

Abstract
Two-photon excitation (2PE) overcomes many challenges in fluorescence microscopy. Compared to confocal microscopy, 2PE microscopy improves depth penetration, owing to the longer excitation wavelength required and to the ability to collect scattered emission photons as a useful signal. It also minimizes photodamage because lower energy photons are used and because fluorescence is confined to the geometrical focus of the laser spot. 2PE is therefore ideal for high-resolution, deep-tissue, time-lapse imaging of dynamic processes in cell biology. Here, we provide examples of important applications of 2PE for in vivo imaging of neuronal structure and signals; we also describe how it can be combined with optogenetics or photolysis of caged molecules to simultaneously probe and control neuronal activity.
Keywords: 2-Photon, Axonal bouton, Calcium imaging, Channelrhodopsin, Confocal microscopy, Dendritic spine, Electroporation, Green fluorescent protein, Optogenetics, Oregon Green BAPTA, Photomultiplier tube, Synaptic plasticity, Turnover, ScanImage, Uncaging
1. Introduction
1.1. Fluorescence Microscopy
Fluorescence microscopy has emerged as the preferred tool for studying the structure and dynamics of biological systems both in vivo and in vitro. In recent decades, we have witnessed unprecedented technological advances in molecular and cellular imaging, which was fueled in large part by the discovery of fluorescent molecules that can be used to image cellular structure and the dynamics of organelles or even single proteins [1]. The sensitivity of fluorescence detection for these molecules (e.g., fluorescent proteins from a variety of aquatic invertebrate species such as jellyfish, synthetic fluorescent dyes, and quantum dots) is exquisite [2]. Chemists and molecular biologists have also modified fluorescent proteins such that individual proteins can be used to monitor changes in intracellular calcium, pH, protein–protein interactions, or the function of single enzymes such as kinases or the ubiquitin proteasome system [1].
In parallel with these discoveries, rapid advances in fluorescence microscopy techniques have led to creative new ways to study biological processes [3]. For example, confocal microscopy allowed scientists to visualize these fluorescent molecules with improved spatial resolution, fluorescence lifetime imaging microscopy (FLIM) and Forster resonance energy transfer (FRET) led them to study protein–protein interactions and fluorescence recovery after photobleaching (FRAP) made it possible to investigate protein turnover and trafficking, to name a few. In addition, the diffraction resolution limit of light microscopy was recently broken with novel methods, such as stimulated emission depletion (STED), bringing us to a new era of super-resolution microscopy [4-6].
1.2. Two-Photon Excitation Microscopy
Besides the discovery of fluorescent proteins, perhaps the most significant advance in bio-imaging of the last 25 years was the invention, in 1990, of two-photon excitation (2PE) microscopy [7]. Since then, 2PE has become widely popular for in vivo imaging of neuronal structure because of its superior depth penetration and reduced photobleaching compared to confocal or epifluorescence microscopy [8]. Imaging deeper with 2PE is achieved by means of excitation with an infrared laser and optics that collect much of the scattered emitted light, while maintaining excellent spatial resolution. An added bonus is that 2PE greatly decreases photodamage (such as photobleaching and phototoxicity) and is therefore more compatible with experiments requiring prolonged continuous tissue illumination (e.g., calcium imaging). Both advantages make 2PE ideally suited to chronic in vivo imaging in the intact brain.
Single-photon excitation requires the absorption of a high energy photon to excite an orbital electron of a fluorophore to a vibrationally and electronically excited state (excitation; Fig. 1a). When this electron relaxes to its ground state, it emits a photon of light of a different wavelength than the excitation photon (fluorescence emission). In 2PE this process is achieved by the quasisimultaneous absorption of two photons: the first one excites the electron to a virtual intermediate state, and the second one completes its excitation to reach the final excited state.
Fig. 1.

Principles of 2PE microscopy. (a) Jablonski energy diagram showing the electron excitation process in single- (left) and two-photon (right) fluorescence microscopy. Single-photon excitation (left) requires the absorption of a high energy photon to excite an orbital electron of a fluorophore to a vibrationally and electronically excited state (excitation). When this electron relaxes to its ground state, it emits a photon of light of a different wavelength than the excitation photon (fluorescence emission). In 2-photon excitation (2PE; right), this process is achieved by the quasi-simultaneous absorption of two photons; the first one excites the electron to a virtual intermediate state, and the second one completes the excitation to reach the electronic excited state. (b) Confocal versus 2PE microscope systems. In confocal systems (left), the presence of a pinhole right before the photodetector rejects the photons emitted from outside the focus (e.g., photon #6), as well as those scattered on their way to the PMT (e.g., photon #4). Only unscattered photons coming from the focal plane are able to pass through the pinhole (e.g., photon #5) and contribute to the signal. In 2PE systems (right) there is no need for a pinhole since photons contributing to the signal come only from the geometrical focus of the excitation spot (e.g., photon #5), even if they were scattered on their path to the PMT (e.g., photon #4)
The main shortcoming of single-photon excitation is its inefficient fluorescence excitation: because light absorption occurs throughout much of the specimen, a pinhole in front of the detector is required in confocal microscopy to reject fluorescent photons emanating from outside the focus (Fig. 1b). Unfortunately, the pinhole similarly rejects in-focus photons that subsequently scatter, and as a result only unscattered photons contribute to the signal. This inefficiency demands high laser power for imaging, which creates unwanted photodamage [9] and limits imaging depth to the free mean path of visible light (≤100 μm in biological tissue [10]).
In 2PE, two lower energy photons are absorbed simultaneously to excite the fluorophore. Because of the steep dependence of absorption rate on photon concentration (light intensity), fluorescence is confined to the geometrical focus of the laser excitation spot, which provides inherent optical sectioning. A pinhole is not required because all emitted photons (regardless of how much they scatter on their path to the detector) convey a useful signal (Fig. 1b). Greater tissue penetration is possible in 2PE (compared to confocal microscopy) thanks to the longer excitation wavelength used and to the ability to collect scattered emission photons as a useful signal. These properties of 2PE also limit photodamage since the absorption is confined to a tiny focal volume, and fewer excitation events are required to achieve the same signal due to the improved collection efficiency. In addition, because 2PE microscopy requires photons of lower energy than those used in one-photon excitation, this reduces photodamage further.
2. Materials
2.1. Instrumentation: Laser and Microscope
Although commercial systems are available for multiphoton microscopy, some users may prefer to custom build their 2PE microscope (Fig. 2). Because the hardware and software needed for laser beam scanning and data acquisition in 2PE and confocal microscopy are so similar, commercial confocal systems can sometimes be converted into a two-photon microscope [11]. For further reading on configuring a 2PE system, please refer to the following resources [3, 8, 9, 12].
Fig. 2.
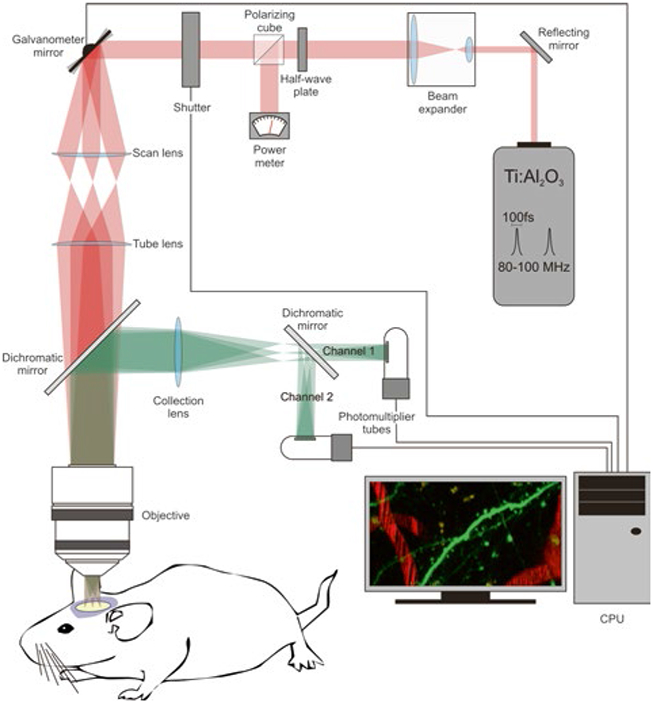
Components of a 2PE microscope. The basic components of a 2PE microscope include the following elements: the two-photon laser (Ti:sapphire laser), a beam expander (the goal is to fill the back aperture of the objective lens), a halfwave plate combined with a polarizing cube to control and measure the laser power, a fast shutter to control exposure, scanning mirrors (vertical and horizontal, in this example they are closed-loop galvanometer mirrors), a scan lens, a tube lens, a dichromatic mirror, the objective, a collection lens, the photomultiplier tubes, and a CPU to integrate and control the image acquisition process
Mode-locked lasers are well matched to the requirements for efficient 2PE (short pulse width of 50–100 fs and high repetition rates of ~100 MHz). In particular, tunable femtosecond Ti:Al2O3 (titanium:sapphire) lasers are commonly used in most laboratories because the spectral range (690–1,050 nm) is sufficient to excite a wide variety of available fluorophores. Fixed wavelength mode-locked lasers in the 1,000–1,250 nm range (e.g., Nd:YLF, Yb:KYW, or Cr:forsterite) can also be used, for example, to image red-shifted fluorescent proteins [13], and they are more affordable than tunable lasers. Longer wavelengths of ~1,300 nm can be achieved when an optical parametric oscillator (OPO) is coupled to a standard Ti:Al2O3 laser; this configuration may be desirable to achieve deeper tissue penetration [14] or multicolor imaging [12]. Others have reached record depths of imaging using regenerative amplifiers as the excitation source, which lower the laser repetition rate and thereby increase the yield of nonlinear optical processes [10, 15].
In addition to the laser, the main components in a standard 2PE microscope are a beam expander, a fast shutter, scanning mirrors (closed loop or resonant), photomultiplier tubes (PMT), and the different optical components (e.g., scan and tube lenses, reflecting and dichroic mirrors, objective lens), needed to guide the laser beam to the sample and the emitted light to the PMTs (Fig. 2).
2.2. Image Acquisition and Image Processing Tools
For custom microscopes several image acquisition and image processing tools have been developed:
ScanImage [16] is an image acquisition application for controlling laser scanning microscopes. It was developed in MATLAB (The MathWorks, Inc.) and is currently used by >150 laboratories worldwide (https://openwiki.janelia.org/wiki/display/ephus/ScanImage). This versatile software uses simple graphical user interfaces to operate the scan mirrors (scan speed, zoom, rotation) and define the properties of the images to be acquired (e.g., number of slices in a stack, step size), as well as settings to carry out uncaging experiments.
Another helpful application is ImageJ (http://rsbweb.nih.gov/ij/) and some of its plug-in collections (www.macbiophotonics.ca/imagej; http://www.uhnresearch.ca/facilities/wcif/imagej/), which are used for image processing and analysis.
Neurolucida (MicroBrightField, Inc.) and its confocal image stack module is a commercial software that can be used to trace and analyze complete axonal and dendritic arbors of neurons within image stacks acquired with 2PE and extract useful information (e.g., length, branching nodes, branch order, complexity index, etc.) to characterize the neuron imaged. Standard tools for analysis of calcium imaging data (see Subheading 3.4) are still being developed [17], and individual labs use custom routines usually written in MATLAB.
3. Methods
3.1. Making the Brain Visible for 2PE Microscopy: Fluorescent Labeling of Cells and Tissues
Both synthetic dyes and fluorescent proteins are available to image the structure of neurons and glia in the brain (e.g., tracing the dendrites and axons of particular neuronal types) or to record neuronal activity with calcium imaging. Commonly used synthetic dyes for imaging neuronal structure with 2PE include Lucifer yellow, Alexa fluor, DiI, and fluorescein. Intravascular injection of fluorescent dextrans permits the study of blood flow dynamics at the level of capillaries in the superficial layers of the cortex [18-21]. In addition, methoxy X-O4 is a fluorescent compound that crosses the blood-brain barrier and binds to amyloid plaques in mouse models of Alzheimer disease [22]. Synthetic calcium indicator dyes include Fluo-4, Fura-2, and OGB-1. Suforhodamine 101, a red dye that labels glia [23], is commonly used in calcium imaging experiments to distinguish neurons from glia.
A wide array of fluorescent proteins also exists over a rainbow palette of colors that allow an increasing number of applications [24, 25]. For example, EGFP, YFP, mCherry, and td-Tomato are frequently used for imaging neuronal structure with 2PE, while TNXXL, YC3.60, and GCaMP6 have been developed as genetically encoded fluorescent calcium indicator proteins [26-29] (see Note 1). For in vivo imaging of brain cells, specific cell subsets can be labeled with fluorescent proteins through an array of genetic methods (alone or combined). Below we discuss briefly the most commonly used methods for labeling cells for imaging with 2PE:
Filling cells with synthetic dyes: For live imaging of neural structure with 2PE microscopy, cells can be labeled with fluorescent dyes using Diolistics [30], by direct injection of dyes into the brain or by filling them with a particular dye during whole-cell recordings [31]. Similarly, synthetic calcium dyes can be introduced directly into cells or electroporated using a patch pipette [32]. Alternatively, the acetoxy-methyl ester (AM) variants of dyes like Fluo-4 or OGB-1 can be bulk-loaded into the brain, which allows for recording the activity of large ensembles of neurons [33-35].
Transgenic and gene targeting (see Note 2): Mice engineered to express fluorescent proteins under the control of specific promoters (e.g., thyl promoter; [36]) have been used for chronic imaging neuronal structure in vivo for a decade [37, 38]. The use of conditional gene expression (e.g., Cre-loxP, Flp-FRT) and inducible systems (e.g., Tet-ON/Tet-OFF) provides further spatial and temporal control of gene expression [39].
Transfection methods: In the context of live cell imaging with 2PE, nucleic acids (DNA or shRNA constructs) are most commonly introduced in neurons using plasmid electroporation or viral infection. These methods can be used in Cre mice for conditional gene activation to obtain higher labeling specificity [39]. For in vivo electroporation, the delivery of a plasmid into neurons of living mice is achieved by injecting a DNA solution into the brain (or into a single cell with patch-clamp electrophysiology) followed by short electric pulses that permeabilize cell membranes temporarily and allow plasmid entry [40]. For in utero electroporation, the DNA is injected into the cerebral ventricles in mouse embryos in order to target subpopulations of neuron precursors (e.g., layer 2/3 pyramidal neurons of the cerebral cortex) [41-43]. Recombinant viruses (e.g., adenoviruses, lentiviruses, herpes viruses) are increasingly used for gene delivery into neurons, typically via stereotaxic intracranial injections targeted to a particular brain region [44-46].
3.2. Cranial Window for In Vivo 2PE Microscopy
Two different surgical preparations, glass-covered cranial window and thinned skull, have been developed to get optical access to the brain and image structure and functionality of labeled neurons [47-50]. The cranial window preparation requires the removal of a piece of skull (leaving the dura intact), and then the craniotomy is covered with a tiny glass coverslip (Fig. 3). The thinned skull preparation requires mechanical thinning of the superficial layers of the skull, preserving a thin layer of the bone that allows imaging through it. There is also a variant of the thinned skull approach, in which the skull is thinned, polished, and ultimately reinforced with a layer of cyanocrylate glue and a cover glass [51]. With any of these methods, one can achieve sufficient spatial resolution with 2PE microscopy to detect individual dendritic spines or axonal boutons.
Fig. 3.

Cranial window surgery for in vivo imaging with 2PE microscopy. The location of the cranial window is selected based on anatomical landmarks or functional imaging (1). In this case, the window was placed over the left barrel cortex. Under anesthesia, the skull is exposed, and a circular portion of the bone is gently carved with a pneumatic drill (2). Next, the bone flap is removed with small forceps (3) taking care not to damage the underlying meninges and vasculature (4). A glass coverslip is gently placed over the craniotomy (5). The edges of the glass window are sealed with cyanoacrylate glue and dental cement (6). A small well is also made around the window with dental acrylic to accommodate the objective lens and a drop of water for imaging. A titanium bar is then embedded in the dental acrylic (7), which can later be used to attach the mouse on to the microscope stage. The cortical vasculature can be seen under the microscope objective (7). Please refer to Mostany R and C Portera-Cailliau (2008) for a video of the procedure
The main advantage of the glass-covered cranial window method is that it allows for multiple imaging sessions to be conducted for longitudinal imaging of the same neuronal or glial processes within large fields of view. The cranial window approach (but not the thin skull preparation) also makes it possible to perform certain manipulations of the brain before sealing the craniotomy with a cover glass (e.g., electrode implantation, direct intracortical pharmacology, bolus loading of calcium dyes, viral injections). In addition, craniotomies are necessary for implanting gradient refractive index lenses (GRIN) in experiments that require imaging of deep brain structures [52] (see Note 3). The main drawback of the cranial window method is that it is more technically demanding as only the best preparations remain optically transparent for weeks or months, and even the slightest perturbation of the dura mater will cause a quick worsening of the quality of the imaging. Because the skull is never breached with the thinned skull technique, the incidence of infection and inflammation is minimal, and the success rate is much higher [53]. One downside of transcranial imaging is that skull thinning, which has to be repeated before every imaging session, can only been done a limited number of times (<3), or else the quality of imaging deteriorates. In this regard, the polished and reinforced preparation avoids the bone inflammation that results from repeated thinning [51].
3.3. Imaging Neuronal Structure with 2PE Microscopy
The structure and function of different cell types in the brain that express fluorescent dyes or proteins can be imaged in vivo using 2PE microscopy allowing the study of dynamic anatomical and functional changes as a result of learning and memory or in response to sensory inputs from the environment, or how they compensate for or degenerate in disease. The extraordinary resolution of the images acquired with 2PE microscopy makes it possible to observe tiny neuronal structures, such as spines and axonal boutons (~1 μm in diameter), and even quantify the turnover and trafficking of synaptic proteins (e.g., PSD95, Ras) within these structures [13, 54]. In the last decade, several studies that imaged synaptic structure in vivo with 2PE were able to record changes in synapses that were either associated with sensory experience and learning motor tasks (reviewed in [55, 56]), or triggered by stroke [18]. Chronic 2PE microscopy in vivo can also be used to image neuroglial [57] and neurovascular [51] interactions, blood flow dynamics [21, 19], and amyloid plaques [58, 59], among others. By imaging neurons in the intact brain of living mice, such longitudinal, high-resolution imaging studies of dendritic and axonal segments have provided valuable information about dynamic aspects of synapses that could not previously be examined using histological studies in fixed tissue (Fig. 4).
Fig. 4.

Chronic high-resolution imaging of dendritic spines with in vivo 2PE microscopy. High-resolution images of dendritic spines acquired with in vivo 2PE microscopy before and after stroke. The day of imaging is shown in the lower right-hand corner. Shown is an apical dendritic segment from a layer 5 pyramidal neuron in peri-infarct cortex before (green) and after (red) unilateral permanent middle cerebral artery occlusion (MCAO) in mice. All images are maximum intensity projections (4–7 slices, 1.5 μm apart). A few examples of always present spines (yellow arrowheads), gained spines (green arrowheads), and lost spines (red arrowheads) are shown. Blue asterisks at +4 d post MCAO denote transient dendritic swelling after stroke. See Mostany et al. [18] for details
3.4. Recording Neuronal Signals with 2PE Microscopy
3.4.1. Calcium Imaging
When neurons fire action potentials, the intracellular concentration of calcium ions rises. One can record neuronal activity optically by using fluorescent dyes (or proteins) that respond to binding of calcium by changing their spectral properties (e.g., their fluorescent intensity increases or decreases, or their excitation/emission spectra change). Calcium imaging using 2PE microscopy is an ideal tool for interrogating large ensembles of neurons in the intact brain [32] because it offers advantages over traditional single unit recordings using microelectrodes. First, with calcium imaging one can record signals from hundreds (potentially thousands) of neurons simultaneously. When combined with mouse genetics to label individual subpopulations of neurons, one can also be certain of which cell types the calcium signals are being recorded from. Second, calcium imaging is less invasive, and circuits can be recorded without penetrating electrodes that might disrupt normal activity.
For calcium imaging, one can use 2PE microscopy to record calcium transients in single dendritic spines [60, 61] or to monitor the spontaneous or evoked activity of large ensembles of neurons simultaneously with single cell resolution [62-64] (Fig. 5).
Fig. 5.

Calcium imaging of neuronal ensemble activity with 2PE microscopy. (a) Typical field of view of layer 2/3 neurons (green) and glia (yellow) stained with the calcium indicator dye Oregon Green BAPTA-1AM and imaged with in vivo 2PE microscopy. Sulforhodamine 101 (a red dye) was used to stain glia. The image is a single frame (~120 μm below the dura) in a representative calcium imaging movie (3 min, 3.9 frames per second) from a 14-day-old mouse. (b) Calcium traces showing the relative changes in fluorescence intensity over the baseline fluorescence (ΔF/F) of 5 different layer 2/3 neurons from a representative calcium movie. The upward deflections represent spiking events within those neurons
Unfortunately, two-photon calcium imaging suffers from important drawbacks, such as poor temporal resolution and low signal-to-noise ratio [17, 65]. Newer generation genetically encoded calcium indicators with improved signal-to-noise will overcome some of these limitations. For example, although synthetic indicators are superior for detecting single spikes, some of the newer genetically encoded indicators (YC3.60, GCaMP6f) may detect action potentials quite reliably [66]. Importantly, recent developments in faster scanning (e.g., acousto-optical deflectors), parallelization of 2PE (e.g., multifocal multiphoton microscopy), and improved photodetectors suggest that over the next decade optical probing of neural activity with calcium imaging could eventually be an excellent alternative to electrophysiology, offering a less invasive approach to record action potential firing in large ensembles of identifiable neurons in three dimensions (see Subheading 4, step 2).
3.4.2. Voltage-Sensing Proteins
Another way to optically probe neuronal activity is to image changes in membrane potential. For this purpose several voltage-sensing proteins (e.g., FLaSh, VSFP2, SPARC, Flare, Opto-patch) have been designed that work well with 2PE microscopy and could in theory be useful to monitor the activity of thousands of individual neurons simultaneously [67-69]. These sensors are usually FRET based and can report both subthreshold changes in membrane potential and spiking activity of neurons. Unfortunately, most current approaches for voltage sensing with genetically encoded or synthetic indicators have poor spatial resolution and very low signal-to-noise ratio, requiring the averaging of many stimuli to detect responses.
3.5. Combining 2PE Microscopy with Optogenetics, Glutamate Uncaging
3.5.1. Optogenetics
Another advantage of 2PE microscopy is that it can easily be combined with optogenetics and be used to uncage neurotransmitters for temporally and spatially precise manipulation and probing of neuronal activity within intact neural circuits.
This technique enables researchers to silence or stimulate genetically specified classes of neurons (or other electrically excitable cells) with exquisite temporal precision using light-sensitive molecules [70, 71]. For example, channelrhodopsin-2 (ChR2) is a light-activated cation channel that depolarizes neurons, whereas halorhodopsin (NphR) and archaerhodopsin-3 (Arch) are a chloride channel and an outward proton pump, respectively, that enable almost complete silencing of neurons. Using optogenetic tools, cell type-specific and minimally invasive photostimulation has revealed causal relationships between activity of neuronal populations and animal behavior. In addition to evoked behaviors, electrophysiological recordings (e.g., patch-clamp or “optrodes”) [72] or calcium imaging [73] can be used as the readout of neuronal activity. Thus, by combining 2PE microscopy with calcium dyes or voltage-sensitive dyes, subsets of neurons expressing optogenetic sensors could be specifically modulated with light to reveal and mimic patterns of connectivity with phenomenal temporal and spatial precision [74]. In the future, as improved red-shifted calcium indicator dyes become available, it will be easier to combine optogenetics with two-photon calcium imaging. Although optogenetic manipulations are by enlarge done with single-photon excitation, 2PE of ChR2 is possible in vivo [75], and this option may be preferable when precise stimulation of individual synapses or portions of circuits is desired.
3.5.2. Two-Photon Uncaging
The photorelease of caged, biochemically inert effector molecules (e.g., nucleotide, neurotransmitter, second messenger) can be used to control molecular interactions in cells [76]. With two-photon uncaging, the absorption energy is used to release the caged molecule, typically a neurotransmitter, from its protective chemical group, in order to mimic normal synaptic activity. The photoactivation of caged neurotransmitters has achieved synapse-specific resolution with the use of 2PE in brain tissue. By combining 2PE and uncaging (e.g., of MNI-glutamate), postsynaptic receptors of individual spines can be stimulated with excellent temporal and spatial resolution while structural and activity changes are assessed. These techniques have been successfully applied to the study of postsynaptic signaling and local circuit mapping at the level of individual dendritic spines [77-79]. An exciting recent advance was the successful stimulation of identified dendritic spines in vivo, in adult mice, using two-photon uncaging [80].
3.6. Future Directions
The invention of 2PE microscopy has revolutionized fluorescence imaging over the last 2 decades, thanks to the vision of physicists, chemists, engineers, and biologists working towards a common goal. Over the next few years, we will continue to witness unprecedented advances that lead to additional improvements in this powerful tool. In particular, developments that will enhance our ability to image deeper in the brain and with better temporal resolution in awake behaving mice will be especially useful.
3.6.1. Deeper and Brighter Imaging
Light scattering of both the near-infrared excitation wavelengths used in 2PE microscopy and of the emitted fluorescence imposes limits on how deep one can image [81]. As a result, in vivo calcium imaging with conventional 2PE has been restricted to the superficial layers of the neocortex. A variety of approaches have been established to increase the depth range for in vivo calcium imaging, including the use of high numerical aperture objectives and longer wavelengths (e.g., with fixed-wavelength laser sources or OPOs), using adaptive optics and wavefront optimization to correct for spherical aberrations and lensing effects [82, 83] or implementing ultrashort-pulsed regenerative amplifiers [15]. An alternative approach to deeper imaging is to improve on the collection of scattered fluorescent emissions that are not transmitted through the objective and would otherwise be a wasted signal, for example, by using a ring of optical fibers around the objective [84]. A different approach entirely is to use microendoscopes to reach deep brain structures, such as the hippocampus [52, 85]. In addition, signal-to-noise ratios will improve as better fluorophores become available. For calcium imaging this is a critical issue, because when faster scanning is implemented (see below), the pixel dwell time of the laser will be reduced, resulting in less signal from the indicator. Major efforts are underway to design better genetically encoded calcium indicators to solve these problems [28].
3.6.2. Faster Imaging
A major problem with calcium imaging is that it is hard to relate changes in fluorescence of the indicator molecule to neuronal spiking both in terms of determining the exact numbers of action potentials and especially the timing between them. This is due to limitations in the signal-to-noise ratio of most indicators (see above) and in the speed of acquisition. Recent solutions have been put forward to improve image acquisition speed. For example, spatiotemporal multiplexing of multibeam scanning with 2PE can improve temporal resolution and make it possible to record neurons at different depths simultaneously [86]. Alternatively, one can record only useful signals from cell bodies using targeted path scanning [87] or acousto-optical deflectors [88, 89]. Lastly, there is considerable effort in the field to shape the excitation beam to minimize or eliminate scanning, in order to rapidly excite large numbers of cells in parallel without significantly compromising the pixel dwell time. These novel geometries include the use of Bessel beams [90] and light sheets [91] to achieve thin 2PE planes perpendicular to the emission path or the implementation of spatial light modulators that shape the laser beam into an arbitrary light pattern, which allows for simultaneous imaging (or uncaging) at different locations [92] (see Note 4). Further developments in years to come will no doubt bring us closer to our goal of being able to record, with millisecond precision, the firing of thousands of neurons, distributed over a volume of brain tissue in behaving animals.
3.6.3. 2PE Microscopy in Behaving Animals
An important goal of neuroscientists is to link changes in the structure and function of neuronal circuits to changes in behavior. It is therefore critically important to study these phenomena in the intact behaving animal. In the case of calcium imaging of neuronal activity, this has been accomplished by recording in head-restrained mice while they navigate virtual environments [93] or by miniaturizing 2PE microscopes into portable devices that allow investigators to image freely moving animals [84, 94].
4. Notes
GECIs come in two varieties: single-fluorophore sensors and sensors involving FRET between two proteins. A good example of single-fluorophore GECIs is the GCaMP family of sensors, which is composed of a circularly permuted EGFP molecule that is flanked on one side by the calcium-binding protein calmodulin and on the other by the calmodulin-binding peptide M13. FRET-based sensors (e.g., the yellow cameleon dye YC 3.60) require a nonradiative energy transfer between an excited donor fluorophore (e.g., enhanced cyan fluorescent protein) and an acceptor fluorophore (e.g., circularly permuted Venus protein). The two fluorophores are connected by a linker sequence that is composed of a calcium-binding protein (e.g., calmodulin-M13, troponin); upon calcium binding, a conformational change in the linker protein brings the distance between the donor and acceptor fluorescent proteins to less than 10 nm, which is necessary for FRET to occur. This is reflected by a decrease in the fluorescence intensity of the donor protein (blue) and an increase in the acceptor fluorescence (green), and the calcium signal is therefore expressed as the ratio of the two.
Additional information on specific gene expression patterns in available mouse lines can be found in Allen Brain Atlas (http://www.alleninstitute.org), Genepaint (http://www.genepaint.org), GENSAT (http://www.gensat.org), or The Jackson Laboratory (http://jaxmice.jax.org).
The microendoscopy approach to image deep structures relies on the use of GRIN lenses, which use internal variations in the refractive index (as opposed to curved refractive surfaces of conventional lenses) to guide light. In essence, GRIN lenses act as an optical relay that projects the scanning pattern of the 2PE microscope to a focal plane deep inside the tissue sample. A wide array of microendoscopes is available with varying physical length, optical working distance and numerical apertures, and field of views. An important shortcoming of microendoscopy is that it is associated with some degree of tissue damage because it relies on the insertion of a GRIN lens into the brain.
Unfortunately, because these light-sheet approaches are currently limited in their depth penetration, it will be challenging to adapt them for live imaging in the intact rodent brain.
Acknowledgments
This work was supported by the Stein Oppenheimer Endowment Award and by grants from the US National Institutes of Health (5R01HD054453 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development and 5RC1NS068093 from the National Institute of Neurological Disorders and Stroke).
References
- 1.Miyawaki A (2011) Proteins on the move: insights gained from fluorescent protein technologies. Nat Rev Mol Cell Biol 12:656–668 [DOI] [PubMed] [Google Scholar]
- 2.Eigen M, Rigler R (1994) Sorting single molecules: application to diagnostics and evolutionary biotechnology. Proc Natl Acad Sci U S A 91:5740–5747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilt BA, Burns LD, Wei Ho ET et al. (2009) Advances in light microscopy for neuroscience. Annu Rev Neurosci 32:435–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hell SW, Wichmann J (1994) Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 19:780–782 [DOI] [PubMed] [Google Scholar]
- 5.Hell SW (2003) Toward fluorescence nanoscopy. Nat Biotechnol 21:1347–1355 [DOI] [PubMed] [Google Scholar]
- 6.Sigrist SJ, Sabatini BL (2012) Optical super-resolution microscopy in neurobiology. Curr Opin Neurobiol 22:86–93 [DOI] [PubMed] [Google Scholar]
- 7.Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248:73–76 [DOI] [PubMed] [Google Scholar]
- 8.Svoboda K, Yasuda R (2006) Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron 50:823–839 [DOI] [PubMed] [Google Scholar]
- 9.Denk W, Svoboda K (1997) Photon upmanship: why multiphoton imaging is more than a gimmick. Neuron 18:351–357 [DOI] [PubMed] [Google Scholar]
- 10.Theer P, Hasan MT, Denk W (2003) Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett 28:1022–1024 [DOI] [PubMed] [Google Scholar]
- 11.Nikolenko V, Nemet B, Yuste R (2003) A two-photon and second-harmonic microscope. Methods 30:3–15 [DOI] [PubMed] [Google Scholar]
- 12.Entenberg D, Wyckoff J, Gligorijevic B et al. (2011) Setup and use of a two-laser multiphoton microscope for multichannel intravital fluorescence imaging. Nat Protoc 6:1500–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray NW, Weimer RM, Bureau I et al. (2006) Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol 4:e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobat D, Durst ME, Nishimura N et al. (2009) Deep tissue multiphoton microscopy using longer wavelength excitation. Nat Protoc 17:13354–13364 [DOI] [PubMed] [Google Scholar]
- 15.Mittmann W, Wallace DJ, Czubayko U et al. (2011) Two-photon calcium imaging of evoked activity from L5 somatosensory neurons in vivo. Nat Neurosci 14:1089–1093 [DOI] [PubMed] [Google Scholar]
- 16.Pologruto TA, Sabatini BL, Svoboda K (2003) ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grewe BF, Helmchen F (2009) Optical probing of neuronal ensemble activity. Curr Opin Neurobiol 19(5):520–529 [DOI] [PubMed] [Google Scholar]
- 18.Mostany R, Chowdhury TG, Johnston DG et al. (2010) Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci 30:14116–14126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mostany R, Portera-Cailliau C (2008) A method for 2-photon imaging of blood flow in the neocortex through a cranial window. J Vis Exp. (12). pii: 678. doi: 10.3791/678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaffer CB, Friedman B, Nishimura N et al. (2006) Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol 4:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleinfeld D, Mitra PP, Helmchen F et al. (1998) Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci U S A 95:15741–15746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klunk WE, Bacskai BJ, Mathis CA et al. (2002) Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61: 797–805 [DOI] [PubMed] [Google Scholar]
- 23.Nimmerjahn A, Kirchhoff F, Kerr JN et al. (2004) Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 1:31–37 [DOI] [PubMed] [Google Scholar]
- 24.Shaner NC, Steinbach PA, Tsien RY (2005) A guide to choosing fluorescent proteins. Nat Methods 2:905–909 [DOI] [PubMed] [Google Scholar]
- 25.Chudakov DM, Matz MV, Lukyanov S et al. (2010) Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90:1103–1163 [DOI] [PubMed] [Google Scholar]
- 26.Lutcke H, Murayama M, Hahn T et al. (2010) Optical recording of neuronal activity with a genetically-encoded calcium indicator in anesthetized and freely moving mice. Front Neural Circ 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian L, Hires SA, Mao T et al. (2009) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods 6:875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Looger LL, Griesbeck O (2012) Genetically encoded neural activity indicators. Curr Opin Neurobiol 22:18–23 [DOI] [PubMed] [Google Scholar]
- 29.Mank M, Santos AF, Direnberger S et al. (2008) A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat Methods 5:805–811 [DOI] [PubMed] [Google Scholar]
- 30.Gan WB, Grutzendler J, Wong WT et al. (2000) Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron 27:219–225 [DOI] [PubMed] [Google Scholar]
- 31.Portera-Cailliau C, Pan DT, Yuste R (2003) Activity-regulated dynamic behavior of early dendritic protrusions: evidence for different types of dendritic filopodia. J Neurosci 23:7129–7142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grienberger C, Konnerth A (2012) Imaging calcium in neurons. Neuron 73:862–885 [DOI] [PubMed] [Google Scholar]
- 33.Garaschuk O, Milos R, Konnerth A (2006) Targeted bulk-loading of fluorescent indicators for two-photon brain imaging in vivo. Nat Protoc 1:380–386 [DOI] [PubMed] [Google Scholar]
- 34.Golshani P, Portera-Cailliau C (2008) In vivo 2-photon calcium imaging in layer 2/3 of mice. J Vis, Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacLean J, Yuste R (2005) Imaging action potentials with calcium indicators: practical guide. In: Yuste R, Konnerth A (eds) Imaging neurons a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp 351–355 [Google Scholar]
- 36.Feng G, Mellor RH, Bernstein M et al. (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51 [DOI] [PubMed] [Google Scholar]
- 37.Grutzendler J, Kasthuri N, Gan WB (2002) Long-term dendritic spine stability in the adult cortex. Nature 420:812–816 [DOI] [PubMed] [Google Scholar]
- 38.Trachtenberg JT, Chen BE, Knott GW et al. (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794 [DOI] [PubMed] [Google Scholar]
- 39.Jefferis GS, Livet J (2012) Sparse and combinatorial neuron labelling. Curr Opin Neurobiol 22:101–110 [DOI] [PubMed] [Google Scholar]
- 40.Judkewitz B, Rizzi M, Kitamura K et al. (2009) Targeted single-cell electroporation of mammalian neurons in vivo. Nat Protoc 4:862–869 [DOI] [PubMed] [Google Scholar]
- 41.Saito T, Nakatsuji N (2001) Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol 240:237–246 [DOI] [PubMed] [Google Scholar]
- 42.Dixit R, Lu F, Cantrup R et al. (2011) Efficient gene delivery into multiple CNS territories using in utero electroporation. J Vis Exp. (52). pii: 2957. doi: 10.3791/2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cruz-Martin A, Crespo M, Portera-Cailliau C (2010) Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30:7793–7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atasoy D, Aponte Y, Su HH et al. (2008) A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci 28:7025–7030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lowery RL, Majewska AK (2010) Intracranial injection of adeno-associated viral vectors. J Vis Exp. (45). pii: 2140. doi: 10.3791/2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schultz BR, Chamberlain JS (2008) Recombinant adeno-associated virus transduction and integration. Mol Ther 16:1189–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holtmaat A, Bonhoeffer T, Chow DK et al. (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4:1128–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mostany R, Portera-Cailliau C (2008) A craniotomy surgery procedure for chronic brain imaging. J Vis Exp. (12). pii: 680. doi: 10.3791/680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cruz-Martin A, Portera-Cailliau C (2010) In vivo imaging of axonal and dendritic structures in developing cortex. In: Sharpe J, Wong R (eds) Imaging in developmental biology: a laboratory manual, 1st edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang G, Pan F, Parkhurst CN et al. (2010) Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc 5:201–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drew PJ, Shih AY, Driscoll JD et al. (2010) Chronic optical access through a polished and reinforced thinned skull. Nat Methods 7:981–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung JC, Mehta AD, Aksay E et al. (2004) In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J Neurophysiol 92:3121–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu HT, Pan F, Yang G et al. (2007) Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci 10:549–551 [DOI] [PubMed] [Google Scholar]
- 54.Yasuda R, Harvey CD, Zhong H et al. (2006) Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci 9:283–291 [DOI] [PubMed] [Google Scholar]
- 55.Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658 [DOI] [PubMed] [Google Scholar]
- 56.Yu X, Zuo Y (2011) Spine plasticity in the motor cortex. Curr Opin Neurobiol 21:169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318 [DOI] [PubMed] [Google Scholar]
- 58.Spires TL, Meyer-Luehmann M, Stern EA et al. (2005) Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci 25:7278–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai J, Grutzendler J, Duff K et al. (2004) Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci 7:1181–1183 [DOI] [PubMed] [Google Scholar]
- 60.Sabatini BL, Svoboda K (2000) Analysis of calcium channels in single spines using optical fluctuation analysis. Nature 408:589–593 [DOI] [PubMed] [Google Scholar]
- 61.Chen X, Leischner U, Rochefort NL et al. (2011) Functional mapping of single spines in cortical neurons in vivo. Nature 475:501–505 [DOI] [PubMed] [Google Scholar]
- 62.Cossart R, Aronov D, Yuste R (2003) Attractor dynamics of network UP states in the neocortex. Nature 423:283–288 [DOI] [PubMed] [Google Scholar]
- 63.Kerr JN, Greenberg D, Helmchen F (2005) Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A 102:14063–14068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohki K, Chung S, Ch'ng YH et al. (2005) Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature 433:597–603 [DOI] [PubMed] [Google Scholar]
- 65.Gobel W, Helmchen F (2007) In vivo calcium imaging of neural network function. Physiology 22:358–365 [DOI] [PubMed] [Google Scholar]
- 66.Hendel T, Mank M, Schnell B et al. (2008) Fluorescence changes of genetic calcium indicators and OGB-1 correlated with neural activity and calcium in vivo and in vitro. J Neurosci 28:7399–7411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perron A, Mutoh H, Akemann W et al. (2009) Second and third generation voltage-sensitive fluorescent proteins for monitoring membrane potential. Front Neural Circ 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahrens KF, Heider B, Lee H et al. (2012) Two-photon scanning microscopy of in vivo sensory responses of cortical neurons genetically encoded with a fluorescent voltage sensor in rat. Front Neural Circ 6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chanda B, Blunck R, Faria LC et al. (2005) A hybrid approach to measuring electrical activity in genetically specified neurons. Nat Neurosci 8:1619–1626 [DOI] [PubMed] [Google Scholar]
- 70.Fenno L, Yizhar O, Deisseroth K (2011) The development and application of optogenetics. Annu Rev Neurosci 34:389–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bernstein JG, Garrity PA, Boyden ES (2012) Optogenetics and thermogenetics: technologies for controlling the activity of targeted cells within intact neural circuits. Curr Opin Neurobiol 22:61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deisseroth K (2011) Optogenetics. Nat Methods 8:26–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang YP, Oertner TG (2007) Optical induction of synaptic plasticity using a light-sensitive channel. Nat Methods 4:139–141 [DOI] [PubMed] [Google Scholar]
- 74.Peron S, Svoboda K (2011) From cudgel to scalpel: toward precise neural control with optogenetics. Nat Methods 8:30–34 [DOI] [PubMed] [Google Scholar]
- 75.Papagiakoumou E, Anselmi F, Begue A et al. (2010) Scanless two-photon excitation of channelrhodopsin-2. Nat Methods 7:848–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ellis-Davies GC (2009) Basics of photoactivation. Cold Spring Harb Protoc. pdb top55. [DOI] [PubMed] [Google Scholar]
- 77.Matsuzaki M, Ellis-Davies GC, Nemoto T et al. (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4:1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sobczyk A, Scheuss V, Svoboda K (2005) NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci 25:6037–6046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ashby MC, Isaac JT (2011) Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron 70:510–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Noguchi J, Nagaoka A, Watanabe S et al. (2011) In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J Physiol 589:2447–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oheim M, Beaurepaire E, Chaigneau E et al. (2001) Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J Neurosci Methods 111:29–37 [DOI] [PubMed] [Google Scholar]
- 82.Rueckel M, Mack-Bucher JA, Denk W (2006) Adaptive wavefront correction in two-photon microscopy using coherence-gated wavefront sensing. Proc Natl Acad Sci U S A 103:17137–17142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Booth MJ (2007) Adaptive optics in microscopy. Philos Transact A Math Phys Eng Sci 365:2829–2843 [DOI] [PubMed] [Google Scholar]
- 84.Engelbrecht CJ, Gobel W, Helmchen F (2009) Enhanced fluorescence signal in nonlinear microscopy through supplementary fiber-optic light collection. Opt Express 17:6421–6435 [DOI] [PubMed] [Google Scholar]
- 85.Flusberg BA, Cocker ED, Piyawattanametha W et al. (2005) Fiber-optic fluorescence imaging. Nat Methods 2:941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cheng A, Goncalves JT, Golshani P et al. (2011) Simultaneous two-photon calcium imaging at different depths with spatiotemporal multiplexing. Nat Methods 8:139–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lillis KP, Eng A, White JA et al. (2008) Two-photon imaging of spatially extended neuronal network dynamics with high temporal resolution. J Neurosci Methods 172:178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grewe BF, Langer D, Kasper H et al. (2010) High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat Methods 7:399–405 [DOI] [PubMed] [Google Scholar]
- 89.Duemani Reddy G, Kelleher K, Fink R et al. (2008) Three-dimensional random access multiphoton microscopy for functional imaging of neuronal activity. Nat Neurosci 11: 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Planchon TA, Gao L, Milkie DE et al. (2011) Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat Methods 8:417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Truong TV, Supatto W, Koos DS et al. (2011) Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat Methods 8:757–760 [DOI] [PubMed] [Google Scholar]
- 92.Nikolenko V, Watson BO, Araya R et al. (2008) SLM microscopy: scanless two-photon imaging and photostimulation with spatial light modulators. Front Neural Circ 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dombeck DA, Harvey CD, Tian L et al. (2010) Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci 13:1433–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kerr JN, Nimmerjahn A (2012) Functional imaging in freely moving animals. Curr Opin Neurobiol 22:45–53 [DOI] [PubMed] [Google Scholar]