

Summary
Type I and II interferons (IFNs) stimulate pro-inflammatory programs critical for immune activation, but also induce immune-suppressive feedback circuits that impede control of cancer growth. Here, we sought to determine how these opposing programs are differentially induced. We demonstrated that the transcription factor Interferon Regulatory Factor 2 (IRF2) was expressed by many immune cells in the tumor in response to sustained IFN signaling. CD8+ T cell-specific deletion of IRF2 prevented acquisition of the T cell exhaustion program within the tumor, and instead enabled sustained effector functions that promoted long-term tumor control, and increased responsiveness to immune-checkpoint and adoptive cell therapies. The long-term tumor control by IRF2-deficient CD8+ T cells required continuous integration of both IFN-I and IFN-II signals. Thus, IRF2 is a foundational feedback molecule that redirects IFN signals to suppress T cell responses and represents a potential target to enhance cancer control.
Keywords: interferon regulatory factor 2, IRF2, type I interferon, interferon gamma, CD8+ T cells, T cell exhaustion, cancer, immunotherapy, adoptive cell transfer, CyTOF
Graphical Abstract
How interferons switch from pro-inflammatory to immunosuppressive programs that facilitate cancer growth has remained enigmatic. Lukhele et al. establish that the transcription factor Interferon Regulatory Factor 2 attenuates and redirects interferon signalling to program CD8+ T cell exhaustion to avert tumor control.
Introduction
Type I interferons (IFN-I; IFNα/β) and type II interferon (IFN-II, IFNγ) have long been recognized for their immune stimulatory activities (Alspach et al., 2019; Lukhele et al., 2019). Yet, IFN-Is and IFN-II are also emerging as central regulators of both the chronic immune activation and the suppression that drive cancer progression (Boukhaled et al., 2021; Snell et al., 2017). The efficacy of many types of anti-cancer therapies, including checkpoint blockades, are associated with increased IFN signaling (Boukhaled et al., 2021; Snell et al., 2017). However, elevated and sustained IFN-I signaling also drives T cell exhaustion (Boukhaled et al., 2021; Budhwani et al., 2018; Minn, 2015; Snell et al., 2017), a process culminating in the attenuation of CD8+ T cell function through distinct transcriptional, epigenetic and metabolic reprogramming (McLane et al., 2019; Wherry, 2011). In addition, IFN-II increases PDL1 expression on the surface of tumor cells. PDL1 binds to PD1 on activated CD8+ T cells in the tumor microenvironment (TME), driving their apoptotic cell death (Dong et al., 2002). Thus, a paradox emerges wherein IFN signaling is critical for the induction and maintenance of CD8+ T cell activity, but simultaneously abolishes that same activity. Yet, how IFNs exert these distinct functions and the CD8+ T cell intrinsic pathways they induce to reroute proinflammatory to suppressive signals remain ill-defined.
IFN-Is signal through a dimeric IFNAR1/IFNAR2 receptor that activates the kinases Jak1 and Tyk2 to initiate STAT1 and STAT2 phosphorylation (among other pathways) to induce expression of hundreds of IFN-I stimulated genes (ISGs), including interferon regulatory factors (IRFs) (Lukhele et al., 2019). On the other hand, IFN-II signals through its IFNγ receptor (composed of IFNγR1 and IFNγR2 chains) activate Jak1 and Jak2 that, in turn, phosphorylate STAT1 (Alspach et al., 2019). STAT1 then homodimerizes to form gamma-activated factors that translocate into the nucleus to induce ISG expression including the IRFs. By differentially inducing and antagonizing IRFs, IFN-I and IFN-II signaling triggers a broad range of immunologic programs (Antonczyk et al., 2019). Central to these outcomes is the interplay between IRF1 and IRF2, known for their positive and negative regulation, respectively, of IFN-I and IFN-II signaling. IRF1 is activated by IFN-II [as well as IFN-I and nuclear factor-kappa β (NF-κβ)] to induce the pro-inflammatory and immune stimulatory functions critical to prevent tumor growth (Drew et al., 1995a; Harada et al., 1993). IRF2 is constitutively expressed in many immune cells and is upregulated in response to either IFN-I or IFN-II (Harada et al., 1989; Taniguchi and Takaoka, 2001). Although the exact functions of IRF2 are mechanistically unclear, one important role is limiting immune activation and resultant autoimmunity (Hida et al., 2000). IRF2 antagonizes IRF1 by competing for binding to the same promoter elements of IFN-I and IFN-II-inducible genes (Harada et al., 1989), and by inhibiting nuclear translocation of IRF1 (Wang et al., 2007). IRF2 also interacts with NF-κβ (Chae et al., 2008; Drew et al., 1995b), STAT1 (Rouyez et al., 2005), IRF8 (Bovolenta et al., 1994; Sharf et al., 1995), and IRF9 (Hida et al., 2000; Tanaka et al., 1993), factors that influence the ability of immune cells to control tumors. While the transcriptional role of IRF2 is largely repressive, IRF2 activates gene transcription in certain contexts (Vaughan et al., 1995; Vaughan et al., 1998; Yamamoto et al., 1994), for example, IRF2 cooperates with IRF1 to induce TLR3 in HeLa cells (Ren et al., 2015). Thus, IRF2 balances IFN stimulation by differentially inducing and antagonizing key transcriptional regulators.
Increased IRF2 expression by tumor cells themselves generally correlates with development and progression of many human cancers, potentially through repressing cancer cell intrinsic IFN signaling (Chen et al., 2021a; Mei et al., 2017; Sakai et al., 2014; Wang et al., 2007; Yi et al., 2013). In high IRF2 expressing tumors such as esophageal cancers, IRF2 promotes tumor survival by inhibiting transcription of the IFNγR, thereby enhancing tumor-intrinsic resistance to IFN-II (Wang et al., 2008). Conversely, some tumor types downregulate IRF2 to evade immune targeting. For example, IRF2 directly represses PDL1 expression and activates components of the MHC-I pathway, both of which increase susceptibility to T cell mediated killing (Kriegsman et al., 2019; Yan et al., 2020). Overall, these studies underscore an important role of IRF2 expression by tumor cells themselves, however, how IRF2 expression in the immune cell compartment influences their responses to IFN-I and II (collectively referred to as IFNs) and ability to control tumors is unknown.
Herein, we identified IRF2 as a central regulator of CD8+ T cell exhaustion in cancer. Deletion of IRF2 intrinsically programmed CD8+ T cells to resist exhaustion and maintain anti-tumor functions in the otherwise suppressive TME, thereby enabling long-term tumor control and increased responsiveness to immune checkpoint blockade. Compared to IRF2-sufficient CD8+ T cells, adoptive transfer of IRF2-deficient CD8+ T cells provided superior ability to control established tumors. In the absence of IRF2, the CD8+ T cell exhaustion signature normally observed within the tumor was instead replaced with a program of functional cytotoxic T cells. Similarly, the suppressive effects of IFN-I and IFN-II signaling were prevented in IRF2-deficient CD8+ T cells, and instead IFNs enhanced and sustained CD8+ T cell function for long-term tumor control. Thus, IRF2 is a CD8+ T cell-intrinsic nexus that translates signals from the inflammatory TME to adjust gene expression, attenuate cell activation and transcriptionally program T cell exhaustion to prevent tumor control.
Results
IRF2 is expressed across immune subsets and its deficiency enables tumor control
To determine the immune cell expression of IRF2 within the TME, we subcutaneously (s.c.) inoculated IRF2-sufficient wild-type (WT) mice with MC38 colorectal adenocarcinoma cells and isolated total tumor-infiltrating cells 14 days after tumor initiation. Cells were then analyzed by mass cytometry (CyTOF) with a panel identifying all major and most minor immune cell populations (Table S1). IRF2 was widely expressed across immune cells from mouse MC38 tumors (Fig 1A). Levels of IRF2 were minimally changed in tumor-infiltrating B cells, macrophages and dendritic cells (DCs) compared to their splenic counterparts, with the spleen being largely immunologically unaffected by the MC38 tumor growth. In contrast, CD4+ and CD8+ T tumor-infiltrating lymphocytes (TILs) showed significant IRF2 upregulation compared to their splenic counterparts (Fig 1B), suggestive that IRF2 levels in T cells are increased within the TME. Similarly, human melanoma tumor-infiltrating immune cells possessed a broad IRF2 expression pattern (Fig 1C), indicating conserved immune-wide IRF2 expression within the mouse and human TME.
Figure 1. IRF2 expression across immune subsets and IRF2 deficiency enables tumor control.
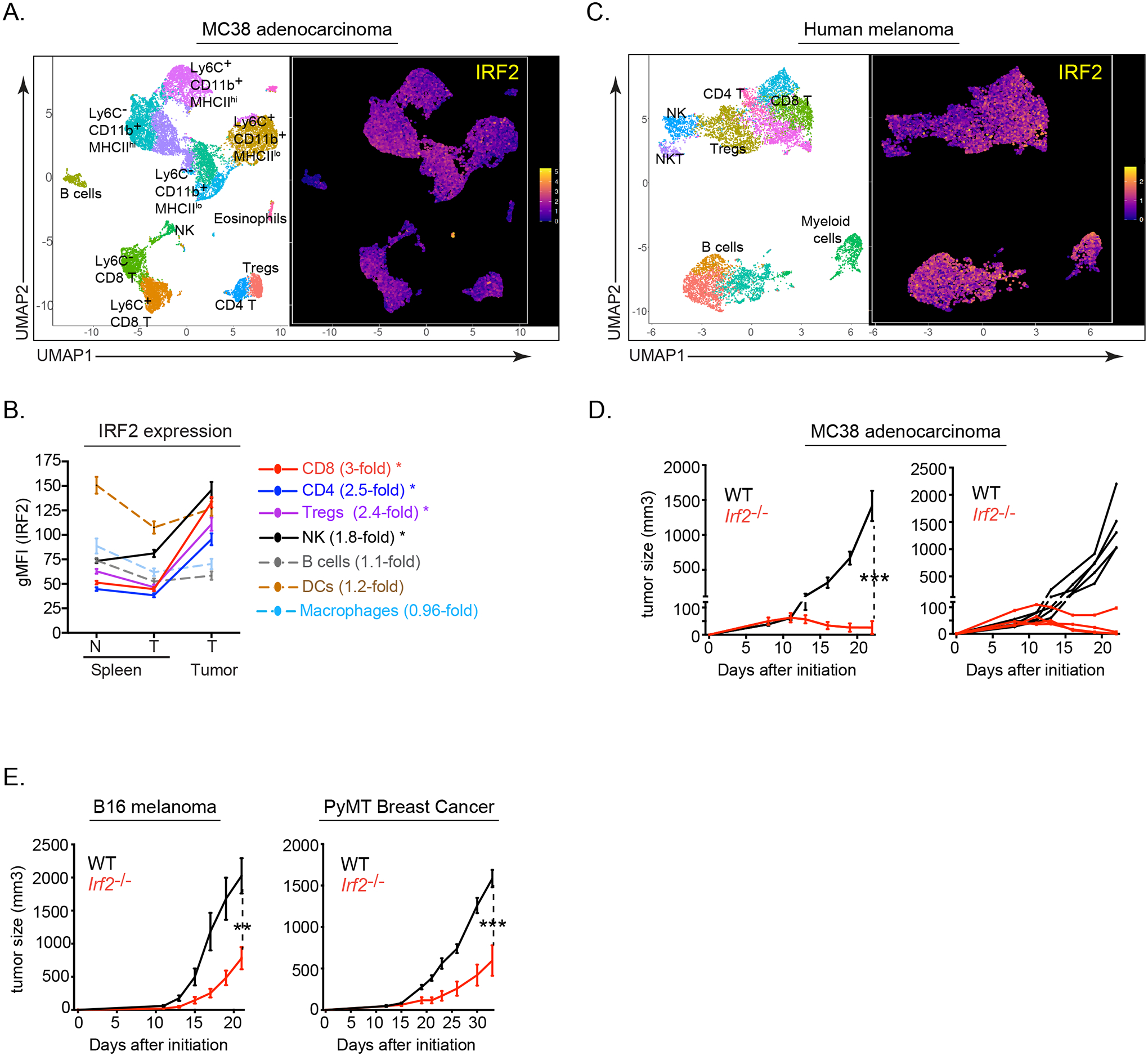
(A) UMAP plots of CyTOF data showing IRF2 expression in PhenoGraph-defined CD45+ tumor-infiltrating immune cell clusters from mouse MC38 adenocarcinoma tumors.
(B) Graph showing IRF2 expression (gMFI) in the spleens of naïve (N) mice or from mice with MC38 tumors (T), as well as from tumor-infiltrating immune cells. Numbers next to the cell type indicate the fold change between IRF2 expression in the tumor compared to the spleens from those same mice. * p<0.01.
(C) UMAP plots of CyTOF data showing IRF2 expression and distribution in PhenoGraph-defined CD45+ tumor-infiltrating immune cell clusters from human melanoma tumors.
(D) Tumor growth kinetics of wildtype (WT, black) and Irf2−/− (red) mice following implantation with MC38 tumor cells. Longitudinal line graphs show the average tumor volumes +/− standard error from the mean (SEM; left) and the tumor volumes of the individual mice (right).
(E) Average tumor volumes ± SEM of wildtype (WT, black) and Irf2−/− (red) mice following implantation with B16-F10 cells or PyMT cells..
Data are representative of at least two independent experiments containing 5 or more mice per group in each experiment. A total of 5 human melanoma tumors were assessed for IRF2 expression., *p<0.01, ** p<0.001, *** p< 0.0001. One-way ANOVA for multiple comparisons used for tumor growth kinetics.
To investigate the immune-mediated role of IRF2 in tumor control, WT mice or IRF2-deficient (Irf2−/−) mice (Matsuyama et al., 1993) were given MC38 tumor cells. The tumors grew similarly through 11 days in both the WT and Irf2−/− mice (Fig 1D), indicating normal tumor initiation in the absence of IRF2. However, while the MC38 tumors progressed in WT mice, and all reached endpoint by 25 days, the tumors were controlled in Irf2−/− mice, with some mice exhibiting no detectable tumors and all mice surviving the duration of the 50-day experimental period (Fig 1D, S1A). Like MC38 tumors, Irf2−/− mice had prolonged survival and enhanced control of minimally immunogenic B16-F10 melanoma and an orthotopic polyoma middle T antigen (PyMT) breast tumor (Fig 1E), indicating that the absence of IRF2 enables control of diverse tumor types.
Tumor control required CD8+ T cell intrinsic IRF2 expression
To identify the specific cell type(s) that mediated tumor control in the absence of IRF2, we depleted CD4+ or CD8+ T cell subsets in Irf2−/− mice, based on their increased expression of IRF2 within the tumor (Fig 1B). CD4+ T cell depletion in Irf2−/− mice prior to MC38 implantation to some extent abrogated tumor control, although tumor control was also enhanced in CD4-depleted WT mice (Fig S1B), likely reflecting the beneficial effects of Treg cell depletion. In contrast, CD8+ T cell depletion prior to MC38 initiation abolished the tumor control observed in isotype antibody treated Irf2−/− mice (Fig 2A), underscoring a key role of CD8+ T cells in enabling tumor control in the Irf2−/− mice. Further, CD8+ T cell depletion in Irf2−/− mice 3 weeks after MC38 tumor injection (at a time after tumors in WT mice had reached endpoint) rapidly led to rebound of tumor growth (Fig 2B), indicating that CD8+ T cells actively and continually maintained the long-term tumor control in Irf2−/− mice. The number of CD8+ TILs was similar between WT and Irf2−/− CD8+ T cells (Fig S1C), indicating comparable expansion/maintenance of the CD8+ TILs. To directly test the role of IRF2 deficiency within the CD8+ T cells, we created Irf2 floxed mice and crossed them with CD8-E8iii-Cre mice, to generate mice that only lack IRF2 expression in the CD8+ T cells (termed CD8-IRF2cKO mice; Fig S1D). The lack of IRF2 expression in CD8-IRF2cKO mice did not affect the expansion or survival of CD8+ TILs as their numbers were similar to WT (Fig S1E). The CD8-IRF2cKO mice efficiently controlled MC38 tumors in a manner like Irf2−/− mice (Fig 2C), indicating that Irf2-deletion in CD8+ T cells specifically enabled the long-term tumor control.
Figure 2. Tumor control required CD8+ T cell-intrinsic IRF2 expression.
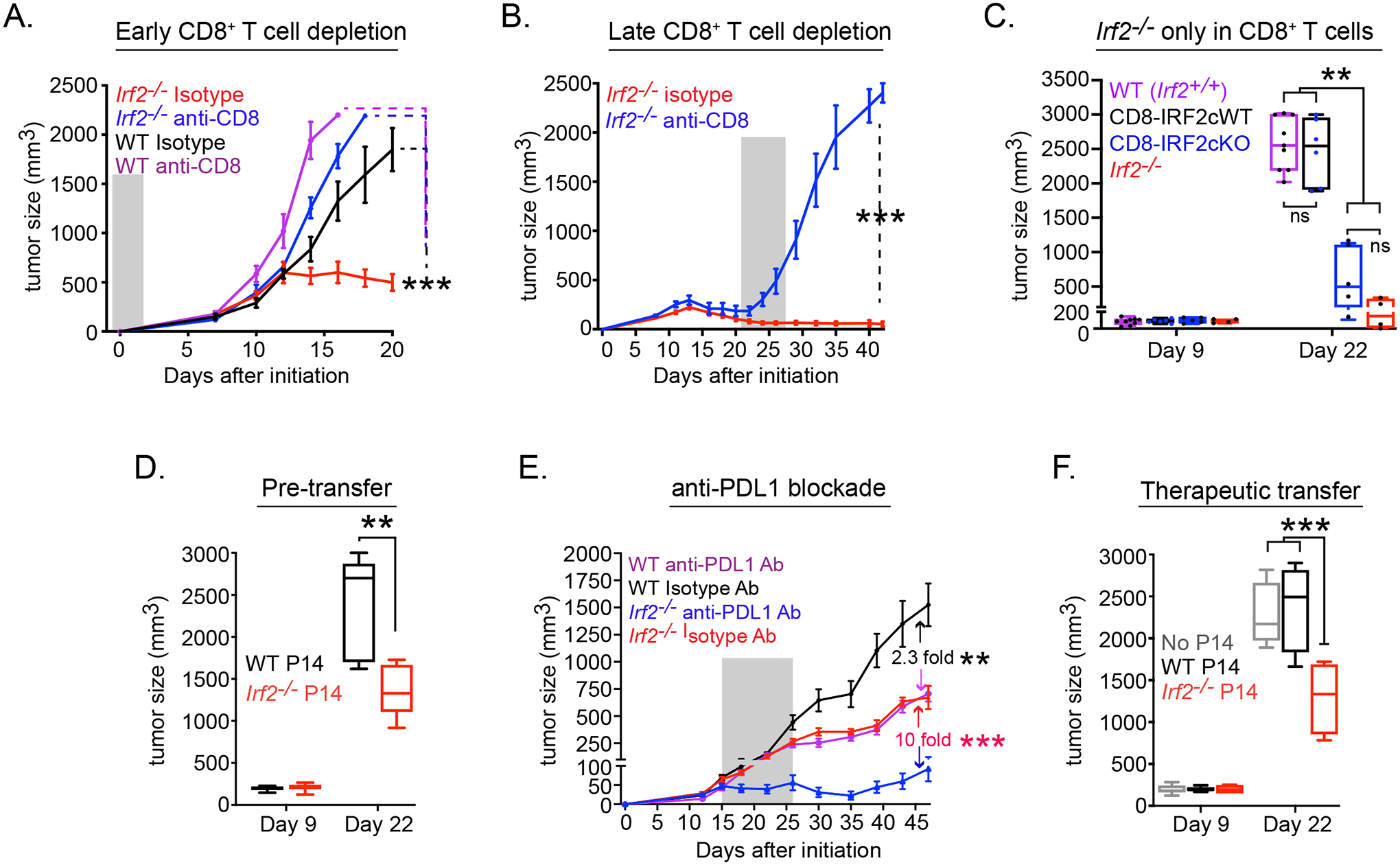
Tumor growth kinetics in WT or Irf2−/− mice that received isotype control or anti-CD8 depleting antibody either (A) one day before (early CD8+ T cell depletion) or (B) 21 days after (late CD8+ T cell depletion) MC38 initiation. For late depletion, only Irf2−/− mice were used since WT mice had already reached endpoint by day 21. Shaded region indicates duration of antibody treatment.
(C) MC38 tumor growth in WT control (i.e., Irf2+/+, purple), CD8-IRF2cWT (i.e., Irf2+/+ CD8Cre+ mice, black), CD8-IRF2cKO (IRF2-deficient only in CD8+ T cells; blue), or Irf2−/− (red) mice.
(D) Tumor size after WT mice received 2×105 naïve WT (black) or Irf2−/− (red) P14 T cells one day prior to receiving MC38-GP tumor.
(E) Tumor growth in WT or Irf2−/− mice with orthotopic PyMT breast tumor cells that were treated with isotype or anti-PDL1 blocking antibody beginning on day 15 after tumor implantation. Number in graph indicates the fold change in tumor size between the isotype vs. anti-PDL1 treatment for WT or Irf2−/− mice. Shaded region indicates duration of antibody treatment.
(F) MC38-GP tumoresize after WT mice received 2×105 pre-activated WT (black) or Irf2−/− (red) P14 T cells (i.v.) on day 9 after tumor initiation.
Data are representative of at least two independent experiments. Error bars represent mean ± SEM. ** p<0.001, *** p< 0.0001. One-way ANOVA for multiple comparisons used for tumor growth kinetics.
To further test the direct role of IRF2 in CD8+ T cells toward tumor control, we engineered MC38 cells to stably express the MHC-I Db-restricted lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP)33–41 epitope (referred to as MC38-GP tumor cells). We adoptively transferred naïve WT or Irf2−/− LCMV-GP33–41-specific (i.e., tumor-specific) CD8+ P14 transgenic T cells into WT mice prior to MC38-GP initiation. While the tumors grew initially alike between WT and Irf2−/− P14 CD8+ T cell recipients (like WT and Irf2−/− mice), mice that received the Irf2−/−P14 T cells exhibited enhanced tumor control (Fig 2D). Both the WT and Irf2−/−P14 T cells maintained similar tumor infiltration prior to divergence in tumor sizes (Fig S1F). Further, whereas all mice receiving WT P14 T cells reached endpoint by day 21, the mice that received Irf2−/− P14 T cells were all alive at day 21, and 60% were still alive at 25 days (Fig S1G), indicating that a small fraction of Irf2−/− tumor-specific CD8+ T cells can effectively inhibit tumor growth.
Enhanced immune checkpoint blockade and adoptive cell therapy by IRF2−/− CD8 T cells
The improved CD8+ T cell mediated tumor control in Irf2−/− mice suggested that these cells may be more amenable to CD8+ T cell enhancing immune therapy. To test this, we treated mice with established PyMT breast tumors with anti-PDL1 blocking antibody. Although PyMT breast tumors are controlled better in Irf2−/− compared to WT mice, the tumors still progress, allowing the opportunity for therapeutic intervention to control established tumors. Anti-PDL1 treatment was initiated at day 15, at a time after T cell priming and when tumors in WT and Irf2−/− mice had reached ~50mm3 but were still comparable in size. PDL1 blockade in WT mice induced a 2-fold reduction in PyMT tumor growth, whereas a 10-fold reduction was observed in Irf2−/− mice (Fig 2E, S1H). The enhanced efficacy of PDL1 blockade was also observed in the CD8-IRF2cKO mice (Fig S1I), indicating that IRF2-deficiency within CD8+ T cells enhances the efficacy of anti-PDL1 immunotherapy.
We next tested the therapeutic benefit of adoptively transferring WT or Irf2−/− tumor-specific CD8+ P14 T cells to control established tumors. Naïve WT or Irf2−/− P14 T cells were stimulated ex vivo for 24 hours with anti-CD3/CD28 antibodies and 2×105 cells were transferred into WT mice harboring established MC38-GP tumors. Transfer of this very low amount of WT P14 T cells had no effect on tumor growth (Fig 2F). On the other hand, transfer of Irf2−/−CD8+ P14 T cells into mice with established tumors effectively impeded tumor growth (Fig 2F), demonstrating the efficacy of adoptively transferred Irf2−/− CD8+ T cells to control growth of established tumors at much lower numbers than required for WT CD8+ T cells.
IRF2-deficient CD8+ T cells resist exhaustion and maintain functionality in the TME
To understand how IRF2-deficiency within the CD8+ T cells facilitated long-term tumor control, we analyzed MC38-infiltrating CD8+ T cells from WT and Irf2−/− mice by CyTOF (Table S1). Analyses were performed at day 12 after MC38 implantation, when tumor sizes were similar between WT and Irf2−/− mice. PhenoGraph-based clustering of WT and Irf2−/− CD8+ T cells yielded 5 clusters (Fig 3A). In WT CD8+ T cells, cluster (c)3 comprised most cells (64% WT vs. 27% Irf2−/−), whereas c4 (5% WT vs 14% Irf2−/−) and c5 (6% WT vs 39% Irf2−/−) made up the majority in the Irf2−/− CD8+ T cells (Fig 3A, S2A). To define these clusters, we compared the protein expression in each cluster relative to its expression in the other clusters (Fig 3B, 3C). The proportionally dominant WT CD8+ T cell cluster (c3) expressed high levels of Helios, Lag3, Tim3, CD39 and PD1 (Fig 3B, 3C), indicating a terminally differentiated/exhausted subset predominating within the tumors of WT mice. Conversely, the proportionally dominant clusters (c4, c5) in Irf2−/− CD8+ T cells, expressed lower levels of these inhibitory receptors, in combination with increased expression of the activation-induced proteins SLAMF1 (CD150) and in c5, Ki67 (a protein expressed in cycling cells) (Fig 3B, 3C), suggesting that c4 and c5 are activated but have not differentiated into an exhausted state. Consistent with a generally enhanced activation profile, even within the same clusters, the Irf2−/− CD8+ T cells exhibited increased expression of CD80, SLAMF1, Blimp1, Ki67 and CD25 [a protein that associates with an enhanced effector phenotype (Kalia et al., 2010)] compared to their WT counterparts (Fig 3B, S2B). A small, but significant, increase in cellular activation was also observed in Irf2−/− CD8+ T cells within the spleens and lymph nodes of naïve mice (Fig S2C), indicating a role of IRF2 in suppressing immune activation, likely to self or commensal antigens (Gao et al., 2012; Hida et al., 2000). The proportions of non-activated, naïve/central memory TCF1+ cells (c2, TCF1+PD1−CD39−) were comparable between the WT and Irf2−/− CD8+ TILs (Fig 3A, S2D). Similarly, the proportions of TCF1+ PD1+ CD8+ T cells that have been shown to be capable of self-renewing as well as generating terminally differentiated cytotoxic T cells (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016; Wu et al., 2016) were also comparable between the WT and Irf2−/− CD8+ T cells (Fig S2D), indicating that IRF2 deficiency does not deplete the TCF1+ stem-like population (Miller et al., 2019; Siddiqui et al., 2019).
Figure 3. IRF2-deficient CD8+ T cells resist exhaustion and maintain functionality in the TME.
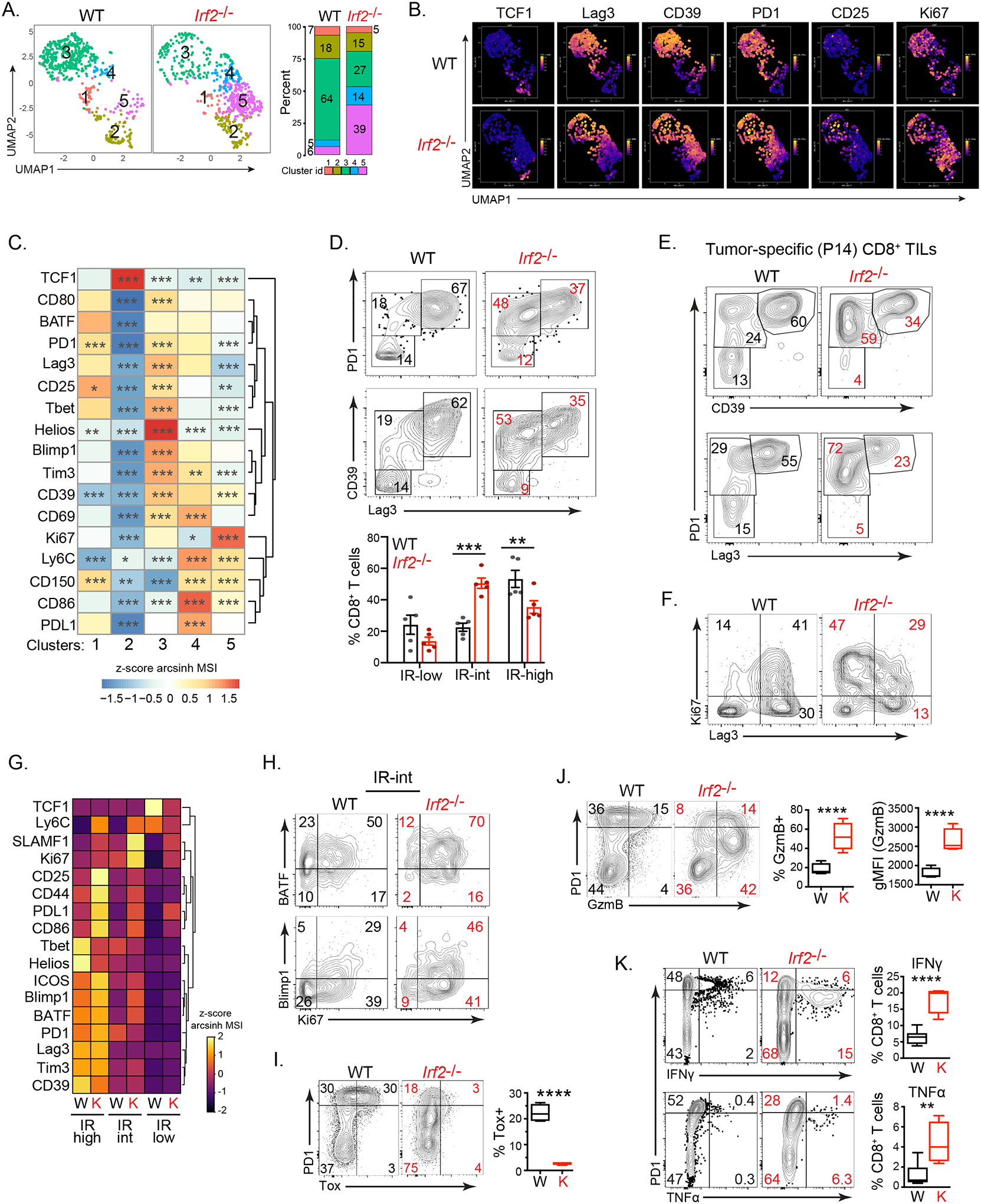
(A) UMAP plots of CyTOF data showing PhenoGraph-defined clusters of WT and Irf2−/−CD8+ TILs on day 12 after MC38 initiation. The bar graph depicts the proportion of each cluster in WT and Irf2−/− mice.
(B) UMAP plots show the single-cell expression of the indicated protein in CD8+ TILs from panel A.
(C) The heatmap represents relative expression (normalized z-scores of the arcsinh transformed mean signal intensity; MSI) of the indicated protein in each cluster from panel A compared to the other clusters combined using Wilcoxon rank-sum test.
(D) Eexpression of the inhibitory receptors (IR) CD39, PD1 and Lag3 in WT and Irf2−/−CD8+ TILs. Numbers in the plots show the percent of cells in each gate. The graph compares proportions of CD8+ T cells expressing low, intermediate or high levels of IRs combined from 4 independent experiments. Error bars represent SEM.
(E) Expression of CD39, PD1 and Lag3 in WT and IRF2−/− tumor-specific CD8+ P14 T cells from mice implanted with MC38-GP tumors. Numbers show percent of cells in each gate.
(F) Ki67 expression in WT and Irf2−/−CD8+ TILs. Numbers indicate the percent of cells in each quadrant.
(G) Heatmap depicting expression (z-score of median) of the indicated protein in IR-low, IR-int and IR-hi WT (W) or Irf2−/− (K) CD8+ TILs.
(H) BATF, Blimp1 and Ki67 expression by IR-int WT and Irf2−/−CD8+ TILs. Numbers indicate the percent of cells in each quadrant.
(I and J) PD1, Tox and GzmB expression in WT and Irf2−/−CD8+ TILs. Graphs indicate the proportions of cells expressing and the per-cell expression levels (gMFI) of the indicated protein.
(K) Flow plots show IFNγ and TNFα production in ex vivo GP33–41 peptide stimulated CD8+ TILs on day 12 after MC38-GP initiation. Graphs indicate the proportions of cells expressing IFNγ and TNFα.
Data are representative of at least three independent experiments. In each experiment, tumors from 4–7 mice were pooled from WT or Irf2−/− mice to obtain sufficient numbers of CD8+ TILs for analysis. * p<0.05, ** p<0.01, *** p<0.001, **** p< 0.0001.
Dividing the CD8+ TILs based on their expression of the inhibitory receptors (IRs) PD1, Lag3 and CD39 revealed a dominant PD1hi, CD39hi, Lag3hi exhausted-phenotype population in WT mice (Fig 3D). Conversely, this IR-high population was diminished in the Irf2−/− CD8+ T cells, and instead an IR-intermediate (IR-int) population +expressing lower levels of PD1, Lag3 and CD39 dominated (Fig 3D). A similar phenotype was observed following adoptive transfer of WT and Irf2−/− tumor-specific P14 T cells into otherwise WT mice (Fig 3E), indicating the cell intrinsic role of IRF2 in CD8+ TILs. The IR-int population of CD8+ T cells in Irf2−/− mice and in adoptively transferred Irf2−/− tumor-specific P14 T cells also expressed the highest levels of Ki67, consistent with this population consisting of an activated, but not exhausted fraction (Fig 3F, 3G, S2E). Overall, Ki67 expression was increased in CD8+ TILs from Irf2−/− mice and Irf2−/− tumor-specific P14 T cells (Fig 3F, S2E, S2F). In particular, a larger proportion of the Irf2−/− IR-int population co-expressed Ki67 with BATF and Blimp1 (Fig 3H, Fig S2G, S2H), proteins that are associated with sustained effector function (Chen et al., 2021b; Shin et al., 2009; Xin et al., 2015). Similarly, the BATF interacting partner, IRF4, that cooperatively limits T cell exhaustion to favor robust effector functions in the tumor (Seo et al., 2021) and in chronic viral infections (Grusdat et al., 2014; Xin et al., 2015), was highly expressed in the Irf2−/− IR-int population (Fig S2I). Throughout the different IR-expressing populations, the Irf2−/− CD8+ T cells expressed increased levels of multiple activation proteins (Fig 3G), indicating that even within the phenotypically similar populations, the IRF2-deficient CD8+ T cells exhibited increased activation and proliferation, and decreased exhaustion profiles.
The expression of the exhaustion “master-regulator” Tox was almost completely absent in the Irf2−/− CD8+ T cells (Fig 3I). In WT CD8+ T cells, Tox expression was largest in the PD-1hi population, consistent with these being the most exhausted (Khan et al., 2019) (Fig 3I). However, even within the PD-1hi subset of Irf2−/− CD8+ T cells, Tox expression was minimal (Fig 3I), indicating an overall abrogation of the exhaustion master-regulator Tox in the global absence of IRF2. The intermediate IR expression and diminished Tox suggested that deletion of IRF2 may permit CD8+ T cells to retain functionality within the TME. Consistent with this hypothesis, a large proportion of Irf2−/− CD8+ T cells expressed the cytolytic protein granzyme B (GzmB) specifically within the IR-int population and did so at higher single cell expression levels (Fig 3J). To further probe sustained functionality, WT and Irf2−/− TILs were stimulated ex vivo with tumor-specific GP33–41 peptide on day 12 after MC38-GP tumor initiation (when tumor sizes were similar). Compared to WT mice, a larger proportion of Irf2−/− CD8+ TILs produced IFNγ and TNFα, with the increase specifically within the IR-int cells (Fig 3K). Consistent with the retained functionality, the tumor-specific Irf2−/− CD8+ T cells co-expressed TNFα and IFNγ, unlike their WT CD8+ T cell counterparts (Fig S2J). Thus, Irf2−/− CD8+ T cells exhibit lower expression levels of IRs, increased expression of cytotoxic molecules, and elevated polyfunctional cytokine production within the TME.
IRF2 is preferentially expressed in activated and ISG-producing CD8+ T cells from mouse and human tumors
To further understand the relationship between IRF2 expression and cellular activation, we analyzed IRF2 distribution in CD8+ TILs. When CD8+ T cells were clustered based on the upper and lower third of IRF2 expression, distinct enrichment patterns emerged (Fig 4A, S3A, S3B). The IRF2-low cells were enriched in TCF1+ clusters (c5, c7, c8) that also expressed low amounts of most activation-induced proteins (Fig 4A–C, S3B), consistent with a less activated state. In contrast, c4, c6 and almost exclusively c1, were enriched in the IRF2-high fraction and expressed the highest levels of CD44, CD39, CD69, PD1, Ki67, BATF, GzmB, Helios, and Tbet (Fig 4A–C, S3B). The IRF2-high subsets also expressed increased levels of the ISG Protein Kinase R (PKR), suggesting that IRF2 expression is linked to the strength of IFN-I signaling (Fig 4C). Spearman correlation analysis in all CD8+ TILs (not divided into IRF2 high or low subsets) further revealed positive correlations of IRF2 with CD44, PD1, CD39, PKR, Tbet, BATF and Ki67, and a negative association with TCF1 and CD62L, markers of naïve CD8+ T cells (Fig S3C). Thus, increasing IRF2 expression correlates with heightened expression of activation, cycling and ISG proteins in the TME.
Figure 4.
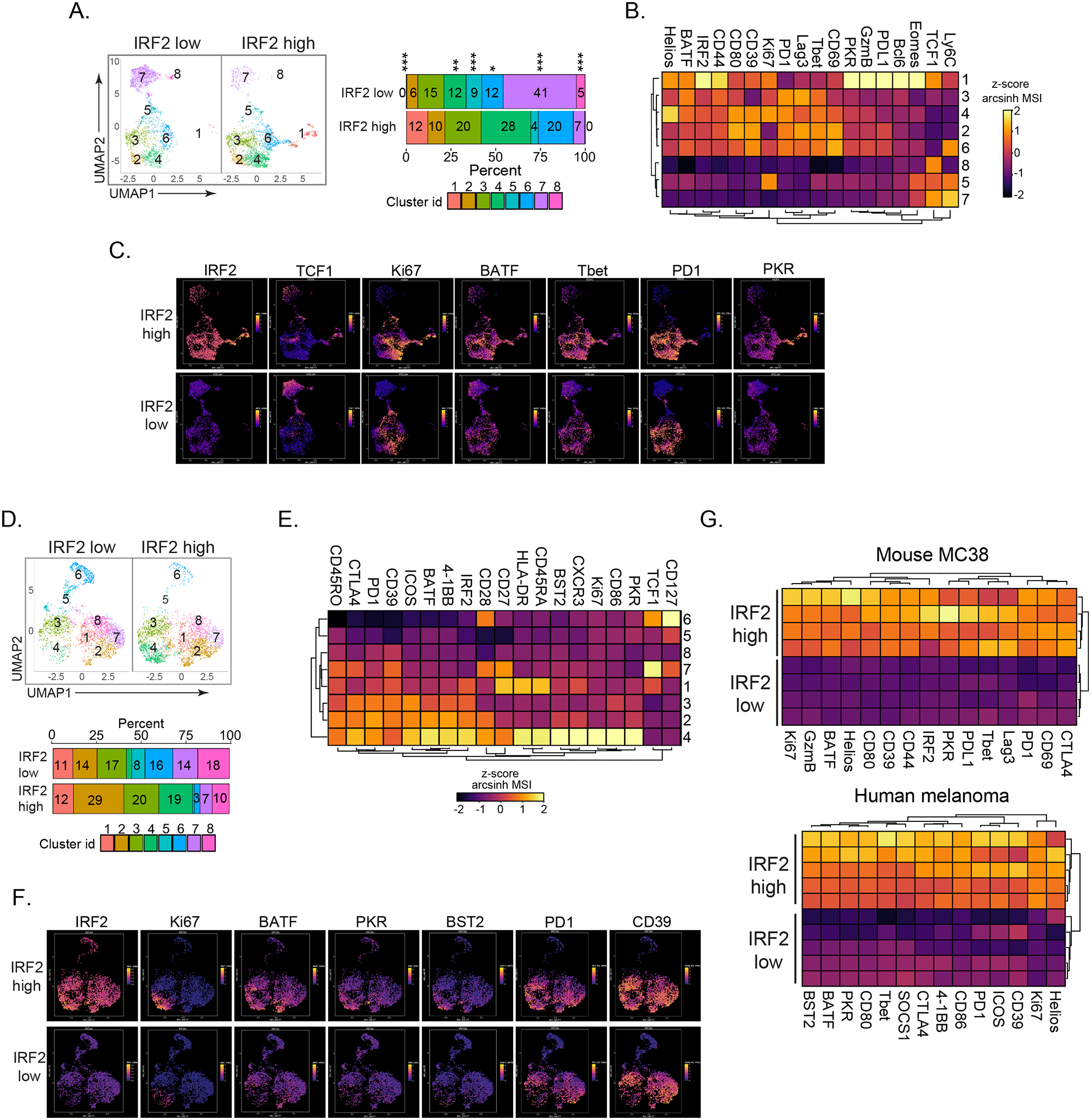
IRF2 is highly expressed in activated and ISG-producing mouse and human CD8+ TILs.
CD8+ TILs were divided into IRF2 high (upper 30%) and low (lower 30%) levels of IRF2 expression.
(A) PhenoGraph-defined clusters divided into IRF2 high and IRF2 low CD8+ T cells from mouse MC38 tumors (day 14). Bar graph depicts the proportion of each cluster in their respective groups.
(B) Heatmap represents expression of the indicated protein in each cluster.
(C) Expression and distribution of the indicated protein in the IRF2 high and low clusters of MC38-infiltrating CD8+ T cells.
(D-F) IRF2 distribution in human melanoma tissue biopsies.
(D) CD8+ TILs were divided into IRF2 high and low fractions and then clustered as in panel A. Shown is one representative tumor. Bar graph depicts the proportion of each cluster in their respective groups.
(E) Heatmap represents expression of the indicated protein in each cluster.
(F) Expression and distribution of the indicated protein in the IRF2 high and low clusters of CD8+ TILs.
(G) Heatmaps compare arcsinh transformed z-score of the MSI of the indicated protein in the IRF2 high and low CD8+ TILs in mouse MC38 tumors (top) and human melanoma (bottom). Each row represents CD8+ T cells from a different tumor.
The mouse MC38 data are representative of 3 independent experiments, each with at least 4 mice. * p<0.05, ** p<0.01, *** p< 0.0001. Unpaired, two-tailed Student’s t-test used to analyze significance of cluster proportions between IRF2 high and IRF2 low groups.
Human melanoma-infiltrating CD8+ T cells also exhibited differential distribution of IRF2 among clusters (Fig 4D). When CD8+ TILs were divided into the upper and lower third of IRF2 expression (Fig 4D, S3D), the TCF1+ cells (the naïve c6, and potentially the progenitor-like c7) and another less activated c8 were enriched with the IRF2-low subset (Fig 4D, 4E, S3E). On the other hand, increased levels of activation (CD39, PD1, ICOS, BATF), cycling (Ki67) and ISG (PKR, Bst2) proteins were consistently observed in the IRF2-high subset of human melanomas (Fig 4E, 4F). The cluster with the highest amount of IRF2 (c4), is also the most activated/terminally differentiated, and is almost entirely absent from the IRF2-low fraction (Fig 4D, 4E, S3E). Indeed, in total melanoma-infiltrating CD8+ T cells, expression of these activation proteins positively correlated with IRF2 expression (Fig S3F). Overall, in both mouse and human CD8+ TILs, IRF2-high expressing cells were enriched for co-expression of all activation-induced proteins measured (Fig 4G+, with IRF2 highest expressed in the most activated subsets.
Transcriptional profiling revealed sustained effector programming and resistance to exhaustion
To decipher how IRF2 transcriptionally programs CD8+ TILs, we performed combined single-cell (sc)RNA-seq plus antibody (Ab) staining on CD45-enriched tumor-infiltrating cells from WT and Irf2−/− mice. The Ab staining enabled identification of CD8+ T cells by protein expression, as RNA often under-represents the CD8+ T cell population (Mair et al., 2020), and the ability to further exclude cell doublets. Seurat-based clustering of the WT and Irf2−/− CD8+ TILs resolved 5 clusters (Fig 5A, S4A, Table S2). Cluster 0 (accounting for half of the WT cells) was largely absent in the Irf2−/− cells, while c3 accounted for half of the Irf2−/− CD8+ TILs and was almost absent in WT CD8+ TILs (Fig 5A, S4A). The abundance of c1, c2, and c4 were largely comparable between WT and Irf2−/− cells (Fig 5A). Cluster 1 expressed cell cycle and survival associated genes Mki67, Birc5, Ube2c, Pclaf, Stmn1 and Rrm2 (Fig 5B). Similarly, the TCF1+ c2 was largely comprised of naïve CD8+ T cells [Tcf7+, Sell+, Lef1+, Cx3cr1−, Pdcd1−, Entpd1−, Havcr2−, cd44lo, Itgal− (encoding Cd11a), and Icos−] and potentially some Tcf7+, Satb1+ regenerative, stem-like cells, and was equally present in both the WT and Irf2−/− CD8+ TILs (Fig 5A, 5B, Table S2), suggesting that Irf2−/− does not skew the differentiation of the TCF1+ populations. Cluster 4 are γδ T cells that were present at the same frequencies in WT and Irf2−/− CD8+ TILs (Fig 5A, 5B).
Figure 5: Transcriptional, epigenetic and gene-binding profiling.
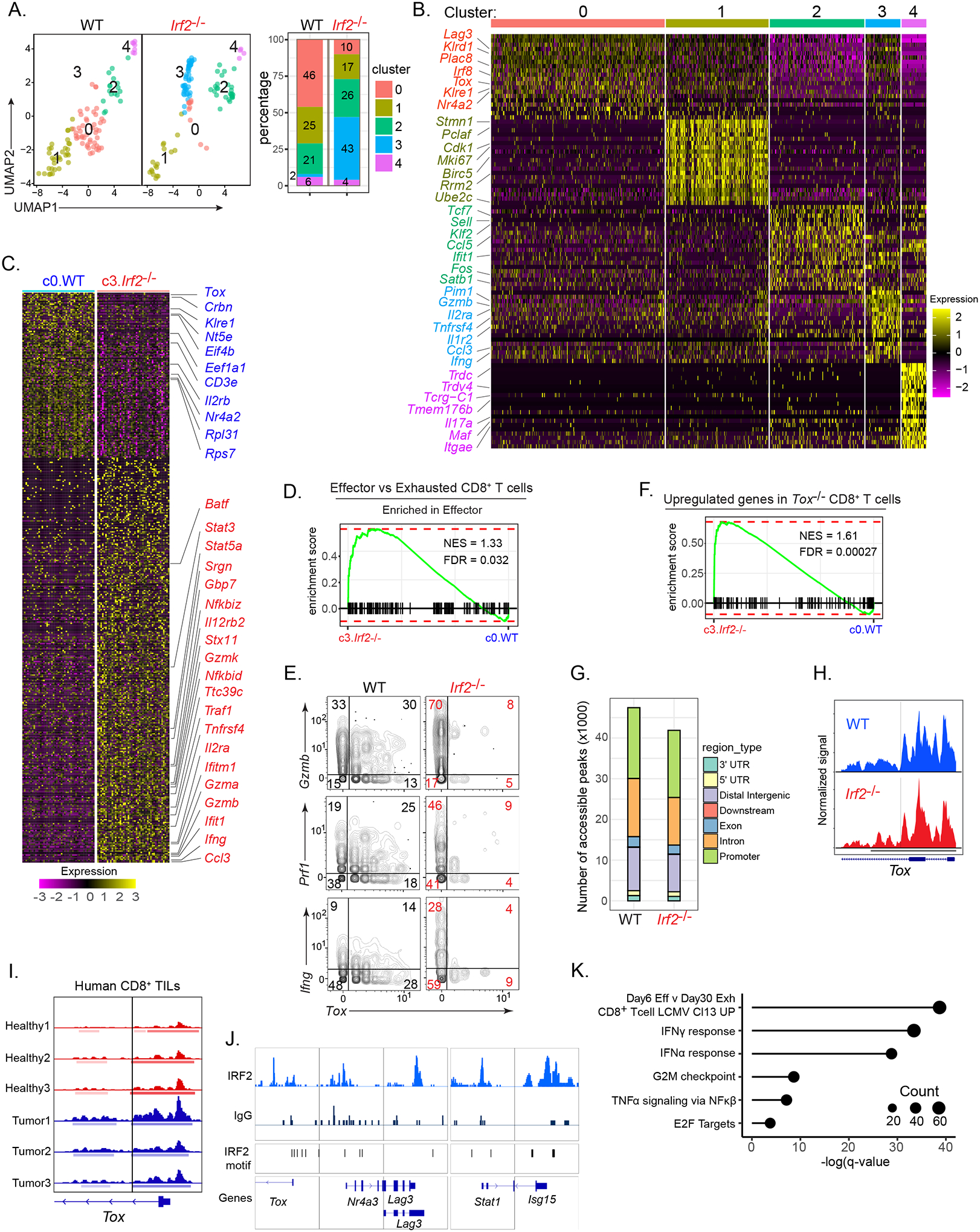
(A) WT and Irf2−/−CD8+ TILs derived from scRNA-seq data and clustered using Seurat. Bar graph depicts the proportion of each cluster within their respective group.
(B) Heatmap of top 20 up-regulated genes defining the clusters.
(C) Heatmap of differentially expressed genes between WT cluster 0 (c0.WT) and Irf2−/− cluster 3 (c3.Irf2−/−).
(D) GSEA plot showing enrichment of c0.WT and c3.Irf2−/− CD8+ T cells in gene signatures of effector vs exhausted CD8+ T cell pathway (from ImmuneSigDB).
(E) 2D plots showing Gzmb, Ifng, Prf1 and Tox RNA expression by WT and Irf2−/−CD8+ TILs.
(F) GSEA plot showing enrichment of c3.Irf2−/− CD8+ T cells in the gene signature of Tox-deficient CD8+ T cells, from (Khan et al., 2019).
(G) scATAC-seq analysis indicating the number of accessible peaks in each region of WT and Irf2−/−CD8+ TILs.
(H) Open chromatin state at IRF2-binding sites in the Tox promoter of WT (blue) and Irf2−/− (red) CD8+ TILs. Solid vertical line represents predicted IRF2 motif with a p<0.00005.
(I) ATAC-seq plot indicating open chromatin state at IRF2-binding sites in the Tox promoter of CD8+ T cells isolated from peripheral blood lymphocytes obtained from (red peaks) three healthy donors; and (blue peaks) PD1hi CD8+ TILs from 2 melanoma patients (tumor 1 and 2) and 1 lung cancer patient (tumor 3). Solid vertical lines represent predicted IRF2 motifs with a p<0.00005.
(J) Representative alignments of CUT&Tag peaks depicting IRF2 and IgG control antibody binding to the indicated loci of in vitro activated CD8+ T cells from the spleen and lymph nodes of WT mice.
(K) Selective list of pathways (and their respective adjusted P value) based on the genes that interact with IRF2. The size of each dot indicates the number of genes in that pathway.
Almost exclusively within the WT CD8+ TIL population, c0 (c0.WT) was defined by genes associated with immune dysfunction. These genes included Tox, Nr4a2 [which functions with Tox to drive exhaustion (Chen et al., 2019; Seo et al., 2019)], Irf8 (Mognol et al., 2017), Nt5e [encoding CD73 (Briceno et al., 2021)], Klre1 [which negatively regulates cytotoxicity (Westgaard et al., 2003)], Crbn [which is associated with decreased CD8+ T cell activation and effector function (Hesterberg et al., 2020)], and Il2rb [which has been shown to drive terminal exhaustion in chronic viral infections (Beltra et al., 2016)] (Fig 5B, 5C, Table S3). In contrast, the predominating Irf2−/− CD8+ TIL c3 (c3.Irf2−/−) exhibited increased expression of cytotoxic genes (Gzma, Gzmb, Gzmk, Stx11, Srgn), inflammatory cytokines and receptors [Ccl3, il2ra, Il12rb2, Tnfrsf4 (encoding OX40)], NFkb-signaling factors (Traf1, Nfkbid, Nfkbiz, Nfkbia), factors that sustain effector functionality [Batf (Chen et al., 2021b; Grusdat et al., 2014; Xin et al., 2015)], Ttc39c [(encoding Bach2) (Yao et al., 2021)] and numerous ISGs (e.g., Bst2, Ifit1, Ifitm1/2, Isg15, Slfn1, Irf7, CD274, Gbps2–7) (Fig 5B, 5C, Table S3). Genes associated with protein translation (Eif4b, Eef1a1, Eef1b2) and ribosomal assembly were decreased in the Irf2−/− CD8+ TILs (Fig 5C, Table S3), suggesting the heightened effector cell response phase associated with suppressed protein translation (Araki et al., 2017; Wherry et al., 2007). Further, the c3.Irf2−/− CD8+ TILs had an overall gene signature enriched in T cell effector function, compared to the exhaustion programming observed in c0.WT CD8+ TILs (Fig 5D). The WT CD8+ TILs also expressed some of the cytotoxic and immune-stimulatory genes, such as Gzmb, Prf1 and Ifng, however they did so at reduced levels compared to the Irf2−/− cells (both in proportions and at a single-cell expression) and with a large fraction of the WT cells co-expressing Tox (Fig 5E). Since the RNA expression is from the cells directly ex vivo (i.e., no in vitro stimulation), the increased levels of Ifng, Prf1 and Gzmb RNAs represent increased production of these anti-tumor factors by the Irf2−/− CD8+ TILs.
Consistent with the lack of Tox protein and gene expression, c3.Irf2−/− CD8+ TILs were enriched in the signature of genes upregulated in Tox-deficient CD8+ T cells (Khan et al., 2019) (Fig 5F+). To further understand how IRF2 epigenetically regulates CD8+ TILs, we performed ATAC-seq on CD8+ TILs from WT vs Irf2−/− mice or from CD8-IRF2cWT and CD8-IRF2cKO mice. Overall, the number of accessible peaks was largely comparable between CD8+ TILs from WT and Irf2−/− mice, as well as from CD8-IRF2cWT and CD8-IRF2cKO mice (Fig 5G, Fig S4B, Table S4), indicating that IRF2 does not generally affect chromatin accessibility in this context. Further, several of the DEGs identified by scRNA-seq, including Tox, exhibited similar chromatin accessibility between Irf2+/+ and Irf2−/− CD8+ TILs (Fig 5H). Despite the similar chromatin accessibility, many of the DEGs contain IRF2 binding sites within open chromatin regions (Fig 5H, Table S5), suggesting that IRF2 regulates expression of these genes rather than acting as an epigenetic regulator. A similar open chromatin state at IRF2-binding sites in the Tox promoter was observed in ATAC-seq data (Philip et al., 2017) from human PD1+ CD8+ TILs from melanoma and lung cancer patients (Fig 5I). In human naïve CD8+ T cells, the chromatin at the IRF2 binding sites in the Tox promoters were also open (Fig 5I), suggesting potential IRF2 binding to the Tox promoter in the naïve state as well. To directly probe for gene targets of IRF2, we performed Cleavage Under Targets & Tagmentation (CUT&Tag) (Kaya-Okur et al., 2019) on in vitro activated CD8+ T cells from the spleen and lymph nodes of WT (Irf2+/+) mice. Consistent with many DEGs containing IRF2 binding sites (Fig 5H, Table S5), IRF2 interacted with many genes including those associated with immune dysfunction (Tox, Nr4a3, Lag3, Ctla4), immune-stimulatory and pro-inflammatory functions (Stat1, Traf2, Nfkbid, Nfkbie, Ifnab, Tbx21, Prdm1, Cd3e, Il12rb1), protein translation (Eif2ak4, Eif1a, Eif3e, Eif3h, Eif4e3), as well as numerous ISGs (Isg15, Isg20, Ifit1, Bst2, Usp18, CD274, Gbps2–10) (Fig 5J, Table S6). Given the diverse nature or functions of the IRF2 target genes, we determined whether IRF2 binding genes were also enriched in motifs for other transcription factors (TFs). HOMER motif analysis indicated that the IRF2 target genes were enriched for TF motifs involved in immune activation (IRF1, ISRE, BATF, AP-1, PRDM1, IRF4) as well as repression of effector responses including Fli1 (Chen et al., 2021c) (Fig S4C, Table S6). Thus, instead of epigenetically regulating genes, IRF2 (independently and/or through transcription factor complexes) interacts with genes enriched in a variety of pathways including IFN signaling, TNFα/NF-κβ signaling, immune exhaustion, and protein translation (Fig 5K), ultimately resulting in inhibition of CD8+ T cell responses.
Network and pathway profiling identifies transcriptional modifications mediated by IRF2
To understand the changes in transcriptional networks, we performed the upstream regulator analysis using IPA on WT and Irf2−/−CD8+ TILs. Comparing the dominant clusters in WT (c0) and Irf2−/− (c3) CD8+ TILs, the upstream regulator analysis predicted IFN-related pathways (IFNγ, IFNα, IFNβ, IRF1, IRF3, IRF7, Stat1, Stat4) to be most enriched in Irf2−/− CD8+ T cells were (Fig 6A, Table S7), indicating enhanced IFN-I and IFN-II. This was further complemented by decreased regulation by the SOCS1 pathway (a negative regulator of IFN-II signaling; Fig 6A). Regulators for other pro-inflammatory and immune-activating pathways were also predicted to be activated in the Irf2−/− cells, including CD3 (TCR), cytokines involved in sustained T cell function (IL2, IL21, IL12), NF-κβ (CHUK, NFKBIA, NFKB complex; IKBKB), mTOR (LARP1, RICTOR, MTORC1) and Toll-like receptors (TLR3, TLR4, TLR7, TLR9, MyD88) (Fig 6A, Table S7). In contrast, negative regulators of proinflammatory signaling pathways were predicted to be decreased in Irf2−/− CD8+ TILs, including networks regulated by inhibitory receptors and suppressive cytokines (CTLA4, IL10RA, TGFBR2), by CITED2 [which inhibits NF-κβ activation, IFN responses and pro-inflammatory activity (Lou et al., 2011; Pong Ng et al., 2020)]; by BCL6 [which can antagonize GzmB expression to limit CD8+ T cell cytolytic activity (Yoshida et al., 2006)]; as well as Tox stimulated pathways. In line with the reduced expression of translation-related molecules (Fig 5C), upstream regulators for networks controlling cellular proliferation and metabolism (MLXIPL,MYCN, MYC, Irgm1) were inhibited in the Irf2−/− cells (Fig 6A, Table S7).
Figure 6. IRF2 re-routes transcriptional networks and programming.
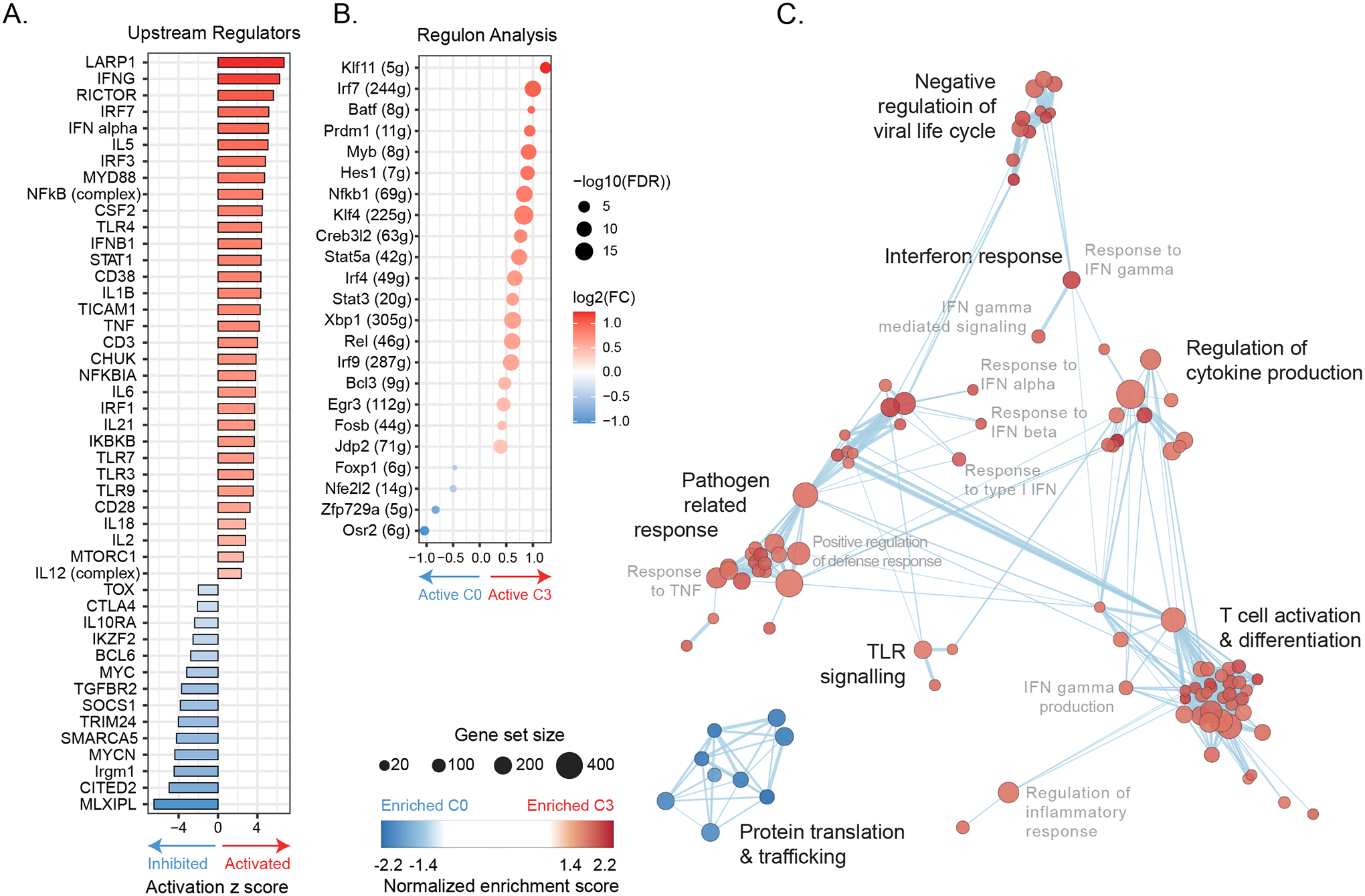
(A) Bar graph depicts z-scores of IPA-predicted upstream regulator molecules from the DEG dataset comparing c3.Irf2−/− to c0.WT CD8+ TILs. Upstream regulators predicted to be most enriched (Activated) in c3.Irf2−/− CD8+ T cells are shown in red and those most activated in c0.WT in blue.
(B) SCENIC-based fold changes in average regulon activity indicating whether a regulatory network is more active (red) or inhibited (blue) in c3.Irf2−/− vs c0.WT tumor infiltrating CD8+ T cells.
(C) Enrichment map showing biological processes enriched in up-regulated genes in c0.WT (blue) and up-regulated genes in c3.Irf2−/− (red) CD8+ TILs.
To further probe changes in TF activity networks in WT and Irf2−/− CD8+ T cells, we used SCENIC (single-cell regulatory network interference and clustering) (Aibar et al., 2017), an algorithm that pairs each TF to its putative direct binding target(s) [regulon(s)] within the same cell, indicating its activity level and whether the transcription factor and its targets are expressed in the same direction. Quantification of the fold change in average regulon activity indicated that the increased gene expression by CyTOF and scRNA-seq of multiple transcription factors in Irf2−/− CD8+ TILs corresponded to a similar increase in their target RNAs, including Nfkb1, Rel, BATF, IRF4, Stat3, Stat5a, and pathways involved in ISG expression (IRF7, IRF9) (Fig 6B). The target regulons of the Nfkb1 regulator Bcl3 also exhibited increased activity, potentially promoting survival, and enhancing effector responses (Jaiswal et al., 2021; Mitchell et al., 2001; Valenzuela et al., 2005). The increased activity of both BATF and IRF4 regulons in c3.Irf2−/− cells provided further downstream evidence for their role in countering exhaustion in the absence of IRF2, and their increased activity is consistent with the decreased levels of Tox observed in IRF2-deficient CD8+ T cells (Chen et al., 2021b; Grusdat et al., 2014; Seo et al., 2021; Xin et al., 2015). Thus, the individual gene expression changes in the Irf2−/− CD8+ TILs translate to transcriptional network programs that sustain effector function.
While the pathways enriched in the elevated genes in c0.WT CD8+ TILs involved protein translation and trafficking, many of the transcriptional pathways defining the c3.Irf2−/− CD8+ TILs were related to antiviral responses, T cell activation and differentiation, and responses to IFN-I and IFN-II signaling (Fig 6C, Table S7). The increase in the IFN-I, IFN-II and viral-related response signaling pathways in the Irf2−/− CD8+ TILs (Fig 6C, Table S7) was consistent with the role of IRF2 as a transcriptional repressor of IFN signaling (Harada et al., 1989; Lukhele et al., 2019; Matsuyama et al., 1993), but was unexpected to also be associated with sustained CD8+ T cell function since increased/ongoing IFN signaling drives immune suppression (Snell et al., 2017).
Both IFN-I and IFN-II signaling are constitutively required for tumor control in IRF2-deficient mice
We next sought to determine the role of IFN signaling in modulating tumor control in IRF2 deficiency. Treatment with IFNβ increased IRF2 expression in mouse CD8+ T cells above its constitutive expression levels (Fig 7A), and many (but not all) IFN-I and IFN-II signaling-associated molecules were increased at the protein level in Irf2−/− CD8+ T cells (Fig 7B), demonstrating that in vivo in the TME, IRF2 restricts the level of IFN-I and IFN-II signaling by CD8+ T cells. To next determine whether IFNs continue to increase IRF2 expression in the context of cancer, we used anti-IFNAR and anti-IFNγ antibodies to respectively block IFN-I and IFN-II signaling in WT mice with established MC38-tumors. Blocking either IFN-I or IFN-II signaling alone, and particularly in combination, substantially reduced IRF2 levels in CD8+ T cells (Fig 7C). Thus, IFNs induce and actively sustain the heightened IRF2 expression by CD8+ T cells, which subsequently feeds-back to limit IFN signaling.
Figure 7. IFN-I and IFN-II are required for long-term tumor control in Irf2−/− mice.
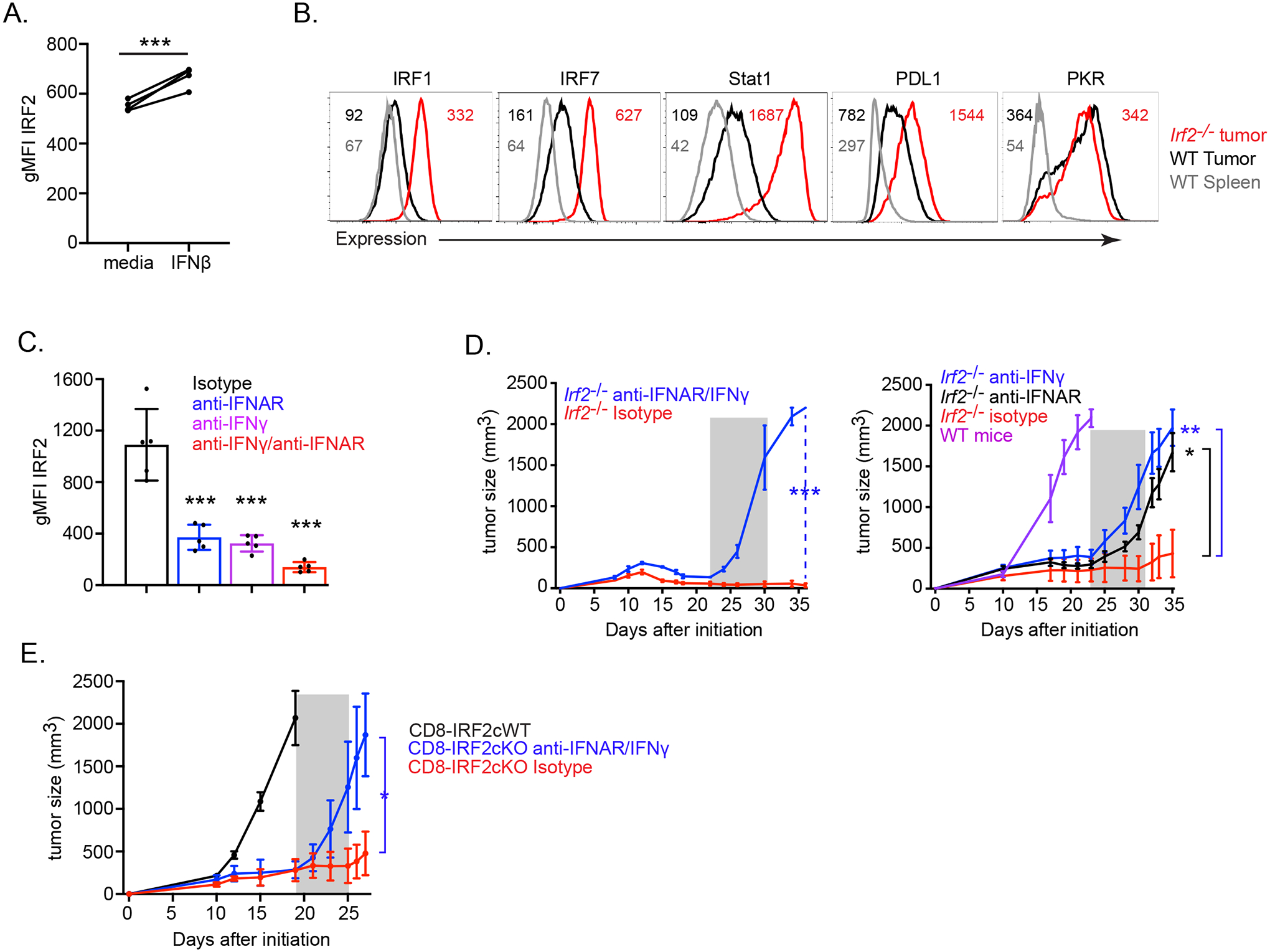
(A) IRF2 expression (gMFI) following media control and IFNβ stimulation of naïve WT mouse CD8+ T cells. *** p< 0.0001, paired Student’s t-test.
(B) Expression of IFN-I and IFN-II signaling-associated molecules in WT and Irf2−/− CD8+ T cells from the spleen and tumor of mice on day 12 after MC38 initiation. Data are representative of two independent experiments, each with at least 5 mice per group pooled.
(C) Graph shows IRF2 expression (gMFI) in dLNs of MC38 tumor-bearing WT mice following treatment beginning at day 9 with either isotype control, anti-IFNAR blocking, anti-IFNγ blocking, or dual (anti-IFNγ and IFNAR) blocking antibodies. Data are representative of two independent experiments, each with at least 5 mice per treatment condition. *** p< 0.0001, unpaired, two-tailed Student’s t-test.
(D) Tumor growth kinetics of (left) MC38 tumor-bearing Irf2−/− mice treated with either isotype (red line), or a combination of anti-IFNγ and anti-IFNAR (blue line) antibodies. Antibody treatments were initiated at 22 days post tumor implantation, after WT mice had reached endpoint. (Right) Tumor growth kinetics of MC38 tumor-bearing Irf2−/− mice treated with either isotype (red), anti-IFNγ (blue) or anti-IFNAR (black) blocking antibodies. Antibody treatments were initiated at 23 days and after WT mice (purple) had reached endpoint. Shaded area indicates duration of antibody treatment. Data are representative of two independent experiments, each with at least 5 mice per treatment condition.
(E) Tumor growth kinetics of MC38 tumor-bearing CD8-IRF2cKO mice treated with either isotype (red line) or a combination of anti-IFNγ and anti-IFNAR (blue line) antibodies, beginning at 19 days post tumor implantation, after CD8-IRF2cWT mice (black) had reached endpoint. Shaded area indicates duration of antibody treatment. Data are representative of three independent experiments.
* p<0.05, ** p<0.01, *** p< 0.0001. One-way ANOVA for multiple comparisons used for tumor growth kinetics.
We next determined whether the long-term tumor control in Irf2−/− mice required continual interferon stimulation. We blocked IFN-I and IFN-II signaling in Irf2−/− mice with established MC38-tumors (at a time after the tumors in WT mice had reached endpoint). Blockade of IFNAR alone, IFNγ alone, or IFNAR and IFNγ together three weeks after MC38-tumor initiation in Irf2−/− mice led to rapid rebound of tumor growth and diminished survival in all treated animals, while isotype antibody recipients continued to suppress tumor growth (Fig 7D). To directly test if this dependence on continual interferon stimulation required IRF2 deficiency specifically within the CD8+ T cells, we blocked IFN-I and IFN-II signaling in CD8-IRF2cKO mice with established MC38-tumors. IFNAR and IFNγ blockade also induced rapid tumor recrudescence in CD8-IRF2cKO mice (Fig 7E). Thus, IRF2 is a CD8+ T cell-intrinsic feedback inhibitor that translates IFN signals from the TME to transcriptionally program T cell exhaustion and, consequently, prevents long-term tumor control.
Discussion
How IFNs molecularly switch from pro-inflammatory to suppressive outcomes has long been a subject of interest. Here, we identified IRF2 as a keystone factor that translated IFN signals within the TME to temper inflammation. This feedback inhibition then suppressed the CD8+ T cell response and allowed tumor escape from immune control. In essence, IRF2 re-routed IFN induced transcriptional programming from pro- to anti- inflammatory signaling. In the absence of IRF2, these exhaustion promoting functions of IFNs were bypassed and CD8+ T cells instead retained high levels of anti-tumor activity and were able to effectively control tumor growth within the otherwise suppressive TME. That the sustained IFN-I signaling would continue to promote robust CD8+ T cell effector function is counter-intuitive based on the established role of prolonged IFN signaling to instead switch to drive immune suppression and CD8+ T cell exhaustion in multiple chronic disease contexts. The fact that this entire shift in the IFN programming focuses down to a single molecule was also unexpected, given the diversity of the ISGs driven by IFN signaling. The similarity between IRF2 expression and distribution in mouse and human CD8+ TILs implies a conserved role of IRF2. Thus, IRF2 is a foundational feedback mechanism that quells IFN-induced inflammatory reactions, and as a result, is a central regulator of T cell exhaustion in cancer.
The highly inflammatory conditions and prolonged IFN signaling within the TME necessitate counter measures to prevent excessive immunopathology, and tumors can co-opt these mechanisms to silence T cells for their own growth. Exposed to these inflammatory conditions, a wide variety of immune cells in the tumor exhibit IRF2 expression. Our study establishes that the failure of the immune response to control a variety of tumor types was highly dependent on IRF2 expression within the CD8+ TILs. Deletion of IRF2 only in CD8+ T cells was sufficient to induce long-term tumor control, and to enhance multiple immunotherapy-based strategies to control established tumors. While our data certainly does not exclude or negate potential IRF2-mediated contributions from other cell types within the tumor, it indicates that the attenuation of IFN signals by IRF2 is a CD8+ T cell intrinsic inflection point skewing from effector to exhausted functional states. These observations consolidate the complex roles of IFN-Is in tumorigenesis (Boukhaled et al., 2021; Lukhele et al., 2019; Snell et al., 2017) wherein IFNs stimulate antitumor immune responses while secondarily amplifying IRF2 expression to suppress and deactivate the functional CD8+ T cells to drive cancer progression. In essence, deleting IRF2 uncouples the sustained benefit of IFN signaling from the negative feedback inhibition IFN signaling also generates, placing IRF2 as the key lynchpin in the opposing regulation of IFN signaling.
Our study identifies IRF2 as a key factor that regulates the specific transcriptional programs that drive exhaustion. To this end, several transcription factors that directly mediate and counter T cell exhaustion are differentially regulated in the absence of IRF2 expression, including Blimp1, BATF, IRF4, NFAT, Nr4a and Tox. In conjunction, the expression of cytotoxic genes, proinflammatory cytokines as well as factors that sustain effector CD8+ T cells functions were enhanced following deletion of IRF2. It is interesting that in the absence of IRF2, the CD8+ T cells lose the Tox gene expression signature and resist exhaustion but resist the overstimulation-induced cell death in the TME. Their sustained presence is likely due at least in part to the fact that they still express intermediary levels of inhibitory receptors (Odorizzi et al., 2015; Scott et al., 2019), as well as increased expression of factors that sustain antigen stimulated (Grusdat et al., 2014; Xin et al., 2015) and tumor infiltrating T cells, such as BATF and IRF4 (Seo et al., 2021). Thus, IRF2 regulates a multi-directional response that instills exhaustion, while also suppressing the expression of multiple transcriptional nodes that sustain effector function in times of chronic antigen stimulation.
Resistance to checkpoint inhibitors and adoptive cell therapies remain a significant challenge. Based on its negative regulatory role in CD8+ T cells, deletion of IRF2 synergized with anti-PDL1 blockade to durably control a relatively non-immunogenic PyMT model of breast cancer. The efficacy of PDL1 blockade heavily relies on the ability to amplify CD8+ T cell responses, and although there are likely several mechanisms sensitizing tumors to immunotherapy, the enhanced functional state of IRF2-deficient CD8+ T cells and their diminished expression of inhibitory receptors situates them ready-to-respond to immunotherapy. In addition, the ability to avert immune exhaustion and retain their superior effector functions under inflammatory conditions makes Irf2−/− CD8+ T cells strong candidates for adoptive CAR T cell therapies at cell numbers well below those required for WT CD8+ T cells. Thus, modulating IRF2 represents a new target for multiple therapeutic modalities aimed to enhance tumor-specific T cell functions and control tumor growth.
Limitations of the study
Our study identifies IRF2 as a key factor that negatively regulates IFN signaling, immune activation, and CD8+ T cell function, ultimately impeding the ability to control multiple mouse tumors. Even though IRF2 expression is similar in mouse and human melanoma TILs, an immune function of IRF2 expression in human cancers and in particular in CD8+ T cells remains to be directly tested. Further, there is a need to evaluate the suitability of Irf2−/− CD8+ T cells for adoptive CAR T cell therapies against human cancers. In addition, the exact mechanisms through which IRF2 synergizes with immune-checkpoint therapies remains unknown, yet this property may be a new strategy to fight less immunogenic tumors. Lastly, cancer control is a complex process, involving a variety of IRF2-expressing cell types that are also capable of IFN signaling. It is likely that IRF2 in these other cells also contribute to the anti-tumor immune response and the cancer control observed. As such, how IRF2 influences their function within the TME will have to be evaluated to fully understand the interplay between inflammation, immunosuppression and for the development of novel antitumor immunotherapies.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Brooks (dbrooks@uhnresearch.ca).
Materials availability
Further information and material requests should be addressed to David Brooks (dbrooks@uhnresearch.ca).
Data and code availability
Single-cell RNA-seq, ATAC-Seq and CUT&Tag data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
All original codes (sc-RNA-Seq, scATAC-Seq and CUT&Tag analyses codes) have been deposited at the BrooksLab repository in github (https://github.com/diala-ar/BrooksLab) and are publicly available as of the date of publication. DOIs are listed in the key resources table
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Y(89)-anti-Lag3 (Clone C9B7W) | Biolegend | Cat# 125202; AB_961187 |
| 115In-anti-Ly6C (Clone HK14) | Biolegend | Cat#128001; RRID: AB_1134213 |
| 141Pr-anti-CD44 (Clone IM7) | Biolegend | Cat#103001; RRID: AB_312952 |
| 142Nd-anti-CXCR5 (Clone L138D7) | Biolegend | Cat#145502; RRID: AB_2561955 |
| 142Nd-anti-Siglec-F (Clone E50-2440) | BD Biosciences | Cat#552125; RRID: AB_394340 |
| 143Nd-anti-IRF2 (EPR4644(2)) | Abcam | Cat#ab229443; RRID: NA |
| 144Nd-anti-Granzyme B (Clone GB11) | ThermoFisher | Cat#MA1-80734; RRID: AB_931084 |
| 145Nd-anti-CTLA-4 (Clone UC10-4B9) | Biolegend | Cat#106302; RRID: AB_313251 |
| 146Nd-anti-EOMES (Clone Dan11mag) | eBioscience | Cat#14-4875-82; RRID: AB_11042577 |
| 147Sm-anti-CD45.2 (Clone 104) | Biolegend | Cat#109802; RRID: AB_313439 |
| 148Nd-anti-CD11b (Clone M1/70) | Fluidigm | Cat#3148003B; RRID: AB_2814738 |
| 148Nd-anti-ICOS (Clone C398.4A) | Fluidigm | Cat#3148019B; RRID: AB_2756435 |
| 149Sm-anti-CD69 (Clone H1.273) | Biolegend | Cat#104502; RRID: AB_313105 |
| 150Nd-anti-Ly6G (Clone 1A8) | Biolegend | Cat#127601; RRID: AB_1089179 |
| 150Nd-anti-CD73 (Clone TY/11.8) | Biolegend | Cat#127202; RRID: AB_1089066 |
| 151Eu-anti-CD25 (Clone 3C7) | Fluidigm | Cat#3151007B; RRID: AB_2827880 |
| 152Sm-anti-CD86 (Clone GL-1) | Biolegend | Cat#105007; RRID: AB_313150 |
| 153Eu-anti-CD8a (Clone 53-6.7) | Fluidigm | Cat#3153012B; RRID: AB_2885019 |
| 153Eu-anti-CD45.1 (Clone A20) | Fluidigm | Cat#3153002B; RRID:AB_268784 |
| 154Sm-anti-BATF (Clone D7C5) | Fluidigm | Cat#3154012A; RRID: AB_2687838 |
| 155Gd-anti-CD103 (Clone 2 × 107) | Biolegend | Cat#121401; RRID: AB_535944 |
| 155Gd-anti-CD103 (Clone M290) | BD Biosciences | Cat#553699; RRID: AB_394995 |
| 156Gd-anti-PD-L1 (Clone M1H5) | eBioscience | Cat#14-5982-82; RRID: AB_467781 |
| 158Gd-anti-RORɣt (Clone Q31-378) | BD Biosciences | Cat#562197; RRID: AB_10894594 |
| 159Tb-anti-CD39 (Clone 24DMS1) | eBioscience | Cat#14-0391-82; RRID: AB_1210501 |
| 160Gd-anti-CD4 (Clone rm4-5) | Biolegend | Cat#100505; RRID: AB_312708 |
| 161Dy-anti-T-bet (Clone 4B10) | Fluidigm | Cat#3161014B; RRID: AB_2858233 |
| 162Dy-anti-Foxp3 (Clone MF-14) | Biolegend | Cat#126401; RRID: AB_1089120 |
| 163Dy-anti-Tim-3 (Clone B8.2C12) | Biolegend | Cat#134002; RRID: AB_1626128 |
| 163Dy-anti-Bcl6 (Clone K112.91) | BD Biosciences | Cat#561520; RRID: AB_10713172 |
| 164Dy-anti-CD62L (Clone MEL-14) | Fluidigm | Cat#3164003B; RRID: AB_2885021 |
| 165Ho-anti-TCRb (Clone H57-597) | Biolegend | Cat#109201; RRID: AB_313424 |
| 166Er-anti-B220 (Clone RA3-6B2) | Invitrogen | Cat#14-0452-82; RRID: AB_467254 |
| 166Er-anti-Blimp1 (Clone ROS195G) | Biolegend | Cat# 648202; RRID:AB_2300132 |
| 167Er-anti-TCF-1 (Clone S33-966) | BD Biosciences | Cat#624084; RRID: NA |
| 168Er-anti-Bcl6 (Clone K112.91) | BD Biosciences | Cat#561520; RRID: AB_10713172 |
| 168Er-anti-PKR (Clone EPR19374) | Abcam | Cat#ab224887; RRID:N/A |
| 169Tm-anti-SLAM (Clone TC15-12F12.2) | Biolegend | Cat#115901; RRID: AB_313680 |
| 170Er-anti-NK1.1 (Clone PK136) | Fluidigm | Cat#3170002B; RRID: AB_2885023 |
| 171Yb-anti-CD80 (Clone 16-10A1) | Fluidigm | Cat#3171008B; RRID: AB_2885024 |
| 172Yb-anti-Ki67 (Clone B56) | BD Biosciences | Cat#550609; RRID: AB_393778 |
| 173Yb-anti-Helios (Clone 22F6) | Biolegend | Cat#137202; RRID: AB_10900638 |
| 174Yb-anti-CD11c (Clone N418) | Biolegend | Cat#117301; RRID: AB_313770 |
| 174Yb-anti-Lag3 (Clone C9B7W) | Fluidigm | Cat#3174019B; RRID:N/A |
| 175Lu-anti-PD1 (Clone RMP-30) | Biolegend | Cat#109101; RRID: AB_313418 |
| 176Yb-anti-Thy1.2 (Clone 53-21) | ThermoFisher | Cat#14-0902-82; RRID: AB_467379 |
| 209Bianti-MHC II (Clone M5/114.15.2) | Fluidigm | Cat#3209006B; RRID: AB_2885025 |
| Y(89)-anti-human CD45 (Clone HI30) | Fluidigm | Cat#3089003B; RRID:N/A |
| Cd(111)-anti-human CD80 (Clone BB1) | BD Biosciences | Cat#555681; RRID:AB_396033 |
| Cd(112)-anti-human CD45RO (Clone UCHL1) | Biolegend | Cat#304202; RRID: AB_314418 |
| Cd(114)-anti-human Helios (Clone 22F6) | Biolegend | Cat#137202; RRID:AB_10900638 |
| In(115)-anti-human SOCS1 (Clone 4H1) | EMD Millipore | Cat#04-002; RRID:AB_612104 |
| In(116)-anti-human Ki67 (Clone Ki-67) | Biolegend | Cat#350502; RRID:AB_10662385 |
| Pr(141)-anti-human CD45RA (Clone HI100) | Biolegend | Cat#304102; RRID:AB_314406 |
| ND(142)-anti-human HLA-DR (Clone L243) | Biolegend | Cat#307602; RRID:AB_314680 |
| ND(143)-anti-IRF2 (Clone EPR4644(2)) | Abcam | Cat#ab229443; RRID:N/A |
| ND(144)-anti-human CD33 (Clone WM53) | Biolegend | Cat#303402; RRID:AB_314346 |
| ND(145)-anti-human CXCR3 (Clone G025H7) | Biolegend | Cat#353706; RRID:AB_10962912 |
| ND(146)-anti-human CD8a (Clone RPA-T8) | Biolegend | Cat#301053; RRID:AB_2562810 |
| Sm(147)-anti-human CD4 (Clone RPA-T4) | Biolegend | Cat#300541; RRID:AB_2562809 |
| ND(148)-anti-human ICOS (Clone DX29) | BD Biosciences | Cat#557801; RRID:AB_396877 |
| Sm(149)-anti-human FoxP3 (Clone 236A-E7) | ThermoFisher | Cat#14-477782; RRID:AB_467556 |
| ND(150)-anti-human CD103 (Clone B-Ly7) | ThermoFisher | Cat#14-103882; RRID:AB_467412 |
| Eu(151)-anti-human CD39 (Clone A1) | Biolegend | Cat#328221; RRID:AB_2563747 |
| Sm(152)-anti-human CD11c (Clone Bu15) | Biolegend | Cat#337221; RRID:AB_2562834 |
| Eu(153)-anti-human CD3 (Clone UCHT1) | Biolegend | Cat#300443; RRID:AB_2562808 |
| Gd(155)-anti-human CD303 (Clone 201A) | Biolegend | Cat#354215; RRID:AB_2563739 |
| Gd(156)-anti-human CD14 (Clone M5E2) | Biolegend | Cat#301843; RRID:AB_2562813 |
| Gd(158)-anti-human CD27 (Clone O323) | Biolegend | Cat#302839; RRID:AB_2562817 |
| Tb(159)-anti-human CD19 (Clone HIB19) | Biolegend | Cat#302247; RRID:AB_2562815 |
| Gd(160)-anti-human CD25 (Clone M-A251) | Biolegend | Cat#356102; RRID:AB_2561752 |
| Dy(161)-anti-human Tbet (Clone 4B10) | Biolegend | Cat#644802; RRID:AB_1595503 |
| Dy(162)-anti-human CD28 (Clone CD28.2) | Biolegend | Cat#302937; RRID:AB_2563737 |
| Dy(163)-anti-human CD137 (Clone 4B4-1) | Biolegend | Cat#309802; RRID:AB_314781 |
| Dy(164)-anti-human CD86 (Clone 2331) | BD Biosciences | Cat#555655; RRID:AB_396010 |
| Ho(165)-anti-human PD1 (Clone EH12.2H7) | Biolegend | Cat#329941; RRID:AB_2563734 |
| Er(166)-anti-human Tim3 (Clone F38-2E2) | Biolegend | Cat#345019; RRID:AB_2563790 |
| Er(167)-anti-human PKR (Clone 6H3A10) | Novus Biological | Cat#NBP2-37242; RRID:N/A |
| Er(168)-anti-human CD73 (Clone AD2) | Biolegend | Cat#344002; RRID:AB_2154067 |
| Tm(169)-anti-human CTLA4 (Clone 14D3) | ThermoFisher | Cat#14-152982; RRID:AB_467512 |
| Er(170)-anti-human CD317 (Clone RS38E) | Biolegend | Cat#348402; RRID:AB_10588013 |
| Yb(171)-anti-human Granzyme B (Clone GB11) | ThermoFisher | (Special order) |
| Yb(172)-anti-human CD127 (Clone EBioRDR5) | ThermoFisher | Cat#14-1278-8; RRID:N/A |
| Yb(173)-anti-human NKP46 (Clone 9 × 102) | Biolegend | Cat#331902; RRID:AB_1027637 |
| Yb(174)-anti-human TCF-1 (Clone S33-966) | BD Biosciences | (Special order) |
| Lu(175)-anti-human CD274 (Clone 29E.2A3) | Biolegend | Cat#329719; RRID:AB_2565429 |
| Yb(176)-anti-human Lag3 (Clone 7H2C65) | Biolegend | Cat#369202; RRID:AB_2616877 |
| Bi(209)-anti-human CD11b (Clone ICRF44) | Fluidigm | Cat#3209003B; RRID:N/A |
| APC anti-mouse/human CD11b (clone M1/70) | Biolegend | Cat#101211; RRID: AB_312794 |
| APC/Fire 750 anti-mouse CD11c (clone N418) | Biolegend | Cat#117351; RRID: AB_2572123 |
| Brilliant Violet 421 anti-mouse CD11c (clone N418) | Biolegend | Cat#117343; RRID: AB_2563099 |
| PerCP/Cyanine5.5 anti-mouse CD45.1 (clone A20) | Biolegend | Cat#110728; RRID:AB_893346 |
| APC anti-mouse CD45.2 (clone 104) | Biolegend | Cat#109814; RRID:AB_389211 |
| PerCP/Cyanine5.5 anti-mouse CD45.2 (clone 104) | Biolegend | Cat#109828; RRID:AB_893350 |
| APC/Fire 750 anti-mouse CD4 (clone GK1.5) | Biolegend | Cat#100459; RRID: AB_2572110 |
| Brilliant Violet 605 anti-mouse CD4 (clone GK1.5) | Biolegend | Cat#100547; RRID:AB_11125962 |
| Brilliant Violet 711 anti-mouse CD8α (clone 53-6.7) | Biolegend | Cat#100747; RRID:AB_11219594 |
| PerCP/Cyanine5.5 anti-mouse CD8α (clone 53-6.7) | Biolegend | Cat#100734; RRID:AB_2075238 |
| PerCP/Cyanine5.5 anti-mouse B220 (clone RA3-6B2) | Biolegend | Cat#103236; RRID:AB_893354 |
| Alexa Fluor 700 anti-mouse B220 (clone RA3-6B2) | Biolegend | Cat#103231; RRID:AB_493716 |
| Brilliant Violet 421 anti-mouse Tcrβ (clone H57-597) | Biolegend | Cat#109229; RRID:AB_10933263 |
| PerCP/Cyanine5.5 anti-mouse Tcrβ (clone H57-597) | Biolegend | Cat#109228; RRID:AB_1575173 |
| Brilliant Violet 421 anti-mouse FoxP3 (clone MF-14) | Biolegend | Cat#126419; RRID:AB_2565933 |
| Alexa Fluor 488 anti-mouse FoxP3 (clone MF-14) | Biolegend | Cat#126406; RRID:AB_1089113 |
| PerCP/Cyanine5.5 anti-mouse NK1.1 (clone PK136) | Biolegend | Cat#108728; RRID:AB_2132705 |
| Pacific Blue anti-mouse Ly6C (clone HK1.4) | Biolegend | Cat#128014; RRID:AB_1732079 |
| PerCP/Cyanine5.5 anti-mouse Ly6G (clone 1A8) | Biolegend | Cat#127616; RRID:AB_1877271 |
| FITC anti-mouse Granzyme B (clone GB11) | Biolegend | Cat#515403; RRID:AB_2114575 |
| Pacific Blue anti-mouse Granzyme B (clone GB11) | Biolegend | Cat#515407; RRID:AB_2562195 |
| PE anti-mouse TCF1 (clone S33-966) | BD Bioscience | Cat#564217; RRID:AB_2687845 |
| APC anti-mouse Tbet (clone 4B10) | Biolegend | Cat#644814; RRID:AB_10901173 |
| FITC anti-mouse Ki67 (clone 35/Ki-67RUO) | BD Bioscience | Cat#556026; RRID:AB_396302 |
| FITC anti-mouse CD44 (clone IM7) | Biolegend | Cat#103006; RRID:AB_312957 |
| Brilliant Violet 421 anti-mouse CD25 (clone PC61) | Biolegend | Cat#102043; RRID:AB_2562611 |
| PECy7 anti-mouse CD62L (clone MEL-14) | Biolegend | Cat#104418; RRID:AB_313103 |
| Brilliant Violet 421 anti-mouse PDL1 (clone 10F.9G2) | Biolegend | Cat#124315; RRID:AB_10897097 |
| PECy7 anti-mouse CD39 (clone (24DMS1) | eBioscience | Cat#25-0391-82; RRID:AB_1210766 |
| APC anti-mouse Lag3 (clone C9B7W) | Biolegend | Cat#125210; RRID:AB_10639727 |
| Brilliant Violet 786 anti-mouse PD1 (clone 29F.1A12) | Biolegend | Cat#135225; RRID:AB_2563680 |
| PE anti-mouse Tox (clone txrx10) | eBioscience | Cat#12-6502-82; RRID:AB_10855034 |
| PE anti-mouse BATF (clone D7C5) | Cell Signaling | Cat#27120; RRID:AB_2798938 |
| PE anti-mouse IRF4 (clone IRF4.3 × 104) | Biolegend | Cat#646403; RRID:N/A |
| Alexa Fluor 488 anti-mouse IRF1 (clone D5E4) | Cell Signaling | Cat#14028; RRID:AB_2798366 |
| PE anti-mouse IRF1 (clone D5E4) | Cell Signaling | Cat#12732; RRID:AB_2798008 |
| PE anti-mouse IRF7 (clone MNGPKL) | eBioscience | Cat#12-5829-80; RRID:AB_2572628 |
| anti-mouse PKR (clone EPR19374) | Abcam | Cat#ab184257; RRID:AB_2916271 |
| Alexa Fluor 647 anti-mouse Stat1 (clone 1/stat1) | BD Bioscience | Cat#558560; RRID:AB_647143 |
| Alexa Fluor 647 anti-mouse Blimp1 (clone 5 × 107) | Biolegend | Cat#150004; RRID:AB_2565618 |
| PE anti-mouse Blimp1 (clone 5 × 107) | Biolegend | Cat#150005; RRID:AB_2565991 |
| Alexa Fluor 647 donkey anti-Rabbit IgG (clone Poly4064) | Biolegend | Cat#406414; RRID:AB_2563202 |
| anti-IRF2 antibody (clone [EPR4644(2)]) | Abcam | Cat#ab124744; RRID:AB_10974405 |
| APC anti-mouse IFNγ (clone XMG1.2) | Biolegend | Cat#505810; RRID:AB_315404 |
| Brilliant Violet TNFα anti-mouse (clone MP6-XT22) | Biolegend | Cat#506328; RRID:AB_2562902 |
| InVivoMAb anti-mouse IFNAR-1 (clone MAR1-5A3) | BioXcell | Cat#BE0241; RRID:AB_2687723 |
| InVivoMAb anti-mouse IFNγ (clone XMG1.2) | BioXcell | Cat#BE0055; RRID:AB_1107694 |
| InVivoMAb anti-mouse PDL1 (clone 10F.9G2) | BioXcell | Cat#BE0101; RRID:AB_10949073 |
| InVivoMAb anti-mouse CD8α (clone 2.43) | BioXcell | Cat#BE0061; RRID:AB_1125541 |
| InVivoMAb anti-mouse CD4 (clone GK1.5) | BioXcell | Cat#BE0003; RRID:AB_1107642 |
| InVivoMAb rat IgG2b isotype control (clone LTF-2) | BioXcell | Cat#BE0090; RRID:AB_1107780 |
| InVivoMAb IgG1 isotype control (clone MOPC-21) | BioXcell | Cat#BE0083; RRID:AB_1107784 |
| Ms CD4 Olgo AMM2056 (clone GK1.5) | BD Bioscience | Cat#940471; RRID:AB_2876318 |
| Ms CD8b Olgo AMM2189 (clone H35-17.2) | BD Bioscience | Cat#940360; RRID:AB_2876231 |
| Ms TCRβ CHN Olgo AMM2021 (clone H57-597) | BD Bioscience | Cat#940125, RRID:AB_2876014 |
| Ms CD3e Olgo AMM2001 (clone 145-2C11) | BD Bioscience | Cat#940107; RRID:AB_2875997 |
| Biological samples | ||
| Human melanoma samples | Princess Margaret Cancer Biobank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Cell-ID Cisplatin | Fluidigm | Cat#201064 |
| Cell-ID Intercalator-Iridium—125 mM | Fluidigm | Cat#201192A |
| Zombie Aqua Fixable Viability Kit | Biolegend | Cat#423102 |
| Mouse IL-2 Recombinant Protein | GIBCO | Cat#PMC0025 |
| Brefeldin A | Sigma | Cat#B7651-5MG |
| DNase I | Sigma | Cat#DN25-1G |
| Cisplatin | BioVision | Cat#1550 |
| Collagenase I | ThermoFisher | Cat#17018029 |
| Fixation Buffer | Biolegend | Cat#420801 |
| Intracellular Staining Permeabilization Wash Buffer (10X) | Biolegend | Cat#421002 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| EasySep Mouse CD8+ T cell Isolation Kit | STEMCELL | Cat#19853 |
| 20-Plex Pd Barcoding Kit | Fluidigm | Cat#201060 |
| Mouse CD45+ (TIL) Microbeads | Miltenyi Biotec | Cat#110-021-618 |
| Fc block | ThermoFisher | Cat#16-9161-73 |
| Recombinant mouse IFNβ | R & D Systems | Cat#12401-1 |
| Deposited data | ||
| ATAC-seq data from human tumor-infiltrating PD1+ CD8+ T cells | (Philip et al., 2017) | GEO accession#: GSE89308 |
| Transcriptional features of Tox-deficient CD8+ T cells | Khan et al., (2019) | GEO accession#: GSE131871 |
| CUT&TAG | This manuscript | GEO accession#: GSE199177 |
| scATAC-Seq | This manuscript | GEO accession#: GSE199177 |
| scRNA-Seq | This manuscript | GEO accession#: GSE199177 |
| Original codes | This manuscript | https://github.com/diala-ar/BrooksLab |
| Experimental models: Cell lines | ||
| MMTV-PyMT cell line | Dr. Christopher PAGE (University Health Network) | N/A |
| PyMG tumor cells | Generated by our lab | N/A |
| B16-F10 tumor cells | Dr. Tracy McGaha (University Health Network) | N/A |
| MC38 tumor cells | Dr. Daniel de Carvalho (University Health Network) | N/A |
| MC38-GP cells | Generated by our lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J mice | Jackson laboratory | Cat#000664 |
| LCMV-GP33-specific CD8 TCR transgenic (P14) mice | Bred in our facility | N/A |
| Mouse: Irf2−/− | Provided by Dr. Tak Mak at the University Health Network (Matsuyama et al., 1993), and bred in our facility | N/A |
| Mouse: Irf2+/+ | Bred in our facility | N/A |
| Irf2fll/fl mice | generated and bred in our facility | N/A |
| CD8α-Cre (E8iii-Cre) transgenic mice (C57BL/6-Tg(CD8α-cre)1Itan/J, Stock No. 008766 | The Jackson Laboratory | Stock no. 008766 |
| Software and algorithms | ||
| Flow Jo version 9 and 10 | BD FLOWJO | http://www.flowjo.com/ |
| BD FACSsuite v1.4.0.7047 | BD Biosciences | https://www.bdbiosciences.com/ |
| Cytobank | Cytobank, Inc | https://www.cytobank.org |
| Graphpad Prism v8 and v9 | GraphPad Software, Inc | https://www.graphpad.com/ |
| R | R Core Team | https://www.r-project.org/ |
| UMAP | (McInnes et al., 2018) | RRID:SCR_018217 |
| PhenoGraph | (Levine et al., 2015) | RRID:SCR_016919 |
| diffcyt R | (Weber et al., 2019) | https://bioconductor.org/packages/release/bioc/html/diffcyt.html |
| HOMER v4.8 findMotifsGenome.pl | (Duttke et al., 2019) | N/A |
| Bedtools v2.27.1 getfasta tool | (Quinlan and Hall, 2010) | N/A |
| Ingenuity Pathway Analysis software | Qiagen | N/A |
| Cytoscape | (Shannon et al., 2003) | N/A |
| SeqGeq software | BD Biosciences | N/A |
| BD RhapsodyTM WTA Analysis pipeline | BD Biosciences | https://www.sevenbridges.com |
| SEACR v1.3 | (Meers et al., 2019) | https://github.com/FredHutch/SEACR |
| bowtie v2.4.1 | https://bowtie-bio.sourceforge.net/index.shtml | |
| cellranger-atac count (v 2.0.0) | 10X genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Integrative genomics viewer (IGV) | https://igv.org/ | |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 mice were purchased from The Jackson Laboratory or the breeding colony at the Princess Margaret Cancer Center, University Health Network (PMCC, UHN). Irf2−/− mice (Matsuyama et al., 1993) were kindly provided by Dr. Tak Mak at the PMCC (UHN). Briefly, the Irf2−/− were generated by replacing exon 3 of the Irf2 gene (which encodes amino acids 30 – 63 and is part of the DNA-binding domain of the protein) with a neomycin resistance cassette. The Irf2fll/fl mice (described below) were crossed with CD8α-Cre (E8iii-Cre) transgenic mice (C57BL/6-Tg(CD8α-cre)1Itan/J, Stock No. 008766, The Jackson Laboratory) to delete IRF2 from peripheral CD8α-expressing T cells. LCMV-GP33-specific CD8+ TCR transgenic (P14) mice have been described previously (Brooks et al., 2006). Mice were housed under specific pathogen–free conditions at the PMCC (UHN). Mouse handling conformed to the experimental protocols approved by the OCI Animal Care Committee at the PMCC (UHN). Experiments were performed using sex and age matched male and/or female mice. Mice used were between 7 and 10 weeks old at the initiation of each experiment.
Generation of IRF2 floxed mice
Construction of mIrf2 conditional targeting vector:
A targeting construct was designed to conditionally delete the 3rd exon of the mouse interferon regulatory factor 2 gene. To generate the genomic fragments for the 5’ and 3’ homology arms, we designed primers based on mouse mIrf2 genomic sequence (GenBank Accession No. NC_000074) to use in PCR from mouse C57BL/6 genomic DNA (Jackson Laboratory, Bar Harbor, Maine). In brief, PCR primers 5’- GCA CTT AGC GAT CGC AGC TGC TCC TTG GAC CAA TGA CCT T −3’(Irf2 AsiSI sense) and 5’- AAG TTA AAT CGA TAG AAG ACT CCT GGC GCA TGC TCA GTC −3’ (Irf2 ClaI antisense) were used to amplify a 4497 bp 5’ homology-arm fragment (corresponding to mIrf2 intron 2 sequence) from 200 ng of C57Bl/6J genomic DNA using the PfuUltra II fusion HS DNA polymerase (Agilent Technologies, Santa Clara CA). Using the same PCR conditions, a 4555 bp 3’ homology-arm fragment (encompassing exon 4, exon 5, and part of intron 5 of mIrf2) was amplified from C57Bl/6J Genomic DNA using the PCR primers 5’- TGG ACC AGT TTA AAC ATA TTG GAA GCT CGT CTC TGC −3’ (Irf2 PmeI sense) and 5’- ATT TAT GCG GCC GCT CAC TTC CTG GAT GAA CAT GGC −3’ (Irf2 NotI antisense). PfuUltra II fusion HS DNA polymerase was also used to amplify a 1146 bp fragment from C57Bl/6J genomic DNA spanning the targeted mIrf2 exon 3 using the primers 5’ TTC TGG TCT TAA TTA ACT TTA GCA GGA CTA GGA TTA CAG 3’ (mIrf2 Ex3 PacI sense) and 5’ AAT ATG ATT AAT TAA AAG GTC CAC ATC TAA AGA TAT CTC C 3’ (mIrf2 Ex3 PacI antisense).
The resulting PCR products were gel-purified using the Nucleospin® Gel and PCR Clean-up system (Machery-Nagel Gmbh & Co., Germany), and TA-overhangs were added via a 20-minute incubation with Taq DNA polymerase (New England BioLabs, Ipswich, MA) at 68°C in the presence of 1mM dNTPs and 1X Buffer. The fragments were then TA-cloned into pCR2.1-TOPO (Invitrogen, San Diego, CA) and subcloned into a modified pBluescript II KS (Stratagene, La Jolla, CA) vector containing a PGK-neomycin cassette flanked by both LOXP and FRT sequences. A diphtheria toxin (DTA) gene was inserted 3’ of the long arm to negatively select against non-homologous targeting. Insert sequence was validated using fluorescent dideoxy-nucleotide sequencing and automated detection (ABI/Perkin Elmer, Forest City, CA).
Targeted disruption of the murine Irf2 gene in ES cells:
The mIrf2 conditional targeting vector (25 μg) was linearized with NotI restriction endonuclease at the 3’ end of the 3’homology-arm and electroporated into C57Bl6/N ES cells (NIH Knockout Mouse Project (KOMP) repository; University of California Davis) using a Bio-Rad Gene Pulser, 0.34 kV, and 0.25 mF. ES cell culture was carried out as previously described (Hakem et al., 1996). After G418 selection (250 μg/ml), homologous recombinants were identified by 5’ and 3’ flanking PCR and confirmed by sequencing following published protocols (Hakem et al., 1996). Homologous recombination at the 5’ homology-arm was confirmed by Terra™ PCR Direct (Takara Bio, Mountain View, CA, USA) amplification of a 4657 bp fragment using the primers Irf2 5PCR Sense: 5’ GCC AGG CCA TTT GTT TAG GAA TGC AGG AG −3’, in the flanking sequence of mIrf2 intron 2, and the vector-specific primer PCRA antisense: 5’- CGA CGG TCA ACG AGC AGT CCA GCG TAT CC −3’. Homologous recombination at the 3’ homology-arm was confirmed by Terra™ PCR Direct amplification of a 4858 bp fragment using the vector-specific primer PCRB sense: 5’- GCT TGA CTC GCT ACG TGG ATC GTC GAA C −3’ and the mIrf2 intron 5’ flanking primer: Irf2 3PCR antisense 5’- AAA CCC ACC GGC CTG ATA CAC GTT CTA C −3’.
Generation of Irf2 flox mice:
Chimeric mice were produced by microinjection of independent mIrf2 +/− ES cell clones into E3.5 C57BL/6J blastocysts and transferred to pseudopregnant foster mothers. Chimeric males were mated with C57Bl/6J females (Jackson Laboratory). Germ line transmission of the mutant allele was confirmed by PCR analysis of tail DNA from mice with an agouti coat color. The PGK-Neo cassette was removed from mouse Irf2+/− mice by crossing with Flp-deleter mice (Jackson Laboratory stock #009086) (Farley et al., 2000) and PCR genotyping and sequence validation of recombination at the FRT sites.
Human melanoma samples
All human tissue specimens were obtained through protocols approved by the institutional review board (University Health Network Research Ethics Board). Surgical specimens were obtained from the Princess Margaret Cancer Biobank. Written informed consent was obtained from all donors.
Tumor models, cell culture and tumor injections
MC38 tumor cells derived from C57BL/6 murine colon adenocarcinoma were a generous gift from Dr. Daniel de Carvalho at the PMCC (UHN) and were cultured in McCoy 5A medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin, 1% streptomycin and 1% L-glutamine. To generate the MC38-GP cells, MC38 cells were stably transduced with pMIG retroviral vector expressing the LCMV GP1–100 sequence and a Puromycin resistance gene. Puromycin was used to select for successfully transduced cells. To select single cell MC38-GP clones, cells were sorted and grown from a single cell per well in McCoy 5A medium supplemented with 10% FBS, 1% penicillin and streptomycin and 1% L-glutamine and puromycin. 2×105 MC38 or MC38-GP tumor cells were injected subcutaneously (s.c.) into one hind leg of each mouse. B16-F10 tumor cells derived from C57BL/6 murine B16-F10 skin melanoma were generously provided by Dr. Tracy McGaha at the PMCC (UHN) and cultured in DMEM medium (Gibco) supplemented with 10% FBS, 1% penicillin, 1% streptomycin and 1% L-glutamine. 1×105 B16-F10 tumor cells were injected s.c. into one hind leg of each mouse. The MMTV-PyMT cell line was a generous gift from Dr. Christopher Page at the PMCC (UHN) and was cultured in McCoy 5A medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin, 1% streptomycin and 1% L-glutamine. 1×106 PyMT cells were injected into a single mammary gland fat pad of each mouse.
METHOD DETAILS
P14 T cell adoptive transfer
LCMV-GP33–41-specific CD8+ P14 T cells were isolated from the spleens and lymph nodes of C57BL/6 and Irf2−/− transgenic mice by negative selection (Stem Cell Technologies). For pre-tumor transfer, 2×105 naïve P14 T cells were transferred i.v. in the retro-orbital sinus. For therapeutic transfer experiments, P14 T cells were activated for 24 hours using plate-bound anti-CD3 (clone 2C11, 10μg/mL) and soluble anti-CD28 (1μg/mL) antibodies. Cells were then counted and 2×105 P14 cells were transferred i.v. in the retro-orbital sinus nine days following tumor implantation.
Isolation of tumor-infiltrating cells
To isolate immune cells from the tumor, tumors were cut into small fragments and digested in RPMI media containing collagenase I (100U/mL, ThermoFisher), DNaseI (10ug/mL, Sigma) and 2% FBS in a gentleMACS dissociator (Cat#130–093-235 Miltenyi Biotec) using the gentleMACS program 37c_m_TDK_1. Cells were filtered through a 70μm pre-separation filters (Cat#130–095-823 Miltenyi Biotec) and subjected to red blood cell lysis. CD45+ tumor infiltrating lymphocytes were isolated from the digested single-cell suspensions using Mouse CD45 (TIL) Microbeads (Cat#110–021-618 Miltenyi Biotec), autoMACS columns (Cat#130–021-101 Miltenyi Biotec) and an autoMACS Pro Separator (Miltenyi Biotec) following the manufacturer’s instructions. Purified CD45+ single-cell suspensions from tumors from at least 5 mice per condition were pooled before staining.
Time-of-Flight mass cytometry (CyTOF)
Antibodies directly conjugated to metal tags were purchased from Fluidigm. Purified unconjugated antibodies were labeled with metal tags at the SickKids-UHN Flow and Mass Cytometry Facility using the MaxPar Antibody Labeling Kit from Fluidigm. CyTOF staining was performed as previously described(Snell et al., 2021). Briefly, mouse single cell suspensions (up to 5×106 cells) were pulsed with 12.5 mM Cisplatin (BioVision) in PBS for 1 min at room temperature (RT) prior to quenching with CyTOF staining media (Mg+/Ca+ HBSS containing 2% FBS (Multicell), 10mM HEPES (Corning), and FBS underlay. Cells were then resuspended in CyTOF staining media containing Fc block (prepared in house) for 15 mins at 4°C and a 2x CyTOF staining media containing metal-tagged surface antibodies (Table S1) was added for an additional 30 min at 4°C. Cells were then fixed and permeabilized using the eBioscience Foxp3 / Transcription Factor Staining set (ThermoFisher) according to manufacturer’s instructions. Cells were then stained with metal tagged intracellular antibodies (Table S1) prepared in the permeabilization buffer from the same kit. All antibody concentrations were used at concentrations previously determined by titration. Cells were then incubated overnight in PBS (Multicell) containing 0.3% (w/v) saponin, 1.6% (v/v) paraformaldehyde (Polysciences Inc) and 50 nM Iridium (Fluidigm). Human melanoma samples were stained in a similar manner except the Fc block (ThermoFisher, catalog #16–9161-73) was for 10 min at RT. Cells were analyzed on a Fluidigm Helios or CyTOF-2 mass cytometer instruments at the SickKids-UHN Flow and Mass cytometry Facility and at the PMCC Tumor Immunotherapy Profiling Facility. FCS data files were manually de-barcoded and analyzed using Cytobank 6.2 (Cytobank, Inc), FlowJo software (v.9 or v.10, Treestar) and the GraphPad Prism v8 or v9.
Heatmaps were generated from arcsinh-transformed median of spectral indices (MSI) values, plotted in R using the viridis color package and the ggplots package. The CyTOF data was clustered and analyzed using the Phenograph(Levine et al., 2015) algorithm together with the R implementation of UMAP. An equal number of cells was used for clustering, determined based on the lowest common denominator between WT and Irf2−/− tumor infiltrating CD8+ T cells.
Flow cytometry and intracellular cytokine stimulation
Single-cell suspensions of cells from the indicated tissue or tumor were stained with a zombie aqua viability stain (Biolegend) at 4°C and then surface stained using antibodies against targeted molecules CD45 (30-F11), CD45.2 (104), CD45.1 (A20), CD8α (53–6.7), CD4 (GK1.5), Tcrβ (H57–597), PD1 (29F.1A12), Lag3 (C9B7W), CD39 (24DMS1), PDL1 (10F.9G2), B220 (RA3–6B2), CD62L (MEL-14), CD25 (PC61), CD86 (GL-1), CD11c (3.9), CD11b (M1/70), CD44 (IM7), NK1.1 (PK136), Ly6C (HK1.4), Ly6G (1A8). Cells were then washed in PBS and staining buffer, then fixed and permeabilized using the eBioscience Foxp3 / Transcription Factor Staining Kit (ThermoFisher) according to manufacturer’s instructions. Cells were then stained with intracellular antibodies [Granzyme B (GB11), Ki67 (35/Ki-67RUO), Tbet (4B10), TCF1 (S33–966), Tox (txrx10), BATF (D7C5), IRF4 (IRF4.3E4), IRF1 (D5E4), Stat (1/stat1), PKR (EPR19374, IRF7 (MNGPKL), Blimp1 (5E7), FoxP3 (MF-14, FJK-16s), IRF2 (EPR4644(2))] prepared in the permeabilization buffer from the same kit, for 30 min at 37°C. Samples were then washed and, where necessary, the intracellular staining was repeated using secondary fluor-tagged antibodies.
Single-cell suspensions of tumor cells were counted and restimulated with 2 mg/ml of MHC class I-restricted LCMV peptide GP33–40 in standard complete T cell medium (RPMI supplemented with 10% FBS, 1% penicillin and streptomycin, HEPES, 1% Sodium pyruvate, 1% non-essential amino acids, 1% L-glutamine and 2-mercaptoethanol) containing 50 U/mL recombinant murine IL-2 and 1 mg/mL brefeldin A (Sigma), at 37°C. After 5 hours, cells were stained with a zombie aqua viability stain (Biolegend) and then stained for surface molecules using antibodies against CD45.2 (104), CD8α (53–6.7), CD4(GK1.5), Tcrβ (H57–597) antibodies. Cells were then fixed and permeabilized (Biolegend cytokine staining kit) and then stained with anti-IFNγ (XMG1.2) and anti-TNFα (MP6-XT22), both from BioLegend. Cells cultured similarly but in the absence of the GP33–40 peptide served as negative controls when quantifying cytokine production. Samples were then washed and resuspended in FACS staining buffer and acquired using a FACSVerse or FACSLyric analyzer (BD Biosciences). Data was analyzed using FlowJo software (v.9 or v.10, Treestar) and the GraphPad Prism v8 or v9.
IFNβ stimulation of mouse cells
Single-cell suspensions of splenocytes from naïve mice were counted and incubated with 100U/mL IFNβ for 8 hours in T cell RPMI medium supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin and 1% L-glutamine. Cells were then surface stained for CD8 (53–6.7), CD4 (GK1.5), B220 (RA3–6B2) and then intracellularly stained for FoxP3 (MF-14) and IRF2 [EPR4644(2), Abcam].
In vivo antibody treatments
All in vivo antibodies were purchased from BioXcell, diluted in PBS and injected intraperitoneally (i.p.). CD8+ and CD4+ T cells were depleted by administering 250μg of anti-CD8α (2.43) antibody or anti-CD4(GK1.5) one day prior to tumor injection. For late CD8+ T cell depletion, 250μg of anti-CD8α (2.43) antibody or isotype control (LTF-2) was injected from 3 weeks after MC38 tumor injection, for a total of three anti-CD8α treatments (every 2 days). To block PDL1 in vivo, 500μg of anti-PDL1 (10F.9G2) or isotype control (LTF-2) antibody treatment was initiated i.p. at 15 days after PyMT tumor cell initiation (when tumors in WT and Irf2−/− mice had reached ~50mm3 but were still comparable in size) and then every 3 days for a total of 5 treatments. To block IFN-I signaling in established MC38-tumors, mice were treated every 2 days with 500μg of anti-IFNAR (MAR1–5A3) of isotype control (mouse IgG1) for the first 2 treatments and 250μg for subsequent treatments. To block IFNγ signaling, 500μg of anti-IFNγ (XMG1.2) was administered every 2 days. Isotype control antibodies were injected in a similar fashion.
Cell Sorting and single-cell RNA Sequencing
Single-cell suspensions of tumor cells were counted and stained with a zombie aqua viability dye (Biolegend) and then surface stained with anti-CD45.2 (A20) antibody. Stained cells were washed and resuspended in RPMI medium supplemented with 10% FBS, 1% penicillin and streptomycin and 1% L-glutamine. At least 8 mice were pooled per group and CD45+ tumor-infiltrating cellsimmune cells were FACSorted on a Moflo Astrios (Beckman Coulter). Sorted cells were labelled with sample multi-plexing antibodies and AbSeq Ab-Oligos (antibody-oligonucleotides) using the BD Mouse Immune Single-Cell Multiplexing Kit (Cat. No. 633793). Whole transcriptome Analysis (WTA), AbSeq and Sample Tag libraries from approximately 32,000 cells were constructed using the BD Rhapsody™ WTA Amplification Kit (Cat. No. 633801, Becton, Dickinson and Company, Franklin Lakes, New Jersey) from cDNA encoded on beads after cell capture on the BD Rhapsody™ Single-Cell Analysis System, using the manufacturer’s recommended protocol. Briefly, the WTA library was amplified via random priming while in parallel, AbSeq and Sample Tag libraries were amplified by PCR after denaturation from cell capture beads. All libraries underwent index PCR to add full-length Illumina sequencing adapters and indices. Libraries were evaluated for quantity and quality on an Agilent Bioanalyzer, pooled and sequenced on an Illumina NovaSeq6000 instrument targeting approximately 2 billion reads on an S2 flowcell for paired-end 2×100 bp sequencing and 20% PhiX spike-in. Library preparation and sequencing were performed by The Centre for Applied Genomics, The Hospital for Sick Children, Toronto, Canada.
Seven Bridges pipeline:
Sequence reads of tumor (WT and Irf2−/−) cells were aligned to the GRCm38 mouse genome and demultiplexed using the standard BD Rhapsody™ WTA Analysis pipeline on the Seven Bridges server (https://www.sevenbridges.com). Cells annotated as multiplets or undetermined by the pipeline were excluded. All bioinformatics analyses were performed using customized scripts in R (v4.1.1) unless mentioned otherwise.
Seurat analyses:
Data was further analysed using the R Seurat (v 4.0.4) package (Hao et al., 2021). To filter out low quality cells and poorly expressed genes, we applied the following quality control steps: First, cells with <500 read counts, with >25% mitochondrial reads, and expressing <700 and >7000 genes in Tumor WT were removed. Second, cells with <500 read counts, with >20% mitochondrial reads, and expressing <700 and >6000 genes in Tumor IRF2−/− were excluded. Third, genes expressed in <10 cells in each condition were also excluded. Finally, doublets were removed using the Chord R package (v1.0.0). Gene expression (RNA assay) data was then normalized using the logNormalize method and the antibody-derived tags (ADTs) assay data using the CLR method. RNA assay was scaled and adjusted by regressing out cell cycle genes and mitochondrial proportion. PCA was performed on most variable genes. To cluster cells, we constructed a weighted nearest neighbor (WNN) graph using the FindMultiModalNeighbors function with the first 20 principal components (PCs) for the RNA assay and 12 PCs for the ADT assay. CD8+ T cells from both WT and Irf2−/− were identified using both RNA and ADT and reclustered (0.4 resolution). Markers of each cluster were identified for RNA and ADT assays using the FindAllMarkers functions with default parameters. Differential gene expression between clusters and conditions (WT and Irf2−/−) were performed using the FindMarkers function without filtering any gene with the following parameters: logfc.threshold=0, min.pct=0, min.diff.pct=0. The gene list was used to rank the genes for the GSEA analyses and to identify differentially expressed genes (DEGs). DEGs were identified by filtering out genes with an FDR> 0.05, with an absolute value of log2 fold change <0.25 and expressed in less than 1% in either of the two compared subpopulations. RNA gene expression was also analyzed using SeqGeq software (v.1.6 or 1.7, BD Biosciences).
Gene set enrichment analysis (GSEA):
We performed a GSEA analysis to test whether EFFECTOR_VS_EXHAUSTED_CD8_TCELL (UP and DN) from C7 of MSigDB (Liberzon et al., 2011; Subramanian et al., 2005) and DEGs in TOX−/− CD8+ T cells were enriched for up- or down-regulated genes in c3.Irf2−/− vs c0.WT. The up- and down-regulated genes in TOX−/− gene sets were taken from (Khan et al., 2019) and filtered to keep genes with adjusted P-value < 10−5. DEGs in c3.Irf2−/− vs c0.WT were identified by the FindMarkers function, as mentioned previously, without filtering any genes, and were ranked by sign(log2FC) * -log10(pval), where log2FC is the log2 fold change of average of gene expression in IRF2−/− and average in WT and pval represents the P-value calculated using Wilcoxon rank-sum test. Enriched pathways were identified using the fgsea R package (v1.18.0) with 1,000 permutations. the following parameters: minSize=5, maxSize=500, nPermSimple=100000, eps=0, nproc=1 and the Hallmark gene sets taken from
Enrichment Map:
To identify biological processes enriched in c0.WT and c3.Irf2−/− cells, we performed a GSEA analysis for RNA assay data and Gene Ontology Biological Processes gene sets using the GSEA application with default parameters. An Enrichment Map of the GSEA results was generated using the Enrichment Map Visualization tool from the GSEA application. Gene sets included in the Enrichment Map have a P-value <0.005 and an FDR <0.05 and a cutoff of 0.5 for the overlap coefficient used as a similarity metric between the different gene sets. Enrichment Map was illustrated in Cytoscape (Shannon et al., 2003).
Upstream regulator analysis:
Predicted upstream regulator analysis of differentially expressed genes of c3.Irf2−/− vs c0.WT was performed using the Ingenuity Pathway Analysis software (Qiagen) according to the developer’s instructions.
SCENIC analyses:
We assessed regulon activity in c0.WT and c3.Irf2−/− CD8+ T cells by performing an analysis of the gene regulatory network using the pyscenic implementation of SCENIC (Aibar et al., 2017) consisting of three steps: 1) identifying regulons based of TF-targets co-expression using GRN, 2) pruning regulons to keep direct targets of TFs using RcisTarget, and finally 3) assessing regulon activity score (RAS) for each regulon on cellular level using AUCell. Due to the stochastic nature of GRN, SCENIC might generate slightly different results on each run. To define regulons in a robust manner, we ran the first two steps of SCENIC using arboreto_with_multiprocessing and ctx functions from pyscenic library 100 times with default parameters. Results of the 100 runs were aggregated as follows: TFs and TF-target pairs occurring 70 out of 100 times were retained. Regulons were redefined using the retained TFs and TF-target pairs. Then RAS scores were generated and binarized using aucell and binarize functions from the pyscenic library. RAS scores of the two clusters were compared using the Wilcoxon rank-sum test in R and p-values were adjusted using the FDR method. We retained regulons having an FDR < 0.05, an absolute value of log2 fold change (c3.Irf2−/− / c0.WT) > 0.25 and regulons active in at least 10% of the cells of either of the two compared clusters.
ATAC-seq
Single-cell suspensions of WT, Irf2−/−, CD8-IRF2cWT and CD8-IRF2cKO MC38-infiltrating CD8+ T cells were isolated using autoMACS (Miltenyi Biotec), washed and treated with DNase I, and nuclei isolated according to the 10x Genomics nuclei isolation for single cell ATAC sequencing protocol. ATAC libraries were prepared and sequenced (to a target depth of 25,000 reads per cell) on the Novaseq6000 at the Princess Margaret Genomics Center, following the 10x Genomics sequencing workflow and protocol.
Reads alignment and quantification:
base calls were generated using Illumina RTA v3.4.4 as bcl files which were then converted into fastq files using bcl2fastq v2.20 with default parameters. Reads in fastq files were aligned using cellranger-atac count (v 2.0.0) to the reference mm10 genome (i.e., refdata-cellranger-arc-mm10–2020-A-2.0.0). The chemistry parameter of cellranger-atac count function was set to ARC-v1, since only scATAC-Seq data was analysed from the multiome data.
Peaks calling, Quality control, and normalization:
R (v 4.1.3) and the Signac (v 1.6.0) R package were used for scATAC-Seq data analyses. Reads occurring in less than 10 cells and less than 200 genes were excluded from further analyses. Peaks were called using MACS2 v2.2.7.1 (Zhang et al., 2008) with the following parameters: format=‘BEDPE’, effective.genome.size=‘mm’, cleanup=F. Reads occurring in genomic blacklist regions as defined in (Amemiya et al., 2019) were identified. The following criteria were used to exclude outlier cells from subsequent analyses: 1) reads within peak < 2000 in CD8-IRF2cWT and CD8-IRF2cKO, and 800 in Irf2−/− and WT; 2) reads within peaks > the 99th percentile of reads within peaks in each sample; 3) percentage of unique reads occurring within peaks < 35% in CD8-IRF2cKO, 37% in CD8-IRF2cWT and 30% in Irf2−/− and WT, 4) ratio of reads in genomic blacklist region > 0.1 in CD8-IRF2cWT and CD8-IRF2cKO and 0.12 in Irf2−/− and WT, 5) nucleosome signal > 4, and 6) TSS enrichment score < 2. Term frequency-inverse document frequency (TF-IDF) was used for normalization using the Signac R package.
Peaks annotation and inference of IRF2 motifs:
The TFBStools (v 1.32.0) R package in combination with position weight matrices (PWM) taken from the JASPAR2020 (v 0.99.10) database were used to infer occurrence of IRF2 motifs within each peak. Promoters were predicted using the GenomicFeatures (v 1.46.1) R package as 3000 bases upstream and 3000 bases downstream the transcription start site. Peaks that occur at least once in at least 10 cells were considered accessible. The following parameters were used to annotate peaks using the annotatePeak function in the ChIPseeker (v 1.30.0) R package: tssRegion=c(−3000, 3000), level=‘gene’, genomicAnnotationPriority=c(“Promoter”, “5UTR”, “3UTR”, “Exon”,”Intron”, “Downstream”, “Intergenic”). Finally, peaks were visualized using the CoveragePlot function from the R Signac package.
Human melanoma samples from published ATAC-seq data:
Triplicates of ATAC-seq data from PD1hi CD8+ T cells of a melanoma patient and from CD8+/CD45RA+ peripheral blood lymphocytes of healthy donors were obtained from published ATAC-seq data (Philip et al., 2017). Samples were aligned to the mm10 genome using bowtie v2.4.1, and the resulting bam files were further preprocessed using samtools v1.10 and picard v2.22.1. Peaks were called using MACS2 v2.2.7.1 (Zhang et al., 2008) with the following parameters: ‘-q 0.01 -nomodel --shift −100 --extsize 200’. Genome-wide IRF2 transcription binding site accessibility was predicted at a p-cutoff of 0.0005 using the TFBStools v1.28.0 R package (Tan and Lenhard, 2016) in coordination with position frequency matrices obtained via the JASPAR2020 v0.99.10 database (Fornes et al., 2020). Bedgraph files from each sample and a bed file of all predicted IRF2 binding sites were visualized using IGV v2.10.3. Additionally, pre-processed normalized counts for the ATAC data were obtained from GSE89308. Peak annotations were cross validated using the ChIPSeeker v1.26.2 R package (Yu et al., 2015). Differential accessibility was inferred using DESeq2 v1.30.1 (Love et al., 2014) using the apeglm v1.12.0 R package (Zhu et al., 2019) for the effect size shrinkage estimations. Finally, all gene-set enrichment analysis and over-representation analyses were done using the clusterProfiler v3.18.1 R package (Yu et al., 2012). Two type of gene sets were created for GSEA, one was comprised of transcription factors and all genes with their predicted binding site within their promoter region (TSS +/− 1kb), and the other comprised the standard mSigDB gene sets from the msigdb v7.4.1 R package (Liberzon et al., 2015).
CUT&Tag
Naïve CD8+ T cells were isolated from the spleens and lymph nodes of C57BL/6 by negative selection (Stem Cell Technologies). Isolated CD8+ T cells were activated for 3 days using 5μg/mL plate-bound anti-CD3 (16–0031-86 Invitrogen) and 2μg/mL soluble anti-CD28 (16–0281-86 Invitrogen) antibodies in 10% RPMI complete media containing 100U/ml rhIL2 (200–02 PeproTech). Nuclei were isolated and CUT&Tag performed using the protocol from Epicypher (EpiCypher® CUTANA™ Direct-to-PCR CUT&Tag Protocol v1.6 Revised: 11.04.2021). Prior to sequencing, samples were normalized based on their DNA concentrations and pooled. A high-output 75-cycle Nextseq kit [NextSeq 500/550 High Output Kit v2.5 (75 Cycles), 20024906 Illumina] was used for paired-end sequencing, giving each sample 10–12 million reads.
Data analysis:
Samples were aligned to the mm10 genome using bowtie v2.4.1 with the following parameters: ‘--local --very-sensitive --no-mixed --no-discordant --phred33 -I 10 -X 700’ and preprocessed using samtools v1.10. A bedgraph file was generated by scaling the reads by a scaling factor defined as 1,000,000/number_of_aligned_reads. Regions overlapping the ENCODE mm10 blacklist regions, as defined by Amemiya et al. (Amemiya et al., 2019) were then removed. Peaks were called using SEACR v1.3 (Meers et al., 2019) with a signal_threshold of 1 and run in `stringent` and `norm` mode. IRF2 motifs were then mapped to peaks using the catalogue of predicted IRF2 motifs in the mm10 genome described above. By calculating the proportion of peaks containing an IRF2 motif in the IRF2 sample, we established a minimum quantile-based cut-off of 0.81 for the total-signal which corresponded to the largest increase in IRF2-motif containing peaks. We did a similar analysis for peaks that were found to overlap between IRF2 and the IgG control; we calculated a maximum quantile-based cut-off of 0.82 for the IgG sample which corresponded to the largest drop off of IRF2-containing motifs in overlapping peaks. Using this refined IRF2 catalogue of peaks, we then performed an over-representation analysis of the HALLMARK and C7:IMMUNESIGDB categories of msigdbr v7.4.1 (Liberzon et al., 2015) using clusterProfiler v4.2.2 (Wu et al., 2021)]. Significant HALLMARK and IMMUNESIGDB “exhausted” related pathways were extracted at a BH-corrected p-value of 0.05. The DNA sequences corresponding to the IRF2 peaks were derived using the bedtools v2.27.1 getfasta tool (Quinlan and Hall, 2010), which was then used as an input for the HOMER v4.8 findMotifsGenome.pl (Duttke et al., 2019) tool with a size parameter of 200. Analysis of transcription factor motifs enriched in IRF2 target peaks was performed using HOMER and ranked based on their Target/Background scores. The Target/Background ratio reflects the enrichment of motifs in the target peaks divided by the enrichment of those motifs in a random HOMER simulated k-mer (k=3 nucleotides) background, i.e., the number of target-peaks that have the indicated motif divided by the number of background-peaks that have the same motif.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical parameters are described in the figure legends. In all figures, error bars indicate the standard error of the mean (SEM). Student’s t-tests (two-tailed, unpaired) for comparing two conditions and One-way ANOVA (two-tailed, unpaired) for multiple comparisons were performed using GraphPad Prism v8 or v9. For survival assays, log-rank analysis (Mantel-Cox test) was performed using GraphPad Prism software v8 or v9.
Supplementary Material
Table S2. scRNA-seq cluster-defining genes, Related to Figure 5.
Table S3. Differentially expressed genes for WT cluster 0 and Irf2−/− cluster 3, Related to Figure 5.
Table S4. Genes exhibiting differential chromatin accessibility in CD8+ TILs in WT vs Irf2−/− mice, and CD8-IRF2cWT vs CD8-IRF2cKO mice, Related to Figure 5.
Table S5. Genes containing IRF2 motif(s) in their promoter regions, Related to Figure 5.
Table S6. IRF2 bound and TF motifs enriched in IRF2 target genes by CUT&TAG, Related to Figure 5.
Table S7. IPA-predicted upstream regulators for WT cluster 0 and Irf2−/− cluster 3, and Gene Ontology Biological Processes, Related to Figure 6.
Highlights.
IRF2 expression by CD8+ TILs drives T cell exhaustion
IRF2 drives the suppressive program induced by interferons
IRF2-deletion enables CD8+ T cell-driven control of multiple tumor types
IRF2 deficiency boosts efficacy of adoptive cell and immune checkpoint therapies
Acknowledgements
We thank past and present members of the Brooks laboratory. This work was supported by the Canadian Institutes of Health Research (CIHR) Foundation Grant FDN148386, the National Institutes of Health (NIH) grant AI085043, and the Scotiabank Research Chair to (DGB); CIHR 162114 and NCI CA255670 grants (T.L.M.); NIH grant AI148403 (WC); and the Helena Lam Fellowship in Cancer Research (SL.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alspach E, Lussier DM, and Schreiber RD (2019). Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, Wesoly J, and Bluyssen HAR (2019). Direct Inhibition of IRF-Dependent Transcriptional Regulatory Mechanisms Associated With Disease. Front Immunol 10, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Morita M, Bederman AG, Konieczny BT, Kissick HT, Sonenberg N, and Ahmed R (2017). Translation is actively regulated during the differentiation of CD8(+) effector T cells. Nat Immunol 18, 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltra JC, Bourbonnais S, Bedard N, Charpentier T, Boulange M, Michaud E, Boufaied I, Bruneau J, Shoukry NH, Lamarre A, and Decaluwe H (2016). IL2Rbeta-dependent signals drive terminal exhaustion and suppress memory development during chronic viral infection. Proc Natl Acad Sci U S A 113, E5444–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhaled GM, Harding S, and Brooks DG (2021). Opposing Roles of Type I Interferons in Cancer Immunity. Annual Review Pathology 16, 167–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovolenta C, Driggers PH, Marks MS, Medin JA, Politis AD, Vogel SN, Levy DE, Sakaguchi K, Appella E, Coligan JE, and et al. (1994). Molecular interactions between interferon consensus sequence binding protein and members of the interferon regulatory factor family. Proc Natl Acad Sci U S A 91, 5046–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briceno P, Rivas-Yanez E, Rosemblatt MV, Parra-Tello B, Farias P, Vargas L, Simon V, Cardenas C, Lladser A, Salazar-Onfray F, et al. (2021). CD73 Ectonucleotidase Restrains CD8+ T Cell Metabolic Fitness and Anti-tumoral Activity. Front Cell Dev Biol 9, 638037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, McGavern DB, and Oldstone MB (2006). Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Invest 116, 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhwani M, Mazzieri R, and Dolcetti R (2018). Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front Oncol 8, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae M, Kim K, Park SM, Jang IS, Seo T, Kim DM, Kim IC, Lee JH, and Park J (2008). IRF-2 regulates NF-kappaB activity by modulating the subcellular localization of NF-kappaB. Biochemical and biophysical research communications 370, 519–524. [DOI] [PubMed] [Google Scholar]
- Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, and Rao A (2019). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Chen S, Feng W, Li Z, Luo Y, and Zhu X (2021a). A STING-related prognostic score predicts high-risk patients of colorectal cancer and provides insights into immunotherapy. Ann Transl Med 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, Zheng S, Burns R, Taparowsky EJ, and Cui W (2021b). BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nat Immunol 22, 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Arai E, Khan O, Zhang Z, Ngiow SF, He Y, Huang H, Manne S, Cao Z, Baxter AE, et al. (2021c). In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280 e1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8, 793–800. [DOI] [PubMed] [Google Scholar]
- Drew PD, Franzoso G, Becker KG, Bours V, Carlson LM, Siebenlist U, and Ozato K (1995a). NF kappa B and interferon regulatory factor 1 physically interact and synergistically induce major histocompatibility class I gene expression. J Interferon Cytokine Res 15, 1037–1045. [DOI] [PubMed] [Google Scholar]
- Drew PD, Franzoso G, Carlson LM, Biddison WE, Siebenlist U, and Ozato K (1995b). Interferon regulatory factor-2 physically interacts with NF-kappa B in vitro and inhibits NF-kappa B induction of major histocompatibility class I and beta 2-microglobulin gene expression in transfected human neuroblastoma cells. Journal of neuroimmunology 63, 157–162. [DOI] [PubMed] [Google Scholar]
- Duttke SH, Chang MW, Heinz S, and Benner C (2019). Identification and dynamic quantification of regulatory elements using total RNA. Genome Res 29, 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, and Dymecki SM (2000). Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28, 106–110. [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao PS, Leung DY, Rafaels NM, Boguniewicz M, Hand T, Gao L, Hata TR, Schneider LC, Hanifin JM, Beaty TH, et al. (2012). Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J Invest Dermatol 132, 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grusdat M, McIlwain DR, Xu HC, Pozdeev VI, Knievel J, Crome SQ, Robert-Tissot C, Dress RJ, Pandyra AA, Speiser DE, et al. (2014). IRF4 and BATF are critical for CD8(+) T-cell function following infection with LCMV. Cell Death Differ 21, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. (1996). The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 85, 1009–1023. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, and Taniguchi T (1989). Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 58, 729–739. [DOI] [PubMed] [Google Scholar]
- Harada H, Kitagawa M, Tanaka N, Yamamoto H, Harada K, Ishihara M, and Taniguchi T (1993). Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and −2. Science 259, 971–974. [DOI] [PubMed] [Google Scholar]
- Hesterberg RS, Beatty MS, Han Y, Fernandez MR, Akuffo AA, Goodheart WE, Yang C, Chang S, Colin CM, Alontaga AY, et al. (2020). Cereblon harnesses Myc-dependent bioenergetics and activity of CD8+ T lymphocytes. Blood 136, 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, Sato T, Hirose S, Shirai T, Taki S, and Taniguchi T (2000). CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity 13, 643–655. [DOI] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal H, Ciucci T, Wang H, Tang W, Claudio E, Murphy PM, Bosselut R, and Siebenlist U (2021). The NF-kappaB regulator Bcl-3 restricts terminal differentiation and promotes memory cell formation of CD8+ T cells during viral infection. PLoS Pathog 17, e1009249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, and Ahmed R (2010). Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32, 91–103. [DOI] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegsman BA, Vangala P, Chen BJ, Meraner P, Brass AL, Garber M, and Rock KL (2019). Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J Immunol 203, 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Sun S, Chen W, Zhou Y, Huang Y, Liu X, Shan Y, and Wang C (2011). Negative feedback regulation of NF-kappaB action by CITED2 in the nucleus. J Immunol 186, 539–548. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukhele S, Boukhaled GM, and Brooks DG (2019). Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin Immunol 43, 101277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair F, Erickson JR, Voillet V, Simoni Y, Bi T, Tyznik AJ, Martin J, Gottardo R, Newell EW, and Prlic M (2020). A Targeted Multi-omic Analysis Approach Measures Protein Expression and Low-Abundance Transcripts on the Single-Cell Level. Cell Rep 31, 107499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A, and et al. (1993). Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97. [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Meers MP, Tenenbaum D, and Henikoff S (2019). Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Z, Wang G, Liang Z, Cui A, Xu A, Liu Y, Liu C, Yang Y, and Cui L (2017). Prognostic value of IRF-2 expression in colorectal cancer. Oncotarget 8, 38969–38977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ (2015). Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol 36, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TC, Hildeman D, Kedl RM, Teague TK, Schaefer BC, White J, Zhu Y, Kappler J, and Marrack P (2001). Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat Immunol 2, 397–402. [DOI] [PubMed] [Google Scholar]
- Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, Hogan PG, Rao A, and Trifari S (2017). Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A 114, E2776–E2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi PM, Pauken KE, Paley MA, Sharpe A, and Wherry EJ (2015). Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med 212, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong Ng H, Kim GD, Ricky Chan E, Dunwoodie SL, and Mahabeleshwar GH (2020). CITED2 limits pathogenic inflammatory gene programs in myeloid cells. FASEB J 34, 12100–12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Cui K, Zhang Z, and Zhao K (2015). Division of labor between IRF1 and IRF2 in regulating different stages of transcriptional activation in cellular antiviral activities. Cell Biosci 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouyez MC, Lestingi M, Charon M, Fichelson S, Buzyn A, and Dusanter-Fourt I (2005). IFN regulatory factor-2 cooperates with STAT1 to regulate transporter associated with antigen processing-1 promoter activity. Journal of immunology 174, 3948–3958. [DOI] [PubMed] [Google Scholar]
- Sakai T, Mashima H, Yamada Y, Goto T, Sato W, Dohmen T, Kamada K, Yoshioka M, Uchinami H, Yamamoto Y, and Ohnishi H (2014). The roles of interferon regulatory factors 1 and 2 in the progression of human pancreatic cancer. Pancreas 43, 909–916. [DOI] [PubMed] [Google Scholar]
- Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, Lopez-Moyado IF, Georges RO, Zhang W, Onodera A, et al. (2019). TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A 116, 12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, Gonzalez-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, Rao A, and Hogan PG (2021). BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol 22, 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharf R, Azriel A, Lejbkowicz F, Winograd SS, Ehrlich R, and Levi BZ (1995). Functional domain analysis of interferon consensus sequence binding protein (ICSBP) and its association with interferon regulatory factors. The Journal of biological chemistry 270, 13063–13069. [DOI] [PubMed] [Google Scholar]
- Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, and Wherry EJ (2009). A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110. [DOI] [PubMed] [Google Scholar]
- Snell LM, McGaha TL, and Brooks DG (2017). Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol 38, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell LM, Xu W, Abd-Rabbo D, Boukhaled G, Guo M, Macleod BL, Elsaesser HJ, Hezaveh K, Alsahafi N, Lukhele S, et al. (2021). Dynamic CD4(+) T cell heterogeneity defines subset-specific suppression and PD-L1-blockade-driven functional restoration in chronic infection. Nat Immunol 22, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G, and Lenhard B (2016). TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics 32, 1555–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Kawakami T, and Taniguchi T (1993). Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol Cell Biol 13, 4531–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, and Takaoka A (2001). A weak signal for strong responses: interferon-alpha/beta revisited. Nature reviews. Molecular cell biology 2, 378–386. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Valenzuela JO, Hammerbeck CD, and Mescher MF (2005). Cutting edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol 174, 600–604. [DOI] [PubMed] [Google Scholar]
- Vaughan PS, Aziz F, van Wijnen AJ, Wu S, Harada H, Taniguchi T, Soprano KJ, Stein JL, and Stein GS (1995). Activation of a cell-cycle-regulated histone gene by the oncogenic transcription factor IRF-2. Nature 377, 362–365. [DOI] [PubMed] [Google Scholar]
- Vaughan PS, van der Meijden CM, Aziz F, Harada H, Taniguchi T, van Wijnen AJ, Stein JL, and Stein GS (1998). Cell cycle regulation of histone H4 gene transcription requires the oncogenic factor IRF-2. The Journal of biological chemistry 273, 194–199. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu D, Chen P, Koeffler HP, Tong X, and Xie D (2008). Negative feedback regulation of IFN-gamma pathway by IFN regulatory factor 2 in esophageal cancers. Cancer research 68, 1136–1143. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu DP, Chen PP, Koeffler HP, Tong XJ, and Xie D (2007). Involvement of IFN regulatory factor (IRF)-1 and IRF-2 in the formation and progression of human esophageal cancers. Cancer research 67, 2535–2543. [DOI] [PubMed] [Google Scholar]
- Westgaard IH, Dissen E, Torgersen KM, Lazetic S, Lanier LL, Phillips JH, and Fossum S (2003). The lectin-like receptor KLRE1 inhibits natural killer cell cytotoxicity. J Exp Med 197, 1551–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ (2011). T cell exhaustion. Nat Immunol 12, 492–499. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. [DOI] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, Esplugues E, Wen R, Wang D, Parish IA, et al. (2015). A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Rep 13, 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Lamphier MS, Fujita T, Taniguchi T, and Harada H (1994). The oncogenic transcription factor IRF-2 possesses a transcriptional repression and a latent activation domain. Oncogene 9, 1423–1428. [PubMed] [Google Scholar]
- Yan Y, Zheng L, Du Q, Yan B, and Geller DA (2020). Interferon regulatory factor 1 (IRF-1) and IRF-2 regulate PD-L1 expression in hepatocellular carcinoma (HCC) cells. Cancer Immunol Immunother 69, 1891–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Lou G, Sun HW, Zhu Z, Sun Y, Chen Z, Chauss D, Moseman EA, Cheng J, D’Antonio MA, et al. (2021). BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8(+) T cells. Nat Immunol 22, 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y, Wu H, Gao Q, He HW, Li YW, Cai XY, Wang JX, Zhou J, Cheng YF, Jin JJ, et al. (2013). Interferon regulatory factor (IRF)-1 and IRF-2 are associated with prognosis and tumor invasion in HCC. Annals of surgical oncology 20, 267–276. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sakamoto A, Yamashita K, Arguni E, Horigome S, Arima M, Hatano M, Seki N, Ichikawa T, and Tokuhisa T (2006). Bcl6 controls granzyme B expression in effector CD8+ T cells. Eur J Immunol 36, 3146–3156. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. scRNA-seq cluster-defining genes, Related to Figure 5.
Table S3. Differentially expressed genes for WT cluster 0 and Irf2−/− cluster 3, Related to Figure 5.
Table S4. Genes exhibiting differential chromatin accessibility in CD8+ TILs in WT vs Irf2−/− mice, and CD8-IRF2cWT vs CD8-IRF2cKO mice, Related to Figure 5.
Table S5. Genes containing IRF2 motif(s) in their promoter regions, Related to Figure 5.
Table S6. IRF2 bound and TF motifs enriched in IRF2 target genes by CUT&TAG, Related to Figure 5.
Table S7. IPA-predicted upstream regulators for WT cluster 0 and Irf2−/− cluster 3, and Gene Ontology Biological Processes, Related to Figure 6.
Data Availability Statement
Single-cell RNA-seq, ATAC-Seq and CUT&Tag data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
All original codes (sc-RNA-Seq, scATAC-Seq and CUT&Tag analyses codes) have been deposited at the BrooksLab repository in github (https://github.com/diala-ar/BrooksLab) and are publicly available as of the date of publication. DOIs are listed in the key resources table
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Y(89)-anti-Lag3 (Clone C9B7W) | Biolegend | Cat# 125202; AB_961187 |
| 115In-anti-Ly6C (Clone HK14) | Biolegend | Cat#128001; RRID: AB_1134213 |
| 141Pr-anti-CD44 (Clone IM7) | Biolegend | Cat#103001; RRID: AB_312952 |
| 142Nd-anti-CXCR5 (Clone L138D7) | Biolegend | Cat#145502; RRID: AB_2561955 |
| 142Nd-anti-Siglec-F (Clone E50-2440) | BD Biosciences | Cat#552125; RRID: AB_394340 |
| 143Nd-anti-IRF2 (EPR4644(2)) | Abcam | Cat#ab229443; RRID: NA |
| 144Nd-anti-Granzyme B (Clone GB11) | ThermoFisher | Cat#MA1-80734; RRID: AB_931084 |
| 145Nd-anti-CTLA-4 (Clone UC10-4B9) | Biolegend | Cat#106302; RRID: AB_313251 |
| 146Nd-anti-EOMES (Clone Dan11mag) | eBioscience | Cat#14-4875-82; RRID: AB_11042577 |
| 147Sm-anti-CD45.2 (Clone 104) | Biolegend | Cat#109802; RRID: AB_313439 |
| 148Nd-anti-CD11b (Clone M1/70) | Fluidigm | Cat#3148003B; RRID: AB_2814738 |
| 148Nd-anti-ICOS (Clone C398.4A) | Fluidigm | Cat#3148019B; RRID: AB_2756435 |
| 149Sm-anti-CD69 (Clone H1.273) | Biolegend | Cat#104502; RRID: AB_313105 |
| 150Nd-anti-Ly6G (Clone 1A8) | Biolegend | Cat#127601; RRID: AB_1089179 |
| 150Nd-anti-CD73 (Clone TY/11.8) | Biolegend | Cat#127202; RRID: AB_1089066 |
| 151Eu-anti-CD25 (Clone 3C7) | Fluidigm | Cat#3151007B; RRID: AB_2827880 |
| 152Sm-anti-CD86 (Clone GL-1) | Biolegend | Cat#105007; RRID: AB_313150 |
| 153Eu-anti-CD8a (Clone 53-6.7) | Fluidigm | Cat#3153012B; RRID: AB_2885019 |
| 153Eu-anti-CD45.1 (Clone A20) | Fluidigm | Cat#3153002B; RRID:AB_268784 |
| 154Sm-anti-BATF (Clone D7C5) | Fluidigm | Cat#3154012A; RRID: AB_2687838 |
| 155Gd-anti-CD103 (Clone 2 × 107) | Biolegend | Cat#121401; RRID: AB_535944 |
| 155Gd-anti-CD103 (Clone M290) | BD Biosciences | Cat#553699; RRID: AB_394995 |
| 156Gd-anti-PD-L1 (Clone M1H5) | eBioscience | Cat#14-5982-82; RRID: AB_467781 |
| 158Gd-anti-RORɣt (Clone Q31-378) | BD Biosciences | Cat#562197; RRID: AB_10894594 |
| 159Tb-anti-CD39 (Clone 24DMS1) | eBioscience | Cat#14-0391-82; RRID: AB_1210501 |
| 160Gd-anti-CD4 (Clone rm4-5) | Biolegend | Cat#100505; RRID: AB_312708 |
| 161Dy-anti-T-bet (Clone 4B10) | Fluidigm | Cat#3161014B; RRID: AB_2858233 |
| 162Dy-anti-Foxp3 (Clone MF-14) | Biolegend | Cat#126401; RRID: AB_1089120 |
| 163Dy-anti-Tim-3 (Clone B8.2C12) | Biolegend | Cat#134002; RRID: AB_1626128 |
| 163Dy-anti-Bcl6 (Clone K112.91) | BD Biosciences | Cat#561520; RRID: AB_10713172 |
| 164Dy-anti-CD62L (Clone MEL-14) | Fluidigm | Cat#3164003B; RRID: AB_2885021 |
| 165Ho-anti-TCRb (Clone H57-597) | Biolegend | Cat#109201; RRID: AB_313424 |
| 166Er-anti-B220 (Clone RA3-6B2) | Invitrogen | Cat#14-0452-82; RRID: AB_467254 |
| 166Er-anti-Blimp1 (Clone ROS195G) | Biolegend | Cat# 648202; RRID:AB_2300132 |
| 167Er-anti-TCF-1 (Clone S33-966) | BD Biosciences | Cat#624084; RRID: NA |
| 168Er-anti-Bcl6 (Clone K112.91) | BD Biosciences | Cat#561520; RRID: AB_10713172 |
| 168Er-anti-PKR (Clone EPR19374) | Abcam | Cat#ab224887; RRID:N/A |
| 169Tm-anti-SLAM (Clone TC15-12F12.2) | Biolegend | Cat#115901; RRID: AB_313680 |
| 170Er-anti-NK1.1 (Clone PK136) | Fluidigm | Cat#3170002B; RRID: AB_2885023 |
| 171Yb-anti-CD80 (Clone 16-10A1) | Fluidigm | Cat#3171008B; RRID: AB_2885024 |
| 172Yb-anti-Ki67 (Clone B56) | BD Biosciences | Cat#550609; RRID: AB_393778 |
| 173Yb-anti-Helios (Clone 22F6) | Biolegend | Cat#137202; RRID: AB_10900638 |
| 174Yb-anti-CD11c (Clone N418) | Biolegend | Cat#117301; RRID: AB_313770 |
| 174Yb-anti-Lag3 (Clone C9B7W) | Fluidigm | Cat#3174019B; RRID:N/A |
| 175Lu-anti-PD1 (Clone RMP-30) | Biolegend | Cat#109101; RRID: AB_313418 |
| 176Yb-anti-Thy1.2 (Clone 53-21) | ThermoFisher | Cat#14-0902-82; RRID: AB_467379 |
| 209Bianti-MHC II (Clone M5/114.15.2) | Fluidigm | Cat#3209006B; RRID: AB_2885025 |
| Y(89)-anti-human CD45 (Clone HI30) | Fluidigm | Cat#3089003B; RRID:N/A |
| Cd(111)-anti-human CD80 (Clone BB1) | BD Biosciences | Cat#555681; RRID:AB_396033 |
| Cd(112)-anti-human CD45RO (Clone UCHL1) | Biolegend | Cat#304202; RRID: AB_314418 |
| Cd(114)-anti-human Helios (Clone 22F6) | Biolegend | Cat#137202; RRID:AB_10900638 |
| In(115)-anti-human SOCS1 (Clone 4H1) | EMD Millipore | Cat#04-002; RRID:AB_612104 |
| In(116)-anti-human Ki67 (Clone Ki-67) | Biolegend | Cat#350502; RRID:AB_10662385 |
| Pr(141)-anti-human CD45RA (Clone HI100) | Biolegend | Cat#304102; RRID:AB_314406 |
| ND(142)-anti-human HLA-DR (Clone L243) | Biolegend | Cat#307602; RRID:AB_314680 |
| ND(143)-anti-IRF2 (Clone EPR4644(2)) | Abcam | Cat#ab229443; RRID:N/A |
| ND(144)-anti-human CD33 (Clone WM53) | Biolegend | Cat#303402; RRID:AB_314346 |
| ND(145)-anti-human CXCR3 (Clone G025H7) | Biolegend | Cat#353706; RRID:AB_10962912 |
| ND(146)-anti-human CD8a (Clone RPA-T8) | Biolegend | Cat#301053; RRID:AB_2562810 |
| Sm(147)-anti-human CD4 (Clone RPA-T4) | Biolegend | Cat#300541; RRID:AB_2562809 |
| ND(148)-anti-human ICOS (Clone DX29) | BD Biosciences | Cat#557801; RRID:AB_396877 |
| Sm(149)-anti-human FoxP3 (Clone 236A-E7) | ThermoFisher | Cat#14-477782; RRID:AB_467556 |
| ND(150)-anti-human CD103 (Clone B-Ly7) | ThermoFisher | Cat#14-103882; RRID:AB_467412 |
| Eu(151)-anti-human CD39 (Clone A1) | Biolegend | Cat#328221; RRID:AB_2563747 |
| Sm(152)-anti-human CD11c (Clone Bu15) | Biolegend | Cat#337221; RRID:AB_2562834 |
| Eu(153)-anti-human CD3 (Clone UCHT1) | Biolegend | Cat#300443; RRID:AB_2562808 |
| Gd(155)-anti-human CD303 (Clone 201A) | Biolegend | Cat#354215; RRID:AB_2563739 |
| Gd(156)-anti-human CD14 (Clone M5E2) | Biolegend | Cat#301843; RRID:AB_2562813 |
| Gd(158)-anti-human CD27 (Clone O323) | Biolegend | Cat#302839; RRID:AB_2562817 |
| Tb(159)-anti-human CD19 (Clone HIB19) | Biolegend | Cat#302247; RRID:AB_2562815 |
| Gd(160)-anti-human CD25 (Clone M-A251) | Biolegend | Cat#356102; RRID:AB_2561752 |
| Dy(161)-anti-human Tbet (Clone 4B10) | Biolegend | Cat#644802; RRID:AB_1595503 |
| Dy(162)-anti-human CD28 (Clone CD28.2) | Biolegend | Cat#302937; RRID:AB_2563737 |
| Dy(163)-anti-human CD137 (Clone 4B4-1) | Biolegend | Cat#309802; RRID:AB_314781 |
| Dy(164)-anti-human CD86 (Clone 2331) | BD Biosciences | Cat#555655; RRID:AB_396010 |
| Ho(165)-anti-human PD1 (Clone EH12.2H7) | Biolegend | Cat#329941; RRID:AB_2563734 |
| Er(166)-anti-human Tim3 (Clone F38-2E2) | Biolegend | Cat#345019; RRID:AB_2563790 |
| Er(167)-anti-human PKR (Clone 6H3A10) | Novus Biological | Cat#NBP2-37242; RRID:N/A |
| Er(168)-anti-human CD73 (Clone AD2) | Biolegend | Cat#344002; RRID:AB_2154067 |
| Tm(169)-anti-human CTLA4 (Clone 14D3) | ThermoFisher | Cat#14-152982; RRID:AB_467512 |
| Er(170)-anti-human CD317 (Clone RS38E) | Biolegend | Cat#348402; RRID:AB_10588013 |
| Yb(171)-anti-human Granzyme B (Clone GB11) | ThermoFisher | (Special order) |
| Yb(172)-anti-human CD127 (Clone EBioRDR5) | ThermoFisher | Cat#14-1278-8; RRID:N/A |
| Yb(173)-anti-human NKP46 (Clone 9 × 102) | Biolegend | Cat#331902; RRID:AB_1027637 |
| Yb(174)-anti-human TCF-1 (Clone S33-966) | BD Biosciences | (Special order) |
| Lu(175)-anti-human CD274 (Clone 29E.2A3) | Biolegend | Cat#329719; RRID:AB_2565429 |
| Yb(176)-anti-human Lag3 (Clone 7H2C65) | Biolegend | Cat#369202; RRID:AB_2616877 |
| Bi(209)-anti-human CD11b (Clone ICRF44) | Fluidigm | Cat#3209003B; RRID:N/A |
| APC anti-mouse/human CD11b (clone M1/70) | Biolegend | Cat#101211; RRID: AB_312794 |
| APC/Fire 750 anti-mouse CD11c (clone N418) | Biolegend | Cat#117351; RRID: AB_2572123 |
| Brilliant Violet 421 anti-mouse CD11c (clone N418) | Biolegend | Cat#117343; RRID: AB_2563099 |
| PerCP/Cyanine5.5 anti-mouse CD45.1 (clone A20) | Biolegend | Cat#110728; RRID:AB_893346 |
| APC anti-mouse CD45.2 (clone 104) | Biolegend | Cat#109814; RRID:AB_389211 |
| PerCP/Cyanine5.5 anti-mouse CD45.2 (clone 104) | Biolegend | Cat#109828; RRID:AB_893350 |
| APC/Fire 750 anti-mouse CD4 (clone GK1.5) | Biolegend | Cat#100459; RRID: AB_2572110 |
| Brilliant Violet 605 anti-mouse CD4 (clone GK1.5) | Biolegend | Cat#100547; RRID:AB_11125962 |
| Brilliant Violet 711 anti-mouse CD8α (clone 53-6.7) | Biolegend | Cat#100747; RRID:AB_11219594 |
| PerCP/Cyanine5.5 anti-mouse CD8α (clone 53-6.7) | Biolegend | Cat#100734; RRID:AB_2075238 |
| PerCP/Cyanine5.5 anti-mouse B220 (clone RA3-6B2) | Biolegend | Cat#103236; RRID:AB_893354 |
| Alexa Fluor 700 anti-mouse B220 (clone RA3-6B2) | Biolegend | Cat#103231; RRID:AB_493716 |
| Brilliant Violet 421 anti-mouse Tcrβ (clone H57-597) | Biolegend | Cat#109229; RRID:AB_10933263 |
| PerCP/Cyanine5.5 anti-mouse Tcrβ (clone H57-597) | Biolegend | Cat#109228; RRID:AB_1575173 |
| Brilliant Violet 421 anti-mouse FoxP3 (clone MF-14) | Biolegend | Cat#126419; RRID:AB_2565933 |
| Alexa Fluor 488 anti-mouse FoxP3 (clone MF-14) | Biolegend | Cat#126406; RRID:AB_1089113 |
| PerCP/Cyanine5.5 anti-mouse NK1.1 (clone PK136) | Biolegend | Cat#108728; RRID:AB_2132705 |
| Pacific Blue anti-mouse Ly6C (clone HK1.4) | Biolegend | Cat#128014; RRID:AB_1732079 |
| PerCP/Cyanine5.5 anti-mouse Ly6G (clone 1A8) | Biolegend | Cat#127616; RRID:AB_1877271 |
| FITC anti-mouse Granzyme B (clone GB11) | Biolegend | Cat#515403; RRID:AB_2114575 |
| Pacific Blue anti-mouse Granzyme B (clone GB11) | Biolegend | Cat#515407; RRID:AB_2562195 |
| PE anti-mouse TCF1 (clone S33-966) | BD Bioscience | Cat#564217; RRID:AB_2687845 |
| APC anti-mouse Tbet (clone 4B10) | Biolegend | Cat#644814; RRID:AB_10901173 |
| FITC anti-mouse Ki67 (clone 35/Ki-67RUO) | BD Bioscience | Cat#556026; RRID:AB_396302 |
| FITC anti-mouse CD44 (clone IM7) | Biolegend | Cat#103006; RRID:AB_312957 |
| Brilliant Violet 421 anti-mouse CD25 (clone PC61) | Biolegend | Cat#102043; RRID:AB_2562611 |
| PECy7 anti-mouse CD62L (clone MEL-14) | Biolegend | Cat#104418; RRID:AB_313103 |
| Brilliant Violet 421 anti-mouse PDL1 (clone 10F.9G2) | Biolegend | Cat#124315; RRID:AB_10897097 |
| PECy7 anti-mouse CD39 (clone (24DMS1) | eBioscience | Cat#25-0391-82; RRID:AB_1210766 |
| APC anti-mouse Lag3 (clone C9B7W) | Biolegend | Cat#125210; RRID:AB_10639727 |
| Brilliant Violet 786 anti-mouse PD1 (clone 29F.1A12) | Biolegend | Cat#135225; RRID:AB_2563680 |
| PE anti-mouse Tox (clone txrx10) | eBioscience | Cat#12-6502-82; RRID:AB_10855034 |
| PE anti-mouse BATF (clone D7C5) | Cell Signaling | Cat#27120; RRID:AB_2798938 |
| PE anti-mouse IRF4 (clone IRF4.3 × 104) | Biolegend | Cat#646403; RRID:N/A |
| Alexa Fluor 488 anti-mouse IRF1 (clone D5E4) | Cell Signaling | Cat#14028; RRID:AB_2798366 |
| PE anti-mouse IRF1 (clone D5E4) | Cell Signaling | Cat#12732; RRID:AB_2798008 |
| PE anti-mouse IRF7 (clone MNGPKL) | eBioscience | Cat#12-5829-80; RRID:AB_2572628 |
| anti-mouse PKR (clone EPR19374) | Abcam | Cat#ab184257; RRID:AB_2916271 |
| Alexa Fluor 647 anti-mouse Stat1 (clone 1/stat1) | BD Bioscience | Cat#558560; RRID:AB_647143 |
| Alexa Fluor 647 anti-mouse Blimp1 (clone 5 × 107) | Biolegend | Cat#150004; RRID:AB_2565618 |
| PE anti-mouse Blimp1 (clone 5 × 107) | Biolegend | Cat#150005; RRID:AB_2565991 |
| Alexa Fluor 647 donkey anti-Rabbit IgG (clone Poly4064) | Biolegend | Cat#406414; RRID:AB_2563202 |
| anti-IRF2 antibody (clone [EPR4644(2)]) | Abcam | Cat#ab124744; RRID:AB_10974405 |
| APC anti-mouse IFNγ (clone XMG1.2) | Biolegend | Cat#505810; RRID:AB_315404 |
| Brilliant Violet TNFα anti-mouse (clone MP6-XT22) | Biolegend | Cat#506328; RRID:AB_2562902 |
| InVivoMAb anti-mouse IFNAR-1 (clone MAR1-5A3) | BioXcell | Cat#BE0241; RRID:AB_2687723 |
| InVivoMAb anti-mouse IFNγ (clone XMG1.2) | BioXcell | Cat#BE0055; RRID:AB_1107694 |
| InVivoMAb anti-mouse PDL1 (clone 10F.9G2) | BioXcell | Cat#BE0101; RRID:AB_10949073 |
| InVivoMAb anti-mouse CD8α (clone 2.43) | BioXcell | Cat#BE0061; RRID:AB_1125541 |
| InVivoMAb anti-mouse CD4 (clone GK1.5) | BioXcell | Cat#BE0003; RRID:AB_1107642 |
| InVivoMAb rat IgG2b isotype control (clone LTF-2) | BioXcell | Cat#BE0090; RRID:AB_1107780 |
| InVivoMAb IgG1 isotype control (clone MOPC-21) | BioXcell | Cat#BE0083; RRID:AB_1107784 |
| Ms CD4 Olgo AMM2056 (clone GK1.5) | BD Bioscience | Cat#940471; RRID:AB_2876318 |
| Ms CD8b Olgo AMM2189 (clone H35-17.2) | BD Bioscience | Cat#940360; RRID:AB_2876231 |
| Ms TCRβ CHN Olgo AMM2021 (clone H57-597) | BD Bioscience | Cat#940125, RRID:AB_2876014 |
| Ms CD3e Olgo AMM2001 (clone 145-2C11) | BD Bioscience | Cat#940107; RRID:AB_2875997 |
| Biological samples | ||
| Human melanoma samples | Princess Margaret Cancer Biobank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Cell-ID Cisplatin | Fluidigm | Cat#201064 |
| Cell-ID Intercalator-Iridium—125 mM | Fluidigm | Cat#201192A |
| Zombie Aqua Fixable Viability Kit | Biolegend | Cat#423102 |
| Mouse IL-2 Recombinant Protein | GIBCO | Cat#PMC0025 |
| Brefeldin A | Sigma | Cat#B7651-5MG |
| DNase I | Sigma | Cat#DN25-1G |
| Cisplatin | BioVision | Cat#1550 |
| Collagenase I | ThermoFisher | Cat#17018029 |
| Fixation Buffer | Biolegend | Cat#420801 |
| Intracellular Staining Permeabilization Wash Buffer (10X) | Biolegend | Cat#421002 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| EasySep Mouse CD8+ T cell Isolation Kit | STEMCELL | Cat#19853 |
| 20-Plex Pd Barcoding Kit | Fluidigm | Cat#201060 |
| Mouse CD45+ (TIL) Microbeads | Miltenyi Biotec | Cat#110-021-618 |
| Fc block | ThermoFisher | Cat#16-9161-73 |
| Recombinant mouse IFNβ | R & D Systems | Cat#12401-1 |
| Deposited data | ||
| ATAC-seq data from human tumor-infiltrating PD1+ CD8+ T cells | (Philip et al., 2017) | GEO accession#: GSE89308 |
| Transcriptional features of Tox-deficient CD8+ T cells | Khan et al., (2019) | GEO accession#: GSE131871 |
| CUT&TAG | This manuscript | GEO accession#: GSE199177 |
| scATAC-Seq | This manuscript | GEO accession#: GSE199177 |
| scRNA-Seq | This manuscript | GEO accession#: GSE199177 |
| Original codes | This manuscript | https://github.com/diala-ar/BrooksLab |
| Experimental models: Cell lines | ||
| MMTV-PyMT cell line | Dr. Christopher PAGE (University Health Network) | N/A |
| PyMG tumor cells | Generated by our lab | N/A |
| B16-F10 tumor cells | Dr. Tracy McGaha (University Health Network) | N/A |
| MC38 tumor cells | Dr. Daniel de Carvalho (University Health Network) | N/A |
| MC38-GP cells | Generated by our lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J mice | Jackson laboratory | Cat#000664 |
| LCMV-GP33-specific CD8 TCR transgenic (P14) mice | Bred in our facility | N/A |
| Mouse: Irf2−/− | Provided by Dr. Tak Mak at the University Health Network (Matsuyama et al., 1993), and bred in our facility | N/A |
| Mouse: Irf2+/+ | Bred in our facility | N/A |
| Irf2fll/fl mice | generated and bred in our facility | N/A |
| CD8α-Cre (E8iii-Cre) transgenic mice (C57BL/6-Tg(CD8α-cre)1Itan/J, Stock No. 008766 | The Jackson Laboratory | Stock no. 008766 |
| Software and algorithms | ||
| Flow Jo version 9 and 10 | BD FLOWJO | http://www.flowjo.com/ |
| BD FACSsuite v1.4.0.7047 | BD Biosciences | https://www.bdbiosciences.com/ |
| Cytobank | Cytobank, Inc | https://www.cytobank.org |
| Graphpad Prism v8 and v9 | GraphPad Software, Inc | https://www.graphpad.com/ |
| R | R Core Team | https://www.r-project.org/ |
| UMAP | (McInnes et al., 2018) | RRID:SCR_018217 |
| PhenoGraph | (Levine et al., 2015) | RRID:SCR_016919 |
| diffcyt R | (Weber et al., 2019) | https://bioconductor.org/packages/release/bioc/html/diffcyt.html |
| HOMER v4.8 findMotifsGenome.pl | (Duttke et al., 2019) | N/A |
| Bedtools v2.27.1 getfasta tool | (Quinlan and Hall, 2010) | N/A |
| Ingenuity Pathway Analysis software | Qiagen | N/A |
| Cytoscape | (Shannon et al., 2003) | N/A |
| SeqGeq software | BD Biosciences | N/A |
| BD RhapsodyTM WTA Analysis pipeline | BD Biosciences | https://www.sevenbridges.com |
| SEACR v1.3 | (Meers et al., 2019) | https://github.com/FredHutch/SEACR |
| bowtie v2.4.1 | https://bowtie-bio.sourceforge.net/index.shtml | |
| cellranger-atac count (v 2.0.0) | 10X genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Integrative genomics viewer (IGV) | https://igv.org/ | |