

Abstract
Time-lapse in vivo imaging of neuronal structures is critical for understanding the assembly of neural circuits during development. Imaging developing neurons in vivo can be performed with relative ease in lower vertebrates, but ideally, one would also like to image the developing mammalian brain. In vivo chronic imaging of mice is particularly desirable because of the availability of transgenic lines that model human neuropsychiatric disease or those that allow cell- or region-specific expression of fluorescent proteins (e.g., green fluorescent protein [GFP], channelrhodopsins, and genetically encoded calcium indicators). Unfortunately, although chronic imaging of neural structures in adult mice that express GFP is now commonplace, similar approaches in neonatal mice face several additional challenges. First, the small size of the animal complicates the cranial window surgery. Second, there is a tendency for dams to cannibalize pups with head caps. Third, the head cap can impede the normal growth of the skull in neonates, which can limit the duration of imaging. Here, we describe a method for implanting chronic glass-covered cranial windows in the skulls of early postnatal mice through which axonal and dendritic structures can be imaged in vivo over a period of hours or even days.
MATERIALS
It is essential that you consult the appropriate Material Safety Data Sheets and your institution′s Environmental Health and Safety Office for proper handling of equipment and hazardous material used in this protocol.
Reagents
Agarose (low-melting-point)
Animals
To image L2/3 callosal axons, we used male and female C57BL/6 wild-type mice transfected with a pCAG-EGFP plasmid (Addgene plasmid 11150; Matsuda and Cepko 2004) by in utero electroporation (Saito and Nakatsuji 2001) at embryonic day 16.
Cyanoacrylate glue (e.g., Krazy Glue)
Dental cement (e.g., Lang Dental Mfg Co., Inc.)
Ethanol (70%)
Isoflurane (Baxter)
Lidocaine/epinephrine solution (1%/1:100,000) (Hospira 0409-3178)
Paraformaldehyde (4%) (Fisher Scientific)
Saline (sterile) (Hudson RCI)
Surgical foam (e.g., Surgifoam from Ferrosan Medical Devices)
Equipment
Air canister
Anesthesia vaporizer and regulator (e.g., Surgivet, Inc.)
Blanket (water-recirculating) (Gaymar Industries, Inc.)
Cotton swab applicators (e.g., Henry Schein Medical)
Coverslips (round, glass, #1, 3-mm-diameter, custom-made)
Sterilize before use.
Dissection microscope (Stemi 2000, Zeiss)
Electric hair trimmer (Wahl) (optional; see Step 3)
Fiber-optic lamp (Dolan-Jenner Industries)
Forceps (fine)
Iridectomy scissors
Microscope (two-photon, equipped with image acquisition/analysis software)
The in vivo images of GFP-expressing L2/3 neurons shown here (Figs. 1 and 2) were acquired with a custom-built two-photon microscope, using a Ti:sapphire laser (Chameleon Ultra II, Coherent) running at ~910 nm. The objective (40×, 0.8 numerical aperture [NA]), scan lens, and trinoc were from Olympus, and the photomultiplier tube was from Hamamatsu; for a detailed description of the microscope, see Cruz-Martin et al. (2010). Image acquisition was achieved with ScanImage software (Pologruto et al. 2003) written in MATLAB (MathWorks) and analysis was performed using Neurolucida (MBF Bioscience, Inc.).
FIGURE 1.
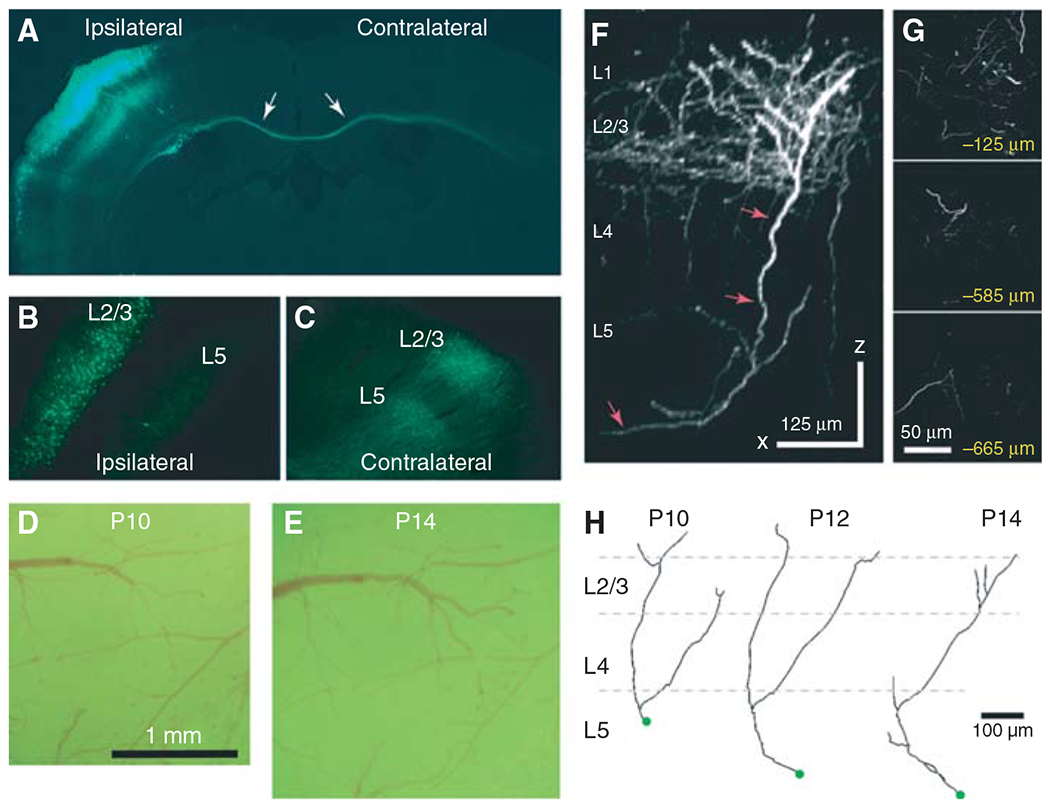
High-resolution long-term imaging of neocortical axons in GFP-electroporated mice. (A) Low-magnification fluorescence photomicrograph of a coronal section through the brain of a mouse electroporated with GFP at embryonic day 16 and perfused at P15; the same animal was imaged in vivo (D–G). Note in (A) a cluster of L2/3 pyramidal neurons in the hemisphere ipsilateral to the side of electroporation and a bundle of axons coursing across the corpus callosum (arrows). (B) Higher magnification view of the ipsilateral hemisphere. GFP expression is restricted to L2/3 neurons and their processes. (C) Callosal axons can be seen in the contralateral hemisphere forming dense arborizations in L5 and L2/3. (D,E) Photomicrographs of dural vessels taken through the cranial window the day after the window surgery (i.e., P10; D) and at P14 (E). The window should remain pristine (i.e., blood free, without inflammation) for the duration of the experiment. (F) Side view of a stack of images obtained with in vivo two-photon imaging at P14 (x–z projection, 136 slices, 5 μm apart). Axons branch extensively in L2/3 at this age. The Neurolucida reconstruction of one of these axons is shown in (H). This mouse was imaged daily between P10 and P14. (G) Images from the same stack at different z-planes below the dura. (H) Tracings of the axon marked with arrows in (F) at P10–P14. Note pruning of axonal branches in L1 and more elaborate branching in L2/3 and L5 over time.
FIGURE 2.
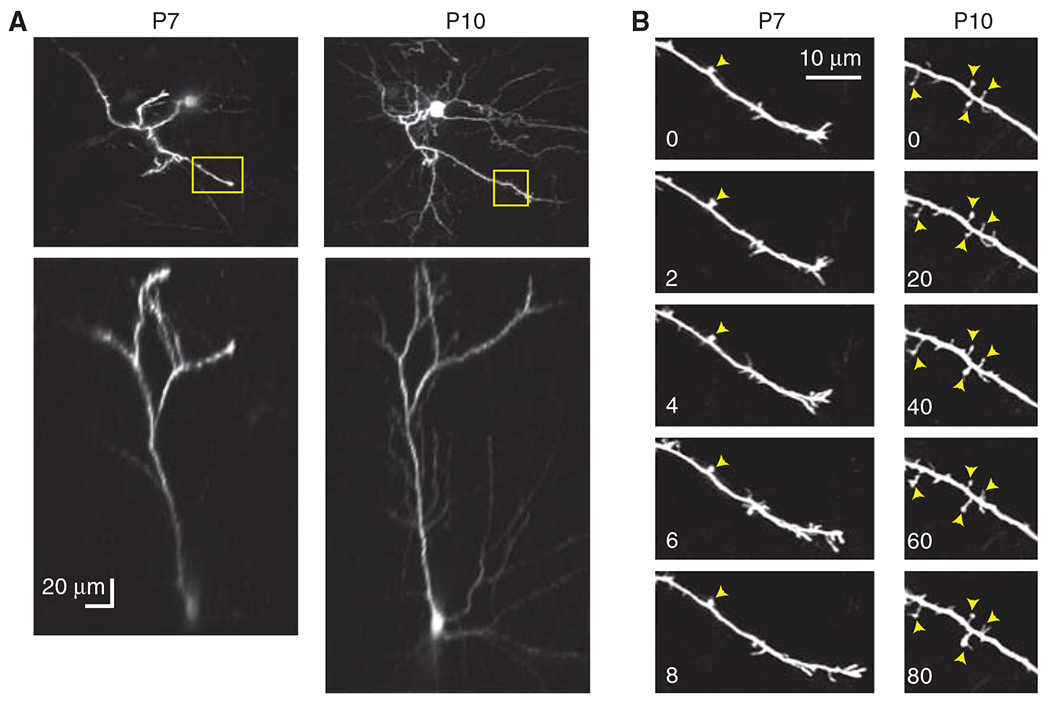
Chronic in vivo two-photon imaging of early dendritic protrusions. Mice were electroporated at embryonic day 16 with GFP. (A) Top- and side-view projections of in vivo two-photon images of the apical dendritic arbor of the same L2/3 pyramidal neuron at P7 and P10 (85–100 slices, 3 μm apart). (B) High-resolution time-lapse imaging of early dendritic protrusions in an apical dendrite (yellow boxes in A). Time stamps are in minutes. Yellow arrowheads point to “always present” protrusions. Note that protrusions are less motile and have longer lifetimes at older ages.
Pneumatic dental drill (Henry Schein)
Scalpel (equipped with #15 blades)
Stereotaxic apparatus (Stoelting Co.)
Temperature-control device (e.g., Thermopad from Harvard Apparatus)
Titanium bar (0.125 × 0.375 × 0.05 in.)
Vibratome (Leica Microsystems)
METHOD
Cranial Window Attachment
This technique is similar to one described previously for adult mice (Mostany and Portera-Cailliau 2008b; Holtmaat et al. 2009). Surgeries can be performed on mice as early as postnatal day (P) 3.
Anesthetize a mouse with 5% isoflurane. Keep the animal anesthetized throughout the surgery using a maintenance dose of 1.5% isoflurane.
Place the mouse on a stereotaxic apparatus with blunt ear bars. Rest the mouse on a warm water-recirculating blanket.
- Disinfect the skin by swabbing with 70% ethanol.For older mice (>P8), use a hair trimmer to expose the scalp before disinfecting the skin.
Using sterilized iridectomy scissors or a scalpel, cut a small (e.g., 6 × 8-mm for a P7 mouse) oval piece of skin from over the target area for the window. As you cut the skin, apply one to two drops of lidocaine/epinephrine solution to reduce pain and bleeding of the wound edges.
Scrape the periosteum over the exposed skull with a scalpel to achieve a dry surface.
- Apply a thin layer of cyanoacrylate glue over the entire surface of the skull, except over a circular region where the window will be placed. Allow the glue to dry completely (e.g., 5 min).Avoid putting glue across several bony sutures, because this will prevent later skull growth.
- Using a pneumatic dental drill, carefully carve out a round portion of the skull, 2 mm in diameter. Use an air canister or a drop of saline to blow/wash away the bone dust. Stop when the grooves start to crack.Note that the bone is quite soft (i.e., barely ossified) before P7.
- Using the tips of the fine forceps, remove the skull overlying the cortex with the utmost care so as not to damage the underlying dura.Sometimes it is best to drill slightly deeper along a portion of the window, and then tear the skull using forceps. The dry cyanoacrylate around the window prevents the rip from extending beyond the intended edges of the window.
Irrigate the dura copiously with sterile saline. Use small pieces of surgical foam (~4 mm2) to stop any superficial bleeding of dural vessels.
Gently place a round cover glass (3 mm in diameter) over the exposed dura. Dry the skull around the window, leaving a small meniscus of saline under the glass.
- Gently press down on the glass coverslip. Apply fresh cyanoacrylate glue to the edges of the window to seal it in place.Avoid allowing large air bubbles to collect under the window.
Apply dental cement around the glass and to the edges of the skin.
- Stop the flow of anesthesia. Place the mouse into a recovery cage resting on a warm blanket for ≥60 min after surgery before returning it to its dam.See Troubleshooting and Discussion.
Imaging
For acute (i.e., nonsurvival) experiments, imaging can begin right away. However, for chronic time-lapse experiments over several days, it is best to begin imaging the day after surgery to ensure the pup survives the initial period with its dam. Imaging sessions should last ≤2 h, to avoid prolonged anesthesia exposure.
14. Anesthetize the mouse with isoflurane. Keep the animal anesthetized throughout the procedure using a maintenance dose of 1%–1.5% isoflurane.
15. Attach a titanium bar to the skull with dental cement. Allow the cement to harden for ≥10 min.
16. Secure the mouse to the stage of the microscope using the titanium bar. Keep the mouse warm with a temperature-control device.
- 17. Begin imaging. For studies of large-scale growth and pruning of axons, acquire several slightly overlapping image stacks (up to 40 per animal) at low magnification. Using a 40× objective and a 1× ScanImage zoom, this corresponds to an ~360 × 370 μm field of view (for a 512 × 512-pixel image). Alternatively, for high-magnification imaging of axon growth cones or spine/bouton dynamics, select several small regions of interest (ROIs) at 5× ScanImage zoom measuring ∼78 × 79 μm.Note that at early ages, neuronal processes can be sparsely labeled (e.g., in GFP-M mice; Feng et al. 2000), but over subsequent days, the labeling can become quite dense. Therefore, for long-term imaging experiments, select areas with an initially low density of labeled processes.Avoid selecting ROIs under blood vessels (even small ones) because these tend to grow over subsequent days and obscure the image.See Troubleshooting.
- 18. Select random ROIs along the axon/dendritic shaft, or center them on tips of growing processes. To simplify analysis, collect short image stacks of more than 20 optical sections at 1- or 5-μm steps for 5× and 1× magnification, respectively.For chronic imaging, identify the ROIs over several imaging sessions by using the surface vasculature as landmarks (Holtmaat et al. 2009).See Troubleshooting.
- 19. To achieve close to identical fluorescence levels across imaged regions and across imaging sessions, adjust the laser intensity as needed, especially at deeper focal planes or to compensate for increased GFP expression or for darkening of a particular ROI resulting from growing blood vessels.With good window preparations, one can collect high-resolution images of fluorescent processes within 300–450 μm below the pial surface.
Tracing of Axons and Analysis
- 20. Trace imaged dendrites or axons in three dimensions within tiled stacks using the confocal module of Neurolucida.From these tracings, one can derive data regarding process length (growth and retraction over time), branch density, and tortuosity.
Brain Reconstruction with En-Bloc Imaging and Slicing
Retrospective reconstructions of selected axonal or dendritic arbors using brain reconstruction with en-bloc imaging and slicing can be used to confirm their identities. Following several cycles of serial imaging and slicing, we have been able to follow processes from layer 1 all the way down to the thalamus (Portera-Cailliau et al. 2005).
21. After the last imaging session, perfuse the mouse intracardially with 4% paraformaldehyde.
22. Remove the brain. Postfix overnight.
23. Cut the hemisphere that was imaged in vivo. Immerse in low-melting-point agarose. Mount on the well of a vibratome slicer that can be mounted on the stage of the two-photon microscope.
- 24. Image the same neuronal structures in the same orientation as during in vivo imaging and as deep as possible (e.g., up to 800 μm in a single set of image stacks).Previously imaged axons or dendrites can be identified using blood vessel imprints on the dura as landmarks that can be compared with photographs of the same vessels through the window or by using injections of fluorescent beads as fiducial points.
25. Slice off the most superficial 300–600 μm of brain tissue. Reimage the structure of interest, always keeping track of the location of the imaged processes.
TROUBLESHOOTING
Problem (Step 13): The dam does not cannibalize the pups, but she does not take care of them.
Solution: This typically happens with inexperienced dams and, therefore, can be solved by using experienced surrogate dams or those known to care for pups with head caps. Note, however, that rejection of pups can also be caused by the head cap being too large or by an unusual scent on the pup (e.g., dental cement).
Problem (Step 17): Window turns opaque, or fluorescence intensity is much dimmer 1–2 d after it was implanted.
Solution: The most likely reasons for this problem are infection, bone growth, or excessive inflammation. Follow sterile techniques as much as possible throughout the procedure. Be sure to sterilize the glass coverslips. Blowing off bone dust and irrigating with saline will help reduce inflammation, bone growth, and infection. If necessary, use carprofen (5 mg/kg) for 1–3 d post-operatively to prevent/reduce inflammation. Others have used a single preoperative injection of dexamethasone (20 μL of a 4% solution) to reduce cerebral edema (Gray et al. 2006). A related problem can be excessive edema triggered by rough handling of the brain surface during surgery. As swelling resolves, the brain sinks below the glass. This can be prevented by using very delicate movements throughout the procedure and by reducing the total surgery time to ~45 min.
Problem (Step 17): There is movement artifact during imaging; that is, the images “bounce.”
Solution: This occurs when there is too much space between the brain surface and the glass coverslip, which can occur because insufficient pressure was applied to the glass while applying the cyanoacrylate glue or because the brain sank over time. It could also be a sign that the head cap has partially detached from the skull. Try adding a drop of low-melting-point agarose to the brain surface just before placing the coverslip, to fill gaps between the edges of the skull and the brain.
Problem (Step 18): There is blood under the window after several days of imaging, or the head cap breaks apart during imaging.
Solution: This usually occurs within the first 24 h of imaging and is more likely to happen with the youngest mice. It is often caused by the dam grooming the pup (in neonates) or by the pup itself trying to scratch the head cap (in older pups). It is also possible that the skull surface was not dry enough when the glue or dental cement was applied, allowing the head cap to break off from the skull. Sometimes the bleeding will stop spontaneously (if it is caused by bruising of the dura), and the hematoma can be resorbed within a day or two. We avoid including mice with significant bleeding (>5% of the window) in experiments because it is not clear what effects bleeding will have on the structural dynamics being studied. A large hematoma or a sudden increase in motion artifact during imaging usually signifies the window has detached from the skull, at least partially. This invariably means the end of the experiment because one does not wish to reimplant the cranial window in the middle of an imaging experiment.
DISCUSSION
Impact of Window Surgery on Neural Development
Provided the mice survive, cranial window surgery itself seems innocuous to them: They recover from it readily, gain weight, and behave indistinguishably from their unoperated littermates. However, head caps can restrict head growth in the youngest mice. Care must be taken to implant very small windows that do not cross multiple bony sutures. When windows are implanted before P5, mice can only be imaged for 2–3 d because, beyond that point, animals do not gain weight as fast as their unoperated littermate controls. In contrast, when windows are implanted after P8, mice grow normally. As a control, we have imaged fixed brains that had never undergone surgery or in vivo imaging at various postnatal ages and found that the density of branch points was not different from that of experimental mice (Portera-Cailliau et al. 2005), suggesting that there were no deleterious effects caused by the surgery or chronic imaging.
Postoperative Care to Minimize Cannibalism
Take these steps to reduce the incidence of cannibalism by the dam.
Because of its strong scent, avoid using betadine for skin prepping. If the animal protocol demands the use of betadine, desensitize the dam by dabbing her snout with a cotton swab soaked with betadine periodically before the surgery, starting the night before.
Try to minimize the size of the window. Large head caps can restrict bone growth and are more likely to lead to cannibalism. It is best not to place the titanium bar (used to secure the mouse to the microscope stage) at the time of the window surgery. Dams are more likely to aggressively groom pups with large head caps, particularly if they have a metal bar. Instead, implant the windows 1 d before imaging, and attach the bar the same day of the first imaging session.
Allow the pup to recover for ≥60 min after surgery before returning it to its dam, to ensure the animal is fully awake and can fend for itself if groomed aggressively.
Reduce the size of the litter to three or four pups. This helps to minimize competition for access to the dam for nursing. Perform multiple surgeries in one sitting because it might help the dam accept the pups if there are many of them with head caps.
Carefully monitor the cage; tap on it every time the dam attempts to groom the head cap area.
Use experienced surrogate dams, especially if one can be identified that is particularly good at accepting pups with head caps. Rub the pups with the surrogate dam’s urine before returning them to the dam’s cage.
Applications of the Method
We routinely use chronic high-resolution in vivo imaging through glass-covered cranial windows in early postnatal mice to study neuronal structure (e.g., of GFP-expressing cells) or function (e.g., calcium imaging) (Golshani et al. 2009; Cruz-Martin et al. 2010). This method can be used to monitor the growth and branching of axons over multiple days (Fig. 1) or the dynamics of early dendritic protrusions over minutes to hours (Fig. 2). Both figures show images of L2/3 neurons transfected with GFP by in utero electroporation at embryonic day 16.
This method has also been used for chronic in vivo imaging of the growth and pruning of thalamocortical and Cajal–Retzius axons from P4 to P19 in the mouse neocortex (Portera-Cailliau et al. 2005) and to image the dynamics of PSD-95 clusters in layer (L) 2/3 pyramidal neurons between P10 and P21 (Gray et al. 2006). A similar approach used GFP expression for acute imaging of mouse dendritic spine dynamics in L2/3 pyramidal neurons that had been transfected with Sindbis virus injections after the second postnatal week (Lendvai et al. 2000). In addition to imaging neuronal structures, the techniques described could be used to image other fluorescent structures (e.g., glia, pathological protein aggregates) or to image blood flow using fluorescent dextran conjugates injected systemically (Kleinfeld et al. 1998; Mostany and Portera-Cailliau 2008a).
Advantages and Disadvantages
Chronic in vivo imaging of the mouse neocortex through a cranial window during early postnatal development offers unique advantages for studying neuronal structure and function. Although time-lapse imaging can be achieved in cultured preparations, one ultimately wishes to know how neurons behave within intact circuits in the living animal. An important benefit of this preparation is that the same structures can be imaged before and after a manipulation (e.g., stroke, sensory deprivation, and learning a behavioral task) so that each animal serves as its own control, reducing variability or sample bias.
Unfortunately, this technique is not without its shortcomings. In the youngest mice, long-term imaging over weeks is not possible. Another problem is that deep brain structures (>700–1000 μm below the pial surface) are not readily accessible to in vivo two-photon microscopy. Future improvements in microscopy chemistry and molecular biology will generate the tools and probes necessary to make it even easier to image the developing brain and to record the assembly of neuronal circuits with improved spatial resolution and cell specificity.
ACKNOWLEDGMENTS
We are grateful to Dr. Rob Weimer, Dr. Anthony Holtmaat, and Dr. Karel Svoboda for their help, advice, and support. This research is supported by grants from the National Institute for Childhood and Developmental Disorders (National Institutes of Health), the Dana Foundation, FRAXA, and research grant No. 1-FY-06-357 from the March of Dimes Foundation.
REFERENCES
- Cruz-Martin A, Crespo M, Portera-Cailliau C. 2010. Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30: 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. 2000. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41–51. [DOI] [PubMed] [Google Scholar]
- Golshani P, Gonçalves JT, Khoshkhoo S, Mostany R, Smirnakis S, Portera-Cailliau C. 2009. Rapid desynchronization of network activity in developing neocortex is internally mediated. J Neurosci 29: 10890–10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. 2006. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol 4: e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow D, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Lee W- CA, Knott G, et al. 2009. Long-term high resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4: 1128–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfeld D, Mitra PP, Helmchen F, Denk W. 1998. Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci 95: 15741–15746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. 2000. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404: 876–881. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. 2004. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci 101: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C. 2008a. A method for 2-photon imaging of blood flow in the neocortex through a cranial window. J Vis Exp 12. doi: 10.3791/678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C. 2008b. A craniotomy surgery procedure for chronic brain imaging. J Vis Exp 12. doi: 10.3791/680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. 2003. ScanImage: Flexible software for operating laser scanning microscopes. Biomed Eng Online 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Weimer RM, De Paola V, Caroni P, Svoboda K. 2005. Diverse modes of axon elaboration in the developing neocortex. PLoS Biol 3: e272. doi: 10.1371/journal.pbio.0030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. 2001. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol 240: 237–246. [DOI] [PubMed] [Google Scholar]