

SUMMARY
Although KRAS has long been considered undruggable, direct KRASG12C inhibitors have shown promising initial clinical efficacy. However, the majority of patients still fail to respond. Adaptive feedback reactivation of RAS-mitogen-activated protein kinase (MAPK) signaling has been proposed by our group and others as a key mediator of resistance, but the exact mechanism driving reactivation and the therapeutic implications are unclear. We find that upstream feedback activation of wild-type RAS, as opposed to a shift in KRASG12C to its active guanosine triphosphate (GTP)-bound state, is sufficient to drive RAS-MAPK reactivation in a KRASG12C-independent manner. Moreover, multiple receptor tyrosine kinases (RTKs) can drive feedback reactivation, potentially necessitating targeting of convergent signaling nodes for more universal efficacy. Even in colorectal cancer, where feedback is thought to be primarily epidermal growth factor receptor (EGFR)-mediated, alternative RTKs drive pathway reactivation and limit efficacy, but convergent upstream or downstream signal blockade can enhance activity. Overall, these data provide important mechanistic insight to guide therapeutic strategies targeting KRAS.
In brief
Ryan et al. provide evidence that feedback reactivation of the RAS-MAPK pathway through wild-type NRAS and HRAS, as opposed to a shift in KRASG12C to its active GTP-bound state, can drive adaptive resistance to KRASG12C inhibitors in a KRASG12C-independent manner.
Graphical Abstract
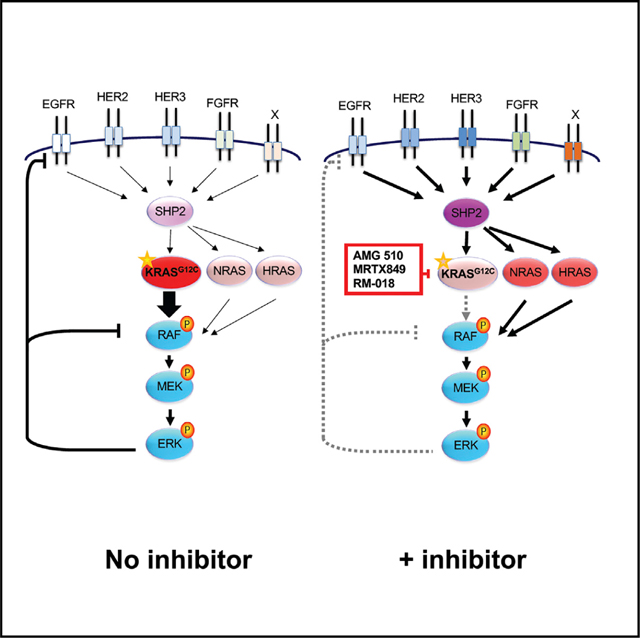
INTRODUCTION
RAS is the most frequently mutated oncogene in cancer and previously considered to be an undruggable target (Cox et al., 2014; Ryan and Corcoran, 2018). RAS family members (including KRAS, NRAS, and HRAS) act as a binary switch, cycling from a guanosine diphosphate (GDP)-bound “OFF” state to a guanosine triphosphate (GTP)-bound “ON” state, transmitting exogenous signals to downstream pathways within cells. Mutations at the G12 locus push KRAS into primarily a GTP-bound ON state and are GAP insensitive. However, the KRASG12C mutation undergoes intrinsic hydrolysis, allowing the protein to cycle back to the GDP-bound OFF state more rapidly than other G12 mutations. Recently, direct inhibitors of the KRASG12C mutation were developed to exploit this cycling of the KRASG12C mutation, and clinical-stage inhibitors AMG 510 (sotorasib) and MRTX849 (adagrasib), among others, function by locking the KRASG12C protein in the GDP-bound inactive state by accessing the Switch-II pocket and covalently binding to the mutant cysteine residue (Kim et al., 2020; Lito et al., 2016; Moore et al., 2020; Ostrem et al., 2013). Both clinical-stage inhibitors have demonstrated efficacy and tolerability in patients as single agents, with AMG 510 being approved by the US Food and Drug Administration (FDA) for non-small cell lung cancer (NSCLC) (Author Anonymous, 2020, 2021; Hong et al., 2020; Johnson et al., 2020). Overall, NSCLC patients respond to AMG 510 and MRTX849 at a higher rate than colorectal cancer (CRC) patients, 37% and 45% versus 10% and 22%, respectively (Fakih et al., 2022; Johnson et al., 2020; Riely et al., 2021; Skoulidis et al., 2021; Weiss et al., 2021). However, even in NSCLC, the majority of patients do not respond to therapy, and many patients with an objective response eventually progress on therapy, with onset of clinical resistance characterized by diverse mechanisms primarily converging on the RAS mitogen-activated protein kinase (MAPK) pathway (Awad et al., 2021; Tanaka et al., 2021).
Defining mechanisms of resistance to KRASG12C inhibition will be critical to improving this class of inhibitors and identifying clinical combination strategies. Preclinically, multiple upstream nodes and parallel pathways have been identified as potential drivers of resistance and combination strategies for KRASG12C GDP-state inhibitors (Fedele et al., 2021; Hallin et al., 2020; Lou et al., 2019; Misale et al., 2019; Molina-Arcas et al., 2019; Ryan et al., 2020). Many combination strategies converge on receptor tyrosine kinase (RTK) signaling in a context-dependent manner, and preclinical models demonstrate dependency on RTK (epidermal growth factor receptor [EGFR], human EGFR related [HER], fibroblast growth factor receptor [FGFR], and insulin growth factor receptor [IGFR]) or convergent SHP2 signaling in the presence of KRAS inhibition. However, the exact mechanism by which RTK signaling leads to RAS-MAPK pathway reactivation in the presence of KRASG12C inhibitors is unclear. One proposed mechanism is that increased RTK signaling shifts KRASG12C toward the GTP-bound active state, rendering GDP-state inhibitors, such as the preclinical inhibitor ARS-1620, unable to efficiently bind and block signaling through of KRASG12C (Lou et al., 2019; Xue et al., 2020). In addition, our previous work identified RTK-driven wild-type (WT) RAS activation as a potential driver of adaptive feedback across diverse KRASG12C mutant models, leaving the outstanding question of which mechanism of RAS feedback, KRASG12C reactivation or WT RAS activation, plays a dominant role in RAS-MAPK pathway reactivation.
Furthermore, as multiple RTKs have been implicated in driving adaptive feedback, it is unclear whether one or more specific RTKs drive feedback in a dominant manner in specific cancer types. For example, in CRC harboring a KRASG12C mutation, EGFR signaling has been proposed as the dominant driver of downstream RAS-MAPK reactivation in the presence of KRASG12C inhibitors and is a dependency that differs from NSCLC (Amodio et al., 2020). However, other studies have suggested that signaling from multiple RTKs can drive RAS-MAPK reactivation and that targeting a single RTK may not be universally effective. Deciphering between these possibilities will be key to developing effective combination strategies to overcome resistance.
Here, we evaluate the relative contributions of KRAS and WT RAS (NRAS and HRAS) to adaptive feedback resistance to both KRASG12C GDP-bound inactive-state and KRASG12C GTP-bound active-state inhibitors in a panel of CRC and non-CRC KRASG12C-mutant models. We observe that feedback reactivation of RAS-MAPK signaling is largely driven by an activation of WT NRAS and HRAS and that levels of active KRAS remain suppressed in the presence of KRASG12C inhibition. We also observe that loss of WT RAS function, either through gene knockdown or upstream pharmacologic inhibition via SHP2 or EGFR, can abrogate adaptive feedback reactivation of RAS-MAPK signaling. In KRASG12C CRC models, where feedback reactivation to single-agent KRASG12C inhibitor is most profound, we observed that, while EGFR co-inhibition is effective at suppressing adaptive resistance in many CRC models, we observe evidence of RAS-MAPK reactivation driven by additional RTKs, suggesting that co-targeting of EGFR alone may not be universally effective in CRC. Rather, we find that addition of a downstream inhibitor (e.g., MEK) or targeting a convergent upstream signaling node, such as SHP2, may lead to broader efficacy in CRC. Ultimately, our data suggest that adaptive feedback activation of wild-type RAS, driven by multiple RTKs, is a critical driver of adaptive resistance to KRASG12C inhibition and that combination strategies to block this feedback signaling (as opposed to strategies for enhanced targeting of KRASG12C alone) may be needed to overcome adaptive resistance.
RESULTS
Adaptive feedback reactivation of RAS-MAPK is observed despite sustained suppression of active KRAS by KRASG12C inhibitors
While initial clinical activity of the KRASG12C inhibitors sotorasib (AMG 510) and adagrasib (MRTX849) has been encouraging, the majority of KRASG12C CRC and NSCLC patients fail to respond to therapy. Thus, it is imperative to elucidate the vulnerabilities of current KRASG12C GDP-state inhibitors to guide the development of strategies to improve clinical efficacy. Prior studies from our group and others demonstrated that KRASG12C GDP-state inhibitors are susceptible to adaptive feedback of the key effector MAPK pathway downstream of mutant KRAS. However, much of the previous work characterizing adaptive feedback was performed with preclinical GDP-state inhibitors, such as ARS-1620, which are less potent than clinical inhibitors, such as AMG 510 and MRTX849. To determine whether adaptive feedback reactivation of RAS-MAPK signaling is observed in the presence of more potent clinical KRASG12C GDP-state inhibitors, we treated eight established and patient-derived KRASG12C-mutant cell models (including NSCLC, CRC, and pancreatic) with ARS-1620, AMG 510, or MRTX849 at their approximate 90% inhibitory concentration (IC90) over a time course treatment from 4 to 72 h (Figures 1A, S1A, and S1B). Despite showing effective inhibition of the MAPK pathway at 4 h, comparable rapid and robust reactivation of RAS-MAPK signaling was observed in the presence of each of the three inhibitors, such that phospho-ERK levels returned to an average of ~75% of baseline levels by just 72 h (Figure 1B). These data suggest that adaptive feedback is likely to be a key driver of resistance, even to more potent clinical KRASG12C inhibitors.
Figure 1. KRASG12C inactive GDP-state inhibitors are prone to adaptive feedback reactivation of the MAPK pathway that is not driven by KRAS.

(A) KRAS-G12C mutant cell lines were treated with AMG 510 (100 nM) for 0, 4, 24, 48, and 72 h. Blot analysis was performed for phospho- (p)MEK, pERK, pRSK, pAKT, and total MYC with GAPDH as a loading control.
(B) Densitometry of pERK normalized to GAPDH for blots in (A) and cell lines treated with ARS-1620 (10 μM) or MRTX849 (100 nM) for 4, 24, 48, and 72 h; results represent an average of pERK across all eight cell lines).
(C and G) Cell lines were treated with 10 μM ARS-1620 or 100 nM AMG 510 for 4, 24, 48, or 72 h either refreshed at each time point or not refreshed throughout the time course, and lysates were subject to a RAF-RBD pull-down and blot analysis of KRAS, NRAS, HRAS, and total RAS as well as pERK, pRSK, and GAPDH for input samples.
(D and H) Densitometry of pERK normalized to GAPDH for blots in (C) and (G).
(E and I) Densitometry analysis of KRAS-GTP levels normalized to input KRAS and GAPDH loading control for blots in (C) and (G).
(F and J) LC/MS analysis of ARS-1620 (10 μM) or AMG 510 (100 nM) drug levels in media over time incubated either alone at 37°C or with the H358 cell line.
As discussed above, two potential mechanisms have been proposed for how adaptive feedback reactivation can lead to RAS-MAPK reactivation despite the presence of KRASG12C inhibitor. First, increased RTK signaling may shift a greater fraction of KRASG12C into its active, GTP-bound state, to which current inhibitors of the inactive GDP-bound state are less able to bind. Second, increased RTK signaling can potentially activate WT RAS present in cancer cells, leading to downstream RAS-MAPK reactivation by effective “bypass” of KRASG12C altogether. Elucidating the respective contributions of each potential mechanism to adaptive resistance is critical, as the first mechanism might be surmountable by developing novel KRASG12C inhibitors capable of binding and inhibiting the active GTP-bound form of KRASG12C-GTP, while the second mechanism achieves resistance through a KRASG12C-independent mechanism that would not be surmountable by a KRASG12C inhibitor alone and may require combination strategies. Thus, determining whether one or both of these mechanisms play a dominant role in driving adaptive resistance will be key to designing future therapeutic strategies.
To determine the relative contributions of each potential mechanism to adaptive RAS-MAPK pathway reactivation, we performed isoform-specific RAS pull-downs to determine levels of the active GTP-bound states of each RAS isoform over time during treatment with either ARS-1620 or AMG 510. Interestingly, we observed that the suppression of KRAS-GTP achieved by each inhibitor was sustained over the 72-h period, and we did not detect a notable “rebound” in KRAS-GTP levels corresponding to the robust rebound in downstream MAPK signaling, measured by P-ERK levels, suggesting that the adaptive rebound in RAS-MAPK signaling may not be driven primarily by reactivation of KRASG12C in the presence of inhibitor due to an increased shift toward its active, GTP-bound form (Figures 1C–1E and 1G–1I). Notably, this finding contrasts with some prior studies (Xue et al., 2020), which have reported a rebound in KRAS-GTP levels over time following treatment with the preclinical KRASG12C inhibitor ARS-1620. One potential explanation for these distinct results may relate to differences in the experimental approaches used. In our studies, we replace media and drug every 24 h and 4 h prior to lysis to avoid any potential influence of drug instability. However, in some prior studies (Xue et al., 2020), drug is added at the start of the time course experiment only, such that a potential rebound in KRASG12C activity could be observed if drug instability led to a decrease in inhibitor concentration over time.
To distinguish between these two possibilities, we treated cells with either ARS-1620 (10 mM) or AMG 510 (100 nM) for a 72-h time course either refreshing or not refreshing drug for each time point prior to performing isoform-specific RAS-GTP pull-down to determine levels of active RAS. In cell lines treated with ARS-1620, KRAS-GTP levels rebounded only when drug was not refreshed, while phospho-ERK levels rebounded regardless of whether ARS-1620 was refreshed or not refreshed (Figures 1D and 1E). In comparison, AMG 510 was able to sustain KRAS-GTP inhibition over 72 h in all cell models, irrespective of whether drug was refreshed. A rebound in phospho-ERK levels was observed over time in both treatment conditions, despite lack of rebound in active KRAS-GTP levels (Figures 1G–1I).
To confirm whether potential drug instability of ARS-1620 in media culture conditions might be contributing to the observation of KRAS-GTP rebound over time when drug is not refreshed, we assessed drug levels of either ARS-1620 or AMG 510 by liquid chromatography (LC)-mass spectrometry (MS) over time incubated at 37°C in tissue culture media alone or in media incubated on the H358 cell line for 4 to 72 h. Interestingly, ARS-1620 levels dropped dramatically over time, by 75% over the course of 72 h and in media alone and by >95% when incubated with the H358 cell line. In contrast with ARS-1620, AMG 510 drug levels remained stable in media alone or media incubated with the H358 cell line over the course of 72 h (Figures 1F and 1J). Collectively, these results suggest that instability of ARS-1620 leading to a rapid decline in inhibitor concentration may explain the observation of KRAS-GTP rebound in some conditions and suggest that, if levels of either inhibitor are maintained, a notable rebound in KRASG12C activity is not observed.
Activation of wild-type RAS can sustain MAPK signaling in KRASG12C cells treated with KRASG12C inhibitors
Since we did not see clear evidence of KRAS-GTP rebound over 72 h following treatment with KRASG12C GDP-state inhibitors, we next evaluated whether a rebound in KRAS activity might be observed over longer periods of time. We treated cells for 7 days with multiple concentrations of ARS-1620 and AMG 510 prior to isoform-specific RAS pull-down. Both ARS-1620 and AMG 510 (with fresh media and inhibitor replaced every 24 h) were able to sustain KRAS inhibition over 7 days, yet phospho-ERK levels rebounded in all treatment conditions (Figure 2A). Importantly, while durable KRAS-GTP suppression was seen out to 7 days, levels of the active GTP-bound forms of both WT NRAS and HRAS increased by 7 days in all three cell lines treated with either ARS-1620 or AMG 510 (Figures 2A–2C). A similar increase in active GTP-bound WT NRAS and HRAS was observed in 72-h experiments (Figures 1C and 1G). Interestingly, an increase in total KRAS protein levels was observed over time following inhibitor treatment, as observed previously, yet KRAS-GTP levels remained suppressed. We did not observe consistent changes in total HRAS and NRAS protein levels, despite observing a consistent increase in NRAS-GTP and HRAS-GTP levels. Broadly, across eight cell models, KRAS inhibition as measured by active RAS pull-down was maintained in the presence of AMG 510, while NRAS- and HRAS-GTP levels increased over time (Figures 2D and S1C), mirroring our previously published results with ARS-1620 (Ryan et al., 2020). Similar to AMG 510, MRTX849 also sustained KRAS suppression in multiple cell models with increases in NRAS- and HRAS-GTP levels, suggesting a potential shared mechanism of bypass signaling driven by WT RAS activation and not an on-target loss of inhibitor efficacy on KRASG12C over time (Figure S1D).
Figure 2. Wild-type RAS drives adaptive feedback reactivation of the RAS MAPK pathway.

(A) SW1463, MIA PaCa-2, and H358 cell lines were treated with 1 or 10 μM ARS-1620 or 0.1 or 0.3 μM AMG 510 for 4 h or 7 days with drug refreshed every 2 days, and lysates were subject to a RAF-RBD pull-down and blot analysis of KRAS, NRAS, HRAS, and total RAS as well as pERK, pRSK, and GAPDH for input samples.
(B) Densitometry analysis of KRAS-GTP levels normalized to input KRAS and GAPDH loading control for blots in (A).
(C) Densitometry analysis of KRAS-, NRAS-, and HRAS-GTP levels normalized to input KRAS and GAPDH loading control for blots in (A).
(D and E) Densitometry analysis of KRAS-GTP, NRAS-GTP, and HRAS-GTP levels normalized to input RAS and GAPDH loading control of blots of cell lines treated with AMG 510 (100 nM) for 4, 24, 48, or 72 h in Figure S1C.
(F and H) SW1463, MIA PaCa-2, and H358 cell lines were subject to siRNA knockdown of NRAS, HRAS, and NRAS and HRAS and treated with AMG 510 (100 nM) or RM-018 (100 nM) for 24, 48, and 72 h. Blot analysis was performed for pMEK, pERK, pRSK, pAKT, and total NRAS, HRAS, KRAS, and MYC with GAPDH as a loading control.
(G and I) Densitometry of pERK normalized to GAPDH for blots in (F) and (H); results represent an average of pERK across three cell lines.
Statistical significance was evaluated by Student’s t test, where *p < 0.05 and **p < 0.01. ns, not significant.
To assess whether feedback activation of WT NRAS and HRAS might play a critical functional role in adaptive reactivation of RAS-MAPK signaling following treatment with KRASG12C inhibitors, we measured the degree of RAS-MAPK feedback reactivation following small interfering RNA (siRNA)-mediated knockdown of NRAS, HRAS, or both. We utilized siRNA-mediated knockdown, as cells did not tolerate simultaneous genetic ablation of both NRAS and HRAS through CRISPR-CAS9 editing. Transient knockdown of NRAS, HRAS, or both WT RAS proteins abrogated rebound in phospho-ERK levels when treated with AMG 510 for 4–72 h (Figures 2F and 2G). In all cell lines, the greatest reduction in phospho-ERK was seen with loss of both WT RAS proteins, and loss of NRAS appeared to have a greater effect on phospho-ERK rebound than loss of HRAS. The lack of RAS-MAPK rebound observed in the absence of WT NRAS and HRAS suggests that WT RAS activation may be a major driver of RAS-MAPK feedback reactivation in this setting.
To further delineate potential mechanisms of KRASG12C-dependent versus KRASG12C-independent pathway reactivation, we utilized the KRASG12C active-state inhibitor RM-018, which is able to covalently bind and inhibit the active GTP-bound form of KRASG12C (Schulze et al., 2019; Tanaka et al., 2021). If feedback reactivation of RAS-MAPK signaling was driven primarily or exclusively through KRASG12C reactivation via an increased shift toward its active GTP-bound state, this active-state inhibitor would be predicted to prevent pathway reactivation. However, we also observed adaptive reactivation of RAS-MAPK pathway over time following treatment with RM-018 (Figures 2H and 2I). Importantly, this feedback reactivation appeared dependent on signaling through WT NRAS and/or HRAS, as knockdown of NRAS and HRAS abrogated feedback in the presence of RM-018 in all cell lines tested, as measured by phospho-ERK (Figures 2G and 2H), mirroring results with AMG 510. Collectively, these data suggest that activation of WT RAS isoforms may play a dominant role in driving adaptive feedback reactivation in a KRASG12C-independent manner.
Upstream or downstream vertical inhibition strategies enhance the activity of KRASG12C inhibitors
Since our data suggest that bypass signaling occurring through WT RAS can be a key mediator of adaptive resistance to both KRASG12C GDP- or GTP-state inhibitors, we sought to further define the mechanisms by which feedback signaling is propagated. In the presence of either the inactive-state inhibitor AMG 510 or the active-state inhibitor RM-018 over 72 h, a sustained suppression of KRAS-GTP was observed, but reactivation of P-ERK accompanied by increased activation of NRAS and HRAS was observed (Figures 3A and 3B). Prior work by our group and others has shown that RTK-mediated feedback signaling into RAS can be attenuated by targeting a convergent signaling node, such as SHP2, which integrates signals from multiple RTKs into RAS (Fedele et al., 2021; Hallin et al., 2020; Lou et al., 2019; Ryan et al., 2020). When cells were treated with either inactive- or active-state inhibitors in combination with the SHP2 inhibitor RMC-4550 for a 72-h time course, we observed that upstream inhibition of SHP2 was able to block the increase in NRAS-GTP and HRAS-GTP levels observed following KRASG12C inhibition and mitigated reactivation of P-ERK. These findings support that signals upstream of SHP2, presumably RTK-mediated, are driving WT RAS activation and RAS-MAPK rebound in the presence of either active- or inactive-state KRASG12C inhibitors.
Figure 3. Vertical combination strategies abrogate adaptive response to KRASG12C inhibition.

(A) MIA PaCa-2 cells were treated with AMG 510 (100 nM) or RM-018 (100 nM) alone or in combination with the SHP2 inhibitor RMC-4550 (1 μM) for 4, 24, 48, or 72 h, and lysates were subject to a RAF-RBD pull-down and blot analysis of KRAS, NRAS, HRAS, and total RAS as well as pERK, pRSK, and GAPDH for input samples.
(B) Densitometry analysis of KRAS-GTP levels normalized to input KRAS and GAPDH loading control (bar) for blots and pERK normalized to GAPDH loading control (line) in (A). Data represent combined densitometry for MIA PaCa-2 in (A) and SW1463 and H358 in Figure S2A.
(C) Densitometry analysis of KRAS-GTP levels to input KRAS and GAPDH loading control and densitometry analysis of KRAS-GTP, NRAS-GTP, and HRAS-GTP levels normalized to input RAS and GAPDH loading control of blots of cell lines treated with AMG 510 alone or in combination with RMC-4550 or the MEK inhibitor trametinib (10 nM) in Figure S2C.
(D and E) Densitometry analysis of pERK normalized to loading control GAPDH for blots of KRAS12C mutant non-CRC and CRC subjected to indicated treatments in in Figures S2D and S2E.
(F and G) Densitometry analysis of KRAS-GTP, NRAS-GTP, and HRAS-GTP levels normalized to input RAS and GAPDH loading control of blots of KRASG12C-mutant CRC cell lines treated with AMG 510 alone or in combination with RMC-4550 or the EGFR inhibitor panitumumab (30 μg/mL) for 4 or 48 h in Figure S3A.
(H) Quantification of crystal violet stain of CRC cell lines treated with AMG 510 (100 nM), RMC-4550 (1 μM), panitumumab (30 μg/mL), trametinib (10 nM), or a combination for 10–14 days in Figure S3B.
While these studies suggest that the rebound in P-ERK over time following KRASG12C inhibition is not KRASG12C dependent and can occur in the presence of active- or inactive-state inhibitors, we did observe a marked difference in the degree of initial KRASG12C and P-ERK achieved. Interestingly, the active-state inhibitor RM-018 achieved a greater degree of initial KRAS-GTP suppression (>95%) compared with AMG 510 (~75%) by 4 h and a similarly greater degree of P-ERK suppression (Figures 3A and 3B). In addition, the maximal degree of KRAS-GTP inhibition achieved throughout the 72-h time course appeared greater with RM-018 than with AMG 510. Notably, co-inhibition of upstream signalling with the SHP2 inhibitor RMC-4550 did not further reduce KRAS-GTP in the presence of RM-018 but was able to further reduce KRAS-GTP levels in the presence of AMG 510, to levels comparable to those achieved with RM-018 alone. These data suggest that initial inhibition of KRASG12C activity by active-versus inactive-state inhibitors may be differentially affected by upstream signaling, which could alter the balance of KRASG12C in its active GTP-bound state. Thus, while feedback reactivation of RAS-MAPK signaling may occur in a KRASG12C-independent manner in the presence of active-state inhibitors, inhibition of the active state of KRASG12C could still provide advantages in terms of the depth and kinetics of target suppression.
Assessing the role of upstream inhibition in abrogating adaptive feedback, we noted that SHP2 co-inhibition increased the degree of KRAS-GTP inhibition in both short-term (4 h) and long-term (7 days) conditions and abrogated the induction of NRAS- and HRAS-GTP levels by AMG 510 long term in the NSCLC lung cell line H358, pancreatic ductal adenocarcinoma (PDAC) cell line MIA PaCa2, and CRC cell line SW1463 (Figures 3C, 3D, and S2C). Interestingly, in contrast to upstream inhibition, downstream inhibition with trametinib reduced only phospho-ERK levels while increasing WT RAS-GTP levels long term across all cell models (Figures 3C and S2C). Thus, while vertical inhibition with a downstream inhibitor may aid in suppressing downstream MAPK output, it may also increase upstream feedback into KRAS and WT RAS.
To investigate the efficacy of clinically relevant KRASG12C inhibitor combinations more broadly across tumor types, we treated KRASG12C non-CRC and CRC cell models with AMG 510 in combination with the EGFR monoclonal antibody (mAb) panitumumab, the SHP2 inhibitor RMC-4550, and the MEK inhibitor trametinib and measured adaptive rebound in phosphor-ERK (Figures 3D, 3E, S2D, and S2E). RMC-4550 and trametinib were broadly effective in abrogating phospho-ERK across both non-CRC and CRC cell models, while panitumumab was only effective in the CRC cell line subset, consistent with prior data suggesting that EGFR is a major driver of adaptive feedback in CRC (Corcoran et al., 2012; Amodio et al., 2020). Similar to the GDP-state inhibitor AMG 510, a SHP2 inhibitor combination was also able to reduce rebound in phospho-ERK in cell lines treated with the active GTP-state inhibitor RM-018 (Figures 3A, 3B, and S2C). In a panel of five KRASG12C CRC models, 4 and 48 h treatment with AMG 510 in combination with panitumumab or RMC-4550 reduced KRAS-GTP levels further than KRASG12C inhibition alone, with the most pronounced reduction of KRAS-GTP at 4 h. In addition, upstream inhibition with panitumumab or RMC-4550 also reduced the adaptive increase in levels of NRAS-GTP and HRAS-GTP after 48 h, demonstrating a dual mechanism of upstream inhibition combined with KRASG12C inhibition in CRC (Figures 3F, 3G, and S3A). In CRC, panitumumab and RMC-4550 were equivalent in enhancing KRASG12C reduction in cell growth over time, and trametinib also showed a similar reduction in growth (Figures 3H and S3B) Globally, upstream inhibition with SHP2 is effective in abrogating pERK rebound, while upstream inhibition with either EGFR or SHP2 is comparably effective in KRASG12C mutant CRC in reducing total RAS activity, phospho-ERK rebound, and in vitro growth.
KRASG12C CRC models display varied sensitivity to EGFR or MEK inhibitor combinations in vivo
Since CRC is believed to exert one of the more robust adaptive feedback responses among cancer types, we focused our subsequent studies on CRC models. EGFR has been identified as a major driver of adaptive resistance to KRASG12C inhibitors in CRC with EGFR combinatorial strategies for KRASG12C CRC patients under clinical evaluation (Amodio et al., 2020). In BRAFV600E CRC, EGFR combinations demonstrate efficacy in overcoming resistance to BRAF inhibitors in a preclinical setting and in the clinic (Corcoran et al., 2012; Prahallad et al., 2012; Tabernero et al., 2021). As clinical trials with KRASG12C inhibitor combinations are still in the nascent stages for CRC, modeling the most efficient inhibitor combinations preclinically may predict response in the clinic. Our in vitro experiments demonstrate that doublet combinations of either EGFR or SHP2 with KRASG12C inhibition display similar suppression of MAPK signaling and growth, yet a pool of residual phospho-ERK remains under each condition (Figure 3E). Triplet combination therapies have shown promise preclinically and have produced increased response rates clinically for BRAFV600E CRC (Corcoran et al., 2018; Hazar-Rethinam et al., 2018; Kopetz et al., 2019). Thus, we evaluated whether a triplet therapy of both upstream and downstream inhibition in combination with KRASG12C inhibitors might display superior inhibition of RAS-MAPK activity compared with doublet therapy. In a panel of five KRASG12C CRC models, a triplet combination of the EGFR inhibitor panitumumab or SHP2 inhibitor RMC-4550 with AMG 510 and MEK inhibitor trametinib showed superior MAPK inhibition than doublet combinations as measured by phospho-ERK levels (Figures 4A–4D, S4A, and S4B).
Figure 4. EGFR and MEK doublet and triplet therapies enhance the efficacy of KRASG12C inhibition in vivo.

(A and C) B8182 and F3008 PDX-derived KRASG12C-mutant CRC lines were treated with AMG 510 alone or in combination with panitumumab, RMC-4550, or trametinib for 24, 48, and 72 h. Blot analysis was performed for pMEK, pERK, pRSK, pAKT, and total MYC with GAPDH as a loading control.
(B and D) Densitometry of pERK normalized to GAPDH for blots in (A) and (C) and Figures S4A and S4B.
(E) Indicated KRASG12C CRC PDX models were treated daily with AMG 510 (100 mg/kg) and trametinib (1 mg/kg) and twice weekly with panitumumab (0.5 mg) alone or in combination for 21 days.
(F) Waterfall plots of endpoint tumors in (E); statistical significance was evaluated by Mann-Whitney test, where **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Thus, we compared doublet therapies with EGFR or MEK with a triplet combination of EGFR/KRASG12C/MEK inhibition in vivo in a panel of six patient-derived KRASG12C CRC xenograft models. CRC patient-derived xenograft (PDX) models revealed a heterogeneous response to KRASG12C inhibition as a single agent with both resistant models (B8182) and models with a partial response (B8026, C1047, and F3008) to AMG 510 (Figures 3E and 3F). When comparing doublet therapies of either panitumumab or trametinib with AMG 510 alone, a significant difference in response occurred in the B8182, C1144, C1177, and F3008 with both inhibition of EGFR/KRASG12C and KRASG12C/MEK. Triplet therapy of EGFR/KRASG12C/MEK had a significant additional benefit when compared with EGFR/KRASG12C or KRASG12C/MEK doublets in two models (B8026 and B8182). Thus, response to EGFR inhibition when combined with KRASG12C inhibition is heterogeneous in patient-derived CRC models, and the benefit of the triplet combination with MEK is observed in some models.
KRASG12C-mutant CRC RTK expression is heterogeneous and limits efficacy of EGFR inhibitor combinations
Although EGFR has been proposed as a key mediator of adaptive feedback in CRC, one-third of models did not show a significant improvement in efficacy with combined inhibition of KRASG12C and EGFR versus KRASG12C alone. One potential hypothesis to explain this lack of universal benefit could involve signaling contributions from other RTKs. Indeed, reverse-phase protein array (RPPA) of PDX tumors treated with the various inhibitor combinations of AMG 510 revealed induction of phosphorylated levels of several additional RTKs, as well as convergent mediators of RTK signaling, such as SHP2 and GAB2 (Figure 5A). These data suggest EGFR-independent RTK input could constrain the efficacy of KRASG12C/EGFR co-inhibition. To globally assess RTK signaling adaptation over time in response to KRASG12C inhibition, we treated two KRASG12C-mutant CRC models with AMG 510 and relevant therapeutic doublets over time and performed RPPA analyses (Figure 5B). Each cell line had a heterogeneous response to KRASG12C inhibition, with universal suppression of MAPK downstream protein expression (DUSP4 and DUSP6) and an upregulation of multiple RTKs. The SW837 cell line displayed an increase in HER2 phosphorylation and an increase in phosphorylation of the FGFR substrate FRS2α, while the SW1463 cell line showed an increase in phosphorylation of HER3. Both models showed a consistent increase in convergent downstream RTK signaling, with an increase in phosphorylation of the RTK-associated proteins SHP2 and SHC and an increase in total protein levels of GAB2. We next assessed RTK phosphorylation by an RTK array in an expanded panel of KRASG12C CRC models both basally and after treatment with AMG 510 alone or in combination with panitumumab or RMC-4550 for 72 h. All cell models expressed phosphorylated EGFR, which was either expressed highly basally (B8182, SW837, F3008, and SW1463) or increased in expression upon inhibitor treatment (LIM2099) (Figures 5C, S5A, and S5B). Other classes of phosphorylated RTKs expressed in multiple models were HER family receptors (HER2 and HER3) and FGFR family receptors (FGFR1 and FGFR3). However, no two models showed the same pattern of RTKs, highlighting the heterogeneity of KRASG12C CRC and complexity of developing a universal RTK inhibition strategy beyond targeting EGFR. We also assessed the phosphorylation of major classes of RTKs identified through RPPA and RTK arrays, EGFR, HER2, and FGFR3, over time in models treated with either AMG 510 alone or AMG 510 and panitumumab (Figures 5D and S5C). The patient-derived B8182 model expressed high levels of both phospho-HER2 and phospho-FGFR3 over time when treated with AMG 510, which were not affected by concurrent EGFR inhibition. The LIM2099 model showed low phospho-HER2 activation and no change in phospho-FGFR3, while the SW837 cell line showed no increase in phospho-HER2 and an increase in phospho-FGFR3.
Figure 5. Heterogeneous RTK expression limits the durability of EGFR treatment in KRASG12C, and SHP2 inhibition leads to deeper more durable response in vivo.

(A) Combined RPPA analysis of KRASG12C CRC PDX tumors treated with AMG 510 (100 mg/kg), panitumumab (0.5 mg), or trametinib (1 mg/kg) alone or in combination for 3 days.
(B) RPPA analysis of SW837 and SW1463 cell lines treated with the indicated inhibitors for 24 h, 72 h, or 7 days.
(C) RTK array expression analysis of CRC cell lines in Figures S5A and S5B.
(D) B8182, LIM2099, and SW837 cell lines were treated with AMG 510 alone or in combination with panitumumab for 24, 48, or 72 h. Blot analysis was performed for pEGFR, pHER2, pFGFR3, pSHP2, pMEK, pERK, pRSK, pAKT, and total MYC with GAPDH as a loading control.
(E–G) B8182, LIM2099, and SW827 cell lines were treated with AMG 510 alone or in combination with panitumumab or the FGFR inhibitor BGJ398 (1 μM), and densitometry analysis was performed for pERK normalized to GAPDH.
(H–J) B8182, LIM2099, and SW827 cell lines were treated with AMG 510 alone or in combination with panitumumab or RMC-4550, and densitometry analysis was performed for pERK normalized to GAPDH.
(K–M) B8182 PDX, LIM2099, and SW837 xenograft models were treated daily with AMG 510 (100 mg/kg) or RMC-4550 (30 mg/kg) or twice weekly with panitumumab (0.5 mg/kg) alone or in combination for 28, 66, and 35 days, respectively.
(N–P) Waterfall plots of endpoint tumors in xenograft models in (K)–(M).
Statistical significance was evaluated by Mann-Whitney test, where *p < 0.05, **p < 0.01, and ****p < 0.0001.
To address the contributions of additional RTKs to maintaining RAS-MAPK signaling in the presence of KRASG12C and EGFR co-inhibition, we treated KRASG12C CRC models with a triplet of combination of either the FGFR inhibitor BGJ398 or the SHP2 inhibitor RMC-4550 to inhibit upstream signaling (Figures 5E–5J). Collectively, RMC-4550 had a greater effect than BGJ398 in a panel of five KRASG12C-mutant CRC cell lines (Figures 5E–5J, S5D, and S5F). In the B8182 cell line, RMC-4550 has a greater effect on phospho-ERK levels initially than BGJ398 (24 and 48 h), but by 72 h, effect on phospho-ERK was similar with SHP2 or FGFR inhibition, suggesting that bypass feedback to EGFR may partially be driven by FGFR. Interestingly, we did not observe a consistent impact of these combinations on levels of phospho-AKT. In the LIM2099 model, panitumumab has a minimal effect when compared with RMC-4550 with KRASG12C inhibition and there is no difference between the KRASG12C/SHP2 doublet and the KRASG12C/EGFR/SHP2 triplet combination. In this same model, a doublet combination of FGFR had equivalent efficacy when compared with EGFR doublet combination and the triplet combination of KRASG12C/EGFR/FGFR has no added benefit as measured by phospho-ERK rebound. Finally, in the SW837 model, a triplet combination with either KRASG12C/EGFR/SHP2 or KRASG12C/EGFR/FGFR had added benefit over either doublet combination, demonstrating that bypass feedback to EGFR doublet treatment is largely FGFR driven. In all models, RAS activity as measured by phospho-ERK levels was not maximally suppressed by the combination of EGFR and KRASG12C, highlighting the contribution of other RTKs to adaptive feedback signaling in CRC.
SHP2i demonstrates greater efficacy and durability than EGFRi when combined with KRASG12Ci in vivo
Given the diversity of RTKs expressed in CRC beyond EGFR and upregulation of convergent RTK signaling nodes (pSHC, pSHP2, and GAB2) upon treatment with KRASG12C inhibitors, we investigated whether universal inhibition of RTK signaling with the SHP2 inhibitor RMC-4550 would also be equally as active as the EGFR mAb panitumumab in vivo in models of KRASG12C-mutant CRC. Our prior work showed that the preclinical SHP2 inhibitor SHP099 was effective in combination with the preclinical KRASG12C inhibitor in multiple in vitro and in vivo models of KRASG12C-mutant NSCLC, PDAC, and CRC (Ryan et al., 2020). In our current study, SHP2 inhibition showed equipotency as EGFR inhibition across multiple CRC models in abrogating adaptive reactivation of phospho-ERK and further reducing in vitro growth, except for the LIM2099 cell line, where SHP2 showed superior activity in vitro.
To assess the efficacy of SHP2 as a therapeutic combination strategy in KRASG12C-mutant CRC when compared with EGFR, we investigated the combination of KRASG12C inhibition with either SHP2 or EGFR inhibitors in vivo. While AMG 510 alone was able to reduce tumor growth in the B8182, LIM2099, and SW837 models when compared with the vehicle control group, surprisingly, there was a significant difference in efficacy and durability when comparing the AMG 510 and panitumumab treatment arms with the AMG 510 and RMC-4550 treatment arms (Figures 5K–5M). In the B8182 and LIM2099 models, AMG 510 and panitumumab treatment was equally as effective as AMG 510 and RMC-4550 early in the course of treatment; however, the efficacy in the AMG 510 and RMC-4550 treatment arm became significantly better than the AMG 510 and panitumumab arm over time. In the SW837 model, the AMG 510 and RMC-4550 treatment arm was more efficacious than the AMG 510 and panitumumab treatment arm throughout the treatment time course and only the AMG 510 and RMC-4550 arm was significantly different than AMG 510 treatment alone. Collectively, our results demonstrated the therapeutic potential of combining SHP2 inhibitors with KRASG12C inhibitors in KRASG12C CRC and greater efficacy and durability of treatment when compared with an EGFR treatment combination.
DISCUSSION
Despite promising initial activity in clinical trials, the majority of KRASG12C NSCLC and CRC patients still fail to respond to KRASG12C inhibitor monotherapy. As with previous efforts targeting the RAS-MAPK pathway, adaptive feedback reactivation of RAS-MAPK signaling appears to be a major driver of primary resistance. Previously, two potential mechanisms were proposed for how adaptive RAS-MAPK reactivation occurs after KRASG12C inhibition: (1) increased RTK signaling may shift a greater fraction of KRASG12C into its active, GTP-bound state, to which current inactive-state-specific inhibitors are less able to bind, and (2) increased RTK signaling can potentially activate WT RAS present in cancer cells, leading to downstream RAS-MAPK reactivation and bypass of KRASG12C altogether. WT RAS alleles have been shown to be critical in maintaining the growth and signaling of RAS-mutant cell lines, especially from extracellular signals through RTKs and flux through the MAPK pathway (Cheng et al., 2021; Young et al., 2013; Zhou et al., 2016). Determining the relative contributions of each potential resistance mechanism is critical to guide future therapies, as it will dictate whether next-generation or active-state KRASG12C inhibitors alone can solve the problem of adaptive resistance or whether strategies to address KRASG12C-independent resistance are needed.
Our study suggests that KRASG12C-independent feedback reactivation of the RAS-MAPK pathway driven by RTK-mediated activation of WT RAS (specifically, NRAS and HRAS) is a major driver, and perhaps the dominant driver, of adaptive resistance to KRASG12C inhibition. Indeed, even though robust rebound of RAS-MAPK activity was seen over time with all KRASG12C inhibitors evaluated, we observed that all inactive-state-specific inhibitors (AMG 510, MRTX849, and ARS-1620) maintained suppression of the active GTP-bound form of KRASG12C, even out to 7 days. Rather, through isoform-specific pull-down, we observed marked induction of the active GTP-bound forms of HRAS and NRAS (~3-fold on average), corresponding temporally with P-ERK rebound. Our data also suggest that prior studies reporting rebound of KRAS-GTP activity over time (Xue et al., 2020) may be due to the instability of specific KRASG12C inhibitors, such as ARS-1620, which showed a >80% decline in drug concentration over the first 24 h, as KRAS-GTP rebound was not observed if inhibitor was replenished over time. Moreover, we show that knockdown of HRAS and NRAS can abrogate adaptive feedback reactivation, suggesting that WT RAS activation may be the primary driver of adaptive RAS-MAPK reactivation in most models. Finally, we show that even a novel inhibitor of the active GTP-bound state of KRASG12C (RM-018) cannot prevent feedback reactivation of RAS-MAPK alone but that knockdown of NRAS and HRAS was again sufficient to block rebound. Collectively, our results implicate WT RAS activation as a critical mechanism of adaptive feedback resistance to KRASG12C inhibition.
These findings have important implications for developing novel strategies to overcome adaptive resistance. Specifically, because we observe KRASG12C-independent feedback reactivation, next-generation and/or active-state KRASG12C inhibitors alone may not be sufficient to overcome adaptive resistance. Although our data show that active-state inhibitors may have several key advantages, including more rapid and complete suppression of KRASG12C relative to inactive-state inhibitors (Figures 3A and 3B), we still observe RAS-MAPK reactivation over time due to WT RAS activation (Figures 2I and 2J). Rather, our data suggest that therapeutic combinations of KRASG12C inhibitors with upstream or downstream inhibition of the RAS-MAPK pathway may be needed to maintain suppression of RAS-MAPK activity. Accordingly, we observed that either upstream (e.g., SHP2) or downstream (e.g., MEK) inhibition led to more robust and sustained inhibition of RAS-MAPK signaling. Interestingly, although co-inhibition of KRASG12C and MEK led to more robust suppression of P-ERK levels relative to combinations with upstream inhibitors (Figures 3D and 3E), we also observed that this combination led to greater upstream feedback activation of RAS, as evidenced by increased levels of KRAS-GTP, as well as NRAS-GTP and HRAS-GTP (Figure 3C), raising the potential to drive activation of MAPK-independent RAS effector pathways. Early clinical data of combined KRASG12C and MEK inhibition was recently presented, and while the combination appeared to be tolerable, it is not yet clear whether or not this combination will yield improved efficacy (Ramalingam, 2021). Interestingly, our data also demonstrated that triple combinations of KRASG12C inhibitors with upstream and downstream inhibitors (either SHP2 + MEK in all cancers or EGFR + MEK in CRC specifically) demonstrated enhanced RAS-MAPK suppression. In CRC PDX models, triple inhibition of KRASG12C/EGFR/MEK led to significantly improved activity in two of six models relative to KRASG12C/EGFR or KRASG12C/MEK doublets alone but did not markedly enhance activity relative to the doublets in the remaining four of six models. As the tolerability of triple combinations could be more limited based on prior experience (Corcoran et al., 2018), further evaluation is needed to determine whether triplet strategies versus optimized doublet strategies represent the more promising approach.
To study the mechanisms of adaptive feedback in more detail, we focused on CRC, where adaptive feedback is believed to be most robust (Amodio et al., 2020; Corcoran et al., 2012). Indeed, the overall response rate of both AMG 510 and MRTX849 is lower in CRC versus NSCLC, 12% versus 38% and 22% versus 43%, respectively (Fakih et al., 2022; Riely et al., 2021; Skoulidis et al., 2021; Weiss et al., 2021). Preclinical studies have suggested that EGFR-driven adaptive feedback may be a key driver of KRASG12C inhibitor resistance in CRC (Amodio et al., 2020; Hong et al., 2020; Johnson et al., 2020). In addition, early clinical data for combinations of AMG 510 and MRTX849 with anti-EGFR antibodies have demonstrated improved efficacy in CRC patients with confirmed response rates of 28%–39% (Fakih et al., 2021; Weiss et al., 2021). This is similar to experience in BRAFV600E CRC, where targeting EGFR in combination with BRAF led to improved response rates of 20% versus ~5% with BRAF inhibition alone (Kopetz et al., 2015, 2017; Tabernero et al., 2021). However, in each scenario, EGFR antibody combinations only achieve responses in ~20%–30% of patients, suggesting that only a subset of CRCs are truly dependent on EGFR alone as a driver of adaptive resistance and that a majority of CRCs may achieve adaptive resistance through EGFR-independent mechanisms. While our data support that EGFR is a major driver of adaptive resistance in KRASG12C CRC, our phosphorproteomic data reveal that multiple additional RTKs are active or become activated upon KRASG12C inhibitor treatment and can drive EGFR-independent feedback reactivation in many CRC models. These data support the importance of EGFR-independent resistance mechanisms in KRASG12C CRC and highlight the potential need to block adaptive RTK signaling more universally. In line with this hypothesis, we find that co-targeting KRASG12C and SHP2, a convergent signaling node linking multiple RTK signals to RAS, led to more consistent suppression of RAS-MAPK signaling and enhanced durability of response relative to KRASG12C/EGFR combinations across CRC models. Although the tolerability of targeting convergent signaling nodes like SHP2 in combination with KRASG12C inhibitors in patients is not known and is currently under investigation, our data suggest that targeting convergent nodes modulating upstream feedback (such as with SHP2, SOS1, or pan-RAS inhibitors) represent a promising strategy to enhance efficacy that warrant further evaluation. Overall, these data, coupled with recent data detailing acquired resistance to KRASG12C inhibition through multiple alterations converging on RAS-MAPK reactivation (Awad et al., 2021; Tanaka et al., 2021), highlight the importance of achieving and maintaining robust RAS-MAPK inhibition as a critical challenge to improve efficacy in KRASG12C patients.
Limitations of the study
Our study defines the role of WT RAS (NRAS and HRAS) in driving adaptive feedback to both GDP- and GTP-state KRASG12C inhibitors. We describe the role of RTK activation driving NRAS and HRAS in sustaining MAPK signaling and characterize inhibitor combination strategies to tackle WT RAS-driven signaling both in vitro and in vivo. Our study does have limitations, in that we do not capture other RTK-driven pathways, such as the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway as potential drivers of therapeutic resistance to KRASG12C inhibitors. Our study is limited as well in aspects of feasibility and tolerability of proposed inhibitor combinations in the clinic, particularly the triple-inhibitor-therapy strategy in KRASG12C-mutant CRC. We find that WT RAS activation is a limiting factor to KRASG12C inhibitor efficacy when treating with sotorasib and cannot rule out a different pattern of response to other KRASG12C inhibitors, such as adagrasib, which has a higher objective response rate (ORR) clinically than sotorasib. Finally, our study does not rule out the contribution of KRAS reactivation to MAPK signaling over long duration of treatment with KRASG12C inhibitors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contacts, Dr. Ryan Corcoran (rbcorcoran@partners.org) and Dr. Scott Kopetz (skopetz@mdanderson.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
6–8 week-old mice were implanted with human tumor cell lines or patient derived tumors. For animal strain information, please see the key resources table for source of mice. All studies were performed on female mice. All mice were maintained in specific pathogen free housing and were used in accordance with regulations established by the Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC) or University of Texas MD Anderson Cancer Center.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Anti-GAPDH | EMD Millipore | Cat# MAB374 |
| Rabbit monoclonal Anti-HRAS | Proteintech | Cat# 18295-1-AP |
| Mouse monoclonal Anti-KRAS | Sigma | Cat# WH0003845M1 |
| Rabbit monoclonal Anti-MYC | Cell Signaling Technologies | Cat# 5605S |
| Mouse monoclonal Anti-NRAS | Santa Cruz Biotechnologies | Cat# sc-31 |
| Rabbit monoclonal Anti-phospho-AKT S473 | Cell Signaling Technologies | Cat# 4060S |
| Rabbit monoclonal Anti-phospho-EGFR Y1068 | Abcam | Cat# ab5644 |
| Rabbit monoclonal Anti-phospho | ||
| ERK1/2 T202/Y204 | Cell Signaling Technologies | Cat#4370S |
| Rabbit monoclonal Anti-phospho-FGFR3 Y724 | Abcam | Cat#ab155960 |
| Rabbit monoclonal Anti-phospho-HER2 Y1248 | Cell Signaling Technologies | Cat# 2247S |
| Rabbit monoclonal Anti-phospho-MEK S217/S221 | Cell Signaling Technologies | Cat#9154S |
| Rabbit monoclonal Anti-phospho-RSK1 S363/T359 | Abcam | Cat#ab32413 |
| Rabbit monoclonal Anti-phospho-SHP2 Y542 | Cell Signaling Technologies | Cat#3751S |
| Amersham ECL Mouse IgG, | ||
| HRP-linked whole Ab (from sheep) | GE Life Sciences | Cat# NA931-1ML |
| Amersham ECL Rabbit IgG, | ||
| HRP-linked whole Ab | GE Life Sciences | Cat# NA934-1ML |
| Biological samples | ||
| Patient Derived Xenografts (PDX) | University of Texas MD Anderson Cancer Center |
https://www.pdxnetwork.org/md-anderson |
| Chemicals, peptides, and recombinant proteins | ||
| AMG 510 (KRASG12C inhibitor) | MedChemExpress | Cat# HY-114277 |
| ARS-1620 (KRASG12C inhibitor) | Sellechckem | Cat# S8707 |
| BGJ398 (FGFR inhibitor) | Chemietek | Cat# CT-BGJ398 |
| MRTX849 (KRASG12C inhibitor) | MedChemExpress | Cat# HY-130149 |
| Panitumumab (Vectabix, EGFR mAb) | McKesson | Cat #765995 |
| RM-018 (KRASG12C inhibitor) | Revolution Medicines | N/A |
| RMC-4450 (SHP2 inhibitor, in vitro) | Selleckchem | S8718 |
| RMC-4550 (SHP2 inhibitor, in vivo) | Revolution Medicines | N/A |
| Trametinib (MEK inhibitor) | Selleckchem | Cat# S2673 |
| Critical commercial assays | ||
| Active Ras detection kit | Cell Signaling Technologies | Cat# 8821 |
| Human Phospho-Receptor | ||
| Tyrosine Kinase Array Kit | R&D systems | Cat # ARY001B |
| Lipofectamine RNAiMAX | Life Technologies | Cat# 13778100 |
| Thermo ScientificTM SuperSignalTM West | ||
| Femto Chemiluminescent Substrate | Thermo Fisher | Cat# PI34096 |
| Experimental models: Cell lines | ||
| Human B8182 colorectal adenocarcinoma | MD Anderson cell line bank | Katsiampoura et al. (2017)(1) |
| Human F3008 colorectal adenocarcinoma | MD Anderson cell line bank | Katsiampoura et al. (2017)(1) |
| Human LIM-2099 colorectal adenocarcinoma | Millipore Sigma | Cat# 12062002-1VL |
| Human MGH-1088 lung adenocarcinoma | Center for Molecular Therapeutics at the MGH Cancer Center | Crystal et al., 2014 (2) |
| Human MGH-1062 lung adenocarcinoma | Center for Molecular Therapeutics at the MGH Cancer Center | Crystal et al., 2014 (2) |
| Human MIA PaCa-2 pancreatic adenocarcinoma | ATCC | Cat# CRL-1420 |
| Human NCI-H358 lung adenocarcinoma | ATCC | Cat# CRL-5807 |
| Human SW837 colorectal adenocarcinoma | Center for Molecular Therapeutics at the MGH Cancer Center | ATCC, Cat# CCL-235 |
| Human SW1463 colorectal adenocarcinoma | ATCC | Cat# CCL-234 |
| Experimental models: Organisms/strains | ||
| Mouse, Nude, Crl:NU(NCr)-Foxn1nu, 6-8 wk female | Charles River Laboratories | N/A |
| Mouse, NOD/SCID, NOD.CB17-Prkdcscid/NCrCrl 8-week old female mice | Charles River Laboratories | N/A |
| Oligonucleotides | ||
| ON-Target Plus SMART pool Human HRAS | Dharmacon | L-003919-00-0005 |
| ON-Target Plus SMART pool Human NRAS | Dharmacon | L-004142-00-0005 |
| On-Target Plus Control Pool | Dharmacon | D-001810-10-05 |
| Other | ||
| RPPA Arrays | MDA Anderson RPPA core facility |
https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html |
| Software and algorithms | ||
| GeneSys Acquisition Software, Version 1.4.1.1 | Syngene | N/A |
| GraphPad Prism 6 Software | GraphPad | N/A |
| ImageJ | NIH | N/A |
| MORPHEUS software | Broad Institute | https://software.broadinstitute.org/morpheus/ |
METHOD DETAILS
Cell lines and inhibitors
Cell lines were obtained from ATCC, Millipore Sigma or the Center for Molecular Therapeutics at the MGH Cancer Center (Boston, MA) (Crystal et al., 2014), which routinely performs cell line authentication testing by SNP and short tandem repeat analysis, and maintained in DMEM/F12 or RPMI 1640 supplemented with 10% fetal calf serum, and were not cultured longer than 6 months after receipt from cell banks. AMG 510 and MRTX849 used for in vitro and in vivo studies were purchased from MedChemExpress and ARS-1620, RMC-4550, BGJ398, and trametinib used for in vitro studies were purchased from Selleckchem. RM-018 used for in vitro studies and RMC-4550 used in in vivo studies were kindly provided by Revolution Medicines. Panitumumab was obtained from McKesson Pharmaceuticals.
Inhibitor treatment assays
For long-term viability assays, cells were plated at low density 2×102-3×103 in 6-well plates and treated with AMG 510 (100 nM) alone or in combination with RMC-4550 (1 μM), panitumumab (30 μg/ml), or trametinib (10 nM) for 10–14 days with drug refreshed every 2–3 days. Assays were fixed and stained with a crystal violet solution (4% formaldehyde) and plates were scanned using a photo scanner and cell growth was quantified using ImageJ software.
Inhibitor treatment, siRNA treatment and western analyses
Cell lines were treated with AMG 510, ARS-1620, MRTX849 RM-018 (100 nM) alone or in combination with RMC-4550, panitumumab, BGJ398 or trametinib for 4, 24, 48 or 72 h before samples were collected in NP40 lysis buffer. For siRNA knockdown experiments, indicated cell lines were transfected with On-Target Plus non-targeting (NT), NRAS, and HRAS (Dharmacon) in Lipofectamine RNAiMax (Life Technologies) for 6 h and then treated for 24, 48 or 72 h with AMG 510 or RM-018. Whole cell lysates were resolved on 4–12% Bis-Tris gels (Thermo Fisher) and western blotting was performed using antibodies against phospho-SHP2 (Y542), phosphor-MEK (S217/221), phospho-ERK (T202/Y204), MYC, phospho-AKT (S473) (Cell Signaling Technologies), phospho-EGFR (Y1068), phospho-FGFR3 (Y724) phospho-p90 RSK (Abcam), NRAS (Santa Cruz), HRAS (Proteintech) and KRAS and GAPDH (Millipore). Densitometry analysis was performed using ImageJ software.
RAS-GTP pulldown
After indicated inhibitor treatment, RAS activity was assessed by GST-RAF-RBD pulldown (Cell Signaling Technologies), followed by immunoblotting with pan-RAS or RAS isoform–specific antibodies. Pulldown samples and whole-cell lysates were resolved on 4–12% Bis-Tris Gels and western blotting was performed using antibodies against KRAS, NRAS, HRAS, and pan-RAS (Cell signaling). Densitometry analysis was performed using ImageJ software.
LC/MS analysis of drug stability
ARS-1620 or AMG 510 was diluted in DMEM/F12 media with 10% FBS and incubated at 37°C alone or in the presence of the H358 cell line for 4, 24, 48, and 72 h. Cleared media samples were analyzed for ARS-1620 or AMG 510 by LC/tandem MS (MS/MS) methods. The LC/MS/MS sample preparation started with a liquid-phase extraction using a mixture of acetonitrile/Methanol (1:1) followed by centrifugation at 4000 RPM at 4°C for 10 min. The supernatants were injected onto a Shimadzu (Kyoto, Japan) chromatographic separation system equipped with a system controller (model SCL-20A). A Sprite ECHELON C18 4μm 20 × 2.1mm column (New Jersey, USA) was used with the solvent system consisting of solvent A, 0.1% ammonium bicarbonate in water, and solvent B, 0.1% ammonium bicarbonate in methanol. The MS/MS analysis was performed by an ABSciex API 4000 instrument (Applied Biosystems/MDS Sciex, Foster City, CA). The transitions m/z 561.4→m/z 138.3 and m/z 431.2→m/z 334.2 were used for AMG510 or ARS1620 detection. The standard curve was prepared in DMEM/F12 media with 10% FBS with a range from 1.000 to 50,000 ng/ml. The LLOQ of the assay was 1.000 ng/ml.
Xenograft studies
SW837 (5×106) and LIM-2099 (2X106) were injected into 6–8 week old female athymic nude mice (Charles River Laboratories. Treatment of AMG 510 (100mg/kg), RMC-4550 (30 mg/kg), daily oral gavage and panitumumab (0.5 mg) twice weekly by IP injection was initiated when tumor size reached 100–200 mm3 and tumor size was assessed by caliper measurements for 35–66 days. All animal studies were performed through the Institutional Animal Care and Use Committee (IACUC).
PDX cell lines and PDX in vivo studies
Primary human-tumor xenograft models were established as previously described (Katsiampoura et al., 2017). Tumor specimens were obtained from patients with metastatic colorectal cancer at the University of Texas MD Anderson Cancer Center, and all patients provided informed written consent for specimens to be used for research purposes including implantation in xenografts. Samples were obtained with approval of the institutional review board. Xenografts were established in NOD/SCID 8-week old female mice. Once established, PDXs were expanded in 69 athymic nude mice for experiments. After tumors were established with median tumor volume exceeding 200–250 mm3, treatment was commenced via oral gavage with either vehicle control, or drug/combinations as indicated in Figure legends [sotorasib, 100 mg/kg QD PO in 30% captisol, pH 2.2; trametinib, 0.25 mg/kg QD PO in 0.5%CMC/0.5% Tween 80; panitumumab, 0.5 mg, 2xW, IP]. Tumor size and mouse weight were measured twice a week. After 21–28 days, treatment was discontinued, and mice were sacrificed. Tumors from 3 mice per arm were excised (2 hours post treatment), segmented, and immediately flash frozen in liquid nitrogen (for protein, RNA and DNA analysis) or 10% buffered formalin solution (for IHC attaining). Primary human tumors were harvested from established xenograft models and cut into small pieces using sterile surgical instruments. Pieces of tumors were then transferred to dissociation tubes and incubated (5% CO2, 37°C) with dissociation media (0.1 mg Collagenase/Dispase [Millipore Sigma] in 100 μL DNase/RNase -free H2O/10 mL PBS filtered through 0.22 μM syringe filter) for two hours via rotation. Following incubation, supernatant was collected and filtered through 0.7 μM cell strainer (Falcon) into a centrifuge tube. Supernatant was aspirated after centrifugation and cell pellet was resuspended in fresh cell media (RPMI, 10% FBS) before plated into a collagen-coated tissue culture dish (Corning BioCoat). Cell media was replaced the next day to remove residual supernatant before cells were subjected to routine monitoring and cell media replacement every 2–3 days.
RPPA analysis
Cell line and PDX tumor derived samples were collected after indicated treatments in RPPA lysis buffer and processed as previously described(Iadevaia et al., 2010; Tibes et al., 2006). Treatment samples were normalized to untreated controls and heat maps were generated using MORPHEUS software (https://software.broadinstitute.org/morpheus/).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, all numerical data are presented as the mean ± standard error. Statistically significant differences for in vitro experiments were evaluated using two-tailed Student’s t-tests for comparison between two. Statistical significance for xenograft studies was evaluated by Mann-Whitney test. Significance in all cases was recorded as p values (NS when no significant difference, * when p < 0.05, ** when p < 0.01, *** when p < 0.001) and differences were considered statistically significant for p < 0.05. All statistical analyses were conducted using GraphPad Prism.
Supplementary Material
Highlights.
Adaptive RAS-MAPK feedback reactivation occurs following KRASG12C inhibition
RTK-mediated feedback activation of wild-type NRAS and HRAS bypasses KRASG12C
Inhibitor combinations abrogate RAS reactivation, boosting efficacy in KRASG12C CRC
Targeting convergent signaling nodes overcomes adaptive resistance by multiple RTKs
ACKNOWLEDGMENTS
R.B.C. was supported by NIH/NCI U54CA224068, NIH/NCI Gastrointestinal Cancer SPORE P50 CA127003, and a Stand Up To Cancer Colorectal Dream Team Translational Research Grant (SU2C-AACR-DT22-17). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. R.B.C. and S.K. were supported by Cancer Moonshot Administrative Supplement 5U54CA224068-03. S.K. was supported by U54 CA224065 and P30 CA016672. Amgen, Inc. provided study drug and partial study funding. J. Russell Lipford, Agnes Ang, and Karen Rex of Amgen, Inc. contributed to the conception of the work.
DECLARATION OF INTERESTS
R.B.C. has received consulting or speaking fees from Abbvie, Amgen, Array Biopharma/Pfizer, Asana Biosciences, Astex Pharmaceuticals, AstraZeneca, Avidity Biosciences, BMS, C4 Therapeutics, Chugai, Cogent Biosciences, Elicio, Erasca, Fog Pharma, Genentech, Guardant Health, Ipsen, Kinnate Biopharma, LOXO, Merrimack, Mirati Therapeutics, Natera, Navire, Nested Therapeutics, N-of-one/Qiagen, Novartis, nRichDx, Remix Therapeutics, Revolution Medicines, Roche, Roivant, Shionogi, Shire, Spectrum Pharmaceuticals, Symphogen, Syndax, Tango Therapeutics, Taiho, Theonys, Warp Drive Bio, and Zikani Therapeutics; holds equity in Alterome Therapeutics, Avidity Biosciences, C4 Therapeutics, Cogent Biosciences, Erasca, Kinnate Biopharma, Nested Therapeutics, nRichDx, Remix Therapeutics, Revolution Medicines, and Theonys; and has received research funding from Asana, AstraZeneca, Lilly, Novartis, and Sanofi. L.Z. and Y.Z. are employees and hold equity in Eli Lilly. SK. has received consulting or advisory fees from Genentech, EMD Serono, Merck, Holy Stone, Novartis, Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health, Pierre Fabre, Redx Pharma, Ipsen, Daiichi Sankyo, Natera, HalioDx, Lutris, Jacobio, Pfizer, Repare Therapeutics, Inivata, GlaxoSmithKline, Jazz Pharmaceuticals, Iylon, Xilis, Abbvie, Amal Therapeutics, Gilead Sciences, Mirati Therapeutics, Flame Biosciences, Servier, Carina Biotechnology, Bicara Therapeutics, Endeavor BioMedicines, Numab Pharma, and Johnson & Johnson/Janssen.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110993.
REFERENCES
- Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, Arena S, Montone M, Mussolin B, Bian Y, et al. (2020). EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 10, 1129–1139. 10.1158/2159-8290.cd-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Author Anonymous. (2020). Another KRAS Inhibitor Holds Its Own. Cancer Discov. 10, OF2. 10.1158/2159-8290.cd-nb2020-098. [DOI] [PubMed] [Google Scholar]
- Author Anonymous. (2021). FDA Approves First KRAS Inhibitor: Sotorasib. Cancer Discov. 11, OF4. 10.1158/2159-8290.cd-nb2021-0362. [DOI] [PubMed] [Google Scholar]
- Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al. (2021). Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med 384, 2382–2393. 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DK, Oni TE, Thalappillil JS, Park Y, Ting HC, Alagesan B, Prasad NV, Addison K, Rivera KD, Pappin DJ, et al. (2021). Oncogenic KRAS engages an RSK1/NF1 pathway to inhibit wild-type RAS signaling in pancreatic cancer. Proc. Natl. Acad. Sci. U S A 118, e2016904118. 10.1073/pnas.2016904118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, André T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, Hollebecque A, McRee AJ, Siena S, Middleton G, et al. (2018). Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E-mutant colorectal cancer. Cancer Discov. 8, 428–443. 10.1158/2159-8290.cd-17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. (2012). EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235. 10.1158/2159-8290.cd-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, and Der CJ (2014). Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov 13, 828–851. 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. (2014). Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486. 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakih M, Falchook GS, Hong DS, Yaeger R, Chan E, Mather O, Cardona P, Dai T, and Strickler J. (2021). 434P - CodeBreaK 101 subprotocol H: phase Ib study evaluating combination of sotorasib (Soto), a KRASG12C inhibitor, and panitumumab (PMab), an EGFR inhibitor, in advanced KRAS p.G12C-mutated colorectal cancer (CRC). Ann. Oncol 32, S551. 10.1016/j.annonc.2021.08.955. [DOI] [Google Scholar]
- Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, Falchook GS, Han SW, Heinemann V, Muro K, et al. (2022). Sotorasib for previously treated colorectal cancers with KRASG12C mutation (Code-BreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 23, 115–124. 10.1016/S1470-2045(21)00605-7. [DOI] [PubMed] [Google Scholar]
- Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, Mita P, Geer MJ, Hattori T, Koide A, et al. (2021). SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med 218, e20201414. 10.1084/jem.20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71. 10.1158/2159-8290.cd-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazar-Rethinam M, Kleyman M, Han GC, Liu D, Ahronian LG, Shahzade HA, Chen L, Parikh AR, Allen JN, Clark JW, et al. (2018). Convergent therapeutic strategies to overcome the heterogeneity of acquired resistance in BRAFV600E colorectal cancer. Cancer Discov. 8, 417–427. 10.1158/2159-8290.cd-17-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. (2020). KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med 383, 1207–1217. 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadevaia S, Lu Y, Morales FC, Mills GB, and Ram PT (2010). Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer Res. 70, 6704–6714. 10.1158/0008-5472.can-10-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ML, Ou SHI, Barve M, Rybkin II, Papadopoulos KP, Leal TA, Velastegui K, Christensen JG, Kheoh T, Chao RC, and Weiss J. (2020). KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer (CRC) and other solid tumors harboring a KRAS G12C mutation. Eur. J. Cancer 138, S2. 10.1016/S0959-8049(20)31077-7. [DOI] [Google Scholar]
- Katsiampoura A, Raghav K, Jiang ZQ, Menter DG, Varkaris A, Morelli MP, Manuel S, Wu J, Sorokin AV, Rizi BS, et al. (2017). Modeling of patient-derived xenografts in colorectal cancer. Mol. Cancer Therapeut 16, 1435–1442. 10.1158/1535-7163.mct-16-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Xue JY, and Lito P. (2020). Targeting KRAS(G12C): from inhibitory mechanism to modulation of antitumor effects in patients. Cell 183, 850–859. 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, et al. (2015). Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J. Clin. Oncol 33, 4032–4038. 10.1200/jco.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, Wasan H, Ciardiello F, Loupakis F, Hong YS, et al. (2019). Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N. Engl. J. Med 381, 1632–1643. 10.1056/NEJMoa1908075. [DOI] [PubMed] [Google Scholar]
- Kopetz S, McDonough SL, Lenz H-J, Magliocco AM, Atreya CE, Diaz LA, Allegra CJ, Raghav KPS, Morris VK, Wang SE, et al. (2017). Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J. Clin. Oncol 35, 3505. 10.1200/JCO.2017.35.15_suppl.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Solomon M, Li LS, Hansen R, and Rosen N. (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608. 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, Choi ALM, Mitchell DC, Swaney DL, Hann B, et al. (2019). KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal 12, eaaw9450. 10.1126/scisignal.aaw9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, Myers DT, Lee D, Gomez-Caraballo M, Greenberg M, et al. (2019). KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin. Cancer Res 25, 796–807. 10.1158/1078-0432.ccr-18-0368. [DOI] [PubMed] [Google Scholar]
- Molina-Arcas M, Moore C, Rana S, van Maldegem F, Mugarza E, Romero-Clavijo P, Herbert E, Horswell S, Li LS, Janes MR, et al. (2019). Development of combination therapies to maximize the impact of KRASG12C inhibitors in lung cancer. Sci. Transl. Med 11, eaaw7999. 10.1126/scitranslmed.aaw7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Rosenberg SC, McCormick F, and Malek S. (2020). RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov 19, 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, and Bernards R. (2012). Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103. 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Ramalingam SS (2021). A phase 1b study evaluating the safety and efficacy of sotorasib, a KRAS G12C inhibitor, in combination with trametinib, a MEK inhibitor. In KRAS p.G12C-Mutated Solid Tumors. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, pp. 7–10. [Google Scholar]
- Riely GJ, Ou SHI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, Johnson M, Heist RS, Bazhenova L, Barve M, et al. (2021). 99O_PR KRYSTAL-1: activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J. Thorac. Oncol 16, S751–S752. 10.1016/S1556-0864(21)01941-9. [DOI] [Google Scholar]
- Ryan MB, and Corcoran RB (2018). Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol 15, 709–720. 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, Hong CB, and Corcoran RB (2020). Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin. Cancer Res 26, 1633–1643. 10.1158/1078-0432.ccr-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze CJ, Bermingham A, Choy TJ, Cregg JJ, Kiss G, Marquez A, Reyes D, Saldajeno-Concar M, Weller CE, Whalen DM, et al. (2019). Abstract PR10: tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in vivo. Mol. Cancer Therapeut 18, PR10. 10.1158/1535-7163.targ-19-pr10. [DOI] [Google Scholar]
- Skoulidis F, Li BT, Govindan R, Dy GK, Shapiro G, Bauml J, Schuler MH, Addeo A, Kato T, Besse B, et al. (2021). Overall survival and exploratory subgroup analyses from the phase 2 CodeBreaK 100 trial evaluating sotorasib in pretreated KRAS p.G12C mutated non-small cell lung cancer. J. Clin. Oncol 39, 9003. 10.1200/JCO.2021.39.15_suppl.9003. [DOI] [Google Scholar]
- Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, Desai J, Ciardiello F, Loupakis F, Hong YS, et al. (2021). Encorafenib plus cetuximab as a new standard of Care for previously treated BRAF V600Emutant metastatic colorectal cancer: updated survival results and subgroup Analyses from the BEACON study. J. Clin. Oncol 39, 273–284. 10.1200/jco.20.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, Michel AG, Syed MU, Fella KA, Sakhi M, et al. (2021). Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS–MAPK reactivation. Cancer Discov. 11, 1913–1922. 10.1158/2159-8290.cd-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, and Kornblau SM (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Therapeut 5, 2512–2521. 10.1158/1535-7163.mct-06-0334. [DOI] [PubMed] [Google Scholar]
- Weiss J, Yaeger RD, Johnson ML, Spira A, Klempner SJ, Barve MA, Christensen JG, Chi A, Der-Torossian H, Velastegui K, et al. (2021). LBA6 - KRYSTAL-1: adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Ann. Oncol 32, S1294. 10.1016/j.annonc.2021.08.2093. [DOI] [Google Scholar]
- Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, Kim D, Li C, de Stanchina E, Mazutis L, et al. (2020). Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425. 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A, Lou D, and McCormick F. (2013). Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 3, 112–123. 10.1158/2159-8290.cd-12-0231. [DOI] [PubMed] [Google Scholar]
- Zhou B, Der CJ, and Cox AD (2016). The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol 58, 60–69. 10.1016/j.semcdb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request