes profibrotic effects via impaired myofibroblast dedifferentiation [8]. Here, we found that platelet-derived growth factor (PDGF), a specific mitogenic growth factor, induced a downregulation of α-SMA, consistent with myofibroblast dedifferentiation, as reported by Hecker et al. [49]. Interestingly, the effects on PDGF-induced α-SMA suppression were attenuated by BMP4 haploinsufficiency, but enhanced by BMP4 addition, indicating that BMP4 promotes myofibroblast dedifferentiation (supplementary figure S11).
In addition, this study provided novel evidence of the interaction between the BMP4 and TGF-β1 pathways, which induces the fibroblast-to-myofibroblast differentiation. Both BMP4 and TGF-β1 bind to distinct type II serine-threonine kinase receptors, which form a complex with specific type I receptors. The activated receptor complex then transduces signals through phosphorylating Smad2/3 in TGF-β1 signalling and Smad1/5/8 in BMP4 signalling [52]. In a study by Chen et al. [53], reduced p-Smad1/5/8 and elevated p-Smad2/3 levels were observed in the lungs of IPF patients, suggesting that reduced BMP signalling and enhanced TGF-β activity may play critical roles to facilitate the disease development of IPF. Consistently, by using the BLM model of pulmonary fibrosis, Jiang et al. [54] reported that BMP9/BMPR2/Smad1/5/9 signalling was significantly downregulated in BLM-treated lungs and pulmonary microvascular endothelial cells, which contributes to the development of pulmonary fibrosis. In this study, we found a similar reduced levels of p-smad1/5/9 and elevated levels of p-Smad2/3 in the BLM-treated lungs, which were associated with decreased BMP4 expression and augmented TGF-β1 signalling induced by BLM. However, these changes were further augmented by BMP4 haploinsufficiency, but attenuated by BMP4 supplementation. This finding was also validated in TGF-β1-treated cultures of lung fibroblasts, evidenced by decreased level of p-Smad2/3 and increased level of p-Smad1/5/9 in BMP4-treated cells. Our observation is consistent with previous findings that BMP4 inhibited TGF-β1-induced Smad2/3 phosphorylation by activating Smad1/5/9 in retinal pigment epithelium cells [45]. Further study is required to investigate whether BMP4 affects other signalling pathways, including Wnt/β-catenin signalling, involved in the fibroblast-to-myofibroblast transitions.
Cellular senescence, an evolutionarily conserved state of stable replicative arrest, has been proposed as an important theme in IPF and other fibrotic disorders [17]. Cellular senescence, another means of responding to long-term cellular stress, is linked to myofibroblast differentiation and fibrosis [14]. Previous reports have highlighted that the metabolically active, hypersecretory and apoptosis-resistant senescence phenotype of lung fibroblasts are plentifully observed in the lungs of patients with IPF and promotes the release of a variety of fibrotic growth factors, such as TGF-β1 [38, 55]. In this study, TGF-β1 causes cellular senescence in lung fibroblasts, as outlined previously [18, 47]. The downregulation of BMP4 expression promoted the lung fibroblast senescence induced by TGF-β1, and strongly influenced the secretory profile of these fibroblasts. Our results identified that TGF-β1-stimulated lung fibroblasts could secrete multiple SASP factors, which were further enhanced by BMP4 knockdown. Inversely, BMP4 supplementation in lung fibroblasts reduces the expression of senescence markers p16 and p53, consistent with prior evidence that BMP-SMAD-ID signalling axis repressed p16-mediated cell senescence during induced pluripotent stem cell generation [56]. Similar results were observed in BLM-injured BMP4+/− mouse lungs, wherein p16, p21 and p53 senescence biomarker levels were increased. Correspondingly, BMP4 gene therapy in mice with pulmonary fibrosis leads to a significant decrease of senescence-associated protein levels. These findings are concordant with recent publications that BLM induces an obvious molecular signature of senescence in the lung and delivery of rapamycin, a SASP inhibitor, attenuates fibroblast activation and fibrosis in mice following BLM exposure [57, 58]. The suppression of senescence could be explained by the inactivation of Smad2/3 by BMP4, as previous reports have indicated that Smad2/3 acts as a driver of cell senescence in IPF [47]. Since p16 was positively associated with TGF-β1/Smad2/3 and negatively associated with Smad1/5/8 [59], we believe that this beneficial effect of BMP4 might also be correlated to the activation of Smad1/5/9 caused by BMP4.
Autophagy and cellular senescence tend to occur in parallel; moreover, they are believed to reciprocally regulate each other [60]. It has been shown that autophagy correlates with reduced senescence; insufficient autophagy plays a critical role in the accumulation of deleterious cellular components and, therefore, accelerates the development of the senescent phenotype [61]. Among the various targets for autophagic degradation, mitophagy has been widely implicated in cellular senescence in accordance with regulation of ROS; hence, autophagy and mitophagy are required to maintain normal cell function [60]. Mitophagy defects in IPF lung fibroblasts is linked to enhanced deposition of ECM under profibrotic conditions, e.g. exposure to TGF-β1. Insufficient mitophagy and autophagy also cause excessive production of ROS that induce acceleration of fibroblast senescence and myofibroblast differentiation [13, 50], whereas induction of autophagy and treatment with antioxidants reduced fibroblast to myofibroblast differentiation and fibrogenesis, suggesting that this process is driven by increased ROS [62]. In agreement with these studies, we have illustrated that primary lung fibroblasts exposed to TGF-β1 exhibited myofibroblast-like characteristics owing to diminished mitophagy and excessive ROS generation, which is in accordance with previous reports [13, 62]. BMP4 treatment attenuated the TGF-β1-induced mitophagy defects and excessive production of intracellular and mitochondrial ROS. This is apparently consistent with previous observations that BMP4 promotes autophagy activation in hepatocellular carcinoma, myeloid leukaemia cells, HeLa, MCF-7 cells, and some other cell types [63–66]. Moreover, similar results were observed in the BMP4 knockdown model, as well as in mice with AAV9-BMP4. These findings indicate that BMP4 is crucial to initiate mitophagy and improve oxidative stress status. The enhancement of mitophagy and autophagy by BMP4 is also significant because impeding Smad2/3 transcriptional repressor activity promotes autophagy, and activating autophagy can also suppress TGF-β1 induced Smad2/3 signalling transmission [67].
Dual staining with the epithelial marker E-cadherin, the myofibroblast marker α-SMA and the endothelial cell marker CD31 demonstrated the widespread expression of BMP4 in the mouse lung (supplementary figure S12), which is consistent with prior reports of BMP4 expression in the lung epithelial cells, microvasculature and stromal cells [68]. Additionally, the mRNA levels of BMP4 were also decreased in isolated primary alveolar type II (ATII) epithelial cells (supplementary figure S13a) or lung fibroblasts (figure 1h, i) from BLM-treated mouse lungs, as compared to those from control lungs. No significant changes were observed in endothelial cells (supplementary figure S13b). Although our study selectively focused on the beneficial effects of BMP4 on lung fibroblasts, we also directly assessed its role in pulmonary epithelial cells (e.g. A549, Beas-2B and primary ATII cells). It has been reported that, in addition to the activation of lung-resident fibroblasts, the origins of the invasive lung myofibroblasts also include EMT of lung epithelial cells [47, 69]. Here, we found that BMP4 inhibited EMT in all aforementioned cell types (supplementary figures S14a–i, l–p, and S15a–f). In addition to EMT, senescence of these lung epithelial cells is also a central phenotype that promotes lung fibrosis. Interestingly, the TGF-β1-induced senescence of A549 cells (supplementary figure S14j and k), Beas-2B cells (supplementary figure S15g and h) and primary ATII cells (supplementary figure S14q) was also attenuated by BMP4.
Given that no cure is currently available for IPF, multiple clinical trials have been carried out to seek possible drugs [70]. For this reason, properties of a particular drug including the appropriate method, times or doses of administration, the concentrations in the lung, and potential side-effects on other organs and tissues should be prioritised. Drug administration delivering directly to the lung through tracheal inhalation rather than systemic delivery could be a better approach for IPF patients. AAV9 vectors are established drug carrier for inhalation due to their safety, their high viral transduction of the lungs and their ability to provide superior duration profiles [29, 30]. These carriers can make various therapeutic targets for delivery to the lung using spray technologies which avoid alveolar exposure and adverse effects [71, 72]. For instance, a unique delivery system with AAV vectors through aerosolised administration of the cystic fibrosis transmembrane conductance regulator has been used to improve pulmonary function in patients with cystic fibrosis [71, 73]. Similarly, our studies showed that AAV9 vectors carrying the BMP4 gene could be efficiently overexpressed in primary ATII cells (supplementary figure S16a) and lung fibroblasts (supplementary figure S16b) following intratracheal injection. The AAV9-based overexpression of BMP4 lasted for ≥6 weeks, as much as 1 year. As a result, intratracheal delivery of AAV9-mediated overexpression of BMP4 protected mice from BLM-induced lung fibrosis. The future of clinical trials focused on AAV9 aerosol-mediated BMP4 gene transfer to the lungs of patients with lung fibrosis deserve further evaluation.
Additionally, another important implication of our work is the need to identify therapeutic strategies that increase BMP4 expression and function in IPF fibroblasts. Further discovery and development of treatment strategies to restore BMP4 expression in IPF may be beneficial in the treatment of lung fibrosis. Nowadays, SB-4 (Eticovo), a selective agonist of the BMP4, has already been approved by health authorities in Europe, Australia, South Korea and Canada, as a subcutaneous therapy option for the treatment of patients with rheumatoid arthritis, but also for the full spectrum approved for its bio-originator etanercept [74]. Since rheumatoid arthritis is a risk factor for development of pulmonary fibrosis, our data support the possibility that SB-4 might be a viable therapy against IPF in clinical settings.
Despite the significant merits described, there are also a number of limitations to our study. For instance, we did not decipher the underlying mechanisms of BMP4 downregulation, and the detailed signalling of BMP4 in regulating senescence and mitophagy in fibroblasts, and other factors, such as inflammatory response and endoplasmic reticulum stress which are involved in myofibroblast accumulation in response to BMP4 treatment. Further studies using cell-type-specific, conditional knockout mice are also warranted to illustrate the potentially cell-type-specific effects of BMP4 in fibrotic lung.
Collectively, our results showed that BMP4 strongly attenuated BLM-induced lung fibrosis in mice and inhibited the differentiation of fibroblasts to myofibroblasts. These antifibrotic effects are probably due to the inhibition of cellular senescence and the enhancement of mitophagy by BMP4 in fibroblasts (figure 8). Hence, the findings described here support the therapeutic potential of BMP4 for clinical treatment of pulmonary fibrosis.
FIGURE 8.

Schematic diagram of the study. Bone morphogenetic protein (BMP)4 ameliorates lung fibrosis by antagonising transforming growth factor (TGF)-β1 signalling, which then attenuates cellular senescence and impaired mitophagy of lung fibroblasts, leading to reduced myofibroblast differentiation and extracellular matrix production. BLM: bleomycin; NOX: NADPH oxidase; mtROS: mitochondrial reactive oxygen species; SASP: senescence-associated secretory phenotype; IL: interleukin; MMP: matrix metalloproteinase; SA-β-gal: senescence-associated β-galactosidase.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02307-2021.Supplement (2.1MB, pdf)
Supplementary video S1 (Controls) ERJ-02307-2021.Video_S1 (2.3MB, mp4)
Supplementary video S2 (BLM+AAV9-GFP) ERJ-02307-2021.Video_S2 (2.3MB, mp4)
Supplementary video S3 (BLM+AAV9-BMP4) ERJ-02307-2021.Video_S3 (2.3MB, mp4)
Shareable PDF
Footnotes
Author contributions: Wenju Lu, Jianxing He, Ruijuan Guan and Jian Wang conceived the project and designed experiments. Ruijuan Guan, Jian Wang and Jingpei Li wrote the manuscript. Wenju Lu, Jingpei Li, Jian Wang, Kai Yang, Dejun Sun and Hongwei Yao edited the manuscript. Jingpei Li collected human lung samples. Ruijuan Guan, Liang Yuan, Ziying Li, Yaowei Fang, Qiuyu Zheng, Lan Wang, Hua Guo, Zhou Cai, Mingjing Ding, Jing Qian, Jingyi Xu, Ran Lin, Jieping Luo and Wei Liu performed experiments. Ruijuan Guan, You Zhou and Yuanyuan Li conducted data analysis. All authors have read and approved the final manuscript.
This article has an editorial commentary: https://doi.org/10.1183/13993003.01702-2022
Conflict of interest: The authors have nothing to disclose.
Support statement: This work was supported by grants from the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01S155), National Natural Science Foundation of China (81770043, 81520108001, 81800072 and 81703792), the National Key R&D Program of China (2016YFC0903700), Natural Science Foundation of Guangdong Province (2018A030310291 and 2016A030311020), Guangzhou Municipal Science and Technology grants (201804010052 and 201607020030), China Postdoctoral Science Foundation (2017M612637 and 2018T110860), Changjiang Scholars and Innovative Research Team in University (grant IRT0961) and the Research Projects of SKLRD (QN-201706, OP-201808, OP-201919). Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Sakai N, Tager AM. Fibrosis of two: epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta 2013; 1832: 911–921. doi: 10.1016/j.bbadis.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu G, Tzouvelekis A, Wang R, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med 2018; 24: 39–49. doi: 10.1038/nm.4447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crystal RG, Bitterman PB, Mossman B, et al. Future research directions in idiopathic pulmonary fibrosis: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med 2002; 166: 236–246. doi: 10.1164/rccm.2201069 [DOI] [PubMed] [Google Scholar]
- 4.Hewitt RJ, Maher TM. Idiopathic pulmonary fibrosis: new and emerging treatment options. Drugs Aging 2019; 36: 485–492. doi: 10.1007/s40266-019-00647-y [DOI] [PubMed] [Google Scholar]
- 5.Somogyi V, Chaudhuri N, Torrisi SE, et al. The therapy of idiopathic pulmonary fibrosis: what is next? Eur Respir Rev 2019; 28: 190021. doi: 10.1183/16000617.0021-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet 2017; 389: 1941–1952. doi: 10.1016/S0140-6736(17)30866-8 [DOI] [PubMed] [Google Scholar]
- 7.Zhao H, Bian H, Bu X, et al. Targeting of discoidin domain receptor 2 (DDR2) prevents myofibroblast activation and neovessel formation during pulmonary fibrosis. Mol Ther 2016; 24: 1734–1744. doi: 10.1038/mt.2016.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato K, Logsdon NJ, Shin YJ, et al. Impaired myofibroblast dedifferentiation contributes to nonresolving fibrosis in aging. Am J Respir Cell Mol Biol 2020; 62: 633–644. doi: 10.1165/rcmb.2019-0092OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB J 2004; 18: 816–827. doi: 10.1096/fj.03-1273rev [DOI] [PubMed] [Google Scholar]
- 10.Zehender A, Huang J, Györfi AH, et al. The tyrosine phosphatase SHP2 controls TGFβ-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat Commun 2018; 9: 3259. doi: 10.1038/s41467-018-05768-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YL, Zhang X, Bai J, et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: potential roles in the inhibition of epithelial-mesenchymal transition and fibroblast activation. Cell Death Dis 2013; 4: e665. doi: 10.1038/cddis.2013.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rafii R, Juarez MM, Albertson TE, et al. A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis 2013; 5: 48–73. doi: 10.3978/j.issn.2072-1439.2012.12.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi K, Araya J, Minagawa S, et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol 2016; 197: 504–516. doi: 10.4049/jimmunol.1600265 [DOI] [PubMed] [Google Scholar]
- 14.Bernard M, Yang B, Migneault F, et al. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 2020; 16: 2004–2016. doi: 10.1080/15548627.2020.1713640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araya J, Kojima J, Takasaka N, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2013; 304: L56–L69. doi: 10.1152/ajplung.00213.2012 [DOI] [PubMed] [Google Scholar]
- 16.Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med 2019; 200: 556–564. doi: 10.1164/rccm.201810-1975TR [DOI] [PubMed] [Google Scholar]
- 17.Schafer MJ, White TA, Iijima K, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017; 8: 14532. doi: 10.1038/ncomms14532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Álvarez D, Cárdenes N, Sellarés J, et al. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 2017; 313: L1164–L1173. doi: 10.1152/ajplung.00220.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15: 482–496. doi: 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 20.Bragdon B, Moseychuk O, Saldanha S, et al. Bone morphogenetic proteins: a critical review. Cell Signal 2011; 23: 609–620. doi: 10.1016/j.cellsig.2010.10.003 [DOI] [PubMed] [Google Scholar]
- 21.Koli K, Myllärniemi M, Vuorinen K, et al. Bone morphogenetic protein-4 inhibitor gremlin is overexpressed in idiopathic pulmonary fibrosis. Am J Pathol 2006; 169: 61–71. doi: 10.2353/ajpath.2006.051263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong Y, Geng Y, Li L, et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med 2015; 212: 235–252. doi: 10.1084/jem.20121878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farkas L, Farkas D, Gauldie J, et al. Transient overexpression of Gremlin results in epithelial activation and reversible fibrosis in rat lungs. Am J Respir Cell Mol Biol 2011; 44: 870–878. doi: 10.1165/rcmb.2010-0070OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Fang Y, Jiang D, et al. Targeting FSTL1 for multiple fibrotic and systemic autoimmune diseases. Mol Ther 2021; 29: 347–364. doi: 10.1016/j.ymthe.2020.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan Q, Ma XY, Liu W, et al. Nascent lung organoids reveal epithelium- and bone morphogenetic protein-mediated suppression of fibroblast activation. Am J Respir Cell Mol Biol 2019; 61: 607–619. doi: 10.1165/rcmb.2018-0390OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moeller A, Ask K, Warburton D, et al. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol 2008; 40: 362–382. doi: 10.1016/j.biocel.2007.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Wang J, Wang Y, et al. Bone morphogenetic protein 4 inhibits liposaccharide-induced inflammation in the airway. Eur J Immunol 2014; 44: 3283–3294. doi: 10.1002/eji.201344287 [DOI] [PubMed] [Google Scholar]
- 28.Wang D, Tai P, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 2019; 18: 358–378. doi: 10.1038/s41573-019-0012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell CL, Vandenberghe LH, Bell P, et al. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J Clin Invest 2011; 121: 2427–2435. doi: 10.1172/JCI57367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zincarelli C, Soltys S, Rengo G, et al. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 2008; 16: 1073–1080. doi: 10.1038/mt.2008.76 [DOI] [PubMed] [Google Scholar]
- 31.Povedano JM, Martinez P, Serrano R, et al. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife 2018; 7: e31299. doi: 10.7554/eLife.31299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuwano K, Kunitake R, Kawasaki M, et al. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1996; 154: 477–483. doi: 10.1164/ajrccm.154.2.8756825 [DOI] [PubMed] [Google Scholar]
- 33.Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann Am Thorac Soc 2016; 13: Suppl. 5, S417–S421. doi: 10.1513/AnnalsATS.201605-341AW [DOI] [PubMed] [Google Scholar]
- 34.van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509: 439–446. doi: 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuliga M, Pechkovsky DV, Read J, et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J Cell Mol Med 2018; 22: 5847–5861. doi: 10.1111/jcmm.13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu B, Ma J, Li J, et al. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat Commun 2020; 11: 2549. doi: 10.1038/s41467-020-16312-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larson-Casey JL, He C, Carter AB. Mitochondrial quality control in pulmonary fibrosis. Redox Biol 2020; 33: 101426. doi: 10.1016/j.redox.2020.101426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporarello N, Meridew JA, Jones DL, et al. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019; 74: 749–760. doi: 10.1136/thoraxjnl-2019-213064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun B, Huo R, Sheng Y, et al. Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension 2013; 61: 352–360. doi: 10.1161/HYPERTENSIONAHA.111.00562 [DOI] [PubMed] [Google Scholar]
- 40.Fan J, Shen H, Sun Y, et al. Bone morphogenetic protein 4 mediates bile duct ligation induced liver fibrosis through activation of Smad1 and ERK1/2 in rat hepatic stellate cells. J Cell Physiol 2006; 207: 499–505. doi: 10.1002/jcp.20593 [DOI] [PubMed] [Google Scholar]
- 41.Zuo WL, Yang J, Strulovici-Barel Y, et al. Exaggerated BMP4 signalling alters human airway basal progenitor cell differentiation to cigarette smoking-related phenotypes. Eur Respir J 2019; 53: 1702553. doi: 10.1183/13993003.02553-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizutani M, Ito Y, Mizuno M, et al. Connective tissue growth factor (CTGF/CCN2) is increased in peritoneal dialysis patients with high peritoneal solute transport rate. Am J Physiol Renal Physiol 2010; 298: F721–F733. doi: 10.1152/ajprenal.00368.2009 [DOI] [PubMed] [Google Scholar]
- 43.Zode GS, Clark AF, Wordinger RJ. Bone morphogenetic protein 4 inhibits TGF-β2 stimulation of extracellular matrix proteins in optic nerve head cells: role of gremlin in ECM modulation. Glia 2009; 57: 755–766. doi: 10.1002/glia.20803 [DOI] [PubMed] [Google Scholar]
- 44.Shu DY, Ng K, Wishart T, et al. Contrasting roles for BMP-4 and ventromorphins (BMP agonists) in TGFβ-induced lens EMT. Exp Eye Res 2021; 206: 108546. doi: 10.1016/j.exer.2021.108546 [DOI] [PubMed] [Google Scholar]
- 45.Yao H, Li H, Yang S, et al. Inhibitory effect of bone morphogenetic protein 4 in retinal pigment epithelial-mesenchymal transition. Sci Rep 2016; 6: 32182. doi: 10.1038/srep32182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pegorier S, Campbell GA, Kay AB, et al. Bone morphogenetic protein (BMP)-4 and BMP-7 regulate differentially transforming growth factor (TGF)-β1 in normal human lung fibroblasts (NHLF). Respir Res 2010; 11: 85. doi: 10.1186/1465-9921-11-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milara J, Ballester B, Montero P, et al. MUC1 intracellular bioactivation mediates lung fibrosis. Thorax 2020; 75: 132–142. doi: 10.1136/thoraxjnl-2018-212735 [DOI] [PubMed] [Google Scholar]
- 48.Jeffery TK, Upton PD, Trembath RC, et al. BMP4 inhibits proliferation and promotes myocyte differentiation of lung fibroblasts via Smad1 and JNK pathways. Am J Physiol Lung Cell Mol Physiol 2005; 288: L370–L378. doi: 10.1152/ajplung.00242.2004 [DOI] [PubMed] [Google Scholar]
- 49.Hecker L, Jagirdar R, Jin T, et al. Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res 2011; 317: 1914–1921. doi: 10.1016/j.yexcr.2011.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zank DC, Bueno M, Mora AL, et al. Idiopathic pulmonary fibrosis: aging, mitochondrial dysfunction, and cellular bioenergetics. Front Med 2018; 5: 10. doi: 10.3389/fmed.2018.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Razdan N, Vasilopoulos T, Herbig U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 2018; 17: e12838. doi: 10.1111/acel.12838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem 2010; 147: 35–51. doi: 10.1093/jb/mvp148 [DOI] [PubMed] [Google Scholar]
- 53.Chen NY, Collum SD, Luo F, et al. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and group III pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2016; 311: L238–L254. doi: 10.1152/ajplung.00142.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang Q, Liu C, Liu S, et al. Dysregulation of BMP9/BMPR2/SMAD signalling pathway contributes to pulmonary fibrosis and pulmonary hypertension induced by bleomycin in rats. Br J Pharmacol 2021; 178: 203–216. doi: 10.1111/bph.15285 [DOI] [PubMed] [Google Scholar]
- 55.Waters DW, Blokland K, Pathinayake PS, et al. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2018; 315: L162–L172. doi: 10.1152/ajplung.00037.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayashi Y, Hsiao EC, Sami S, et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc Natl Acad Sci USA 2016; 113: 13057–13062. doi: 10.1073/pnas.1603668113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aoshiba K, Tsuji T, Kameyama S, et al. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Exp Toxicol Pathol 2013; 65: 1053–1062. doi: 10.1016/j.etp.2013.04.001 [DOI] [PubMed] [Google Scholar]
- 58.Hecker L, Logsdon NJ, Kurundkar D, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 2014; 6: 231ra47. doi: 10.1126/scitranslmed.3008182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tasanarong A, Kongkham S, Khositseth S. Dual inhibiting senescence and epithelial-to-mesenchymal transition by erythropoietin preserve tubular epithelial cell regeneration and ameliorate renal fibrosis in unilateral ureteral obstruction. Biomed Res Int 2013; 2013: 308130. doi: 10.1155/2013/308130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuwano K, Araya J, Hara H, et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig 2016; 54: 397–406. doi: 10.1016/j.resinv.2016.03.010 [DOI] [PubMed] [Google Scholar]
- 61.Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev 2009; 8: 199–213. doi: 10.1016/j.arr.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 62.Sosulski ML, Gongora R, Danchuk S, et al. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFβ1. Aging Cell 2015; 14: 774–783. doi: 10.1111/acel.12357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Gao L, Zheng L, et al. BMP4-mediated autophagy is involved in the metastasis of hepatocellular carcinoma via JNK/Beclin1 signaling. Am J Transl Res 2020; 12: 3068–3077. [PMC free article] [PubMed] [Google Scholar]
- 64.Deng G, Zeng S, Qu Y, et al. BMP4 promotes hepatocellular carcinoma proliferation by autophagy activation through JNK1-mediated Bcl-2 phosphorylation. J Exp Clin Cancer Res 2018; 37: 156. doi: 10.1186/s13046-018-0828-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao X, Liu J, Peng M, et al. BMP4 is involved in the chemoresistance of myeloid leukemia cells through regulating autophagy-apoptosis balance. Cancer Invest 2013; 31: 555–562. doi: 10.3109/07357907.2013.834925 [DOI] [PubMed] [Google Scholar]
- 66.Sheng Y, Sun B, Guo WT, et al. (4-[6-(4-isopropoxyphenyl)pyrazolo [1,5-a]pyrimidin-3-yl] quinoline) is a novel inhibitor of autophagy. Br J Pharmacol 2014; 171: 4970–4980. doi: 10.1111/bph.12821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kong D, Zhang Z, Chen L, et al. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol 2020; 36: 101600. doi: 10.1016/j.redox.2020.101600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dyer LA, Pi X, Patterson C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol Metab 2014; 25: 472–480. doi: 10.1016/j.tem.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Milara J, Ballester B, Safont MJ, et al. MUC4 is overexpressed in idiopathic pulmonary fibrosis and collaborates with transforming growth factor β inducing fibrotic responses. Mucosal Immunol 2021; 14: 377–388. doi: 10.1038/s41385-020-00343-w [DOI] [PubMed] [Google Scholar]
- 70.Kolb P, Upagupta C, Vierhout M, et al. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur Respir J 2020; 55: 1901105. doi: 10.1183/13993003.01105-2019 [DOI] [PubMed] [Google Scholar]
- 71.Moss RB, Rodman D, Spencer LT, et al. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest 2004; 125: 509–521. doi: 10.1378/chest.125.2.509 [DOI] [PubMed] [Google Scholar]
- 72.Flotte TR, Laube BL. Gene therapy in cystic fibrosis. Chest 2001; 120: Suppl. 3, 124S–131S. doi: 10.1378/chest.120.3_suppl.124S [DOI] [PubMed] [Google Scholar]
- 73.Leung K, Louca E, Munson K, et al. Calculating expected lung deposition of aerosolized administration of AAV vector in human clinical studies. J Gene Med 2007; 9: 10–21. doi: 10.1002/jgm.987 [DOI] [PubMed] [Google Scholar]
- 74.Pelechas E, Drosos AA. Etanercept biosimilar SB-4. Expert Opin Biol Ther 2019; 19: 173–179. doi: 10.1080/14712598.2019.1566456 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02307-2021.Supplement (2.1MB, pdf)
Supplementary video S1 (Controls) ERJ-02307-2021.Video_S1 (2.3MB, mp4)
Supplementary video S2 (BLM+AAV9-GFP) ERJ-02307-2021.Video_S2 (2.3MB, mp4)
Supplementary video S3 (BLM+AAV9-BMP4) ERJ-02307-2021.Video_S3 (2.3MB, mp4)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02307-2021.Shareable (236.6KB, pdf)