

ABSTRACT
Intracellular pathogens have evolved various efficient molecular armaments to subvert innate defenses. Cellular ubiquitination, a normal physiological process to maintain homeostasis, is emerging one such exploited mechanism. Ubiquitin (Ub), a small protein modifier, is conjugated to diverse protein substrates to regulate many functions. Structurally diverse linkages of poly-Ub to target proteins allow enormous functional diversity with specificity being governed by evolutionarily conserved enzymes (E3-Ub ligases). The Ub-binding domain (UBD) and LC3-interacting region (LIR) are critical features of macroautophagy/autophagy receptors that recognize Ub-conjugated on protein substrates. Emerging evidence suggests that E3-Ub ligases unexpectedly protect against intracellular pathogens by tagging poly-Ub on their surfaces and targeting them to phagophores. Two E3-Ub ligases, PRKN and SMURF1, provide immunity against Mycobacterium tuberculosis (M. tb). Both enzymes conjugate K63 and K48-linked poly-Ub to M. tb for successful delivery to phagophores. Intriguingly, M. tb exploits virulence factors to effectively dampen host-directed autophagy utilizing diverse mechanisms. Autophagy receptors contain LIR-motifs that interact with conserved Atg8-family proteins to modulate phagophore biogenesis and fusion to the lysosome. Intracellular pathogens have evolved a vast repertoire of virulence effectors to subdue host-immunity via hijacking the host ubiquitination process. This review highlights the xenophagy-mediated clearance of M. tb involving host E3-Ub ligases and counter-strategy of autophagy inhibition by M. tb using virulence factors. The role of Ub-binding receptors and their mode of autophagy regulation is also explained. We also discuss the co-opting and utilization of the host Ub system by M. tb for its survival and virulence.
Abbreviations: APC: anaphase promoting complex/cyclosome; ATG5: autophagy related 5; BCG: bacille Calmette-Guerin; C2: Ca2+-binding motif; CALCOCO2: calcium binding and coiled-coil domain 2; CUE: coupling of ubiquitin conjugation to ER degradation domains; DUB: deubiquitinating enzyme; GABARAP: GABA type A receptor-associated protein; HECT: homologous to the E6-AP carboxyl terminus; IBR: in-between-ring fingers; IFN: interferon; IL1B: interleukin 1 beta; KEAP1: kelch like ECH associated protein 1; LAMP1: lysosomal associated membrane protein 1; LGALS: galectin; LIR: LC3-interacting region; MAPK11/p38: mitogen-activated protein kinase 11; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MAP3K7/TAK1: mitogen-activated protein kinase kinase kinase 7; MAPK8/JNK: mitogen-activated protein kinase 8; MHC-II: major histocompatibility complex-II; MTOR: mechanistic target of rapamycin kinase; NBR1: NBR1 autophagy cargo receptor; NFKB1/p50: nuclear factor kappa B subunit 1; OPTN: optineurin; PB1: phox and bem 1; PE/PPE: proline-glutamic acid/proline-proline-glutamic acid; PknG: serine/threonine-protein kinase PknG; PRKN: parkin RBR E3 ubiquitin protein ligase; RBR: RING-in between RING; RING: really interesting new gene; RNF166: RING finger protein 166; ROS: reactive oxygen species; SMURF1: SMAD specific E3 ubiquitin protein ligase 1; SQSTM1: sequestosome 1; STING1: stimulator of interferon response cGAMP interactor 1; TAX1BP1: Tax1 binding protein 1; TBK1: TANK binding kinase 1; TNF: tumor necrosis factor; TRAF6: TNF receptor associated factor 6; Ub: ubiquitin; UBA: ubiquitin-associated; UBAN: ubiquitin-binding domain in ABIN proteins and NEMO; UBD: ubiquitin-binding domain; UBL: ubiquitin-like; ULK1: unc-51 like autophagy activating kinase 1.
KEYWORDS: Autophagy, E3-Ub ligase, intracellular pathogens, LC3, phagolysosome, ubiquitin-binding receptors, virulence effectors, xenophagy
Introduction
Macroautophagy/autophagy is an evolutionarily conserved cellular homeostatic process across eukaryotes in which cytosolic constituents are enveloped in newly developed membranous vesicles termed phagophores. Autophagy-related (ATG) genes mediate phagophore formation, followed by fusion with the lysosome to degrade autolysosomal contents for recycling [1,2]. Majorly, non-selective autophagy induced by nutritional deprivation results in the recycling of cytosolic components. Nevertheless, autophagy can also be selective wherein specialized cargo; usually, pathogens, protein aggregates, and damaged cellular organelles are targeted for clearance and recycling [3]. This whole process involves the widespread post-translational modification of proteins by a process called ubiquitination. A small protein known as ubiquitin (Ub) is used as a protein modifier [4–6]. Recent advances unveil an exciting and unambiguous role of this process in host-pathogen interaction apart from cellular homeostasis [7]. Bacterial pathogens employ multiple tactics to favorably establish infection for their survival by modulating key host signaling pathways and altering post-translational modifications to dampen the innate immune response evoked by the host [8,9]. Host-induced autophagy against invading intracellular pathogens such as Mycobacterium tuberculosis (M. tb) serves as an innate defense mechanism to control pathogenesis [10–13]. M. tb resides in the phagosome and arrests phagosome maturation by employing its virulence effectors for intracellular survival [14]. However, M. tb manages to escape the phagosome by phagosomal membrane breaching [15,16]. Cytoplasmic M. tb is ubiquitinated by the host E3-Ub ligases, targeting it to the phagophore mediated by Ub-receptors. Consequently, the survival of ubiquitinated M. tb is diminished by autophagosome and lysosome fusion [17,18]. To counter host-induced xenophagy, M. tb exploits several effector proteins to inhibit autophagy-induced killing [12,19–21]. The interaction of the host with the pathogen is highly dynamic, and the host counters the pathogen’s advances through various other adaptations [22]. Pathogens also employ ecological adaptations to survive the hostile environment by forming complex heterochemical structures like biofilms to fend off hostile molecules [23,24]. The evolutionary and environmental pressures in the host niche allow selective protein adaptation in mycobacteria [25–27]. The structural adaptations of these proteins allow it to alter the biochemical machinery of the bacterial cell to thrive in extreme conditions [28,29].
Ub is a 76 amino-acid conserved protein present in all eukaryotes. It is covalently conjugated to the target proteins via an isopeptide bond. The C-terminal glycine of Ub and an ε-amino group of lysine on substrates are involved in covalent isopeptide bond formation [30]. Ubiquitination can be mono, multi-mono, or polyubiquitination, usually designated as the “Ub code”, which determines the fate and altered biological functions of substrate proteins (Figure 1, and Table 1) [31]. Delineating the Ub code would be a present-day marvel as it could reveal the intricacies of cellular physiology to understand the signaling events better and devise novel drug candidates. The diverse topology of Ub linkages enables the transmission of complex physiological signals required for spatiotemporally controlled cellular functions [32]. The compact structures are formed by K11 and K48-linked poly-Ub chains that direct the substrates to the canonical Ub-proteasome pathway for degradation. However, when Ub attaches via Met1 or K63 linkages, these adducts adopt an extended conformation. It enables the reversible recruitment of multi-protein complexes, which are the main non-proteolytic consequence of Ub conjugation events found in immune signaling cascades (Table 1) [5,33,34]. Although previous research mainly focused on the homogenously linked Ub chains, heterogeneously branched Ub chains that perform distinct physiological functions are emerging as a crucial protein modification. Moreover, these types of post-translational modifications involving Ub further expand the specificity, versatility, and efficacy of Ub dependent signaling events (Table 1) [35].
Figure 1.
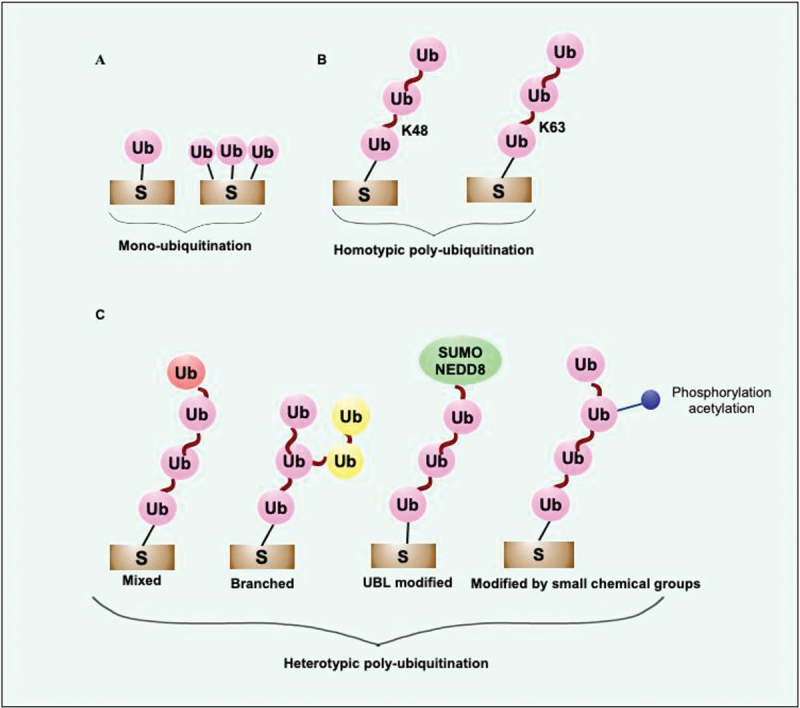
Diverse Ub-signals communicate different biological messages. (a) The diverse Ub-linkages coordinate different biological outcomes such as mono or multi-mono ubiquitination transmit signals for localization, control the activity of ubiquitinated substrates, proteolysis mediated by Ub-proteasome system, and autophagy. (b) K48 and K11 linkages usually transmit signals for degradation of short-lived folded proteins performed by 26S proteasome. K48-linked chains transmit signals for autophagic removal of invading pathogens and misfolded protein aggregates. K63-linked Ub chains transmit signals to remove aggregated proteins and participate in xenophagic clearance of invading cytosolic bacteria by autophagy. Diverse non-degradative processes are regulated by K63 linkages, including activation of DNA damage repair, assembly, and activation of signaling complexes. The main functions of K48- and K63-linked Ub chains are known. However, the crucial roles of newly discovered atypical linkages, including K6, K27, K29, and K33, are less known. Met1-linked linear chain takes part in removing invading pathogens, damaged mitochondria and activates the NFKB1 signaling pathway. (c) Small molecule modifiers of Ub such as acetylation, phosphorylation, and neddylation control various Ub functions. The functions of more complex hetero conjugated Ub are emerging now. S, substrate; Ub, ubiquitin; NEDD8, NEDD8 ubiquitin like modifier.
Table 1.
Functions of various Ub-linkages and the cellular processes involved.
| Type of Ub-linkages | Target | Recognizing molecules | Functions |
|---|---|---|---|
| Monoubiquitination or multi-monoubiquitination | Normally folded proteins | Ub + Ub-binding receptors | Protein interactions, localization, trafficking, activity modulation, DNA repair, endocytosis, transcription, proteasomal degradation |
| K63 poly-Ub chains | Normally folded proteins | Ub + Ub-binding receptors + accessory proteins | NFKB1 signaling, DNA repair, trafficking, endocytosis |
| Branched Ub-chains | Normally folded proteins | Ub + Ub-binding receptors + accessory proteins | APC/cyclosome-mediated proteasomal degradation |
| K11, K48 poly-Ub chains | Normally folded proteins | Ub + Ub-binding receptors | Proteasomal degradation |
| K48 (other linkages?) poly-Ub chains | Misfolded protein aggregates | Ub + Ub-binding receptors | Autophagic degradation |
| K63 poly-Ub chains | Misfolded protein aggregates | Ub + Ub-binding receptors + LC3 + chaperones | Autophagic degradation |
| (other linkages?) | |||
| K63 | Damaged mitochondria | Ub + Ub-binding receptors + LC3 + chaperones | Autophagic degradation including mitophagy, pexophagy, xenophagy, reticulophagy? ribophagy? lipophagy? |
| K6 (?) poly-Ub chains | |||
| K27, K29, K33, Lys6 (?) poly-Ub chains | Normally folded proteins | Ub + Ub-binding receptors + accessary protein | Non-proteolytic processes |
| Met1-linear chain | Normally folded proteins | Ub + Ub-binding receptors + accessory protein | Signaling |
The information provided by specific Ub-linkages “Ub-codes” are interpreted by specialized proteins consisting of UBDs and sometimes involve accessory proteins. The receptors having UBDs recognize the length and type of Ub chains on protein substrates and couple ubiquitinated cargos to subsequent downstream events. Majorly, monoubiquitination or multi-monoubiquitination plays role in protein-protein interaction, localization, endocytosis, proteasomal degradation, DNA repair, transcription, regulation of protein activity, and link ubiquitinated target to autophagy. K48 and K11-linked Ub chains specify proteasomal-mediated degradation of short-lived and misfolded proteins. Conjugation of K48 and K63-linked Ub chains to the misfolded proteins targets them for proteasome or autophagy-mediated degradation. K63-linked Ub chain also directs the autophagic degradation of damaged cellular materials, including mitochondria, and targets intracellular cytosolic pathogens to autophagy. K63-linked chains are involved in assembling many signaling cascades, including NFKB signaling, DNA repair, trafficking, and endocytosis. Met1-linked linear chains target intracellular bacteria and act as assembly signals for downstream NFKB signaling. Compared to atypical Ub linkages, including K6, K27, K29, K33, and branched Ub, the functions of K48 and K63 linkages are known. The diverse functions of atypical Ub linkages and branched Ub are emerging now.
Protein ubiquitination is a three-step, enzymatically catalyzed reversible reaction. E1, an Ub-activating enzyme, catalyzes the formation of high-energy thioester intermediate by using C-terminal glycine of Ub and its active site cysteine in an Mg2+ and ATP-dependent manner. Thereafter, an Ub-conjugating (E2) enzyme’s active site, cysteine, accepts activated Ub from a charged E1. Finally, an E3-Ub ligase interacts with charged E2, substrate protein, and Ub, leading to the formation of Ub adduct to the target protein [6,18]. Like other post-translational modifications, it requires the specialized class of proteases to remove Ub, known as de-ubiquitinating enzymes (Figure 2) [36,37]. Different Ub linkages are recognized by various Ub receptors possessing Ub binding domains (UBDs), generating enormous biological outcomes [38]. UBDs organize in a modular structural element that non-covalently binds to Ub and can distinguish various features of different Ub modifications. UBDs differ in the mode of Ub recognition and structure. Mostly, UBDs form an α-helical structure, zinc fingers, ubiquitin-conjugating (UBC) domains, and pleckstrin homology folds [39]. UBDs mainly encompass a compact three-helix bundle, like UBA1 (ubiquitin like modifier activating enzyme 1), a well-characterized UBD [40]. The UBD of UBA1 and UBA2 form structurally similar folds characterized by a conserved surface hydrophobic patch. The hydrophobic surface patch forms a protein-protein interaction surface that recognizes the hydrophobic surface of Ub formed by five stranded beta-sheet [41]. Other UBDs, including Ub-interacting motifs, ubiquitin-binding motifs, UBAN, GAT, coupling of ubiquitin conjugation to ER degradation domains, C2 (Ca2+-binding) motif, and VHS (Vps27, Hrs, STAM), use α-helices to contact Ub. Various UBDs, such as ubiquitin-binding zinc finger, Npl4 zinc finger domain, zinc finger domain found in TNFAIP3/A20, zinc-finger ubiquitin-binding domain, and Piwi Argonaut and Zwille domains, use zinc fingers to contact or bind to the Ub. The hydrophobic patch on the Ub surface is formed by the amino acids Leu8, Ile44, Val70, and His68. Besides these structurally essential residues, the Ub comprises other functionally critical residues, such as Phe4, Ile36, and Asp58, involved in hydrophobic or hydrogen bond interactions with UBDs. The UBDs can differentiate between poly-Ub linkages by detecting the distance between successive Ub molecules, free C-terminal ends, unanchored chains, and distinctive sequence features of amino acids surrounding the isopeptide bonds [39,42].
Figure 2.
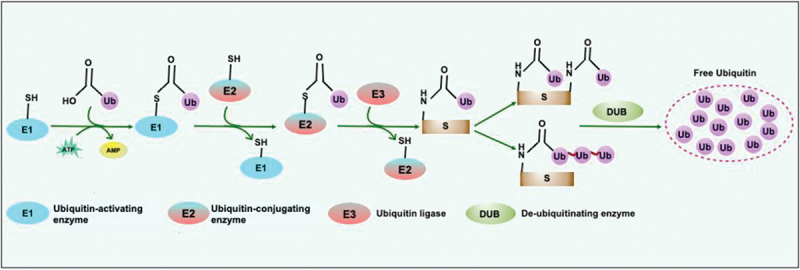
The molecular players of the Ub-conjugation system. Ubiquitination is a three-step enzyme-catalyzed reaction coordinated by E1 or Ub-activating enzyme that catalyzes the ATP and Mg2+ dependent activation of Ub required to form a thioester bond in between catalytic cysteine on the E1 and C terminus glycine of Ub. The transfer of activated Ub from the Ub-conjugating enzyme (E2) to the substrate is mediated by the E3-Ub ligase. The removal and recycling of Ub from substrates are catalyzed by DUBs which maintain the Ub-pool. S, substrate; Ub, ubiquitin; DUB, deubiquitinase.
E3-Ub ligases determine the substrate specificity
The exquisite selectivity and efficiency of the ubiquitination process are attributed to E3-Ub ligases. The E3-Ub ligases are majorly grouped into three major classes: 1). RING E3-Ub ligase; 2). HECT E3-Ub ligase; and 3). RBR E3-Ub ligase [43–45]. All E3-Ub ligases encompass the E2-Ub recognizing domain and are classified according to the structure of this domain and Ub transfer mechanisms. Human genome analysis reveals 700 RING E3-Ub ligase, making them the largest of the three E3-Ub ligase classes [46,47]. RING E3-Ub ligase comprises the RING domain or RING-like domain that consists of cysteine and histidine residues used to coordinate two Zn2+ ions mostly in a cross-brace arrangement. RING E3-Ub ligase provides a docking site for Ub-conjugated E2 enzymes, enhancing the Ub transfer to the substrates. The RING E3-Ub ligase can be monomeric, dimeric, or oligomeric and accordingly use different mechanisms to target the substrate for ubiquitination (Figure 3). The dimerization of the RING E3-Ub ligase is facilitated by the RING domain or mediated through adjacent α-helical regions [48]. Similar to RING E3-Ub ligase, UBOX containing E3-Ub ligase performs related functions. The UBOX E3-Ub ligases consist of a hydrophobic core rather than the structural metal ions [49]. Eight UBOX containing E3-Ub ligase are known in humans and, like RING E3-Ub ligase form monomer, dimer, or oligomer to execute their function (Figure 3a) [50]. The RING domain of E3-Ub ligase does not possess cysteine at the active site, thus do not form high-energy thioester intermediate, unlike HECT and RBR E3-Ub ligases. It implies that the RING E3-Ub ligase function in transferring donor Ub to an acceptor lysine by acting as a scaffold for aligning the catalytic subunits for the nucleophilic attack [43].
Figure 3.
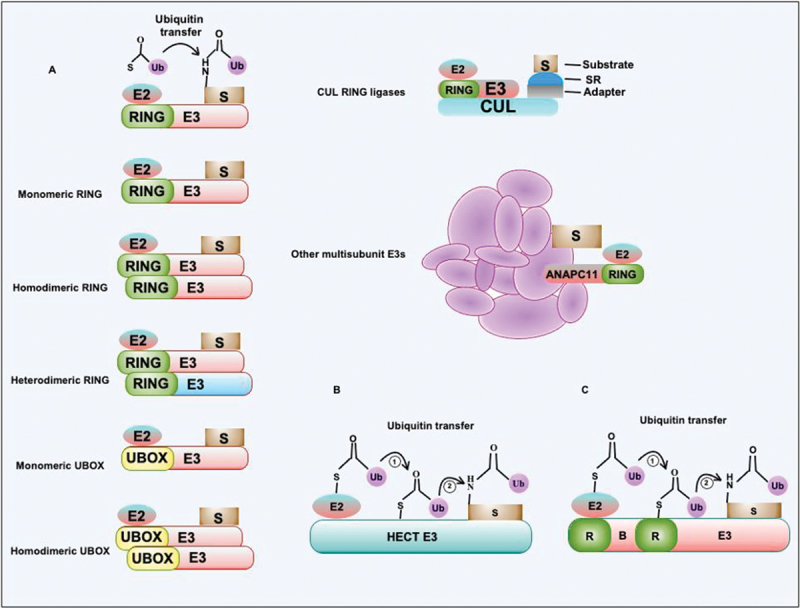
E3-Ub ligase uses a distinct mechanism for the transfer of Ub to substrates. (a) E3-Ub ligase containing the RING domain transfers Ub to target protein substrates from E2-Ub conjugating complex. RING or similar UBOX containing E3-Ub ligases exist in multiple oligomeric states such as monomers, homodimers, and heterodimers. CUL3-E3 RING ligase comprises multiple subunits that coordinate interaction with the substrate. The complex contains adapter protein and substrate-interacting protein together with various CUL isoforms. The anaphase-promoting complex/cyclosome contains multiple subunits that coordinate interaction between target substrates and RING domain-containing E3-Ub ligase together with E2-Ub conjugating enzyme. The role and functions of multi-subunit complex E3-Ub ligases are emerging now. These complexes perform more complex hetero-conjugation of Ub to the substrates. (b and c) The HECT and RBR type E3-Ub ligases transfer Ub from E2-Ub conjugating enzymes to HECT or RING domain conserved cysteines followed by the Ub transfer to target substrates. S, substrate; Ub, ubiquitin; SR, substrate receptor.
HECT E3-Ub ligases are modular proteins containing distinct N- and C-terminal domains. The human genome encodes 28 HECT E3-Ub ligases. The N terminus of HECT E3-Ub ligase possess an E2 binding domain, whereas the C-terminal domain comprises conserved cysteine used in catalysis [51]. In contrast to RING E3-Ub ligase, the HECT E3-Ub ligase conjugates Ub to the target using a two-step reaction mechanism. The first step constitutes the E2-mediated transfer of donor Ub to the catalytic moiety on the HECT domain via a trans-thiolation reaction, thus keeping the high-energy thioester bond. The optimal orientation of the acceptor lysine is thereafter determined by HECT E3-Ub ligase for a nucleophilic attack to form a Ub-E3 ligase thioester bond (Figure 3b) [52]. The substrate specificity and catalysis are governed by the C-terminal end of HECT E3-Ub ligase; however, the exact mechanism remains elusive. The flexible linker region connects the N- and C-terminal domains by rotation and organizes the HECT E3-Ub ligase to facilitate Ub transfer. The RBR E3-Ub ligase comprises a conserved catalytic region found in-between RING1 and RING2 domains. 14 RBR E3-Ub ligases are coded by the human genome containing RING1-IBR-RING2 domains [44,45,53]. The RBR E3-Ub ligase uses a hybrid RING/HECT catalytic mechanism of ubiquitination. The RBR E3-Ub ligase RING1 domain recruits charged E2 and facilitates Ub transfer to RING2, forming a HECT-like thioester intermediate before conjugating Ub to the substrate (Figure 3c). Like the HECT E3-Ub ligase, the RBR E3-Ub ligase also controls the linkage specificity of ubiquitination on the substrate [53].
Ub ligases as weapons of host defense against intracellular pathogens
Xenophagy, also known as selective autophagy of microbes, is an innate defense strategy exploited by the host against diverse pathogens, like viruses, parasites, and intracellular bacteria [54,55]. Wide-ranging Gram-positive and Gram-negative bacteria, such as Shigella flexneri, Salmonella enterica serovar Typhimurium, group A Streptococcus, Francisella tularensis, M. tb, and Listeria monocytogenes, are targeted and restricted by autophagy [56–59]. Despite the significance of autophagy in antibacterial defense, we are far from understanding the mechanistic details of recognizing, targeting, and eliminating such diverse bacterial pathogens using autophagy [3,60]. Although bacteria develop multiple evasion strategies to prevent detection by the host, host cells also employ multiple tactics to recognize and target pathogens [61]. The diverse array of target recognition molecules and receptors play crucial roles at distinct steps of the invasion process for activation of autophagy. Two related innate defense mechanisms have been gaining much traction of late. Bacteria-containing vacuoles that specifically accumulate diacylglycerol are also targeted by selective autophagy [62]. However, sometimes bacteria escape the canonical autophagy pathway. In such cases, host glycans are exposed on the surface of damaged vacuoles containing pathogenic bacteria, which are recognized by LGALS3, LGALS8, and LGALS9 and targeted for autophagy-mediated killing [62–64]. Coating the bacteria or bacteria-associated proteins using poly-Ub chains is another evolutionarily conserved innate host defense strategy of the host for autophagy-driven killing [65–67]. Several host receptor proteins such as SQSTM1, CALCOCO2, NBR1, OPTN, and TAX1BP1 recognize the Ub coat on the bacterial surface and link the ubiquitinated target to the autophagy machinery for autolysosomal degradation [68]. Autophagy receptors use a conserved LIR-motif to interact with Ub-like proteins of the Atg8-family displayed on the phagophore membrane. Each autophagy receptor executes a unique function in antibacterial xenophagy and is required for bacterial growth restriction and killing [66,69,70]. Pertinently, CALCOCO2 and SQSTM1 perform multiple essential functions in xenophagy [66,71]. Earlier works suggest that CALCOCO2 and SQSTM1 can independently be recruited to diverse pathogen surfaces decorated with Ub by the action of various E3-Ub ligases [66,69,72].
The unexpected diversity exhibited by E3-Ub ligases suggests their role in controlling the specificity of the ubiquitination process. Many E3-Ub ligases are implicated in the execution of non-selective autophagy induced by starvation [73–77]. Recently, several E3-Ub ligases such as LRSAM1 (leucine rich repeat and sterile alpha motif containing 1), PRKN, SMURF1, LUBAC (linear ubiquitination assembly complex), and RNF166, are implicated in the innate targeting of the intracellular bacteria for antibacterial autophagy [61,78–81]. LRSAM1 utilizes K6 and K27 ubiquitination to target the autophagy susceptible strains of Shigella, Listeria, Salmonella, and adherent invasive Escherichia coli, but strangely excluding M. tb. PRKN functions in innate resistance to M. tb by conjugating K63 and K48-linked Ub chains [78,82]. RNF166, a RING domain-containing E3-Ub ligase, is involved in the ubiquitination of bacteria or bacteria-containing vacuoles, but the targeted bacterial species have not been identified yet. Interestingly, however, its role in antibacterial autophagy has been reported. RNF166 recruits autophagy receptors CALCOCO2 and SQSTM1, ubiquitinating SQSTM1 using K29 and K33 linkages [61]. SMURF1 specifically decorates K48-linked poly-Ub chains onto M. tb-associated structures and recruits the xenophagy receptor NBR1. SMURF1 coordinates phagophore formation by recruitment of LC3 and LAMP1; its E3-Ub ligase activity and C2 phospholipid-binding domain execute a crucial role in antibacterial xenophagy of M. tb [81]. The LUBAC E3-Ub ligase amplifies poly-Ub chains on S. typhimurium that is already marked with linear poly-Ub chains in the cytosol. The resultant linear poly-Ub chains recruit IKBKG/NEMO (inhibitor of nuclear factor kappa B kinase regulatory subunit gamma) and OPTN to induce xenophagy and activate NFKB1/p50 signaling, independently restricting bacterial survival and proliferation [79]. Interestingly, prkn and smurf1 double-knockout mouse macrophages do not entirely abolish colocalization of M. tb to Ub, implying that other unidentified E3-Ub ligases may also function in inducing antimycobacterial xenophagy [81]. These E3-Ub ligases generate distinct Ub linkages on target proteins that culminate in cellular defense responses against the pathogens. Interestingly, the emerging role of E3-Ub ligases in providing innate defense against diverse intracellular pathogens opens up a new arena to develop novel approaches for targeting them to selective autophagy for enhanced killing. Developing E3-Ub ligase activity modulators that potentiate the ligase activity for increased ubiquitination of intracellular pathogens would be an innovative perspective for ubiquitin-mediated selective autophagic killings.
However, several studies have explored ways to maneuver the ligase activity of diverse E3-Ub ligases to treat various disease conditions. The E3-Ub ligases are proposed as excellent drug targets due to their high specificity in substrate selection. This is especially promising because of the fewer off-targets and lesser side effects [83]. Many of the E3-Ub ligases are targeted to develop selective inhibitors such as MDM2 (MDM2 proto-oncogene) for enhancing the activity of TP53 (tumor protein p53) in preventing various forms of cancers [84]. Therefore, it would be intriguing to develop potent PRKN and SMURF1 agonists that can specifically induce xenophagy mediated killing of M. tb. The novel approach will open an unexplored area for developing selective modifiers/agonists of PRKN and SMURF1 as host-based therapeutics. A recent study shows that overexpression of LGALS8 in macrophages enhances the selective autophagy of M. tb by recruiting autophagy receptor TAX1BP1 supports the notion for developing PRKN- and SMURF1-selective agonists to enhance autophagic degradation of M. tb [85].
Host E3-Ub ligase PRKN and SMURF1 provide innate defense against M. tb
PRKN
PRKN is a newly discovered E3-Ub ligase that belongs to the RBR family of Ub ligases [85]. Phosphorylation of S65 of the UBL domain and Ub leads to the conversion of latent PRKN into the fully active form. The latent form of full-length PRKN’s crystal structure reveals various conserved domains, including RING1, RING2, UBL, RING0, REP, and IBR (Figure 4a). PRKN receives Ub-conjugated E2 at its RING1 domain and transfers Ub to the cysteine of RING2 via a thioester linkage. Dramatic conformational changes are required for its full activation, evidenced by the crystal structures of phosphorylated PRKN and Ub, highlighting the fully operational structure [86,87].
Figure 4.
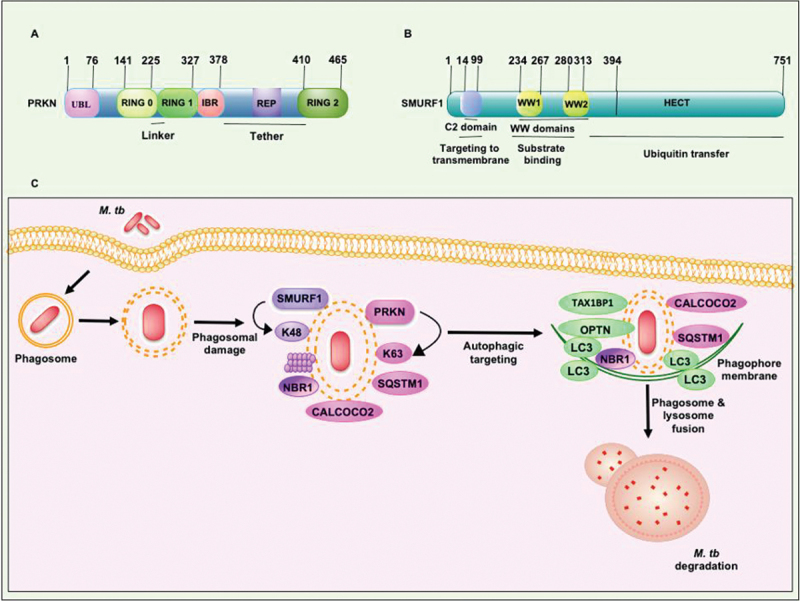
Structural features of PRKN and SMURF1 E3-Ub ligases which execute xenophagy mediated clearance of M. tb. (a) RING1 domain of PRKN consists binding site for Ub-conjugating E2 enzyme. RING2 domain of PRKN contains catalytic cysteine to form a covalent linkage with Ub. The other conserved domains, such as the UBL domain and REP linker region, sandwiched between the RING2 and IBR domains, inhibit RING1 binding to E2. The RING0 domain partially covers the catalytic cysteine residue present in the RING2 domain. (b) The N-terminal C2 domain binds phospholipids and plays an essential role in SMURF1 localization to the membrane. WW domains of SMURF1 are protein interaction domains required for binding with the targets. The transfer of Ub to the protein substrate is governed by the catalytically active C-terminal domain of the HECT family of E3-Ub ligases. The molecular size of both the E3-Ub ligases is shown. (C) PRKN and SMURF1 E3-Ub ligases provide immunity against M. tb. PRKN and SMURF1 ubiquitinate M. tb and its associated membranous structure in the cytosol to recruit autophagy receptors SQSTM1, CALCOCO2, and NBR1. The engagement of LC3 to the phagophore membrane and consequent fusion to the lysosome targets M. tb to xenophagy. REP, repressor element of PRKN.
Several elegant studies demonstrate the essential role played by PRKN. It regulates diverse cellular pathways, including apoptosis, lipid metabolism, and effector cytokine production during infection [88,89]. Mutations and genetic polymorphisms in the PRKN/PARK2 gene increase the susceptibility to Parkinson disease [90]. PRKN also provides immunity against diverse intracellular pathogens such as L. monocytogenes, S. typhimurium, Mycobacterium marinum, Mycobacterium leprae, and M. tb [78]. PRKN catalyzes the K63-linked polyubiquitination to M. tb exposed in the cytosol (or M. tb-associated membranous structure) through the disruption of the phagosomal membrane via the use of a specialized ESX-1 type VII secretion system [78]. K63-linked polyubiquitination recruits autophagy receptors SQSTM1 and CALCOCO2 that engage LC3 to the phagophore [78]. Interestingly, prkn knockout bone marrow-derived macrophages (BMDMs) show reduced colocalization of M. tb with LC3 and ATG12 proteins of autophagy [78]. Moreover, prkn deletion portrays decreased conversion of LC3-I to LC3-II, a hallmark of autophagy progression. The prkn and atg5 knockout BMDMs show increased survival of M. tb inside macrophages (2–2.5-fold). On the contrary, overexpression of PRKN in RAW264.7 macrophages reduces the survival of M. tb. Knockdown of PRKN in the human macrophage cell line U937 also exhibits increased survival and replication inside macrophages [78]. These findings suggest that PRKN plays unequivocal roles in controlling mycobacterial replication and proliferation within macrophages, even though the actual fate of mycobacteria remains unknown (Figure 4c) [78]. Intriguingly, only 30% of intracellular M. tb is ubiquitinated in BMDMs with a considerable portion (~25%) conjugated to K63-linked Ub chains, whereas a minor fraction is conjugated to K48-linked Ub (5%) [78]. These observations suggest that other E3-Ub ligases are also required for conjugation of Ub to the M. tb containing phagosomes along with PRKN.
E3-Ub ligase activity of PRKN is essentially required for the colocalization of Ub to intracellular M. tb as E3-Ub ligase dead mutants cannot conjugate Ub to M. tb [78]. The role of PRKN in providing immunity in vivo against M. tb is confirmed by using wild-type and prkn-deficient mice after a low dose infection [78]. After 3 weeks of infection, prkn-deficient mice show increased (10-fold) bacterial loads compared to wild-type mice in the lung, spleen, and liver. prkn-deficient mice exhibit overwhelming infection and succumb after 85 days, whereas no overt sign of stress is observed in wild-type mice. Moreover, PRKN expression was observed in granulomas and human lung tissues from TB patients that showed colocalization of PRKN to M. tb [78].
Further, it was revealed that PRKN provides innate immunity against various intracellular pathogens in Drosophila melanogaster and Caenorhabditis elegans. The park and odr-1 knockout strains of D. melanogaster and C. elegans show higher bacterial loads and lesser survival [78]. These results implicate that PRKN plays a conserved essential role in providing innate immunity against intracellular pathogens in metazoans [78,91].
SMURF1
SMURF1 is a highly conserved HECT E3-Ub ligase. It organizes in a conserved modular structure and contains two WW domains, a C2 domain at the N terminus and a catalytic HECT domain at the C terminus (Figure 4b) [92]. A long stretch of 35–40 amino acids in the WW domain recognizes substrate through protein-protein interaction. In SMURF1, only one tryptophan residue is conserved in each WW domain. Two WW domains work cooperatively to recognize the substrate [93,94]. SMURF1 catalyzes the ubiquitination of various substrates for 26S proteasome-mediated degradation (Figure 4b). SMURF1 also regulates biologically essential processes that include the pathway involved in bone morphogenesis, the mitogen-activated protein kinase pathway, and the noncanonical WNT pathway. SMURF1 performs multiple functions like regulating autophagy, cell migration, cell polarity, cell growth, and cell cycle regulation [95,96].
SMURF1 contains an E3-Ub ligase and a C2 phospholipid-binding domain. Mutations in the E3-Ub ligase and C2 domains inhibit the colocalization of M. tb to LC3 and Ub, indicating that these domains are required for inducing selective autophagy of M. tb. The C2 domain of SMURF1 is also essential for targeting the M. tb-associated structures within macrophages [81]. A genome-wide siRNA screen identifies SMURF1 as an essential component in targeting Sindbis and Herpes simplex viruses for autophagy-mediated killing [97]. smurf1-deficient mouse embryonic fibroblasts are defective in targeting these viruses to the phagophore; however, these mouse embryonic fibroblasts respond normally to starvation-induced autophagy. A recent study reveals that SMURF1 executes essential functions in the selective bacterial autophagy of M. tb (Figure 4c) [81]. The smurf1 knockout macrophages cannot recruit poly-Ub, NBR1, LC3, the proteasome, and LAMP1 to the M. tb-containing vesicles, therefore, being more permissive to M. tb growth. ESX-1 mediated permeabilization of the phagosome is essential for the recruitment of SMURF1 on M. tb or its associated structures [81]. In contrast to PRKN-mediated K63-linked ubiquitination, SMURF1 conjugates K48-linked Ub chains to M. tb as earlier reported for STAT1 (signal transducer and activator of transcription 1) protein [98]. K48-linked ubiquitination induced by SMURF1 also plays a crucial role in the control of L. monocytogenes infection. The prkn and smurf1 double-knockout macrophages show decreased colocalization of M. tb to Ub and LC3 resulting in enhanced survival [81]. These findings suggest that PRKN and SMURF1 show synergistic function to control M. tb replication in macrophages, mice, and humans. Notably, SMURF1 only recruits autophagy receptor NBR1 to the ubiquitinated M. tb, unlike PRKN, which recruits SQSTM1 and NBR1 [78,81]. Further, SMURF1 also recruits proteasome to the ubiquitinated M. tb dissimilar to PRKN, implying that SMURF1 also has distinct functions in antimycobacterial defense [78,81].
However, similar to PRKN, the infection of M. tb to smurf1-deficient mice exhibit enhanced bacterial load, inflammation, and accelerated mortality [81]. smurf1-deficient mice portray reduced colocalization of M. tb to LC3 and also demonstrated diminished ubiquitination. Additionally, smurf1 knockout BMDMs exhibit decreased targeting of M. tb containing phagophore to the lysosome. smurf1-deficient BMDMs show enhanced replication of M. tb at 24, 48, and 72 h post-infection, suggesting the role of Smurf1 in controlling M. tb replication in murine macrophages. Moreover, to confirm the role of Smurf1, an autophagy-inducing peptide was used, which increases the targeting of M. tb to the lysosome in wild-type but not in smurf1-deficient macrophages. Further, no difference was observed in a mouse model of infection in the bacterial load of wild-type and smurf1-deficient mice in the acute phase of infection. In the chronic phase, smurf1-deficient mice had a higher bacterial load in the lung and spleen [81]. Unlike atg5 knockout mice, mononuclear inflammatory cells were infiltrated to the infected lung rather than polymorphonuclear cells [99]. A higher level of IL17 was observed in smurf1 knockout mice with no pertinent change in other cytokines such as IFNG/IFNγ, IL1A/IL1α, IL1B/IL1β, and IL6. Interestingly, it is revealed that SMURF1 plays a critical role in controlling M. tb replication in humans. Using primary monocytes derived macrophages followed by knockdown of SMURF1 using shRNA promoted a higher bacterial burden. It suggests that SMURF1 executes an essential role in exhibiting selective autophagy against M. tb in humans. Intriguingly, SMURF1 localizes in granuloma and diverse cell types in the human lungs of active TB patients. Furthermore, SMURF1 colocalizes to M. tb in human lungs tissue biopsies suggesting that SMURF1 plays an essential role in providing immunity against M. tb in active tuberculosis patients during the chronic phase of infection [81].
Autophagy receptors play critical roles in targeting intracellular pathogens to phagophores via xenophagy
Ubiquitination of the cargo recruits autophagy receptors containing the LC3-interacting motif or GABARAP-binding region, facilitating the assembly of phagophores around the targeted substrates [100,101]. As discussed above, SQSTM1, CALCOCO2, NBR1, OPTN, and TAX1BP1 also contain UBD that helps in identifying the ubiquitinated cargo and recruitment of phagophore membranes [70]. Cargo recruitment to the phagophore membranes requires interactions between autophagy receptors and LC3/GABARAP proteins embedded in the phagophore membrane [102]. The resulting interaction facilitates the formation of autophagic vacuoles (autophagosomes) that envelope the cargo and consequently direct it to the lysosomes [103].
TAX1BP1 contains several functionally essential domains commonly found in autophagy receptors, including LIR that binds to LC3 and GABARAP (Figure 5) [104]. It also comprises the N-terminal AZI2/NAP1-TBKBP1/SINTBAD interaction domain, the TRAF6 binding region, a dimerization domain, and a C-terminal overlapping Ub and MYO6 (myosin VI) interacting domain [105,106]. Mechanistically, TAX1BP1 functions downstream to SQSTM1 and is required to assemble signaling complexes to the phagosome, as reported with S. typhimurium [107]. TAX1BP1 delivers M. tb to phagophore membranes having LC3, thus functioning in a specific step of autophagic targeting (Table 2) [69].
Figure 5.
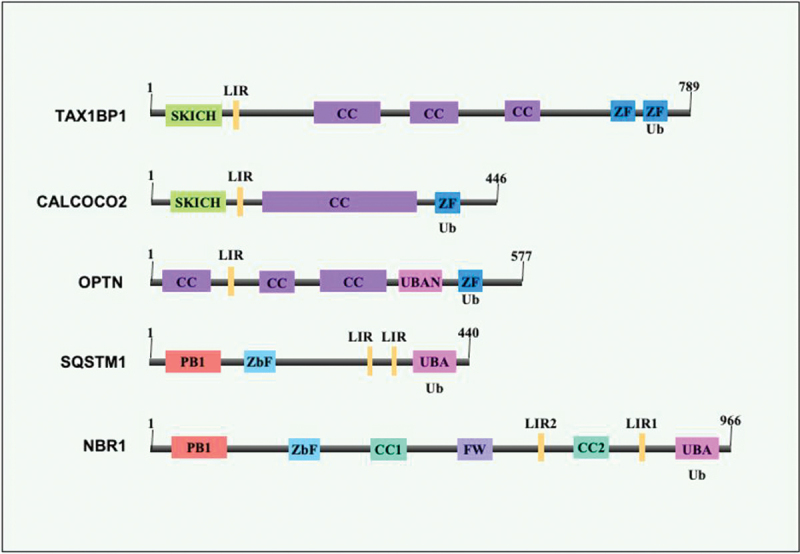
Structural features of xenophagy receptors. The xenophagy receptors (selective autophagy receptors), also known as sequestome-1-like receptors, include TAX1BP1, CALCOCO2, NBR1, SQSTM1, and OPTN. PB1 (dark pink), ZZ type zinc finger domains (light blue and dark blue), coiled-coil (purple), FW domain (light purple), LC3-interacting regions (LIRs) are marked by yellow. UBA domain (light pink) and SKICH domains are marked as light green. The size of the receptors is shown.
Table 2.
List of autophagy receptors involved in xenophagy and their functions.
| Receptors | Process | Tag | Role | References |
|---|---|---|---|---|
| TAX1BP1 | Xenophagy Mitophagy |
Ub | TAX1BP1, modulated by LAMTOR1-LAMTOR2 complex, is recruited to ubiquitinated bacteria along with MYO6 (myosin VI) and helps in elimination of pathogens by xenophagy. | [69,107–109] |
| CALCOCO2 | Mitophagy Xenophagy |
LGALS8, Ub | Targets pathogens to autophagosomes mediated by interaction with LC3C or LGALS8. It also promotes the maturation of autophagosomes by interacting with LC3A, LC3B, and/or GABARAPL2. Moreover, along with MTPAP (mitochondrial poly(A) polymerase) it forms autophagy receptor complex for enhanced removal of damaged mitochondria via mitophagy. | [110,111] |
| OPTN | Autophagy Xenophagy Aggrephagy Mitophagy |
Ub | OPTN acts as an autophagic receptor for the removal of polyubiquitinated substrates. Enhanced binding to Ub chains after phosphorylation by TBK1 promotes mitophagy. OPTN also mediates the degradation of inclusion bodies in Ub-dependent manner. This receptor is also critical for autophagic host defense against intracellular infections. | [112–114] |
| SQSTM1 | Autophagy Xenophagy Mitophagy Aggrephagy Lipophagy Pexophagy |
Ub | SQSTM1 is involved in LC3-mediated linking of ubiquitinated protein aggregates to autophagic machinery. Phosphorylated SQSTM1 promotes Ub conjugation during xenophagy and is important for controlling cytosolic bacteria. Ubiquitination of mitochondrial proteins requires SQSTM1 during mitophagy. It is also involved in organelle quality control like pexophagy as well as lipophagy. | [115,116,117] |
| NBR1 | Mitophagy Xenophagy Aggrephagy Pexophagy |
Ub | NBR1 is a specific receptor involved in pexophagy. It also acts as a receptor for autophagosomal degradation of ubiquitinated targets but is dispensable for mitophagy. NBR1 binds bacterial pathogens and viral particles to mediate their xenophagic degradation. | [81,118,119] |
The autophagy receptors TAX1BP1, CALCOCO2, OPTN, SQSTM1, and NBR1, are involved in the xenophagic clearance of Ub-tagged intracellular cytosolic bacteria. The functions of SQSTM1 in xenophagy and other processes such as mitophagy, aggrephagy, lipophagy, and pexophagy are well-established. The function and roles of other autophagy receptors are emerging now in xenophagy and other “phagy.” Importantly, in autophagic clearance of invading pathogens, these receptors execute non-redundant functions, although they perform redundant functions in other processes such as aggrephagy.
CALCOCO2 contains LIR and unique CLIR regions that interact with LC3A, LC3B, LC3C or GABARAPL2 (Figure 5). CALCOCO2 also contains the N-terminal SKICH domain, coiled-coil domain, C-terminal LGALS8-interacting region, and zinc fingers 1 and 2 (Figure 5). C2H2 zinc finger recognizes mono-Ub, and poly-Ub (K48, K63, and Met1-linked linear Ub chains) to target Ub decorated pathogens for autophagic degradation (Table 2) [120]. CALCOCO2 targets S. typhimurium to the phagophore membrane by simultaneously interacting with LC3C and ubiquitinated cytosolic bacteria or LGALS8 displayed on the damaged vacuoles [63]. CALCOCO2 executes a dual role in the xenophagic clearance of S. typhimurium by targeting it to the phagophore and promoting autolysosome maturation. CALCOCO2 executes a crucial role in xenophagy mediated killing of an array of diverse bacterial pathogens such as L. monocytogenes and Shigella [57,121,122]. Targeting of Shigella to the autophagy pathway is dependent on the coordinated activity of CALCOCO2 and SQSTM1 by regulating the activity of each other [122]. L. monocytogenes recruits CALCOCO2 without the requirement of SQSTM1 activity, implying that different pathogens have evolved different strategies to avoid autophagy [122]. M. tb also recruits CALCOCO2 and SQSTM1 to phagosome in an ESX-1 dependent pathway. BCG, which lacks an ESX-1 type VII secretion system, is not recruited to the phagosome for xenophagy mediated clearance in macrophages [121]. Restoration of ESX-1 in BCG through complementation of the RD1 (region of difference 1) locus of M. tb leads to the increased recruitment of LC3. Further, the knockout of either CALCOCO2 or SQSTM1 leads to the decrease in LC3 colocalization to M. tb [121]. CALCOCO2 also binds with NAP1L1 (nucleosome assembly protein 1 like 1) and TBKBP1/SINTBAD to recruit TBK1, critical for transcriptional induction of type-1 interferons and autophagic innate immune response during M. tb infection [57].
OPTN is an Ub binding, selective autophagy receptor that takes part in xenophagy, aggrephagy, and mitophagy [123–125]. The N terminus of OPTN harbors two coiled-coil domains with an LIR-motif sandwiched between them. The C-terminal domain of OPTN comprises a Ub binding domain UBAN and a conserved zinc finger (Figure 5). UBAN of OPTN is homologous to the regulatory subunit of the IKK complex of the NFKB1 signaling pathway. The UBAN domain selectively binds with M1-linked linear Ub chain and K63-linked conjugates [126,127]. The coiled-coil domain of OPTN forms a stable hetero-tetrameric complex with the C terminus of TBK1, which in turn regulates the function of OPTN in selective autophagy [128,129]. TBK1 phosphorylates the S172 residue of OPTN next to the LIR-motif, enhancing the binding affinities towards Atg8-family proteins. TBK1 also phosphorylates S473 in the UBAN domain of OPTN that enhances the functional efficacy of OPTN in selective autophagy [123,124,130]. The role of OPTN in antimycobacterial defense is recently unraveled [69,108]. It interacts with mycobacterial virulence factors and, after phosphorylation, is colocalized to M. tb in macrophages [69]. TBK1 mediates the phosphorylation of OPTN to increase its affinity for LC3 that promotes lysosomal degradation of L. monocytogenes. Host cells lacking LC3 and OPTN showed increased presence of replicating L. monocytogenes. Moreover, L. monocytogenes subverts or evades the OPTN pathway to achieve persistence in the cytosol or intracellular vacuoles [131]. Gene knockout, knockdown, and overexpression strategies have shown that SQSTM1 and OPTN play essential roles in targeting the M. marinum to phagophores and subsequently to lysosomes [132,133].
SQSTM1 was the first mammalian selective autophagy receptor described [134,135]. Like other xenophagy receptors, SQSTM1 also contains a dimerization domain, an LIR-motif, and a UBD. SQSTM1 also contains nuclear localization signal, zinc finger (ZZ), and KEAP1-interacting region (Figure 5). Polymerization of SQSTM1, mediated by its N-terminal PB1 domain, is essentially required for its selective autophagy function [136,137]. The efficient delivery of cargo to the phagophore depends on the Ub binding of SQSTM1 and its interaction with other effector proteins [138,139]. SQSTM1 takes part in the degradation of N-terminal arginylated protein substrates through macroautophagy [140,141]. SQSTM1 targets viruses like the Sindbis virus for xenophagic elimination [142]. It also targets S. typhimurium together with CALCOCO2 and OPTN for efficient xenophagy mediated clearance. SQSTM1 is reported in xenophagic clearance of other intracellular bacteria such as Listeria, Shigella, M. marinum, and M. tb [108,122,133,143,144]. UBA domain of SQSTM1 interacts with poly-Ub chains linked via K63, and its activity depends on UBA phosphorylation and ubiquitination [145,146]. Several E3-Ub ligases like TRIM50 (tripartite motif containing 50), TRAF6, SMURF2 (SMAD specific E3 ubiquitin protein ligase 2), and KEAP1 directly interact with SQSTM1 to promote ubiquitination of its substrate, thus affecting inclusion body formation and aggrephagy [147–150]. KEAP1-CUL3 E3-Ub ligase ubiquitinates K420 in the UBA domain of SQSTM1, modulating its sequestration and degradation activity [151]. RNF166 also ubiquitinates SQSTM1 at K91 and K189 using atypical Ub-conjugated chains containing K29 and K33 linkages [61]. The role of SQSTM1 in the xenophagic clearance of intracellular bacteria depends on its K29 and K33 Ub linkages (Table 2) [61].
The specific ribosomal proteins and bulk ubiquitinated cytoplasmic proteins are delivered to the autolysosome through the autophagy receptor SQSTM1 and act as mycobactericidal peptides to kill M. tb [152]. Without SQSTM1, the mycobactericidal molecules are not formed, and the autophagic process becomes inefficient, thus leading to the suppression in the elimination of M. tb [152]. SQSTM1 independent autophagy induced in macrophages containing intracellular mycobacteria is governed by the acidification and acquisition of lysosomal hydrolases in the compartments containing mycobacteria in macrophages [152]. This separation of unique functions of SQSTM1 in autophagy conferred by the cytoplasmic proteins of microbicidal properties underscores the unique role that SQSTM1 plays on autophagic organelles. Phagophore formation and progression that contains M. tb is executed by SQSTM1 and ATG5 [152]. ATG5 mediates trafficking of degradative vesicles and MHC-II to mycobacteria-containing autolysosomes, and SQSTM1 is involved in the ubiquitination of M. tb. Mycobacterial antigenic peptides are presented to CD4+ T cells via MHC-II in dendritic cells containing mycobacteria in autolysosome implies that dendritic cells promote the antigen presentation of mycobacterial origin to CD4+ T cells via MHC-II [153].
Further, SQSTM1 also plays a crucial role in regulating antimicrobial immune responses mediated by CGAS (cyclic GMP-AMP synthase), a DNA sensor found in the cytoplasm and the adaptor molecule STING1. SQSTM1, together with TBK1, mediates STING1 degradation via autophagy by phosphorylation and ubiquitination. SQSTM1‐deficient cells are impaired in STING1 degradation leading to overproduction of type-I interferon. STING1 trafficking also depends on SQSTM1, and the absence of SQSTM1 fails to traffic STING1 to phagophores. CGAS‐STING1 pathway mediated sensing of DNA induces activation of TBK1 that phosphorylates IRF3 (interferon regulatory factor 3) for induction of type-1 interferon expression. TBK1 also phosphorylates SQSTM1, which induces STING1 degradation, causing attenuation of the type-1 interferon response, thus regulating the pathway [154].
NBR1 is a selective autophagy receptor whose role in signal transduction is well established. It shows a high similarity to SQSTM1 in its domain organization. It contains the PB1 domain at the N terminus, a coiled-coil domain, LIRs, and a UBA domain at its C-terminal region (Figure 5). The coiled-coil domain mediates NBR1 dimerization. It interacts with SQSTM1 via the PB1 domain, thus linking the receptors together [155–158]. NBR1 also interacts with LC3/GABARAP and Ub independent of receptor SQSTM1. Accumulation of SQSTM1 and NBR1 in inclusion bodies after autophagy inhibition is required for ubiquitination and crosslinking of proteins, resulting in induction of aggrephagy [159]. NBR1 is an evolutionarily conserved autophagy receptor, unlike SQSTM1, which is only present in metazoans [160]. Genetic knockdown and overexpression studies of NBR1 suggest that it is a primary receptor for inducing pexophagy mediated by its UBA, coiled-coil, amphipathic α-helical J, and LIR domains [161]. Similar to SQSTM1 and TAX1BP1, NBR1 also colocalizes to M. tb containing phagosome in macrophages [69]. The association of NBR1 to the M. tb surface-associated Ub is recently reported. PE_PGRS29, a surface-associated Ub binding protein of M. tb, recruits Ub that binds xenophagy receptors, including NBR1 [108]. NBR1 also takes part in the xenophagy mediated clearance of group A Streptococcus and S. typhimurium. Late endosomal/lysosomal adaptor, MAPK, and LAMTOR2 (late endosomal/lysosomal adaptor, MAPK and MTOR activator 2) physically interact with NBR1 and TAX1BP1. This association is required for phagophore recruitment of NBR1 to group A Streptococcus. It is also required to recruit TAX1BP1 to phagophores containing S. typhimurium and group A Streptococcus [109].
Co-option of host Ub system by M. tb to modulate immune defense
The interaction of M. tb with the host and its successful survival inside macrophages is a result of long-term coevolution, culminating in the efficient modulation of the immune system [162,163]. M. tb efficiently manipulates autophagy, apoptosis, and maturation of phagosome to facilitate intracellular survival and pathogenesis [164–168]. Host signaling cascades are amenable pathogen targets due to overall cellular effects and network sharing. Various mycobacterial proteins are known to subdue the host-generated innate and adaptive responses for its survival, thereby magnifying the pathogen’s virulence armor [169–171]. Structural adaptations like protein disorder and the resulting moonlighting function allow interaction with various host signaling pathways to modulate the responses [172,173]. Signaling pathways downstream of NFKB1 and MAP kinases regulate the immunity against invading pathogens by producing immune effector cytokines like TNF/TNFα, IL1B, and IL12, thus modulating the pathogen’s survival inside the host [174,175]. M. tb secreted effectors target different host organelles to effectively manipulate multiple cellular functions [176–178]. Intriguingly, many bacterial effectors mimic the E3-Ub ligase activity of the host, hence co-opting the host Ub system to manipulate the proinflammatory signaling cascade to hijack innate immunity (Figure 6) [7,179]. The ingenious use of the host ubiquitination pathway by M. tb to dampen host innate immunity appears to be a direct outcome of host-pathogen interaction and coevolution [180,181]. Suppression of NFKB1 and MAPKs plays an essential role in the successful survival of M. tb inside macrophages [182]. Unfortunately, mechanistic details of virulence factors mediated suppression of host innate immune signaling remain partially understood [183,184]. We recently unravel that M. tb exploits disorder to order transition of its protein effectors to impact the production of inflammatory cytokines [170]. Nevertheless, M. tb also exploits host Ub via some secreted effectors to induce potent inflammatory responses to increase host tissue pathology for dissemination [185].
Figure 6.
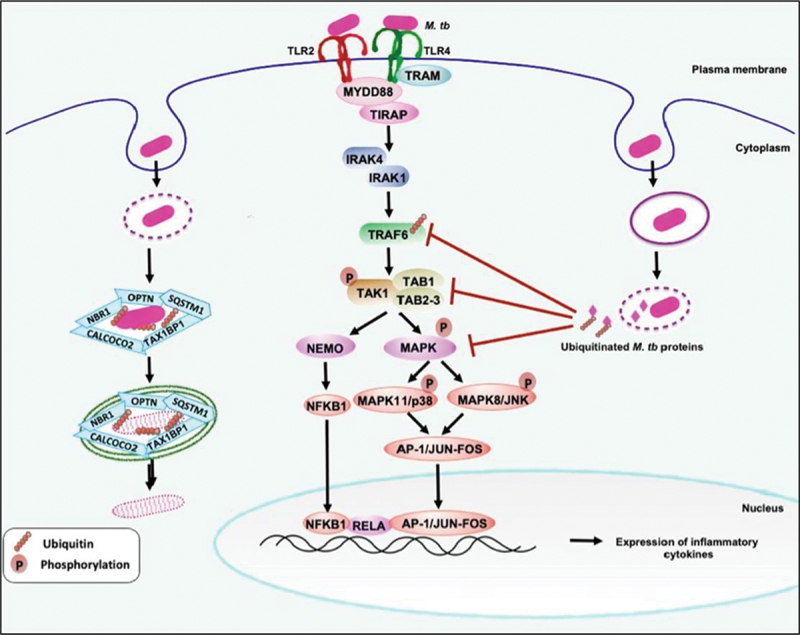
M. tb co-opts the host Ub-system to manipulate and subdue host immunity. The surface immune receptors TLR2-TLR4 recognize M. tb or its associated molecular patterns to initiate downstream signaling events to generate innate immune responses against the pathogen. Downstream to TLRs, the adaptors TRAF6, TAB1-TAK1-binding protein 1, and TAB2-TAB3 sense Ub chains and affect the NFKB1 and MAP kinase signaling to produce proinflammatory cytokines TNF, IL12, IL6, and IL1B. The M. tb secreted effectors (e.g., Mpt53, PtpA, and Rv0222 co-opt the host Ub-system and affect the Ub-mediated activation of kinases MAP3K7 and TRAF6. It ultimately inhibits or activates the NFKB1 and MAPKs signaling and produces innate immune effector cytokines to dampen innate immunity against the pathogen. The host exploits one of the surface Ub-binding M. tb effectors [e.g., PE_PGRS29 (Rv1468c)] to start xenophagy against the pathogen and recruit receptor proteins SQSTM1, NBR1, CALCOCO2, and OPTN.
Mpt53 (DsbE) is a secreted disulfide bond (Dsb)-forming protein of M. tb. It harbors the thioredoxin domain and can oxidize cysteines like other Dsb proteins of M. tb and E. coli [186]. It induces activation of host signaling intermediate MAP3K7/TAK1 by phosphorylation and K63-linked ubiquitination mediated by TRAF6. Mpt53 activates MAP3K7 by inducing the formation of a disulfide bond at C210 that depends on the thioredoxin domain and conserved CXXC motif. Interestingly, Mpt53 further activates MAP3K7 by K63-linked ubiquitination, which is necessarily required for its complete activation. MAP3K7 activation leads to enhanced activity of downstream signaling cascade involving NFKB1, MAPK8/JNK, and MAPK11/p38. The activation of MAPKs results in the production of innate immune system regulatory cytokines TNF, IL6, IL12, and IL1B (Figure 6) [186]. These proinflammatory cytokines play critical roles against M. tb virulence, thus seemingly antithetical effect. The authors suggest that the host uses an evolutionarily conserved Ub system and post-translational modifications to suppress the pathogen’s ability to cause disease. However, the use of Mpt53 to activate MAP3K7 could be an elegant example of an effort by the pathogen to cause an overt-inflammatory response leading to tissue pathology assisting in dissemination.
Recently, the role of another M. tb secreted effector, a low molecular weight tyrosine phosphatase PtpA, is shown to dampen the functions of innate immunity by inhibiting MAPKs and NFKB1 signaling. PtpA contains a unique UBD-like motif that binds to the host Ub leading to the activation of its phosphatase activity. Activated PtpA dephosphorylates phospho-MAPK8 and MAPK11 to inhibit downstream functions, including the production of TNF, IL1B, and IL12 cytokines. PtpA also competitively inhibits K63-linked conjugation of Ub to TAB3 (TGF-beta activated kinase 1 (MAP3K7) binding protein 3) that inhibits activation of NFKB1, manifesting in suppression of proinflammatory cytokine production (Figure 6) [185]. It exemplifies the pathogen’s exploitation of the host Ub system to dampen innate immunity against M. tb by targeting evolutionarily conserved signaling cascades.
Rv0222, a secreted protein of M. tb, also co-opts the host Ub system to subvert the innate host immunity by inhibiting the production of anti-M. tb effector cytokines [182]. ANAPC2 (anaphase promoting complex subunit 2), a host E3-Ub ligase, ubiquitinates Rv0222 at K76 using K11-linked Ub chains. Ubiquitination of Rv0222 promotes its interaction with the host tyrosine phosphatases PTPN6 (protein tyrosine phosphatase non-receptor type 6) and PTPN11 (protein tyrosine phosphatase non-receptor type 11). Rv0222 also interacts with adaptor protein TRAF6, henceforth recruiting tyrosine phosphatases to TRAF6 containing complexes. Interestingly, this recruitment inhibits the K63-linked ubiquitination of TRAF6, thus inhibiting its function. Inhibition of TRAF6 prevents activation of downstream NFKB1 and MAPKs signaling cascades, impeding the production of innate immune effector cytokines (IL1B, IL12, and IL6), compromising anti-tuberculosis immunity (Figure 6). This seminal study reveals an unfamiliar subversion of host-immunity by a virulence effector of M. tb by co-opting the host Ub system [182].
Another recent evidence demonstrates that M. tb PE_PGRS29, a surface-associated protein comprising a UBA domain, interacts with all forms of free-floating Ub using hydrophobic interactions. The interaction between PE_PGRS29 and Ub engages the autophagic receptor NBR1, SQSTM1, OPTN, and CALCOCO2 on bacteria. Recruitment of autophagy receptors targets M. tb to LC3-associated phagophores for autophagic clearance (Figure 6) [108]. This is the first report mentioning the eukaryotic-like UBA domain in bacteria, which interacts with Ub using hydrophobic interactions rather than conjugation of Ub by E3-Ub ligases. The induction of xenophagic clearance by a pathogen-specific protein is likely a strategy to maintain long-term bacterial survival by reducing the inflammatory responses directed against the bacteria. Contrary to the explanation, a host-directed strategy could target the bacteria using a conserved Ub system to mediate autophagic clearance. Another host Ub binding protein, UBQLN1 (ubiquilin 1), also interacts with M. tb secreted and surface proteins to recruit Ub to M. tb or M. tb associated structures [187]. The recruitment of Ub engages autophagy receptor proteins linking degradative LC3-mediated autophagy and clearance. UBQLN1 consists of conserved UBA and UBL domains at its N- and C-termini, though the mechanism of Ub recruitment remains unclear [187].
In an elegant study published recently [176], the authors showed the unusual E1, isopeptidase, and E3-Ub ligase activities employed by M. tb protein PknG to conjugate Ub to the host E2 UBE2L3/UBCH7 (ubiquitin conjugating enzyme E2 L3). PknG, a serine/threonine-protein kinase/STPK similar to the eukaryotic serine/threonine kinases, contains several conserved eukaryotic-like UBL and TPR (translocated promoter region, nuclear basket protein) domains [176,188,189]. M. tb exploits PknG (a secreted protein) to manipulate many crucial host defense strategies to promote M. tb pathogenesis [190]. However, the molecular mechanisms utilized by PknG remain elusive. It is revealed that PknG unusually co-opts the host Ub-machinery to transfer Ub (K48-linkage) to the NFKB1 signaling effectors TRAF2 (TNF receptor associated factor 2) and MAP3K7, inducing degradation of these proteins thus inhibiting proinflammatory cytokine production [176].
M. tb employs its virulence effectors to dampen autophagy
The PE/PPE family is an enigmatic class of mycobacterial proteins specifically found only in pathogenic species [191,192]. Recent studies on PE/PPE proteins have shown their role in modulating cell death pathways. PE6 protein of M. tb possesses the remarkable property of inhibiting autophagy via autophagy master regulator MTOR, inducing the inhibitory phosphorylation of autophagy initiating kinase ULK1, leading to reduced autophagy flux [193]. Many other PE/PPE family proteins (PE_PGRS20, PE_PGRS21, PE_PGRS30, PE_PGRS47, PPE44, and PPE51) were recently identified as an autophagy-inhibitors through the loss of function screening of the M. tb transposon mutant library. The screening identified a total of 16 proteins, of which six belonged to the PE/PPE family [194]. PE/PPE family protein’s inhibitory function was validated using knockout and knock-in strains of M. tb and M. smegmatis. These proteins activate the autophagy master regulator MTOR and inhibit the production of proinflammatory cytokines TNF and IL1B, which play essential roles in mediating the control of M. tb pathogenesis. PE_PGRS47 prevents phagosome maturation and fusion of autophagosome to the lysosome for increased intracellular survival and virulence of M. tb [195]. A recent study demonstrates that PE_PGRS47 and PE_PGRS20 of M. tb effectively inhibit autophagy initiation by physical interaction with RAB1A protein of the host (a small GTPase family protein involved in vesicular transport and fusion to the target membrane). This interaction inhibits the function of autophagy initiating kinase ULK1, thus playing essential roles in the enhanced survival of M. tb inside macrophages. Interestingly, PE_PGRS47 and PE_PGRS20 proteins also inhibit antigen presentation and secretion of proinflammatory cytokines which are critical immune effectors of innate and adaptive immunity [196]. M. tb virulence factor PE_PGRS41, a cell surface localized protein, suppresses autophagic flux via modulating the functions of Atg8-family proteins, a critical effector of phagophore expansion and maturation [197]. These findings suggest that M. tb effectively utilizes PE/PPE family proteins to hamper autophagy and host immunity directed against M. tb for better intracellular survival, virulence, and pathogenesis.
Apart from PE/PPE family, M. tb employs SapM (secreted acid phosphatase of M. tb), which possesses phosphatase activity to inhibit phagophore maturation and acidification of autolysosome. It suppresses autophagy flux in a PI3K-dependent manner. SapM also inhibits RAB family protein GTPase RAB7A (RAB7A, member RAS oncogene family) activity through physical interaction using its C-terminal domain, blocking phagosome-lysosome fusion [198]. M. tb secretory peptidoglycan hydrolase RipA harbors LIR-motif and inhibits autophagy mediated by activation of pro-survival PI3K-AKT1 (AKT serine/threonine kinase 1)-MTOR signaling axis and repression of ULK1 [199]. Heat shock proteins (HSPs) play a pivotal role in M. tb replication inside macrophages and are required for sustained survival in latent M. tb infection [200]. Hsp16.3 of M. tb, an alpha-crystallin-type Hsp, impairs autophagy in latent infection. Enhanced expression of Hsp16.3 inversely affects LC3 protein level disrupting phagophore formation [201]. However, the exact mechanism exploited by Hsp16.3 in autophagy regulation is not fully elucidated. Heparin-binding hemagglutinin (HBHA) of M. tb also dampens autophagy by inhibiting LC3 and BECN1 (beclin 1) expression levels, preventing phagophore maturation [202]. NuoG of M. tb encodes NADH dehydrogenase I subunit G, and its genetic disruption in BCG leads to the enhanced recruitment of LC3 and induction of autophagy. NuoG also inhibits LC3-associated phagocytosis (LAP) by neutralizing CYBB (cytochrome b-245 beta chain)-dependent ROS (reactive oxygen species) production required for efficient conjugation of LC3 to the phagosome and phagosome-to-lysosome fusion (Figure 7) [203]. M. tb Eis (enhanced intracellular survival protein) is an acetyltransferase involved in infection, transmission, and host immune modulation. It downregulates autophagy by inhibition of ROS production in a MAP3K7-dependent manner. Eis also acetylates histone H3 to activate enhanced transcription of IL10, which negatively regulates autophagy by inducing the AKT1-MTOR-RPS6KB/p70S6K pathway [204].
Figure 7.
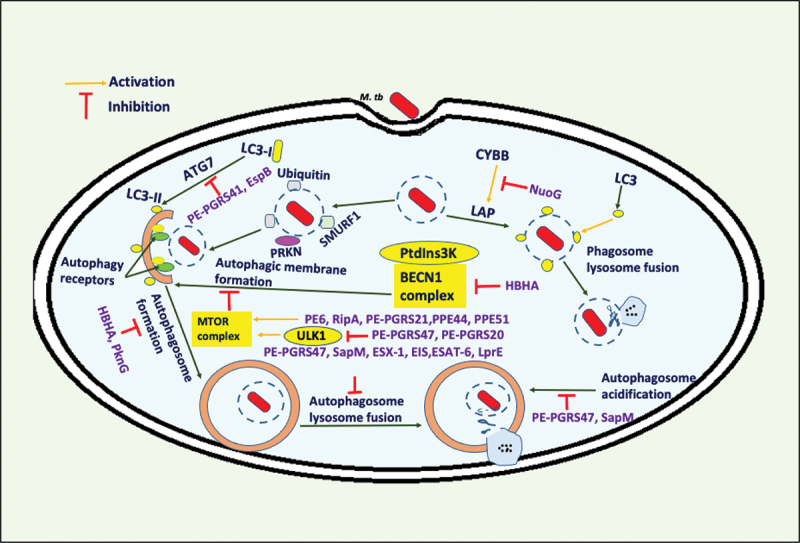
M. tb uses its virulence effector proteins to inhibit autophagy. The M. tb proteins inhibit host-induced autophagy for its efficient intracellular survival inside macrophages. These proteins exhibit divergent mechanisms for the inhibition of autophagy. M. tb utilizes its effectors to dampen autophagy by inhibiting phagophore maturation, autophagosome fusion to the lysosome, and autolysosome acidification. The efficient mechanisms utilized by M. tb culminate in the reduction of autophagy flux. Exploring the interaction between these proteins will shed light on a better understanding of mechanisms of autophagy inhibition by M. tb.
A mycobacterial calcium efflux protein CtpF (Rv1997) inhibits autophagy by activating MTOR. The calcium release from lysosomes activates calmodulin which further activates the MTOR complex to regulate downstream effects [205]. Rv3242c of M. tb encodes a protein possessing phosphoribosyltransferase activity that inhibits NAMPT (nicotinamide phosphoribosyltransferase) of the host. Rv3242c is involved in NAD/nicotinamide adenine dinucleotide biosynthesis in M. tb and regulates autophagy and ROS production. M. smegmatis expressing Rv3242c activates MAPK3 (mitogen-activated protein kinase 3) pathway, inhibiting autophagy in macrophages. Rv3242c also leads to enhanced IL10 production via MAPK11 and MAPK3 pathways, thus, attributing to autophagy inhibition [206]. ESX-1 type VII secretion system associated protein EspB inhibits autophagy by suppressing LC3B expression and phagophore formation. EspB inhibits the expression of IFNGR1 (interferon gamma receptor 1) on macrophages and represses the phosphorylation of IFNGR1 and STAT1. This abolishes IFNG-induced autophagy directed against M. tb [207]. Esat-6 of M. tb, a secretory protein, is a very well-studied effector involved in pathogenesis. Esat-6 regulates MYC (MYC proto-oncogene, bHLH transcription factor) proto-oncogene and MTOR in macrophages to manipulate host innate immune defenses. It also upregulates the expression of SOD2 (superoxide dismutase 2) to mitigate the lethal effects of ROS, inhibiting autophagosome-lysosome fusion for survival inside hostile cellular milieu [208].
Lipoproteins represent an essential class of M. tb virulent proteins, but their exact role in pathogenesis remains elusive. LprE, a membrane-bound putative lipoprotein of M. tb, inhibits autophagy by inhibiting phagolysosomal fusion. LprE mutant of M. tb results in increased expression of LC3, ATG5, and BECN1 proteins. This recruits RAB7A, EEA1 (early endosome antigen 1), and LAMP1 to mature phagosomes, demonstrating its critical role in autophagy inhibition [209]. PknG, the only secreted eukaryotic type serine-threonine kinase of M. tb, is involved in the enhanced intracellular survival by inhibiting phagosome maturation (Figure 7) [190]. PknG interacts with the RAB family of small GTPase RAB14 (RAB14, member RAS oncogene family) employing its TPR domain and α-helix, resulting in an inhibition of its GTPase activity. PknG prevents the hydrolysis of RAB14 bound GTP via physical interaction and inhibits its activation by phosphorylating and inhibiting TBC1D1 (TBC1 domain family member 1), a GTPase activating protein [210]. These results implicate that PknG is a moonlighting protein of M. tb, inhibiting autophagy flux for efficient intracellular survival. These elegant studies demonstrate the diversification of M. tb proteins in tackling host defense responses like autophagy. It would be fascinating to get in-depth insights into the interplay existing among autophagy-inhibiting proteins.
Proteins containing the LIR-motif play essential roles in autophagy regulation at many critical junctures of phagophore biogenesis, extension, and maturation [211,212]. We were interested in exploring the M. tb proteome for the presence of LIR-motif. Moreover, we also tried to decipher whether M. tb effectors already reported having a role in autophagy inhibition comprise this motif. Interestingly, the M. tb proteome analysis revealed that conserved WxxL and xLIR-motifs are present in abundance. A total of 91 proteins are coded by M. tb proteome, which comprises WxxL+xLIR (ILV) motifs that are expected to interact with Atg8-family proteins of the host for autophagy manipulation (Table S1). Further, WxxL and xLIR (WFY) conserved motifs are present in five M. tb proteins. These proteins include Rv1059 (conserved hypothetical protein), Rv1536 (Isoleucyl tRNA synthetase), Rv2182c (1-acylglycerol-3-phosphate O-acyltransferase), Rv2643 (probable arsenic-transport integral membrane protein ArsC), and Rv3038c (conserved hypothetical protein). As discussed above, the M. tb proteins modulate autophagy such as Rv3310, Rv0410c, Rv2031c, Rv3242c, and Rv1477 comprise one WxxL motif each, whereas Rv3151 contain two WxxL motifs. M. tb protein Rv1997 harbors six WxxL motifs, and Rv3881c contains one xLIR-motif (Table S2). These sequence features highlight the integral role of LIR-motifs in autophagy regulation and open an unexplored area in mycobacteriology.
Conclusions and perspective
Autophagy has an established role in the clearance of M. tb as a part of innate immune defense. Disruption of genes involved in autophagy manifests in enhanced survival of M. tb in vitro and in vivo. Induction of autophagy by starvation or rapamycin treatment decreases intracellular survival of M. tb in macrophages [213,214]. However, an increasing body of emerging evidence suggests that loss of canonical autophagy pathway does not correlate with intracellular survival of M. tb [99]. It is, in turn, the inflammatory pathology mediated by polymorphonuclear cells that directly correlates with susceptibility to infection. The ATG5-mediated control seems to be due to alternate autophagic pathways or its role in non-autophagic cellular processes like endocytosis, cell death, and inflammation, as reported earlier [215]. This apparent insignificance of xenophagy in controlling M. tb survival and the recent discovery of a battery of M. tb proteins that dampen autophagy suggests that M. tb has evolved to subvert the host autophagic mechanism. On the contrary, it could be safely speculated that M. tb exploits this process to dampen the inflammation by limiting the cytosolic exposure [99,121]. It could be an evolutionary adaptation to maintain chronic infection by limiting excessive host inflammation to help prolonged survival and greater transmission. Further, rapid clearance of these cytosolic mycobacteria may aid the growth of M. tb in a phagosome that mimics cellular components to avoid host immune surveillance. It has emerged as an efficient survival strategy adopted by intracellular pathogens to enhance survival by residing in vacuolar structures. These complexities in observations warrant detailed studies to delineate this intricate phenomenon with the need to revisit the earlier findings. Moreover, further research is also desired in the frontier areas of M. tb mediated efficient inhibition of host autophagy. Inhibition of the involved virulence effectors using novel therapeutics could complement anti-tuberculosis therapies. The details of LIR and xLIR-containing M. tb proteins could further furnish additional drug targets for the efficient management of tuberculosis disease.
Host-directed therapeutics is an emerging approach for treating tuberculosis by modulating protective immunity. It will be worth exploring the critical roles of targeting the host E3-Ub ligases and their associated molecules in treating tuberculosis. Understanding the role of Ub-associated molecules in autophagy might shed more light on their involvement in controlling intracellular pathogens by generating robust innate immunity. Recent advancements in understanding autophagy regulators in controlling immune responses further enhance the development of promising drug candidates. In particular, exploring the role of molecules/drugs as autophagy-based adjunctive host-directed therapeutics in MDR/XDR tuberculosis treatment would potentially boost the treatment outcomes. Numerous pre-clinical and clinical studies addressing combined usage of conventional treatment and autophagy-adjunctive therapeutics need to be explored. Prospects for killing the M. tb inside the macrophage cells by modulating E3-Ub ligases could be achieved through small molecules or peptides that activate the catalytic activity and interaction with the infecting pathogen. Current approaches like structure-based design and advanced small-molecule screening technologies could be used to develop E3-Ub ligase activators.
DUBs can reversibly remove Ub, leading to enhanced protein stability or attenuation of Ub signaling that contributes to Ub homeostasis and autophagy [216]. DUBs play crucial roles in many aspects of autophagy regulation [217,218]. Various autophagy effectors are regulated by conjugating Ub and its removal using specific DUBs [218–220]. Few DUBs have been linked to modifying poly-Ub chains associated with intracellular pathogens, thus controlling the outcome of xenophagy [218]. Understanding the crosstalk of these antagonistic pathways at the molecular level is still limiting but expected to be valuable for deciphering novel therapeutic interventions against the pathogen [185]. The re-emergence of this dreaded disease due to evolving drug resistance is turning into a significant setback to end tuberculosis [221,222]. Therefore, it is pivotal to identify novel therapeutic targets and develop new vaccines and drugs, even emphasizing rework on century-old BCG to tackle this obnoxious pathogen [223,224].
Supplementary Material
Acknowledgments
MS acknowledges ICMR (Indian Council of Medical Research) for providing a Senior Research Associate fellowship at the National Institute of Pathology (NIOP), New Delhi. NQ is a DHR Young Scientist supported by the Department of Health Research, Ministry of Health and Family Welfare, GoI. AA is thankful to DBT for granting him fellowship (Scientist B) at NIOP. SZ acknowledges the Senior Research fellowship support from CSIR (Council for Scientific and Industrial Research), India GoI. JAS is funded by the Start-up Research Grant from UGC and DST-SERB. NS acknowledges the Senior Research fellowship support from NIOP, New Delhi. JS is an ICMR Postdoctoral Fellow at NIOP. UA is thankful to ICMR for providing him with, research fellowship. IK acknowledges the Senior Research Associate fellowship from DBT. SEH is a JC Bose National Fellow, Department of Science and Technology (DST), GoI and Robert Koch Fellow, Robert Koch Institute, Germany.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- 1.Mizushima N, Levine B, Longo DL.. Autophagy in human diseases. N Engl J Med. 2020. Oct 15;383(16):1564–1576. 10.1056/NEJMra2022774 [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132. [DOI] [PubMed] [Google Scholar]
- 3.Kimmey JM, Stallings CL. Bacterial Pathogens versus Autophagy: implications for Therapeutic Interventions. Trends Mol Med. 2016. Dec;22(12):1060–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016. Apr;26(4):399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67(1):425–479. [DOI] [PubMed] [Google Scholar]
- 6.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22(1):159–180. [DOI] [PubMed] [Google Scholar]
- 7.Lin YH, Machner MP. Exploitation of the host cell ubiquitin machinery by microbial effector proteins. J Cell Sci. 2017. Jun 15; 130(12):1985–1996. 10.1242/jcs.188482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Qian C, Cao X. Post-Translational Modification Control of Innate Immunity. Immunity. 2016. Jul 19;45(1):15–30. 10.1016/j.immuni.2016.06.020 [DOI] [PubMed] [Google Scholar]
- 9.Ribet D, Cossart P. Ubiquitin, SUMO, and NEDD8: key Targets of Bacterial Pathogens. Trends Cell Biol. 2018. Nov;28(11):926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol. 2017. Dec;14(12):963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jo EK. Autophagy as an innate defense against mycobacteria. Pathog Dis. 2013. Mar;67(2):108–118. [DOI] [PubMed] [Google Scholar]
- 12.Chai Q, Wang L, Liu CH, et al. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol Immunol. 2020. Sep;17(9):901–913. DOI: 10.1038/s41423-020-0502-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deretic V, Delgado M, Vergne I, et al. Autophagy in immunity against mycobacterium tuberculosis: a model system to dissect immunological roles of autophagy. Curr Top Microbiol Immunol. 2009;335:169–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vergne I, Chua J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: selective targeting of PI3P-dependent membrane trafficking. Traffic. 2003. Sep;4(9):600–606. [DOI] [PubMed] [Google Scholar]
- 15.Jamwal SV, Mehrotra P, Singh A, et al. Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci Rep. 2016. Mar 16;6(1):23089. 10.1038/srep23089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simeone R, Bobard A, Lippmann J, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 2012. Feb;8(2):e1002507. DOI: 10.1371/journal.ppat.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen RH, Chen YH, Huang TY. Ubiquitin-mediated regulation of autophagy. J Biomed Sci. 2019. Oct 21;26(1):80. 10.1186/s12929-019-0569-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Chai QY, Liu CH. The ubiquitin system: a critical regulator of innate immunity and pathogen-host interactions. Cell Mol Immunol. 2016. Sep;13(5):560–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev. 2019. Jul 1;43(4):341–361. 10.1093/femsre/fuz006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lerner TR, Borel S, Greenwood DJ, et al. Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol. 2017. Mar 6;216(3):583–594. 10.1083/jcb.201603040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romagnoli A, Etna MP, Giacomini E, et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy. 2012. Sep;8(9):1357–1370. DOI: 10.4161/auto.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar A, Rani M, Ehtesham NZ, et al. Commentary: modification of host responses by Mycobacteria. Front Immunol. 2017;8:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Alam A, Rani M, et al. Biofilms: survival and defense strategy for pathogens. Int J Med Microbiol. 2017. Dec;307(8):481–489. DOI: 10.1016/j.ijmm.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Alam A, Grover S, et al. Peptidyl-prolyl isomerase-B is involved in Mycobacterium tuberculosis biofilm formation and a generic target for drug repurposing-based intervention. NPJ Biofilms Microbiomes. 2019;5(1):3. DOI: 10.1038/s41522-018-0075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh Y, Kohli S, Sowpati DT, et al. Gene cooption in mycobacteria and search for virulence attributes: comparative proteomic analyses of Mycobacterium tuberculosis, Mycobacterium indicus pranii and other mycobacteria. Int J Med Microbiol. 2014. Jul;304(5–6):742–748. DOI: 10.1016/j.ijmm.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Saini V, Raghuvanshi S, Khurana JP, et al. Massive gene acquisitions in Mycobacterium indicus pranii provide a perspective on mycobacterial evolution. Nucleic Acids Res. 2012. Nov;40(21):10832–10850. DOI: 10.1093/nar/gks793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohli S, Singh Y, Sharma K, et al. Comparative genomic and proteomic analyses of PE/PPE multigene family of Mycobacterium tuberculosis H(3)(7)Rv and H(3)(7)Ra reveal novel and interesting differences with implications in virulence. Nucleic Acids Res. 2012. Aug;40(15):7113–7122. DOI: 10.1093/nar/gks465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar A, Alam A, Tripathi D, et al. Protein adaptations in extremophiles: an insight into extremophilic connection of mycobacterial proteome. Semin Cell Dev Biol. 2018. Dec;84:147–157. [DOI] [PubMed] [Google Scholar]
- 29.Grover S, Gupta P, Kahlon PS, et al. Analyses of methyltransferases across the pathogenicity spectrum of different mycobacterial species point to an extremophile connection. Mol Biosyst. 2016. May 26;12(5):1615–1625. 10.1039/C5MB00810G [DOI] [PubMed] [Google Scholar]
- 30.Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004. Nov 29;1695(1–3):55–72. 10.1016/j.bbamcr.2004.09.019 [DOI] [PubMed] [Google Scholar]
- 31.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81(1):203–229. [DOI] [PubMed] [Google Scholar]
- 32.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008. Jun;9(6):536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011. Jun 23;12(7):439–452. 10.1038/nrm3143 [DOI] [PubMed] [Google Scholar]
- 34.Corn JE, Vucic D. Ubiquitin in inflammation: the right linkage makes all the difference. Nat Struct Mol Biol. 2014. Apr;21(4):297–300. [DOI] [PubMed] [Google Scholar]
- 35.Haakonsen DL, Rape M. Branching out: improved signaling by heterotypic ubiquitin chains. Trends Cell Biol. 2019. Sep;29(9):704–716. [DOI] [PubMed] [Google Scholar]
- 36.D’Andrea A, Pellman D. Deubiquitinating enzymes: a new class of biological regulators. Crit Rev Biochem Mol Biol. 1998;33(5):337–352. [DOI] [PubMed] [Google Scholar]
- 37.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009. Aug;10(8):550–563. [DOI] [PubMed] [Google Scholar]
- 38.Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81(1):291–322. [DOI] [PubMed] [Google Scholar]
- 39.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009. Oct;10(10):659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dieckmann T, Withers-Ward ES, Jarosinski MA, et al. Structure of a human DNA repair protein UBA domain that interacts with HIV-1 Vpr. Nat Struct Biol. 1998. Dec;5(12):1042–1047. DOI: 10.1038/4220. [DOI] [PubMed] [Google Scholar]
- 41.Mueller TD, Feigon J. Solution structures of UBA domains reveal a conserved hydrophobic surface for protein-protein interactions. J Mol Biol. 2002. Jun 21;319(5):1243–1255. 10.1016/S0022-2836(02)00302-9 [DOI] [PubMed] [Google Scholar]
- 42.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005. Aug;6(8):610–621. [DOI] [PubMed] [Google Scholar]
- 43.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78(1):399–434. [DOI] [PubMed] [Google Scholar]
- 44.Smit JJ, Sixma TK. RBR E3-ligases at work. EMBO Rep. 2014. Feb;15(2):142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber J, Polo S, Maspero E. HECT E3 ligases: a tale with multiple facets. Front Physiol. 2019;10:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.George AJ, Hoffiz YC, Charles AJ, et al. A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front Genet. 2018;9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galdeano C. Drugging the undruggable: targeting challenging E3 ligases for personalized medicine. Future Med Chem. 2017. Mar;9(4):347–350. [DOI] [PubMed] [Google Scholar]
- 48.Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014. Apr;21(4):301–307. [DOI] [PubMed] [Google Scholar]
- 49.Ohi MD, Vander Kooi CW, Rosenberg JA, et al. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat Struct Biol. 2003. Apr;10(4):250–255. DOI: 10.1038/nsb906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vander Kooi CW, Ohi MD, Rosenberg JA, et al. The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry. 2006. Jan 10;45(1):121–130. 10.1021/bi051787e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verdecia MA, Joazeiro CA, Wells NJ, et al. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell. 2003. Jan;11(1):249–259. DOI: 10.1016/S1097-2765(02)00774-8. [DOI] [PubMed] [Google Scholar]
- 52.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009. Jun;10(6):398–409. [DOI] [PubMed] [Google Scholar]
- 53.Spratt DE, Walden H, Shaw GS. RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J. 2014. Mar 15;458(3):421–437. 10.1042/BJ20140006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011. Mar;240(1):92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selleck EM, Orchard RC, Lassen KG, et al. A Noncanonical Autophagy Pathway Restricts Toxoplasma gondii Growth in a Strain-Specific Manner in IFN-gamma-Activated Human Cells. mBio. 2015. Sep 8;6(5):e01157–15. 10.1128/mBio.01157-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joubert PE, Meiffren G, Gregoire IP, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe. 2009. Oct 22;6(4):354–366. 10.1016/j.chom.2009.09.006 [DOI] [PubMed] [Google Scholar]
- 57.Thurston TL, Ryzhakov G, Bloor S, et al. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009. Nov;10(11):1215–1221. DOI: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 58.Case ED, Chong A, Wehrly TD, et al. The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell Microbiol. 2014. Jun;16(6):862–877. DOI: 10.1111/cmi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogawa M, Yoshimori T, Suzuki T, et al. Escape of intracellular Shigella from autophagy. Science. 2005. Feb 4;307(5710):727–731. 10.1126/science.1106036 [DOI] [PubMed] [Google Scholar]
- 60.Jo EK, Yuk JM, Shin DM, et al. Roles of autophagy in elimination of intracellular bacterial pathogens. Front Immunol. 2013;4:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heath RJ, Goel G, Baxt LA, et al. RNF166 Determines Recruitment of Adaptor Proteins during Antibacterial Autophagy. Cell Rep. 2016. Nov 22;17(9):2183–2194. 10.1016/j.celrep.2016.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shahnazari S, Yen WL, Birmingham CL, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010. Aug 19;8(2):137–146. 10.1016/j.chom.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thurston TL, Wandel MP, Von Muhlinen N, et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012. Jan 15;482(7385):414–418. 10.1038/nature10744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jia J, Abudu YP, Claude-Taupin A, et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy. 2019. Jan;15(1):169–171. DOI: 10.1080/15548627.2018.1505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fiskin E, Bionda T, Dikic I, et al. Global Analysis of Host and Bacterial Ubiquitinome in Response to Salmonella Typhimurium Infection. Mol Cell. 2016. Jun 16;62(6):967–981. 10.1016/j.molcel.2016.04.015 [DOI] [PubMed] [Google Scholar]
- 66.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015. Dec;282(24):4672–4678. [DOI] [PubMed] [Google Scholar]
- 67.Khaminets A, Behl C, Dikic I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016. Jan;26(1):6–16. [DOI] [PubMed] [Google Scholar]
- 68.Vainshtein A, Grumati P. Selective Autophagy by Close Encounters of the Ubiquitin Kind. Cells. 2020. Oct 24;9(11):2349. 10.3390/cells9112349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Budzik JM, Swaney DL, Jimenez-Morales D, et al. Dynamic post-translational modification profiling of Mycobacterium tuberculosis-infected primary macrophages. Elife. 2020. Jan 17;9. 10.7554/eLife.51461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma V, Verma S, Seranova E, et al. Selective Autophagy and Xenophagy in Infection and Disease. Front Cell Dev Biol. 2018;6:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verlhac P, Gregoire IP, Azocar O, et al. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe. 2015. Apr 8;17(4):515–525. 10.1016/j.chom.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 72.Judith D, Mostowy S, Bourai M, et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013. Jun;14(6):534–544. DOI: 10.1038/embor.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deng Q, Wang Y, Zhang Y, et al. Pseudomonas aeruginosa Triggers Macrophage Autophagy To Escape Intracellular Killing by Activation of the NLRP3 Inflammasome. Infect Immun. 2016. Jan;84(1):56–66. DOI: 10.1128/IAI.00945-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuang E, Okumura CY, Sheffy-Levin S, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012;8(10):e1003007. DOI: 10.1371/journal.pgen.1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuang E, Qi J, Ronai Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem Sci. 2013. Sep;38(9):453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li F, Zeng J, Gao Y, et al. G9a Inhibition Induces Autophagic Cell Death via AMPK/mTOR Pathway in Bladder Transitional Cell Carcinoma. PLoS One. 2015;10(9):e0138390. DOI: 10.1371/journal.pone.0138390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McEwan DG, Dikic I. Cullins keep autophagy under control. Dev Cell. 2014. Dec 22;31(6):675–676. 10.1016/j.devcel.2014.12.010 [DOI] [PubMed] [Google Scholar]
- 78.Manzanillo PS, Ayres JS, Watson RO, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013. Sep 26;501(7468):512–516. 10.1038/nature12566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noad J, Von der Malsburg A, Pathe C, et al. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-kappaB. Nat Microbiol. 2017. May 8;2(7):17063. 10.1038/nmicrobiol.2017.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Wijk SJL, Fricke F, Herhaus L, et al. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-kappaB and restricts bacterial proliferation. Nat Microbiol. 2017. May 8;2(7):17066. 10.1038/nmicrobiol.2017.66 [DOI] [PubMed] [Google Scholar]
- 81.Franco LH, Nair VR, Scharn CR, et al. The ubiquitin ligase smurf1 functions in selective autophagy of mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017. Jan 11;21(1):59–72. 10.1016/j.chom.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Siqueira MDS, Ribeiro RM, Travassos LH. Autophagy and its interaction with intracellular bacterial pathogens. Front Immunol. 2018;9:935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deng L, Meng T, Chen L, et al. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020. Feb 29;5(1):11. 10.1038/s41392-020-0107-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bielskiene K, Bagdoniene L, Mozuraitiene J, et al. E3 ubiquitin ligases as drug targets and prognostic biomarkers in melanoma. Medicina (Kaunas). 2015;51(1):1–9. DOI: 10.1016/j.medici.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 85.Dove KK, Klevit RE. RING-between-RING E3 ligases: emerging themes amid the variations. J Mol Biol. 2017. Nov 10;429(22):3363–3375. 10.1016/j.jmb.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gladkova C, Maslen SL, Skehel JM, et al. Mechanism of parkin activation by PINK1. Nature. 2018. Jul;559(7714):410–414. DOI: 10.1038/s41586-018-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sauve V, Sung G, Soya N, et al. Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol. 2018. Jul;25(7):623–630. DOI: 10.1038/s41594-018-0088-7. [DOI] [PubMed] [Google Scholar]
- 88.Johnson BN, Berger AK, Cortese GP, et al. The ubiquitin E3 ligase parkin regulates the proapoptotic function of bax. Proc Natl Acad Sci U S A. 2012. Apr 17;109(16):6283–6288. 10.1073/pnas.1113248109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Leseleuc L, Orlova M, Cobat A, et al. PARK2 mediates interleukin 6 and monocyte chemoattractant protein 1 production by human macrophages. PLoS Negl Trop Dis. 2013;7(1):e2015. DOI: 10.1371/journal.pntd.0002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klein C, Westenberger A. Genetics of parkinson’s disease. Cold Spring Harb Perspect Med. 2012. Jan;2(1):a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marin I, Ferrus A. Comparative genomics of the RBR family, including the Parkinson’s disease-related gene parkin and the genes of the ariadne subfamily. Mol Biol Evol. 2002. Dec;19(12):2039–2050. [DOI] [PubMed] [Google Scholar]
- 92.Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004. Mar 15;23(11):1972–1984. 10.1038/sj.onc.1207436 [DOI] [PubMed] [Google Scholar]
- 93.Shearwin-Whyatt L, Dalton HE, Foot N, et al. Regulation of functional diversity within the Nedd4 family by accessory and adaptor proteins. Bioessays. 2006. Jun;28(6):617–628. DOI: 10.1002/bies.20422. [DOI] [PubMed] [Google Scholar]
- 94.Macias MJ, Wiesner S, Sudol M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002. Feb 20;513(1):30–37. 10.1016/S0014-5793(01)03290-2 [DOI] [PubMed] [Google Scholar]
- 95.Wei X, Wang X, Zhan J, et al. Smurf1 inhibits integrin activation by controlling Kindlin-2 ubiquitination and degradation. J Cell Biol. 2017. May 1;216(5):1455–1471. 10.1083/jcb.201609073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cao Y, Zhang L. A Smurf1 tale: function and regulation of an ubiquitin ligase in multiple cellular networks. Cell Mol Life Sci. 2013. Jul;70(13):2305–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Orvedahl A, Sumpter R Jr., Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011. Dec 1;480(7375):113–117. 10.1038/nature10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuan C, Qi J, Zhao X, et al. Smurf1 protein negatively regulates interferon-gamma signaling through promoting STAT1 protein ubiquitination and degradation. J Biol Chem. 2012. May 18;287(21):17006–17015. 10.1074/jbc.M112.341198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kimmey JM, Huynh JP, Weiss LA, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015. Dec 24;528(7583):565–569. 10.1038/nature16451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rogov VV, Stolz A, Ravichandran AC, et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017. Aug;18(8):1382–1396. DOI: 10.15252/embr.201643587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abdrakhmanov A, Gogvadze V, Zhivotovsky B. To eat or to die: deciphering selective forms of autophagy. Trends Biochem Sci. 2020. Apr;45(4):347–364. [DOI] [PubMed] [Google Scholar]
- 102.Shaid S, Brandts CH, Serve H, et al. Ubiquitination and selective autophagy. Cell Death Differ. 2013. Jan;20(1):21–30. DOI: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014. Feb;12(2):101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Whang MI, Tavares RM, Benjamin DI, et al. The ubiquitin binding protein TAX1BP1 mediates autophagasome induction and the metabolic transition of activated t cells. Immunity. 2017. Mar 21;46(3):405–420. 10.1016/j.immuni.2017.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fu T, Liu J, Wang Y, et al. Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc Natl Acad Sci U S A. 2018. Dec 11;115(50):E11651–E11660. 10.1073/pnas.1811421115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ceregido MA, Spinola Amilibia M, Buts L, et al. The structure of TAX1BP1 UBZ1+2 provides insight into target specificity and adaptability. J Mol Biol. 2014. Feb 6;426(3):674–690. 10.1016/j.jmb.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 107.Tumbarello DA, Manna PT, Allen M, et al. The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog. 2015. Oct;11(10):e1005174. DOI: 10.1371/journal.ppat.1005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chai Q, Wang X, Qiang L, et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun. 2019. Apr 29;10(1):1973. 10.1038/s41467-019-09955-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin C, Nozawa T, Minowa-Nozawa A, Toh H, Aikawa C, Nakagawa I. LAMTOR2/LAMTOR1 complex is required for TAX1BP1-mediated xenophagy. Cell Microbiol. 2019;21(4): e12981. 10.1111/cmi.12981 [DOI] [PubMed] [Google Scholar]
- 110.Furuya N, Kakuta S, Sumiyoshi K, Ando M, Nonaka R, Suzuki A, Kazuno S, Saiki S, Hattori N. NDP52 interacts with mitochondrial RNA poly(A) polymerase to promote mitophagy. EMBO Rep. 2018;19(12). 10.15252/embr.201846363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Verlhac P, Viret C, Faure M. Dual function of CALCOCO2/NDP52 during xenophagy. Autophagy. 2015;11(6): 965–966. 10.1080/15548627.2015.1046672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ryan T A, Tumbarello D A. (). Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front Immunol. 2018;9:1024. 10.3389/fimmu.2018.01024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen W, Li H, Chen G, Chern Y, Tu P. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy. 2015;11(4), 685–700. 10.4161/auto.36098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wild P et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039): 228–233. 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. 2005p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsuchiya M, Ogawa H, Koujin T, Mori C, Osakada H, Kobayashi S, Hiraoka Y, Haraguchi T. p62/SQSTM1 promotes rapid ubiquitin conjugation to target proteins after endosome rupture during xenophagy. FEBS Open Bio. 2018;8(3): 470–480. 10.1002/2211-5463.12385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yamada T, Dawson T M, Yanagawa T, Iijima M, Sesaki H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy. 2019;15(11): 2012–2018. 10.1080/15548627.2019.1643185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hafren A, Hofius D.. NBR1-mediated antiviral xenophagy in plant immunity. Autophagy. 2017;13(11): 2000–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Barnett T C, Liebl D, Seymour LM et al. The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe. 2013;14(6): 675–682. 10.1016/j.chom.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xie X, Li F, Wang Y, et al. Molecular basis of ubiquitin recognition by the autophagy receptor CALCOCO2. Autophagy. 2015;11(10):1775–1789. DOI: 10.1080/15548627.2015.1082025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012. Aug 17;150(4):803–815. 10.1016/j.cell.2012.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mostowy S, Sancho-Shimizu V, Hamon MA, et al. p62 and NDP52 proteins target intracytosolic shigella and listeria to different autophagy pathways. J Biol Chem. 2011. Jul 29;286(30):26987–26995. 10.1074/jbc.M111.223610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016. Apr 12;113(15):4039–4044. 10.1073/pnas.1523926113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015. Oct 1;60(1):7–20. 10.1016/j.molcel.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015. Aug 20;524(7565):309–314. 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rahighi S, Ikeda F, Kawasaki M, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009. Mar 20;136(6):1098–1109. 10.1016/j.cell.2009.03.007 [DOI] [PubMed] [Google Scholar]
- 127.Clark K, Nanda S, Cohen P. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol. 2013. Oct;14(10):673–685. [DOI] [PubMed] [Google Scholar]
- 128.Morton S, Hesson L, Peggie M, et al. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett. 2008. Mar 19;582(6):997–1002. 10.1016/j.febslet.2008.02.047 [DOI] [PubMed] [Google Scholar]
- 129.Li F, Xie X, Wang Y, et al. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun. 2016. Sep 13;7(1):12708. 10.1038/ncomms12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li F, Xu D, Wang Y, et al. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy. 2018;14(1):66–79. DOI: 10.1080/15548627.2017.1391970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Puri M, La Pietra L, Mraheil MA, et al. Listeriolysin O regulates the expression of optineurin, an autophagy adaptor that inhibits the growth of listeria monocytogenes. Toxins (Basel). 2017. Sep 5;9(9):273. 10.3390/toxins9090273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Munoz-Sanchez S, van der Vaart M, Meijer AH. Autophagy and Lc3-associated phagocytosis in zebrafish models of bacterial infections. Cells. 2020. Oct 29;9(11):2372. 10.3390/cells9112372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang R, Varela M, Vallentgoed W, et al. The selective autophagy receptors optineurin and p62 are both required for zebrafish host resistance to mycobacterial infection. PLoS Pathog. 2019. Feb;15(2):e1007329. DOI: 10.1371/journal.ppat.1007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007. Aug 17;282(33):24131–24145. 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 135.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007. Dec 14;131(6):1149–1163. 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- 136.Itakura E, Mizushima N. Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. [2011. Jan 10];192(1):17–27.p62 DOI: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kim BW, Kwon DH, Song HK. Structure biology of selective autophagy receptors. BMB Rep. 2016. Feb;49(2):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Clausen TH, Lamark T, Isakson P, et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy. 2010. Apr;6(3):330–344. DOI: 10.4161/auto.6.3.11226. [DOI] [PubMed] [Google Scholar]
- 139.Filimonenko M, Isakson P, Finley KD, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010. Apr 23;38(2):265–279. 10.1016/j.molcel.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang Y, Mun SR, Linares JF, et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat Commun. 2018. Oct 22;9(1):4373. 10.1038/s41467-018-06878-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cha-Molstad H, Yu JE, Feng Z, et al. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun. 2017. Jul 24;8(1):102. 10.1038/s41467-017-00085-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Orvedahl A, MacPherson S, Sumpter R Jr., et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010. Feb 18;7(2):115–127. 10.1016/j.chom.2010.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yoshikawa Y, Ogawa M, Hain T, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009. Oct;11(10):1233–1240. DOI: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 144.Zheng YT, Shahnazari S, Brech A, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009. Nov 1;183(9):5909–5916. 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- 145.Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012. Aug 24;37(2):223–234. 10.1016/j.immuni.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lee Y, Weihl CC. Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy. 2017. Sep 2;13(9):1615–1616. 10.1080/15548627.2017.1339845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fusco C, Micale L, Egorov M, et al. The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS One. 2012;7(7):e40440. DOI: 10.1371/journal.pone.0040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010. Mar;12(3):213–223. DOI: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 149.Lau A, Wang XJ, Zhao F, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010. Jul;30(13):3275–3285. DOI: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009. Jun 12;137(6):1001–1004. 10.1016/j.cell.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee Y, Chou TF, Pittman SK, et al. Keap1/Cullin3 Modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 2017. Apr 4;19(1):188–202. 10.1016/j.celrep.2017.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ponpuak M, Davis AS, Roberts EA, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010. Mar 26;32(3):329–341. 10.1016/j.immuni.2010.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Seto S, Tsujimura K, Horii T, et al. Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS One. 2013;8(12):e86017. DOI: 10.1371/journal.pone.0086017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018. Apr 13;37(8). 10.15252/embj.201797858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lange S, Xiang F, Yakovenko A, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005. Jun 10;308(5728):1599–1603. 10.1126/science.1110463 [DOI] [PubMed] [Google Scholar]
- 156.Whitehouse CA, Waters S, Marchbank K, et al. Neighbor of Brca1 gene (Nbr1) functions as a negative regulator of postnatal osteoblastic bone formation and p38 MAPK activity. Proc Natl Acad Sci U S A. 2010. Jul 20;107(29):12913–12918. 10.1073/pnas.0913058107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lamark T, Perander M, Outzen H, et al. Interaction codes within the family of mammalian phox and bem1p domain-containing proteins. J Biol Chem. 2003. Sep 5;278(36):34568–34581. 10.1074/jbc.M303221200 [DOI] [PubMed] [Google Scholar]
- 158.Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014. Jan 23;53(2):167–178. 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- 159.Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009. Feb 27;33(4):505–516. 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- 160.Svenning S, Lamark T, Krause K, et al. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy. 2011. Sep;7(9):993–1010. DOI: 10.4161/auto.7.9.16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Deosaran E, Larsen KB, Hua R, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013. Feb 15;126(Pt 4):939–952. 10.1242/jcs.114819 [DOI] [PubMed] [Google Scholar]
- 162.Guirado E, Schlesinger LS. Modeling the mycobacterium tuberculosis granuloma - the critical battlefield in host immunity and disease. Front Immunol. 2013;4:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu G, Wang J, Gao GF, et al. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell. 2014. Oct;5(10):728–736. DOI: 10.1007/s13238-014-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000. Feb 15;164(4):2016–2020. [DOI] [PubMed] [Google Scholar]
- 165.Welin A, Eklund D, Stendahl O, et al. Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PLoS One. 2011;6(5):e20302. DOI: 10.1371/journal.pone.0020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ahmad J, Farhana A, Pancsa R, et al. Contrasting function of structured N-terminal and unstructured C-terminal segments of Mycobacterium tuberculosis PPE37 protein. mBio. 2018. Jan 23;9(1). 10.1128/mBio.01712-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Grover S, Sharma T, Singh Y, et al. The PGRS domain of Mycobacterium tuberculosis PE_PGRS protein Rv0297 is involved in endoplasmic reticulum stress-mediated apoptosis through toll-like receptor 4. mBio. 2018. Jun 19;9(3). 10.1128/mBio.01017-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tundup S, Mohareer K, Hasnain SE. Mycobacterium tuberculosis PE25/PPE41 protein complex induces necrosis in macrophages: role in virulence and disease reactivation? FEBS Open Bio. 2014;4(1):822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Arora SK, Alam A, Naqvi N, et al. Immunodominant mycobacterium tuberculosis protein Rv1507A elicits Th1 response and modulates host macrophage effector functions. Front Immunol. 2020;11:1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ahmad J, Khubaib M, Sheikh JA, et al. Disorder-to-order transition in PE-PPE proteins of Mycobacterium tuberculosis augments the pro-pathogen immune response. FEBS Open Bio. 2020. Jan;10(1):70–85. DOI: 10.1002/2211-5463.12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Khubaib M, Sheikh JA, Pandey S, et al. Mycobacterium tuberculosis Co-operonic PE32/PPE65 proteins alter host immune responses by hampering Th1 Response. Front Microbiol. 2016;7:719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Blundell TL, Gupta MN, Hasnain SE. Intrinsic disorder in proteins: relevance to protein assemblies, drug design and host-pathogen interactions. Prog Biophys Mol Biol. 2020. Jul 3;156:34–42. 10.1016/j.pbiomolbio.2020.06.004 [DOI] [PubMed] [Google Scholar]
- 173.Gupta MN, Alam A, Hasnain SE. Protein promiscuity in drug discovery, drug-repurposing and antibiotic resistance. Biochimie. 2020. Aug;175:50–57. [DOI] [PubMed] [Google Scholar]
- 174.Dev A, Iyer S, Razani B, et al. NF-kappaB and innate immunity. Curr Top Microbiol Immunol. 2011;349:115–143. [DOI] [PubMed] [Google Scholar]
- 175.Chen J, Xie C, Tian L, et al. Participation of the p38 pathway in drosophila host defense against pathogenic bacteria and fungi. Proc Natl Acad Sci U S A. 2010. Nov 30;107(48):20774–20779. 10.1073/pnas.1009223107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wang J, Ge P, Lei Z, et al. Mycobacterium tuberculosis protein kinase G acts as an unusual ubiquitinating enzyme to impair host immunity. EMBO Rep. 2021. Jun 4;22(6):e52175. 10.15252/embr.202052175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang J, Ge P, Qiang L, et al. The mycobacterial phosphatase PtpA regulates the expression of host genes and promotes cell proliferation. Nat Commun. 2017. Aug 15;8(1):244. 10.1038/s41467-017-00279-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Puri RV, Reddy PV, Tyagi AK. Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in Guinea pig tissues. PLoS One. 2013;8(7):e70514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Maculins T, Fiskin E, Bhogaraju S, et al. Bacteria-host relationship: ubiquitin ligases as weapons of invasion. Cell Res. 2016. Apr;26(4):499–510. DOI: 10.1038/cr.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ahmed N, Dobrindt U, Hacker J, et al. Genomic fluidity and pathogenic bacteria: applications in diagnostics, epidemiology and intervention. Nat Rev Microbiol. 2008. May;6(5):387–394. DOI: 10.1038/nrmicro1889. [DOI] [PubMed] [Google Scholar]
- 181.Shariq M, Quadir N, Sheikh JA, et al. Post translational modifications in tuberculosis: ubiquitination paradox. Autophagy. 2020. Nov 15;17(3):814–817. 10.1080/15548627.2020.1850009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang L, Wu J, Li J, et al. Host-mediated ubiquitination of a mycobacterial protein suppresses immunity. Nature. 2020. Jan;577(7792):682–688. DOI: 10.1038/s41586-019-1915-7. [DOI] [PubMed] [Google Scholar]
- 183.Dhiman R, Raje M, Majumdar S. Differential expression of NF-kappaB in mycobacteria infected THP-1 affects apoptosis. Biochim Biophys Acta. 2007. Apr;1770(4):649–658. [DOI] [PubMed] [Google Scholar]
- 184.Cho JE, Park S, Cho SN, et al. c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) are involved in Mycobacterium tuberculosis-induced expression of Leukotactin-1. BMB Rep. 2012. Oct;45(10):583–588. DOI: 10.5483/BMBRep.2012.45.10.120. [DOI] [PubMed] [Google Scholar]
- 185.Wang J, Li BX, Ge PP, et al. Mycobacterium tuberculosis suppresses innate immunity by coopting the host ubiquitin system. Nat Immunol. 2015. Mar;16(3):237–245. DOI: 10.1038/ni.3096. [DOI] [PubMed] [Google Scholar]
- 186.Wang L, Liu Z, Wang J, et al. Oxidization of TGFbeta-activated kinase by MPT53 is required for immunity to Mycobacterium tuberculosis. Nat Microbiol. 2019. Aug;4(8):1378–1388. DOI: 10.1038/s41564-019-0436-3. [DOI] [PubMed] [Google Scholar]
- 187.Sakowski ET, Koster S, Portal Celhay C, et al. Ubiquilin 1 promotes IFN-gamma-induced xenophagy of Mycobacterium tuberculosis. PLoS Pathog. 2015. Jul;11(7):e1005076. DOI: 10.1371/journal.ppat.1005076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cowley S, Ko M, Pick N, et al. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Microbiol. 2004. Jun;52(6):1691–1702. DOI: 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 189.Lisa MN, Gil M, Andre-Leroux G, et al. Molecular basis of the activity and the regulation of the eukaryotic-like S/T protein kinase PknG from Mycobacterium tuberculosis. Structure. 2015. Jun 2;23(6):1039–1048. 10.1016/j.str.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 190.Walburger A, Koul A, Ferrari G, et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004. Jun 18;304(5678):1800–1804. 10.1126/science.1099384 [DOI] [PubMed] [Google Scholar]
- 191.Ehtram A, Shariq M, Ali S, et al. Teleological cooption of Mycobacterium tuberculosis PE/PPE proteins as porins: role in molecular immigration and emigration. Int J Med Microbiol. 2021. Apr;311(3):151495. DOI: 10.1016/j.ijmm.2021.151495. [DOI] [PubMed] [Google Scholar]
- 192.Akhter Y, Ehebauer MT, Mukhopadhyay S, et al. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie. 2012. Jan;94(1):110–116. DOI: 10.1016/j.biochi.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 193.Sharma N, Shariq M, Quadir N, et al. Mycobacterium tuberculosis protein PE6 (Rv0335c), a novel TLR4 agonist, evokes an inflammatory response and modulates the cell death pathways in macrophages to enhance intracellular survival. Front Immunol. 2021;12:696491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Strong EJ, Jurcic Smith KL, Saini NK, et al. Identification of autophagy-inhibiting factors of mycobacterium tuberculosis by high-throughput loss-of-function screening. Infect Immun. 2020. Nov 16;88(12). 10.1128/IAI.00269-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Saini NK, Baena A, Ng TW, et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat Microbiol. 2016. Aug 15;1(9):16133. 10.1038/nmicrobiol.2016.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Strong EJ, Ng TW, Porcelli SA, et al. Mycobacterium tuberculosis PE_PGRS20 and PE_PGRS47 proteins inhibit autophagy by interaction with Rab1A. mSphere. 2021. Aug 25;6(4):e0054921. 10.1128/mSphere.00549-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Deng W, Long Q, Zeng J, et al. Mycobacterium tuberculosis PE_PGRS41 Enhances the Intracellular Survival of M. smegmatis within Macrophages via blocking innate immunity and inhibition of host defense. Sci Rep. 2017. Apr 25;7(1):46716. 10.1038/srep46716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hu D, Wu J, Wang W, et al. Autophagy regulation revealed by SapM-induced block of autophagosome-lysosome fusion via binding RAB7. Biochem Biophys Res Commun. 2015. May 29;461(2):401–407. 10.1016/j.bbrc.2015.04.051 [DOI] [PubMed] [Google Scholar]
- 199.Shariq M, Quadir N, Sharma N, et al. Mycobacterium tuberculosis RipA dampens TLR4-mediated host protective response using a multi-pronged approach involving autophagy, apoptosis, metabolic repurposing, and immune modulation. Front Immunol. 2021;12:636644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Meng QL, Liu F, Yang XY, et al. Identification of latent tuberculosis infection-related microRNAs in human U937 macrophages expressing Mycobacterium tuberculosis hsp16.3. BMC Microbiol. 2014. Feb 12;14(1):37. 10.1186/1471-2180-14-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yang L, Zhang C, Zhao Y, et al. Effects of Mycobacterium tuberculosis mutant strain hsp16.3 gene on murine raw 264.7 macrophage autophagy. DNA Cell Biol. 2018. Jan;37(1):7–14. DOI: 10.1089/dna.2016.3599. [DOI] [PubMed] [Google Scholar]
- 202.Zheng Q, Li Z, Zhou S, et al. Heparin-binding hemagglutinin of mycobacterium tuberculosis is an inhibitor of autophagy. Front Cell Infect Microbiol. 2017;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gengenbacher M, Nieuwenhuizen N, Vogelzang A, et al. Deletion of nuoG from the vaccine candidate mycobacterium bovis bcg deltaurec::hly improves protection against tuberculosis. mBio. 2016. May 24;7(3). 10.1128/mBio.00679-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shin DM, Jeon BY, Lee HM, et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010. Dec 16;6(12):e1001230. 10.1371/journal.ppat.1001230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Garg R, Borbora SM, Bansia H, et al. Mycobacterium tuberculosis calcium pump ctpf modulates the autophagosome in an mtor-dependent manner. Front Cell Infect Microbiol. 2020;10:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mohanty S, Jagannathan L, Ganguli G, et al. A mycobacterial phosphoribosyltransferase promotes bacillary survival by inhibiting oxidative stress and autophagy pathways in macrophages and zebrafish. J Biol Chem. 2015. May 22;290(21):13321–13343. 10.1074/jbc.M114.598482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Huang D, Bao L. Mycobacterium tuberculosis EspB protein suppresses interferon-gamma-induced autophagy in murine macrophages. J Microbiol Immunol Infect. 2016. Dec;49(6):859–865. [DOI] [PubMed] [Google Scholar]
- 208.Yabaji SM, Dhamija E, Mishra AK, et al. ESAT-6 regulates autophagous response through SOD-2 and as a result induces intracellular survival of Mycobacterium bovis BCG. Biochim Biophys Acta Proteins Proteom. 2020. Oct;1868(10):140470. DOI: 10.1016/j.bbapap.2020.140470. [DOI] [PubMed] [Google Scholar]
- 209.Padhi A, Pattnaik K, Biswas M, et al. Mycobacterium tuberculosis LprE suppresses TLR2-dependent cathelicidin and autophagy expression to enhance bacterial survival in macrophages. J Immunol. 2019. Nov 15;203(10):2665–2678. 10.4049/jimmunol.1801301 [DOI] [PubMed] [Google Scholar]
- 210.Ge P, Lei Z, Yu Y, et al. M. tuberculosis PknG manipulates host autophagy flux to promote pathogen intracellular survival. Autophagy. 2021. Jun 7;1–19. DOI: 10.1080/15548627.2021.1938912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jacomin AC, Samavedam S, Promponas V, et al. iLIR database: a web resource for LIR motif-containing proteins in eukaryotes. Autophagy. 2016. Oct 2;12(10):1945–1953. 10.1080/15548627.2016.1207016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013. Aug 1;126(Pt 15):3237–3247. 10.1242/jcs.126128 [DOI] [PubMed] [Google Scholar]
- 213.Deretic V. Autophagy in tuberculosis. Cold Spring Harb Perspect Med. 2014. Aug 28;4(11):a018481. 10.1101/cshperspect.a018481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004. Dec 17;119(6):753–766. 10.1016/j.cell.2004.11.038 [DOI] [PubMed] [Google Scholar]
- 215.Ye X, Zhou XJ, Zhang H. Exploring the role of autophagy-related gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front Immunol. 2018;9:2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Csizmadia T, Low P. The Role of Deubiquitinating Enzymes in the Various Forms of Autophagy. Int J Mol Sci. 2020. Jun 12;21(12):4196. 10.3390/ijms21124196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Grumati P, Dikic I. Ubiquitin signaling and autophagy. J Biol Chem. 2018. Apr 13;293(15):5404–5413. 10.1074/jbc.TM117.000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jacomin AC, Taillebourg E, Fauvarque MO. Deubiquitinating enzymes related to autophagy: new therapeutic opportunities? Cells. 2018. Aug 19;7(8):112. 10.3390/cells7080112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jia R, Bonifacino JS. Negative regulation of autophagy by UBA6-BIRC6-mediated ubiquitination of LC3. Elife. 2019. Nov 6;8. 10.7554/eLife.50034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nibe Y, Oshima S, Kobayashi M, et al. Novel polyubiquitin imaging system, PolyUb-FC, reveals that K33-linked polyubiquitin is recruited by SQSTM1/p62. Autophagy. 2018;14(2):347–358. DOI: 10.1080/15548627.2017.1407889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chakaya JM, Marais B, Du Cros P, et al. Programmatic versus personalised approaches to managing the global epidemic of multidrug-resistant tuberculosis. Lancet Respir Med. 2020. Apr;8(4):334–335. DOI: 10.1016/S2213-2600(20)30104-1. [DOI] [PubMed] [Google Scholar]
- 222.Parsa K, Hasnain SE. Proteomics of multidrug resistant Mycobacterium tuberculosis clinical isolates: a peep show on mechanism of drug resistance & perhaps more. Indian J Med Res. 2015. Jan;141(1):8–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Tiberi S, Du Plessis N, Walzl G, et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect Dis. 2018. Jul;18(7):e183–e198. DOI: 10.1016/S1473-3099(18)30110-5. [DOI] [PubMed] [Google Scholar]
- 224.Sheikh JA, Ehtesham NZ, Hasnain SE. Revisiting BCG to control tuberculosis: mucosal delivery and delipidation? Lancet Infect Dis. 2020. Mar;20(3):272–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.