

Abstract
Objective:
The NOTCH1 gene mutation has been identified in bicuspid aortic valve patients. We developed an in vitro model with human induced pluripotent stem cells (iPSCs) to evaluate the role of NOTCH1 in smooth muscle and endothelial cell (EC) differentiation.
Methods:
The iPSCs were derived from a patient with a normal tricuspid aortic valve and aorta. The NOTCH1 gene was targeted in iPSCs with the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 nuclease (Cas9) system. The NOTCH1−/− (NOTCH1 homozygous knockout) and isogenic control iPSCs (wild type) were differentiated into neural crest stem cells (NCSCs) and into cardiovascular progenitor cells (CVPCs). The NCSCs were differentiated into smooth muscle cells (SMCs). The CVPCs were differentiated into ECs. The differentiations of SMCs and ECs were compared between NOTCH1−/− and wild type cells.
Results:
The expression of NCSC markers (SRY-related HMG-box 10 and transcription factor AP-2 alpha) was significantly lower in NOTCH1−/− NCSCs than in wild type NCSCs. The SMCs derived from NOTCH1−/− NCSCs showed immature morphology with smaller size and decreased expression of all SMC-specific contractile proteins. In NOTCH1−/− CVPCs, the expression of ISL1, NKX2.5, and MYOCD was significantly lower than that in isogenic control CVPCs, indicating impaired differentiation from iPSCs to CVPCs. The NOTCH1−/− ECs derived from CVPCs showed significantly lower expression of cluster of differentiation 105 and cluster of differentiation 31 mRNA and protein, indicating a defective differentiation process.
Conclusions:
NOTCH1 is critical in SMC and EC differentiation of iPSCs through NCSCs and CVPCs, respectively. NOTCH1 gene mutations might potentially contribute to the development of thoracic aortic aneurysms by affecting SMC differentiation in some patients with bicuspid aortic valve.
Keywords: NOTCH1, induced pluripotent stem cells, smooth muscle cells, bicuspid aortic valve, thoracic aortic aneurysm
Graphical Abstract

The NOTCH1 knockout displays severely impaired human SMC differentiation.
Central Message
The NOTCH1 gene plays a critical role in neural crest differentiation and subsequent SMC differentiation from human induced pluripotent stem cells.
Bicuspid aortic valve (BAV) is the most common congenital cardiovascular malformation, affecting 0.9% to 2% of people worldwide and approximately 3 million people (2 million adults, 1 million children) in the United States.1,2 The aortic valves of BAV patients are subject to early degeneration and subsequent valve dysfunction. BAV is also frequently found as an underlying cause of proximal aortic dilatation, including aortic root, ascending aorta, and aortic arch, known as BAV aortopathy, which accounts for more morbidity and mortality than all other congenital malformations combined.3 Specifically, dilatation of the proximal aorta occurs in approximately 20% to 84% of BAV patients, which is significantly higher than in patients with tricuspid aortic valves.4 Moreover, BAV patients have a ninefold higher risk of thoracic aortic dissections corresponding with a high risk of mortality.5
Currently, there is no medical treatment to prevent or mitigate thoracic aortic aneurysm development in BAV patients, and the molecular and cellular mechanisms that drive BAV aortopathy are not known. There is a need to further understand the genetic variants involved in BAV aortopathy as well as the pathways involved in aortic valve development.
NOTCH1 was identified through genome-wide linkage analysis6 as the first gene causing nonsyndromic BAV. NOTCH1 encodes for a transmembrane protein responsible for activating intracellular signaling pathways critical in cardiac embryogenesis, aortic and pulmonary valve development, and in the homeostasis of the aorta and other great vessels.6–8 To maintain smooth muscle contractile phenotype, NOTCH1 signaling stimulates the expression of smooth muscle-specific contractile proteins, including smooth muscle α-actin (SMA), calponin 1, smooth muscle 22 α (SM22α), and smooth muscle myosin heavy chain in human aortic smooth muscle cells (SMCs).8 In addition, NOTCH1 functions as the predominant receptor in the endocardial cushion, which forms the semilunar valves through the process of endothelial-to-mesenchymal transformation.9,10
In this study, we modeled the effects of NOTCH1 deficiency on proximal aortic SMCs and aortic valve endothelial cell (EC) differentiation by adapting a well characterized in vitro iPSC differentiation system and Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 nuclease (Cas9) system–mediated gene editing. We hypothesized that depletion of functional NOTCH1 protein would impair human neural crest stem cell (NCSC)–derived SMC differentiation as well as cardiovascular progenitor cell (CVPC)–derived EC differentiation.
METHODS
Patient Selection
This study was approved by the institutional review board at the University of Michigan (date and number of institutional review board approval: January 19, 2012, HUM00054585), and informed consent was obtained from patients. The donor was a patient with a normal NOTCH1 gene and normal trileaflet aortic valve without any aortic aneurysm, which were confirmed using targeted gene sequencing, echocardiogram, computed tomography angiogram of the aorta, and direct operative observation. By knocking out the NOTCH1 gene with CRISPR/Cas9, we revealed the role of the NOTCH1 gene in the differentiation of iPSCs to SMCs and ECs.
iPSC Generation and Culture
The blood samples were obtained through peripheral phlebotomy with heparinized test tubes. The procedure of iPSC derivation was performed according to the methods previously described.11 Peripheral blood mononuclear cells were isolated from human peripheral blood, cultured, and electrotransfected with plasmids containing octamer-binding transcription factor 4 (OCT4), SRY (sex determining region Y)–box 2 (SOX2), Kruppel-like factor 4 (KLF4), and C-MYC (a gift from Dr Xiaobing Zhang, Loma Linda University, Calif). Approximately 30 days post-infection, the colonies were mechanically picked up from the culture dishes and were first cultured with mouse embryonic fibroblasts for 3 passages12 and then transferred to TesRE8 medium (Stemcell Technologies, Vancouver, Canada) on Matrigel-coated (BD Corp, Franklin Lakes, NJ) dishes. iPSCs were passaged every 4 to 6 days with Versene (Life Technologies Corp, Carlsbad, Calif) and iPSCs from passage 25 to passage 35 were used in experiments.
NOTCH1 Single-Guide RNA Design and Electrotransfection of iPSCs
Single-guide RNA (sgRNA) was designed to target NOTCH1 with a CRISPR/Cas9 system for genome engineering. The sgRNA design tool was developed by Dr Feng Zhang and colleagues.13 The sequence of NOTCH1 sgRNA was: 5′-GAGGTGGCTGCGCAGCGACA-3′. The target site was: chr9:+136508236. The quality score was 76. The minimal number of mismatch nucleotides in offsite targets was 3. The sgRNAs were cloned into PX458, which contains SpCas9-2A-enhanced green fluorescent protein, using a previously described method.13 One million iPSCs were electrotransfected with constructed 5 μg of PX458/NOTCH1 sgRNA using Lonza Human Stem Cell Nucleofector Kit 2 with program U-023 on a Nuclefector 2 device (Lonza Ltd, Basel, Switzerland). Another 1 million iPSCs were electrotransfected with PX458 vector as control under the same conditions. Transfected iPSCs were subcloned and the targeted site of NOTCH1 was amplified from each clone and sequenced. Homozygous NOTCH1 mutant and wild type clones were selected and compared for further experiments.
EC Differentiation From iPSC-Derived CVPCs
To differentiate iPSCs into CVPCs, iPSCs were dissociated into single cells with Versene (Life Technologies Corp) and seeded at 2 × 104 cells per cm2 with TesRE8 (Stemcell Technology Inc) medium supplemented with 10 μm Rocki (Y27632; Stemgent Inc, Lexington, Mass).14 When the cells reached a density of 20% to 30%, the medium was changed into the differentiation medium, which contained DMEM/F12 (Life Technologies Corp), L-glutamine (Life Technologies Corp), penicillin/streptomycin (Life Technologies Corp), B27supplement without vitamin A (Life Technologies Corp), 400 μM 1-thioglycerol (Sigma Corp, St Louis, Mo), 50 μg/mL ascorbic acid (Sigma Corp), 25 ng/mL BMP4 (R&D Systems Corp, Minneapolis, Minn), and 6 μM glycogen synthase kinase 3 inhibitor CHIR99021(Sigma Corp). The differentiation medium was refreshed daily for 3 days. To differentiate CVPCs into ECs, CVPCs were dissociated with Accutase (Life Technologies Corp) and seeded at 1 × 104 cells/cm2 on Matrigel (BD Corp)-coated dishes with EC medium containing Stempro34 (Life Technologies Corp), Stempro34 supplement (Life Technologies Corp), penicillin/streptomycin (Life Technologies Corp), L-glutamine (Life Technologies Corp), and 50 ng/mL vascular endothelial growth factor (Peprotech Inc, Rocky Hill, NJ). Medium was refreshed every 2 days for 13 days. Differentiation to CVPCs was tested using Isl-1 immunohisto-chemistry and to ECs using cluster of differentiation 31 (CD31).14
SMC Differentiation From iPSC-Derived NCSCs
To differentiate iPSCs into NCSCs, iPSCs were dissociated into single cells with Versene (Life Technologies Corp) and seeded at 2 × 104 cells/cm2 with TesRE8 (Stemcell Technology Inc) medium supplemented with 10 μM Rocki (Y27632; Stemgent Inc). When the cells reached densities of 20% to 30%, the medium was changed into the differentiation medium, which contained DMEM/F12 (Life Technologies Corp), L-glutamine (Life Technologies Corp), penicillin/streptomycin (Life Technologies Corp), B27 supplement without vitamin A (Life Technologies Corp), 3 μM CHIR99021, 10 μM SB481542, and 200 ng/mL Noggin. After 8 to 10 days, NCSCs were dissociated with Versene (Life Technologies Corp) and seeded on cell culture dishes. When the cells reached densities of 60% to 80%, the medium was changed into the differentiation medium, which contained DMEM/F12 (Life Technologies Corp), L-glutamine (Life Technologies Corp), penicillin/streptomycin (Life Technologies Corp), B27 supplement without vitamin A (Life Technologies Corp), and 2 ng/mL transforming growth factor β1 to differentiate into SMCs. The differentiation medium was refreshed every 2 days for 12 days.
Immunofluorescence Staining and Flow Cytometry
Immunofluorescence staining and flow cytometry were performed as described.15 The following primary antibodies were used: anti-NOTCH1 (Cell Signaling Technology Inc, Danvers, Mass), anti-CD31 (Abcam Inc, Cambridge, Mass), anti-SMA (Sigma Corp) anti-P75 (Advanced Targeting Systems, San Diego, Calif). The following fluorochrome-conjugated secondary antibodies were used: Alexa Fluor 488 goat anti-rabbit immunoglobulin (Ig)G (Molecular Probes, Eugene, Ore), Alexa Fluor 594 goat anti-rabbit IgG (Molecular Probes), and Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes). Slides were mounted with antifade mounting media containing 4′,6-diamidino-2-phenylindole (Prolonggold; Life Technologies Corp), and were observed using a Nikon A1 confocal microscope (Nikon Corp, Tokyo, Japan). In a flow cytometry study, electrotransfected iPSCs were dissociated into single cells with Accutase (Stemcell Technology Inc), and applied to MoFloAstrios (Beckman Coulter Inc, Brea, Calif) flow cytometry machine.
Western Blot Analysis
Whole-cell extracts were prepared using radioimmunoprecipitation assay buffer, were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis gels, and were transferred to acetate cellulose membranes. Primary antibodies used were anti-OCT4 (Abcam Inc), anti-NOTCH1 (Cell Signaling Technology Inc), anti-SMA (Sigma Corp), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:2000; Santa Cruz Inc, Dallas, Tex). Secondary antibodies used were IRDye800CW Donkey anti-Mouse, IRDye680LT Donkey anti-Rabbit, IRDye800CW Donkey anti-Rabbit (all secondary antibodies were from Licor Inc, Lincoln, Neb). A Licor Western blot detection system was used for the dual-color imaging. ImageJ (National Institutes of Health, Bethesda, Md) was used for quantification of bands. Each band was normalized to GAPDH. Experiments were repeated 3 times. All data points and median were plotted.
Quantitative Polymerase Chain Reaction
TRIzol Reagent (Invitrogen, Carlsbad, Calif) was used to purify total RNA. RNA (1 μg) was reverse-transcribed using M-MLV Reverse Transcriptase (Promega Corp, Madison, Wis) and RNasein RNase Inhibitor (Promega Corp). Quantitative polymerase chain reaction (PCR) was carried out with SYBR universal mix (BioRad Corp, Hercules, Calif) and a Stepone Realtime PCR machine (Applied Biosystems, Foster City, Calif). The expression level of genes in each sample was normalized to GAPDH.
Targeted Sequencing of the NOTCH1 Gene in BAV Patients
To identify the NOTCH1 gene mutations in BAV patients, genomic DNA was extracted from peripheral blood of BAV patients (n = 108). Each exon of NOTCH1 gene was amplified using PCR. The PCR products were purified using E.Z.N.A. PCR purification Kit (Omega Bio-tek, Nor-cross, Ga), and Sanger sequenced at the Sequence Core of the University of Michigan Medical School. Sequencing results were reported in ab1 format and opened with Chromas software (version 2.6.5; Technelysium, South Brisbane, Australia). Each peak represented a single nucleotide base in DNA. Each fragment was analyzed using sequence Mutation Surveyor software (version 2.2; Softgenetics, State College, Pa), and confirmed manually.
Statistical Analysis
In this side-by-side comparative study between wild type and NOTCH1 homozygous knockout (NOTCH1−/−) iPSCs, we determined the mutation-associated biological effects using 2 differentiation processes: iPSCs-NCSC-SMCs and iPSCs-CVPCs-ECs. The differentiation experiments were performed at least 3 times. Because we use a cellular model and the difference is so dramatic after knocking out the NOTCH1 gene in human iPSCs, a minimal sample size (n = 3) has sufficient statistical power to determine the significance. Student t test was used to calculate the P value. P <.05 was considered significant. Prism 7 (GraphPad Software, San Diego, Calif) was used for the statistical analysis.
RESULTS
NOTCH1 Is Targeted by CRISPR/Cas9 and sgRNA
We investigated the biological effect of the NOTCH1 gene in human iPSC differentiation to SMCs and ECs. Human iPSCs were generated from peripheral blood mononuclear cells of a donor with a normal trileaflet aortic valve, using nonintegrated DNA vectors containing OCT4, SOX2, C-MYC, and KLF4.16 The pluripotency of iPSCs was confirmed.16 sgRNA targeting NOTCH1 was constructed into PX458 (see Ran et al13; Figure 1, A and B). iPSCs were electrotransfected with PX458, and the homozygous truncated NOTCH1 clone, which harbors a frameshift mutation, was selected from screening using Sanger sequencing (Figure E1, A and B), and selected clones from wild type as well as truncated NOTCH1 groups showed similar stem cell morphology (Figure 1, C). NCSCs were differentiated from both groups of iPSCs and showed typical neural crest morphology (Figure 1, D). In the Western blot assay, normal NOTCH1 expression levels were detected in iPSCs as well as NCSCs of the control group, but were undetectable in the NOTCH1−/− group (Figure 1, E), indicating successful targeting on the NOTCH1 locus. Further, the OCT4 protein level decreased significantly from iPSCs to NCSCs in wild type as well as truncated NOTCH1 groups, indicating efficient exit from pluripotency from iPSCs to NCSCs (Figure 1, E).
FIGURE 1.

NOTCH1 was targeted with CRISPR/Cas9. A, Structure of CRISPR/Cas9 and sgRNA plasmid. B, NOTCH1 protein structure and truncated NOTCH1 after targeting by CRISPR/Cas9 and sgRNA. C, Morphology of wild type (WT) and truncated NOTCH1 (NOTCH1−/−) iPSCs. Scale bars represent 200 μm. D, Morphology of NCSCs with WT and NOTCH1−/− that were differentiated from iPSCs counterparts. Scale bars represent 500 μm. E, Left panel: Western blot of NOTCH1, OCT4, and GAPDH in iPSCs and NCSCs with WT and NOTCH1−/−. Middle and right panels: quantification of Western blot analysis. The signal was normalized to GAPDH and then normalized to WT iPSCs. In the middle panel, **P<.01, NOTCH1−/− iPSCs versus WT iPSCs; and NOTCH1−/− NCSCs versus WT NCSCs separately. In the right panel, **P<.01, WT NCSCs versus WT iPSCs; and NOTCH1−/− NCSC versus NOTCH−/− iPSCs separately. Experiments were repeated 3 times and all data points and median were plotted. U6, U6 promoter; sgRNA, single-guide RNA; CBh, CBh promoter; 2A GFP, 2A green fluorescent protein; EGF, epidermal growth factor; LNR, LIN-12/notch repeat motif; TM, transmembrance domain; ANK, ankyrin repeat; PEST, PEST domain; iPSCs, induced pluripotent stem cells; NCSCs, neural crest stem cells; OCT4, octamer-binding transcription factor 4; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
FIGURE E1.

DNA sequencing result of NOTCH1 homozygous knockout (NOTCH1−/−) cell line. A, Wild type (WT) NOTCH1 sequence, gRNA target site and protospacer adjacent motif (PAM) position. B, Sanger DNA sequencing result of NOTCH1 homozygous (NOTCH1−/−) knockout cell line. The sequence of NOTCH1−/− gene is shown. The vertical black line indicates the position of the deletion of 7 base pairs (bp). The deletion of 7 bp in NOTCH1 results in a frameshift mutation at the 1108th amino acid and a premature stop codon at 1177. The resulting protein lacks NOTCH1 amino acids from residue 1108 to the C-terminus. gRNA, Guide RNA; hNOTCH1, human NOTCH1 gene.
iPSCs With Truncated NOTCH1 Have Impaired SMC Differentiation
iPSCs with truncated NOTCH1 were differentiated into SMCs to dissect the role of NOTCH1 in SMCs differentiation. iPSCs from the wild type as well as the truncated NOTCH1 group were differentiated into neural crest cells using 3 small-molecule inhibitors.17 More than 95% of cells in differentiated cell populations were P75-positive in the immunofluorescence assay, indicating a high differentiation efficiency (Figure 2, A). Quantitative PCR experiments showed SRY-related HMG-box 10 (SOX10) and transcription factor AP-2 alpha (TFAP2A) were significantly elevated in the wild type group, indicating the activation of neural crest cell-specific gene regulatory network in differentiated neural crest cells (Figure 2, B). In the truncated NOTCH1 group, neural crest cells showed significantly lower expression of SOX10 and TFAP2A than the wild type group. Neural crest cells from iPSCs were further differentiated into SMCs. In the control group, more than 95% of differentiated cells were stained positive for SMC contractile protein, SMA; and immunoblotting showed significant elevation of other SMC contractile proteins, calponin 1, transgelin (TAGLN), and myosin 11 (MYH11), indicating that differentiation efficiency to SMCs is high (Figure 2, C and D). Noticeably, SMCs in the truncated NOTCH1 group showed obviously smaller morphology, indicating immature SMC differentiation (Figure 2, C). Western blot assays further confirmed that the expression of the genes of SMC contractile proteins, including MYH11, SMA, CNN1, and TAGLN, were significantly lower in the truncated NOTCH1 group at SMC differentiation day 8 and day 13 (Figure 2, D).
FIGURE 2.
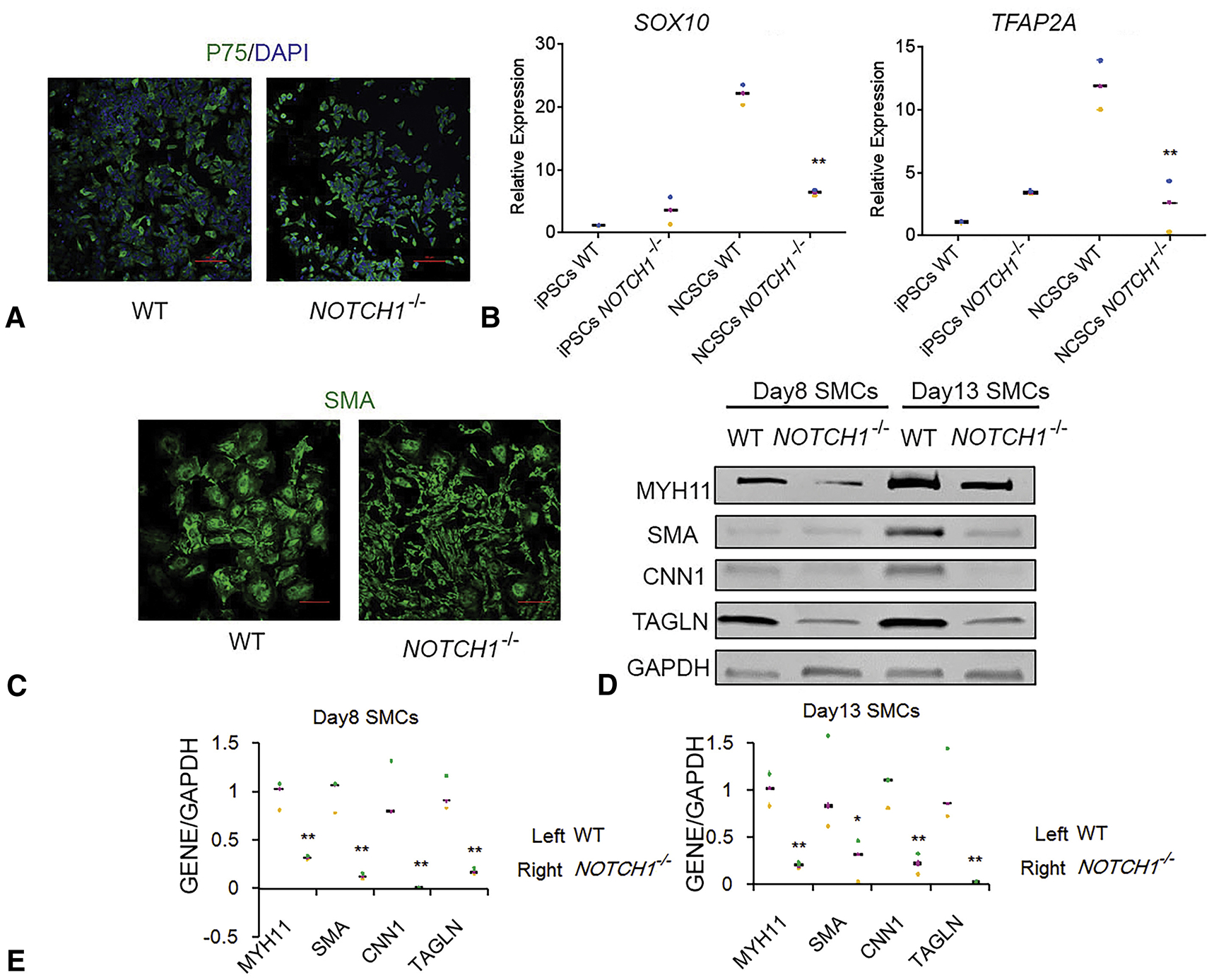
The NOTCH1 knockout shows severely impaired SMC differentiation. A, P75 immunostaining of NCSCs with wild type (WT) and truncated NOTCH1 (NOTCH1−/−) that were differentiated from iPSC counterparts. Scale bars represent 100 μm. B, Quantitative polymerase chain reaction of SOX10 and TFAP2A expression level in iPSCs and NCSCs with WT and NOTCH1−/−. Expression was normalized with GAPDH and further normalized to WT iPSCs. Experiments were repeated 3 times and all data points and median were plotted. **P<.01, NOTCH1−/− NCSCs versus WT NCSCs. C, SMA immunostaining of SMCs that differentiated from NCSCs with WT and NOTCH1−/−. Scale bars represent 100 μm. D, Western blot of expressions of MYH11, SMA, CNN1, TAGLN, and GAPDH in SMCs differentiated from NCSCs with WT and NOTCH1−/−. E, Quantification of Western blot analysis. The signal was normalized to GAPDH and then normalized to WT SMCs. *P<.05; **P<.01, NOTCH1−/− SMCs versus WT SMCs. DAPI, 4′,6-diamidino-2-phenylindole; SOX10, SRY-related HMG-box 10; iPSCs, induced pluripotent stem cells; NCSCs, neural crest stem cells; TFAP2A, transcription factor AP-2 alpha; SMA, smooth muscle α-actin; SMC, smooth muscle cell; MYH11, myosin 11; CNN1, calponin 1; TAGLN, transgelin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
iPSCs With Truncated NOTCH1 Have Impaired Differentiation of ECs
iPSCs with truncated NOTCH1 were differentiated into ECs through CVPCs.18 Quantitative PCR showed islet 1, NKX2.5, and myocardin were significantly lower in the truncated NOTCH1 group, indicating impaired differentiation from iPSCs to CVPCs (Figure 3, A). In the wild type group, CVPCs showed normal expression of NOTCH1 (Figure 3, B), indicating that NOTCH1 plays an essential role in the differentiation of iPSCs to CVPCs. CVPCs from iPSCs were further differentiated into ECs. In the differentiated cells, more than 70% of cells were stained positive for CD31, the marker of ECs (Figure 3, C). Immunofluorescence assay further showed that EC marker CD31 protein levels were significantly lower in the truncated NOTCH1 group at differentiation days 7, 11, and 13 (Figure 3, C). In the whole population of differentiated cells, noticeably, ECs in the truncated NOTCH1 group showed significantly lower expression of cluster of differentiation 105 and CD31, indicating immature EC differentiation (Figure 3, D).
FIGURE 3.

Cardiovascular progenitor cells (CVPCs) and endothelial cells (ECs) differentiation from iPSCs with wild type (WT) and truncated NOTCH1 (NOTCH1−/−). A, Quantitative polymerase chain reaction of ISL1, NKX2.5, and MYOCD expression level in CVPCs with WT and NOTCH1−/−. Expression was normalized with GAPDH and further normalized to WT CVPCs. Experiments were repeated 3 times and all data points and median were plotted. **P<.01, NOTCH1−/− CVPCs versus WT CVPCs. B, NOTCH1 immunostaining of WT CVPCs, which were differentiated from iPSC counterparts. Scale bars represent 100 μm. C, CD31 immunostaining of ECs that differentiated from CVPCs with WTand NOTCH1−/−. Scale bars represent 100 μm. D, Quantitative polymerase chain reaction of CD105 and CD31 expression level in ECs that differentiated from CVPCs with WT and NOTCH1−/−. Expression was normalized with GAPDH and further normalized to WT ECs. Experiments were repeated 3 times and all data points and median were plotted. **P<.01, NOTCH1−/− ECs versus WT ECs. ISL1, islet1; NKX2.5, NK2 homeobox 5; MYOCD, myocardin; DAPI, 4′,6-diamidino-2-phenylindole; CD105, cluster of differentiation 105; CD31, cluster of differentiation 31.
Targeted Gene Sequencing of NOTCH1 in BAV Patients
Virtual translation of coding sequences showed that no missense mutations or nonsense mutations were present in the 108 patients screened. All changes encoded silent mutations.
DISCUSSION
In this study, we knocked out the NOTCH1 gene in human iPSCs with CRISPR/Cas9. We found severely compromised differentiation of: (1) SMCs from NCSC lineage, and (2) ECs from CVPCs. These findings indicate that NOTCH1 gene mutation potentially disrupts differentiation of cells that might drive valvulopathy and aortopathy in BAV patients.
NOTCH1 mutations have been reported to be associated with BAV formation and calcification, but <6% of BAV cases are accounted for by NOTCH1 variation.6,19 The first discovery of NOTCH1-truncated mutations (R1108X and H1505del) in its extracellular domain in 2 families with familial aortic valve disease uncovered the causal genotype-phenotype correlation between functional protein mutations and BAV malformation and calcification.6 Our NOTCH1−/− iPSCs had a frameshift mutation at the 1108th amino acid and a premature stop codon at 1177, which ablated multiple epidermal growth factor repeats and 4 other domains of NOTCH1 (Figure 1, Figure E1). The truncation of NOTCH1 protein created with CRISPR/Cas9 in our study is almost identical to the truncation of NOTCH1 protein in the patients with NOTCH1-truncated mutations (R1108X and H1505del). Because the aortic valve is originated from CVPCs of the second heart field,20–22 we differentiated the iPSCs into CVPCs, then differentiated into ECs to model the process of aortic valve development. Our results indicate the NOTCH1 gene is not only critical for the differentiation of ECs, but also the differentiation of CVPCs from pluripotent stem cells (Figure 3). This is consistent with mice with conditional Notch1 loss-of-function in precardiac mesodermal progenitors exhibiting defective CVPC fate decisions and restrictions on CVPC expansion.23 Therefore, it is possible that the mutations in the NOTCH1 gene in BAV patients could affect processes of differentiation of CVPCs and later ECs, subsequently resulting in aortic valve malformation.
The role of NOTCH1 gene mutations in the aortopathy in BAV patients is unknown. Although there is a longstanding debate on the underlying mechanism of genetic and hemodynamic bases for BAV aortopathy, both lines of studies have shown convincing evidence suggesting that genetic abnormities in vascular walls contribute to the intrinsic susceptibility to hemodynamic shear stress caused by defective aortic valves.24,25 In our previous study, we found that SMCs, which were differentiated from aneurysmal BAV patients’ iPSCs through neural crest cells,16 showed decreased expression of contractile protein myosin heavy chain and impaired contractile function indicating intrinsic aortopathy in aneurysmal BAV patients. However, the cause of decreased contractile protein levels was left unanswered. It was also unknown whether NOTCH1 mutations affect differentiation of the SMCs in BAV patient cells with NOTCH1 mutations. Notch signaling plays critical roles in vascular SMC differentiation throughout the process of cardiovascular systemic development, categorized by 2 processes: construction of the vessel wall and arterial-venous specification.26 Compelling evidence has proven that individually, knockout ligand, receptor, and effectors of the Notch signaling cascade of murine models lead to similar vascular malfunction and striking abnormalities during vascular development.27 In addition, enforced expression of NOTCH intracellular domain in human primary aortic SMCs strengthens the transcriptional activity of SMC contractile genes.8 Several studies identified NOTCH1 missense mutations in 10% to 11% of BAV patients with thoracic aortic aneurysm.28,29 A significantly lower expression of SMC markers, such as SMA, SM22α, calponin, and smoothelin has also been observed in ascending aortic wall tissue samples from BAV patients compared with those from tricuspid aortic valve patients.30,31 Patients with MYH11 and alpha-actin-2 mutations develop thoracic aortic aneurysm and dissection due to malfunction of the mechanics of SMCs.32–34 When the expression of contractile proteins is compromised due to NOTCH1 mutations, one could expect similar impairment of biomechanics of the SMCs and development of aortic aneurysms. Taken together, it is logical to hypothesize that NOTCH1 mutations cause the defective differentiation of SMCs, which contributes to the aortopathy in BAV patients.
The thoracic aortic aneurysm in BAV patients almost exclusively involves the proximal aorta, including the aortic root, ascending aorta, and aortic arch, but spares the aorta distal to the left subclavian artery.35 The SMCs at the proximal aorta arise from NCSCs and the SMCs at the descending thoracic aorta arise from paraxial mesoderm cells.20–22,36 Taking advantage of a well established in vitro vascular SMC differentiation system, by which different lineages of vascular SMCs can be generated with specific embryonic origins that eventually populate different aortic regional sections,20 we detected a role of NOTCH1 on iPSCs-derived NCSC and SMC differentiation. We found that differentiation of NOTCH1−/− iPSCs into the NCSCs was severely compromised. Furthermore, the NOTCH1−/− SMCs differentiated from NCSCs were not mature and expressed significantly decreased levels of all contractile proteins, including SMC myosin heavy chain, SMC actin, calponin, and SM22α. Those findings indicate NOTCH1 is critical in SMC differentiation through the NCSC lineage, which gives rise to all the SMCs in the media of the proximal aorta. In general, Notch-mediated lateral induction and lateral inhibition have been considered important control mechanisms in the development of neural crest domain within ectoderm and subsequent cell fate diversification in murine models.37 Any loss-of-function mutations, such as truncation of NOTCH1, could affect the differentiation and maturation of SMCs in the proximal aorta, causing aortopathy in BAV patients.
CONCLUSIONS
Our initial finding indicated a NOTCH1 defect in iPSCs compromised the efficiency of differentiation of NCSCs as well as CVPCs from pluripotent stem cells, as well as the differentiation of NCSC-derived SMCs and CVPC-derived ECs. Further studies are needed to identify effects of NOTCH1 mutations on NCSC-SMC differentiation in patients and uncover the mechanism of NOTCH1 regulation of neural crest developmental gene networks as well as SMC transcriptional regulatory circuits. The possibility of recapitulating pathophysiologic processes of vascular disease by using in vitro iPSC-derived disease modeling could open opportunities for mechanistic studies and novel targeting drug development for thoracic aortic aneurysm in BAV patients (Video 1).
VIDEO 1.

Discussion of the significance and the role of NOTCH1 in smooth muscle cell and endothelial cell differentiation from human iPSCs and future research direction. Video available at: http://www.jtcvsonline.org/article/S0022-5223(18)30714-1/fulltext.
Limitation
In this study, NOTCH1−/− human iPSCs were established to dissect the role of NOTCH1 in human SMC and EC differentiation. Although we only used 1 donor, we assert that availability and use of an isogenic control (wild type) in iPSC experiments provides powerful advantages to our study of the function of NOTCH1. It is commonly accepted that the results will be the same if additional iPSC cells from independent donors are used. This cell model mimicked the loss-of-function mutations but does not represent the exact NOTCH1 mutations in BAV patients, such as NOTCH1 R1108X, H1505del, or other missense point mutations. We plan to obtain the blood cells from BAV patients with known NOTCH1 mutations, such as R1108X and H1505del, to model BAV and associated thoracic aortic aneurysm. We performed targeted sequencing of the NOTCH1 gene in 108 BAV patients and will continue sequencing more BAV patients for NOTCH1 mutations. We will create the exact point mutations of NOTCH1 identified in BAV with CRISPR/Cas9 technology if those BAV patients with known NOTCH1 mutations are not accessible. We will also examine the heterozygous NOTCH1 knockout, which more closely models BAV patient cells containing a single copy of R1108X or H1505del NOTCH1 mutations. However, the patient population with BAV is a very heterogenous one. The prevalence of NOTCH1 mutations is very low (<6%). NOTCH1 gene mutations might not be the predominant factor driving BAV-associated aortopathy in the general population of BAV patients. Other factors should also be examined, such as the transforming growth factor β pathway.16
Perspective.
The truncation of NOTCH1 protein in induced pluripotent stem cells severely impairs the differentiation of neural crest and subsequent smooth muscle cell differentiation. The results indicate that NOTCH1 mutations in bicuspid aortic valve patients, such as R1108X, could cause aortopathy and subsequent aortic aneurysm in these patients because of impaired differentiation of smooth muscle cells.
Acknowledgments
This work was supported by the AATS Graham Foundation, NIH K08 (5K08HL130614-02), and a McKay Award from the Frankel Cardiovascular Center, University of Michigan. Dr Yang is supported by the HL130614-05, Phil Jenkins and Steve Szatamari Funds.
Abbreviations and Acronyms
- BAV
bicuspid aortic valve
- CD31
cluster of differentiation 31
- CRISPR/Cas9
Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 nuclease (Cas9) system
- CVPCs
cardiovascular progenitor cells
- EC
endothelial cell
- hESCs
human embryonic stem cells
- iPSCs
induced pluripotent stem cells
- MSCs
mesenchymal stem cells
- NOTCH1
human NOTCH1 protein
- NOTCH1 −/−
human NOTCH1 gene homozygous knockout
- NCSCs
neural crest stem cells
- PCR
polymerase chain reaction
- sgRNA
single-guide RNA
- SMA
smooth muscle α-actin
- SM22α
smooth muscle 22 α
- SMC
smooth muscle cell
Footnotes
Conflict of Interest Statement
Authors have nothing to disclose with regard to commercial support.
References
- 1.Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med. 2014;370:1920–9. [DOI] [PubMed] [Google Scholar]
- 2.Itagaki S, Chiang Y, Tang GH. Why does the bicuspid aortic valve keep eluding us? Cardiol Rev. 2016;24:119–30. [DOI] [PubMed] [Google Scholar]
- 3.Michelena HI, Khanna AD, Mahoney D, Margaryan E, Topilsky Y, Suri RM, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA. 2011;306:1104–12. [DOI] [PubMed] [Google Scholar]
- 4.Losenno KL, Goodman RL, Chu MW. Bicuspid aortic valve disease and ascending aortic aneurysms: gaps in knowledge. Cardiol Res Pract. 2012; 2012:145202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol. 1984;53:849–55. [DOI] [PubMed] [Google Scholar]
- 6.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–4. [DOI] [PubMed] [Google Scholar]
- 7.Alva JA, Iruela-Arispe ML. Notch signaling in vascular morphogenesis. Curr Opin Hematol. 2004;11:278–83. [DOI] [PubMed] [Google Scholar]
- 8.Tang Y, Boucher JM, Liaw L. Histone deacetylase activity selectively regulates notch-mediated smooth muscle differentiation in human vascular cells. J Am Heart Assoc. 2012;1:e000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luxan G, D’Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. 2016;118:e1–18. [DOI] [PubMed] [Google Scholar]
- 10.Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental mechanisms of aortic valve malformation and disease. Annu Rev Physiol. 2017;79: 21–41. [DOI] [PubMed] [Google Scholar]
- 11.Su RJ, Baylink DJ, Neises A, Kiroyan JB, Meng X, Payne KJ, et al. Efficient generation of integration-free ips cells from human adult peripheral blood using BCL-XL together with Yamanaka factors. PLoS One. 2013;8:e64496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan Y, et al. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum Mol Genet. 2013;22:4241–52. [DOI] [PubMed] [Google Scholar]
- 13.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang B, Zhou W, Jiao J, Nielsen JB, Mathis MR, Heydarpour M, et al. Protein-altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat Commun. 2017;8:15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao J, Dang Y, Yang Y, Gao R, Zhang Y, Kou Z, et al. Promoting reprogramming by FGF2 reveals that the extracellular matrix is a barrier for reprogramming fibroblasts to pluripotency. Stem Cells. 2013;31:729–40. [DOI] [PubMed] [Google Scholar]
- 16.Jiao J, Xiong W, Wang L, Yang J, Qiu P, Hirai H, et al. Differentiation defect in neural crest-derived smooth muscle cells in patients with aortopathy associated with bicuspid aortic valves. EBioMedicine. 2016;10:282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mica Y, Lee G, Chambers SM, Tomishima MJ, Studer L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 2013;3:1140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Qiu P, Jiao J, Hirai H, Xiong W, Zhang J, et al. Yes-associated protein inhibits transcription of myocardin and attenuates differentiation of vascular smooth muscle cell from cardiovascular progenitor cell lineage. Stem Cells. 2017;35:351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava D, DeWitt N. In vivo cellular reprogramming: the next generation. Cell. 2016;166:1386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha S, Iyer D, Granata A. Embryonic origins of human vascular smooth muscle cells: implications for in vitro modeling and clinical application. Cell Mol Life Sci. 2014;71:2271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung C, Bernardo AS, Pedersen RA, Sinha S. Directed differentiation of embryonic origin-specific vascular smooth muscle subtypes from human pluripotent stem cells. Nat Protoc. 2014;9:929–38. [DOI] [PubMed] [Google Scholar]
- 22.Cheung C, Bernardo AS, Trotter MW, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol. 2012;30:165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol. 2009;11:951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guzzardi DG, Verma S, Fedak PW. Bicuspid aortic valve aortopathy: mechanistic and clinical insights from recent studies. Curr Opin Cardiol. 2017;32:111–6. [DOI] [PubMed] [Google Scholar]
- 25.Girdauskas E, Borger MA, Secknus MA, Girdauskas G, Kuntze T. Is aortopathy in bicuspid aortic valve disease a congenital defect or a result of abnormal hemodynamics? A critical reappraisal of a one-sided argument. Eur J Cardiothorac Surg. 2011;39:809–14. [DOI] [PubMed] [Google Scholar]
- 26.Baeten JT, Lilly B. Notch signaling in vascular smooth muscle cells. Adv Pharmacol. 2017;78:351–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson LM, Gibbons GH. Notch: a mastermind of vascular morphogenesis. J Clin Invest. 2007;117:299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM III. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;134:290–6. [DOI] [PubMed] [Google Scholar]
- 29.Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, Wrobel S, et al. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun. 2006;345: 1460–5. [DOI] [PubMed] [Google Scholar]
- 30.Grewal N, Gittenberger-de Groot AC, Poelmann RE, Klautz RJ, Lindeman JH, Goumans MJ, et al. Ascending aorta dilation in association with bicuspid aortic valve: a maturation defect of the aortic wall. J Thorac Cardiovasc Surg. 2014; 148:1583–90. [DOI] [PubMed] [Google Scholar]
- 31.Ignatieva E, Kostina D, Irtyuga O, Uspensky V, Golovkin A, Gavriliuk N, et al. Mechanisms of smooth muscle cell differentiation are distinctly altered in thoracic aortic aneurysms associated with bicuspid or tricuspid aortic valves. Front Physiol. 2017;8:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renard M, Callewaert B, Baetens M, Campens L, MacDermot K, Fryns JP, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol. 2013;165:314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–93. [DOI] [PubMed] [Google Scholar]
- 34.Milewicz DM, Trybus KM, Guo DC, Sweeney HL, Regalado E, Kamm K, et al. Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler Thromb Vasc Biol. 2017;37:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fazel SS, Mallidi HR, Lee RS, Sheehan MP, Liang D, Fleischman D, et al. The aortopathy of bicuspid aortic valve disease has distinctive patterns and usually involves the transverse aortic arch. J Thorac Cardiovasc Surg. 2008;135: 901–7. 907.e901–2. [DOI] [PubMed] [Google Scholar]
- 36.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cornell RA, Eisen JS. Notch in the pathway: the roles of Notch signaling in neural crest development. Semin Cell Dev Biol. 2005;16:663–72. [DOI] [PubMed] [Google Scholar]