

SUMMARY
CRISPR-Cas13 RNA endonucleases show promise for programmable RNA knockdown. However, sequence-specific binding of Cas13 unleashes non-specific bystander RNA cleavage, or collateral activity, raising concerns for experiments and therapeutic applications. Although robust in cell-free and bacterial environments, collateral activity in mammalian cells remains disputed. We investigate Cas13d collateral activity in a therapeutic context for myotonic dystrophy type 1, caused by a transcribed CTG repeat expansion. We find that, when targeting CUGn RNA in mammalian cells, Cas13d depletes endogenous and transgenic RNAs, interferes with critical cellular processes, and activates stress response and apoptosis. Collateral effects also occur when targeting abundant endogenous transcripts. To minimize collateral activity for repeat-targeting approaches, we introduce GENO, an adeno-associated virus-compatible strategy that leverages guide RNA processing to control Cas13d expression. We argue that thorough assessment of collateral activity is necessary when applying Cas13 in mammalian cells and that GENO illustrates advantages of compact regulatory systems for Cas-based gene therapies.
In brief
Kelley et al. report that, while CRISPR-Cas13d reduces CUGn RNA foci and restores RNA processing in a HeLa model of myotonic dystrophy type 1, Cas13d collateral activity dramatically depletes cellular RNAs and interferes with core processes. They develop a negative autoregulation strategy to enable repeat-targeting therapies by minimizing Cas13d expression.
Graphical Abstract
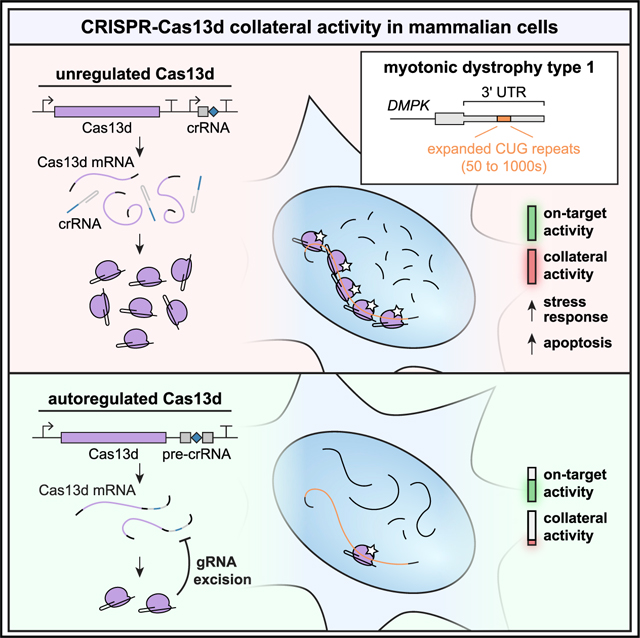
INTRODUCTION
Cas13 is a family of RNA-guided endonucleases capable of sequence-specific binding and cleavage of RNA (Abudayyeh et al., 2016). In class 2 type VI CRISPR systems, Cas13 confers immunity to phage by recognizing a target sequence complementary to the spacer of the crispr RNA (crRNA), or guide RNA (gRNA) (O’Connell, 2019). Upon binding, the two HEPN domains of Cas13 change conformation, forming a dual-R-X4-H catalytic site distal to the RNA-binding cleft with potent ribonuclease (RNase) activity (Zhang et al., 2018, 2019). Its programmable targeting enables broad application of Cas13, as well as nuclease-inactive dCas13 variants and fusions, in eukaryotic cells to reduce expression of RNAs (Abudayyeh et al., 2017; Kushawah et al., 2020; Li et al., 2021b), introduce base edits (Cox et al., 2017; Kannan et al., 2021; Xu et al., 2021) and modifications (Wilson et al., 2020), visualize RNAs (Yang et al., 2019), modulate alternative splicing (Du et al., 2020; Konermann et al., 2018), and capture RNA-protein interactions (Han et al., 2020). In particular, Cas13d is a family of small orthologs especially suited for mammalian use, with efficient adeno-associated virus (AAV) packaging for delivery (Konermann et al., 2018), well-studied determinants of gRNA activity (Wessels et al., 2020), and no protospacer flanking sequence (PFS) constraints (Yan et al., 2018). As a result, Cas13d is a promising candidate for RNA-targeting therapies that avoid risks of permanent genome editing or DNA binding.
Notably, in biochemical and bacterial contexts, sequence-specific binding of Cas13 to the target unleashes non-specific RNase activity capable of cleaving bystander RNAs (Abudayyeh et al., 2016; East-Seletsky et al., 2016). This behavior, often referred to as collateral activity, is a robust feature of all known orthologs (O’Connell, 2019) and has been leveraged to detect nucleic acids at attomolar sensitivity (Gootenberg et al., 2017). Despite this, the extent of collateral activity of Cas13 in mammalian cells remains disputed. Many groups observe no evidence of this behavior in eukaryotic cells in various experiments (Abudayyeh et al., 2017; Huynh et al., 2020; Konermann et al., 2018; Kushawah et al., 2020), and Cas13 has been applied in other studies without mention of collateral effects (Cox et al., 2017; He et al., 2020; Li et al., 2021b; Wessels et al., 2020; Zhou et al., 2020). Yet there is limited but growing evidence that collateral activity depletes RNAs in mammalian cells (Özcan et al., 2021; Wang et al., 2019b, 2021a; Xu et al., 2021) and that Cas13 is toxic in eukaryotes (Buchman et al., 2020), calling into question the utility of Cas13 and presenting substantial risk to Cas13-based therapeutics.
This risk may be magnified in the context of repeat expansion diseases (REDs), in which it can be useful to target the repeats directly (Hu et al., 2009; Lee et al., 2012; Mulders et al., 2009). One example is myotonic dystrophy type 1 (DM1), a multisystemic disease characterized by myotonia, muscle wasting, and hypersomnolence (Ranum and Cooper, 2006). DM1 is caused by a CTG expansion in the 3′ UTR of DMPK (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992), which exerts toxicity primarily through RNA gain-of-function (GOF) mechanisms including sequestration of muscleblind-like (MBNL) RNA-binding proteins (RBPs) (Miller et al., 2000), upregulation of CELF1 (Kuyumcu-Martinez et al., 2007), and repeat-associated non-AUG translation (Zu et al., 2011). In an ensemble of intra- and intermolecular interactions (Jain and Vale, 2017; Krzyzosiak et al., 2012; Querido et al., 2011), MBNL proteins and DMPK mRNAs with CUG expansions cluster into nuclear foci (Miller et al., 2000; Taneja et al., 1995), preventing MBNL from regulating alternative splicing and shifting mRNA isoform ratios (Otero et al., 2021; Wang et al., 2019a). Alleles of >50 CTGs are associated with DM1, yet expansion lengths vary widely and correlate with severity and age of onset (Paulson, 2018). Somatic instability varies repeat length further within a patient’s cells and tissues (Morales et al., 2012), often reaching thousands of repeats in skeletal muscle (Thornton et al., 1994) and brain (Otero et al., 2021). This variation, coupled with differential DMPK expression, produces unique toxicity for each cell within each individual. Therapeutics directly targeting the repeat RNA may best address this complexity by titrating target cleavage rate with repeat length in each cell. While Cas13a can degrade CUGn RNA in patient cells (Zhang et al., 2020), its large size poses significant challenges to delivery. With strong on-target activity, lack of PFS constraints, and efficient AAV packaging (Konermann et al., 2018; Yan et al., 2018), Cas13d with repeat-targeting gRNAs may enable a novel therapeutic platform for DM1 and other REDs with GOF mechanisms. Yet, if present, collateral activity may be exacerbated by repeated protospacers on the target RNA.
Here, we investigate collateral activity of Cas13d in mammalian cells in the context of a CUG-targeting therapy for DM1. In cell culture, we find that Cas13d effectively targets toxic CUG480 RNA and ameliorates MBNL sequestration. However, in human and mouse cells, Cas13d dramatically depletes orthogonal reporter RNAs when targeting CUG480, as well as other transgenic and highly expressed endogenous targets at unique protospacers. To combat collateral activity for repeat-targeting Cas13d, we introduce gRNA excision for negative-autoregulatory optimization (GENO), which uses crRNA processing to attenuate Cas13d expression within AAV packaging constraints. We show that GENO mitigates collateral effects in human cells while retaining modest on-target knockdown of CUGn RNA in patient-derived DM1 myoblasts. These findings shed light on the biochemistry of Cas13d in mammalian cells and illuminate properties desirable in RNA-targeting therapeutics.
RESULTS
Cas13d efficiently reduces toxic CUG480 RNA foci and rescues MBNL-dependent splicing
We first evaluated the potential of a CRISPR-Cas13d platform for DM1 by targeting CUG repeat RNA in HeLa cells and measuring disruption of nuclear RNA foci and rescue of MBNL-mediated alternative splicing. We cotransfected plasmids expressing HA-tagged Ruminococcus flavefaciens Cas13d (RfxCas13d) with two SV40 nuclear localization signals (NLS) and an unfused EGFP marker, a target RNA consisting of exons 11–15 of DMPK with 480 CUG repeats (CUG480), and a CUG-targeting or non-targeting (NT) gRNA (Figure 1A). Since Cas13d does not have strict PFS constraints (Konermann et al., 2018; Yan et al., 2018), we tested gRNAs targeting all three registers of the CUG repeat (Figure 1A; see also Table S1).
Figure 1. Cas13d reduces accumulation of CUGn repeat RNA and rescues MBNL-dependent splicing in a HeLa cell-culture model of DM1.

(A) Diagram of cotransfection experiments to test Cas13d on-target activity and MBNL-dependent splicing. Plasmids encoding Cas13d and EGFP, gRNA, CUG480 target RNA, and an MBNL1 exon 5 splicing minigene are expressed in HeLa cells prior to CUGn RNA FISH and RT-PCR.
(B) gRNAs tested, including three CUG-targeting gRNAs and a non-targeting (NT) control.
(C) Representative FISH images for CUGn RNA (grayscale) in targeting and NT conditions. Cells stained with DAPI (nuclei, cyan) and CellMask (cells, magenta) for segmentation. Scale bars, 10 μm.
(D) Magnified images of CUGn RNA FISH (grayscale) from (C) in targeting and NT conditions. Outlines of nuclei (blue) computed by automatic thresholding. Scale bars, 10 μm.
(E) Median CUGn FISH signal per nucleus, relative to NT. Error bars show 95% confidence interval (CI), estimated by bootstrapping. n > 7 nuclei. *p < 0.05, non-overlapping CI. n.s., not significant (p > 0.05).
(F) Diagram of splicing minigene assay. Sequestration of MBNL by CUGn RNA increases MBNL1 exon 5 inclusion ratio (ψ). CUG-targeting Cas13d aims to rescue ψ to unperturbed levels.
(G) MBNL1 exon 5 ψ for targeting and NT conditions, measured by RT-PCR. Transcriptional inhibition by deactivated Cas9 (dCas9) with matching spacers is also shown. Error bars show standard deviation (SD). n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed Student’s t test.
We performed fluorescence in situ hybridization (FISH) for CUG480 RNA and microscopy to measure disruption of nuclear RNA foci (Figures 1C and 1D). We quantified mean FISH intensity in each transfected nucleus using an automated image analysis pipeline. As expected, we observed a stark reduction of FISH signal when Cas13d was paired with any of the three CUG-targeting gRNAs compared with NT gRNA (Figure 1E, p < 0.05, non-overlapping 95% confidence intervals). Nuclease-inactive dCas13d (Konermann et al., 2018) did not reduce FISH signal with any CUG-targeting gRNA (p > 0.05), indicating that RNA cleavage, rather than binding alone, is required to reduce accumulation of CUG480 RNA in this setting.
To evaluate downstream impacts of CUGn RNA knockdown, we measured MBNL-dependent splicing using a minigene containing MBNL1 exons 4–6 with intervening introns (Figure 1F). Inclusion of MBNL1 exon 5 is suppressed by high concentration of available MBNL in the nucleus but is promoted by its sequestration on CUGn RNA (Gates et al., 2011). We transfected this minigene along with Cas13d, CUG480 target, and gRNA plasmids and measured the ratio of inclusion of MBNL1 exon 5 (ψ) by RT-PCR. We found that targeting CUG480 with Cas13d and any CUG-targeting gRNA strongly reduced ψ compared with NT (Figure 1G, p < 0.01, two-tailed Student’s t test), suggesting that Cas13d robustly restores MBNL-dependent splicing in this model. As expected, we did not observe similar splicing changes when gRNAs were transfected without Cas13d (Figure S1A), indicating that splicing rescue is not a result of endogenous RNA interference.
We also did not observe reduction of j with dCas13d (Figure 1G, p > 0.05, two-tailed t test). To confirm that dCas13d binds the CUG480 RNA, we expressed a dCas13d-EGFP fusion and performed FISH along with immunofluorescence (IF) against MBNL1 and EGFP (Figure S1B). We observed increased colocalization of CUG-targeting dCas13d with nuclear CUG480 foci versus NT (1.25 versus 1.01 enrichment ratio, p < 0.001, two-sided Mann-Whitney U test, Figure S1C), indicating that dCas13d binds CUGn RNA in a sequence-specific manner. Intriguingly, enrichment of MBNL1 in foci was not significantly reduced by CUG-targeting dCas13d (2.86 versus 3.13 enrichment ratio, p > 0.05, Figure S1C), suggesting that although dCas13d may compete with MBNL, these interactions are insufficient to restore splicing in this model.
We also compared Cas13d with nuclease-inactive Streptococcus pyogenes Cas9 (dCas9), which blocks transcription of CUGn RNA only when the gRNA aligns with a CAG protospacer adjacent motif (PAM) (Pinto et al., 2017). As expected, dCas9 rescued splicing only with a gRNA matching the CUG-1 spacer (Figure 1G, p < 0.01, two-tailed t test), whereas gRNAs with AGC or GCA PAMs (CUG-2 and CUG-3, respectively) did not rescue splicing (p > 0.05). In contrast, all three registers supported rescue of splicing by Cas13d, suggesting that its lack of PFS constraints expands the available targeting space for CRISPR therapies.
CUG-targeted Cas13d inhibits EGFP fluorescence
Strangely, although Cas13d reduced CUG480 RNA foci and restored MBNL-mediated splicing, we noticed loss of the EGFP marker expressed from the Cas13d plasmid after transfection of Cas13d, CUG-targeting gRNA, and CUG480 target, but not when either dCas13d or NT gRNA were substituted (Figure S2A). To quantitate this, we transfected CUG480 into HeLa along with Cas13d and gRNA plasmids, and we measured EGFP fluorescence after 20 h. We found that EGFP was reduced by >96% with Cas13d and any CUG-targeting gRNA versus NT (Figure S2B, p < 0.05, two-tailed t test). The same effect was not observed for dCas13d, indicating that loss of EGFP results from Cas13d RNase activity.
To assess whether loss of EGFP simply reflected differences in cell survival, we performed a resazurin cell viability assay 20 h and 44 h after transfection. After 20 h, viability was only slightly reduced when Cas13d was transfected with CUG-1 or CUG-2 gRNA compared with NT (Figure S2C, 8.0% reduction for CUG-1 and 4.8% for CUG-2, p < 0.01, two-tailed t test), indicating that death of transfected cells does not explain the observed loss of EGFP after 20 h. However, a larger reduction in viability was observed after 44 h for all CUG-targeting gRNAs (Figure S2C, >16% reduction for all gRNAs, p < 0.001), suggesting that persistent targeting of CUGn RNA by Cas13d is cytotoxic.
CUG-targeted Cas13d upregulates stress response and apoptosis pathways
We performed RNA sequencing (RNA-seq) to profile pathways disrupted by CUG-targeting Cas13d and compare them with other repeat-targeted technologies (Figure S2D). We transfected HeLa cells with Cas13d and with or without CUG-1 gRNA and sequenced RNA collected after 68 h. For comparison, we also tested dCas9 as well as sequence-matched CUG-targeting and NT short hairpin RNAs (shRNAs), which use RNA interference for knockdown (Paddison et al., 2002) and are popular benchmarks for Cas13 off-targeting (Abudayyeh et al., 2017). Transcripts from 57 genes in the human reference genome contain CUGn repeats longer than the Cas13d spacer (Table S2) and therefore are likely off-targets. We omitted the CUG480 plasmid to enrich for these off-target events. We observed strong correlations of transcripts per million (TPM) estimates between RNA-seq libraries across all conditions (Figure S2E, minimum Pearson’s r of log(TPM) = 0.92, median = 0.98), suggesting low variance introduced during library preparation.
We found that CUG-targeting Cas13d disrupted fewer off-target genes than shRNA (116 versus 443 genes differentially expressed [DE] between targeting and NT conditions, Benjamini-Hochberg false discovery rate [FDR] q < 0.05) but many more genes than dCas9 (3 DE genes, FDR q < 0.05) (Figure S2F). Sixty-nine percent of genes disrupted by CUG-targeting shRNA were downregulated (304 versus 139 genes), consistent with RNA-induced silencing complex-mediated RNA cleavage largely driving DE. In contrast, most genes perturbed by Cas13d were upregulated (83 versus 33 genes), suggesting that a mechanism other than cis RNA cleavage is responsible for most DE genes.
To determine whether differences in off-targeting merely reflect differential knockdown of RNAs containing short CUG repeats, we binned transcripts by length of their longest CUG repeat and calculated the median fold change between targeting and NT conditions. We found a strong relationship between knockdown by shRNA and repeat length for RNAs with short CUG repeats (Figure S2G) and observed a median 35% knockdown of RNAs with CUG repeats >22 nt (Figure S2H). In contrast, both Cas13d and dCas9 exhibited weak knockdown of genes containing short repeats, with very similar profiles between them (Figures S2G and S2H). This suggests that knockdown of endogenous repeat-containing RNAs does not explain the extensive off-target profile of CUG-targeted Cas13d.
We performed gene ontology (GO) analysis using PANTHER (Mi et al., 2019) to investigate pathways enriched in these DE genes. Most processes perturbed by Cas13d were involved in either stress response (48%) or apoptosis signaling (16%) (Figure S2I, enrichment >5, FDR q < 0.05), yet we observed lower enrichment of stress response pathways (18%) and lack of apoptosis signaling with CUG-targeting shRNA (Figure S2J). These results are consistent with Cas13d activation eliciting toxicity beyond what is expected from cis cleavage of off-target RNAs alone.
CUG-targeted Cas13d reduces expression of mCherry reporter in mammalian cells
We hypothesized that loss of EGFP and induction of stress response may result from global depletion of cellular RNAs by Cas13d collateral activity upon activation by CUGn RNA. To assay for collateral activity, we cotransfected a plasmid expressing mCherry as an orthogonal reporter into HeLa along with Cas13d, gRNA, and CUG480, and we measured mCherry in response to CUG480 RNA targeting (Figure 2A). We expected that if collateral activity were extensive, non-specific reduction of RNAs, including mCherry mRNA, would reduce translation and fluorescence Figure 2B). We measured fluorescence after 20 h to minimize the impact of cell viability.
Figure 2. Activation of Cas13d reduces expression of orthogonal reporters in eukaryotic cells.

(A) Diagram of mCherry fluorescence assays to detect Cas13d collateral activity.
(B) Interpretation of fluorescence assays. If collateral activity is weak, EGFP and mCherry are unaffected by Cas13d activation. If strong, both mCherry and EGFPare depleted in trans.
(C) Bulk quantitation of mCherry by plate reader in CUG-targeting and NT conditions, relative to NT. Error bars show SD. n = 5. ***p < 0.001, two-tailed t test.
(D) Representative images of mCherry and EGFP in CUG-targeting and NT conditions. Nuclei stained with DAPI. Scale bars, 10 μm.
(E) Quantitation of mCherry microscopy. Per-cell measurements shown (gray dots) along with the mean (black line). Error bars show SEM. n > 27 cells. *p < 0.05, **p < 0.01, two-sided Mann-Whitney U test.
(F) Bulk mCherry fluorescence when targeting CUG480 RNA with Cas13d in HEK293 and Neuro2a cells. n = 5. ***p < 0.001, two-tailed t test.
(G) Bulk mCherry fluorescence when targeting 24× MS2 hairpin RNA with Cas13d using MS2-targeting gRNAs. A control plasmid (pUC19) without the MS2 target is also included. n = 5. ***p < 0.001, two-tailed t test.
(H) Bulk mCherry fluorescence when targeting puromycin acetyltransferase (pac) RNA at unique sites. n = 5. *p < 0.05, two-tailed t test.
(I) Relationship between MBNL1 exon 5 ψ and mCherry fluorescence for gRNAs targeting unique DMPK sequences in the CUG480 RNA. Gray line indicates ordinary least squares linear regression (Pearson’s r = 0.81). n = 3.
We observed >56% reduction in mCherry fluorescence with Cas13d and any CUG-targeting gRNA relative to NT (Figure 2C, p < 0.001, two-tailed t test) but no reduction with dCas13d, indicating that this effect results from Cas13d RNase activity. To assay fluorescence at the single-cell level, we performed widefield microscopy and quantified mCherry intensity for each cell (Figure 2D). On average, we observed >61% decrease in mCherry with Cas13d, CUG-1 gRNA, and CUG480 target compared with conditions when either dCas13d or NT gRNA were substituted, and a 43% decrease compared with substitution of the target for CUG0 (Figure 2E, p < 0.05, one-sided Mann-Whitney U test). We also observed a smaller reduction in mCherry (44%) between CUG-targeting and NT conditions in the absence of CUG480 target (p < 0.05), likely due to activation by endogenous RNAs. Overall, these results provide strong evidence of abundant Cas13d collateral activity in HeLa when targeting CUGn RNA.
Other researchers observed that collateral activity of Cas13a is dependent on cell type, finding it apparent in human glioma cells but undetectable in non-cancerous HEK293 cells (Wang et al., 2019b). To determine whether collateral activity of Cas13d is specific to HeLa, we performed the same experiment in HEK293 and mouse neuroblastoma Neuro2a cells. We observed similar loss of mCherry in all lines (Figure 2F, p < 0.001, two-tailed t test), suggesting that collateral activity of Cas13d is not cell type dependent.
Collateral inhibition of mCherry occurs when targeting repetitive and unique sequences
To determine whether this behavior is specific to targeting CUG repeats, we designed three gRNAs complementary to MS2 hairpins from a plasmid expressing 24 tandem MS2 repeats (Table S1). We transfected these gRNAs into HeLa along with Cas13d and either the 24× MS2 target plasmid or a control without MS2 hairpins (pUC19). We observed significant loss of mCherry with all three MS2-targeting gRNAs when the 24× MS2 target was present (Figure 2G, average 44% reduction, p < 0.001, two-tailed t test), but not with the control target (p > 0.05). This result shows that collateral activity is not specific to CUGn targets and is only detected when suitable protospacers are expressed.
As repeat expansion RNAs may amplify collateral activity by binding many Cas13d molecules simultaneously, we sought to determine whether collateral activity is also detectable at unique protospacer sequences. We designed three gRNAs targeting puromycin acetyltransferase (pac) at unique sites (Table S1) and transfected them into HeLa along with Cas13d and either a pac-expressing target plasmid or pUC19. We found that mCherry was inhibited for two of the three gRNAs only with the pac target (Figure 2H, average 28% reduction, p < 0.05, two-tailed t test) and not with the control (p > 0.05). This suggests that collateral activity at unique protospacers, although weaker than at repeats, may still be deleterious and should be thoroughly evaluated when using Cas13d for knockdown in mammalian cells.
Rescue of MBNL splicing by DMPK-targeted Cas13d correlates with mCherry inhibition
These results support the hypothesis that collateral activity is a general property of Cas13d, the rate of which is likely a function of biochemical context, concentration of protospacers, and target binding affinity. As cis cleavage by Cas13d is largely independent of flanking sequences (Yan et al., 2018) (apart from a preference for uracil bases [Konermann et al., 2018]), both on-target and collateral RNase activities may be driven primarily by the intracellular KD of the Cas13d:gRNA:target ternary complex and thus may be positively correlated. To test this, we designed eight gRNAs that target unique protospacers within the 3′ UTR of DMPK (all of which are present in the CUG480 target, see Table S1) and cotransfected them into HeLa with Cas13d, CUG480, the MBNL1 exon 5 minigene, and mCherry. We measured both ψ and bulk mCherry fluorescence to probe on-target and collateral activity, respectively. We found a positive correlation between MBNL splicing rescue and mCherry inhibition (Figure 2I, Pearson’s r = 0.81), suggesting that on-target and collateral activities are strongly linked.
Collateral RNase activity occurs when targeting endogenous genes
Our previous experiments focused on overexpressed transfected targets, yet the extent of collateral activity likely depends stoichiometrically on both Cas13d and target concentrations. We therefore sought to measure collateral activity when targeting endogenous genes with genomically encoded Cas13d. We designed gRNAs for six genes across a range of expression levels in HeLa (LDHA, CD63, CD81, LGMN, SYBU, EPOR; see Table S3). In a comprehensive CRISPR screen (Hart et al., 2015), none of these were classified as core fitness genes, and all had low confidence scores for essentiality in HeLa (Bayes factor < −10), making them strong candidates for assaying toxic collateral effects.
To generate a cell line expressing Cas13d, we initially treated HeLa cells with lentivirus carrying Cas13d-T2A-EGFP under an EF1α promoter and isolated GFP-positive cells by flow cytometry, yet we were unableto identify clonallines without large truncations of Cas13d after expansion (Figure S3A). This surprised us, as others have generated lines expressing Cas13a (Wang et al., 2019b) or Cas13d (Wei et al., 2021), and our cassette did not include a gRNA. We instead generated a HeLa line with tetracycline-inducible Cas13d-T2A-EGFP using a piggyBac transposon (Cadiñanos and Bradley, 2007) (Figure 3A). As a reporter for collateral activity, we also integrated a cassette constitutively expressing mCherry. We expanded a clonal line (HeLa-tet:Cas13d-mCherry) and validated expression of Cas13d by western blot and EGFP and mCherry by fluorescence microscopy after incubation with 2 μM doxycycline for 44 h (Figures S3B and S3C).
Figure 3. Targeting endogenous mRNAs with Cas13d activates collateral RNase activity.

(A) Description of collateral activity RNA assay. HeLa-tet:Cas13d-mCherry cells express mCherry constitutively and Cas13d under a doxycycline-inducible promoter. As collateral activity likely depletes transgenic and endogenous RNAs, non-transgenic HeLa cells are spiked in prior to transfection of gRNA and induction of Cas13d. After 44 h, mCherry and GAPDH mRNA are measured by qRT-PCR.
(B) Assay validation using CUG-targeting gRNAs and transfected CUG480. n = 3 transfections, averaged across two qPCR replicates. Error bars show SEM of ΔCq propagated to the plotted ratio. *p < 0.05, **p < 0.01, ***p < 0.001, one-tailed t test of ΔCq.
(C) Ratio of mCherry to GAPDH mRNA when targeting endogenous genes. Target expression was calculated from RNA-seq of untransfected HeLa cells included in our experiment described in Figure S2. n = 3 transfections, averaged across two qPCR replicates. Error bars show SEM of ΔCq propagated to the plotted ratio. Gray line depicts a logistic function fit using non-linear least-squares regression. *p < 0.05, one-tailed t test of ΔCq.
To measure collateral RNase activity at the RNA level, we performed qRT-PCR to compare abundance of mCherry and control (GAPDH) transcripts (Figure 3A). Since we expected collateral activity to degrade RNAs non-specifically (including both mCherry and GAPDH), we mixed HeLa-tet:Cas13d-mCherry cells with non-transgenic HeLa cells at a 1:4 ratio, transfected the coculture with gRNA plasmid, and induced Cas13d for 44 h prior to RNA extraction. As the non-transgenic cells did not express Cas13d or mCherry, they contributed a stable pool of GAPDH mRNAs without influencing mCherry mRNA, enabling measurement of collateral activity by qRT-PCR. To validate this method, we transfected CUG-targeting and NT gRNAs along with CUG480 target and observed reduction of mCherry RNA with all CUG-targeting gRNAs comparable with our fluorescence assays (Figure 3B, average 46% reduction versus NT, p < 0.05, one-tailed t test of ΔCq).
Using this approach, we observed significant reduction of mCherry RNA when targeting two of the six genes in our panel (LDHA and LGMN; Figure 3C, p < 0.05, one-tailed t test of ΔCq) and a negative correlation between target expression level and mCherry RNA abundance (Pearson’s r = −0.87 between [mCherry] and log(target TPM)). These data suggest that although collateral effects are strong when targeting highly expressed genes (e.g., 34% depletion for LDHA), they may be weaker for lowly expressed targets; nevertheless, it is important to screen for them with any target to ensure that non-specific RNA depletion does not confound experiments or cause unintended toxicity.
To investigate whether collateral activity is toxic at endogenous targets, we measured the viability of HeLa-tet:Cas13d-mCherry cells transfected with gRNA plasmids and induced with doxycycline for 44 h. We observed significant reductions in viability for the three most abundant targets (LDHA, CD63, CD81; Figure S3D, p < 0.05, two-tailed t test) and a trend of increasing cell mortality with target expression level. Cell viability did not correlate with depletion of gRNAs targeting these genes in a high-throughput CRISPR screen in HeLa (Hart et al., 2015) (Figure S3E, p > 0.05, β cumulative distribution function), providing further evidence that toxicity results from collateral activity rather than on-target knockdown. Importantly, cell viability may explain part of the reduction of mCherry RNA abundance (Figure 3C), yet both measurements likely reflect the same bystander cleavage behavior. Overall, these results suggest that collateral activity is strongest at repetitive and/or abundant targets due to stoichiometric activation of many Cas13d molecules.
Negative autoregulation by gRNA excision reduces expression of Cas13d
We reasoned that the numerous protospacers on a repetitive target may exacerbate collateral activity by binding many Cas13d:gRNA complexes simultaneously, and that limiting Cas13d expression may reduce collateral activity while maintaining on-target cleavage of CUGn RNA (Figure 4A). We developed an autoregulation strategy, GENO, to reduce and control Cas13d expression by leveraging its crRNA processing activity for self-knockdown (Figure 4B). In GENO, a pre-crRNA containing the spacer flanked by direct repeats is placed in a UTR of the Cas13d mRNA. After transcription and translation, Cas13d excises the gRNA to form the binary complex, cleaving its mRNA and preventing further translation.
Figure 4. Negative autoregulation by gRNA excision reduces Cas13d collateral activity and retains on-target activity at CUGn RNA.

(A) Rationale for Cas13d autoregulation. Overexpression likely produces many activated Cas13d:gRNA complexes for each CUGn target RNA. Minimizing Cas13d expression may greatly reduce collateral activity with minimal loss of on-target activity.
(B) Description of GENO for regulation of Cas13d expression. In GENO, the pre-crRNA is placed in a UTR of the Cas13d mRNA, causing cleavage and degradation during crRNA processing.
(C) Simulation of steady-state concentration of Cas13d:gRNA complex in GENO (solid line) and unregulated (dashed lines) conditions as a function of Cas13d transcription rate (rt, horizontal axis) and gRNA transcription rate in the unregulated design (rt,gRNA, isolines). Autoregulation efficiency ƞGENO is annotated (see Data S1). Translation rate constant kT = 8.3 3 10−3 protein mRNA−1 s−1, crRNA processing rate constant kproc = 1.7 3 10−7 cell mol−1 s−1.
(D) Simulated Cas13d:gRNA concentration as a function of kT (horizontal axis) and kproc (isolines). rt = 0.02 mRNA s−1, rt,gRNA = 0.96 RNA s−1.
(E) Fluorescent western blot comparing Cas13d protein expression from unregulated and GENO plasmids transfected in HeLa. Cas13d is visualized with α-HA antibody (green), and HSP70 is included as a control (pink).
(F) Quantification of western blot. Error bars show SD. n = 3. ***p < 0.001, two-tailed t test.
(G) Ratio of CUG480 RNA to GAPDH mRNA after targeting CUG480 with unregulated or GENO-Cas13d in HeLa, normalized to NT. n = 3 transfections, averaged across two qPCR replicates. Error bars show SEM of ΔCq propagated to the plotted ratio. *p < 0.05, one-tailed t test of ΔCq.
(H) mCherry fluorescence relative to NT 20 h after transfection with unregulated (purple) and GENO (green) Cas13d and CUG-1 gRNA plasmids, CUG480 target, and mCherry, as a function of mass of Cas13d plasmid transfected. Error bars show SD. n = 3. Logistic fit functions shown as solid lines. **p < 0.01, two-tailed t test.
(I) MBNL1 exon 5 ψ measured by RT-PCR. Mass of Cas13d plasmid transfected matches the condition highlighted in (H). Error bars show SD. n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test.
We constructed a dynamical model to describe GENO and proved that GENO strictly reduces Cas13d mRNA and binary complex compared with an unregulated system for all possible transcription, translation, and crRNA processing rates (see Data S1). To estimate how efficiently binary complex concentration is reduced, we performed dynamical simulations across broad ranges of these parameters (Figure S4). From this model, we predicted that GENO robustly reduces binary complex concentration at equilibrium across a wide range of Cas13d transcription rates (Figure 4C), yet this reduction weakens at very high expression, as the concentration in the unregulated design plateaus due to limited gRNA availability. We also predicted that the autoregulation efficiency (ƞGENO, defined as the difference of 1 and the ratio of equilibrium binary complex concentrations in GENO and unregulated designs, see Data S1) increases with Cas13d translation rate (Figure 4D). Interestingly, we found a more complex relationship between ƞGENO and crRNA processing rate, which are positively correlated when translation is fast and negatively correlated when slow (Figure 4D). Overall, these insights suggest that GENO is a simple and robust approach to regulate Cas13d expression in mammalian cells.
To experimentally confirm that GENO reduces Cas13d expression, we transfected plasmids encoding GENO-regulated and unregulated Cas13d into HeLa and performed a western blot (Figure 4E). We found that GENO reduced Cas13d protein by 76% (Figure 4F; p < 0.001, two-tailed t test), validating its utility to regulate expression.
GENO reduces collateral activity and maintains partial on-target rescue
To confirm that knockdown of CUGn RNA is maintained with GENO, we cotransfected the CUG480 target along with unregulated and GENO-regulated Cas13d and measured CUG480 RNA and a control (GAPDH) via qRT-PCR. In both conditions, we found that CUG480 RNA was significantly reduced by CUG-targeting Cas13d versus NT (Figure 4G, p < 0.05, one-tailed t test of ΔCq), with similar knockdown between unregulated and GENO-Cas13d (55% versus 58%).
To evaluate whether GENO reduces collateral activity when targeting CUG RNA, we measured mCherry fluorescence after cotransfecting mCherry with unregulated or GENO-Cas13d, gRNA, and CUG480 target. We screened a range of Cas13d plasmid concentrations (0.5–50 ng per transfection) to estimate the window of therapeutic benefit provided by GENO. With unregulated Cas13d, we observed strong loss of mCherry at all concentrations tested (Figure 4H; p < 0.01, two-tailed t test), underscoring the need for a solution to collateral activity when targeting repeats. We found that GENO mitigated mCherry inhibition to statistically undetectable levels in our assay for all but the two highest concentrations (p > 0.05, two-tailed t test) and increased the half maximal inhibitory concentration (IC50) of mCherry inhibition by 18-fold (from 1.6 ng to 29 ng), producing a wide window within which collateral activity is improved.
To further investigate on-target activity with GENO at lower doses, wechose a Cas13dplasmid concentration (5ng per transfection) at which collateral activity was not detected with GENO and measured MBNL activity using the MBNL1 exon 5 minigene. We found that GENO-Cas13d partially reduced ψ (Figure 4I; 36% of rescue observed without regulation, p < 0.05, two-tailed t test). Overall, we found that GENO preserved moderate on-target activity of Cas13d at CUG RNA while minimizing collateral activity, validating its use to control Cas13d expression. Further optimization will be needed to maximize on-target knockdown while maintaining collateral effects below a tolerable threshold.
GENO reduces AAV-delivered Cas13d expression in human DM1 myoblasts
As a proof of concept for applying autoregulation to an AAV-Cas13d therapy, we investigated whether GENO efficiently regulates expression when delivering Cas13d by AAV and whether GENO-regulated Cas13d reduces CUGn RNA in patient-derived DM1 myoblasts. We synthesized recombinant AAV6 carrying unregulated or GENO-Cas13d with either CUG-1 or NT gRNAs. We chose AAV6 because it efficiently transduces myoblasts and myotubes in culture (Arnett et al., 2014; Pinto et al., 2017), and we used a 0.5-kb cytomegalovirus promoter to keep the AAV genomes within packaging limits. Despite multiple attempts, we were unable to package the unregulated CUG-targeting Cas13d, possibly due to toxicity from its overexpression during virus production. Thus, subsequent experiments focused on GENO-Cas13d paired with CUG-1 or NT gRNAs and unregulated Cas13d with NT gRNA.
We first treated undifferentiated DM1 myoblasts with AAV for 6 days and measured Cas13d protein expression by western blot (Figure 5A). We found that GENO reduced Cas13d production by 87% (p < 0.05, two-tailed t test, n = 3). To measure RNA and protein expression at the single-cell level, we simultaneously performed hybridization chain reaction FISH (HCR FISH) (Choi et al., 2018) to image single molecules of Cas13d mRNA and IF for Cas13d protein (Figure 5B). We detected a mean of 156 diffraction-limited HCR FISH spots per nucleus in cells treated with unregulated Cas13d and NT gRNA and 1.3 spots per nucleus in PBS-treated cells (Figures S5A and S5B), highlighting the sensitivity and specificity of this approach to detect Cas13d RNAs. Additionally, all nuclei in AAV treatment conditions (n = 142) contained >5 HCR FISH spots, indicating nearly 100% transduction efficiency.
Figure 5. AAV-delivered autoregulated Cas13d reduces CUGn RNA accumulation in human DM1 myoblasts.

(A) Fluorescent western blot of Cas13d in AAV-transduced DM1 patient-derived myoblasts. Cas13d is visualized with α-HA antibody (green), and HSP70 is included as a control (pink). Protein ladder (L) is shown.
(B) Representative 20× confocal images of AAV-treated DM1 myoblasts stained for Cas13d mRNA (HCR FISH, magenta) and protein (α-HA IF, yellow). Nuclei stained with DAPI (cyan). Scale bars, 10 μm.
(C) Mean nuclear intensity of Cas13d HCR FISH in unregulated and GENO NT conditions. Dots represent nuclei, black line indicates median. n > 33 nuclei, >3 images. ***p < 0.001, two-sided Mann-Whitney U test. Gray line indicates mean baseline nuclear FISH signal in PBS-treated myoblasts, and gray shaded region indicates SD. n = 26 nuclei, 3 images.
(D) Mean nuclear intensity of α-HA IF in unregulated and GENO NT conditions. Dots represent nuclei, black line indicates median. n > 33 nuclei, >3 images. ***p < 0.001, two-sided Mann-Whitney U test. Gray line and shaded region calculated as in (C).
(E) Representative 40× confocal images of AAV-treated DM1 myoblasts stained for Cas13d mRNA (HCR FISH, magenta) and CUGn RNA (CAG10 FISH probe, grayscale). Nuclei stained with DAPI (cyan). Scale bars, 10 μm.
(F) Mean nuclear intensity of CUGn FISH in GENO targeting and NT conditions. Dots represent nuclei, black line indicates median. n > 43 nuclei, 21 images. *p < 0.05, one-sided Mann-Whitney U test.
(G) Cumulative distribution function (c.d.f.) of FISH spot intensity of individual CUGn RNA foci in GENO targeting (magenta, solid) and NT (gray, solid) conditions. PBS-treated condition is also shown (gray, dashed). n > 745 spots. ***p < 0.001, two-sided Mann-Whitney U test.
We observed that GENO reduced Cas13d mRNA and protein compared with unregulated Cas13d when both were paired with NT gRNA (Figure 5B). By quantifying mean HCR FISH intensity in each nucleus, we found that GENO reduced median Cas13d mRNA expression by 60% after baseline subtraction of the PBS-treated control (Figure 5C, p < 0.001, two-sided Mann-Whitney U test). Similarly, we found an 83% reduction in median Cas13d protein expression by α-HA IF (Figure 5D, p < 0.001). These results confirm that GENO reduces Cas13d expression in human myoblasts and that GENO-Cas13d can be delivered by AAV.
Autoregulated Cas13d reduces CUGn RNA accumulation in human DM1 myoblasts
To evaluate whether GENO-Cas13d can reduce nuclear CUGn RNA foci in patient-derived cells, we transduced DM1 myoblasts with AAV for 6 days and performed both FISH to detect CUGn RNAs from the expanded DMPK allele and HCR FISH for Cas13d mRNA to mark transduced nuclei (Figure 5E). We visualized discrete diffraction-limited spots in both channels using confocal microscopy, and we detected FISH spots and quantified intensities using automated image analysis (see STAR Methods).
After treatment with GENO-Cas13d and CUG-1 gRNA, we found a modest 23% reduction in median CUGn RNA signal per nucleus (Figure 5F, p < 0.05, one-sided Mann-Whitney U test) relative to NT and PBS controls, with no significant difference between these controls (p > 0.05). Although foci number per nucleus was not significantly different (Figure S5C, p > 0.05, two-sided Mann-Whitney U test), we observed a 14% reduction in median foci intensity (Figure 5G, p < 0.001, two-sided Mann-Whitney U test). This suggests that GENO-Cas13d modestly reduces accumulation of CUGn RNAs in DM1 myoblasts by cleaving and dispersing multimeric CUGn RNA structures and initiating decay.
To investigate whether GENO-Cas13d can ameliorate MBNL sequestration, we performed IF for MBNL1 along with FISH for CUGn RNA and HCR FISH for Cas13d mRNA (Figure S5D). For each cell, we quantified the nuclear-to-cytoplasmic ratio of MBNL1, which we expected to decrease upon knockdown of CUGn RNA due to both (1) reduced accumulation of MBNL1 at nuclear foci and (2) a shift toward cytoplasmic localization due to autoregulatory splicing of MBNL1 exon 5 (Gates et al., 2011). We found that GENO-Cas13d with CUG-1 gRNA significantly shifted MBNL1 toward the cytoplasm versus NT (Figure S5E, p < 0.05, one-sided Mann-Whitney U test), but this shift was not statistically significant when compared with the PBS control (p > 0.05), possibly due to confounding effects of AAV transduction and cargo expression. This result suggests that the reduction of CUGn RNA by GENO-Cas13d partially mitigates MBNL sequestration in DM1 myoblasts.
We did not observe a reduction in Cas13d HCR FISH intensity between targeting and NT conditions (Figure S5F), suggesting that collateral activity of GENO-Cas13d in DM1 myoblasts may be weak enough to preserve RNA homeostasis. To investigate this further, we performed HCR FISH for Cas13d mRNA and two unrelated endogenous mRNAs, PPIB and POLR2A (Figure S5G), and counted FISH spots in all imaged nuclei. We did not detect significant shifts in expression of PPIB and POLR2A between any conditions (Figures S5H and S5I, p > 0.05, two-sided Mann-Whitney U test), suggesting that collateral activity of GENO-Cas13d remains effectively mitigated when delivered by AAV to DM1 myoblasts.
DISCUSSION
Cas13 collateral activity in eukaryotic cells is controversial; many groups have found it to be negligible (Abudayyeh et al., 2017; Huynh et al., 2020; Konermann et al., 2018; Kushawah et al., 2020), while evidence of trans cleavage is emerging in certain contexts (Ai et al., 2022; Özcan et al., 2021; Wang et al., 2019b, 2021a; Xu et al., 2021). We observed stark collateral activity of Cas13d in mammalian cells using fluorescence, cell viability, and transcriptome assays, yet there are several plausible reasons why others may have not observed these effects. For one, collateral activity may go undetected by assays that measure relative changes to transcript abundances, e.g., RNA-seq, as RNAs are likely depleted globally at similar rates across genes. Still, we found that collateral activity strong enough to activate stress response and apoptosis pathways can be detected by RNA-seq. We also observed a trend linking total protospacer load and collateral effects, with collateral activity strongest at overexpressed repetitive targets (Figures 2C–2G), weaker at unique sites in overexpressed transgenes (Figure 2H), still weaker at highly expressed endogenous genes (Figure 3C), and undetected at lowly expressed genes (Figure 3C). Therefore, we may not expect reports of collateral activity if unique protospacers in moderately or lowly expressed genes are targeted—a common use case for Cas13. Importantly, our observations do not preclude use of Cas13d for knockdown; rather, we suggest that researchers thoroughly assess trans cleavage when targeting any gene in mammalian cells. Furthermore, we propose a solution for targeting repeat expansion RNAs, which present both extreme challenges and unique opportunities to mitigate collateral activity.
Notably, some of Cas13d’s most successful eukaryotic applications have been cases in which collateral activity may align with research goals rather than detract from or confound them. For example, Cas13d targeting cancer-specific RNAs can induce apoptosis of bladder and pancreatic cancer cells in vitro and halt tumor progression in xenograft models (Jiang et al., 2020; Li et al., 2021a; Zhuang et al., 2021); in this context, extensive collateral activity may augment cancer-specific toxicity. Cas13d has also been used to convert glia to neurons in vivo by targeting Ptbp1 (Zhou et al., 2020) (although these findings are disputed [Hoang et al., 2022; Wang et al., 2021b]). An intriguing hypothesis is that collateral activity may assist cell-type conversion by clearing mRNAs globally, resetting the gene expression state in response to on-target knockdown. Thorough comparison of Cas13d with RNA-targeting tools lacking bystander cleavage (e.g., shRNA) is needed to discern whether collateral activity synergizes with these applications.
To combat collateral activity for RED therapies, we have developed GENO, a simple autoregulation approach that repurposes the innate crRNA processing activity of Cas13. A fundamental motif in natural and synthetic gene networks (Alon, 2006), negative autoregulation stabilizes gene expression by buffering stochasticity (Becskei and Serrano, 2000) and shortens the response time to equilibrium after perturbations (Rosenfeld et al., 2002) such as AAV transduction and initial expression. The simplicity of GENO also unlocks routes to further optimize Cas13d expression. By eliminating the gRNA promoter, GENO reduces the length of the AAV cargo, freeing space for additional regulatory elements. Additionally, our dynamical model predicts the performance of GENO across broad ranges of transcription, translation, and crRNA processing rates (Figure S4), providing a blueprint for further tuning. In DM1 and other REDs (Malik et al., 2021; Sznajder and Swanson, 2019), expansions often exceed thousands of repeats, particularly in severely affected tissues (Otero et al., 2021; Thornton et al., 1994). Thus, we hypothesize that low expression of a repeat-targeting therapy would reduce off-target interactions with RNAs containing short repeats while maintaining on-target activity. Accordingly, our simulations suggest that optimal autoregulation (with weak but consistent Cas13d expression) is achieved when transcription is slow but translation and crRNA processing are fast, yet the optimal concentration will depend on the specific application of GENO to each disease and target tissue. Transcription and translation can be tuned by adjusting promoter and Kozak sequences, and single-base mutations in the pre-crRNA direct repeat affect crRNA processing rate (Zhang et al., 2019), providing a potential lever to adjust this parameter as well. We believe that this theoretical analysis lays groundwork for extending GENO to Cas13-based therapies for other REDs.
Crucially, the feasibility of autoregulation to mitigate collateral activity relies on non-linearity between cis and trans knockdown efficiency driven by the presence of many protospacers on a repetitive target (Figure 4A). To reduce collateral activity at unique targets, other solutions are needed that increase the ratio of cis to trans cleavage for each Cas13d complex. Deep screening of collateral activity across Cas13 orthologs, especially small variants compatible with AAV (Kannan et al., 2021; Xu et al., 2021), may reveal effectors with better selectivity than RfxCas13d, with the caveat that these differences may be target and context dependent. The newly discovered RNA-targeting Cas7–11 (Özcan et al., 2021) is also a promising candidate for therapeutic knockdown. Additionally, protein engineering strategies, such as directed evolution, could generate Cas13d variants with reduced collateral activity. As a hypothetical example, increasing both RNA cleavage and target dissociation rates may improve selectivity by capitalizing on the proximity of the bound target RNA to the catalytic site relative to bystander RNAs. Directed evolution has been used to modify PAM requirements (Kleinstiver et al., 2015a, 2015b) and enhance specificity of Cas9 (Cerchione et al., 2020; Vakulskas et al., 2018), and to improve Cas13a stability in cells (Charles et al., 2021). We believe that this is an encouraging strategy to improve selectivity for more general applications.
For example, as an alternative strategy for DM1, these solutions could enable targeting of unique sequences in DMPK without the trans cleavage we observed with unregulated Cas13d (Figure 2I). Other therapeutic modalities have been tested that target unique DMPK RNA sequences, including anti-sense oligonucleotides (Wheeler et al., 2012) and shRNAs (Langlois et al., 2005), and this approach may improve cis-targeting specificity. Still, we argue that targeting the repeat directly provides important advantages in DM1, including titration of therapeutic potency with repeat length and sparing of mRNAs from the unexpanded allele of DMPK (Angelbello et al., 2019; Pinto et al., 2017), and we show that GENO addresses the major short-comings of repeat-targeting Cas13d.
In some situations, it may be possible to achieve therapeutic goals using nuclease-inactive dCas13d, e.g., by blocking translation initiation (Charles et al., 2021) or interfering with RBPs. We considered applying dCas13d to displace MBNL from CUGn RNA foci and rescue splicing. A recent study showed that a decoy variant of MBNL1 that binds CUGn RNA can release endogenous MBNL1 from foci and correct splicing (Arandel et al., 2022), inspiring confidence in this strategy. However, despite enrichment at nuclear foci (Figure S1C), dCas13d did not displace MBNL sufficiently to affect splicing (Figure 1G), and much of the dCas13d remained unbound (Figure S1B). These observations could merely reflect our overexpression context, whereby CUG480 RNA may fully sequester MBNL and still present unbound sites along the RNA. However, these results may also indicate that the affinity of dCas13d to CUGn RNA is insufficient to displace MBNL. While structures explored by expansion RNAs and RBPs (Zhang and Ashizawa, 2017) may certainly inhibit binding of dCas13, it also remains plausible that the target affinity of dCas13 is weak in general. One study found that dCas13d fused to APEX2 was unable to bind hTR RNA unless also fused to an RNA-binding domain to enhance affinity, even for gRNAs with strong knockdown of hTR by active Cas13d (Han et al., 2020). Although successes in RNA imaging (Abudayyeh et al., 2017; Yang et al., 2019), editing (Cox et al., 2017; Kannan et al., 2021; Xu et al., 2021), and splicing modulation (Du et al., 2020; Konermann et al., 2018) appear to contradict this hypothesis, it is possible that weak binding affinity can often be compensated by overexpression of dCas13. Interestingly, the altruistic and self-preserving goals proposed to explain the evolution of CRISPR-Cas13 systems (Abudayyeh et al., 2016; Meeske et al., 2019) would seem to pressure target affinity in opposing directions, as slow target dissociation, while further suppressing phage by prolonging cis and trans RNA cleavage, likely also reduces the chance of survival after depletion of host RNAs. Further work is needed to characterize the binding affinity of dCas13 orthologs across many target RNAs and to understand the role played by target affinity in CRISPR-Cas13 phage immunity.
Finally, many other challenges lie ahead for Cas-based therapies. Efficient and safe delivery to specific tissues remains a significant hurdle, although recent advances in AAV capsid engineering (e.g., MyoAAV for skeletal muscle [Tabebordbar et al., 2021]) and non-viral delivery (Mitchell et al., 2021) inspire confidence that these obstacles will be overcome. Additionally, immunogenicity of non-human proteins, including CRISPR effectors (Tang et al., 2022), may jeopardize long-term expression of Cas enzymes. Nevertheless, our work here to minimize collateral activity of Cas13d for REDs outlines general principles for optimizing therapeutic cargoes and underscores the need for robust regulatory circuits for gene therapies that precisely target narrow therapeutic windows. We believe that GENO serves as an example for the design of simple and compact engineering solutions to optimize the potency and safety of gene therapies for human disease.
Limitations of the study
In this work, we show collateral activity of CUG-targeted Cas13d in transfected and stable cell lines and apply autoregulation to mitigate this behavior in transfected cells and AAV-transduced patient myoblasts. While we demonstrate these concepts when targeting CUG RNA for DM1, we have not tested GENO-Cas13d at other repeat sequences, and we have not comprehensively studied Cas13d or GENO at a range of target repeat lengths. In addition, while we do show GENO-Cas13d in patient-derived myoblasts, we have not demonstrated efficacy in preclinical animal models, which will be necessary to translate these concepts into therapies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources developed and used in this study should be directed to the lead contact, Eric T. Wang (eric.t.wang@ufl.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request.
Data and code availability
RNA-seq raw read data (FASTQ format) and a kallisto transcript expression table are publicly available through the Gene Expression Omnibus (GEO) at accession no. GSE191329. Raw microscopy images (CZI format) are available upon request.
All code developed for this work was written in Python and is publicly available on GitHub at https://github.com/cpkelley94/geno (https://doi.org/10.5281/zenodo.6807452).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293, Neuro2a, HeLa, and HeLa-derived cell lines were maintained in 10% FBS growth medium (Dulbecco’s modified eagle medium (DMEM) + 10% fetal bovine serum (FBS) + 1% penicillin/streptomycin) in a humidity-controlled 5% CO2 incubator at 37°C. Myoblasts derived from an adult female DM1 patient biopsy (DM-05 (Xia et al., 2013)) were cultured in SkGM-2 skeletal muscle cell growth medium (Lonza, #CC-3245).
METHOD DETAILS
Plasmids and molecular cloning
Plasmids encoding NLS-RfxCas13d-NLS-HA-T2A-EGFP (pXR001) and NLS-dRfxCas13d-NLS-HA-T2A-EGFP (pXR002) under the EF1α promoter were purchased from Addgene (#109049 and #109050, respectively), as was a plasmid encoding a gRNA cloning cassette driven by the U6 promoter (pXR003, #109053) (Konermann et al., 2018). All plasmids in this study were propagated in NEB Stable chemically competent E. coli (New England Biolabs (NEB), #C3040) at 30°C and purified using the Zyppy Plasmid Miniprep kit (Zymo Research, #D4036) or ZymoPURE II Plasmid Midiprep kit (Zymo Research, #D4200). Spacer sequences were cloned into pXR003 by BbsI digestion and Gibson assembly (NEB, #E2611) with synthesized DNA duplexes containing the spacer sequence flanked by 19 bp homology arms (Integrated DNA Technologies (IDT)). Plasmids encoding nuclease-inactive Streptococcus pyogenes Cas9 (dCas9) and gRNAs were utilized from a previous study (Pinto et al., 2017). To match the expression context of Cas13d, dCas9 was cloned into the pXR001 vector by removing Cas13d with BsiWI and NheI and inserting a PCR-amplified dCas9 amplicon by Gibson assembly. CUG-targeting and non-targeting short hairpin RNAs (shRNAs) matching the corresponding Cas13d spacer sequences were cloned into the pLKO.1 vector (Addgene, #10878) by AgeI and EcoRI digestion and ligation of 5′-phosphorylated DNA duplexes using T4 DNA ligase (NEB, #M0202). Target plasmids expressing 0 and 480 CTG repeats in the context of DMPK exons 11–15 (DMPKS and DT480, respectively (Ho et al., 2004)) were gifted by Tom Cooper (Baylor College of Medicine). To study competitive binding between dCas13d and MBNL1 at CUG480 repeat RNA in HeLa cells, a plasmid encoding an NLS-dRfxCas13d-(GGGGS)2-EGFP fusion protein (dCas13d-EGFP) was cloned by inserting fragments encoding dCas13d and EGFP amplified from pXR001 by PCR into the pC035 vector (Addgene, #91925) at the BsiWI and KpnI sites by Gibson assembly.
To investigate collateral activity, a plasmid expressing mCherry under the CMV promoter (pmCherry) was cloned by removing EGFP from pEGFP-C1 (Clontech) with AgeI and BglII and assembling the vector with a synthesized mCherry gene fragment (IDT) using In-Fusion cloning (Takara Bio, #102518). pB-Tet-Cas13d was cloned using Gibson assembly by inserting a fragment PCR-amplified from pXR001 containing NLS-RfxCas13d-NLS-HA-T2A-EGFP into a piggyBac transposon vector (Cadiñanos and Bradley, 2007) expressing the insert in a Tet-On cassette and constitutively expressing puromycin acetyltransferase (pac) and reverse tetracycline-controlled transactivator (rtTA) (Gossen et al., 1995). pB-mCherry was cloned using Gibson assembly by inserting a PCR-amplified mCherry gene from a synthetic gene fragment (IDT) into a piggyBac transposon vector expressing the insert constitutively under an EF1α promoter and constitutively expressing a pac-thymidine kinase (TK) fusion protein.
To implement the gRNA excision negative feedback design, a synthetic DNA fragment containing CUG-1 or NT pre-crRNA (22 nt spacer flanked by two 36 nt direct repeats (DR)) was assembled by annealing two complementary oligonucleotides (IDT Ultramer) and inserted into the transcribed region of pXR001 at the KpnI site by Gibson assembly. Plasmids for recombinant AAV preparation were generated by cloning NLS-RfxCas13d-NLS-HA and gRNA (either separately driven by U6 for the unregulated design or within the 50 UTR of the Cas13d gene as pre-crRNA for negative autoregulation) into a vector containing a CMV expression cassette flanked by AAV2 inverted terminal repeats (ITRs).
Cell transfection
For transient transfection experiments, unless otherwise stated, cells were passaged to 12-well tissue culture plates at a density of 1.5 × 105 cells/well (3.9 × 104 cells/cm2) and transfected with 500 ng plasmid DNA using 2 uL of TransIT-X2 transfection reagent (Mirus Bio, #MIR6005) and 100 uL Opti-MEM I reduced serum medium (ThermoFisher Scientific, #31985088) according to the manufacturer’s protocol. When transfecting multiple plasmids, plasmids were mixed at equimolar concentrations unless otherwise noted. Cells were incubated with transfection reagent for 20 h, after which the culture medium was aspirated and replaced with 10% FBS growth medium. Cells were then incubated for an additional 24 h for 2-day experiments or 48 h for 3-day experiments, followed by fixation or RNA isolation for further analysis.
Fluorescence in situ hybridization (FISH)
Nuclear foci formed by CUGn RNA were visualized by FISH using a previously described protocol (Pinto et al., 2017). Briefly, cells in 4-well coated glass chamber slides (ThermoFisher Scientific, #154917) were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (PFA) in PBS at room temperature (RT) for 10 min, washed 3X with PBS, and permeabilized with ice-cold 70% ethanol in water and stored overnight at −20°C. Cells were washed for 30 min at 30°C in FISH wash buffer (25% formamide, 2X saline sodium citrate (SSC) in water). CAG10 FISH probe labeled with Alexa Fluor 594 (Biosearch Technologies, #SS151541–01) was diluted to a working concentration of 380 ng/mL in FISH hybridization buffer (100 mg/mL dextran sulfate, 1 mg/mL yeast tRNA, 2 mM ribonucleoside vanadyl complex (VRC), 200 ug/mL BSA, 25% formamide, 2X SSC in water) and incubated with cells overnight at 30°C in a humidified chamber. The following day, the probe solution was aspirated and replaced with FISH wash buffer for 30 min at 30°C. For nuclear and whole-cell staining, 1 ng/uL DAPI and 1X CellMask Green Plasma Membrane Stain (ThermoFisher Scientific, #C37608) in PBS was added to the cells for 5 min at RT, followed by three washes with PBS of 5 min each. Slides were mounted with glass #1.5 coverslips in Fluoroshield antifade mounting medium (Sigma-Aldrich, #F6182), sealed with clear nail polish, and stored at −20°C until imaging. Widefield epifluorescence and confocal Airyscan imaging was performed on a Zeiss LSM 880 microscope with a Plan-Apochromat 40x/1.3 Oil DIC M27 objective lens. 10 + images were collected for each condition. For confocal images, Airyscan processing was performed in Zeiss ZEN software (version 2.1 SP3 FP3 black 14.0.20.201).
Image processing and FISH quantitation were performed in Python 3. 3D epifluorescence images in CZI format were separated into FISH, DAPI, and CellMask channels, and each channel was collapsed to 2D by maximum intensity projection along the z-dimension. Nuclei were segmented from the DAPI channel using Cellpose 0.0.2.0 (Stringer et al., 2021). Transfected cells were identified as those with a mean FISH intensity in the nucleus >50% higher than the background intensity, calculated as the median FISH intensity in the region outside the nuclear mask. 8+ transfection-positive nuclei were detected for each condition. The mean FISH signal was calculated for each transfected nucleus in all images for each condition. The median for each condition was calculated, and the 95% confidence interval was estimated by bootstrapping.
Immunofluorescence (IF)
For dCas13d/MBNL1 colocalization experiments using dCas13d-EGFP, IF was performed prior to CUGn RNA FISH. Cells in 4-well chamber slides were washed with PBS and fixed in 4% PFA at RT for 10 min, washed 3X with PBS, and permeabilized with ice-cold 70% ethanol in water and stored overnight at −20°C. Cells were washed 3X with IF wash buffer (PBS +0.1% Tween 20 (PBS-T) + 0.5 mM VRC) and incubated with IF blocking buffer (IF wash buffer +1% BSA) for 30 min at RT. Primary antibodies (mouse α-MBNL1 (MB1a(4A8), 1:4), chicken α-GFP (Abcam, #ab13970, 1:1000)) were mixed in IF blocking buffer and applied to cells for 1 h at RT. Cells were then washed 3X with IF wash buffer. Secondary antibodies (α-mouse Alexa Fluor 647, α-chicken Alexa Fluor 488) were added at a 1:1000 dilution to IF blocking buffer and incubated with cells for 1 h at RT. Cells were washed 3X with PBS and fixed again with 4% PFA for 10 min at RT prior to performing FISH as described above. 2D confocal imaging was performed on a Zeiss LSM 880 microscope with a Plan-Apochromat 40x/1.3 Oil DIC M27 objective lens. 13 images were collected for each condition and saved in CZI format.
In Python 3, images were separated into MBNL1, dCas13d-EGFP, CUGn FISH, and DAPI channels, and nuclei were segmented as previously described. Nuclei containing CUG RNA foci were identified as those in which the 98th percentile pixel intensity in the FISH channel was 20 times higher than the median intensity of the background slide. Nuclei extending beyond the image border were excluded from analysis. For each transfected nucleus, RNA foci were segmented by thresholding at 4 times the median pixel intensity within the nucleus in the FISH channel. For each of the dCas13d-EGFP and MBNL channels, enrichment ratio was calculated by dividing the mean pixel intensity within the foci mask by the mean intensity within the region of the nucleus outside the foci mask.
Minigene splicing assay
To measure MBNL-mediated alternative splicing activity, we used an MBNL-regulated splicing reporter minigene spanning MBNL1 exons 4–6 cloned into the RG6 vector (RG6-MBNL1e5) (Orengo et al., 2011). RG6-MBNL1e5 was cotransfected into HeLa cells with plasmids encoding Cas13d, CUG-targeting or non-targeting gRNA, and CUG480 target. n = 3 transfections per condition. 44 h after transfection, RNA was extracted from cells using 300 uL TRIzol (Zymo Research, #R2050) and purified using the Direct-Zol RNA Miniprep kit (Zymo Research, #R2051) according to the manufacturer’s protocol. 100 ng RNA was reverse-transcribed into cDNA using the iScript Reverse Transcription Supermix (Bio-Rad, #1708841). Minigene isoforms were amplified by Taq PCR from 2 uL cDNA using forward and reverse primers complementary to the vector sequence (RG6_F, RG6_R; 54°C annealing; 28 cycles). Primer sequences are presented in Table S4. Exon 5 inclusion ratios were quantitated from 4 uL of PCR reaction by capillary electrophoresis (Fragment Analyzer, Advanced Analytical 1.1.0.11).
EGFP fluorescence assays
To visualize loss of EGFP expression upon Cas13d targeting, HeLa cells were transfected with plasmids encoding Cas13d and EGFP, CUG-targeting or non-targeting gRNA, and CUG480 target. n = 3 transfections per condition. 20 h after transfection, cells were imaged on an EVOS FL digital inverted fluorescence microscope (Life Technologies, #AMF4300PM) at 10X magnification in phase-contrast and GFP channels. Representative images are shown in Figure S2A.
EGFP quantitation was performed using a plate reader assay. HeLa cells were transfected with identical plasmids as above and plated in a 96-well clear tissue culture plate (Celltreat, #229195). n = 5 transfections per condition, 2×104 cells per well. Untransfected cells were plated as a negative control. After 20 h, growth media was aspirated from the cells and replaced with 100 uL PBS. EGFP fluorescence in each well was measured on a Bio-Tek Cytation 3 plate reader (470 nm excitation, 510 nm emission, 146 gain). Baseline intensity was determined by measuring EGFP fluorescence of untransfected cells, and the mean baseline intensity was subtracted from experimental fluorescence measurements.
Cell viability assay
The PrestoBlue resazurin assay (Invitrogen, #A13261) was used to quantify cell viability in response to CUG-targeting Cas13d. HeLa cells were transfected in a 96-well format with plasmids encoding Cas13d, CUG-targeting or non-targeting gRNA, and CUG480 target. n = 5 transfections per condition, 2×104 cells per well, with a total volume of 100 uL growth medium per well. 5 wells each of untransfected cells and media alone were also plated. 20 h after transfection, 11 uL PrestoBlue reagent was added to each well and mixed gently by pipetting. Cells were incubated with PrestoBlue reagent at 37°C for 30 min before measuring fluorescence on a Bio-Tek Cytation 3 plate reader (550 nm excitation, 590 nm emission, 60 gain). Baseline intensity was calculated as the mean fluorescence of cell-free media and was subtracted from all measurements. Cell viability was calculated by normalizing experimental measurements by the mean fluorescence intensity of untransfected cells.
Cas13d specificity transcriptomic analysis
RNA-seq was performed to screen for transcriptomic off-targets of repeat-targeting approaches (Cas13d, dCas9, and shRNA). CUG-targeted systems and non-targeting controls (no gRNA or non-targeting shRNA) were transfected in triplicate into HeLa cells. CUG480 target plasmid was omitted to enrich for off-target events. After 68 h incubation, RNA was extracted with 300 uL TRIzol and purified using the Direct-zol RNA Miniprep Kit. Ribosomal RNA was depleted from 300 ng total RNA using the NEBNext rRNA Depletion Kit (NEB, #E6310L), and libraries were generated for next-generation sequencing using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, #E7760L) according to the manufacturer protocol. Libraries were multiplexed with Illumina i5 and i7 barcoding primers, pooled at 4 nM, and sequenced in a 2 × 76 bp paired-end format on an Illumina NextSeq 500. >14 million paired end reads were sequenced per library and recorded in FASTQ format.
Transcript expression was quantified by pseudoalignment to the hg19 human reference genome using kallisto 0.43.0 (Bray et al., 2016). Reads were aligned to hg19 using HISAT2 2.0.0-beta (Kim et al., 2019). For each targeting technology (Cas13d, dCas9, shRNA), differential gene expression analysis was performed using DESeq2 1.30.1 (Love et al., 2014) to compare CUG-targeting and non-targeting conditions, with gene counts calculated by htseq-count 0.11.2 (Anders et al., 2015) provided as input. Off-targets were defined as differentially expressed (DE) genes with a false discovery rate (FDR) q < 0.05. For Cas13d and shRNA libraries, gene ontology (GO) analysis was performed using PANTHER (Mi et al., 2019) (annotation version 2021–05-01) to identify biological processes associated with each set of DE genes. GO biological processes with FDR q < 0.05 were considered significant and were assigned to categories according to their descriptions.
For all annotated human transcripts, longest CUGn repeat tract length (in C, U, or G registers) was determined from the NCBI RefSeq reference mRNA sequence. Transcripts were grouped by maximum CUGn length, and median log2 fold-change of TPM between CUGn-targeting and non-targeting conditions was calculated for each transcript group and targeting approach.
Collateral activity mCherry fluorescence assays
A plate reader assay was developed to quantitate bulk mCherry fluorescence in transfected cells. HeLa, HEK293, or Neuro2a cells were transfected in a 96-well format with pXR001, targeting or non-targeting gRNA in pXR003, target or control plasmid, and pmCherry. n = 5 transfections per condition, 2×104 cells per well, with a total volume of 100 uL growth medium per well. 5 wells of untransfected cells were also plated. After 20 h incubation, transfection media was aspirated and replaced with 100 uL PBS. mCherry fluorescence was measured on a Bio-Tek Cytation 3 plate reader (587 nm excitation, 627 nm emission, 202 gain). Baseline intensity was defined as the mean mCherry fluorescence of untransfected cells and was subtracted from experimental measurements.
For single-cell measurement of mCherry expression, HeLa cells were transfected in 4-well coated glass chamber slides with plasmids encoding Cas13d and EGFP, CUG-1 or NT gRNA, CUG480 target or CUG0 control RNA, and mCherry. After 20 h incubation, cells were washed with PBS and fixed in 4% PFA at RT for 10 min. Cells were washed 3X with PBS, and nuclei were stained with 1 ng/uL DAPI in PBS for 5 min at RT, followed by an additional three PBS washes. Slides were mounted with glass #1.5 coverslips in Fluoroshield antifade mounting medium, sealed with clear nail polish, and stored at -20C until imaging. Widefield epifluorescence imaging was performed on a Zeiss LSM 880 microscope with a Plan-Apochromat 40x/1.3 Oil DIC M27 objective lens. 5 + images were collected for each condition and were processed in Fiji v2.0.0-rc-69/1.52p (Schindelin et al., 2012). In each image, all EGFP-positive cells were manually segmented, and total mCherry fluorescence intensity was measured for each cell. >26 cells were measured for each condition. Distributions of mCherry expression were compared between conditions using the two-sided Mann-Whitney U test.
Collateral activity RNA assay
To enable precise measurement of Cas13d collateral RNase activity when targeting transgenic or endogenous RNAs, we developed a HeLa cell line containing genomically integrated cassettes expressing mCherry constitutively and Cas13d and EGFP under a tetracycline-inducible promoter (HeLa-tet:Cas13d-mCherry). First, HeLa cells were transfected in a 12-well format (1.5×105 cells) with 200 ng pB-Tet-Cas13d and 800 ng of plasmid encoding codon-optimized piggyBac transposase (mPB) (Cadiñanos and Bradley, 2007). After 2 days incubation, transfection media was replaced with 10% FBS growth media containing 2 μg/mL puromycin (AG Scientific, #P-1033-SOL) to select for transposon integration. Cells were passaged to a 10-cm dish upon reaching confluency, and puromycin selection was maintained for 2 weeks. 1.5×105 cells were then passaged to a 12-well dish and transfected with 200 ng pB-mCherry and 800 ng mPB plasmid to integrate a constitutively expressing mCherry gene. Once confluent, cells were again passaged to a 10-cm dish. Two weeks after integration, Cas13d and EGFP expression was induced by incubation with 2 mM doxycycline (Sigma, #D3447–500MG) for 2 days, and EGFP-positive and mCherry-positive single cells were sorted into 96-well plates using a BD FACSAria II cell sorter at the University of Florida Interdisciplinary Center for Biotechnology Research (ICBR). Twelve EGFP+ and mCherry + clones were manually identified using an EVOS FL microscope and expanded. The clone exhibiting the strongest expression of both EGFP and mCherry upon addition of doxycycline was chosen for the collateral activity RNA assay.
To perform the assay, HeLa-tet:Cas13d-mCherry and wild-type HeLa cells were mixed at a 1:4 ratio in growth media containing 2 μM doxycycline and plated in a 24-well plate format at a density of 7.5×104 cells/well. Cells were transfected with 125 ng gRNA plasmid and either 125 ng CUG480 plasmid (for CUG-targeting validation experiment) or 125 ng pUC19 as inert carrier DNA (for endogenous gene targeting experiment). n = 3 transfections per condition. After 44 h, RNA was extracted with 300 uL TRIzol and purified using the Direct-zol RNA Miniprep Kit. cDNA was reverse-transcribed from 50 ng RNA using the iScript Reverse Transcription Supermix and diluted 10-fold in water. mCherry and control (GAPDH) expression levels were measured separately by qPCR from 4 uL of diluted cDNA using Taq DNA Polymerase (NEB, #M0270L), dsGreen DNA detection dye (Lumiprobe, #11010), and 200 nM each of forward and reverse primers (mCherry_F/mCherry_R and GAPDH_F/GAPDH_R, respectively). Primer sequences are presented in Table S4 qPCR reactions were performed in duplicate on a C1000 Touch thermocycler (Bio-Rad) using a two-step cycling protocol: initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 45 s dsGreen fluorescence was measured after the extension step of each cycle. Cq values were calculated using Bio-Rad CFX Manager 3.1 software, and the difference between mCherry and GAPDH Cq (ΔCq) was calculated and averaged across the two qPCR replicates. The ratio of mCherry to GAPDH mRNA expression (defined as 2−ΔCq) was plotted for each condition. ΔCq values in targeting and non-targeting conditions were compared using the one-tailed Student’s t test (independent samples, equal variance).
Modeling of negative autoregulation by gRNA excision
An ordinary differential equation (ODE) model was constructed that describes the dynamics of GENO autoregulation of Cas13d expression:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
where R is the concentration of Cas13d mRNA (also containing the pre-crRNA), A is the concentration of Cas13d apoprotein, B is the concentration of Cas13d:gRNA binary complex, rt is the rate of RNA polymerase II transcription of the Cas13d mRNA, kT is the translation rate constant of Cas13d, kproc is the rate constant of crRNA processing, and γi is the degradation rate constant of species i. Transcription is modeled by zero-order kinetics, translation and degradation by first-order kinetics, and crRNA processing by second-order kinetics. Further details of the dynamical model, a reference model for unregulated Cas13d, and analytical proofs are presented in Data S1.
To calculate equilibrium binary complex concentration across ranges of biochemical parameters, simulations were conducted by ODE integration using Python 3. RNA and protein degradation rates were estimated from median half-lives determined from the literature (10 h (Yang et al., 2003) and 36 h (Cambridge et al., 2011), respectively), and the degradation rates of Cas13d apoprotein and binary complex were estimated to be equivalent. In the unregulated reference model, the half-life of free gRNA was estimated as 2 h. For each vector of parameters rt, kT, and kproc, the dynamical model was integrated over 480 h with 5000 timesteps to reach steady state. Autoregulation efficiency ηGENO was calculated for each parameter vector as the difference of unity and the ratio of equilibrium binary complex concentrations between GENO and unregulated conditions (see Data S1).
Western blot
Cas13d protein expression in unregulated and autoregulated conditions was measured by Western blot. HeLa cells were transfected in 12-well format with 500 ng Cas13d plasmid (unregulated or GENO, +100 ng NT gRNA plasmid for unregulated Cas13d). n = 3 transfections per condition. After 44 h incubation, cells were washed with 1 mL PBS, and protein was extracted for 15 min on ice in 150 uL radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific, #89901) containing 2X SIGMAFAST Protease Inhibitor Cocktail (Sigma-Aldrich, #S8830) and 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich, #10837091001). Extractions were transferred to microcentrifuge tubes and centrifuged at 12,000 RCF for 15 min at 4°C. Total protein content in supernatant was measured using the Pierce BCA Protein Assay (Thermo Scientific, #23225). For each sample, 10 μg protein was mixed with 5.4 uL 4X NuPage LDS Sample Buffer (Invitrogen, #NP0008), 0.6 uL 100 mM dithiothreitol (NEB, #B1034A), and water to a total volume of 24 uL. Protein was denatured at 95°C for 5 min prior to loading on a NuPage 4–12% Bis-Tris polyacrylamide gel (#NP0336), with 3 uL Precision Plus Protein All Blue prestained standards (Bio-Rad, #1610373) loaded as a ladder. Gel electrophoresis was performed at 120 V for 45 min in MOPS running buffer (10.46 mg/mL 3-(N-morpholino)propanesulfonic acid (MOPS), 6.06 mg/mL Tris base, 1 mg/mL SDS, 0.3 mg/mL ethylenediaminetetraacetic acid (EDTA) in water). Protein was transferred to a methanol-activated polyvinylidene fluoride (PVDF) membrane (Bio-Rad, #1620264) using the iBlot 2 dry transfer system (ThermoFisher Scientific, #IB21001).
The membrane was incubated in 5 mL SEA BLOCK Blocking Buffer (Thermo Scientific, #37527) for 30 min at RT in a dark container on a rocking mixer. Blocking buffer was removed and replaced with primary antibody solution (rabbit α-HA (C29F4, 1:1000, Cell Signaling Technology) and mouse α-HSP70 (5A5, 1:500, Invitrogen) in 5 mL SEA BLOCK +0.2% Tween 20 (Fisher Bioreagents, #BP152–1)), and the membrane was placed in a 4°C room on a rocking mixer overnight. The next day, the primary antibody solution was removed, and the membrane was washed 3X in PBS-T. After washing, the secondary antibody solution was added (IRDye 800CW donkey α-rabbit and 680RD donkey α-mouse (1:5000 each, LI-COR) in 5 mL SEA BLOCK +0.2% Tween 20) and incubated for 1 h at RT on a rocking mixer. The secondary antibodies were removed, and the membrane was washed 3X in PBS-T. The blot was imaged on an LI-COR Odyssey CLx scanner at 700 and 800 nm excitation wavelengths. Image processing was performed in Fiji, and blot images were inverted for publication.
RT-qPCR
Knockdown of CUG480 RNA by unregulated and regulated Cas13d was measured by qPCR. HeLa cells were transfected in 24-well format with 250 ng Cas13d plasmid (unregulated or GENO, +50 ng gRNA plasmid for unregulated Cas13d) and 100 ng CUG480 plasmid. n = 3 transfections per condition. 44 h after transfection, RNA was extracted from cells using 200 uL TRIzol and purified using the Direct-Zol RNA Miniprep kit. To remove any remaining CUG480 plasmid DNA prior to qPCR, polyadenylated RNA was purified from 250 ng total RNA using Dynabeads Oligo(dT)25 magnetic beads (Invitrogen, #61005) according to the manufacturer’s protocol. Polyadenylated RNA was reverse-transcribed into cDNA using the iScript Reverse Transcription Supermix.
cDNA from CUG480 and GAPDH transcripts was amplified in separate reactions and quantified using the dye-based qPCR approach described above for the collateral activity RNA assay. Primer sequences are presented in Table S4. The difference between CUG480 and GAPDH Cq (ΔCq) was calculated and averaged across two qPCR replicates, and the ratio of expression levels between the transcripts (2−ΔCq) was plotted. ΔCq values in targeting and non-targeting conditions were compared using the one-tailed Student’s t test (independent samples, equal variance).
AAV treatment of DM1 myoblasts
Recombinant AAV constructs were packaged in AAV6 capsids and purified by SignaGen Laboratories (#SL100865, >1013 VG/mL). Production of AAV-CMV-unreg-CUG1 was attempted on three occasions but titers >109 VG/mL could not be obtained. DM1 myoblasts were grown to 50% confluency in 4-well coated glass chamber slides and transduced with AAV at a titer of 2×1010 VG/mL for 6 days prior to fixation and staining.
Hybridization chain reaction FISH (HCR FISH)
To image single molecules of Cas13d mRNA by FISH, HCR v3.0 was performed for signal amplification (Choi et al., 2018). A custom HCR probe set matching the Cas13d CDS mRNA sequence (30 split-initiator probes, B1 amplifier) was designed by Molecular Instruments (Los Angeles, CA). After AAV transduction and culture, myoblasts were fixed in 4% PFA for 10 min at RT, washed 3X with PBS, permeabilized with 0.2% Triton X-100 in PBS for 10 min at RT, and washed again 3X with PBS. If IF was also performed, cells were incubated with IF blocking buffer for 30 min prior to incubation of primary antibody (rabbit α-HA (C29F4, 1:1000)) in IF blocking buffer with cells at 4°C overnight. The next day, cells were washed 3X with IF wash buffer and incubated with secondary antibody (α-rabbit Alexa Fluor 568, 1:1000) in IF blocking buffer for 1 h at RT, followed by three washes in PBS. To cross-link antibodies to antigens prior to HCR FISH, cells were fixed again in 4% PFA for 10 min and washed 3X with PBS.
For HCR FISH, cells were washed twice with 2X SSC and incubated with HCR Probe Hybridization Buffer (Molecular Instruments) for 30 min at 37°C. For each well, 1.2 pmol of Cas13d HCR probe set was mixed with 300 uL HCR Probe Hybridization Buffer and incubated with cells overnight at 37°C in a humidified chamber. If CUGn FISH was also performed, CAG10 Alexa Fluor 594 probe was mixed with the HCR probe solution at 380 ng/mL and prior to overnight incubation. The following day, cells were washed 4X with HCR Probe Wash Buffer (Molecular Instruments) at 37°C and twice with 5X SSC +0.1% Tween 20 (5X SSCT) at RT to remove excess probe, and cells were equilibrated with HCR Amplification Buffer (Molecular Instruments) for 30 min at RT. To prepare amplification hairpins, 2.4 pmol each of B1 h1 and B1 h2 hairpins (Molecular Instruments, 647 nm) were heated in separate tubes to 95°C for 90 s and cooled to RT in a light-protected compartment for 30 min. Hairpins were mixed in 40 uL HCR Amplification Buffer and applied to cells at RT for 2 h. Cells were washed 5 times in 5X SSCT and 3 times in PBS to remove excess hairpins. For nucleus and cytoplasm staining, 1 ng/uL DAPI and 0.1X HCS CellMask Green (ThermoFisher Scientific, #H32714) in PBS was added to the cells for 5 min at RT, followed by three washes with PBS. Slides were mounted on glass #1.5 coverslips using ProLong Diamond antifade mounting medium (ThermoFisher Scientific, #P36965) and stored at −20°C until imaging. 2D confocal imaging was performed on a Zeiss LSM 880 microscope with Plan-Apochromat 40x/1.3 Oil DIC M27 and Plan-Apochromat 20x/0.8 M27 objective lenses. All fields of view were chosen randomly in the DAPI channel.
For cells labeled with Cas13d HCR FISH and α-HA IF, 3–5 images per condition were collected at 20x magnification. All images were processed in Python 3 to analyze HCR FISH and IF signal intensities per nucleus. Images in CZI format were loaded and channels were separated into 2D arrays. Nuclei were identified by thresholding the DAPI channel using a modified Otsu’s method (Otsu, 1979). Within each nucleus, the mean pixel intensity in each of the HCR FISH and IF channels was calculated. Nuclear measurements were pooled across all images within each condition.
For cells labeled with Cas13d HCR FISH and CUGn FISH, 21 images per condition were collected at 40x magnification and processed using Python 3 to measure FISH intensities and detect diffraction-limited spots. Nuclei were thresholded as above. Mean nuclear HCR FISH and CUGn FISH signal intensities were calculated for each nucleus. In both FISH channels, spots were detected using the Laplacian of Gaussian method (Lindeberg, 1998) (thresholds of 0.6% for HCR FISH and 0.2% for CUGn FISH, Gaussian standard deviation of 2 px). For each detected CUGn FISH spot, spot intensity was calculated by convolving a Gaussian kernel (standard deviation of 2 px) with the FISH channel image and measuring the pixel brightness at the centroid of the spot in the blurred image. Per-nucleus and per-spot measurements were pooled across all images within each condition.
For cells labeled with α-MBNL1 IF, CUGn FISH, and Cas13d HCR FISH, 8 images per condition were collected at 40x magnification, and MBNL1 expression and localization was quantified using Python 3. Images were loaded, channels were separated, and nuclei were thresholded as previously. Cells were segmented by thresholding the MBNL1 channel using a modified Li’s method (Li and Tam, 1998). Cells with nuclei that extend beyond the border of the image were excluded from analysis. For each cell, the mean pixel intensity in the MBNL1 channel in the nucleus and the cytoplasm was calculated, and the ratio was calculated and displayed.
For cells labeled with HCR FISH for PPIB, POLR2A, and Cas13d mRNA, 6 images per condition were collected at 40x magnification and processed with Python 3 to detect diffraction-limited spots. Nuclei were thresholded as previously, and PPIB and POLR2A HCR FISH spots in each nucleus were detected using the Laplacian of Gaussian method (0.4% threshold). Per-nucleus measurements were pooled across images within each condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments are provided in the corresponding figure legends and Results sections of this manuscript. A statistical significance threshold of p < 0.05 was used for all tests conducted in this study, except to identify differentially expressed genes and GO categories in RNA-seq analysis, for which the Benjamini-Hochberg false discovery rate (FDR) correction was applied prior to significance thresholding. Student’s t tests and Mann-Whitney U tests were performed in Python 3 using statistical functions provided in scipy.stats.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal α-MBNL1 (MB1a(4A8)) | MilliporeSigma | Cat#MABE70; RRID:AB_2618248 |
| Chicken polyclonal α-GFP | Abcam | Cat#ab13970; RRID:AB_300798 |
| Alexa Fluor 647 conjugated goat α-mouse secondary | Life Technologies | Cat#A21235; RRID:AB_2535804 |
| Alexa Fluor 488 conjugated goat α-chicken secondary | Life Technologies | Cat#A11039; RRID:AB_2534096 |
| Rabbit monoclonal α-HA (C29F4) | Cell Signaling Technology | Cat# 3724S; RRID:AB_1549585 |
| Mouse monoclonal α-HSP70 (5A5) | Invitrogen | Cat#MA3007; RRID:AB_325455 |
| IRDye 800CW donkey α-rabbit secondary | LI-COR | Cat#926-32213; RRID:AB_621848 |
| IRDye 680RD donkey α-mouse secondary | LI-COR | Cat#926-68072; RRID:AB_10953628 |
| Alexa Fluor 568 conjugated donkey α-rabbit secondary | Invitrogen | Cat#A10042; RRID:AB_2534017 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB Stable chemically competent E. coli | New England Biolabs | Cat#C3040 |
| AAV-CMV-unreg-NT | This paper | N/A |
| AAV-CMV-GENO-CUG1 | This paper | N/A |
| AAV-CMV-GENO-NT | This paper | N/A |
|
| ||
| Chemicals peptides and recombinant proteins | ||
|
| ||
| Fluoroshield antifade mounting medium | Sigma-Aldrich | Cat#F6182 |
| ProLong Diamond antifade mounting medium | ThermoFisher Scientific | Cat#P36965 |
| 4′,6-diamidino-2-phenylindole (DAPI) | Invitrogen | Cat#D1306 |
| HCS CellMask Green cell stain | ThermoFisher Scientific | Cat#H32714 |
| T4 DNA ligase | New England Biolabs | Cat#M0202 |
| TransIT-X2 transfection reagent | Mirus Bio | Cat#MIR6005 |
| Paraformaldehyde (PFA) | Electron Microscopy Sciences | Cat#15710 |
| Ribonucleoside vanadyl complex | New England Biolabs | Cat#S1402S |
| Puromycin | AG Scientific | Cat#P-1033-SOL |
| Doxycycline | Sigma | Cat#D3447-500MG |
| TRI Reagent | Zymo Research | Cat#R2050-1-200 |
| dsGreen DNA detection dye | Lumiprobe | Cat#11010 |
| SIGMAFAST Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#S8830 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat#10837091001 |
| 4X NuPage LDS Sample Buffer | Invitrogen | Cat#NP0008 |
| Dithiothreitol (DTT) | New England Biolabs | Cat#B1034A |
| SEA BLOCK Blocking Buffer | Thermo Scientific | Cat#37527 |
|
| ||
| Critical commercial assays | ||
|
| ||
| PrestoBlue resazurin cell viability assay | Invitrogen | Cat#A13261 |
| Taq 2X master mix | New England Biolabs | Cat#M0270L |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | Cat#M0530L |
| Gibson assembly master mix | New England Biolabs | Cat#E2611 |
| Zyppy Plasmid Miniprep kit | Zymo Research | Cat#D4036 |
| Direct-Zol RNA Miniprep kit | Zymo Research | Cat#R2051 |
| iScript Reverse Transcription Supermix | Bio-Rad | Cat#1708841 |
| NEBNext rRNA Depletion Kit | New England Biolabs | Cat#E6310L |
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina |
New England Biolabs | Cat#E7760L |
| Pierce BCA Protein Assay | Thermo Scientific | Cat#23225 |
| Dynabeads Oligo(dT)25 magnetic beads | Invitrogen | Cat#61005 |
|
| ||
| Deposited data | ||
|
| ||
| GEO (RNA-seq of repeat-targeting Cas13d, dCas9 and shRNA in HeLa) | This paper | GSE191329 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: HeLa | ATCC | CCL-2 |
| Human: HEK293 | ATCC | CRL-1573 |
| Human: DM-05 adult DM1 patient-derived myoblasts | (Xia et al., 2013) | N/A |
| Mouse: Neuro2a | ATCC | CCL-131 |
| Human: HeLa-tet:Cas13d-mCherry | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| gRNA spacer sequences, see Table S1 | This paper | N/A |
| PCR primers, see Table S1 | This paper | N/A |
| Alexa Fluor 594 conjugated CAG10 FISH probe | Biosearch Technologies | Cat#SS151541-01 |
| Cas13d mRNA HCR FISH probe set, B1 amplifier | Molecular Instruments | N/A |
| PPIB mRNA HCR FISH probe set, B2 amplifier | Molecular Instruments | N/A |
| POLR2A mRNA HCR FISH probe set, B5 amplifier | Molecular Instruments | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pXR001 (NLS-RfxCas13d-NLS-HA-T2A-EGFP) | (Konermann et al., 2018) | Addgene #109049 |
| pXR002 (NLS-dRfxCas13d-NLS-HA-T2A-EGFP) | (Konermann et al., 2018) | Addgene #109050 |
| pXR003 (RfxCas13d gRNA cloning plasmid) | (Konermann et al., 2018) | Addgene #109053 |
| pXR-dSpCas9 | This paper | N/A |
| pCR-Blunt II-TOPO- U6-(CAG)6 gRNA | (Pinto et al., 2017) | N/A |
| pLKO.1 (shRNA cloning plasmid) | Addgene #10878 | |
| DMPKS (CUG0 target plasmid) | Gift from Thomas Cooper | N/A |
| DT480 (CUG480 target plasmid) | (Ho et al., 2004) | Addgene #80413 |
| dCas13d-EGFP (NLS-dRfxCas13d-(GGGGS)2-EGFP) | This paper | N/A |
| pmCherry | This paper | N/A |
| pB-Tet-Cas13d | This paper | N/A |
| pB-mCherry | This paper | N/A |
| pXR001-GENO-CUG1 | This paper | N/A |
| pXR001-GENO-NT | This paper | N/A |
| RG6-MBNL1e5 | (Orengo et al., 2011) | N/A |
| mPB (PiggyBac transposase) | (Cadiñanos and Bradley, 2007) | N/A |
| pUC19 | New England Biolabs | Cat#N3041S |
|
| ||
| Software and algorithms | ||
|
| ||
| Code generated in this study | This paper | https://github.com/cpkelley94/geno |
| Python 3.6 | Python Software Foundation | https://www.python.org/ |
| ZEN v2.1 SP3 FP3 black 14.0.20.201 | ZEISS | https://www.micro-shop.zeiss.com/en/us/softwarefinder/software-categories/zen-black/ |
| Cellpose 0.0.2.0 | (Stringer et al., 2021) | https://www.cellpose.org/ |
| kallisto 0.43.0 | (Bray et al., 2016) | https://pachterlab.github.io/kallisto/ |
| HISAT2 2.0.0-beta | (Kim et al., 2019) | https://daehwankimlab.github.io/hisat2/ |
| DESeq2 1.30.1 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| htseq-count 0.11.2 | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/ |
| PANTHER gene ontology analysis, annotation v.2021-05-01 | (Mi et al., 2019) | http://www.pantherdb.org/ |
| Fiji v2.0.0-rc-69/1.52p | (Schindelin et al., 2012) | https://imagej.net/software/fiji/ |
| Bio-Rad CFX Manager 3.1 | Bio-Rad | https://www.bio-rad.com/en-us/sku/1845000-cfx-manager-software?ID=1845000 |
|
| ||
| Other | ||
|
| ||
| Cytation 3 plate reader | Bio-Tek | N/A |
| NextSeq 500 | Illumina | N/A |
| C1000 Touch thermocycler | Bio-Rad | N/A |
| Odyssey CLx scanner | LI-COR | N/A |
| EVOS FL digital inverted fluorescence microscope | Life Technologies | Cat#AMF4300PM |
| LSM 880 confocal microscope with Airyscan | ZEISS | N/A |
| Fragment Analyzer | Advanced Analytical | 1.1.0.11 |
| iBlot 2 dry transfer system | ThermoFisher Scientific | Cat#IB21001 |
Highlights.
CRISPR-Cas13d robustly degrades CUG repeat RNAs toxic in myotonic dystrophy type 1
Cas13d collateral activity dramatically depletes off-target RNAs in mammalian cells
Negative autoregulation minimizes collateral activity at repetitive target RNAs
Compact regulation strategies improve gene therapies by precisely controlling dose
ACKNOWLEDGMENTS
We are grateful to all members of the Wang lab for their continuous support and collaborative environment, and in particular Ona McConnell and Dr. Ryan Hildebrandt for assistance with cell-culture experiments. We thank the University of Florida Interdisciplinary Center for Biotechnology Research for flow cytometry services. We also thank Yuxi Ai and Prof. Jeremy Wilusz (University of Pennsylvania) for our collaborative discussions. Finally, we thank Geno the Yorkshire terrier, the eponym for our autoregulation approach. This work was supported by NIH R01 AG058636 and the NIH P50 NS048843 Senator Paul D. Wellstone Muscular Dystrophy Specialized Research Center award to E.T.W. C.P.K. is supported by the NSF Graduate Research Fellowship.
Footnotes
DECLARATION OF INTERESTS
E.T.W. has consulted for Expansion Therapeutics, Entrada Therapeutics, Design Therapeutics, Triplet Therapeutics, and Faze Medicines. E.T.W. is a co-founder of, shareholder in, and adviser to Kate Therapeutics. Financial support for research has been provided to the lab of E.T.W. at the University of Florida by Expansion Therapeutics, Entrada Therapeutics, and Kate Therapeutics. E.T.W. and C.P.K. are inventors on a patent application filed by the University of Florida related to this work.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111226.
REFERENCES
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573. 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284. 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai Y, Liang D, and Wilusz JE (2022). CRISPR/Cas13 effectors have differing extents of off-target effects that limit their utility in eukaryotic cells. Nucleic Acids Res. gkac159. 10.1093/nar/gkac159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. (2006). An Introduction to Systems Biology: Design Principles of Biological Circuits (CRC Press; ). [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinforma. Oxf. Engl 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelbello AJ, Rzuczek SG, Mckee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET, and Disney MD (2019). Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. USA 116, 7799–7804. 10.1073/pnas.1901484116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arandel L, Matloka M, Klein AF, Rau F, Sureau A, Ney M, Cordier A, Kondili M, Polay-Espinoza M, Naouar N, et al. (2022). Reversal of RNA toxicity in myotonic dystrophy via a decoy RNA-binding protein with high affinity for expanded CUG repeats. Nat. Biomed. Eng 6, 207–220. 10.1038/s41551-021-00838-2. [DOI] [PubMed] [Google Scholar]
- Arnett AL, Konieczny P, Ramos JN, Hall J, Odom G, Yablonka-Reuveni Z, Chamberlain JR, and Chamberlain JS (2014). Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev 1, 14038. 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becskei A, and Serrano L. (2000). Engineering stability in gene networks by autoregulation. Nature 405, 590–593. 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, and Hudson T. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Buchman AB, Brogan DJ, Sun R, Yang T, Hsu PD, and Akbari OS (2020). Programmable RNA targeting using CasRx in flies. CRISPR J. 3, 164–176. 10.1089/crispr.2020.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadiñanos J, and Bradley A. (2007). Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 35, e87. 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge SB, Gnad F, Nguyen C, Bermejo JL, Krüger M, and Mann M. (2011). Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. J. Proteome Res 10, 5275–5284. 10.1021/pr101183k. [DOI] [PubMed] [Google Scholar]
- Cerchione D, Loveluck K, Tillotson EL, Harbinski F, DaSilva J, Kelley CP, Keston-Smith E, Fernandez CA, Myer VE, Jayaram H, et al. (2020). SMOOT libraries and phage-induced directed evolution of Cas9 to engineer reduced off-target activity. PLoS One 15, e0231716. 10.1371/journal.pone.0231716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles EJ, Kim SE, Knott GJ, Smock D, Doudna J, and Savage DF (2021). Engineering improved Cas13 effectors for targeted post-transcriptional regulation of gene expression. [Google Scholar]
- Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Dev. Camb. Engl 145, dev165753. 10.1242/dev.165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F. (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Jillette N, Zhu JJ, Li S, and Cheng AW (2020). CRISPR artificial splicing factors. Nat. Commun 11, 2973. 10.1038/s41467-020-16806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JHD, Tjian R, and Doudna JA (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YH, Pizzuti A, Fenwick RG, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, and de Jong P. (1992). An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258. 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Gates DP, Coonrod LA, and Berglund JA (2011). Autoregulated splicing of muscleblind-like 1 (MBNL1) Pre-mRNA. J. Biol. Chem 286, 34224–34233. 10.1074/jbc.M111.236547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Freundlieb S, Bender G, Müller G, Hillen W, and Bujard H. (1995). Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769. 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- Han S, Zhao BS, Myers SA, Carr SA, He C, and Ting AY (2020). RNA-protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc. Natl. Acad. Sci. USA 117, 22068–22079. 10.1073/pnas.2006617117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526. 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- He B, Peng W, Huang J, Zhang H, Zhou Y, Yang X, Liu J, Li Z, Xu C, Xue M, et al. (2020). Modulation of metabolic functions through Cas13d-mediated gene knockdown in liver. Protein Cell 11, 518–524. 10.1007/s13238-020-00700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Charlet-B N, Poulos MG, Singh G, Swanson MS, and Cooper TA (2004). Muscleblind proteins regulate alternative splicing. EMBO J. 23, 3103–3112. 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T, Kim DW, Appel H, Pannullo NA, Leavey P, Ozawa M, Zheng S, Yu M, Peachey NS, and Blackshaw S. (2022). Genetic loss of function of Ptbp1 does not induce glia-to-neuron conversion in retina. Cell Rep. 39, 110849. 10.1016/j.celrep.2022.110849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K, Wu J, Bezprozvanny I, and Corey DR (2009). Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol 27, 478–484. 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh N, Depner N, Larson R, and King-Jones K. (2020). A versatile toolkit for CRISPR-Cas13-based RNA manipulation in Drosophila. Genome Biol. 21, 279. 10.1186/s13059-020-02193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, and Vale RD (2017). RNA phase transitions in repeat expansion disorders. Nature 546, 243–247. 10.1038/nature22386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Li H, Liu X, Zhang J, Zhang W, Li T, Liu L, and Yu X. (2020). Precise and efficient silencing of mutant KrasG12D by CRISPR-CasRx controls pancreatic cancer progression. Theranostics 10, 11507–11519. 10.7150/thno.46642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S, Altae-Tran H, Jin X, Madigan VJ, Oshiro R, Makarova KS, Koonin EV, and Zhang F. (2021). Compact RNA editors with small Cas13 proteins. Nat. Biotechnol 10.1038/s41587-021-01030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, and Joung JK (2015a). Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol 33, 1293–1298. 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, et al. (2015b). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485. 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, and Hsu PD (2018). Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676, e14. 10.1016/j.cell.2018.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, and Kozlowski P. (2012). Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 40, 11–26. 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushawah G, Hernandez-Huertas L, Abugattas-Nuñez Del Prado J, Martinez-Morales JR, DeVore ML, Hassan H, Moreno-Sanchez I, Tomas-Gallardo L, Diaz-Moscoso A, Monges DE, et al. (2020). CRISPR-Cas13d induces efficient mRNA knockdown in animal embryos. Dev. Cell 54, 805–817, e7. 10.1016/j.devcel.2020.07.013. [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang G-S, and Cooper TA (2007). Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 28, 68–78. 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois M-A, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J, Rossi JJ, and Lee NS (2005). Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. J. Biol. Chem 280, 16949–16954. 10.1074/jbc.M501591200. [DOI] [PubMed] [Google Scholar]
- Lee JE, Bennett CF, and Cooper TA (2012). RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 109, 4221–4226. 10.1073/pnas.1117019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CH, and Tam PKS (1998). An iterative algorithm for minimum cross entropy thresholding. Pattern Recognit. Lett 19, 771–776. 10.1016/S0167-8655(98)00057-9. [DOI] [Google Scholar]
- Li C, Cao Y, Zhang L, Li J, Wang J, Zhou Y, Wei H, Guo M, Liu L, Liu C, et al. (2021a). CRISPR-CasRx targeting LncRNA LINC00341 inhibits tumor cell growth in vitro and in vivo. Front. Mol. Biosci 8, 638995. 10.3389/fmolb.2021.638995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li X, Xue W, Zhang L, Yang L-Z, Cao S-M, Lei Y-N, Liu C-X, Guo S-K, Shan L, et al. (2021b). Screening for functional circular RNAs using the CRISPR-Cas13 system. Nat. Methods 18, 51–59. 10.1038/s41592-020-01011-4. [DOI] [PubMed] [Google Scholar]
- Lindeberg T. (1998). Feature detection with automatic scale selection. Int. J. Comput. Vis 30, 79–116. 10.1023/A:1008045108935. [DOI] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, and O’Hoy K. (1992). Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 255, 1253–1255. 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Malik I, Kelley CP, Wang ET, and Todd PK (2021). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol 22, 589–607. 10.1038/s41580-021-00382-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeske AJ, Nakandakari-Higa S, and Marraffini LA (2019). Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature 570, 241–245. 10.1038/s41586-019-1257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc 14, 703–721. 10.1038/s41596-019-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, and Swanson MS (2000). Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19, 4439–4448. 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, and Langer R. (2021). Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov 20, 101–124. 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, Wilson RH, Adam B, del Valle G, Brian R, et al. (2012). Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet 21, 3558–3567. 10.1093/hmg/dds185. [DOI] [PubMed] [Google Scholar]
- Mulders SAM, van den Broek WJAA, Wheeler TM, Croes HJE, van Kuik-Romeijn P, de Kimpe SJ, Furling D, Platenburg GJ, Gourdon G, Thornton CA, et al. (2009). Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 106, 13915–13920. 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MR (2019). Molecular mechanisms of RNA targeting by cas13containing type VI CRISPR-cas systems. J. Mol. Biol 431, 66–87. 10.1016/j.jmb.2018.06.029. [DOI] [PubMed] [Google Scholar]
- Orengo JP, Ward AJ, and Cooper TA (2011). Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol 69, 681–690. 10.1002/ana.22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero BA, Poukalov K, Hildebrandt RP, Thornton CA, Jinnai K, Fujimura H, Kimura T, Hagerman KA, Sampson JB, Day JW, et al. (2021). Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep. 34, 108634. 10.1016/j.celrep.2020.108634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N. (1979). A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern 9, 62–66. 10.1109/TSMC.1979.4310076. [DOI] [Google Scholar]
- Özcan A, Krajeski R, Ioannidi E, Lee B, Gardner A, Makarova KS, Koonin EV, Abudayyeh OO, and Gootenberg JS (2021). Programmable RNA targeting with the single-protein CRISPR effector Cas7–11. Nature 597, 720–725. 10.1038/s41586-021-03886-5. [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, and Conklin DS (2002). Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 16, 948–958. 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson H. (2018). Repeat expansion diseases. Handb. Clin. Neurol 147, 105–123. 10.1016/B978-0-444-63233-3.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto BS, Saxena T, Oliveira R, Méndez-Gómez HR, Cleary JD, Denes LT, McConnell O, Arboleda J, Xia G, Swanson MS, et al. (2017). Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell 68, 479–490, e5. 10.1016/j.molcel.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querido E, Gallardo F, Beaudoin M, Ménard C, and Chartrand P. (2011). Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell Sci 124, 1703–1714. 10.1242/jcs.073270. [DOI] [PubMed] [Google Scholar]
- Ranum LPW, and Cooper TA (2006). RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci 29, 259–277. 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, and Alon U. (2002). Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol 323, 785–793. 10.1016/s0022-2836(02)00994-4. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer C, Wang T, Michaelos M, and Pachitariu M. (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106. 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
- Sznajder ŁJ, and Swanson MS (2019). Short tandem repeat expansions and RNA-mediated Pathogenesis in myotonic dystrophy. Int. J. Mol. Sci 20, E3365. 10.3390/ijms20133365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, Krunnfusz A, Tavakoli S, Widrick JJ, Messemer KA, et al. (2021). Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 184, 4919–4938, e22. 10.1016/j.cell.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja KL, McCurrach M, Schalling M, Housman D, and Singer RH (1995). Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol 128, 995–1002. 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XE, Tan SX, Hoon S, and Yeo GW (2022). Pre-existing adaptive immunity to the RNA-editing enzyme Cas13d in humans. Nat Med 28, 1372–1376. 10.1038/s41591-022-01848-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CA, Johnson K, and Moxley RT (1994). Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann. Neurol 35, 104–107. 10.1002/ana.410350116. [DOI] [PubMed] [Google Scholar]
- Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J, et al. (2018). A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med 24, 1216–1224. 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Treacy D, Eichinger K, Struck A, Estabrook J, Olafson H, Wang TT, Bhatt K, Westbrook T, Sedehizadeh S, et al. (2019a). Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet 28, 1312–1321. 10.1093/hmg/ddy432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhou J, Wang Q, Wang Y, and Kang C. (2021a). Rapid design and development of CRISPR-Cas13a targeting SARS-CoV-2 spike protein. Theranostics 11, 649–664. 10.7150/thno.51479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-L, Serrano C, Zhong X, Ma S, Zou Y, and Zhang C-L (2021b). Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell 184, 5465–5481, e16. 10.1016/j.cell.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu X, Zhou J, Yang C, Wang G, Tan Y, Wu Y, Zhang S, Yi K, and Kang C. (2019b). The CRISPR-cas13a gene-editing system induces collateral cleavage of RNA in glioma cells. Adv. Sci. Weinh. Baden-Wurtt. Ger 6, 1901299. 10.1002/advs.201901299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Lotfy P, Faizi K, Kitano H, Hsu PD, and Konermann S. (2021). Deep Learning of Cas13 Guide Activity from High-Throughput Gene Essentiality Screening. 10.1101/2021.09.14.460134. [DOI]
- Wessels H-H, Méndez-Mancilla A, Guo X, Legut M, Daniloski Z, and Sanjana NE (2020). Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol 38, 722–727. 10.1038/s41587-020-0456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, and Thornton CA (2012). Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488, 111–115. 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Chen PJ, Miao Z, and Liu DR (2020). Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol 38, 1431–1440. 10.1038/s41587-020-0572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Santostefano KE, Goodwin M, Liu J, Subramony SH, Swanson MS, Terada N, and Ashizawa T. (2013). Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell. Reprogramming 15, 166–177. 10.1089/cell.2012.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Zhou Y, Xiao Q, He B, Geng G, Wang Z, Cao B, Dong X, Bai W, Wang Y, et al. (2021). Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat. Methods 18, 499–506. 10.1038/s41592-021-01124-4. [DOI] [PubMed] [Google Scholar]
- Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, and Scott DA (2018). Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Mol. Cell 70, 327–339, e5. 10.1016/j.molcel.2018.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, and Darnell JE (2003). Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863–1872. 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L-Z, Wang Y, Li S-Q, Yao R-W, Luan P-F, Wu H, Carmichael GG, and Chen L-L (2019). Dynamic imaging of RNA in living cells by CRISPR-cas13 systems. Mol. Cell 76, 981–997, e7. 10.1016/j.molcel.2019.10.024. [DOI] [PubMed] [Google Scholar]
- Zhang N, and Ashizawa T. (2017). RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev 44, 17–29. 10.1016/j.gde.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Ye Y, Ye W, Perčulija V, Jiang H, Chen Y, Li Y, Chen J, Lin J, Wang S, et al. (2019). Two HEPN domains dictate CRISPR RNA maturation and target cleavage in Cas13d. Nat. Commun 10, 2544. 10.1038/s41467-019-10507-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Konermann S, Brideau NJ, Lotfy P, Wu X, Novick SJ, Strutzenberg T, Griffin PR, Hsu PD, and Lyumkis D. (2018). Structural basis for the RNA-guided ribonuclease activity of CRISPR-cas13d. Cell 175, 212–223, e17. 10.1016/j.cell.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bewick B, Xia G, Furling D, and Ashizawa T. (2020). A CRISPR-cas13a based strategy that tracks and degrades toxic RNA in myotonic dystrophy type 1. Front. Genet 11, 594576. 10.3389/fgene.2020.594576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Su J, Hu X, Zhou C, Li H, Chen Z, Xiao Q, Wang B, Wu W, Sun Y, et al. (2020). Glia-to-Neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 181, 590–603, e16. 10.1016/j.cell.2020.03.024. [DOI] [PubMed] [Google Scholar]
- Zhuang C, Zhuang C, Zhou Q, Huang X, Gui Y, Lai Y, and Yang S. (2021). Engineered CRISPR/Cas13d sensing hTERT selectively inhibits the progression of bladder cancer in vitro. Front. Mol. Biosci 8, 646412. 10.3389/fmolb.2021.646412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MAC, et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 108, 260–265. 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq raw read data (FASTQ format) and a kallisto transcript expression table are publicly available through the Gene Expression Omnibus (GEO) at accession no. GSE191329. Raw microscopy images (CZI format) are available upon request.
All code developed for this work was written in Python and is publicly available on GitHub at https://github.com/cpkelley94/geno (https://doi.org/10.5281/zenodo.6807452).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.