

Abstract
Introduction:
Frequent and severe bleeding events (SBE) in patients with inherited qualitative platelet disorders Bernard-Soulier Syndrome (BSS) and Glanzmann Thrombasthenia (GT) can lead to secondary iron deficiency anemia (IDA). SBE are primarily treated with platelet transfusions or recombinant activated factor VII (rFVIIa) infusions. The impact of IDA on bleeding management and disease outcomes is understudied.
Aim:
To evaluate bleeding management, outcomes, and any association with IDA in pediatric patients with BSS and GT.
Methods:
Retrospective chart-review of pediatric patients with BSS or GT followed at a single hemophilia treatment center between 2007 and 2019.
Results:
We identified 14 patients with BSS (n = 2) or GT (n = 12). Patients received rFVIIa (7%), platelet transfusions (7%), or a combination of both (57%) for SBE. Eleven patients (79%) had IDA requiring oral and/or intravenous iron replacement and 50% required red blood cell transfusions. Due to recurrent SBE and refractory IDA, three patients (21%) received rFVIIa prophylaxis at 90 μg/kilogram 2–3 times/week for ≥15 months. Patients initiated on rFVIIa prophylaxis had a median baseline hemoglobin of 9.8 g/dL (min-max: 8.0–10.7 g/dL) compared to 11.7 g/dL (8.4–13.8 g/dL) for patients treated on-demand. Following initiation of rFVIIa prophylaxis, median hemoglobin and ferritin increased by 1.3 g/dL (0.7–2.5 g/dL) and 14.6 ng/mL (0.2–42.9 ng/mL), respectively, and bleeding rates were reduced by 7–78%.
Conclusion:
IDA is a known complication of recurrent bleeding events in individuals with inherited bleeding disorders. Routine monitoring for IDA may help improve bleeding management and reduce bleed burden in BSS/GT.
Keywords: Glanzmann Thrombasthenia, Bernard-Soulier Syndrome, iron deficiency anemia, inherited blood coagulation disorder, pediatrics, platelet glycoprotein
1 |. INTRODUCTION
Bernard-Soulier Syndrome (BSS) and Glanzmann Thrombasthenia (GT) are rare autosomal recessive platelet function disorders that primarily result in recurrent mucocutaneous bleeding.1–3 The prevalence of BSS and GT are estimated to be approximately 1 in 1 million, with higher disease prevalence reported in areas with increased consanguinity.1–3 BSS is associated with moderate macrothrombocytopenia and is caused by defects in platelet glycoprotein (GP) genes GP1BA/GP1BB (encoding GPIb) or GP9 (encoding GPIX). These mutations lead to quantitative or qualitative defects of the GPIb-IX-V complex resulting in dysregulated platelet adhesion to von Willebrand factor (VWF) and platelet activation.3–5 GT is characterized by mutations in integrin genes ITGA2B (encoding GPIIb) or ITGB3 (encoding GPIIIa), which alter the function or expression of the GPIIb/IIIa complex and result in abnormal platelet aggregation.2,3,6
The bleeding phenotype of patients with BSS or GT ranges from mild to severe and includes easy bruising, epistaxis, mucosal bleeding, and increased bleeding following trauma or surgery.1–3 Heavy menstrual bleeding (HMB) is common in adolescent and adult females.7 The current treatment paradigm consists of on-demand hemostatic management of acute bleeding episodes or short-term prophylactic treatment prior to planned surgical or dental interventions.2,8–10 Anti-fibrinolytic (AF) agents, such as aminocaproic acid or tranexamic acid, platelet transfusion, and recombinant activated factor VII (rFVIIa) infusion comprise the primary treatment modalities utilized in both BSS and GT.2,3,8–13 Historically, platelet transfusion has been the mainstay of therapy for severe bleeds unresponsive to supportive measures or AF agents. Unfortunately, platelet transfusion carries known risks of transfusion reactions, including transfusion-related infections and alloimmunization resulting in platelet refractoriness (i.e., development of GPIb, GPIIb/IIIa, or HLA-antibodies), which may occur in up to 50% of patients.3 Recombinant FVIIa is approved by the U.S. Food and Drug Administration and European Medicines Agency for the treatment of surgical and non-surgical bleeding in patients with BSS/GT with platelet refractoriness or alloimmunization.14,15 Patients with GT achieve effective hemostasis with rFVIIa infusion for acute bleeding events or surgical procedures with or without platelet refractoriness or alloimmunization.12,13,16,17 Individuals with BSS have also demonstrated significant reduction in or cessation of bleeding following rFVIIa administration.4,5,18
Patients with frequent or severe bleeding events (SBE) are at increased risk of developing iron deficiency anemia (IDA) from chronic blood loss.19 In the pediatric population, IDA has been implicated in reduced stature, reduced growth velocity, altered immune function, and impaired cognitive function.19–23 Rates of iron deficiency with and without anemia and the impact of IDA on bleeding management in BSS and GT are poorly characterized.24 The ideal frequency of monitoring and therapeutic approach to iron deficiency in this population is also unclear.25 While limited case reports describe the use of short-term prophylactic rFVIIa as a method to prevent bleeding events not controlled by other therapies,26 a standardized indication for long-term prophylactic rFVIIa has not been defined. This study aims to describe the impact of bleeding phenotype and IDA on disease management and outcomes of a pediatric cohort with BSS or GT, including the effect of long-term prophylactic rFVIIa infusions on associated bleeding complications in a subset of individuals.
2 |. MATERIALS AND METHODS
A single institution retrospective review of electronic medical records for patients with BSS or GT seen between January 1, 2007 and December 31, 2019 at the Hemophilia of Georgia Center for Bleeding and Clotting Disorders of Emory University Pediatric Hemophilia Treatment Center at the Aflac Cancer and Blood Disorders Center of Children’s Hospital of Atlanta in Georgia was performed. Children’s Healthcare of Atlanta IRB approval was obtained (protocol no. 00000456), and all research was conducted according to the principles outlined in the Declaration of Helsinki. Inclusion criteria included female and male patients <21 years of age at the time of diagnosis with BSS or GT confirmed by flow cytometric platelet glycoprotein expression. Individuals with GP1BB hemizygosity associated with 22q11 deletion syndrome were excluded.
The primary aims of this study were to describe the frequency and hemostatic management of bleeding events and to evaluate the impact of bleeding events on measures of iron status and bleeding prevention strategies. Treatment modality for bleeds were identified as AF agent, platelet transfusion, or rFVIIa infusion. For each patient, the annualized bleeding rate (ABR) and rate of SBE were determined. An SBE was defined as a bleeding event requiring: 1) hemostatic intervention with platelet transfusion or rFVIIa or 2) medical provider evaluation in a clinical setting (i.e., emergency department, hospital, or clinic). Patient red blood cell (RBC) indices, including hemoglobin, hematocrit, mean corpuscular volume, and red cell distribution width were recorded, in addition to iron indices of iron, total iron binding capacity, iron saturation, and ferritin.
2.1 |. Statistical analysis
Differences in hemoglobin or ferritin concentrations between iron treatment therapies were determined by Mann Whitney U test. A one-way ANOVA with Tukey’s correction for multiple comparisons was used to compare differences in the hemoglobin and ferritin concentrations of patients with BSS and GT receiving on-demand rFVIIa compared to those on prophylactic rFVIIa. The relationship between median hemoglobin or ferritin concentration and ABR were determined by Pearson’s correlation coefficient. Statistical analyses were performed with Prism 6.0 (GraphPad Software, La Jolla, CA). A P value < 0.05 was considered statistically significant.
3 |. RESULTS
3.1 |. Summary of patient demographics
We identified 14 patients with BSS (n = 2) or GT (n = 12) (Table 1). The cohort had a median age of 1.7 years at diagnosis (range: 2 months to 13 years) and was 50% male and 50% female. All patients had abnormal platelet aggregation studies prompting analysis of platelet glycoprotein expression by flow cytometry. Both patients with BSS had reduced antibody binding to GPIb and 11 of 12 patients with GT had reduced antibody binding to GPIIb/IIIa on flow cytometry. Further testing of the one GT patient with normal GPIIb/IIIa binding revealed dysfunctional GPIIb/IIIa receptors. The majority of the cohort (93%) did not have GP1BA/GP1BB, GP9, ITGA2B, or ITGB3 gene analysis performed. One patient with GT (patient 8) was found to have compound heterozygous nonsense variants c.1750C>T (p.Arg584*) and c.2671C>T (p.Gln891*) in ITGA2B.
TABLE 1.
Summary of patient characteristics
| Patient no. | Age at diagnosis | Sex | Age at data extraction | Diagnosis | ABR (events/year) | Primary bleeding manifestations | Primary treatment modality | rFVIIa Prophylaxis | Duration of rFVIIa prophylaxis (months) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 6 y | F | 7 y | GT | 3.3 | Epistaxis | Anti-fibrinolytics | - | - |
| 2 | 7 m | M | 8 y | BSS | 17.3 | Epistaxis | On-demand rFVIIa & platelets | Yes | 16 |
| 3 | 18 m | F | 7 y | GT | 2.2 | Oral bleeding | On-demand rFVIIa & platelets | Yes | 15 |
| 4 | 22 m | M | 2 y | GT | 3.0 | Epistaxis & Trauma-induced hematomas | On-demand rFVIIa & platelets | - | - |
| 5 | 18 m | M | 22 y | GT | 1.9 | Epistaxis&GI bleeding | On-demand rFVIIa & platelets | - | - |
| 6 | 2 y | F | 9 y | BSS | 1.1 | Epistaxis | On-demand rFVIIa & platelets | - | - |
| 7 | 13 y | F | 19 y | GT | 4.4 | Epistaxis, HMBa, & Hemarthrosis | On-demand rFVIIa & platelets | Yes | 31 |
| 8 | 2 m | F | 5 y | GT | 0.8 | Epistaxis | Anti-fibrinolytics | - | - |
| 9 | 19 m | F | 12 y | GT | 1.2 | Epistaxis, Oral bleeding &HMBa | On-demand rFVIIa & platelets | - | - |
| 10 | 11 y | M | 18 y | GT | 2.2 | Epistaxis & Oral bleeding | On-demand rFVIIa | - | - |
| 11 | 11 y | M | 26 y | GT | 0.1 | Epistaxis | Anti-fibrinolytics | - | - |
| 12 | 18 m | M | 18 y | GT | 4.1 | Epistaxis & Oral bleeding | Anti-fibrinolytics | - | - |
| 13 | 13 y | F | 17 y | GT | 1.2 | HMB | On-demand platelets | - | - |
| 14 | 15 m | M | 16 y | GT | 7.3 | Epistaxis, GI bleeding, & Intracranial hemorrhage | On-demand rFVIIa & platelets | - | - |
Abbreviations: ABR, annualized bleeding rate; BSS, Bernard-Soulier Syndrome; GT, Glanzmann Thrombasthenia; HMB, heavy menstrual bleeding; m, month; No., number; rFVIIa, recombinant activated factor VII; y, year.
Hormonal therapies for menstrual suppression were utilized as adjunct therapies for HMB, including combined oral contraceptive pill (patients 7 & 9), intramuscular medroxyprogesterone (patients 7 & 9), or levonorgestrel intrauterine device (patient 7).
3.2 |. Bleeding phenotype and treatment modalities
Bleeding manifestations included epistaxis (79% of patients), oral bleeding (29%), gastrointestinal bleeding (14%), trauma-induced hematomas (7%), and knee hemarthrosis (7%) (Table 1). One patient was found to have an intracranial hemorrhage at 3 years of age during a hospitalization for epistaxis. Of the seven female patients, three (43%) had reached menarche and experienced HMB as a primary bleeding manifestation. The median ABR was 2.2 (min-max 0.1–17.3 events/year). Approximately 93% of patients had at least one SBE (min-max 0–26 SBE).
Treatment strategies for bleeding events included AF agents alone (29%), rFVIIa infusions (7%), platelet transfusion (7%), or a combination of platelets and rFVIIa (57%) (Table 1). For most bleeding episodes, AF agents were utilized as adjunct therapy with rFVIIa infusions and/or platelet transfusion. Two of the menstruating patients with GT utilized various hormonal therapies (i.e., combined oral contraceptive pill, intramuscular medroxyprogesterone injection, or intrauterine device) for menstrual suppression in addition to the primary treatment modalities described above. There were no reported thrombotic events.
3.3 |. Monitoring and treatment of IDA
The frequency of monitoring for IDA varied widely, with an increased frequency of monitoring in individuals with a higher number of bleeding episodes. RBC indices were obtained on all patients at least once annually during the study period. Clinical testing of RBC indices ranged from 1–39 tests/year for the cohort with a median of 4.5 tests/year. Iron indices were less frequently tested, with recordings documented for only nine patients (64%). Frequency of monitoring iron indices ranged from 0 to 3.2 tests/year with a median of 1 test/year.
RBC and iron indices for each individual patient are summarized in Table 2. The cohort had a median hemoglobin of 10.9 g/dl (min-max: 8.0–13.8 g/dl) and median ferritin of 11.5 ng/ml (min-max: 5.4–19.9 ng/ml). Eleven patients (79%) were diagnosed with IDA based on RBC or iron indices (Figure 1A). Eight patients (57%) received oral iron replacement alone, three patients (21%) received a combination of oral and intravenous iron replacement, and three patients (21%) received no treatment (Figure 1B and Table 3). Seven patients (50%) from the cohort required packed RBC transfusions due to severe anemia in the setting of an acute or recent SBE (Figure 1C). There were no significant differences in median hemoglobin or ferritin concentrations in patients receiving oral or parenteral iron replacement compared to those not receiving these therapies (Figure 2A–B and D–E). In contrast, patients who received a pRBC transfusion had a significantly lower median hemoglobin level of 10.4 g/dl compared to a median hemoglobin of 13.2 g/dl in non-transfused patients (P = 0.003, Figure 2C). Although not statistically significant, a trend towards lower ferritin levels was observed in those who did versus did not receive pRBC transfusion (median ferritin 9.5 ng/dl vs. 15.5 ng/dl, P = 0.7, Figure 2F).
TABLE 2.
Hematologic indices of all patients with BSS and GT*
| Patient no. | Hemoglobin (g/dL) | Hematocrit (%) | MCV (fl) | RDW (%) | Iron (μg/dL) | TIBC (μg/dL) | Iron saturation (%) | Ferritin (ng/ml) |
|---|---|---|---|---|---|---|---|---|
| 1 | 11.1 (9.5–12.2) | 33.4 (28.6–36.3) | 91.0 (85.3–92.3) | 13.4 (13.2–13.6) | 76 (43–108) | 393 (381–405) | 19 (11–27) | 15.5 (14.2–16.8) |
| 2 | 10.7 (10.4–10.9) | 31.5 (31.1–32.1) | 75.2 (74.4–75.8) | 16.0 (15.4–16.3) | 52 (29–79) | 381 (339–413) | 14 (7–20) | 19.9 (12.4–26.3) |
| 3 | 10.6 (10.3–11.7) | 32.8 (32.3–35.0) | 82.1 (78.0–83.3) | 15.3 (14.4–15.9) | 33 (33) | 481 (481) | 7 (7) | 10.9 (10.9) |
| 4 | 11.0 (7.4–12.4) | 32.4 (25.4–39.3) | 79.7 (65.3–80.6) | 14.6 (13.2–18.4) | 65 (20–116) | 492 (403–543) | 15 (4–26) | 5.4 (4.5–50.7) |
| 5 | 8.4 (7.6–9.1) | 25.6 (23.2–27.7) | 81.8 (79.0–84.4) | 17.0 (16.5–17.3) | 42 (14–95) | 409 (332–447) | 12 (4–34) | 8.0 (4.1–24.0) |
| 6 | 12.7 (11.6–13.1) | 36.5 (34.0–38.3) | 80.2 (77.7–81.0) | 13.4 (13.0–13.6) | - | - | - | - |
| 7 | 8.0 (7.3–9.1) | 28.2 (26.1–30.4) | 79.4 (74.5–81.2) | 18.4 (17.5–18.8) | 15 (12–17) | 445 (409–468) | 3 (2–5) | 6.8 (5.4–12.2) |
| 8 | 10.6 (9.2–12.0) | 32.5 (27.4–34.6) | 80.5 (73.9–89.7) | 12.9 (12.3–14.7) | 48 (48) | 318 (318) | 14 (14) | 18.5 (18.5) |
| 9 | 10.2 (9.7–10.5) | 30.5 (29.9–32.6) | 77.3 (76.0–79.2) | 15.8 (14.6–16.6) | 44 (11–231) | 293 (273–437) | 15 (3–83) | 16.8 (6.2–27.9) |
| 10 | 13.2 (12.8–13.9) | 39.2 (37.1–42.0) | 84.5 (82.9–85.3) | 13.2 (12.9–13.4) | - | - | - | - |
| 11 | 13.8 (13.5–17.1) | 39.8 (37.8–47.5) | 84.6 (79.0–87.9) | 12.8 (12.3–13.3) | - | - | - | - |
| 12 | 13.3 (9.8–14.0) | 39.1 (30.9–41.2) | 75.3 (66.3–77.7) | 17.0 (15.3–18.0) | 38 (11.4–91.0) | 548 (511–664) | 7 (2–18) | 7.9 (3.7–14.4) |
| 13 | 13.5 (13.5) | 41.6 (41.6) | 91.0 (91.0) | 14.6 (14.6) | - | - | - | - |
| 14 | 10.4 (10.0–10.8) | 30.4 (29.6–31.4) | 82.9 (78.6–85.4) | 16.2 (16.0–16.5) | - | - | - | - |
| Median Values of Cohort | 11.1 | 33.4 | 81.8 | 14.6 | 46 | 401 | 15 | 11.7 |
Data presented as medians (95% confidence intervals).
FI femtoliters (1×10–15); MCV mean corpuscular volume; RDW, red cell distribution width; TIBC, total iron binding content.
Hematologic indices presented represent baseline values for all individuals.
FIGURE 1.
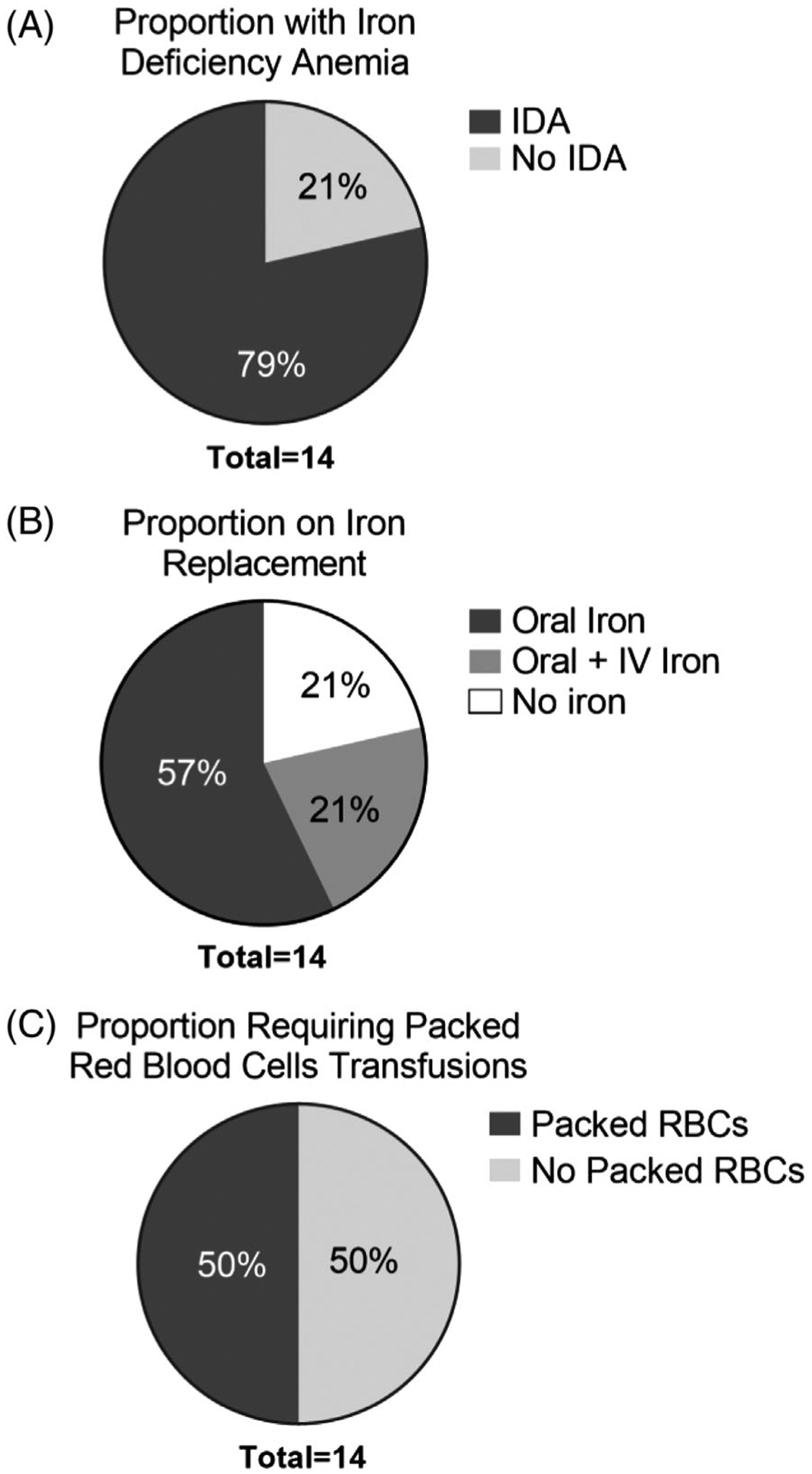
Distribution of iron deficiency anemia and iron replacement therapies. The proportion of patients with iron deficiency anemia (A) is depicted. The proportion of individuals in the entire cohort who received oral and/or parental iron replacement (B). Proportion of the entire cohort that received packed red blood cell (pRBC) transfusions is shown (C)
TABLE 3.
Summary of iron replacement history for BSS/GT patients
| Patient no. | Sex, diagnosis | IDA | Oral iron replacement?a | IV iron replacement?b | Iron replacement therapies prescribedc |
|---|---|---|---|---|---|
| 1 | F, GT | - | - | - | No iron replacement. |
| 2 | M, BSS | Yes | Yes | Yes | Oral iron: multiple administrations over 4 years. IV iron: Due to persistent IDA, recurrent bleeding symptoms, & oral iron intolerance, oral iron transitioned to IV iron infusions alone every 4 weeks if hemoglobin <10 g/dL or ferritin <15 ng/ml. |
| 3 | F, GT | Yes | Yes | - | Oral iron: multiple administrations 1–4 months in duration. |
| 4 | M, GT | Yes | Yes | - | Oral iron: multiple administrations 3–6 months in duration. |
| 5 | M, GT | Yes | Yes | - | Oral iron: multiple administrations 15–30 months in duration. |
| 6 | F, BSS | Yes | Yes | - | Oral iron: multiple administrations 1–3 months in duration. |
| 7 | F, GT | Yes | Yes | Yes | Oral iron: multiple administrations over 7 years. IV iron: Due to persistent IDA from recurrent bleeding symptoms, IV iron replacementwas initiated after 3 years of attempted oral iron replacement alone. IV iron infused monthly for 4 years then as needed. |
| 8 | F, GT | Yes | Yes | - | Oral iron: multiple administrations 1–3 months in duration. |
| 9 | F, GT | Yes | Yes | Yes | Oral iron:multiple administrations 2 – 42 months in duration. IV iron: One time IV iron infusion during a hospitalization for an acute bleeding event. |
| 10 | M, GT | - | - | - | No iron replacement. |
| 11 | M, GT | - | - | - | No iron replacement. |
| 12 | M, GT | Yes | Yes | - | Oral iron: 36 months in duration. |
| 13 | F, GT | Yes | Yes | - | Oral iron: unspecified duration. |
| 14 | M, GT | Yes | Yes | - | Oral iron: unspecified duration. |
Abbreviations: ABR, annualized bleeding rate; BSS, Bernard-Soulier Syndrome; GT, Glanzmann Thrombasthenia; HMB, heavy menstrual bleeding; IDA, Iron Deficiency Anemia; IV, intravenous.
Ferrous sulfate was the most commonly used formulation for first-line oral iron replacement. In few instances, other formulations utilized included ferrous fumarate, Carbonyl Iron, and Iron PS Complex-D4. Dosing ranged from 3–6 mg elemental iron/kg/day.
Iron Sucrose was the only formulation used for IV iron replacement. Dosing ranged from 5–7 mg elemental iron/kg/infusion.
Description of oral iron replacement regimens reflects the regimen prescribed by the medical provider per the medical record but may not accurately reflect home medication adherence.
FIGURE 2.
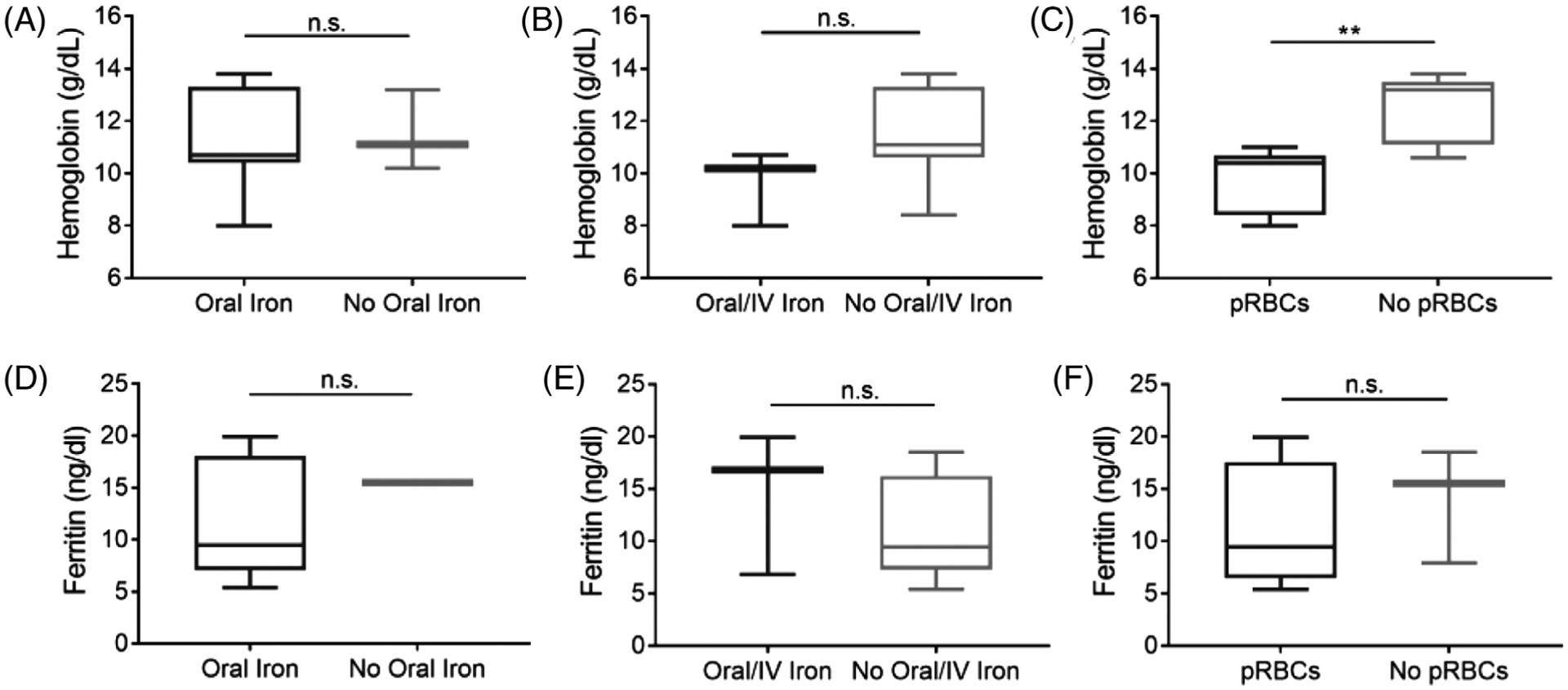
Hemoglobin and ferritin concentrations by iron replacement therapies. Box and whisker plots of hemoglobin levels in individuals with BSS or GT who did and did not receive oral iron replacement alone (A), oral/IV iron replacement (B), or pRBC transfusions (C). Ferritin levels of individuals with BSS or GT who did and did not receive oral iron replacement alone (D), oral/IV iron replacement (E), or pRBC transfusions (F). IV, intravenous and packed red blood cells (pRBC). Data are presented as medians with 5–95th percentiles. Differences in hemoglobin or ferritin concentrations between iron treatment therapies were determined by Mann Whitney U test, **P < 0.01
3.4 |. Disease outcomes and prophylaxis
Ten patients (71%) utilized on-demand therapy with AF agents, rFVIIa, and/or platelet transfusion with adequate bleeding control during the study period. However, due to recurrent prolonged SBE and chronic refractory IDA, three patients (21%) were initiated on long-term rFVIIa prophylaxis for bleeding prevention (Table 1). Individuals on rFVIIa prophylaxis consisted of patient 2 (BSS with recurrent epistaxis), patient 3 (GT with recurrent oral bleeding), and patient 7 (GT with refractory epistaxis, hemarthrosis, and HMB). These 3 patients received rFVIIa prophylaxis at 90 μg/kilogram (kg) 2–3 times per week for 15–31 months during the study period. Patient 2 required biweekly platelet transfusions prior to initiation of and concomitant to thrice-weekly rFVIIa prophylaxis to achieve adequate bleeding control and prevent frequent hospitalizations due to SBE. One patient (Patient 7) on rFVIIa prophylaxis required a central venous access device for prophylactic rFVIIa infusions. There were no reported adverse events associated with rFVIIa prophylaxis, including thrombosis or central venous line infections. One patient (Patient 3) on twice-weekly rFVIIa prophylaxis discontinued prophylaxis 5 months prior to the end of this study period per parental preference to reduce the frequency of peripheral infusions and preserve peripheral access until onset of menarche. Another patient (Patient 14) with significant bleeding manifestations, including recurrent epistaxis and intracranial hemorrhage associated with new onset of seizures, underwent successful allogeneic cord blood hematopoietic stem cell transplant (HSCT) as curative therapy at age 3 years. The patient’s post-HSCT course was complicated by graft versus host disease affecting the skin that ultimately resolved. This patient maintained 100% donor chimerism with normalization of platelet aggregation studies and antibody binding to GPIIb/IIIa on platelet glycoprotein expression profile >10 years post-HSCT.
The ABR of patients 2, 3, and 7 on rFVIIa prophylaxis decreased by 78%, 55%, and 7%, respectively (Figure 3A). RBC and iron indices for patients on rFVIIa prophylaxis prior to and after initiation of prophylaxis are detailed in Table 4. Patients on rFVIIa prophylaxis had a median hemoglobin of 9.8 g/dL (min-max 8.0–10.7 g/dL) compared to a median hemoglobin of 11.7 g/dL (min-max 8.4–13.8 g/dL) for patients treated solely with on-demand therapy (Figure 3B). Hemoglobin levels increased by a median of 1.3 g/dL (min-max 0.7–2.5 g/dL) after initiating rFVIIa prophylaxis. Patients receiving rFVIIa prophylaxis had a median ferritin of 12.5 ng/ml (min-max 5.4–18.5 ng/ml), similar to the median ferritin of 12.0 ng/ml (min-max 6.8–19.9 ng/ml) for patients treated with on-demand therapy (Figure 3C). The ferritin concentration increased by a median 14.6 ng/ml (min-max 0.2–42.9 ng/ml) for patients on rFVIIa prophylaxis. There was no correlation between hemoglobin or ferritin concentrations and ABR for the cohort (Figure 3D–E).
FIGURE 3.
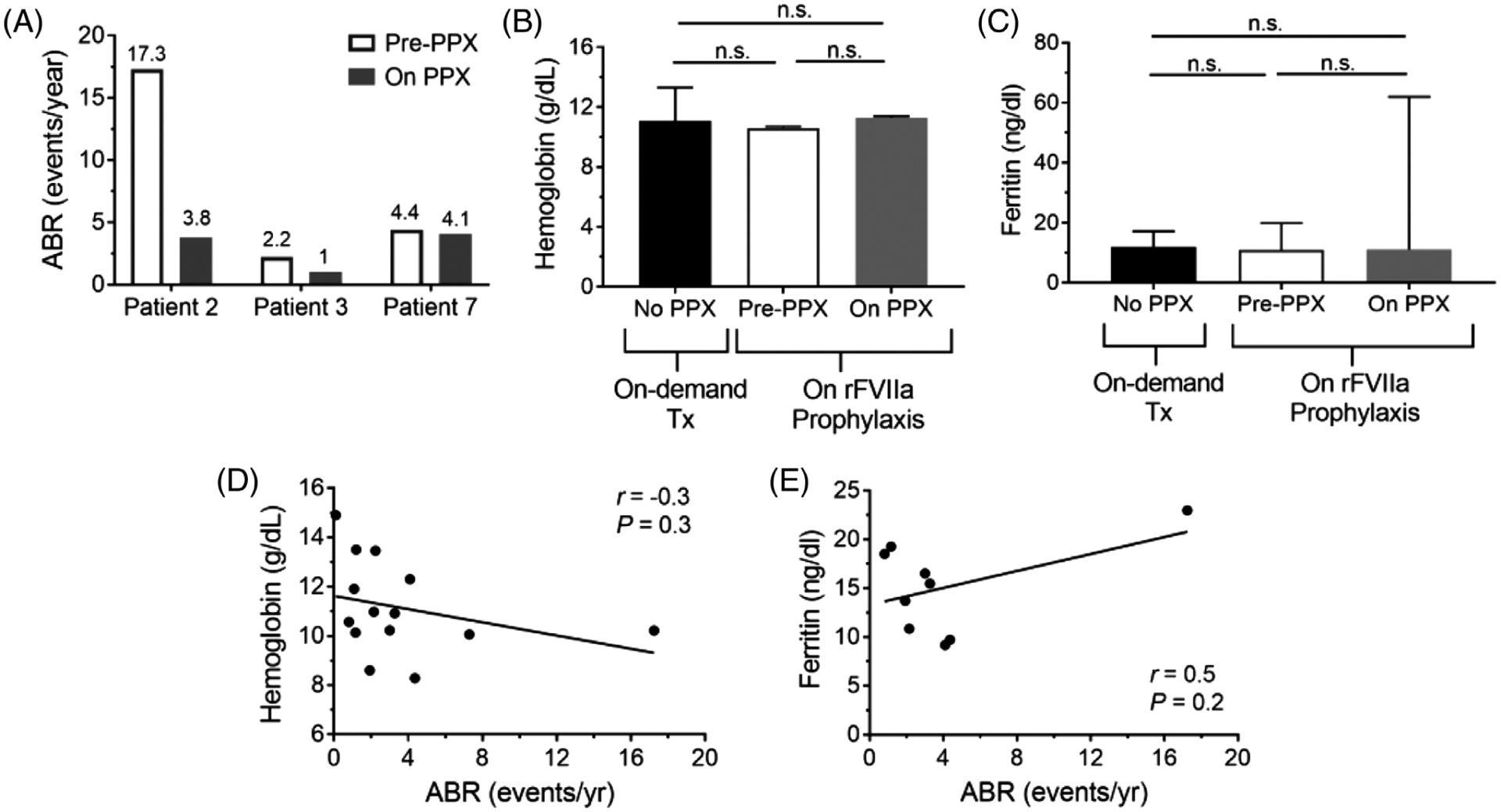
Bleeding rates, hemoglobin, and ferritins of pediatric patients with BSS and GT with and without rFVIIa prophylaxis. Annualized bleeding rates (ABR) of the three individuals on rFVIIa infusions prior to and on prophylaxis (A). Hemoglobin (B) and ferritin (C) concentrations of patient cohort with BSS or GT. Data are presented as medians ± interquartile range. Correlation of median hemoglobin (D) or median ferritin (E) concentrations with ABRs as events/year are shown. Differences in hemoglobin and ferritin concentrations between individuals with and without rFVIIa prophylaxis were determined by one-way ANOVA with Tukey’s correction for multiple comparisons. The relationship between median hemoglobin or ferritin concentration and ABR were determined by Pearson’s correlation coefficient
TABLE 4.
Hematologic indices of BSS/GT patients on rFVIIa prophylaxis for bleeding prevention
| Patient 2 | Patient 3 | Patient 7 | ||||
|---|---|---|---|---|---|---|
| Pre-PPX | On PPX | Pre-PPX | On PPX | Pre-PPX | On PPX | |
| Hemoglobin (g/dL) | 10.7 (10.4–10.9) | 11.4 (11.0–12.0) | 10.6 (10.3–11.7) | 11.3 (8.7–12.3) | 8.0 (7.3–9.1) | 10.5 (9.2–11.3) |
| Hematocrit (%) | 31.5 (31.1–32.1) | 34.1 (32.8–35.6) | 32.8 (32.3–35.0) | 34.3 (26.3–37.3) | 28.2 (26.1–30.4) | 35.5 (30.1–38.0) |
| MCV (fl) | 75.2 (74.4–75.8) | 77.6 (76.1–79.0) | 82.1 (78.0–83.3) | 84.4 (83.2–85.8) | 79.4 (74.5–81.2) | 82.1 (76.7–83.1) |
| Ferritin (ng/ml) | 19.9 (12.4–26.3) | 62 (62) | 10.9 (10.9) | 11.1 (11.1) | 6.8 (5.4–12.2) | 8.4 (6.5–15.8) |
| Iron (μg/dL) | 52 (29–79) | 37 (26–46) | 33 (33) | 52 (52) | 15 (12–17) | 21 (9–62) |
| TIBC (μg/dL) | 381 (339–413) | 358 (345–373) | 481 (481) | 406 (406) | 445 (409–468) | 401 (370–433) |
| Iron saturation (%) | 14 (7–20) | 10 (7–13) | 7 (7) | 13 (13) | 3 (2–5) | 6 (2–17) |
Abbreviations: MCV, mean corpuscular volume; PPX, prophylaxis.
Data presented as medians (95% confidence intervals).
4 |. DISCUSSION
In this study, we report a cohort of pediatric patients with BSS and GT with considerable variability in bleeding phenotype and describe the association between bleeding management and outcomes with IDA. All patients had a minimum of one mucocutaneous bleeding event and the most frequent bleeding manifestation observed was epistaxis, occurring in 79% of patients. The patients were treated with a variety of hemostatic agents including AF agents, rFVIIa infusion, platelet transfusion, or a combination of rFVIIa and platelets for acute bleeds, which is consistent with published treatment regimens.2,3,8,9,13,17
A range of bleeding severity in patients with BSS and GT has been previously described, but a quantifiable method for identifying patients with a more severe bleeding phenotype is limited by the rarity of these diseases and a lack of rapid point-of-care platelet function testing to evaluate hemostatic response to therapy.3 The clinical severity of BSS or GT may correlate with underlying genetic phenotype, but this association is still being elucidated in international cohorts as routine geno-typing outside of a research study is not feasible in many healthcare settings.5,6,27,28 Variation in clinical manifestations and disease outcomes may be further impacted by socioeconomic factors, including access to clinical resources and availability of treatments modalities for bleeding management.2,19 Bleeding manifestations can additionally fluctuate with age, paralleling the developmental stages of pediatric and adult patients2,6. HMB is the most common bleeding manifestation for girls and women with rare bleeding disorders.29 Three of our seven female patients had reached menarche, and all three had HMB. Two menarchal patients required and trialed multiple hormonal therapies for menstrual regulation. The rarity of these diagnoses highlights the importance of national and international registries for data collection on disease manifestation and long-term outcomes in this patient population, which may help inform optimal therapeutic approaches and efficacy.8,9,12
In other inherited bleeding disorders such as hemophilia A, hemophilia B, and rare bleeding disorders due to clotting factor deficiencies, the benefits of prophylaxis with factor concentrates to prevent bleeding symptoms has been extensively described and advised as standard of care for individuals with severe bleeding.30,31 In contrast, bleeding management in BSS or GT has been limited primarily to on-demand platelet transfusions, on-demand rFVIIa administration, or short-term rFVIIa prophylaxis in the post-surgical or severe bleed setting.26 One study by Andıc et al. reported a reduction in epistaxis and oral bleeding events corresponding to significantly improved quality of life in a 28-year-old male with GT after a 5-month period of low dose rFVIIa prophylaxis at 20 μg/kg weekly.26 In our patient cohort, we identified a subset of three patients with frequent SBE recalcitrant to on-demand treatment and secondary chronic refractory IDA that ultimately prompted high dose prophylactic rFVIIa infusions at 90 μg/kg twice or thrice weekly. These patients had a reduction in their ABRs by 7–78% following initiation of rFVIIa prophylaxis. Although one patient (Patient 7) had a 7% reduction in ABR on rFVIIa prophylaxis, the patient had a 66% reduction in platelet transfusion needs, cessation of HMB in combination with intrauterine device placement, and a substantial improvement in quality of life. This is the first study to describe the bleeding outcomes of high dose rFVIIa prophylaxis in pediatric patients with BSS or GT, suggesting feasibility of this therapeutic approach. Importantly, no thromboembolic events were reported with rFVIIa prophylaxis.
Although IDA is a known complication of frequent bleeding events in individuals with inherited bleeding disorders, the prevalence of IDA and recommendations for frequency of monitoring have not been established in this patient population. In this cohort, 79% of patients had IDA during the study period. The frequency of testing iron indices varied tremendously with some patients never having iron indices obtained. Serum ferritin concentration is the predominant measure used for monitoring iron status. The optimal ferritin threshold for diagnosis of iron deficiency remains a subject of debate. The World Health Organization advises a threshold of <12 μg/L for children and <15 μg/L for non-pregnant women of child-bearing age.32 A similar ferritin threshold for iron deficiency has been described in young girls and women with HMB.33,34 However, a recent study by Mei Z. et al. suggests thresholds for iron deficiency of 20 μg/L for children and 25 μg/L for non-pregnant women between 15–49 years of age are more clinically relevant based on serial cross-sectional National Health and Nutrition Examination Surveys (NHANES).35 Based on this recommendation, all 14 patients with BSS/GT in our cohort would meet the threshold definition for iron deficiency. Despite the risk of frequent bleeding symptoms and limited prophylactic options, the optimal ferritin concentration to maintain in pediatric patients with rare inherited bleeding disorders is not well-defined.
Routine monitoring of RBC and iron indices is a critical part of the medical management of this patient population that may help to supplement evaluation of bleeding phenotype, aid in treatment optimization, and help to identify patients who necessitate escalated treatment intervention to limit bleeding and its associated sequelae. In this study, the patients on rFVIIa prophylaxis had a lower baseline hemoglobin and ferritin than those with adequate bleeding control treated with on-demand therapies, although this was not statistically significant given the small sample size and limited iron indices recorded. Others have proposed that aggressive and early treatment of IDA during critical developmental phases through the pediatric timeframe may prevent long-term neurologic deficits and protect growth velocity.20,21 In light of limited prophylactic options, rFVIIa prophylaxis may represent a feasible approach to bleeding management in individuals with BSS or GT with severe bleeding phenotypes and refractory bleeding-associated complications. Further prospective, multi-institutional, and international clinical studies and data registries are warranted to evaluate the optimal dosing regimen, hemostatic efficacy, and long-term risks of this approach.
5 |. CONCLUSION
This study supports the current recognition that the bleeding phenotype of patients with BSS and GT vary by individual. All patients had mucocutaneous bleeding as their primary bleeding manifestation with epistaxis representing the predominant bleeding symptom. Routine monitoring for iron deficiency with or without anemia may allow for opportunities to optimize health and aid in bleeding management of patients with inherited qualitative platelet disorders, particularly during the critical neurocognitive developmental phases of childhood. Three patients in our cohort with more severe bleeding phenotype and refractory IDA showed clinical improvement after initiation of rFVIIa prophylaxis. This study highlights the importance of developing and investigating novel agents and treatment approaches for individuals with these rare congenital qualitative platelet disorders to reduce disease burden.
ACKNOWLEDGMENTS
Supported by NHLBI K99 grant HL150595 (G.B.), Hemophilia of Georgia Clinical Scientist Development Award (G.B.), NHLBI K99 grant HL150626 (C.L.M.), Hemophilia of Georgia Center for Bleeding & Clotting Disorders of Emory Hemostasis Research Award (G.B. and C.L.M.).
Funding information
National Heart, Lung, and Blood Institute, Grant/Award Numbers: K99HL150595, K99HL150626; Hemophilia of Georgia
Footnotes
DISCLOSURES
The authors stated that they have no interests which might be perceived as posing a conflict or bias. A.L. and C.L.M have no conflicts of interests to disclose. G.B. has participated in advisory boards for Bayer, Genentech, and Octapharma and has received honoraria from Bio Products Laboratory and Kedrion for educational programming.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Solh T, Botsford A, Solh M. Glanzmann’s thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options. J Blood Med 2015;6:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poon MC, Di Minno G, d’Oiron R, Zotz R. New Insights Into the Treatment of Glanzmann Thrombasthenia. Transfus Med Rev 2016;30:92–99. [DOI] [PubMed] [Google Scholar]
- 3.Grainger JD, Thachil J, Will AM. How we treat the platelet glycoprotein defects; Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults. Br J Haematol 2018;182:621–632. [DOI] [PubMed] [Google Scholar]
- 4.Andrews RK, Berndt MC. Bernard-Soulier syndrome: an update. Semin Thromb Hemost 2013;39:656–662. [DOI] [PubMed] [Google Scholar]
- 5.Boeckelmann D, Hengartner H, Greinacher A, et al. Patients with Bernard-Soulier syndrome and different severity of the bleeding phenotype. Blood Cells Mol Dis 2017;67:69–74. [DOI] [PubMed] [Google Scholar]
- 6.Nurden AT, Pillois X, Wilcox DA. Glanzmann thrombasthenia: state of the art and future directions. Semin Thromb Hemost 2013;39:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadowaki S, Makino S, Mohri Y, Awaguni H, Shinozuka J, ImashukuS. Difficulty in controlling heavy menstrual bleeding at menarche in a patient with Glanzmann’s thrombasthenia. Blood Coagul Fibrinolysis 2021;32:155–158. [DOI] [PubMed] [Google Scholar]
- 8.Di Minno G, Zotz RB, d’Oiron R, et al. The international, prospective Glanzmann Thrombasthenia Registry: treatment modalities and outcomes of non-surgical bleeding episodes in patients with Glanzmann thrombasthenia. Haematologica 2015;100:1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zotz RB, Poon MC, Di Minno G, D’Oiron R, Glanzmann Thrombasthenia Registry I. The International Prospective Glanzmann Thrombasthenia Registry: Pediatric Treatment and Outcomes. TH Open 2019;3:e286–e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valera MC, Kemoun P, Cousty S, Sie P, Payrastre B. Inherited platelet disorders and oral health. J Oral Pathol Med 2013;42:115–124. [DOI] [PubMed] [Google Scholar]
- 11.Orsini S, Noris P, Bury L, et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. Haematologica 2017;102:1192–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poon MC, d’Oiron R, Zotz RB, et al. The international, prospective Glanzmann Thrombasthenia Registry: treatment and outcomes in surgical intervention. Haematologica 2015;100:1038–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajpurkar M, Chitlur M, Recht M, Cooper DL. Use of recombinant activated factor VII in patients with Glanzmann’s thrombasthenia: a review of the literature. Haemophilia 2014;20:464–471. [DOI] [PubMed] [Google Scholar]
- 14.United States Food and Drug Administration (FDA) Approved Blood Products: Recombinant Coagulation Factor VIIa (NovoSeven RT), 2018. https://www.fda.gov/vaccines-blood-biologics/approved-blood-products/novosevenrt.
- 15.European Medicines Agency Approved Products: Activated eptacof alfa (Novoseven), 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/novoseven.
- 16.Recht M, Rajpurkar M, Chitlur M, et al. Independent adjudicator assessments of platelet refractoriness and rFVIIa efficacy in bleeding episodes and surgeries from the multinational Glanzmann’s thrombasthenia registry. Am J Hematol 2017;92:646–652. [DOI] [PubMed] [Google Scholar]
- 17.Poon MC. The use of recombinant activated factor VII in patients with Glanzmann’s Thrombasthenia. Thromb Haemost 2021;121:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poon MC, d’Oiron R. Alloimmunization in congenital deficiencies of platelet surface glycoproteins: focus on Glanzmann’s Thrombasthenia and Bernard-Soulier’s Syndrome. Semin Thromb Hemost 2018;44:604–614. [DOI] [PubMed] [Google Scholar]
- 19.Kongalappa S, Reddy JM, Durugappa T, D’Souza F, Subramanian S, Prakash A. Glanzmann Thrombasthenia in children: experience from a tertiary care center in Southern India. J Pediatr Hematol Oncol 2019;41:e68–e71. [DOI] [PubMed] [Google Scholar]
- 20.Halterman JS, Kaczorowski JM, Aligne CA, Auinger P, Szilagyi PG. Iron deficiency and cognitive achievement among school-aged children and adolescents in the United States. Pediatrics 2001;107:1381–1386. [DOI] [PubMed] [Google Scholar]
- 21.Baker RD, Greer FR, Committee on Nutrition American Academy of P. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0–3 years of age). Pediatrics 2010;126:1040–1050. [DOI] [PubMed] [Google Scholar]
- 22.Hassan TH, Badr MA, Karam NA, et al. Impact of iron deficiency anemia on the function of the immune system in children. Medicine (Baltimore) 2016;95:e5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allali S, Brousse V, Sacri AS, Chalumeau M, de Montalembert M. Anemia in children: prevalence, causes, diagnostic work-up, and long-term consequences. Expert Rev Hematol 2017;10:1023–1028. [DOI] [PubMed] [Google Scholar]
- 24.VanderMeulen H, Sholzberg M. Iron deficiency and anemia in patients with inherited bleeding disorders. Transfus Apher Sci 2018;57:735–738. [DOI] [PubMed] [Google Scholar]
- 25.Scaramellini N, Capecchi M, Artoni A, La Marca S, Cappellini MD, Motta I. Ferric carboxymaltose for sub-acute and chronic iron deficiency anemia in inherited platelet function defects. Intern Emerg Med 2021;16:505–507. [DOI] [PubMed] [Google Scholar]
- 26.Andic N, Oguz N, Gunduz E, Kiraz Bulduk T, Uskudar Teke H. Weekly low-dose recombinant factor VIIa prophylaxis in Glanzmann thrombasthenia. Blood Coagul FIbrinolysis 2021;32:349–351. [DOI] [PubMed] [Google Scholar]
- 27.Savoia A, Kunishima S, De Rocco D, et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat 2014;35:1033–1045. [DOI] [PubMed] [Google Scholar]
- 28.Nurden AT, Pillois X, Fiore M, et al. Expanding the mutation spectrum affecting αIIbβ3 integrin in Glanzmann Thrombasthenia: screening of the ITGA2B and ITGB3 Genes in a Large International Cohort. Hum Mutat 2015;36:548–561. [DOI] [PubMed] [Google Scholar]
- 29.Siboni SM, Spreafico M, Calò L, et al. Gynaecological and obstetrical problems in women with different bleeding disorders. Haemophilia 2009;15:1291–1299. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia 2013;19:e1–47. [DOI] [PubMed] [Google Scholar]
- 31.Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood 2015;125:2052–2061. [DOI] [PubMed] [Google Scholar]
- 32.WHO Guidelines Approved by the Guidelines Review Committee. WHO guideline on use of ferritin concentrations to assess iron status in individuals and populations. Geneva: World Health Organization, 2020. [PubMed] [Google Scholar]
- 33.Rae C, Furlong W, Horsman J, et al. Bleeding disorders, menorrhagia and iron deficiency: impacts on health-related quality of life. Haemophilia 2013;19:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Bourgeois T, Klima J, Berlan ED, Fischer AN, O’Brien SH. Iron deficiency and fatigue in adolescent females with heavy menstrual bleeding. Haemophilia 2013;19: 225–230. [DOI] [PubMed] [Google Scholar]
- 35.Mei Z, Addo OY, Jefferds ME, Sharma AJ, Flores-Ayala RC, Brittenham GM. Physiologically based serum ferritin thresholds for iron deficiency in children and non-pregnant women: a US National Health and Nutrition Examination Surveys (NHANES) serial cross-sectional study. Lancet Haematol 2021;8:e572–e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.