

Abstract
Elucidating the earliest pathogenic steps in cancer development is fundamental to improving its early detection and prevention. Ovarian high-grade serous carcinoma, a highly aggressive cancer, originates from the fallopian tube epithelium through a precursor stage, serous tubal intraepithelial carcinoma (STIC). In this study, we performed spatial transcriptomic analysis to compare STICs, carcinoma, and their matched normal fallopian tube. Several differentially expressed genes in STICs and carcinomas were involved in cancer metabolism and detected in a larger independent transcriptomic dataset of ovarian high-grade serous carcinomas. Among these, insulin-like growth factor binding protein-2 (IGFBP2) was found to undergo DNA hypomethylation and be increased at the protein level in STICs. Pyrosequencing revealed an association of IGFBP2 expression with the methylation state of its proximal enhancer, and 5-azacytidine treatment increased IGFBP2 expression. In postmenopausal fallopian tubes where most STICs are detected, IGFBP2 immunoreactivity was detected in all 38 proliferatively active STICs but was undetectable in morphologically normal tubal epithelia, including those with TP53 mutations. In premenopausal fallopian tubes, IGFBP2 expression was limited to the secretory epithelium at the proliferative phase, and estradiol treatment increased IGFBP2 expression levels. IGFBP2 knockdown suppressed the growth of IGFBP2-expressing tubal epithelial cells via inactivation of the AKT pathway. Taken together, demethylation of the proximal enhancer of IGFBP2 drives tumor development by maintaining the increased IGFBP2 required for proliferation in an otherwise estrogen-deprived, proliferation-quiescent, and postmenopausal tubal microenvironment.
Keywords: IGFBP2, serous tubal intraepithelial carcinoma, STIC, high-grade ovarian serous carcinoma, tumor initiation, transcriptome, estradiol, methylation, biomarkers and intervention
Introduction
Elucidating pathogenic steps in early cancer development is fundamental to identifying biomarkers for early detection and to exploring effective strategies for cancer prevention. Both tasks represent an unmet need in cancers that are usually not amenable to routine screening and primary prevention. Ovarian cancer is one such example – the malignancy is located deep in the pelvis, not readily detected clinically until late-stage disease, and clinically aggressive. Currently, there are few effective approaches for intercepting its progression from a non-invasive precursor stage to an advanced incurable stage.
Ovarian cancer is unique among human cancers because it is unlikely to develop de novo. As supported by a wealth of epidemiologic, clinical, pathological, and molecular studies, the emerging paradigm in the genesis of high-grade serous carcinoma (HGSC), the most common ovarian cancer, is that HGSC initially arises in fallopian tubes through precursor lesions that then progress to disseminated disease involving adjacent ovaries, peritoneal soft tissues, and lymph nodes (1,2). TP53 mutation is the earliest known molecular genetic change, and its missense or nonsense mutations occur in almost all precursor lesions and HGSCs (3–5). TP53 mutation is apparently insufficient to drive malignant transformation as evidenced by the presence of a TP53 mutated lesion in the fallopian tube, termed “p53 signature,” containing a minute stretch of clonally derived, morphologically normal-appearing tubal epithelium (about 12 to 50 cells) histologically indistinguishable from the background of TP53 wild-type epithelium. Most of those p53 signature lesions are not proliferative and may remain clinically indolent, most likely representing an evolutionary dead-end.
However, some TP53 mutated clones may continue proliferating and accumulate chromosomal alterations and additional sequence mutations, leading to genetic heterogeneity that provides a platform for selection and evolution to HGSC. Those lesions are collectively known as serous tubal intraepithelial carcinoma (STIC) and are histologically characterized by highly atypical cells with enlarged nuclei, mitosis, and apoptosis manifesting as an in situ lesion of HGSC. Since STIC and p53 signatures are minuscule and not grossly visible, these lesions can only be microscopically diagnosed in formalin-fixed and paraffin-embedded tissue sections.
Based on a mathematical model and a clinical correlation study, a STIC may take an extended time, often decades, to develop after the first hit, TP53 mutation, but a shorter time to progress (approximately 6–7 years) to HGSC (5,6). Genetic predisposition including mutations in BRCA1 and BRCA2 accelerates the progression of STIC to HGSC. In a multi-center study after extensively examining surgically removed fallopian tubes, STIC and p53 signatures were detected, respectively, in 10% and 27% of fallopian tubes from BRCA1/2 carriers undergoing prophylactic salpingo-oophorectomy (7).
To better understand the mechanisms contributing to tumor initiation in HGSC, researchers have compared molecular alterations among STICs, p53 signatures, and matched normal-appearing fallopian tube epithelium to depict the genetic and epigenetic landscape in tumor initiation (5,8,9). Moreover, STIC also expressed several HGSC protein biomarkers (10–16). Despite this progress, the genome-wide transcriptomic alterations in STICs remain elusive, mainly due to challenges in performing transcriptomic assays in such miniscule lesions in formalin-fixed and paraffin-embedded tissues. Here, we report spatial profiles of transcriptomes in STICs, corresponding normal fallopian tube epithelium and stroma, and associated HGSCs. The data reveal the earliest changes in gene expression during the development of HGSC and identify a novel role of Insulin-like Growth Factor Binding Protein-2 (IGFBP2) in initiating HGSC.
Materials and Methods
Tissue acquisition and preparation
All formalin-fixed and paraffin-embedded tissues were retrieved from the pathology archival files and the gynecologic tissue repository at the Johns Hopkins Medical Institutions and the Inova Fairfax Hospital, Fairfax, Virginia. This study was conducted in accordance with the ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and was approved by the local institutional review board. Written informed consent for collection of tissue for research was obtained from all study participants and all samples were de-identified. We used morphological criteria assisted by immunohistochemistry to diagnose STICs and p53 signatures as previously reported (3). A total of 76 precursor lesions including 15 p53 signatures and 61 STICs were available for this study. Among them, 59 (77.63%) were incidental findings without concurrent ovarian HGSC. For each lesion, normal-appearing tubal epithelium was available for study. After serial sectioning, 13 STICs with sufficient STIC cells remained and were available for transcriptomic profiling, while the remaining cases were used for immunohistochemistry. We also included three HGSC tissues (from two patients) with concurrent STIC where the HGSC involvement of fallopian tubes was minimal.
Small segments of fallopian tubes from premenopausal women undergoing salpingo-oophorectomy for benign indications were analyzed and the segments were comprised of the remaining tissue following routine sampling for pathologic diagnosis. Portions of the tubes were sectioned, opened, and incubated with MEBM (Lonza, CC3153) with/without 50 nM estradiol (E2) overnight. After incubation, the tissue segments were fixed in 10% neutral buffered formalin and embedded in paraffin for immunohistochemistry. The rest of the fallopian tube was opened and incubated with trypsin at 37°C for 15 minutes followed by neutralization with RPMI-1640, 10% FBS. The epithelium was harvested by gently scraping mucosal surface using a cell scraper. The cells were passed through 100 μm cell strainers to remove tissue debris then were suspended in soft gel with or without 50 nM E2 to grow spheroids.
Spatial Transcriptomic Profiling
The GeoMx Human Whole Transcriptome assay (Nanostring, Inc., RRID: SCR_021660) was employed to measure mRNA expression levels on formalin-fixed and paraffin-embedded tissues. This assay was based on using a DNA barcoding technology for mRNA detection via a UV photocleavable linker. Unstained slides (5 μm) from 13 STIC lesions, 3 HGSCs, and their matched normal-appearing tubal tissues. Individual tissue sections were labeled with a cocktail of probes and 3 fluorescently labeled morphology markers: a pan-cytokeratin antibody with a green fluorescent dye, a p53 antibody with a red fluorescent dye, and DAPI to detect nuclei. Areas of interest (AOI) were selected based on fluorescence imaging. In addition to epithelium, we also selected stroma immediately underlying the lesions for transcriptomic analysis. UV light illuminated each AOI to release the barcodes from that region for subsequent counting, and the barcodes were then mapped back to distinct regions of interest.
We used 3rd quadrant (Q3; Nanostring, Inc.) normalization of all raw data because it consistently outperformed other methods, including Negative Probe, which over-normalizes small regions, and Housekeeper Normalization, which was susceptible to inconsistency in experiments investigating disparate sample types that often had different expression levels. All data analyses were performed using R software. For each comparison, we first selected genes with at least a 2-fold change in mean expression levels. We performed two-sample t-test on log2 transformed gene expression data to select the top genes with the smallest p-values. In volcano plots, only genes with p <0.05 were included. The top upregulated and downregulated genes were indicated by different colors. The heatmap was constructed using log2 transformation of the top 70 genes with the smallest p-values.
Immunohistochemistry (IHC)
Immunohistochemistry was performed using monoclonal antibodies against IGFBP2, p53, and Ki-67 (Supplemental Table S1). Antigen retrieval and immunostaining followed previous reports (17). The staining patterns in both normal-appearing fallopian tube epithelium and precursor lesions including STICs and p53 signatures were recorded. H-score was used to semi-quantitatively measure IGFPB2 immunoreactivity in epithelial cells using the formula: H-score = Ʃ (intensity score) × (% of lesion cells at a given intensity level) × 100. Intensity was scored as 0, 1, 2, or 3 by two investigators. Rare discrepancies were resolved by review and discussion for a consensus. The p53 staining pattern was interpreted i) as a missense mutation pattern when intense and diffuse immunoreactivity in epithelial cells was present; ii) as a null mutation pattern when the signal was virtually undetectable (with a positive internal control); and iii) as a wild-type pattern when weak focal staining was present. The percentage of Ki-67-positive nuclei was determined among lesions and control epithelium.
Cell Culture, drug treatment, and lentivirus transduction
Cell lines were cultured with RPMI-1640 (Gibco, 11875093), 10% heat-inactivated fetal bovine serum (FBS, Sigma, F4135), and 1% penicillin-streptomycin (Gibco). All the cell lines were confirmed to be mycoplasma negative by the universal mycoplasma detection kit (ATCC, 30–1012K). To determine the effect of DNA demethylation on the expression of IGFBP2, we treated fallopian tube epithelial (FTE) cell lines expressing a low level of IGFBP2, including FT194 (RRID: CVCL_A4AW), FT241, and FT2821A (18,19), with 0.5 μM 5-Azacytidine (5-AZA, Sigma, A2385) for 24 hours. Control cells were left untreated. Both floating and attached cells were collected for RNA and protein expression analysis. To perform immunocytochemistry of IGFBP2 on cell blocks, we harvested cells treated with 0.5 μM 5-AZA for 24 hours. Those cells were fixed with 10% neutral buffered formalin, and the cell pallets were then embedded in paraffin and sectioned onto unstained plus slides for IGFBP2 staining. To assess the effect of E2 on the regulation of IGFBP2 expression, FT194 and FT241 cells were cultured in RPMI-1640 medium supplemented with 1% penicillin/streptomycin and 50 nM E2 (Sigma, E2758) for 48 hours.
We knocked down IGFBP2 using Mission Lentiviral Transduction Particles targeting the IGFBP2 coding sequence or Nontarget shRNA lentiviral particles as controls (Sigma). Two sets of IGFBP2 shRNA with target sequences CCAGTTCTGACACACGTATTT and ACAGTGCAAGATGTCTCTGAA were used in this study. Cells with high IGFBP2 expression, including FT2821J (18,19), OVCAR3 (RRID: CVCL_0465), and PEA2 (RRID: CVCL_2683), were incubated with viral stock supplemented with 8 μg/ml Polybrene (Sigma, H9268) for 24–72 hours. For organoid culture, the primary FTE cells were transduced without polybrene supplement for 72 hours. After transduction, cells were harvested using TrypLE Express Enzyme (Gibco, 1264–013) and cultured in VitroGel ORGANOID-1 (TheWell, VHM04–1, see below).
Pyrosequencing
A total of 7 samples including 4 normal fallopian tube tissues, one STIC, and two HGSCs were analyzed by pyrosequencing. After laser capture microdissection, DNA was extracted using QIAamp FFPE DNA tissue kit (Qiagen, 56404) followed by bisulfite-conversion using the EZ DNA Methylation-Lightning Kit (Zymo, D5031) according to the manufacturer’s instructions. DNA sequence upstream (Chr2: 216631996–216633220, hg38) from the IGFBP2 coding region was obtained from the UCSC genome browser (20). The average methylation of the CpG sites was quantified by pyrosequencing using the PyroMark PCR Kit (Qiagen, 978703) and PyroMark Q96 MD (Qiagen). In brief, PCR products were denatured, washed, and annealed to a sequencing primer. The pyrosequencing reaction started at the 3′-end of the sequencing primer. Each sample well received nucleotides (A, T, G, and C) one at a time. If the added base was complementary to the base in the PCR product, light emission occurred through an enzymatic cascade. The light intensity was recorded and presented graphically in a pyrogram. The performance of each pyrosequencing assay was evaluated using standards consisting of 0–100% bisulfite-converted CpG-Methylated HeLa Genomic DNA (New England BioLabs) mixed with bisulfite-converted unmethylated Human Male Genomic DNA (Promega). The same assays were also performed on cell lines to confirm demethylation after 5-AZA treatment. Supplemental Table S2 listed the primers used for pyrosequencing.
Subcutaneous xenograft tumor model
All animal-related procedures were approved by the Johns Hopkins University Animal Care and Use Committee. OVCAR3 and PEA2 cells transduced with lentivirus with shRNA against IGFBP2 or control shRNA (107 cells) were mixed 1:1 with Matrigel (Corning, 356234) and subcutaneously injected into the flank subcutaneous tissues in each of 4–6-week-old nu/nu female mice (n=10, Jackson lab, RRID: IMSR_JAX:002019). The tumor size was measured periodically using calipers. Tumor volume was calculated using the formula: TV= 4/3 π (length/2) (width/2)2.
Statistical analysis
To validate our findings, we compared expression changes of the current Nanostring data to results from previously published data (GSE114493) (21). Post-alignment normalization of the RNAseq data and differential expression analysis was performed, as previously described (21) using Voom in the limma package from Bioconductor (22) (23). For biological assays, data were presented as mean ± one standard error. Unpaired two-tailed Student t-test was used to evaluate the statistical significance of the difference between two groups unless otherwise stated. Differences were considered significant for p-value < 0.05. Figures and tables were prepared using standard R functions or Prism software.
Data availability
The data generated in this study are available within the article and its supplemental data files. The raw data of the spatial transcriptomics were provided by the Nanostring bioinformatic team through a service agreement and are available in Supplemental Table S3.
Results
Transcriptomic analysis in the development of high-grade serous carcinomas
This study intended to discover differentially expressed genes in STIC as compared to normal fallopian tube epithelium (NFTE). We focused on studying incidental STICs; therefore only a few concurrent HGSCs were available for comparison. GeoMx human whole transcriptome assays were performed on 13 STICs, 3 HGSCs, and 12 samples of adjacent NFTE derived from 7 patients. Stromal components obtained 100 μm directly underneath each epithelial segment of STIC and NFTE were also analyzed. In total, we analyzed 49 AOIs (Fig. 1A–E). All AOIs showed a sequencing saturation above the 50% cutoff, which approximated a read depth of 100 reads/μm2, as recommended by the manufacturer (Fig. 1F). To adjust counts among different samples for meaningful comparison, we used Q3 to normalize the raw data (Fig. 1G). In total, we assayed 18,676 genes across the entire set of AOIs with 11,941 genes expressed in 10% of the AOIs and 3,645 genes expressed in 50% of the AOIs (Fig. 1H). In three of those STICs of which the lesion cells were abundant, we selected two AOIs from different regions in each STIC. Using log2 transformation of gene expression, we computed the ratio of variable (Rvar) for individual genes between the intra-STIC gene expression variance to that of inter-STIC gene expression across all lesions. A ratio Rvar <1 implies gene expressions across AOIs within the same STIC was closer to each than expressions across different STICs. We have observed Rvar <1 in 95% (17745/18677), 79% (14827/18677), and 70% (13100/18677) of genes from the three STICs, respectively (Fig.1I). We noted that one STIC lesion had a remarkably high expression level in genes expressing CD45 and CD4 in the STIC-associated stroma. Indeed, review on the H&E section showed an intense lymphocytic infiltrate underneath the STIC lesion but not in adjacent NFTE from the same patient or in STIC lesions from the remaining cases that had a background level of CD45 expression by Nanostring (Fig. 1J). This finding supports the robustness of the transcriptomic profiling technique and analysis.
Figure 1. Transcriptomic analysis in ovarian cancer precursors.
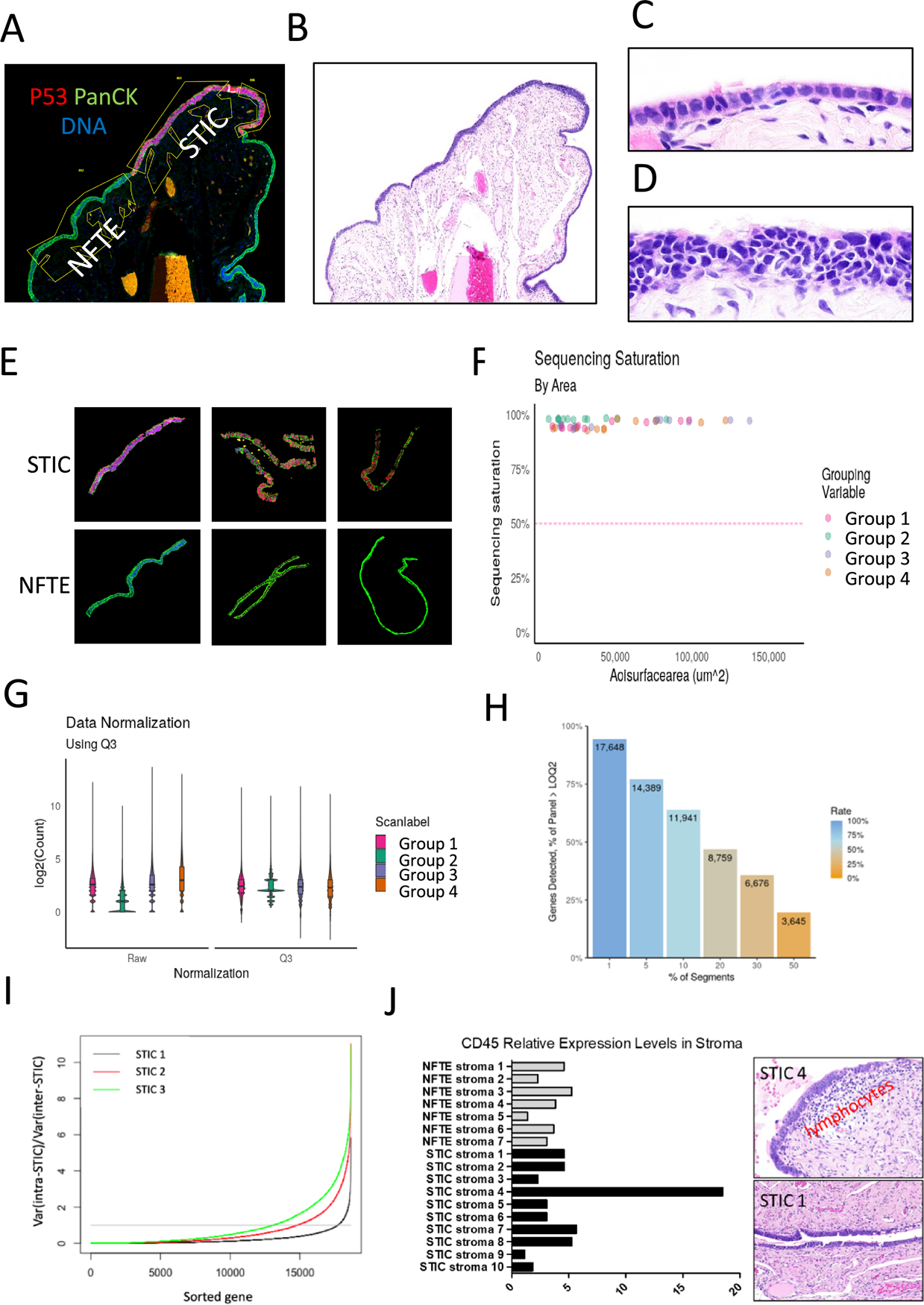
(A) A representative image of human fallopian tube stained with p53, pan-cytokeratin, and DAPI. The specimen contains a STIC, which is labeled by red immunofluorescence with an anti-p53 antibody while all epithelial cells are labeled by green immunofluorescence with a pan-cytokeratin antibody. (B) The corresponding H&E staining at the same magnification. (C) Image from B of normal fallopian tube epithelium with H&E staining under a higher magnification. (D) Image from B of STIC with H&E staining under a higher magnification. (E) Examples of STIC and normal fallopian tube epithelium (NFTE) from cases analyzed. (F) Sequencing saturation by areas of interest (AOI). Each dot in the plot represents one sample. Samples were grouped for transcriptomic profiling. (G) Data from F normalized to the third quartile (Q3) to account for differences in cellularity, AOI size, etc. (H) Percentage of genes detected across 49 AOIs. A total of 18,676 genes were assayed. (I) Comparison of intra-STIC and inter-STIC gene expression variances. The X-axis is the number of genes analyzed by Nanostring. The Y-axis indicates the ratio of the intra-STIC gene expression variance from two different AOIs with the same lesion to the gene expression variance across all lesions (inter-STIC) for individual genes. The horizontal (grey) line indicates the ratio of one. (J) Relative CD45 expression in STIC stroma (left panel). STIC Case 4 shows a high expression level and histologically it contains an increased lymphocyte density in the stroma (right upper panel). In contrast, another STIC, has a very low CD45 level and a background lymphocyte density in stroma (right lower panel). Both STICs are associated with concurrent high-grade serous carcinomas.
Analyses of the transcriptomic data between STIC and NFTE from the same specimens identified 82 genes showing greater than 2-fold changes in expression levels. Among them, changes in expression of 39 genes were significant (p< 0.01) (Fig. 2A–B and Supplemental Table S4). KEGG pathway analysis in the STIC specific genes (as compared to NFTE) shows gene enrichment in several pathways, most notably the PI3K-Akt signaling pathway (Supplemental Fig. S1 and Table S5). In comparing stromal components of STIC and NFTE, we identified 60 genes showing greater than 2-fold changes in expression levels. Among these, only 3 genes were significant (p< 0.01) (Fig. 2C–D). We also compared STICs to HGSCs and found 1,526 genes with greater than 2-fold changes in expression levels, 1,068 genes with greater than 3-fold changes, and among them 128 genes showed significant changes (p< 0.01) (Fig. 2E–F).
Figure 2. The most differentially expressed genes between tissue groups.
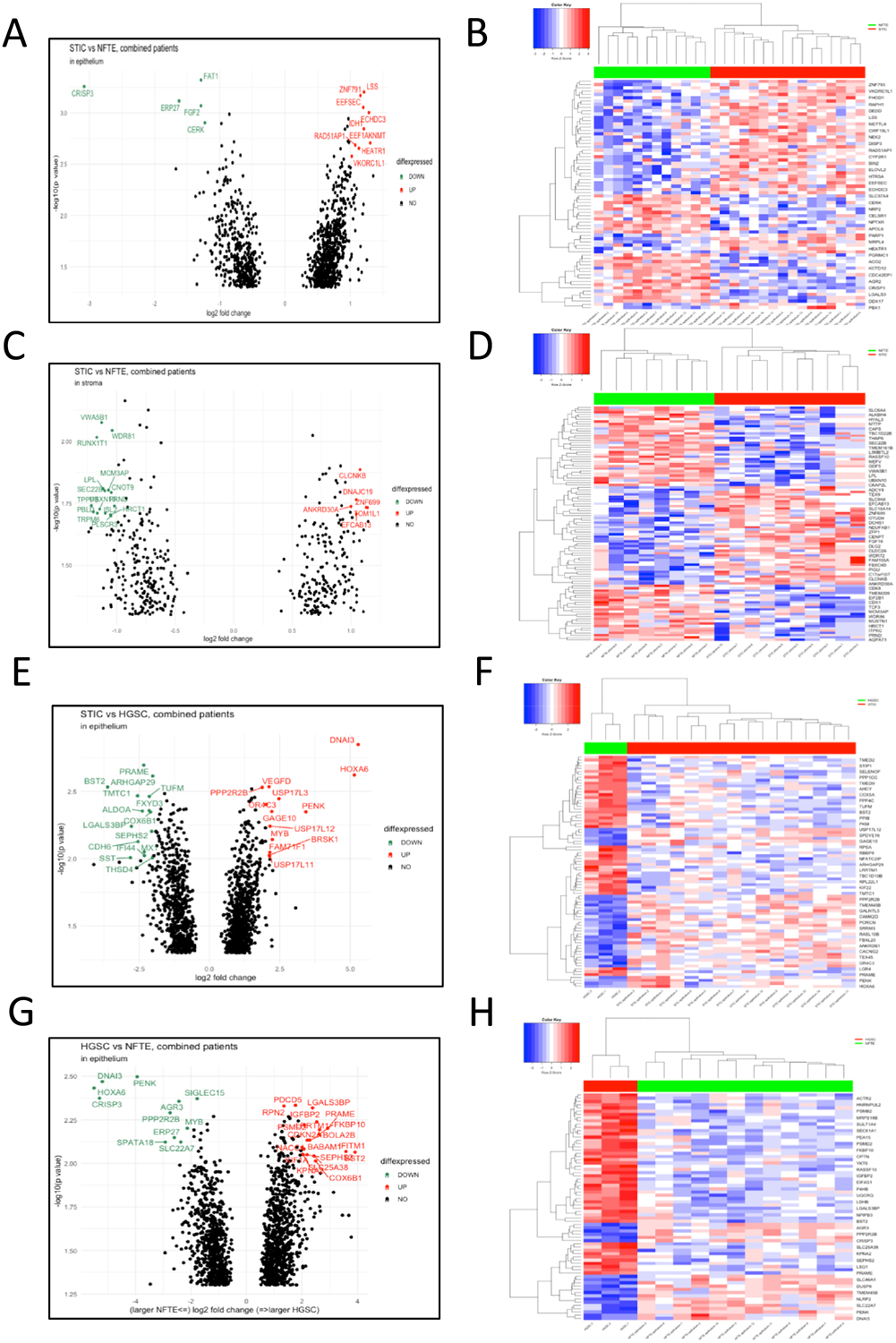
Volcano plots and heat maps showing genes that are either upregulated or downregulated between two groups. (A-B) Differentially expressed genes between STICs and normal fallopian tube epithelium (NFTE). (C-D) Differentially expressed genes in associated stroma between STIC and NFTE samples. (E-F) Differentially expressed genes between STICs and HGSC. (G-H) Differentially expressed genes between HGSC and NFTE.
Because only three concurrent HGSCs were available for Nanostring, we used our pre-existing RNA sequence data from 96 HGSCs and NFTEs to determine if we could detect differentially expressed genes in STICs and HGSCs from this current study in HGSCs but not in NFTEs from that previous study (21). We found that most (57/76) top candidate genes (Fig. 2G–H) identified here were differentially expressed in the previous dataset along the same trend in HGSC as compared to NFTE (adjusted p < 0.05) (Supplemental Fig. S2) (21). Based on the lowest p-value and availability of antibodies that were suitable for staining FFPE tissues, we chose 4 genes including two upregulated genes (IGFBP2 and PRAME) and two downregulated genes (FGF2 and CRISP3) for immunohistochemical validation. Alongside the HGSCs, we intended to determine if changes in expression also occurred in STICs. As expected, our results showed prominent IGFBP2 and PRAME immunoreactivity in representative STICs but not in adjacent NFTE. We also observed FGF2 and CRISP3 staining in NFTE but not in STICs or HGSCs (Supplemental Fig. S3).
Upstream DNA hypomethylation of IGFBP2 in STIC and ovarian cancer
In addition to molecular genetic changes, DNA hypomethylation is another common attribute of developing cancer as tumor cells alter transcriptional activities in several cancer-related genes such as PD-L1 (24,25). Thus, we studied whether the overexpressed genes in STICs and HGSCs were associated with DNA hypomethylation in the upstream regulatory regions of the corresponding genes. Based on the MethylationEPIC dataset of methylation profiles in NFTE, STICs, and HGSCs (26), we assessed correlations between DNA methylation and gene expression levels. We used the following criteria to define differentially methylated regions: 1) presence of CpG sites within 1500 b.p. from the transcription start site; 2) adjusted p-value < 0.05; 3) NFTE β-value > 0.7 or < 0.3; 4) at least 2 CpG sites fulfilling the above criteria. In total, 10 out of the 111 differentially expressed genes identified by transcriptomic analysis fulfilled the criteria, including MEST, PRR9, FGF2, LUC7L3, IGFBP2, PRAME, SEPHS2, PENK, SIGLEC15, and SLC2A7. Figure 3A–B and Supplemental Fig. S4 show the DNA methylation status in the regulatory elements of representative IGFBP2, FGF2 and PRAME. Among these genes, IGFBP2 and PRAME were of particular interest as upregulation of their protein levels were validated in STICs. Since IGFBP2 has been reported to be associated with ovarian cancer metastasis and invasion (27), we further characterized it in this study.
Figure 3. Hypomethylation of CpG sites upstream of IGFBP2 in precursor lesions is associated with IGFBP2 overexpression.
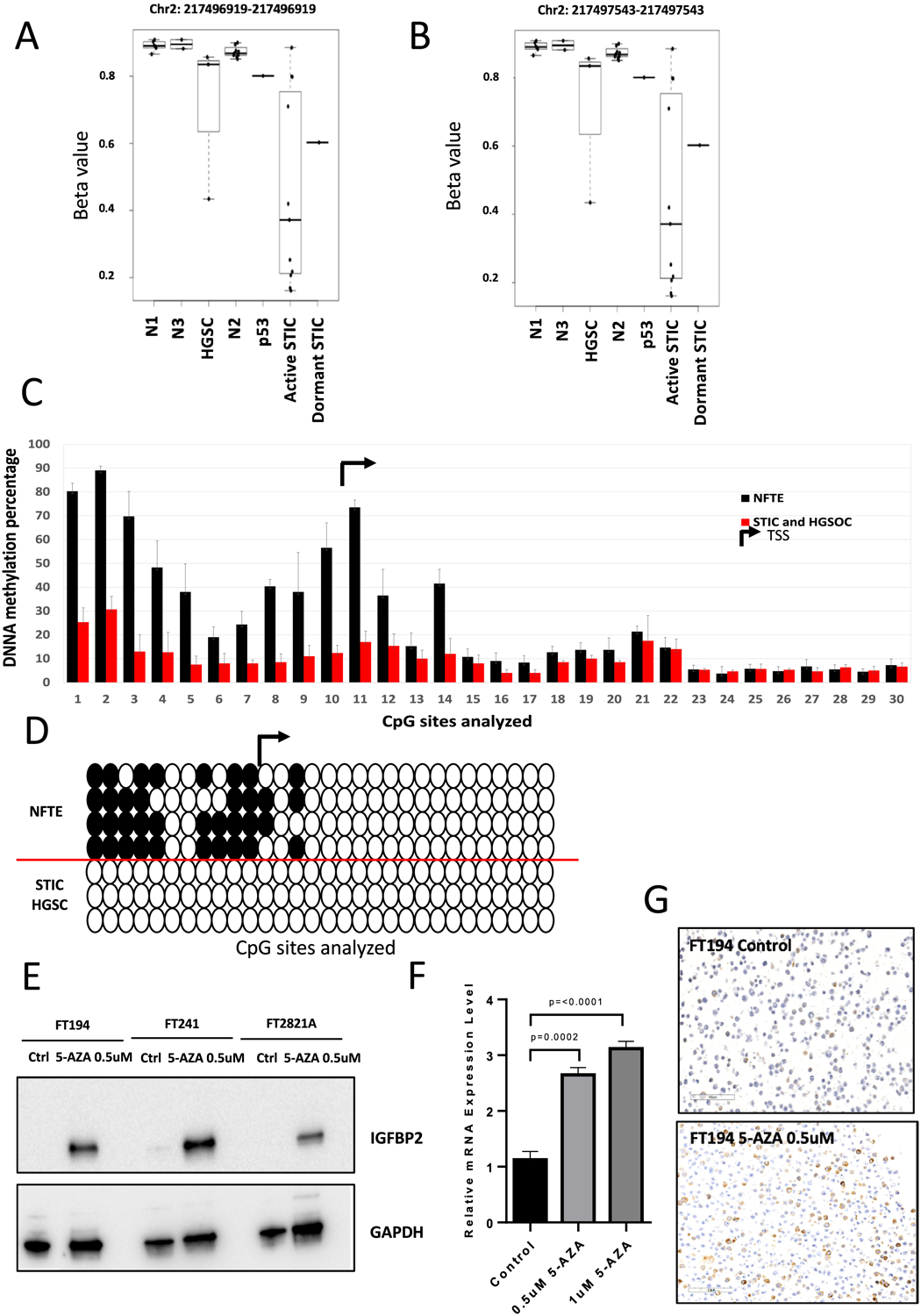
(A-B) DNA methylation status of the two CpG sites upstream of IGFBP2 identified by MethylationEPIC array in microdissected NFTE, precursor lesions, and HGSC. Each dot represents one sample. N1: NFTE immediately proximal to STIC; N2: NFTE distal to STIC; N3: NFTE from the contralateral fallopian tube; p53: p53 signature. (C) Pyrosequencing analysis showing DNA methylation status upstream of IGFBP2. TSS, transcription starting site. (D) Methylation of 40% in total CpG sites is used as a cutoff. White circles indicate unmethylated cytosines and black circles indicate methylated cytosines within CpG sites. (E) Immunoblots showing IGFBP2 expression levels in cellular extracts of FT194, FT241, and FT2821A cells after treatment with 5-AZA for 24 hours. GAPDH is used as a loading control. (F) qRT-PCR of IGFBP2 in FT194 after treated with 0.5 μM or 1 μM 5-AZA for 24 hours. (G) Immunostaining of IGFBP2 in FT194 cells after treatment with 0.5 μM 5-AZA for 24 hours.
We sought to determine the extent of DNA hypomethylation upstream of IGFBP2 in detail. Laser-microdissection was performed to enrich epithelial cells from one STIC, two HGSCs, and 4 NFTE specimens. IGFBP2 immunostaining was used to specifically locate the areas with high IGFBP2 expression for microdissection. We performed pyrosequencing on the 7 samples following bisulfite conversion and sequenced 11 CpG sites within 679 b.p. upstream of the transcription start site (Chr2: 216632150–216632827, hg38), which overlaps the region identified as proximal enhancers by ENCODE. In accordance with the results of MethylationEPIC array data, the CpG sites within this proximal enhancer region showed significant hypomethylation in STICs and HGSCs compared to NFTE. We also sequenced 19 CpG sites downstream of the transcription start site (including 14 CpG sites within a CpG island, Chr2: 216632828–216633181, hg38, total length 354 base pairs) and these CpG sites resided in regions of promoter-like signature, to rule out the possibility that upregulation of IGFBP2 expression was caused by promoter hypomethylation (28). None of these CpG sites showed significant methylation changes in STICs and HGSCs compared to NFTE (Fig. 3C–D).
To further support the notion that DNA hypomethylation was responsible for transcriptional upregulation, we treated NFTE cell lines (FT194, FT241, FT2821A) having low or undetectable levels of intrinsic IGFBP2 expression with 0.5 μM 5-AZA, and confirmed DNA demethylation by pyrosequencing (Supplemental Fig. S5). Twenty-four hours after the treatment, we conducted semi-quantitative RT-PCR, Western blotting, and immunostaining, which showed a significant increase in both mRNA and protein levels of IGFBP2 (Fig. 3E–G). These data suggest that DNA hypomethylation in the proximal enhancer was associated with IGFBP2 overexpression.
IGFBP2 expression in fallopian tube tissues, precursors, and high-grade serous carcinomas
To determine the prevalence of IGFBP2 overexpression in tubal lesions and HGSCs, we applied immunohistochemistry on formalin-fixed and paraffin-embedded tissue sections. A total of 61 STICs, 15 p53 signatures, 30 HGSCs, and 55 normal fallopian tubes were evaluated using the same protocol. The levels of IGFBP2 expression in the lesional tissues were compared to normal fallopian tube epithelium on the same sections if present. We also stained sections with p53 and Ki-67 antibodies to help locate tubal lesions and assess their proliferative activity. Proliferatively active STICs (hereinafter “active STIC”) were those with a Ki-67 labeling index at least 5 times greater than that in surrounding normal epithelium. Those STICs showing proliferative activity that was similar to the surrounding normal epithelial level of Ki-67 labeling index were termed proliferatively dormant STICs (hereinafter “dormant STIC”) or serous tubal intraepithelial lesions (STIL). We found intense and diffuse immunoreactivity of IGFBP2 in 36 (94.74%) of 38 proliferatively active STICs, 2 (7.41%) of 27 dormant STICs, and none of 15 p53 signatures. Staining results together with clinicopathological features and representative photomicrographs are summarized in Table 1 and shown in Fig. 4A–C. Figure 4D shows H-scores of IGFBP2 staining on different types of tissues. The great majority of proliferatively active STICs and high-grade serous carcinomas have significantly higher H-scores than NFTE. There were 4 cases showing STICs containing both proliferatively active and dormant areas. In those cases, we found a higher staining intensity of IGFBP2 in the regions with a higher Ki-67 labeling index compared to the adjacent dormant counterpart with a low Ki-67 index (Fig. 4E). IGFBP2 immunoreactivity was undetectable in tubal epithelium associated with concurrent STICs or HGSCs in postmenopausal women, and we extended this finding to normal appearing fallopian tubes without any lesions in younger premenopausal women. Interestingly, we observed positive IGFBP2 staining in secretory epithelial cells in most premenopausal fallopian tube samples. This result raises the possibility that IGFBP2 expression in normal premenopausal fallopian tubes is estrogen dependent.
Table 1.
The expression of IGFBP2 in ovarian cancer precursor lesions and associated patient characteristics.
| Specimen | Age | Histology | p53 mutation pattern | IGFBP2 staining | ki67% | Concurrent HGSOC | Germline mutations |
| FTE-1 | 67 | p53 signature | Missense | Negative | 2 | No | |
| FTE-2 | 56 | p53 signature | Missense | Negative | NA | No | |
| FTE-3 | 46 | p53 signature | Missense | Negative | 1 | No | |
| FTE-4 | 46 | p53 signature | Missense | Negative | 1 | No | |
| FTE-5 | 46 | p53 signature | Missense | Negative | 1 | No | |
| FTE-6 | 46 | p53 signature | Missense | Negative | 0 | No | |
| FTE-7 | 73 | p53 signature | Missense | Negative | 1.2 | No | |
| FTE-8 | 73 | p53 signature | Missense | Negative | 2 | No | |
| FTE-9 | 73 | p53 signature | Missense | Negative | 1 | No | |
| FTE-10 | 58 | p53 signature | Missense | Negative | 5.3 | No | Lynch syndrome |
| FTE-11 | 70 | p53 signature | Missense | Negative | 1 | No | |
| FTE-12 | 70 | p53 signature | Missense | Negative | 2 | No | |
| FTE-13 | 72 | p53 signature | Missense | Negative | 4.3 | Yes | |
| FTE-14 | 60 | p53 signature | Missense | Negative | NA | No | |
| FTE-15 | 52 | p53 signature | Missense | Negative | 1 | No | |
| FTE-16 | 71 | Dormant/Active STIC* | Missense | Positive/positive | 7/35 | No | |
| FTE-17 | 50 | Dormant/Active STIC* | Missense | Negative/positive | 1/43 | No | |
| FTE-18 | 44 | Dormant/Active STIC* | Missense | Negative/positive (weak) | 5.2/40 | No | VUS, ATM(p.V455M), RAD50(p.R1260H) |
| FTE-19 | 65 | Dormant/Active STIC* | Nonsense | Negative/positive (weak) | NA | No | |
| FTE-20 | 46 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-21 | 47 | Dormant STIC | Missense | Negative | 4 | No | |
| FTE-22 | 46 | Dormant STIC | Missense | Negative | 6 | Yes | |
| FTE-23 | 72 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-24 | 73 | Dormant STIC | Missense | Negative | 1 | No | |
| FTE-25 | 73 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-26 | 58 | Dormant STIC | Missense | Negative | 6 | No | Lynch syndrome |
| FTE-27 | 58 | Dormant STIC | Missense | Negative | 2 | No | Lynch syndrome |
| FTE-28 | 51 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-29 | 72 | Dormant STIC | Missense | Negative | 1 | Yes | |
| FTE-30 | 55 | Dormant STIC | Missense | Negative | 1 | No | BRCA, type? |
| FTE-31 | 60 | Dormant STIC | Missense | Positive | 5 | No | |
| FTE-32 | 68 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-33 | 66 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-34 | 69 | Dormant STIC | Missense | Negative | 1 | Yes | |
| FTE-35 | 69 | Dormant STIC | Missense | Negative | 2 | Yes | |
| FTE-36 | 58 | Dormant STIC | Missense | Negative | 1 | No | |
| FTE-37 | 58 | Dormant STIC | Missense | Negative | 1 | No | |
| FTE-38 | 58 | Dormant STIC | Missense | Negative | 0 | No | |
| FTE-39 | 58 | Dormant STIC | Missense | Negative | No | ||
| FTE-40 | 73 | Dormant STIC | Missense | Negative | 3 | No | |
| FTE-41 | 73 | Dormant STIC | Missense | Negative | 2 | No | |
| FTE-42 | 73 | Dormant STIC | Missense | Negative | 3 | No | |
| FTE-43 | 46 | Active STIC | Missense | Positive | 74 | No | |
| FTE-44 | 65 | Active STIC | Missense | Positive | 95 | No | BRCA2 |
| FTE-45 | 65 | Active STIC | Missense | Positive | 96 | No | BRCA2 |
| FTE-46 | 66 | Active STIC | Missense | Positive | NA | Yes | |
| FTE-47 | 76 | Active STIC | Missense | Positive | NA | No | |
| FTE-48 | 58 | Active STIC | Nonsense | Positive (weak) | 84 | No | |
| FTE-49 | 71 | Active STIC | Missense | Positive | 12 | No | |
| FTE-50 | 71 | Active STIC | Nonsense | Positive | 53 | No | |
| FTE-51 | 71 | Active STIC | Nonsense | Positive | 55 | No | |
| FTE-52 | 56 | Active STIC | Missense | Positive (weak) | NA | No | |
| FTE-53 | 75 | Active STIC | Missense | Positive | 62 | No | |
| FTE-54 | 75 | Active STIC | Missense | Positive | 69 | No | |
| FTE-55 | 71 | Active STIC | Missense | Positive (weak) | 17 | No | BRCA1 |
| FTE-56 | 71 | Active STIC | Missense | Positive | 29 | No | |
| FTE-57 | 71 | Active STIC | Missense | Positive | 23 | No | |
| FTE-58 | 57 | Active STIC | Missense | Positive | NA | Yes | |
| FTE-59 | 63 | Active STIC | Missense | Positive | NA | Yes | |
| FTE-60 | 57 | Active STIC | Missense | Positive | 44 | Yes | |
| FTE-61 | 57 | Active STIC | Missense | Positive | 52 | Yes | |
| FTE-62 | 57 | Active STIC | Missense | Positive | 47 | Yes | |
| FTE-63 | 79 | Active STIC | Missense | Positive | 20 | No | |
| FTE-64 | 46 | Active STIC | Nonsense | Positive (weak) | 62 | No | BRCA1 |
| FTE-65 | 46 | Active STIC | Nonsense | Positive (weak) | 78 | No | BRCA1 |
| FTE-66 | 44 | Active STIC | Missense | Positive | 25 | No | VUS, ATM(p.V455M), RAD50(p.R1260H) |
| FTE-67 | 41 | Active STIC | Missense | Positive | 81 | No | BRCA2 |
| FTE-68 | 51 | Active STIC | Nonsense | Negative | 23 | No | |
| FTE-69 | 72 | Active STIC | Missense | Positive | 42 | Yes | |
| FTE-70 | 72 | Active STIC | Missense | Positive | 31 | Yes | |
| FTE-71 | 62 | Active STIC | Missense | Positive | 40 | Yes | BRCA1 |
| FTE-72 | 85 | Active STIC | Nonsense | Positive | 70 | Yes | |
| FTE-73 | 85 | Active STIC | Nonsense | Positive | 78 | Yes | |
| FTE-74 | 67 | Active STIC | Nonsense | Positive | 33 | No | |
| FTE-75 | 65 | Active STIC | Missense | Negative | 21 | No | |
| FTE-76 | 59 | Active STIC | Missense | Positive | 52 | Yes |
EEC: endometrial endometrioid carcinoma; MMMT: malignant mixed mesenchymal tumor; ESC: endometrial serous carcinoma; BCC: basal cell carcinoma; NA: not available.
dormant SITC continuous with active STIC.
Figure 4. Immunohistochemistry of IGFBP2 and correlation with DNA methylation.
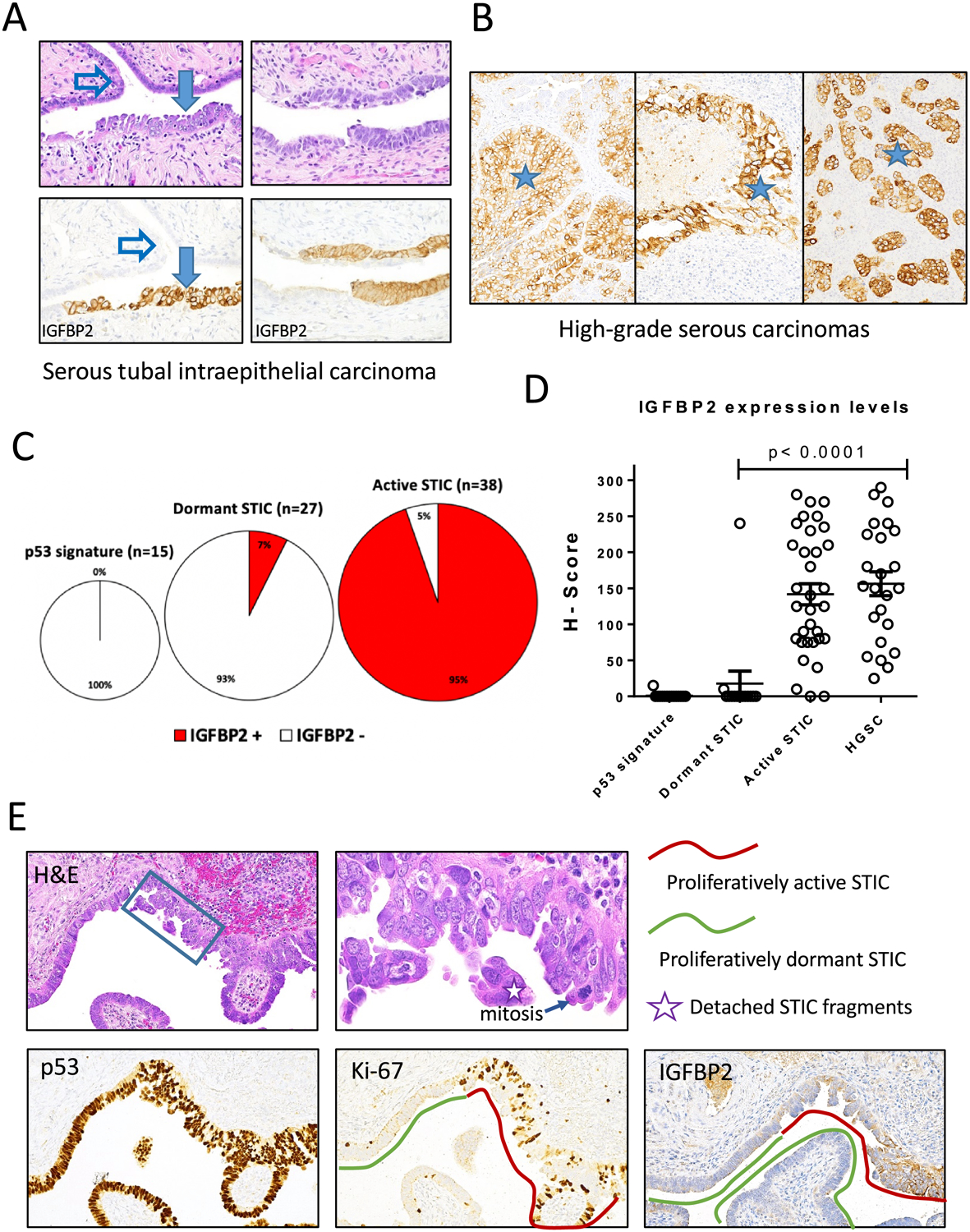
(A-B) Examples showing IGFBP2 immunostaining in STIC (A) and HGSC (B) but not adjacent normal fallopian tube epithelium (NFTE). Solid arrow, STIC; open arrow, NFTE; asterisks, high-grade serous carcinomas in three different cases. (C) Summary of IGFBP2 staining in p53 signature, dormant STIC, and active STIC. Number and percentage of positive cases are shown. (D) Scatter plot of IGFBP2 immunostaining H-scores of various lesions. Each symbol represents an individual lesion. (E) An example of STIC containing both proliferatively active and dormant areas. IGFBP2 immunostaining is positive in proliferatively active STIC (Ki-67 positive).
Estrogen enhances IGFBP2 expression in normal fallopian tube epithelium
To test the hypothesis that estrogen enhances IGFBP2 expression, we first correlated IGFBP2 staining intensity in tubal epithelial cells with menstrual cycle and with the postmenopausal state. In premenopausal fallopian tubes, we found that IGFBP2 expression in secretory tubal epithelial cells was more prominent in the proliferative phase than in the secretory phase, while in postmenopausal fallopian tubes immunoreactivity was undetectable. However, the signal intensity of IGFBP2 in proliferative phase was much weaker than active STIC (Fig. 5A–B and 4D). As E2 plays a key role in the cyclic changes in female reproductive organs, we next examined if the expression of IGFBP2 was regulated by E2. After treating FT194 and FT241 tubal epithelial cells with E2 for 24 hours, we observed an increased expression of ER-α, the predominant isoform of estrogen receptor (ER) in endometrium and fallopian tubes. Strikingly, the expression of IGFBP2 was also increased following E2 treatment (Fig. 5C). We obtained fresh normal fallopian tube tissue fragments from salpingo-oophorectomy (with or without hysterectomy) specimens performed for benign reasons including leiomyoma in pre-menopausal women. After treating the fallopian tube tissues ex vivo with E2, secretory epithelium became positive for IGFBP2 whereas the vehicle-treated tissues did not (Fig. 5D).
Figure 5. Estrogen regulates IGFBP2 expression in normal premenopausal fallopian tube epithelium.
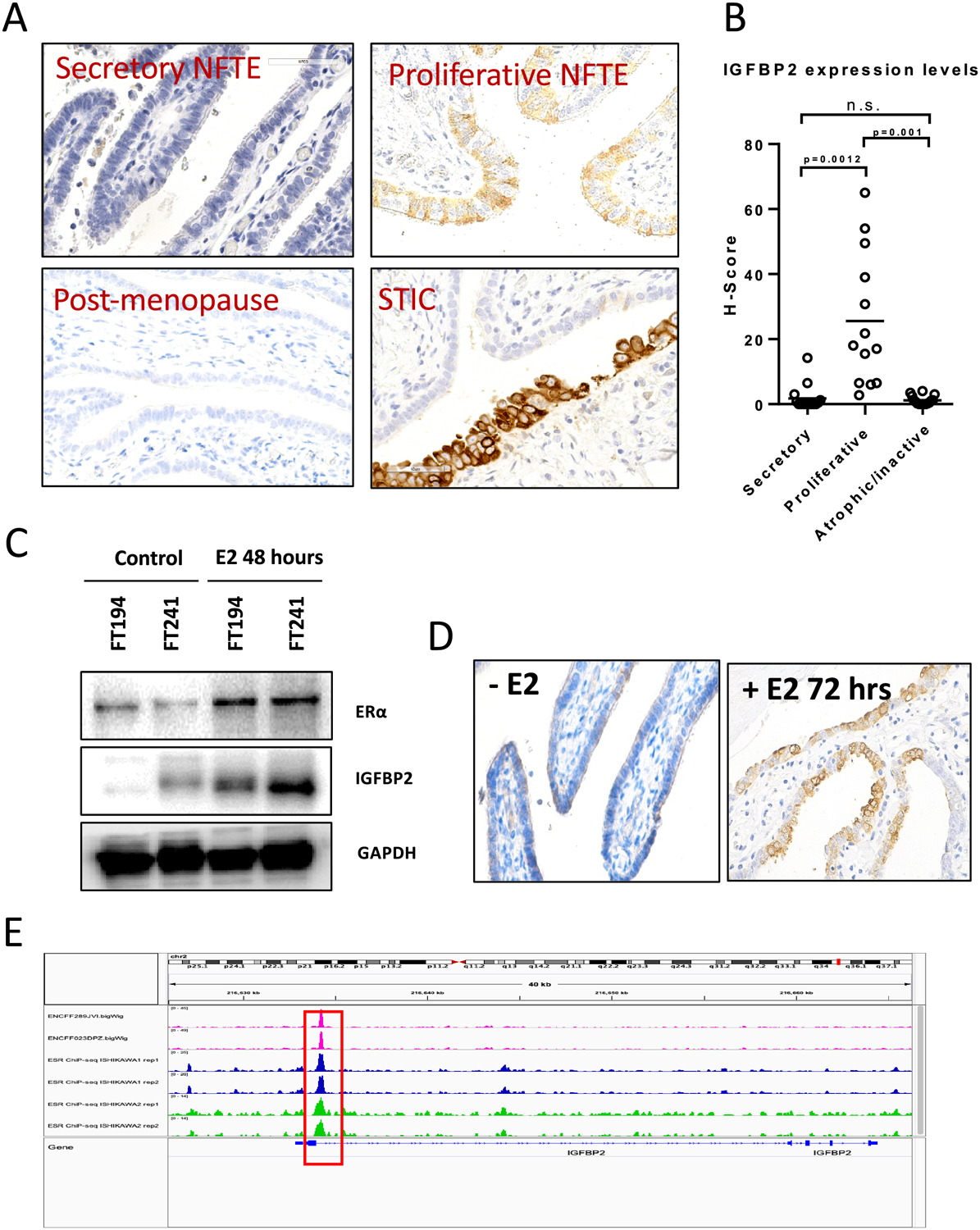
(A) IGFBP2 immunoreactivity in fallopian tubes from women at different menstrual phases (secretory/proliferative/postmenopausal) and in STIC. (B) H-score of IGFBP2 expression levels at different menstrual phases. (C) Immunoblots for ER-alpha and IGFBP2 in cellular extracts of FT194 and FT241 cell lines after treatment with 50 nM estradiol (E2) for 48 hours. GAPDH is used as a loading control. (D) IGFBP2 expression increases in freshly harvested human fallopian tube tissue after E2 treatment. (E) Chromatin immunoprecipitation-sequencing data for ER-α on T47D and Ishikawa cell lines showing ESR1 peaks (red squares) located within 1 k.b. downstream of the TSS of IGFBP2. The data were obtained from the ENCODE database.
We then retrieved data from ENCODE pertaining to chromatin-immunoprecipitation sequencing (ChIP-seq) using an anti-ESR1 antibody on Ishikawa and T47 cell lines to assess the potential transcriptional regulation of IGFBP2 by ESR1. The ENCODE data showed that ESR1 peaks were located 1 k.b. downstream of the transcription start site within the presumed promoter region of IGFBP2 (Fig. 5E). These data suggest E2 regulates the expression of IGFBP2 in premenopausal normal fallopian tube epithelium.
Knockdown of IGFBP2 suppresses proliferation and spheroid formation by normal fallopian tube epithelium
To determine whether IGFBP2 expression was essential for cell survival and growth, we knocked down IGFBP2 in one fallopian tube epithelial cell line (FT2821J) and two HGSC (OVCAR3 and PEA2) cell lines that expressed IGFBP2. Using immunoblot analysis, we confirmed that shRNA knockdown of IGFBP2 suppressed IGFBP2 expression (Fig. 6A). In all three cell groups, IGFBP2 knockdown led to lower proliferative activity in 2-D cell culture systems (Fig. 6B–C).
Figure 6. Knockdown of IGFBP2 decreases Akt phosphorylation and suppresses the proliferation and spheroid formation of normal fallopian tube epithelium.
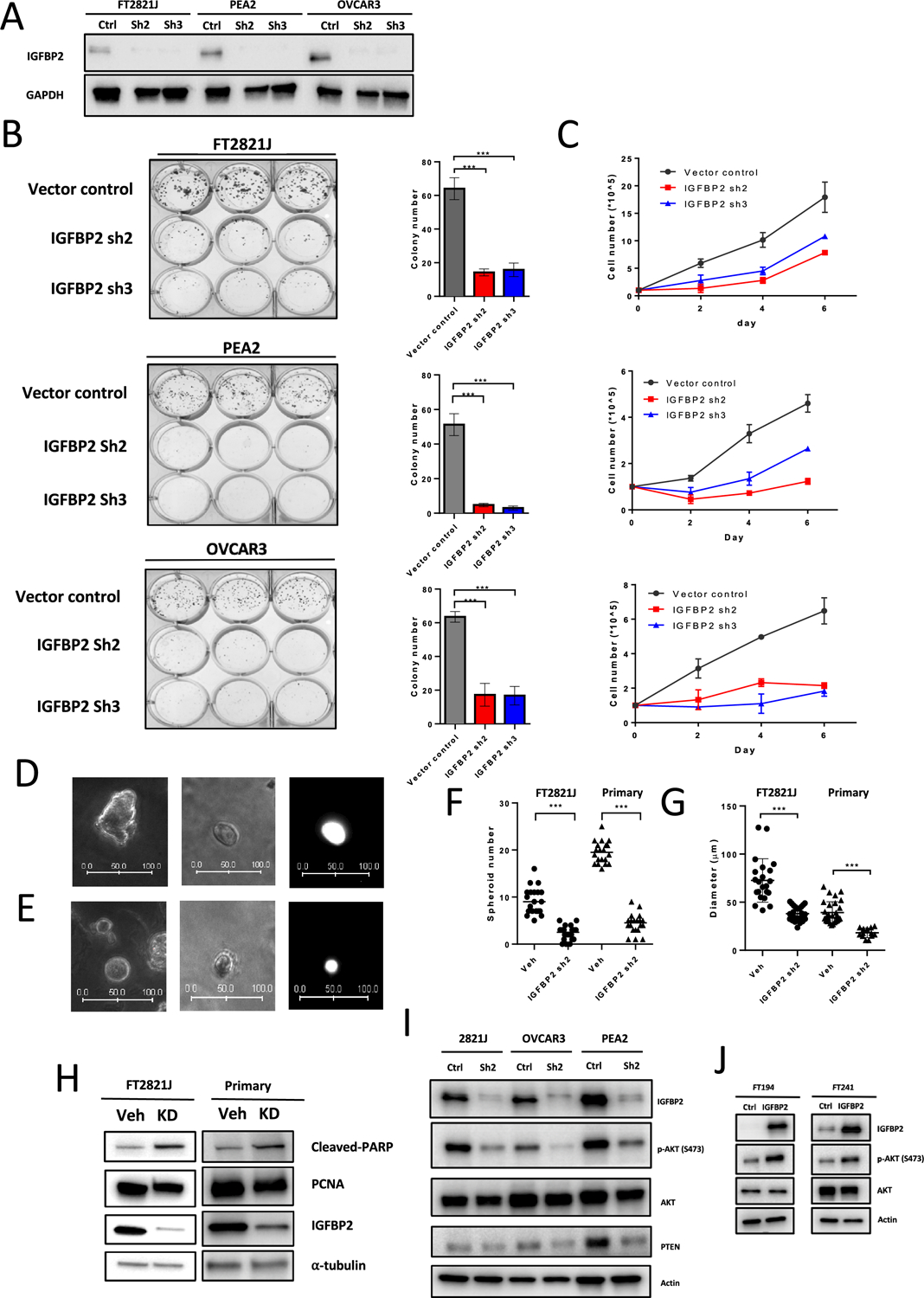
(A) Immunoblots of IGFBP2 from FT2821J, PEA2, and OVCAR3 cell lines after IGFBP2 shRNA lentivirus knockdown. (B) (left) Colony formation assays in FT2821J, PEA2, and OVCAR3 cells after IGFBP2 knockdown. (right) Quantification of data from left panel. (C) Cell proliferation assays after IGFBP2 knockdown in FT2821J, PEA2, and OVCAR3 cells. (D-E) Representative images of FT2821J spheroids. (left) Vehicle control. (middle) IGFBP2 knockdown. (right) GFP fluorescence image of the spheroid shown in the middle panel. Scale bar units, μm. (F-G) Scatter plots showing the number (F) and diameter (G) of spheroids after knockdown of IGFBP2 in FT2821J and primary FTE cells. (H) Immunoblots showing expression of cleaved-PARP, PCNA, IGFBP2, and α-tubulin (loading control) in extracts from FT2821J cells and primary cultures of fallopian tube epithelium after treatment with IGFBP2 shRNA lentivirus for 3 days. (I) Knockdown of IGFBP2 in IGFBP2-high cell lines reduces Akt phosphorylation but less PTEN expression. (J) Overexpression of IGFBP2 induces Akt phosphorylation in IGFBP2-low FTE cell lines (FT194 and FT241).
To better simulate the in vivo condition, we performed 3D culture and mouse xenograft tumor model. Based on 3D spheroid cultures generated from freshly harvested human fallopian tube epithelial cells or FT2821J cells, we also observed smaller sizes and numbers of spheroids in knockdown groups compared to controls (Fig. 6D–G). Western blot analysis also showed reduced PCNA and increased cleaved PARP expression after IGFBP2 knockdown (Fig. 6H). Silencing of IGFBP2 significantly inhibited OVCAR3 tumor growth in mice as compared to control cells which were transduced with lentivirus carrying nontarget shRNA (Supplemental Fig. S6A). The growth inhibitory effect rendered by IGFBP2 inactivation was consistent with that in previous reports (29,30). Western blot was performed to confirm the knockdown of IGFBP2 before the xenograft injection (Supplemental Fig. S6B). We next sought to determine which pathways downstream of IGFBP2 contribute to cell proliferation and stemness. Western blot analysis confirmed decreased Akt phosphorylation after knockdown of IGFBP2 (Fig. 6I) and increased Akt phosphorylation after overexpression of IGFBP2 (Fig. 6J), while p-p38/p-ERK/p-JUNK remained unchanged (data not shown). Western blot on PTEN was also performed as IGFBP2 has been reported to negatively regulate PTEN (31,32). Interestingly, PTEN expression did not increase upon IGFBP2 knockdown. This result suggests that expression of IGFBP2 activates the Akt pathway and contributes to cell proliferation through a mechanism other than downregulating PTEN in our model.
Discussion
The emerging paradigm of HGSC genesis from fallopian tube epithelium has provided investigators an opportunity to study the earliest molecular changes in fallopian tube precursor lesions. Unlike other common neoplastic diseases, very few recurrent somatic mutations in cancer-driver genes have been detected and functionally validated in the development of STIC, with the exception of mutations in TP53 and in genes involved in homologous recombination-mediated DNA double strand break repair including BRCA1 and BRCA2 (5,8,9). This finding indicates that mechanisms other than somatic mutation play a critical role in HGSC initiation. For example, epigenetic changes such as DNA methylation and chromatin modification can also contribute to aberrantly expressed driver genes that activate the signaling pathways that may transform tubal epithelial cells (33,34). In this study, we first performed spatial transcriptomic analysis in STIC epithelium and underlying stroma to identify aberrantly expressed cancer-related genes in comparison to normal tubal epithelium, and we determined whether epigenetic regulation of their expression took place. We then focused on one aberrantly expressed gene, IGFBP2, to explore its roles in HGSC development.
Nanostring digital spatial profiling analysis in STICs and HGSCs demonstrates the difference of global mRNA profiles as compared to normal fallopian tube epithelium. As expected, we observe that the two different regions within a STIC more resemble each other than with other STICs. We also identified several cancer-related genes that were either upregulated or downregulated in epithelial cells compared to normal-appearing epithelium. The majority of the differentially expressed genes in STIC and HGSC can be verified in RNAseq data from an independent set of HGSCs and normal fallopian tube cases (21). We were able to validate representative genes including FGF2, CRISP3, PRAME, and IGFBP2 using an orthogonal approach, immunohistochemistry, on selected STIC and HGSC samples. Loss of FGF2 has been reported during malignant transformation of mammary duct epithelium (35) and has also been shown to have a causal role in branching morphogenesis in breast cancer cell lines (35,36). In normal tissues, CRISP3 was expressed in neutrophils and exocrine tissues including salivary glands, pancreas, and prostate (37). It is likely that downregulation of FGF2 and CRISP3 expression in STICs is related to their loss of secretory differentiation or cell lineage. On the other hand, PRAME is a cancer-related gene expressed in testis that has been reported to be expressed in many cancers (38), likely due to promoter hypomethylation (39–41). Activation of PRAME has been reported to be oncogenic through binding to retinoid acid receptor, preventing target gene transcription (42). In contrast to epithelial cells, we only identify few genes of interest in the stroma underlying STIC lesions, raising the possibility that stromal microenvironment is mostly similar between STIC and normal fallopian tube epithelium. Our results may help generate experimentally testable hypotheses regarding initiation of ovarian HGSC.
Our pre-existing RNAseq transcriptomic data in HGSC and normal fallopian tube epithelium as well as our methylomic data in STIC, HGSC, and normal fallopian tube epithelium allowed us to further identify a focused set of genes that are upregulated in STIC and HGSC due to gene hypomethylation and which are functionally associated with pathogenesis of ovarian cancer. As a result, we selected an upregulated gene, IGFBP2, for functional characterization in STIC development. Our results showed that the proximal enhancer of IGFBP2 regulated IGFBP2 gene expression in STIC and HGSC. Thus, IGFBP2, like human cancer antigens, can result from re-expression of genes due to epigenetic relaxation (43–45). Along this line, autoantibodies against IGFBP2 were detected in serum from patients with lung, breast, and colorectal cancers (46–48), and a DNA vaccine targeting the N-terminus of IGFBP2 has been recently proposed (49), indicating that IGFBP2 is a tumor antigen.
Our findings have biological implications in tumor initiation of HGSC. In premenopausal women, fallopian tube epithelial cells are repeatedly exposed to follicular fluid during ovulation. The insulin-like growth factor (IGF) axis has been proposed to play an important role in mediating the effects of follicular fluid on tubal epithelium. This is evidenced by a previous study showing that binding of IGF-2 from follicular fluid to IGF-1R receptor on fallopian tube epithelial cell lines promotes stemness through activation of the downstream AKT/mTOR/NANOG pathway (50,51). In addition, IGFBP2 in the follicular fluid binds to IGF-2, prolonging its half-life and thus its biological effects (50,52,53).
In the current study, we report that IGFBP2 is also expressed in fallopian tube secretory epithelial cells from premenopausal but not postmenopausal women, and such local effect may further enhance the IGF activity in tubal epithelium. Because of its well-established proliferation-promoting role, IGF-related signaling helps tubal epithelial cells regenerate and substitute for those that are severely damaged by follicular fluid-derived genotoxic factors, mainly reactive oxygen species (50,54,55) However, the tubal epithelium that survives can accumulate molecular genetic alterations including somatic mutations like TP53 mutation and DNA copy number gain/loss if cellular proliferation resumes. It has been estimated that tumor initiation from the first hit, mostly likely a TP53 mutation to form a STIC, takes decades of slow progression from that initial lesion at a young age to the patient’s early 50s when cyclic ovulations cease, and serum estrogen levels sharply decrease.
As ER expression continues even after menopause in gynecologic tissues including fallopian tube epithelium (56,57), loss of IGFBP2 expression in the fallopian tube epithelium of postmenopausal women is likely due to decreased serum estrogen levels. Indeed, our in vitro and ex vivo studies showing that IGFBP2 expression in tubal secretory epithelial cells depends on estradiol is consistent with our observation that, in all specimens analyzed, postmenopausal fallopian tubes lose IGFBP2 expression. A similar finding has also been reported in endometrial stromal cells in which estradiol increases IGFBP2 expression (58,59). While estrogen contributes to the proliferative activity in tubal epithelium in premenopausal women, the greatly reduced estrogen level in postmenopausal women does not support the proliferative activity in those extant precursor lesions, acting as a natural barrier for tumor progression. Therefore, in order for those STIC cells to progress to HGSC, STIC cells need to acquire new molecular re-wiring that stimulates their proliferation and clonal expansion. From this perspective, we identified a new pathogenic mechanism in HGSC in which epigenetic alterations re-activate IGFBP2 expression which in turn locally promotes cellular proliferation for tumor progression. This mechanism allows STIC and HGSC cells to exhibit estrogen-independent growth in postmenopausal women.
IGFBP2 has been suggested to participate in cancer-driving signaling in a number of cancer types in addition to ovarian cancer (27,60,61). Targeting IGFBP2 and its pathway has emerged as a new avenue for cancer therapy. Pharmacological targeting of the HIF1α-IGFBP2 axis impairs IGF1-AKT signaling and suppresses the growth and metastasis of relapsed anaplastic Wilms tumor (29). OGX-225, an antisense oligonucleotide that targets IGFBP2 mRNA, decreases the proliferation of breast cancer cells (62). Moreover, a human single chain recombinant monoclonal antibody that targets the C-terminus of IGFBP2 and interferes with its binding to integrin inhibits migration and invasion of glioma cells (63), and neutralizing antibodies against IGFBP2 lead to decreased activation of downstream oncogenic signaling pathways and decreased tumor growth (64).
A number of limitations in this study should be pointed out. The technical platform we used for transcriptomic profiling is not ideal for studying very small lesions (less than 100 lesional cells) including minute STICs and p53 signatures. Neither is it suitable for addressing possible intra-STIC heterogeneity, which will require methods with single cell resolution. Based on the current data, we are not able to correlate molecular changes and clinical outcomes in those women with incidental STICs. A large consortium-type study involving collection of tissue materials accompanied by long-term follow-up information would be required. Second, the effects of 5-AZA used in this study were global and not specific to the proximal enhancer of IGFBP2. Our results shown here cannot rule out the possibility of 5-AZA having an indirect (or non-specific) impact on IGFBP2 through global demethylation. Third, it should not be construed that IGFBP2 overexpression is sufficient to sustain the growth and expansion of STICs. For that, it will be necessary to perform functional validation and characterization of other STIC-upregulated genes and to determine how they collaborate to contribute to tumor initiation. Lastly, it should be noted that we only focused on STIC as the origin of HGSC in this study; we did not explore the transcriptomic alterations in other unusual non-STIC precursors such as ovarian surface or cortical inclusion cysts showing dysplasia (1).
In summary, we report that the STIC transcriptomic landscape is populated with several differentially expressed cancer-associated genes that distinguish STICs from normal tubal epithelium. This data set may help, in conjunction with the TCGA ovarian cancer data, various research projects aimed at elucidating the pathophysiology of ovarian cancer and identification of biomarkers for early detection and chemoprevention. In particular, we demonstrate that IGFBP2 upregulation likely plays a pivotal role in propelling tumor progression in ovarian cancer precursor lesions, and we explain how epigenetic regulation of the IGFBP2 during ovarian cancer initiation accounts for estrogen-independent growth of STIC cells.
Supplementary Material
Significance:
Spatial transcriptomics of the earliest precursor lesions of ovarian cancer reveals a role of IGFBP2 in propelling tumor initiation, providing new insights into ovarian cancer development.
Acknowledgements
We gratefully acknowledge Dr. Ronny Drapkin from University of Pennsylvania and Dr. Wei Zhang from Wake Forest University School of Medicine for providing fallopian tube epithelial cell lines and IGFBP2 plasmid, respectively. We thank Dr. Zheng-Cheng Yu at Johns Hopkins for assisting with the mice xenograft experiment. This study was supported by grants: Breakthrough Cancer Foundation (BTC-IOC), US Department of Defense Ovarian Cancer Research Program (OC210403), NIH/NCI (P50CA228991, R01CA260628, R01CA215483, U2C CA271891), the Honorable Tina Brozman Foundation, and Richard W. TeLinde Endowment from the Johns Hopkins University.
Footnotes
Authors’ Disclosures: The authors declare no potential conflicts of interest.
References
- 1.Shih IM, Wang Y, Wang TL. The Origin of Ovarian Cancer Species and Precancerous Landscape. Am J Pathol 2021;191:26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. The American journal of surgical pathology 2007;31:161–9 [DOI] [PubMed] [Google Scholar]
- 3.Vang R, Gupta M, Wu LS, Yemelyanova AV, Kurman RJ, Murphy KM, et al. Diagnostic reproducibility of hydatidiform moles: ancillary techniques (p57 immunohistochemistry and molecular genotyping) improve morphologic diagnosis. Am J Surg Pathol 2012;36:443–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhn E, Kurman RJ, Vang R, Sehdev AS, Han G, Soslow R, et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma- evidence supporting the clonal relationship of the two lesions. J Pathol 2012;226:421–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu RC, Wang P, Lin SF, Zhang M, Song Q, Chu T, et al. Genomic landscape and evolutionary trajectories of ovarian cancer precursor lesions. J Pathol 2019;248:41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu NY, Fang C, Huang HS, Wang J, Chu TY. Natural history of ovarian high-grade serous carcinoma from time effects of ovulation inhibition and progesterone clearance of p53-defective lesions. Mod Pathol 2020;33:29–37 [DOI] [PubMed] [Google Scholar]
- 7.Visvanathan K, Shaw P, May BJ, Bahadirli-Talbott A, Kaushiva A, Risch H, et al. Fallopian Tube Lesions in Women at High Risk for Ovarian Cancer: A Multicenter Study. Cancer Prev Res (Phila) 2018;11:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun 2017;8:1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckert MA, Pan S, Hernandez KM, Loth RM, Andrade J, Volchenboum SL, et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov 2016;6:1342–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn E, Kurman RJ, Soslow RA, Han G, Sehdev AS, Morin PJ, et al. The diagnostic and biological implications of laminin expression in serous tubal intraepithelial carcinoma. Am J Surg Pathol 2012;36:1826–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karst AM, Levanon K, Duraisamy S, Liu JF, Hirsch MS, Hecht JL, et al. Stathmin 1, a marker of PI3K pathway activation and regulator of microtubule dynamics, is expressed in early pelvic serous carcinomas. Gynecol Oncol 2011;123:5–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novak M, Lester J, Karst AM, Parkash V, Hirsch MS, Crum CP, et al. Stathmin 1 and p16(INK4A) are sensitive adjunct biomarkers for serous tubal intraepithelial carcinoma. Gynecologic oncology 2015;139:104–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sehdev AS, Kurman RJ, Kuhn E, Shih Ie M. Serous tubal intraepithelial carcinoma upregulates markers associated with high-grade serous carcinomas including Rsf-1 (HBXAP), cyclin E and fatty acid synthase. Mod Pathol 2010;23:844–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen EY, Mehra K, Mehrad M, Ning G, Miron A, Mutter GL, et al. Secretory cell outgrowth, PAX2 and serous carcinogenesis in the Fallopian tube. The Journal of pathology 2010;222:110–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roh MH, Yassin Y, Miron A, Mehra KK, Mehrad M, Monte NM, et al. High-grade fimbrial-ovarian carcinomas are unified by altered p53, PTEN and PAX2 expression. Mod Pathol 2010;23:1316–24 [DOI] [PubMed] [Google Scholar]
- 16.Quick CM, Ning G, Bijron J, Laury A, Wei TS, Chen EY, et al. PAX2-null secretory cell outgrowths in the oviduct and their relationship to pelvic serous cancer. Mod Pathol 2012;25:449–55 [DOI] [PubMed] [Google Scholar]
- 17.Vang R, Visvanathan K, Gross A, Maambo E, Gupta M, Kuhn E, et al. Validation of an algorithm for the diagnosis of serous tubal intraepithelial carcinoma. Int J Gynecol Pathol 2012;31:243–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung JG, Stoeck A, Guan B, Wu RC, Zhu H, Blackshaw S, et al. Notch3 interactome analysis identified WWP2 as a negative regulator of Notch3 signaling in ovarian cancer. PLoS Genet 2014;10:e1004751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song G, Chen L, Zhang B, Song Q, Yu Y, Moore C, et al. Proteome-wide Tyrosine Phosphorylation Analysis Reveals Dysregulated Signaling Pathways in Ovarian Tumors. Mol Cell Proteomics 2019;18:448–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome research 2002;12:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intraepithelial carcinoma. Nat Commun 2017;8:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article3 [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee A, Rodger EJ, Ahn A, Stockwell PA, Parry M, Motwani J, et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018;4:312–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeifer GP. Defining Driver DNA Methylation Changes in Human Cancer. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pisanic TR 2nd, Wang Y, Sun H, Considine M, Li L, Wang TH, et al. Methylomic Landscapes of Ovarian Cancer Precursor Lesions. Clin Cancer Res 2020;26:6310–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee EJ, Mircean C, Shmulevich I, Wang H, Liu J, Niemisto A, et al. Insulin-like growth factor binding protein 2 promotes ovarian cancer cell invasion. Mol Cancer 2005;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012;13:484–92 [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Nelson MV, Bailey C, Zhang P, Zheng P, Dome JS, et al. Targeting the HIF-1alpha-IGFBP2 axis therapeutically reduces IGF1-AKT signaling and blocks the growth and metastasis of relapsed anaplastic Wilms tumor. Oncogene 2021;40:4809–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patil SS, Gokulnath P, Bashir M, Shwetha SD, Jaiswal J, Shastry AH, et al. Insulin-like growth factor binding protein-2 regulates beta-catenin signaling pathway in glioma cells and contributes to poor patient prognosis. Neuro Oncol 2016;18:1487–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perks CM, Vernon EG, Rosendahl AH, Tonge D, Holly JM. IGF-II and IGFBP-2 differentially regulate PTEN in human breast cancer cells. Oncogene 2007;26:5966–72 [DOI] [PubMed] [Google Scholar]
- 32.Foulstone EJ, Zeng L, Perks CM, Holly JM. Insulin-like growth factor binding protein 2 (IGFBP-2) promotes growth and survival of breast epithelial cells: novel regulation of the estrogen receptor. Endocrinology 2013;154:1780–93 [DOI] [PubMed] [Google Scholar]
- 33.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science 2017;357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun 2018;9:3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korah R, Das K, Lindy ME, Hameed M, Wieder R. Coordinate loss of fibroblast growth factor 2 and laminin 5 expression during neoplastic progression of mammary duct epithelium. Human pathology 2007;38:154–60 [DOI] [PubMed] [Google Scholar]
- 36.Liu D, Buluwela L, Ali S, Thomson S, Gomm JJ, Coombes RC. Retroviral infection of the FGF2 gene into MCF-7 cells induces branching morphogenesis, retards cell growth and suppresses tumorigenicity in nude mice. Eur J Cancer 2001;37:268–80 [DOI] [PubMed] [Google Scholar]
- 37.Udby L, Calafat J, Sorensen OE, Borregaard N, Kjeldsen L. Identification of human cysteine-rich secretory protein 3 (CRISP-3) as a matrix protein in a subset of peroxidase-negative granules of neutrophils and in the granules of eosinophils. J Leukoc Biol 2002;72:462–9 [PubMed] [Google Scholar]
- 38.Lezcano C, Muller AM, Frosina D, Hernandez E, Geronimo JA, Busam KJ, et al. Immunohistochemical Detection of Cancer-Testis Antigen PRAME. Int J Surg Pathol 2021;29:826–35 [DOI] [PubMed] [Google Scholar]
- 39.Wang C, Gu Y, Zhang K, Xie K, Zhu M, Dai N, et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun 2016;7:10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Barger CJ, Eng KH, Klinkebiel D, Link PA, Omilian A, et al. PRAME expression and promoter hypomethylation in epithelial ovarian cancer. Oncotarget 2016;7:45352–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woloszynska-Read A, James SR, Link PA, Yu J, Odunsi K, Karpf AR. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 42.Epping MT, Wang L, Edel MJ, Carlee L, Hernandez M, Bernards R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 2005;122:835–47 [DOI] [PubMed] [Google Scholar]
- 43.Bell RE, Golan T, Sheinboim D, Malcov H, Amar D, Salamon A, et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res 2016;26:601–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohan S, Baylink DJ. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J Endocrinol 2002;175:19–31 [DOI] [PubMed] [Google Scholar]
- 45.Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol 2013;14:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Jiang T, Zhang J, Zhang B, Yang W, You G, et al. Elevated serum antibodies against insulin-like growth factor-binding protein-2 allow detecting early-stage cancers: evidences from glioma and colorectal carcinoma studies. Ann Oncol 2012;23:2415–22 [DOI] [PubMed] [Google Scholar]
- 47.Park KH, Gad E, Goodell V, Dang Y, Wild T, Higgins D, et al. Insulin-like growth factor-binding protein-2 is a target for the immunomodulation of breast cancer. Cancer Res 2008;68:8400–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Ying X, Han S, Wang J, Zhou X, Bai E, et al. Autoantibodies against insulin-like growth factorbinding protein-2 as a serological biomarker in the diagnosis of lung cancer. Int J Oncol 2013;42:93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cecil DL, Liao JB, Dang Y, Coveler AL, Kask A, Yang Y, et al. Immunization with a Plasmid DNA Vaccine Encoding the N-Terminus of Insulin-like Growth Factor Binding Protein-2 in Advanced Ovarian Cancer Leads to High-level Type I Immune Responses. Clin Cancer Res 2021;27:6405–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsu CF, Huang HS, Chen PC, Ding DC, Chu TY. IGF-axis confers transformation and regeneration of fallopian tube fimbria epithelium upon ovulation. EBioMedicine 2019;41:597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chu T, Shih IM. Follicular fluid has more to offer: Insulin-like growth factor axis on ovarian carcinogenesis. EBioMedicine 2019;41:30–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allard JB, Duan C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front Endocrinol (Lausanne) 2018;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer 2014;14:329–41 [DOI] [PubMed] [Google Scholar]
- 54.Huang HS, Chu SC, Hsu CF, Chen PC, Ding DC, Chang MY, et al. Mutagenic, surviving and tumorigenic effects of follicular fluid in the context of p53 loss: initiation of fimbria carcinogenesis. Carcinogenesis 2015;36:1419–28 [DOI] [PubMed] [Google Scholar]
- 55.Bahar-Shany K, Brand H, Sapoznik S, Jacob-Hirsch J, Yung Y, Korach J, et al. Exposure of fallopian tube epithelium to follicular fluid mimics carcinogenic changes in precursor lesions of serous papillary carcinoma. Gynecol Oncol 2014;132:322–7 [DOI] [PubMed] [Google Scholar]
- 56.Brodowska A, Grabowska M, Bittel K, Ciecwiez S, Brodowski J, Szczuko M, et al. Estrogen and Progesterone Receptor Immunoexpression in Fallopian Tubes among Postmenopausal Women Based on Time since the Last Menstrual Period. Int J Environ Res Public Health 2021;18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S, Dai X, Gao Y, Shen F, Ding J, Chen Q. The positivity of estrogen receptor and progesterone receptor may not be associated with metastasis and recurrence in epithelial ovarian cancer. Sci Rep 2017;7:16922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giudice LC, Milkowski DA, Fielder PJ, Irwin JC. Characterization of the steroid-dependence of insulin-like growth factor-binding protein-2 synthesis and mRNA expression in cultured human endometrial stromal cells. Hum Reprod 1991;6:632–40 [DOI] [PubMed] [Google Scholar]
- 59.Chan TW, Pollak M, Huynh H. Inhibition of insulin-like growth factor signaling pathways in mammary gland by pure antiestrogen ICI 182,780. Clin Cancer Res 2001;7:2545–54 [PubMed] [Google Scholar]
- 60.Wang H, Rosen DG, Wang H, Fuller GN, Zhang W, Liu J. Insulin-like growth factor-binding protein 2 and 5 are differentially regulated in ovarian cancer of different histologic types. Mod Pathol 2006;19:1149–56 [DOI] [PubMed] [Google Scholar]
- 61.Haschemi R, Kobelt D, Steinwarz E, Schlesinger M, Stein U, Bendas G. Insulin-like Growth Factor Binding Protein-2 (IGFBP2) Is a Key Molecule in the MACC1-Mediated Platelet Communication and Metastasis of Colorectal Cancer Cells. Int J Mol Sci 2021;22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.So AI, Levitt RJ, Eigl B, Fazli L, Muramaki M, Leung S, et al. Insulin-like growth factor binding protein-2 is a novel therapeutic target associated with breast cancer. Clin Cancer Res 2008;14:6944–54 [DOI] [PubMed] [Google Scholar]
- 63.Patil SS, Railkar R, Swain M, Atreya HS, Dighe RR, Kondaiah P. Novel anti IGFBP2 single chain variable fragment inhibits glioma cell migration and invasion. J Neurooncol 2015;123:225–35 [DOI] [PubMed] [Google Scholar]
- 64.Phillips LM, Zhou X, Cogdell DE, Chua CY, Huisinga A, K RH. et al. Glioma progression is mediated by an addiction to aberrant IGFBP2 expression and can be blocked using anti-IGFBP2 strategies. J Pathol 2016;239:355–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplemental data files. The raw data of the spatial transcriptomics were provided by the Nanostring bioinformatic team through a service agreement and are available in Supplemental Table S3.