

SUMMARY
Mitochondrial dysfunction, a hallmark of aging, has been associated with the onset of aging phenotypes and age-related diseases. Here, we report that impaired mitochondrial function is associated with increased glutamine catabolism in senescent human mesenchymal stem cells (MSCs) and myofibroblasts derived from patients suffering from Hutchinson-Gilford progeria syndrome. Increased glutaminase (GLS1) activity accompanied by loss of urea transporter SLC14A1 induces urea accumulation, mitochondrial dysfunction, and DNA damage. Conversely, blocking GLS1 activity restores mitochondrial function and leads to amelioration of aging hallmarks. Interestingly, GLS1 expression is regulated through the JNK pathway, as demonstrated by chemical and genetic inhibition. In agreement with our in vitro findings, tissues isolated from aged or progeria mice display increased urea accumulation and GLS1 activity, concomitant with declined mitochondrial function. Inhibition of glutaminolysis in progeria mice improves mitochondrial respiratory chain activity, suggesting that targeting glutaminolysis may be a promising strategy for restoring age-associated loss of mitochondrial function.
Graphical Abstract
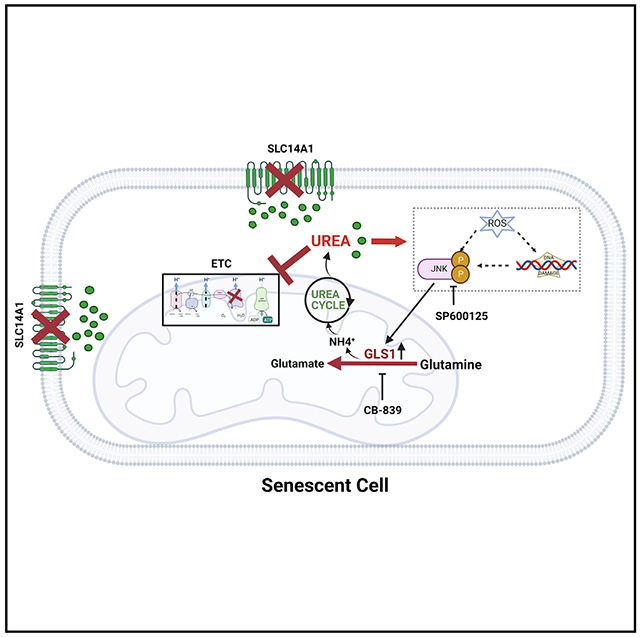
In brief
Choudhury et al. report that senescent cells exhibit enhanced glutaminolysis and loss of urea transporter, increasing urea production and accumulation. Increased intracellular urea severely impairs mitochondrial function and exacerbates the aging phenotype. Inhibiting glutaminolysis by blocking GLS1 significantly improves mitochondrial function in senescent cells and progeria mice.
INTRODUCTION
Aging is considered a principal cause of major human pathologies including atherosclerosis, type 2 diabetes,1 skin sagging and loss of elasticity,1 sarcopenia,2 and neurodegenerative diseases.3 Mitochondrial dysfunction, one of the major hallmarks of aging, has been implicated in loss of stem cell function and development of inflammatory phenotype.4–7 Various senescence models have shown deterioration of mitochondrial oxidative phosphorylation (OXPHOS), as dysfunctional mitochondria with severe defects in respiratory chain complexes accumulate in senescent cells.8–12 Impaired mitochondrial function is a driving force for dysregulated glucose metabolism and insulin resistance that may result in age-related diseases, like type 2 diabetes.13,14 Cells manifesting defective mitochondria resulting in loss of ATP production may undergo metabolic rewiring to adapt to their bioenergetic needs.15–17
Mesenchymal stem cells (MSCs) are extensively used in regenerative medicine and cell therapy. Mitochondrial metabolism plays a crucial role in the regenerative capacity and differentiation potential of MSCs into different lineages such as osteoblasts, adipocytes, and chondrocytes, which require the regulation of mitochondrial dynamics and function.18 Furthermore, several studies reported mitochondrial transfer between MSCs and other cells as an important mechanism for restoring cellular bioenergetics and repairing damaged tissues.19 These studies highlight the importance of understanding MSC metabolism and alterations or impairments associated with aging.
Recently, we discovered that major hallmarks associated with cellular senescence in mesenchymal stem cells, progeroid fibroblasts, and skeletal muscle cells were reversed by ectopic expression of the pluripotency factor, NANOG.20–22 These include restoration of myogenic differentiation potential, synthesis of extracellular matrix and re-formation of the actin cytoskeleton. Here, we report that senescent MSCs with impaired mitochondria and glycolysis activate signaling pathways that trigger glutamine catabolism. While this metabolic rewiring fuels the TCA cycle to meet bioenergetic demands, it comes with unwanted consequences related to increased reactive oxygen species (ROS) and DNA damage. Blocking the JNK signaling pathway mediating this rewiring improved mitochondrial function and cellular respiration, suggesting potentially druggable means for restoring the function of senescent MSC.
RESULTS
Cellular senescence is accompanied by increased mitochondrial impairment and decreased glucose consumption
We hypothesized that the state of senescence of MSCs is accompanied by metabolic reprogramming to provide senescent MSCs with the energy required for survival. To address this hypothesis, we employed several models of cellular senescence including replicative senescent MSCs and fibroblasts from patients suffering from Hutchison-Gilford Progeria syndrome (HGPS), an established model of premature aging.23,24 In previous reports from our laboratory, expression of the pluripotency factor NANOG reversed the hallmarks of cellular senescence such as expression of SA-β-gal, proliferation, and DNA damage as evidenced by the levels of γ-H2AX,21 as well as restoring the ability of senescent MSCs for extracellular matrix synthesis, myogenic differentiation capacity, and contractile function.20,25 These results suggested that NANOG can be employed as a means of reversing the hallmarks of cellular aging and to study the metabolic reprogramming in “rejuvenated” stem cells.
Since declined mitochondrial dysfunction is known to accompany cellular aging, we first examined the protein expression of cytochrome c oxidase (COX IV), the major regulation site for mitochondrial respiration and cytochrome c, which catalyzes the final step in mitochondrial electron transport chain. Specifically, senescent MSCs (S) showed significantly reduced mitochondrial membrane potential (Figures 1A and 1B), cytochrome c expression (Figures 1C and 1D), and COX IV expression (Figures 1E–1G) compared with young proliferating MSCs (Y) or senescent MSCs expressing NANOG (SN). It has been reported that dysfunctional electron transport chain (ETC) leads to ROS accumulation.26 Indeed, we observed high ROS accumulation in S cells, which was suppressed by NANOG (Figures S1A and S1B).
Figure 1. Impaired mitochondrial function in senescent mesenchymal stem cells restored by NANOG.
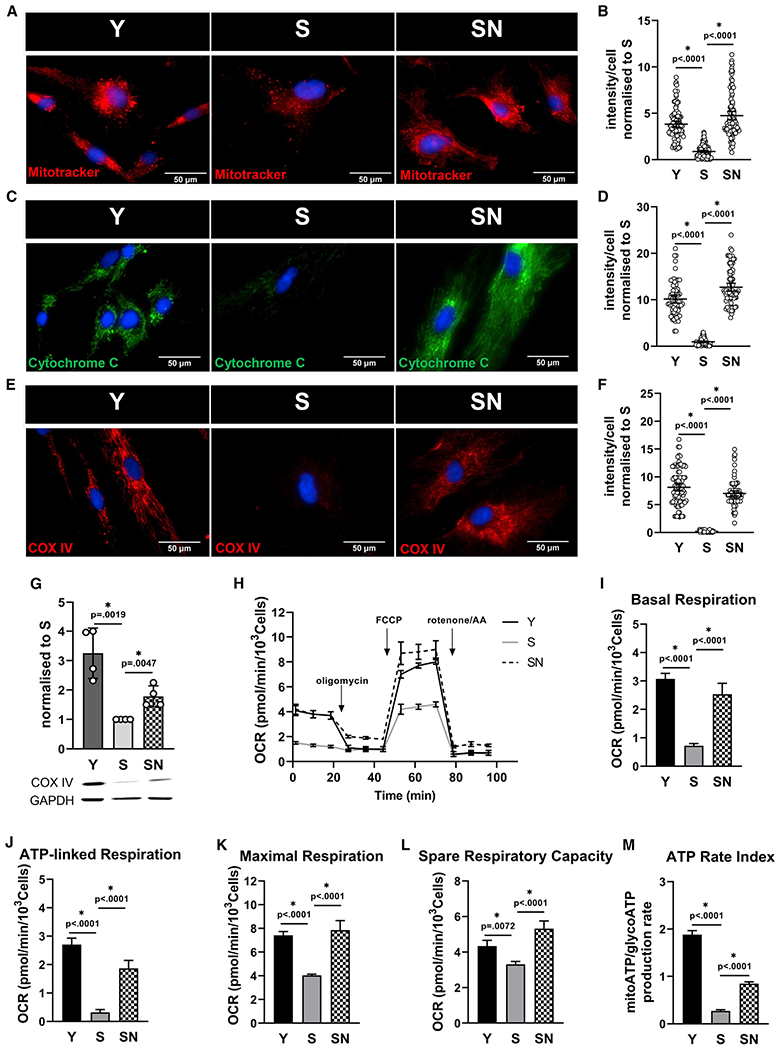
(A) Representative images of Mitotracker Red live stain of Y, S, and SN cells; scale bar represents 50 μm.
(B) Quantification of Mitotracker Red intensity per cell; data shown as mean ± 95% confidence interval (CI) for >100 cells.
(C) Immunostaining for cytochrome c; scale bar represents 50 μm.
(D) Quantification of cytochrome c intensity per cell; data shown as mean ± 95% CI for >100 cells.
(E) Immunostaining for mitochondrial complex IV (COX IV); scale bar represents 50 μm.
(F) Quantification of COX IV intensity per cell; data shown as mean ± 95% CI for >100 cells.
(G) Western blot for COX IV and quantification (n = 3 independent experiments). GAPDH served as loading control.
(H) Measurement of oxygen consumption rate (OCR) using Seahorse extracellular flux analyzer.
(I–L) Basal respiration (I), ATP-linked respiration (J), maximal respiration (K), and spare respiratory capacity (L) calculated from the OCR data in (H).
(M) ATP rate assay using Seahorse extracellular flux analyzer, demonstrating mitochondrial ATP production rate significantly reduced in S cells.
To further analyze the mitochondrial respiratory activity, we measured the oxygen consumption rate using the Seahorse analyzer. We found striking deficiency in basal and ATP-linked respiration with senescence (Figures 1H–1J). Similar results were observed for maximal and spare respiratory capacity (Figures 1K and 1L), which indicate the capacity of cells to respond to energetic demands and their mitochondrial fitness. In agreement, the ATP rate assay using Seahorse analyzer confirmed a significant decrease of mitochondrial ATP production rate in senescent stem cells (Figure 1M). Loss of mitochondrial function has been associated with increased phosphorylation of pyruvate dehydrogenase (PDH), the gatekeeper enzyme linking glycolysis to mitochondrial tricarboxylic acid (TCA) cycle, leading to its inactivation. Indeed, we observed significantly higher phosphorylation of PDH E1a subunit in S cells, indicating impaired connectivity of glycolysis to oxidative phosphorylation (Figures S1C and S1D).
Interestingly, mitochondrial dysfunction in senescent stem cells could be restored by ectopic expression of NANOG (SN), a pluripotency-associated transcription factor that has been shown to restore the senescent phenotype, differentiation capacity, and the generation of the extracellular matrix of aged MSCs.20,21,25 NANOG restored the expression of all three critical mitochondrial components including COX IV, cytochrome c, and mitochondrial membrane potential (Figures 1A–1G) and led to reduced phosphorylation of PDH E1a subunit (Figures S1C and S1D). Further, the impaired mitochondrial respiratory capacity of S cells was fully restored by NANOG (Figures 1H–1L), and mitochondrial ATP production was significantly improved (Figure 1M).
Given that senescent cells demonstrated impaired mitochondrial respiration, we examined whether they exhibited increased glucose uptake and glycolysis to meet their cellular bioenergetic demand. Surprisingly, glucose uptake was also lower in S cells as evidenced by uptake of the fluorescent glucose analog 2-NBDG (Figures S1E and S1F). Decreased glucose uptake was accompanied by significantly reduced expression of glucose transporters GLUT3 and GLUT4 (Figures S1G and S1H) and diminished insulin sensitivity (Figure S1I). Glycostress test using Seahorse analyzer further showed that S cells exhibited decreased glycolysis and glycolytic capacity (Figures S1J–S1L). Interestingly, S cells demonstrated similar ATP production rate when compared with Y and SN cells (Figure S1M). On the other hand, senescent cells expressing NANOG showed not only improved mitochondrial function (Figures 1A–1L) but also increased glucose uptake and glycolytic capacity similar to Y cells (Figures S1E–S1L), suggesting that NANOG could induce metabolic reprogramming in senescent MSCs. Collectively, these data show that senescent MSC exhibited impaired mitochondrial function and reduced glucose uptake and glycolysis as well as insulin resistance, suggesting that they might be using another carbon source to meet their metabolic demands.
Increased glutamine consumption leads to increased ATP, lactate, and pyruvate in senescent MSCs
Since aged stem cells showed decreased glucose uptake and the mitochondrial ETC was also severely compromised, we hypothesized that S cells might be using a different carbon source as a fuel to meet their bioenergetic demands. Despite having a dysfunctional ETC, senescent stem cells exhibited higher production of TCA cycle intermediates α-ketoglutarate (α-KG), glutamate, oxaloacetate (OAA), and citrate (Figure 2A), prompting us to hypothesize that they might be utilizing amino acids as an energy source. To test this hypothesis, we fed the cells with cocktails of amino acids, with different entry points into the TCA cycle converting into pyruvate, α-KG, succinate, or OAA, respectively. Surprisingly, the cocktail of amino acids converting into α-KG generated the highest increase in ATP compared with glucose in S cells, but not in Y or SN cells (Figure 2B). When cells were fed with individual amino acids converting into α-KG, only glutamine induced significant increase in ATP concentration in S cells (Figure 2C). Since glutamine is converted to glutamate by glutaminase (GLS), we examined the expression and activity levels of GLS1. Interestingly, GLS1 was highly expressed in senescent stem cells at both the mRNA (Figure 2D) and protein level (Figures 2E and 2F). Also, GLS enzyme activity was higher in S cells even when in glucose-only medium and further increased upon addition of glutamine, with the highest increase experienced by S cells (Figure 2G).
Figure 2. Increased glutamine consumption by senescent MSCs.
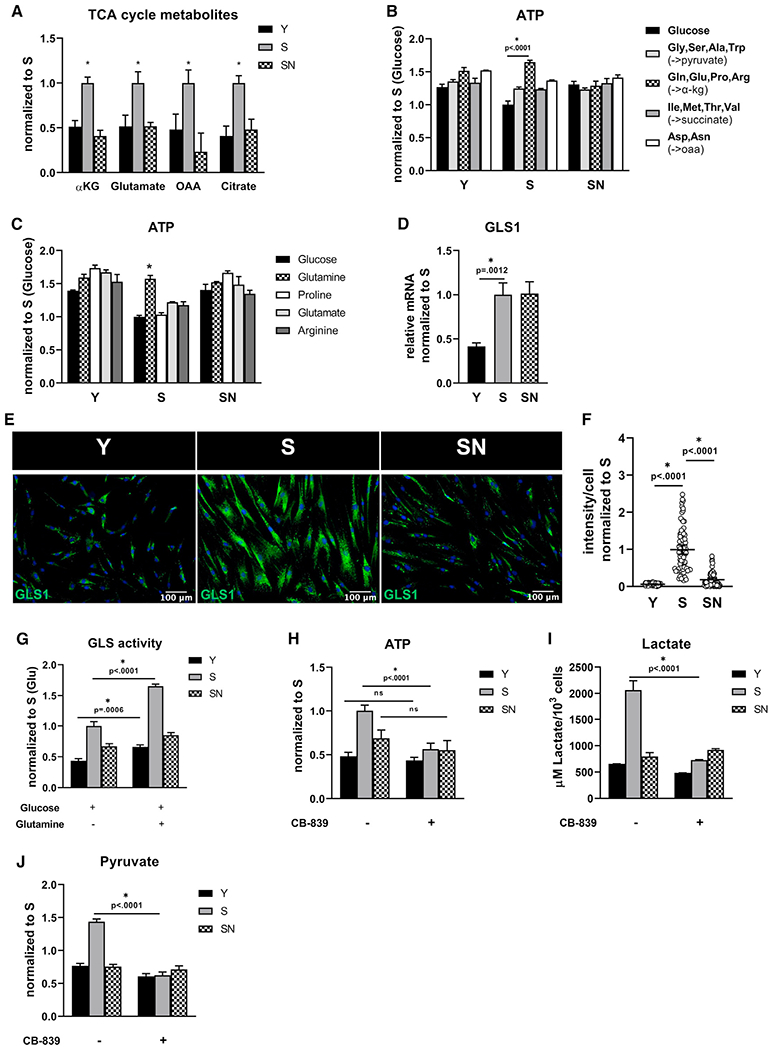
(A)Y, S, and SN cells were starved for 30 min and then stimulated with medium containing 25 mM glucose and 4 mM glutamine, followed by measurement of TCA cycle intermediates α-ketoglutarate, glutamate, oxaloacetate, and citrate and after 24hr.
(B) Intracellular ATP concentration measured after cells were fed with glucose (5 mM) alone or glucose with different cocktails of amino acids (5 mM) with different entry points into the TCA cycle.
(C) Intracellular ATP measured after cells were fed with glucose (5 mM) alone or glucose with the indicated amino acid (5 mM).
(D) Quantification of gene expression of GLS1 via quantitative RT-PCR normalized to S and internally normalized to RPL32 cycle number.
(E) Immunostaining for GLS1; scale bar represents 100 μm.
(F) Quantification of GLS1 intensity per cell; data shown as mean ± 95% CI for >100 cells.
(G) Glutaminase activity in medium containing only glucose (25 mM) or both glucose (25 mM) and glutamine (4 mM).
(H) Intracellular ATP measured in the presence or absence of GLS1 inhibitor, CB-839 (4 μM) at 24 h after treatment.
(I) Extracellular lactate concentration measured in the presence or absence of CB-839.
(J) Intracellular pyruvate concentration measured in the presence or absence of CB-839. Data in bar graphs represented as means ± SD.
To further test the dependence of aged stem cells on glutamine, S cells were treated with a specific GLS1 inhibitor, CB-839, which significantly decreased ATP concentration only in S but not in Y or SN cells (Figure 2H). Furthermore, the ATP concentration was higher in S cells even when cells were starved (no glutamine or glucose in media) and was diminished by treatment with CB-839, suggesting that endogenous glutamine reserves might be used by senescent MSCs in the absence of exogenous glutamine (Figure S2A). Surprisingly, aged stem cells exhibited higher extracellular acidification rate (ECAR), a measure of lactate efflux (Figure S2B), which decreased after treatment with CB-839 (Figure S2C) with no change in mitochondrial respiration (Figure S2D). In agreement, the levels of lactate and pyruvate (Figures 2I and 2J) were higher in S cells and decreased significantly with CB-839.
Notably, NANOG reversed the TCA metabolites concentration, GLS1 expression and activity, the levels of ATP, ECAR, lactate, and pyruvate (Figures 2A, 2E–2J, S2A, and S2B), and turned S cells independent of glutamine (CB-839 had no effect, Figures 2H–2J and S2A) similar to Y cells. Collectively, these data show that S cells rewired their metabolism to use glutamine as the primary energy source, while NANOG-driven rejuvenation decreased glutamine dependence similar to the state of young proliferating stem cells.
The decline in mitochondrial function is associated with accumulation of urea
Next, we examined whether increased glutamine usage by senescent MSCs resulted in higher urea production, a byproduct of amino acid catabolism. Indeed, intracellular ammonium and urea content were higher in aged stem cells (Figure 3A). In agreement, arginase 2 (ARG2), the major enzyme in the urea cycle, was also highly upregulated in S cells (Figures 3B and 3C). Other amino acids also led to increased urea production but to a lesser extent than glutamine (Figure S3A). Increasing glutamine concentration increased urea production (Figure S3B), while inhibiting glutamine breakdown by CB-839 led to significantly decreased urea and ammonia levels in S cells (Figures S3C and S3D). In addition, the human solute carrier family 14 member 1 (SLC14A1), which encodes the type-B urea transporter (UT-B) was diminished in S cells as revealed through RNA-seq analysis and further confirmed by RT-PCR (Figure 3D), suggesting that urea accumulation inside cells may possibly compromise mitochondrial function and lead to DNA damage, two of the major hallmarks of aging.
Figure 3. Urea accumulation causes mitochondrial dysfunction.
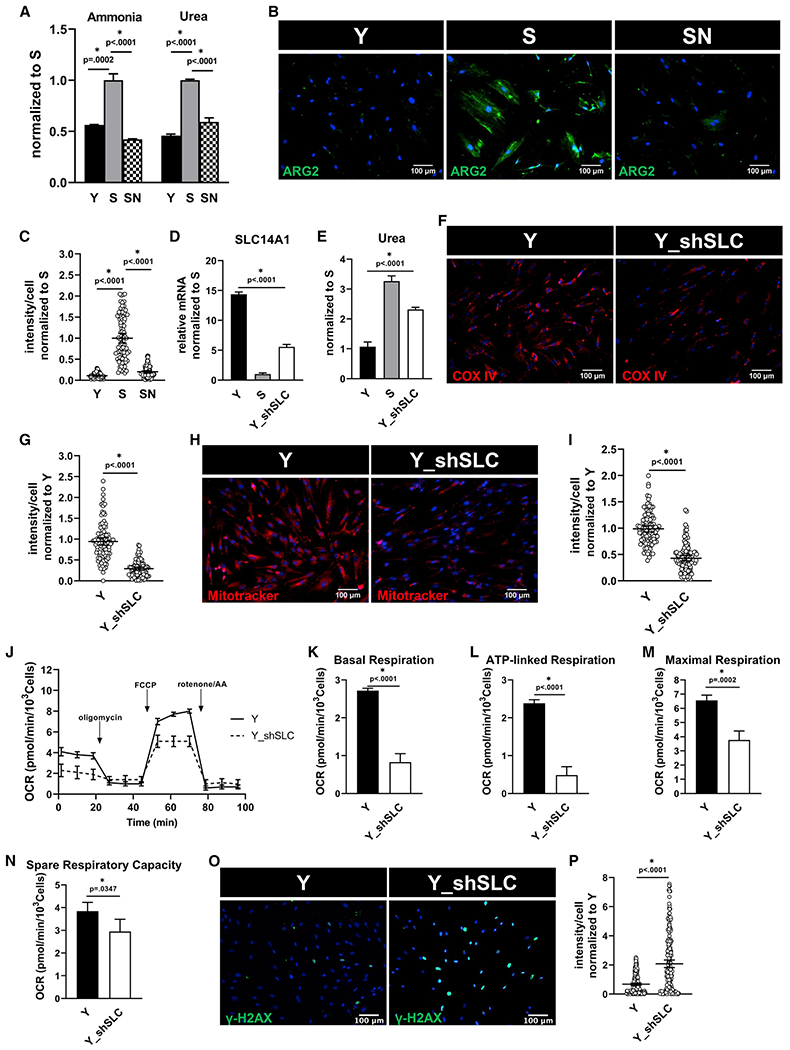
(A) Intracellular ammonia and urea concentration measured in Y, S, and SN cells. Data normalized to S and represented as mean ± SD.
(B) Immunostaining for urea cycle enzyme, arginase-2 (ARG-2); scale bar represents 100 μm.
(C) Quantification of ARG-2 intensity per cell normalized to S; data shown as mean ± 95% CI for >100 cells.
(D) Quantification of gene expression of SLC14A1 via quantitative RT-PCR normalized to S and internally normalized to RPL32 cycle number. Y_shSLC denotes Y cells with SLC14A1 knockdown.
(E) Intracellular urea concentration measured in Y, S, and Y_shSLC cells, normalized to Y cells.
(F) Immunostaining for COX IV; scale bar represents 100 μm.
(G) Quantification of COX IV intensity per cell normalized to Y; data shown as mean ± 95% CI for >100 cells.
(H) Mitotracker Red live stain images; scale bar represents 100 μm.
(I) Quantification of Mitotracker Red intensity per cell; data shown as mean ± 95% CI for >100 cells.
(J) Measurement of oxygen consumption (OCR) using Seahorse extracellular flux analyzer.
(K–N) Basal respiration (K), ATP-linked respiration (L), maximal respiration (M), and spare respiratory capacity (N) rates calculated from the OCR data in (L).
(O) Immunostaining for phosphorylated form of histone H2AX (γ-H2AX), a measure of DNA damage; scale bar represents 100 μm.
(P) Quantification of γ-H2AX intensity per cell, normalized to Y; data shown as mean ± 95% CI for >100 cells.
To address this hypothesis, we knocked down SLC14A1 in Y cells (Y_shSLC) to mimic the loss in SLC14A1 expression in S cells (Figure 3D). Indeed, SLC14A1 knockdown led to increased concentration of intracellular urea (Figure 3E) compared with Y, albeit to a lesser extent than S cells, which had even lower SLC14A1 levels (Figure 3D). SLC14A1 loss also led to loss of COX IV (Figures 3F and 3G), loss of mitochondrial membrane potential (Figures 3H and 3I), and accumulation of ROS (Figures S3E and S3F), indicating dysfunctional mitochondria. In agreement, loss of the urea transporter compromised mitochondrial respiration as shown by Seahorse analysis of cellular energetics (Figures 3J–3N) and led to significantly increased phosphorylated histone variant H2AX, γ-H2AX in Y_shSLC (Figures 3O and 3P), an indicator of DNA damage and a key hallmark of aging.27 Similar findings were observed upon treatment of cells with 1 mM urea (Figures S3G–S3N). Taken together, these findings show that increased amino acid catabolism in combination with downregulation of the urea transporter in aged cells led to accumulation of intracellular urea, which in turn caused mitochondrial dysfunction, oxidative stress, and DNA damage. Furthermore, we employed a second model of cellular senescence, namely fibroblasts from a patient suffering from HGPS, an established disease model of premature aging.28,29 HGPS is an autosomal dominant genetic disease that affects the nuclear envelope and is characterized with symptoms of accelerated aging,29,30 including cardiovascular disease.31 Similar to the culture senescence model, we applied a Tet-On system to express NANOG in HGPS cells,20 which enables nuclear expression of NANOG in the presence of doxycycline (Dox) (Figure S4A). Notably, expression of NANOG led to amelioration of cellular aging hallmarks, γ-H2AX indicating DNA damage and SA-β-gal in HGPS myofibroblasts (Figures S4B–S4E), in agreement with a previous report from our laboratory.21 As healthy control, we used fibroblasts from the patient’s father, who did not carry the mutation. Interestingly, HGPS fibroblasts showed higher glutaminase activity (Figure S4F) and urea accumulation (Figure S4G). This was accompanied by decreased COX IV protein expression and higher ROS accumulation (Figures S4H–S4J). Although HGPS cells did not show significant changes in the basal and ATP-linked respiration (Figures S4K–S4M), they did show significant decrease in maximal and spare respiratory capacity as evidenced by Seahorse analysis (Figures S4N and S4O). Notably, expression of NANOG rewired all these metabolic indicators back to the levels of healthy cells (Figures S4A–S4O), in agreement with restoring the aging hallmarks.
Urea accumulation was associated with mitochondrial malfunction in vivo
Progeroid syndromes are due to genetic disorders affecting the nuclear envelop and DNA repair mechanisms, and they mimic physiological aging.32 The lamin A knockin (LAKI) model of progeria results in accumulation of the truncated form of lamin A (progerin),33 which normally accumulates over time with aging and induces nuclear defects in old individuals.34 Indeed, LAKI mice displayed aging phenotypes including the accumulation of senescence-associated enzyme SA-β-Gal in their skin (Figures S5A and S5B), compared with wild-type (WT) mice.
Similar to our in vitro findings, skin tissues of LAKI mice exhibited significantly upregulated GLS1 expression (Figures 4A and 4B), elevated glutaminase activity (Figure 4D), and increased accumulation of urea (Figure 4E). They also exhibited reduced COX IV expression (Figures 4A and 4C) and elevated ROS (Figure 4F) compared with WT mice. Given the importance of mitochondrial function for the heart, we also compared the heart tissues of LAKI vs. WT mice. In LAKI hearts, there was a trend of increased GLS1 expression (Figures 4G and 4H), but it was not statistically significant, and GLS1 activity remained unaffected (Figure 4J); however, similar to skin, LAKI hearts exhibited high accumulation of urea (Figure 4K), as well as significantly reduced expression of COX IV (Figures 4G and 4I) and elevated levels of ROS (Figure 4L), compared with WT hearts.
Figure 4. Increased GLS1 expression and activity were associated with urea accumulation and mitochondria dysfunction in LAKI mouse tissues.
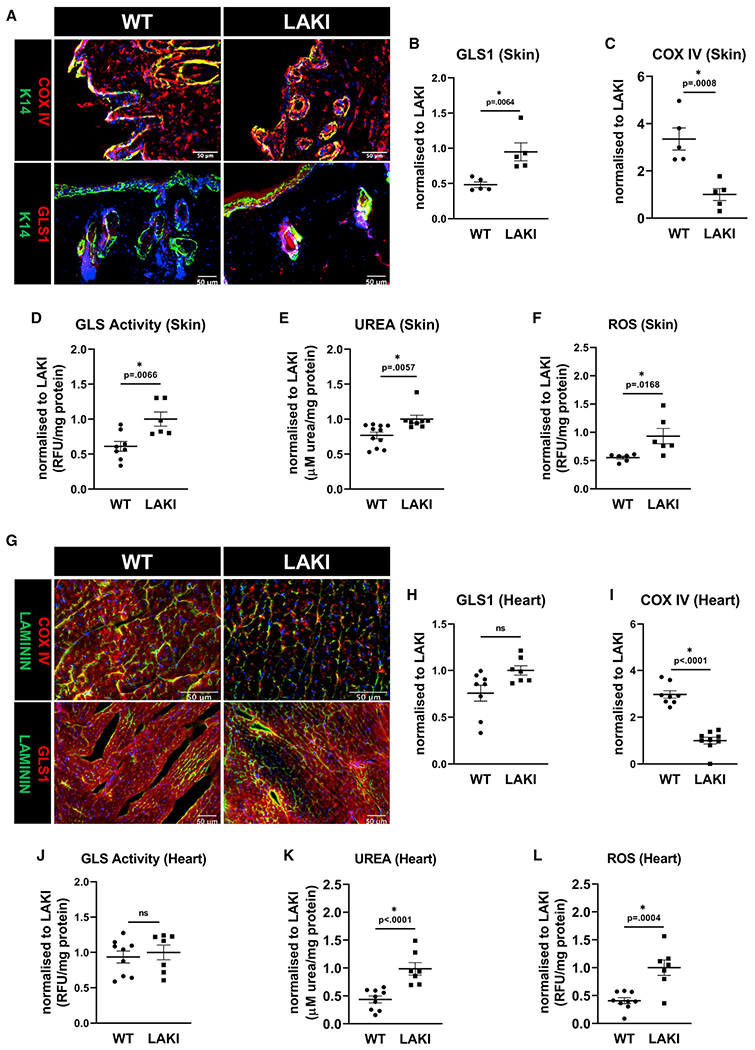
Representative immunostaining images (A) and quantification of GLS1 (B) and COX IV staining (C) in skin. GLS activity (D), urea accumulation (E), and ROS level (F) were measured in the skin of WT and heterozygous LAKI progeria mouse (LMNAG609G/+) at the age of 10 months.
(G–I) Representative immunostaining images (G) and their quantification for GLS1 (H) and COX IV (I) in the heart tissue. Measurements of GLS activity (J), urea accumulation (K), and ROS levels (L). Data shown as mean ± SE for each cohort of animals (each bubble represents one mouse, n = 5–8). * designates statistical significance compared with LAKI (p < 0.05).
In addition, we investigated naturally aged mice. We defined mice of age between 19 and 24 months old as old, and 4-month-old mice as young. Similar to LAKI mice, old mice exhibited increased GLS1 expression and activity and high levels of urea, as well as significantly decreased COX IV expression and elevated ROS, compared with young, in both skin (Figures S5C–S5H) and heart (Figures S5I–S5N) tissues. Overall, these results confirm the in vitro results showing increased level of urea, ROS accumulation, and mitochondria dysfunction in aged mouse tissues.
Increased JNK activity enhances glutamine catabolism and mitochondrial impairment in senescent cells
Next, we sought to elucidate the signaling pathway affecting GLS1 expression and glutamine catabolism. To this end, we investigated the potential connection between MAPK pathways (ERK, P38, and JNK), which have been shown to be associated with age-associated mitochondrial dysfunction.35 First, we examined the phosphorylation status of ERK, P38, and JNK and observed significant upregulation of p-JNK in S cells (Figures S6A–S6D). Interestingly, pharmacological inhibition of JNK by SP600125 led to decreased GLS1 expression both at the mRNA (Figure 5A) and protein levels (Figures 5B and 5C), and this was accompanied by decreased GLS activity (Figure 5D) and intracellular urea concentration (Figure 5E) to the levels of Y cells. Inhibition of JNK was significantly more effective in decreasing both GLS activity and urea concentration than inhibition of P38, while ERK inhibition had no effect (Figures S6E and S6F). Surprisingly, JNK inhibition restored COX IV expression (Figure 5F), which was accompanied by increased mitochondrial respiratory capacity in S cells in a dose-dependent manner (Figures S6G–S6K).
Figure 5. JNK regulates GLS and suppresses mitochondrial function in senescent cells.
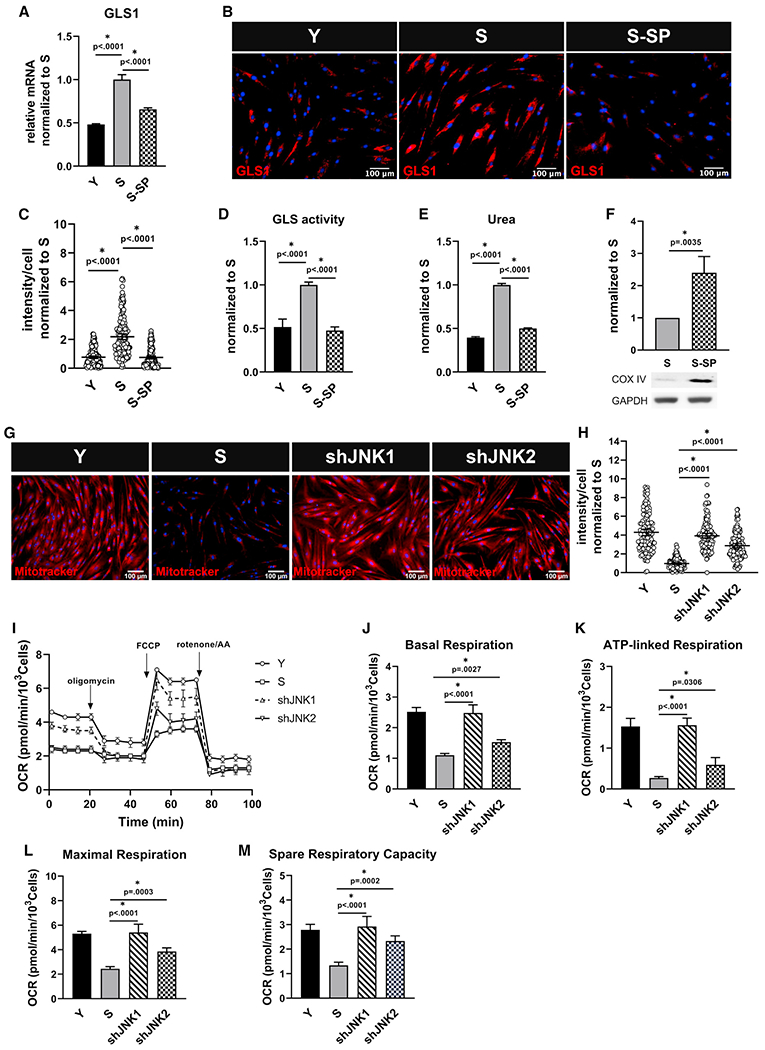
Senescent MSCs were treated with SP600125 (5 μM), and (A) GLS1 expression was measured via quantitative RT-PCR normalized to S and internally normalized to RPL32 cycle number; data shown as mean ± SD.
(B) Immunostaining for GLS1; scale bar represents 100 μm.
(C) Quantification of GLS1 intensity per cell, normalized to S; data shown as mean ± 95% CI for >200 cells.
(D) Glutaminase activity measurement in Y, S, and S cells treated with SP600125, normalized to S; data shown as mean ± SD.
(E) Intracellular urea concentration, normalized to S; data shown as mean ± SD.
(F) Western blot for COX IV and quantification, normalized to S (n = 3 independent experiments). GAPDH served as loading control.
(G) Live stain images of Mitotracker Red showing upregulation of mitochondrial membrane potential in senescent cells after knocking down JNK1 or JNK2; scale bar represents 100 μm.
(H) Quantification of Mitotracker Red intensity per cell; data shown as mean ± 95% CI for >100 cells.
(I) Measurement of oxygen consumption (OCR) using Seahorse extracellular flux analyzer in Y, S, shJNK1, and shJNK2 cells.
(J–M) Basal respiration (J), ATP-linked respiration (K), maximal respiration (L), and spare respiratory capacity (M) rates calculated from the OCR data in (I).
To identify which of the two JNK isoforms, JNK1 or JNK2, mediated these effects, we knocked down JNK1 or JNK2 individually in S cells (Figure S6L). Indeed, both JNK1 and JNK2 knockdown led to decreased GLS1 activity (Figure S6M) and suppressed urea accumulation (Figure S6N). Interestingly, although knockdown of either JNK1 or JNK2 increased mitochondrial membrane potential (Figures 5G and 5H), JNK1 knockdown was more effective in restoring mitochondrial respiration, as evidenced by complete restoration of basal, ATP-linked, and maximal respiration as well as spare respiratory capacity of S cells (Figures 5I–5M). Taken together, these results suggest that in senescent MSCs activated JNK upregulated GLS1 transcription, leading to increased glutamine catabolism, urea accumulation, and impaired mitochondrial function.
GLS1 inhibition restores mitochondrial function and decreases DNA damage
Our results implicate urea accumulation due to glutamine catabolism in impairing mitochondrial function in senescent stem cells. Therefore, we examined whether blocking GLS1 activity could restore mitochondrial function and reverse the hallmarks of senescence. Indeed, treatment of senescent MSCs with either BPTES or CB-839 for 10 days suppressed urea production (Figure 6A), decreased ROS accumulation significantly (Figure S7A), and restored mitochondrial membrane potential (Figures 6B and 6C), without loss of cell viability (Figures S7B and S7C). Interestingly, senescent MSCs exhibited decreased intracellular pH that increased upon GLS1 inhibition (Figures S7D and S7E), in agreement with increased viability. In agreement, GLS1 inhibition significantly increased basal, ATP-linked, and maximal respiration and fully restored the spare respiratory capacity of senescent cells (Figures 6D–6H). Interestingly, the extent of increase in mitochondrial respiration depended on the CB-839 concentration (Figures S7F–S7H). CB-839 treatment did not affect COX IV expression (Figure S7I) but increased the insulin sensitivity of senescent stem cells (Figure S7J). Notably, BPTES or CB-839 treatment significantly decreased γ-H2AX foci in the nuclei of senescent cells, indicating reduced DNA damage (Figures 6I and 6J) and reduced the mRNA expression of key senescence-associated inflammatory genes, particularly IL1α and IL1β (Figures 6K–6N).
Figure 6. Inhibition of GLS1 improves mitochondrial function in senescent mesenchymal stem cells.
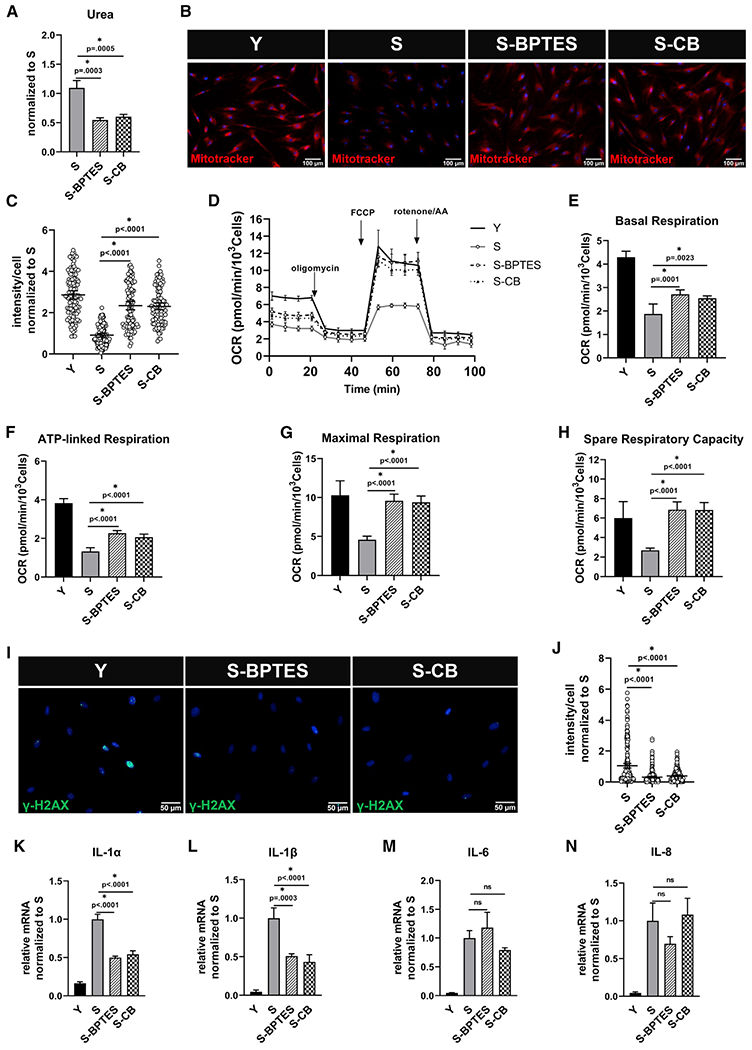
Senescent MSCs were treated with GLS1 inhibitor, BPTES (10 μM), or CB-839 (4 μM) or for 10 days. (A) Intracellular urea concentration measurement, normalized to S; data shown as mean ± SD.
(B) Representative images of Mitotracker Red live stain of Y, S, S-BPTES, and S-CB cells; scale bar represents 100 μm.
(C) Quantification of Mitotracker Red intensity per cell, normalized to S; data shown as mean ± 95% CI for >200 cells.
(D) Measurement of oxygen consumption rate (OCR) using Seahorse extracellular flux analyzer.
(E–H) Basal respiration (E), ATP-linked respiration (F), maximal respiration (G), and spare respiratory capacity (H) calculated from the OCR data in (D).
(I) Immunostaining for the phosphorylated form of histone H2AX (γ-H2AX); scale bar represents 50 μm.
(J) Quantification of γ-H2AX intensity per cell, normalized to S; data shown as mean ± 95% CI for >100 cells.
(K–N) Quantification of gene expression of inflammatory cytokines via quantitative RT-PCR normaliszed to S and internally normalized to RPL32 cycle number.
Furthermore, knocking down GLS1 in S cells (Figures S7K–S7M) led to marked decrease in intracellular urea concentration (Figure S7N), and this was accompanied by increased mitochondrial membrane potential (Figures S7O and S7P) and improved mitochondrial ATP-linked respiration and spare respiratory capacity (Figures S7Q–S7S). Interestingly, although blocking glutaminolysis did not reverse expression of SA-β-Gal (Figure S7T) or cell cycle inhibitors (p53, p21, p16) in senescent MSCs (Figures S7U–S7W), it did improve mitochondrial function and decreased DNA damage and ROS accumulation, suggesting overall improved cellular function. Collectively, these results implicate GLS1 as a potential target for restoring mitochondrial function in aged stem cells.
GLS1 inhibition improved mitochondrial function in LAKI mice
We also examined the effect of GLS1 inhibition on a mice model of accelerated aging. To this end, we injected CB-839 intraperitoneally in LAKI mice, three times per week for 1 month. At the end of the experiment, mitochondria were isolated from the skin and heart tissues, and mitochondrial respiratory chain activity was evaluated (Figures 7A and 7F). Although no significant change was observed in complex II activity (Figures 7B and 7G), complex IV activity was significantly improved with CB-839 treatment in both skin (Figure 7C) and heart tissue (Figure 7H). Improved mitochondrial function was concomitant with decreased urea concentration (Figures 7D and 7I) and decreased ROS level (Figures 7E and 7J). Overall, our data confirm the in vitro results showing that GLS1 inhibition restored impaired mitochondrial function and suggest that blocking glutamine catabolism by the anti-cancer drug CB-839 might have potential benefits in reversing age-associated mitochondrial dysfunction.
Figure 7. GLS1 inhibition improves mitochondrial function in LAKI mouse tissues.
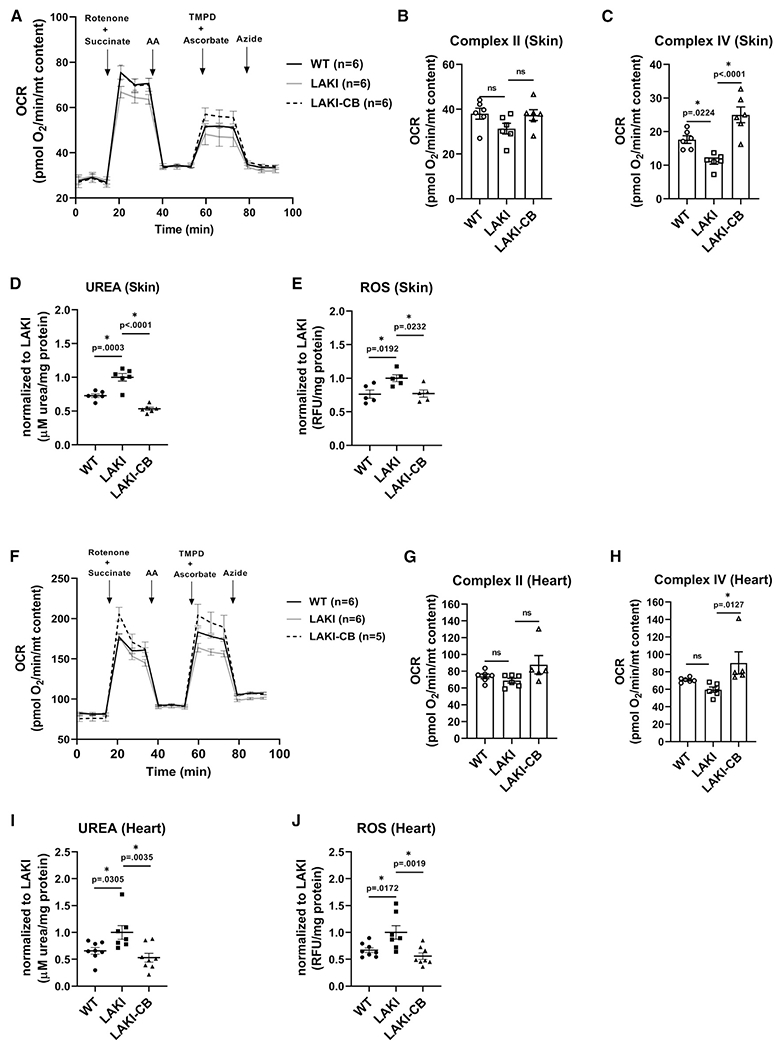
(A) Measurement of oxygen consumption rate of isolated mitochondria from skin of WT, heterozygous LAKI progeria mouse (LMNAG609G/+) and LAKI mice treated with CB-839 (LAKI-CB); data normalized to mitochondrial content.
(B and C) Complex II and complex IV activities calculated from the OCR data in (A).
(D and E) Measurement of urea concentration and ROS level in skin tissue; data normalized to LAKI.
(F) Measurement of oxygen consumption rate of isolated mitochondria from heart tissues, normalized to mitochondrial content.
(G and H) Complex II and complex IV activities calculated from the OCR data in (F).
(I and J) Measurement of urea concentration and ROS level in heart tissue; data normalized to LAKI. Data shown as mean ± SE for each cohort of animals (each bubble represents one mouse, n = 5–8). * designates statistical significance compared with LAKI (p < 0.05).
DISCUSSION
Although significant progress has been made into understanding the mechanisms underlying age-associated cellular dysfunction, many aspects related to impaired metabolism remain unknown.36,37 Understanding metabolic impairments is critical for understanding aging and developing strategies to prevent or delay age-related diseases. In this paper, we focused on delineating the metabolic rewiring associated with mitochondrial dysfunction that we observed in senescent MSCs. We found that glycolysis and mitochondrial respiration were compromised in senescent MSCs, which rewired their metabolism to use glutamine as a carbon source. Increased glutamine catabolism led to intracellular urea accumulation, which further deteriorated mitochondrial function and induced senescence. Conversely, blocking glutamine deamination by inhibiting GLS1 partially restored mitochondrial function and decreased hallmarks of cellular senescence. Notably, similar results were also observed with progeria cells derived from patients and mice suffering from HGPS as well as physiologically aged mice, further supporting the physiological significance of metabolic rewiring in aging.
Cellular senescence and loss of stem cell activity has been associated with decline in mitochondrial function.12,38,39 Indeed, we observed severe mitochondrial defects, such as loss of COX IV, in senescent MSCs, leading to significantly diminished ATP production from ETC. Surprisingly, despite nonfunctional ETC, senescent MSCs maintained higher total ATP levels and demonstrated higher lactate production, compared with their non-senescent counterparts, suggesting use of alternative carbon sources to maintain high energy level. Indeed, senescent MSC exhibited higher glutaminase activity than proliferating cells. Inhibition of GLS1 activity led to decreased levels of TCA cycle intermediates as well as glycolytic/gluconeogenic metabolites such as pyruvate and lactate, indicating the role of glutamine in increasing lactate efflux. Increased lactic acid in blood, urine, cerebrospinal fluid, and in tissues has previously been reported in patients exhibiting defects in oxidative phosphorylation (OXPHOS).40 Based on our observations, we surmised that elevated amino acid catabolism might be leading to such a phenomenon.40 Furthermore, intracellular ATP concentration decreased significantly in aged MSCs by inhibiting GLS1, further implicating glutamine as a primary energy source in senescent cells. Increased glutamine anaplerosis has been reported in several cells carrying mitochondrial DNA (mtDNA) mutations and exhibiting severe OXPHOS defects.15,16 In idiopathic pulmonary fibrosis, glutaminolysis has been shown to promote apoptosis resistance through epigenetic regulation of XIAP and survivin, members of the inhibitor of apoptosis family of proteins.41 Indeed, a-ketoglutarate derived from glutamate is a cofactor of JMJD3 histone demethylase, which was shown to bind to XIAP and survivin promoters in a glutamine-dependent manner. Since senescent cells are known to be resistant to apoptosis,40,42–44 it is possible that metabolic rewiring from glycolysis to glutaminolysis may serve to promote resistance to apoptosis and survival. A recent study demonstrated increased dependence of oncogene-induced senescent cells on glutamine for their survival, as blocking GLS1 led to increased mortality of aged cells17 that was attributed to drastic increase in intracellular acidosis. In contrast, in our system, we observed a small but significant increase in pH upon GLS1 inhibition (Figures S7B and S7C) and no significant change in cell viability (Figures S7D and S7E). This discrepancy may be attributed to the different models of cellular senescence. Indeed, the effects of senolytic drugs were shown to be not only cell-type dependent,45–48 but also dependent on the mechanism of senescence induction with DNA-damage-induced, oncogene-induced, and replicative senescence expressing varying levels of anti-apoptotic BCL family proteins.49 It will be interesting to compare the anti-apoptotic, metabolic, or other pathways of various senescence models to delineate the mechanisms responsible for the differential responses to GLS1 inhibition.
JNK kinases were originally identified by their ability to phosphorylate c-Jun in response to stress induced by a variety of stimuli such as TNF-α44 or UV radiation.50 JNK was shown to be a negative regulator of mitochondrial metabolic function, as its activation was linked to phosphorylation of PDH, likely by pyruvate dehydrogenase kinase-2, in the aged rat brain.51,52 Interestingly, JNK isoforms may differ in their role regulating cellular metabolism. For example, JNK1 was shown to negatively regulate aerobic glycolysis by phosphorylating PDH in hepatocellular carcinoma,53 while JNK2 was shown to positively regulate aerobic glycolysis in myeloma cancer cells.54 Interestingly, we discovered that senescent MSCs had higher levels of phosphorylated JNK, and that blocking JNK kinase activity by SP600125 or shRNA decreased GLS1 expression both at the mRNA and protein levels. This was accompanied by decreased GLS activity, glutaminolysis, and urea accumulation, ultimately leading to restoration of mitochondrial respiratory activity. Although knocking down JNK1 or JNK2 decreased GLS1 activity and urea levels, JNK1 knockdown was more effective than JNK2 in restoring mitochondrial respiration. JNK1 knockdown was also more effective than the GLS inhibitor, CB-839, suggesting that JNK1 might be affecting mitochondrial function through more than one metabolic pathway. It is well known that senescent cells secrete pro-inflammatory cytokines (senescence-associated secretory phenotype, SASP) that affect their neighbors and contribute to chronic inflammation.52,55 It would be interesting to examine whether JNK activation in senescent cells is the result of SASP signaling, providing a link between inflammation and metabolic rewiring in senescence.
Glutamine turns to glutamate that is subsequently converted to α-ketoglutarate fueling the TCA cycle and, in the process, generating ammonia and urea, which is secreted out of the cell via membrane transporters. In the body, urea is eliminated through body fluids, mainly urine and also through blood and saliva.56 Several publications reported a highly active urea cycle in the brain of patients suffering from Alzheimer’s disease, indicating potential involvement in neurodegenerative diseases associated with aging.57,58 The importance of the urea transporter in cardiac physiology was first shown when its knockdown led to urea accumulation in the heart, severely compromising cardiac electrophysiology.59 Remarkably, our findings clearly revealed high urea accumulation in senescent cells and aged tissues from progeria mice including the heart and skin. In addition to increased amino acid breakdown, senescent MSCs showed decreased expression of the urea transporter further increasing intracellular urea accumulation and exacerbating the aging phenotype, as evidenced by impaired mitochondrial function, increased oxidative stress, and DNA damage. Furthermore, treatment with urea or silencing of the urea transporter in young MSC induced aging-related hallmarks such as mitochondrial dysfunction and DNA damage. Conversely, inhibiting glutamine catabolism decreased urea, partially restoring mitochondrial function and decreasing senescence hallmarks, indicating the importance of antibiotic-antimycotic (AA) catabolism in the process of cellular senescence.
In summary, senescent stem cells exhibit impaired glycolysis and mitochondrial function, including loss of ATP via the ETC. To compensate and survive, MSCs rewire their metabolism to use amino acids, such as glutamine, as their primary energy source. In addition to lactate overproduction, such metabolic reprogramming results in high levels of urea accumulation causing mitochondrial dysfunction and DNA damage. Targeting glutamine metabolism and signaling pathways regulating it as well as associated metabolites and by-products might be a promising strategy to reverse or delay cellular senescence.
Limitations of the study
While GLS1 is highly expressed in the skin and heart tissues of both aged and progeria mice, it is not clear whether this observation can be generalized to all tissues. Furthermore, while inhibition of GLS1 by CB-839 significantly improved mitochondrial function and decreased ROS levels in the progeria mouse heart and skin tissues, it remains unclear whether CB-839 can improve health and extend lifespan.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Stelios T. Andreadis (sandread@buffalo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Original microscopy (.czi) and Seahorse analyzer data (.asyr) are available upon request.
This paper does not report original code.
Any additional information to reanalyze the data reported in this paper may be directed to and will be fulfilled by lead contact Stelios T. Andreadis (sandread@buffalo.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Experimental animals
Lamin-A Knocked In (LAKI, C57BL/6, LmnaG609G/G609G)33 mice were kindly donated by Dr. Dudley Lamming at the University of Wisconsin-Madison School of Medicine Madison, Wisconsin, USA.62 Mixed gender of mice at the age of 10 months were used in the study, when the heterozygous progeria mice (LmnaG609G/+) show signs of aging. The mice were housed in a temperature-controlled animal holding room (20.5° to 23.9°C) with 12-hour light/12-hour dark cycle between 6:00 and 18:00 standard time. Food and water were provided ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University at Buffalo.
Murine tissue samples
Heart and skin tissues from aged mice (19–24 months) or young mice (4 months) of mixed genders and C57BL/6 strain were obtained from the tissue bank of the National Institute Aging. Part of each tissue was stored frozen in liquid nitrogen for protein, RNA and mitochondrial isolation, and part was prepared for OCT embedding.
Cell lines and cell culture
Human Hair Follicle derived-Mesenchymal Stem Cells (hHF-MSCs here in denoted as MSCs) from a 73-year-old donor60 were isolated as previously characterized with respect to their proliferation and differentiation capacity.23 Human dermal fibroblasts from patients suffering from Hutchinson-Gilford Progeria Syndrome (HGPS; HGADFN167) and from a healthy donor (HGADFN168, father of HGADFN167) were obtained from the Progeria Research Foundation (Peabody, MA).
METHOD DETAILS
Treatment of mice with CB-839
Heterozygous Progeria mice (LmnaG609G/+) of mixed gender, at the age of 10 months were intraperitoneally injected with CB-839 (10mg/kg) or vehicle control (5% DMSO in corn oil), three times a week for one month followed by tissue collection.
Skin and heart tissues were harvested, and part of each tissue was snap frozen in liquid nitrogen for protein, RNA or mitochondria isolation, and the remaining was immersed in embedding medium immediately (OCT, Sakura Finetek, Torrance, CA) and frozen in 2-methylbutane (Sigma-Aldrich) chilled with dry ice. Embedded tissue sections (10μm thick) were cut using the freezing microtome (CM1950, Leica) and then placed on positively charged glass slides (Stellar Scientific, Baltimore, MD), and stored at −80°C.
Cell culture
Human MSCs and human fibroblasts were cultured in high glucose Dulbecco’s modified Eagle Medium (DMEM; Invitrogen, Grand Island NY) supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals, Flowery Branch, GA) and 1% (v/v) Antibiotic-Antimycotic (AA, Gibco). On the other hand, the medium for starvation condition was Dulbecco’s modified Eagle Medium (DMEM, Gibco, Grand Island, NY) depleted of glucose and 10% (v/v) fetal bovine serum. In general, MSCs at passage 6–7 were characterized as young cells (Y), while MSCs at passage p13-16 were characterized as aged or senescent cells (S). Human fibroblast, HGPS and the parental control fibroblasts were used at passages 8–10. To generate cells expressing NANOG, we use a tetracycline-regulatable system which enables NANOG expression upon DOX treatment as previously described.21 To evaluate the effect of NANOG on senescence phenotype, cells were induced upon treatment with 1 μg/mL DOX (Sigma Aldrich, St. Louis, MO) to overexpress NANOG for 9–11 days.
Plasmids constructs and cell transduction
Gene knockdown was performed using the shLVDP vector, developed in our laboratory.63 The shRNA sequences have been listed in the key resource table. Briefly, complementary oligos containing sequences targeting JNK1, JNK2,61 SLC14A1 or GLS1 were mixed at 50μM each and annealed as previously described.63 The annealed products were cloned downstream of the H1 promoter in the shLVDP. A scramble sequence without any homology to the human genome (confirmed by BLAST) was used as control. For lentivirus production, three plasmids (lentiviral vector, psPAX2, and pMD2.G) were used to transfect 293T cells, using the standard calcium phosphate precipitation method. The virus was harvested 24h post-transfection, filtered through a 0.45μm filter (Corning, Corning NY), and pelleted by centrifugation (50,000g at 4°C for 2h). Finally, the pellet was resuspended in fresh DMEM and stored at −80°C until use.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cytochrome C | Invitrogen | Cat# MA5-11823; RRID: AB_10987059 |
| COX-IV | Abcam | Cat# ab33985; RRID: AB_879754 |
| PDHA1 (phospho S293) | Abcam | Cat# ab92696; RRID: AB_10711672 |
| KGA/GAC | Proteintech | Cat# 12855-1-AP; RRID: AB_2110381 |
| Phospho-Histone H2A.X (Ser139) | Cell Signaling | Cat# 9718; RRID: AB_2118009 |
| Tomm20 | Abcam | Cat# ab186735; RRID: AB_2889972 |
| Cytokeratin 14 | Invitrogen | Cat# PA5-32460; RRID: AB_2549929 |
| Laminin | Abcam | Cat# ab11575; RRID: AB_298179 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat# A11008; RRID: AB_143165 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A11012; RRID: AB_2534079 |
| Goat anti-mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat# A11001; RRID: AB_2534069 |
| Goat anti-mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A11005; RRID: AB_2534073 |
| SAPK/JNK | Cell Signaling | Cat# 9252; RRID: AB_2250373 |
| Phospho-SAPK/JNK (Thr183/Tyr185) | Cell Signaling | Cat# 9255; RRID: AB_2307321 |
| P38 MAPK | Cell Signaling | Cat# 8690; RRID: AB_10999090 |
| Phospho-p38 MAPK (Thr180/Tyr182) | Cell Signaling | Cat# 4511; RRID: AB_2139682 |
| P44/42 MAPK (Erk1/2) | Cell Signaling | Cat# 4695; RRID: AB_390779 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Cat# 4370; RRID: AB_2315112 |
| JNK1 | Santa Cruz Biotechnology | Cat# sc-1648; RRID: AB_675868 |
| JNK2 | Cell Signaling | Cat# 9258; RRID: AB_2141027 |
| NANOG | R&D Systems | Cat# AF1997; RRID: AB_355097 |
| GAPDH | Cell Signaling | Cat# 5174; RRID: AB_10622025 |
| Anti-rabbit IgG HRP linked | Cell Signaling | Cat# 7074; RRID: AB_2099233 |
| Anti-mouse IgG HRP linked | Cell Signaling | Cat# 7076; RRID: AB_330924 |
| Antibody dilutions and application, see Table S2 | ||
| Biological samples | ||
| Heart and skin tissues from aged mice (19–24 months) or young mice (4 months) of mixed genders (C57BL/6 strain) | National Institute on Aging | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Mitotracker Red | Invitrogen | Cat# M7512 |
| DCFDA/H2DCFDA-Cellular ROS Assay Kit | Abcam | Cat# ab113851 |
| 2-NBDG | Invitrogen | Cat# N13195 |
| SYBR green PCR mix | Applied Biosystems | Cat# 4309155 |
| Antimycin | Sigma | Cat# A8674 |
| Rotenone | Sigma | Cat# R8875 |
| Oligomycin | Sigma | Cat# O4876 |
| TMPD | Sigma | Cat# 87890 |
| Succinate | Sigma | Cat# S9512 |
| L-Ascorbic Acid | Fisher Scientific | Cat# A61100 |
| Sodium Azide | Sigma | Cat# S8032 |
| 2-DG | Sigma | Cat# D8375 |
| Glucose | Agilent technologies | Cat# 103577-100 |
| Pyruvate | Agilent technologies | Cat# 103578-100 |
| Glutamine | Agilent technologies | Cat# 103579-100 |
| Seahorse XF DMEM Medium | Agilent technologies | Cat# 103575-100 |
| Seahorse XFe96 FluxPak | Agilent technologies | Cat# 102416-100 |
| CB-839 (GLS inhibitor) | Selleckchem | Cat# S7655 |
| BPTES (GLS inhibitor) | Selleckchem | Cat# S7753 |
| Corn Oil | Selleckchem | Cat# S6701 |
| M.O.M Immunodetection kit | Vector Laboratories | Cat# BMK-2202 |
| Hoechst 33342 | Invitrogen | Cat# 62249 |
| Critical commercial assays | ||
| Luminescent ATP detection Assay Kit | Abcam | Cat# ab113849 |
| Lactate-Glo Assay | Promega | Cat# J5021 |
| Glutamate Assay Kit | Abcam | Cat# ab83389 |
| Alpha Ketoglutarate Assay Kit | Abcam | Cat# ab83431 |
| PicoProbe Glutaminase Assay Kit | Biovision | Cat# K455-100 |
| Pyruvate Assay Kit | Abcam | Cat# ab65342 |
| Ammonia Assay Kit | Abcam | Cat# ab83360 |
| Urea Assay Kit | Abcam | Cat# ab83362 |
| DCF ROS/RNS Assay Kit | Abcam | Cat# ab238535 |
| Senescence Detection Kit | Abcam | Cat# ab65351 |
| Experimental models: Cell lines | ||
| Human: Mesenchymal stem cells isolated from hair follicle | Bajpai et al.60 | N/A |
| Human: dermal fibroblasts from patients suffering from Hutchison-Gilford Progeria Syndrome with classic mutation | The Progeria Research Foundation | HGADFN167 |
| Human: dermal fibroblasts from father of HGPS patient without mutation | The Progeria Research Foundation | HGADFN168 |
| Experimental models: Organisms/strains | ||
| Mouse: LAKI (C57BL/6, LmnaG609G/+) | Provided by Dr. Dudley Lamming | N/A |
| Oligonucleotides | ||
| shRNA targeting human SLC14A1 GGGCTCTGAGTATATAACTGT | This paper | N/A |
| shRNA targeting human JNK1 GGGCCTACAGAGAGCTAGTTCTTAT | You et al.61 | N/A |
| shRNA targeting human JNK2 GCCAACTGTGAGGAATTATGTCGAA | You et al.61 | N/A |
| shRNA targeting human GLS1_1 GGAGCAATTGTTGTGACTTCA | This paper | N/A |
| shRNA targeting human GLS1_2 GCATTCCTGTGGCATGTATGA | This paper | N/A |
| Primers for qPCR, see Table S1 | N/A | N/A |
| Software and algorithms | ||
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Biorender | Biorender | https://biorender.com/ |
| Zen 3.0 (blue edition) | Carl Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| Microsoft Publisher | Microsoft | N/A |
| Seahorse Wave Desktop | Agilent technologies | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
Senescence-associated β-galactosidase assay (SA-β- gal)
SA-β-gal activity was detected using a kit according to the manufacturer’s recommendations (Abcam). Fixed stained cells or tissue slides were then visualized and photographed using a Zeiss Axio Observer Z1 inverted microscope with an ORCA-ER CCD camera (Hamamatsu, Japan). For cells in culture, the percentage of SA-β-gal positive cells was determined from multiple fields of view containing approximately 200 cells/sample. For tissue sections, we measured the fraction of the SA-β-gal positive area over the total tissue area in each tissue sample.
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA was extracted using RNeasy Plus Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. The cDNA was produced with OneStep RT-PCR kit (Qiagen). Real-time PCR was performed with SYBR Green Supermix (Bio-Rad) using the primer pairs as listed in Table S1.
Western blot analysis
For Western Blot analysis, cells were lysed on ice and lysates were centrifuged at 15,000 x g for 10 min at 4°C. Supernatants were collected, and the amount of protein in each sample was determined by Bradford assay. Subsequently, 20μg of total protein was loaded onto 4% to 20% Tris-Glycine SDS-PAGE gels. After electrophoresis, proteins were transferred to PVDF membrane (Bio-Rad Laboratories, Hercules, CA) and the expression level of the indicated proteins was then detected using the antibodies. The primary antibody dilutions are listed in Table S2 (see key resource table for details). The protein bands were detected using horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and SuperSignal West Pico PLUS chemiluminescence substrate (Invitrogen). ChemiDoc MP imaging system (Bio-rad) was then used to visualize protein bands and the images were analyzed using the Image Lab software (Bio-Rad).
Immunostaining
Immunostaining was performed, according to the manufacturer’s recommendations. All primary antibodies were used at 1:200 dilution in 5% (v/v) goat serum (Invitrogen)/PBS. The following primary antibodies were used: rabbit anti-p-PDHA, rabbit anti-KGA/GAC, rabbit anti-phospho-Histone H2A.X and rabbit anti-TOMM20. The primary antibody dilutions are listed in Table S2 (see key resource table for details). Subsequently, samples were incubated with secondary antibody at 1:200 dilution in 5% (v/v) goat serum/PBS for 1h at room temperature. Nuclei were counterstained with 12.5μg/mL Hoechst 33342 (Invitrogen) for 5 min at room temperature. The results were analyzed using Image J software and expressed as a ratio of fluorescence intensity to the cell number.
Immunohistochemistry
The tissue sections at −80°C were fixed in 10% buffered formalin (Sigma-Aldrich) for 10min followed by several washes in PBS. Then the slides were permeabilized with pre-cold methanol (Sigma-Aldrich) for 6 min at −20°C and rinsed in PBS. For COX IV staining, samples were first processed with the M.O.M kit and following the manufacture’s protocols (Vector Laboratories, Burlingame, CA) to reduce non-specific endogenous mouse Ig staining. The next day, the samples were stained with Alexa Fluor 568 conjugated goat anti-mouse secondary antibody at 1:400 dilution in 5% (v/v) goat serum/PBS for 1h at room temperature. Next, tissues slides were incubated with Cytokeratin 14 primary antibody (skin) or laminin primary antibody (heart), dissolved in 5% (v/v) goat serum/PBS overnight at 4°C. Subsequently, slides were incubated in Alexa Fluor 488 conjugated goat anti-rabbit secondary antibody at 1:200 dilution. The primary antibody dilutions are listed in Table S2 (see key resource table for details).
Seahorse assay
We used Seahorse extracellular flux (XFe96) analyzer (Agilent technologies, Santa Clara, CA) to measure the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), which are measurements of mitochondrial respiration and glycolysis. MSCs were seeded at 10,000 cells/well of XFe96 seahorse culture plates. After 24 hours, cells were rinsed and switched into Seahorse Base Medium (XF DMEM medium, #103575, Agilent technologies). For mitostress test, the medium was supplemented with 10mM glucose and 1mM pyruvate to maintain cells’ viability. Glycolysis, glycolytic capacity and glycolytic reserve were measured after sequential injection of 5mM glucose, 1μM oligomycin and 50mM 2-DG, in Seahorse Base Medium. All calculations were performed according to the manufacturer protocol using the measurement values.
Metabolic assays
In vitro, MSCs or progeria fibroblasts were seeded in 96-well white plates (3,000 cell/cm2). The next day, cells were rinsed with PBS followed by starvation in DMEM depleted of glucose and pyruvate medium for 15 min and then incubated in high glucose DMEM supplemented overnight with various metabolites and/or inhibitors as indicated. Assays to measure cellular ATP concentration (Abcam), intracellular urea concentrations (Abcam), glutaminase activity (Biovision), reactive oxygen species level (Abcam), and intracellular intermediate concentrations including TCA intermediates (Abcam), glycolytic intermediates (Abcam) were conducted based on manufacturer’s instructions.
Mitochondrial isolation and respirometry of frozen mitochondrial sample
Mitochondria were isolated from heart and skin tissues according to the protocol described by Acin-Prez et al.64 Briefly, twenty milligrams of frozen tissue were homogenized in 200 μL of ice-cold mitochondrial assay (MAS) buffer. The homogenate was centrifuged at 1000×g for 5 min at 4°C. The pellet was discarded, and the resulting supernatant was centrifuged again at 10,000× g for 10 min at 4°C. The mitochondrial pellet was resuspended in 100μL of MAS buffer and protein concentration was assessed by Bradford assay using BSA as standard. The mitochondrial extract was used in respirometry and metabolic assays. Mitochondrial respiratory chain oxygen consumption (OCR) was measured using Seahorse extracellular flux (XFe96) analyzer (Agilent technologies, Santa Clara, CA) as described previously.64 Briefly, heart mitochondria (1μg/well) and skin mitochondria (10μg/well) were loaded onto Seahorse microplates and different substrates were used for assessing Complex II and IV activities: 2μM Rotenone and 2.5mM Succinate for Complex II; 0.5mM TMPD (tetra methyl phenylene diamine) and 1mM Ascorbic acid for Complex IV; 5μM Antimycin A and 50mM sodium azide were added to inhibit mitochondrial Complex III and Complex IV, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance for each experiment was assessed by one-way or two-way ANOVA followed by Tukey’s multiple comparison test, using GraphPad Prism 8. The data were represented as mean ± 95% confidence interval (CI) of one representative experiment, where each bubble represents one cell; or mean ± standard error for multiple independent experiments and in-vivo experiments. Each experiment was repeated at least three times with triplicate samples for each condition to ensure reproducibility of the results.
Supplementary Material
Highlights.
Overactivated glutaminolysis leads to increased urea production in senescent cells
Loss of urea transporter leads to urea accumulation causing mitochondrial dysfunction
JNK pathway positively regulates GLS1 expression
Inhibiting GLS1 improves mitochondrial function and ameliorates aging hallmarks
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health, R01AG052387 and R01AG068250 and from the New York Stem Cell Science (NYSTEM) (Contract #C30290GG) to S.T.A. Figures were created using Microsoft Publisher and schematics using Biorender (Biorender.com; publication and licensing rights agreement MB24JBPCQV).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111744.
DECLARATION OF INTERESTS
The authors declare that they do not have financial or nonfinancial competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a gender minority in their field of research. We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Gong Z, and Muzumdar RH (2012). Pancreatic function, type 2 diabetes, and metabolism in aging. Int. J. Endocrinol 2012, 320482. 10.1155/2012/320482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metter EJ, Conwit R, Tobin J, and Fozard JL (1997). Age-associated loss of power and strength in the upper extremities in women and men. J. Gerontol. A Biol. Sci. Med. Sci 52, B267–B276. 10.1093/gerona/52a.5.b267. [DOI] [PubMed] [Google Scholar]
- 3.Mrak RE, Griffin ST, and Graham DI (1997). Aging-associated changes in human brain. J. Neuropathol. Exp. Neurol 56, 1269–1275. 10.1097/00005072-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 4.van Deursen JM (2014). The role of senescent cells in ageing. Nature 509, 439–446. 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufour E, and Larsson NG (2004). Understanding aging: revealing order out of chaos. Biochim. Biophys. Acta 1658, 122–132. 10.1016/j.bbabio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 7.Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, Perez-Garcia A, Kiourtis C, Dasgupta N, Lei X, et al. (2020). Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34, 428–445. 10.1101/gad.331272.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emelyanova L, Preston C, Gupta A, Viqar M, Negmadjanov U, Edwards S, Kraft K, Devana K, Holmuhamedov E, O’Hair D, et al. (2018). Effect of aging on mitochondrial energetics in the human atria. J. Gerontol. A Biol. Sci. Med. Sci 73, 608–616. 10.1093/gerona/glx160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu EPK, Reinhold J, Yu H, Starks L, Uryga AK, Foote K, Finigan A, Figg N, Pung YF, Logan A, et al. (2017). Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler. Thromb. Vasc. Biol 37, 2322–2332. 10.1161/ATVBAHA.117.310042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soro-Arnaiz I, Li QOY, Torres-Capelli M, Meléndez-Rodríguez F, Veiga S, Veys K, Sebastian D, Elorza A, Tello D, Hernansanz-Agustín P, et al. (2016). Role of mitochondrial complex IV in age-dependent obesity. Cell Rep. 16, 2991–3002. 10.1016/j.celrep.2016.08.041. [DOI] [PubMed] [Google Scholar]
- 11.Boveris A, and Navarro A (2008). Brain mitochondrial dysfunction in aging. IUBMB Life 60, 308–314. 10.1002/iub.46. [DOI] [PubMed] [Google Scholar]
- 12.Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. 10.15252/embj.201592862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JA, Wei Y, and Sowers JR (2008). Role of mitochondrial dysfunction in insulin resistance. Circ. Res 102, 401–414. 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, and Shulman GI (2003). Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300, 1140–1142. 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motori E, Atanassov I, Kochan SMV, Folz-Donahue K, Sakthivelu V, Giavalisco P, Toni N, Puyal J, and Larsson NG (2020). Neuronal metabolic rewiring promotes resilience to neurodegeneration caused by mitochondrial dysfunction. Sci. Adv 6, eaba8271. 10.1126/sciadv.aba8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, Valsecchi F, Primiano G, Calder EL, Carelli V, et al. (2018). Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 27, 1007–1025.e5. 10.1016/j.cmet.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johmura Y, Yamanaka T, Omori S, Wang TW, Sugiura Y, Matsumoto M, Suzuki N, Kumamoto S, Yamaguchi K, Hatakeyama S, et al. (2021). Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270. 10.1126/science.abb5916. [DOI] [PubMed] [Google Scholar]
- 18.Li Q, Gao Z, Chen Y, and Guan MX (2017). The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 8, 439–445. 10.1007/s13238-017-0385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spees JL, Olson SD, Whitney MJ, and Prockop DJ (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 103, 1283–1288. 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mistriotis P, Bajpai VK, Wang X, Rong N, Shahini A, Asmani M, Liang MS, Wang J, Lei P, Liu S, et al. (2017). NANOG reverses the myogenic differentiation potential of senescent stem cells by restoring ACTIN filamentous organization and SRF-dependent gene expression. Stem Cell. 35, 207–221. 10.1002/stem.2452. [DOI] [PubMed] [Google Scholar]
- 21.Rong N, Mistriotis P, Wang X, Tseropoulos G, Rajabian N, Zhang Y, Wang J, Liu S, and Andreadis ST (2019). Restoring extracellular matrix synthesis in senescent stem cells. FASEB J. 33, 10954–10965. 10.1096/fj.201900377R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shahini A, Choudhury D, Asmani M, Zhao R, Lei P, and Andreadis ST (2018). NANOG restores the impaired myogenic differentiation potential of skeletal myoblasts after multiple population doublings. Stem Cell Res. 26, 55–66. 10.1016/j.scr.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 23.Bajpai VK, and Andreadis ST (2012). Stem cell sources for vascular tissue engineering and regeneration. Tissue Eng. Part B Rev 18, 405–425. 10.1089/ten.TEB.2011.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mistriotis P, and Andreadis ST (2013). Hair follicle: a novel source of multipotent stem cells for tissue engineering and regenerative medicine. Tissue Eng. Part B Rev 19, 265–278. 10.1089/ten.TEB.2012.0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahini A, Mistriotis P, Asmani M, Zhao R, and Andreadis ST (2017). NANOG restores contractility of mesenchymal stem cell-based senescent microtissues. Tissue Eng. Part A 23, 535–545. 10.1089/ten.TEA.2016.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JE, Westrate LM, Wu H, Page C, and Voeltz GK (2016). Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143. 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma V, Singh P, Pandey AK, and Dhawan A (2012). Induction of oxidative stress, DNA damage and apoptosis in mouse liver after subacute oral exposure to zinc oxide nanoparticles. Mutat. Res 745, 84–91. 10.1016/j.mrgentox.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, and Kennedy BK (2008). Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol. Biol. Cell 19, 5238–5248. 10.1091/mbc.E08-05-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burtner CR, and Kennedy BK (2010). Progeria syndromes and ageing: what is the connection? Nat. Rev. Mol. Cell Biol 11, 567–578. [DOI] [PubMed] [Google Scholar]
- 30.Prokocimer M, Barkan R, and Gruenbaum Y (2013). Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 12, 533–543. 10.1111/acel.12070. [DOI] [PubMed] [Google Scholar]
- 31.Brassard P, and Gustafsson F (2016). Exercise intolerance in heart failure: did we forget the brain? Can. J. Cardiol 32, 475–484. 10.1016/j.cjca.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 32.Carrero D, Soria-Valles C, and López-Otín C (2016). Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells. Dis. Model. Mech 9, 719–735. 10.1242/dmm.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osorio FG, Navarro CL, Cadiñanos J, López-Mejía IC, Quirós PM, Bartoli C, Rivera J, Tazi J, Guzmán G, Varela I, et al. (2011). Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med 3, 106ra107. 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- 34.Scaffidi P, and Misteli T (2006). Lamin A-dependent nuclear defects in human aging. Science 312, 1059–1063. 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javadov S, Jang S, and Agostini B (2014). Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol. Ther 144, 202–225. 10.1016/j.pharmthera.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herranz N, and Gil J (2018). Mechanisms and functions of cellular senescence. J. Clin. Invest 128, 1238–1246. 10.1172/JCI95148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon SM, Hong SM, Lee YK, Min S, and Yoon G (2019). Metabolic features and regulation in cell senescence. BMB Rep. 52, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papa L, Djedaini M, and Hoffman R (2019). Mitochondrial role in stemness and differentiation of hematopoietic stem cells. Stem Cells Int. 2019, 4067162. 10.1155/2019/4067162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye G, Xie Z, Zeng H, Wang P, Li J, Zheng G, Wang S, Cao Q, Li M, Liu W, et al. (2020). Oxidative stress-mediated mitochondrial dysfunction facilitates mesenchymal stem cell senescence in ankylosing spondylitis. Cell Death Dis. 11, 775. 10.1038/s41419-020-02993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smeitink JA, Zeviani M, Turnbull DM, and Jacobs HT (2006). Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 3, 9–13. 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Franco R, and Cidlowski JA (2009). Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 16, 1303–1314. 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 42.Wang E (1995). Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 55, 2284–2292. [PubMed] [Google Scholar]
- 43.Salminen A, Ojala J, and Kaarniranta K (2011). Apoptosis and aging: increased resistance to apoptosis enhances the aging process. Cell. Mol. Life Sci 68, 1021–1031. 10.1007/s00018-010-0597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, and Woodgett JR (1994). The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369, 156–160. [DOI] [PubMed] [Google Scholar]
- 45.Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, and Robbins PD (2017). The clinical potential of senolytic drugs. J. Am. Geriatr. Soc 65, 2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, et al. (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdelgawad IY, Agostinucci K, Ismail SG, Grant MK, and Zordoky BN (2022). EA. hy926 cells and HUVECs share similar senescence phenotypes but respond differently to the senolytic drug ABT-263. Cells 11, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deryabin PI, Shatrova AN, and Borodkina AV (2021). Apoptosis resistance of senescent cells is an intrinsic barrier for senolysis induced by cardiac glycosides. Cell. Mol. Life Sci 78, 7757–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et al. (2016). Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun 7, 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devary Y, Gottlieb RA, Lau LF, and Karin M (1991). Rapid and preferential activation of the c-jun gene during the mammalian UV response. Mol. Cell Biol 11, 2804–2811. 10.1128/mcb.11.5.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Q, Lam PY, Han D, and Cadenas E (2009). Activation of c-Jun-N-terminal kinase and decline of mitochondrial pyruvate dehydrogenase activity during brain aging. FEBS Lett. 583, 1132–1140. 10.1016/j.febslet.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Q, Lam PY, Han D, and Cadenas E (2008). c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem 104, 325–335. 10.1111/j.1471-4159.2007.04957.x. [DOI] [PubMed] [Google Scholar]
- 53.Iansante V, Choy PM, Fung SW, Liu Y, Chai JG, Dyson J, Del Rio AA-O, D’Santos C, Williams R, Chokshi S, et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat. Commun 6 7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbarulo A, Iansante V, Chaidos A, Naresh K, Rahemtulla A, Franzoso G, Karadimitris A, Haskard DO, Papa S, and Bubici C (2013). Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene 32, 4231–4242. 10.1038/onc.2012.448. [DOI] [PubMed] [Google Scholar]
- 55.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, and Campisi J (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weiner ID, Mitch WE, and Sands JM (2015). Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin. J. Am. Soc. Nephrol 10, 1444–1458. 10.2215/CJN.10311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansmannel F, Sillaire A, Kamboh MI, Lendon C, Pasquier F, Hannequin D, Laumet G, Mounier A, Ayral AM, DeKosky ST, et al. (2010). Is the urea cycle involved in Alzheimer’s disease? J. Alzheimers Dis 21, 1013–1021. 10.3233/JAD-2010-100630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicolas C, Jaisson S, Gorisse L, Tessier FJ, Niquet-Léridon C, Jacolot P, Pietrement C, and Gillery P (2019). Carbamylation and glycation compete for collagen molecular aging in vivo. Sci. Rep 9, 18291. 10.1038/s41598-019-54817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meng Y, Zhao C, Zhang X, Zhao H, Guo L, Lu B, Zhao X, and Yang B (2009). Surface electrocardiogram and action potential in mice lacking urea transporter UT-B. Sci. China C Life Sci 52, 474–478. 10.1007/s11427-009-0047-y. [DOI] [PubMed] [Google Scholar]
- 60.Bajpai VK, Mistriotis P, and Andreadis ST (2012). Clonal multipotency and effect of long-term in vitro expansion on differentiation potential of human hair follicle derived mesenchymal stem cells. Stem Cell Res. 8, 74–84. 10.1016/j.scr.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.You H, Padmashali RM, Ranganathan A, Lei P, Girnius N, Davis RJ, and Andreadis ST (2013). JNK regulates compliance-induced adherens junctions formation in epithelial cells and tissues. J. Cell Sci 126, 2718–2729. 10.1242/jcs.122903. [DOI] [PubMed] [Google Scholar]
- 62.Shahini A, Rajabian N, Choudhury D, Shahini S, Vydiam K, Nguyen T, Kulczyk J, Santarelli T, Ikhapoh I, Zhang Y, et al. (2021). Ameliorating the hallmarks of cellular senescence in skeletal muscle myogenic progenitors in vitro and in vivo. Sci. Adv 7, eabe5671. 10.1126/sciadv.abe5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alimperti S, Lei P, Tian J, and Andreadis ST (2012). A novel lentivirus for quantitative assessment of gene knockdown in stem cell differentiation. Gene Ther. 19, 1123–1132. 10.1038/gt.2011.208. [DOI] [PubMed] [Google Scholar]
- 64.Acin-Perez R, Benador IY, Petcherski A, Veliova M, Benavides GA, Lagarrigue S, Caudal A, Vergnes L, Murphy AN, Karamanlidis G, et al. (2020). A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 39, e104073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original microscopy (.czi) and Seahorse analyzer data (.asyr) are available upon request.
This paper does not report original code.
Any additional information to reanalyze the data reported in this paper may be directed to and will be fulfilled by lead contact Stelios T. Andreadis (sandread@buffalo.edu).