

Abstract
We previously showed that apremilast, an FDA-approved PDE4 inhibitor, selectively alters behavioral responses to ethanol and certain GABAergic drugs in a PKA-dependent manner in C57BL6/J mice. Here, we investigated if PKA phosphorylation of β3 GABAA receptor subunits is involved in apremilast regulation of ethanol, propofol, or diazepam responses. Apremilast prolonged rotarod ataxia and loss of the righting reflex by ethanol and propofol in wild-type mice, but not in β3-S408A/S409A knock-in mice. In contrast, apremilast hastened recovery from the ataxic and sedative effects of diazepam in both genotypes. These findings suggest that apremilast modulation of ethanol and propofol behaviors in wild-type mice is mediated by β3 subunit phosphorylation, whereas its actions on diazepam responses involve a different mechanism. The PKA inhibitor H-89 prevented apremilast modulation of ethanol-induced ataxia. Apremilast sensitized wild-type males to ethanol-induced ataxia and decreased acute functional tolerance (AFT) in females but had no effect in β3-S408A/S409A mice of either sex. These results could not be attributed to genotype differences in blood ethanol clearance. There were also no baseline genotype differences in ethanol consumption and preference in two different voluntary drinking procedures. However, the ability of apremilast to reduce ethanol consumption was diminished in β3-S408A/S409A mice. Our results provide strong evidence that PKA-dependent phosphorylation of β3 GABAA receptor subunits is an important mechanism by which apremilast increases acute sensitivity to alcohol, decreases AFT, and decreases ethanol drinking.
Keywords: PDE4 inhibitor apremilast, β3 GABAA receptor subunit, PKA phosphorylation, acute functional tolerance to ethanol, two-bottle choice ethanol consumption
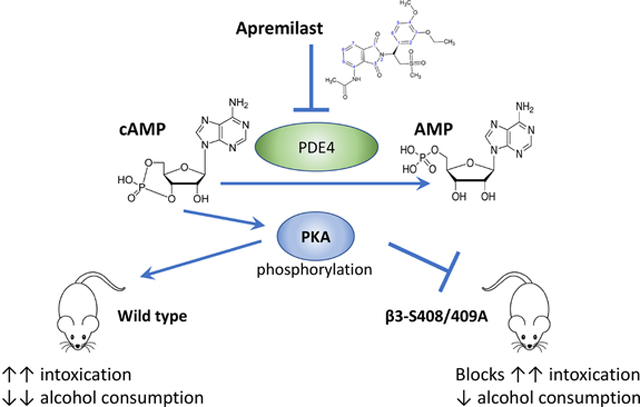
Gracphical Abstract
PKA phosphorylation of GABAA β3 subunits is a mechanism by which apremilast increases alcohol intoxication and decreases alcohol consumption.
1. INTRODUCTION
Phosphodiesterases (PDEs) catalyze the hydrolysis of the second messengers cAMP and cGMP, thereby limiting the activation of protein kinase A (via cAMP) and protein kinase G (via cGMP) (Logrip, 2015). An extensive literature indicates that cAMP signaling regulates behavioral responses to alcohol (Newton and Messing, 2006). PDE4 is highly expressed in reward-responsive brain regions (Logrip, 2015) and has been implicated in alcohol and drug dependence (Wen et al., 2018). Several different PDE4 inhibitors decrease alcohol seeking and consumption in rodents (Blednov et al., 2014; Franklin et al., 2015; Hu et al., 2011; Liu et al., 2017; Wen et al., 2012). We found that apremilast, an FDA-approved PDE4 inhibitor used to treat psoriasis, also decreases voluntary ethanol drinking in male and female mice (Blednov et al., 2018b). Apremilast also prolongs acute intoxicating effects of ethanol, which is a response commonly associated with reduced ethanol consumption in rodents (Blednov et al., 2018a). Importantly, in a recent human laboratory study, apremilast suppressed alcohol drinking in non-treatment-seeking individuals with moderate to severe alcohol use disorder (AUD) (Grigsby et al., 2021).
Although it is well established that PKA modulates behavioral responses to alcohol and other drugs of abuse (Lee and Messing, 2008), the mechanisms by which PDE4 inhibitors act to reduce alcohol drinking are not known. Most mechanistic investigations have focused on PKA-mediated phosphorylation of NMDA receptors, DARPP-32, and CREB (Newton and Messing, 2006). One study examined PKA regulation of GABAA receptors in alcohol responses, reporting that intracerebroventricular administration of the PKA activator Sp-cAMP increased the sedative-hypnotic effects of ethanol and the GABAA receptor agonist muscimol (Kumar et al., 2012). Sp-cAMP also altered trafficking of receptors containing α1 and α4 subunits (Bohnsack et al., 2016; Carlson et al., 2016). The PKA substrates mediating these effects have not been identified.
Over 20 years ago, it was established that PKA phosphorylates the large intracellular loops of β1 and β3 subunits of GABAA receptors, resulting in opposite effects on channel modulation. Activation of PKA increases the amplitude of GABA-stimulated currents in HEK293 cells transfected with α1β3 or α1β3γ2 subunits, but this effect is eliminated in receptors containing β3 subunits with alanine mutations at S408 and S409 (McDonald et al., 1998). PKA also phosphorylates the large intracellular loop of the β1 subunit at S409, which in contrast, reduces the amplitude of GABA-stimulated currents in HEK293 cells transfected with α1β1 or α1β1γ2 subunits. This effect is eliminated in cells transfected with β1 subunits with an alanine mutation at S409 (Moss et al., 1992). These findings are supported by our study in Xenopus oocytes, showing that a phosphomimetic (aspartate) mutation at the PKA site in β1 decreases GABA-stimulated currents in receptors containing α1 or α3 (but not α2) and γ2 subunits, whereas a phosphomimetic mutation at the PKA site in β3 increases currents in receptors containing α1 or α2 (but not α3) and γ2 subunits (Blednov et al., 2020).
These functional studies further informed our investigation of PKA-dependent mechanisms by which apremilast regulates responses to ethanol and GABAergic drugs in vivo (Blednov et al., 2020). In C57BL/6J mice, apremilast prolonged the acute sedative-hypnotic and ataxic effects of ethanol, propofol, zolpidem, and loreclezole; apremilast did not alter sedation and ataxia induced by etomidate or gaboxadol, and it unexpectedly shortened the duration of sedation and ataxia by diazepam (Blednov et al., 2020). The differential effects of apremilast were attributed to likely preferential actions of these drugs on different GABAA receptor subunits. The PKA inhibitor H-89 blocked the ability of apremilast to alter responses to ethanol, propofol, and diazepam (Blednov et al., 2020). Together with earlier work showing apremilast modulation of ethanol drinking and related behaviors (Blednov et al., 2018a; Blednov et al., 2018b), our collective findings suggested that apremilast regulates responses to ethanol and certain GABAergic drugs by increasing PKA-dependent phosphorylation of β3 subunits of GABAA receptors. To test this hypothesis, it was necessary to use a knock-in mouse model since there are no selective modulators to target the β3 subunit and since most GABAergic drugs lack sufficient subunit specificity. In the current study, we used β3-S408A/S409A knock-in (hereafter designated as β3-S408/409A) mice as a genetic tool to prevent PKA phosphorylation of β3 subunits (Vien et al., 2015) and compared the effects of apremilast on behavioral responses to ethanol, propofol, or diazepam. Our results provide strong evidence that PKA-dependent β3 subunit phosphorylation is an important mechanism for apremilast regulation of ethanol consumption and other acute behaviors that influence drinking.
2. MATERIALS AND METHODS
2.1. Mice
β3-S408/409A heterozygous mice on a C57BL/6J background (Vien et al., 2015) were crossed with C57BL/6J mice from a colony maintained in the Animal Resources Center at the University of Texas at Austin. Mice were group-housed by sex (4 to 5 per cage) in temperature- and humidity-controlled rooms with free access to food and water using a 12-h light/dark cycle (lights on at 7:00 AM). Experiments began when the mice were 2 to 3 months old. Mice were allowed to adapt to the testing rooms for about one week before testing. Experiments were approved by the Institutional Animal Care and Use Committee at The University of Texas at Austin and comply with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Both male and female mice were tested in all procedures. The number of animals used in each experiment is reported in the figure captions.
2.2. Drug Administration
Ethanol (100% stock, Aaper Alcohol and Chemical, Shelbyville, KY) solutions were prepared in 0.9% saline (20%, v/v) for i.p. injection. Apremilast (Toronto Research Chemicals Inc., North York, ON, Canada) was freshly prepared as a suspension in saline with 3–4 drops of Tween-80. Apremilast (20 mg/kg, p.o.) was administered once daily at 0.05 ml/10 g body weight 1 h before experiments. We previously showed that this dose produces robust inhibition of ethanol drinking without altering total fluid intake (Blednov et al., 2018b). This time frame was chosen based on our work showing peak levels of apremilast in plasma, liver, and brain 1 h after administration (Blednov et al., 2018b). Since apremilast (20 mg/kg) can transiently decrease spontaneous locomotor activity (Blednov et al., 2018b), this time frame also prevents it from potentially confounding behavioral responses to ethanol. Diazepam and propofol were purchased from Sigma-Aldrich (St. Louis, MO) and freshly prepared in 0.9% saline with 3–4 drops of Tween-80 and injected i.p. at 0.1 ml/10 g body weight. The propofol suspension was also sonicated for 10 min. H-89 was purchased from Tocris Bioscience (Minneapolis, MN) and freshly prepared in 0.9% saline with 3–4 drops of Tween-80 and injected at 0.05 ml/10 g body weight. H-89 (10 mg/kg, s.c.) was given 30 min after apremilast (20 mg/kg, p.o.) and 30 min before ethanol (2 g/kg, i.p.). To study apremilast effects on ethanol consumption in the intermittent drinking procedure, saline (p.o.) or apremilast (5, 10, 15, 20 and 25 mg/kg, p.o.) was administered every-other-day 1 h before beginning the drinking session. Each apremilast dose was given for two drinking days. Before apremilast treatment, all mice were injected with saline for two drinking days.
2.3. Rotarod Ataxia
Mice were trained on a fixed-speed rotarod (Economex; Columbus Instruments, Columbus, OH) at 10 rpm, and training was complete when they were able to remain on the rotarod for 60 s (basal level). Every 15 min after drug injection, each mouse was placed on the rotarod and latency to fall was measured until the mouse could once again remain on the rotarod for 60 s. Saline (p.o.) or apremilast (20 mg/kg, p.o.) was given once 1 h before i.p. injection of ethanol (2 g/kg), diazepam (7 mg/kg), or propofol (35 mg/kg). These doses elicit mild ataxia in mice and are similar to our previous study of apremilast modulation of ethanol and GABAergic drugs (Blednov et al., 2020). To study PKA inhibition, mice were given apremilast (20 mg/kg, p.o.) 1 h before rotarod testing and then treated with saline (s.c.) or the PKA inhibitor H-89 (10 mg/kg, s.c.) 30 min before injection of ethanol (2 g/kg, i.p.). We previously showed that this dose of H-89 blocked the ability of apremilast to modulate responses to ethanol and GABAergic drugs (Blednov et al., 2020). We originally chose this PKA inhibitor and dose based on the ability to prevent ethanol-induced increases in α1 GABAA subunit expression by PKA in rats (Kumar et al., 2012).
2.4. Loss of the Righting Reflex
Sedative-hypnotic doses of ethanol, propofol, and diazepam were used to measure duration of the loss of righting reflex (LORR). When mice became ataxic, they were placed in the supine position in V-shaped plastic troughs until they were able to right themselves three times within 30 s. The duration of the LORR was defined as the time elapsed between being placed in the supine position until recovering the righting reflex. Saline (p.o.) or apremilast (20 mg/kg, p.o.) was injected once 1 h before injection of sedative-hypnotic doses of ethanol (3.6 g/kg, i.p.), propofol (120 mg/kg, i.p.), or diazepam (50 mg/kg, i.p.), the same doses used in our previous apremilast studies (Blednov et al., 2020; Blednov et al., 2018a).
2.5. Acute Functional Tolerance
Acute functional tolerance (AFT) to the motor ataxic effect of ethanol was measured using the two-dose method (Erwin and Deitrich, 1996). Ethanol-naïve mice were trained to balance on a fixed-speed rotarod (10 rpm) for a 60-s period. After basal training, half of the mice received saline (p.o.) and the other half received apremilast (20 mg/kg, p.o.). One hour later, mice were injected with ethanol (1.75 g/kg, i.p.) and placed back on the rotarod until they fell off. Mice were tested in 5-min intervals until they regained the ability to balance on the rotarod for 60 s. Once this was achieved (t1), a retro-orbital blood sample was collected to measure blood ethanol concentration (BEC1). Mice were then immediately given a second ethanol injection (2 g/kg, i.p.). After losing the ability to remain on the rotarod, mice were tested in 5-min intervals until they regained the ability to balance for 60 s (t2). Then a second blood sample was collected to measure BEC2. AFT was defined as the difference in BEC at t2 and t1 (BEC2 - BEC1). BEC measurements are described in the section on blood ethanol clearance.
2.6. Two-Bottle Choice Ethanol Drinking
In the two-bottle choice (2BC) procedure, mice had 24-h access to one bottle of ethanol and one bottle of water. Mice were offered a series of increasing concentrations of ethanol (3%, 6%, 9%, 12%, and 15% v/v) with each concentration available for four days. The placement of the ethanol bottle was alternated with each drinking session to control for side preferences, and mice were weighed every four days. Ethanol and water intake (g/kg body weight/24h) were calculated for each mouse, and values were averaged over four days at each concentration.
2.7. Two-Bottle Choice Every-Other-Day Ethanol Drinking
During 2BC-EOD drinking, a procedure that induces high voluntary alcohol consumption in mice (Melendez, 2011; Rosenwasser et al., 2013), mice had access to a bottle of 15% (v/v) ethanol and a bottle of water during 24-h drinking sessions offered every-other-day. Water only was available on non-drinking days. Measurements of ethanol intake, preference for ethanol, and total fluid intake were averaged over two drinking days with different bottle positions. 2BC-EOD drinking was studied in two different experiments, and the data were combined.
Mice from the second experiment were then used to study the effects of apremilast on 2BC-EOD drinking. Ethanol (15%) consumption was measured for at least four drinking days to ensure stable levels of drinking (i.e., < 10% variation in average intake on days 1–2 and days 3–4). Ethanol intake was then measured after saline injection for two drinking days, and mice were assigned to treatment and control groups based on similar levels of ethanol intake and preference. Beginning on the third drinking day, mice were injected once daily with either saline (p.o.) or increasing doses of apremilast (5, 10, 15, 20 and 25 mg/kg, p.o.). Each dose of apremilast was given for two drinking days, and drinking measures were averaged. Data were normalized to the average intake of saline-treated mice of the same genotype across drinking days 3–12.
2.8. Blood Ethanol Clearance
Ethanol clearance from blood was measured over 4 h after injection of ethanol (2 g/kg and 4 g/kg, i.p.). Retro-orbital blood samples (~20 μl) were collected in capillary tubes and centrifuged for 6 min at 3,100 × g using a Haematospin 1400 centrifuge (Analox Instruments, London, UK). Plasma samples were stored at −20°C until BECs were determined in 5-μl aliquots using an AM1 Alcohol Analyzer (Analox Instruments). The machine was calibrated every 15 samples using an industry standard, and BECs were determined using commercially available reagents according to the manufacturer’s instructions. Samples were averaged from duplicate runs and expressed as mg/dl.
2.9. Statistical Analysis
Data are reported as mean ± S.E.M values. The number of animals used in each study are reported in the figure captions. Statistical analysis was performed using Prism 9 (GraphPad Software, Inc., La Jolla, CA). Data from male and female mice were analyzed separately by two-way ANOVA or two-way ANOVA with repeated measurements and Tukey’s multiple comparisons post-hoc tests. To measure development of AFT, Wilcoxon signed rank tests were used to compare experimental data with a theoretical median of 0.
3. RESULTS
3.1. PKA phosphorylation of β3 subunits is required for apremilast modulation of ethanol and propofol ataxia but not diazepam ataxia
We previously hypothesized that apremilast prolongs recovery from ethanol- or propofol-induced rotarod ataxia by enhancing phosphorylation of β3-containing GABAA receptors and accelerates recovery from diazepam-induced ataxia by enhancing phosphorylation of β1-containing GABAA receptors (Blednov et al., 2020). To test this hypothesis, we examined rotarod ataxia in β3-S408/409A mice, which have mutations that render β3 subunits resistant to PKA-mediated phosphorylation. Apremilast (20 mg/kg, p.o.) prolonged ethanol (2 g/kg, i.p.) ataxia in wild-type mice but accelerated recovery in β3-S408/409A mice (Fig. 1A). Analysis of the area over the recovery curve showed a significant treatment × genotype interaction in male (F1,24 = 124.8, p < 0.0001; Fig. 1B) and female (F1,25 = 171.1, p < 0.0001; Fig. 1C) mice. In the framework of our hypothesis, these results suggest that apremilast modulation of ethanol ataxia in wild-type animals primarily depends on phosphorylation of β3-containing GABAA receptors (which increases inhibitory GABA-stimulated currents), while phosphorylation of β1-containing receptors (which decreases inhibitory GABAergic signaling) is unmasked in the mutant mice.
Figure 1. Apremilast modulation of ethanol- and propofol-induced ataxia, but not diazepam-induced ataxia, is disrupted in β3-S408/409A knock-in mice.
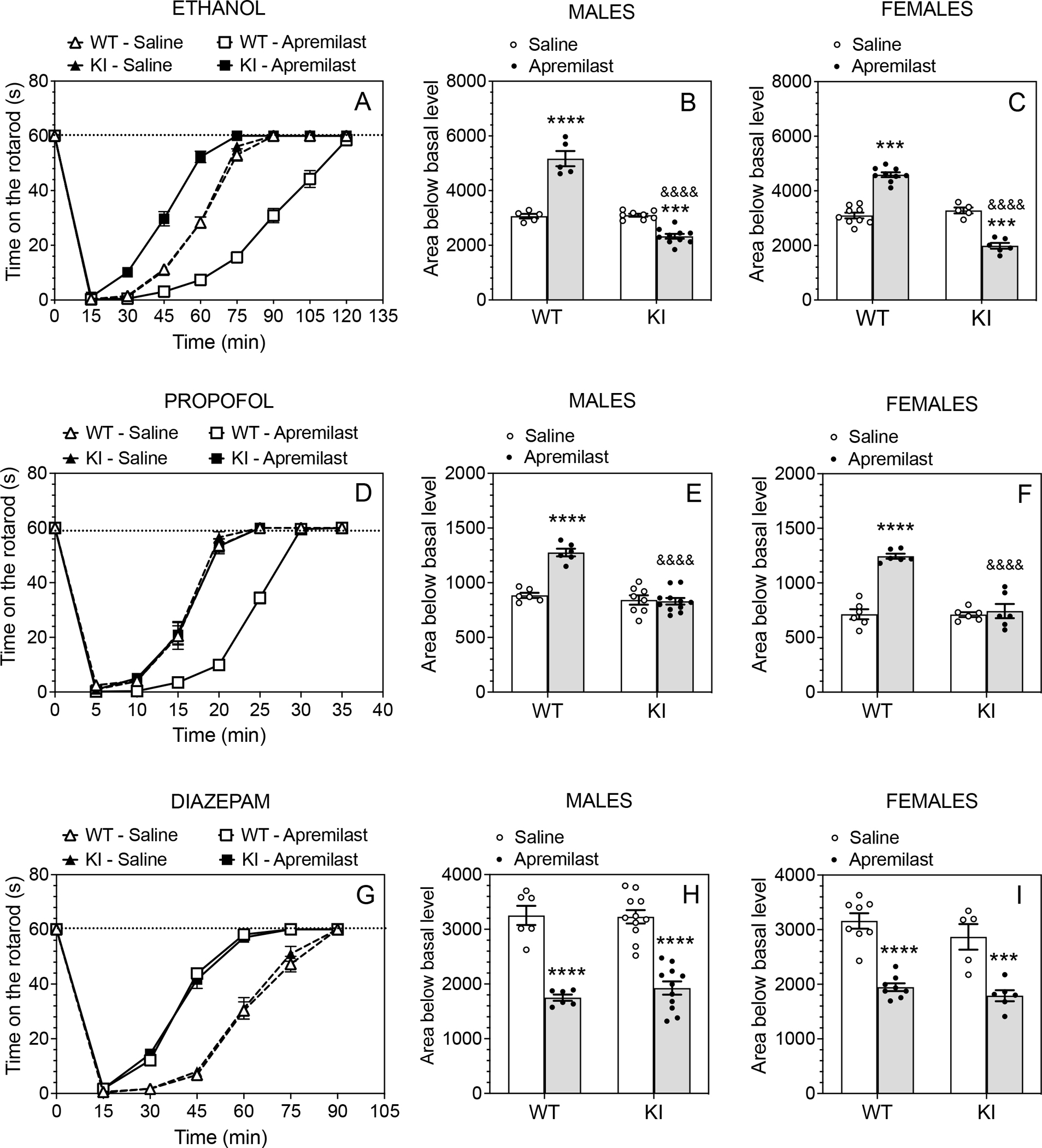
Time on the rotarod in saline (p.o.)- vs. apremilast (20 mg/kg, p.o.)-pretreated wild-type (WT) and β3-S408/409A knock-in (KI) mice after i.p. injection of (A) ethanol, 2g/kg (WT: n = 14 per group; KI: n = 13–16 per group), (D) propofol, 35 mg/kg (WT: n = 12 per group; KI: n = 14–17 per group), or (G) diazepam, 7 mg/kg (WT: n = 14 per group; KI: n = 16–17 per group). Data from male and female mice were combined. (B, C) Area below basal level and above the recovery curve for ethanol-induced ataxia in males (WT: n = 5 per group; KI: n = 8–10 per group) and females (WT: n = 9 per group; KI: n = 5–6 per group). (E, F) Area below basal level and above the recovery curve for propofol-induced ataxia in males (WT: n = 6 per group; KI: n = 8–11 per group) and females (n = 6 per group and per genotype). (H, I) Area below basal level and above the recovery curve for diazepam-induced ataxia in males (WT: n = 6 per group; KI: n = 11 per group) and females (n = 8 per group; KI: n = 5–6 per group). *** p < 0.001, **** p < 0.0001, apremilast- vs. saline-treated group of the same genotype; &&&& p < 0.0001, apremilast-treated groups of WT vs. KI mice (Tukey’s multiple comparisons post-hoc tests).
Apremilast significantly prolonged recovery from ataxia induced by propofol (35 mg/kg, i.p.) in wild-type mice but had no effect in β3-S408/409A mice (Fig. 1D). There were significant effects of apremilast treatment on recovery from ataxia in both male (Fig. 1E) and female (Fig. 1F) mice, and the areas of the recovery curves showed a significant treatment × genotype interaction in both sexes (males: F1,27 = 31.72, p < 0.0001; females: F1,20 = 33.76, p < 0.0001).
In contrast to ethanol and propofol ataxia, apremilast accelerated recovery from diazepam (7 mg/kg, i.p.) ataxia in both genotypes (Fig. 1G). Apremilast decreased the area of the recovery curve in males (F1,30 = 107.3, p < 0.0001; Fig. 1H) and in females (F1,23 = 70.05, p < 0.0001; Fig.1I) without a difference between genotypes in either sex. Thus, the mechanism of apremilast modulation of diazepam ataxia is independent of β3 phosphorylation.
3.2. PKA activation is required for apremilast modulation of ethanol ataxia in both wild-and β3-S408/409A mice
Because apremilast inhibits PDE4 and increases activation of PKA, we tested the PKA inhibitor H-89 (Hidaka et al., 1984) on apremilast modulation of ethanol ataxia in wild-type and β3-S408/409A mice. One hour before rotarod testing, mice were given apremilast (20 mg/kg, p.o.) and were then treated with H-89 (10 mg/kg, s.c.) or saline (s.c.) 30 min before ethanol (2 g/kg, i.p.). In both sexes, H-89 completely prevented the ability of apremilast to either prolong ethanol ataxia in wild-type mice or to shorten the duration of ataxia in β3 mutant mice (Fig. 2A). There were no genotype differences between males (Fig. 2B) or females (Fig. 2C) after H-89 treatment. As shown for the areas of the recovery curves, there were significant effects of H-89 treatment (males: F1,20 = 17.60, p < 0.001; females: F1,20 = 26.12, p < 0.0001), genotype (males: F1,20 = 157.0, p < 0.0001; females: F1,20 = 247.1, p < 0.0001), and treatment × genotype interaction (males: F1,20 = 208.3, p < 0.0001; females: F1,20 = 263.7, p < 0.0001). These results show that PKA activation is required for apremilast regulation of ethanol ataxia in both genotypes.
Figure 2. The PKA inhibitor H-89 prevents the ability of apremilast to alter ethanol-induced ataxia in wild-type and β3-S408/409A knock-in mice.
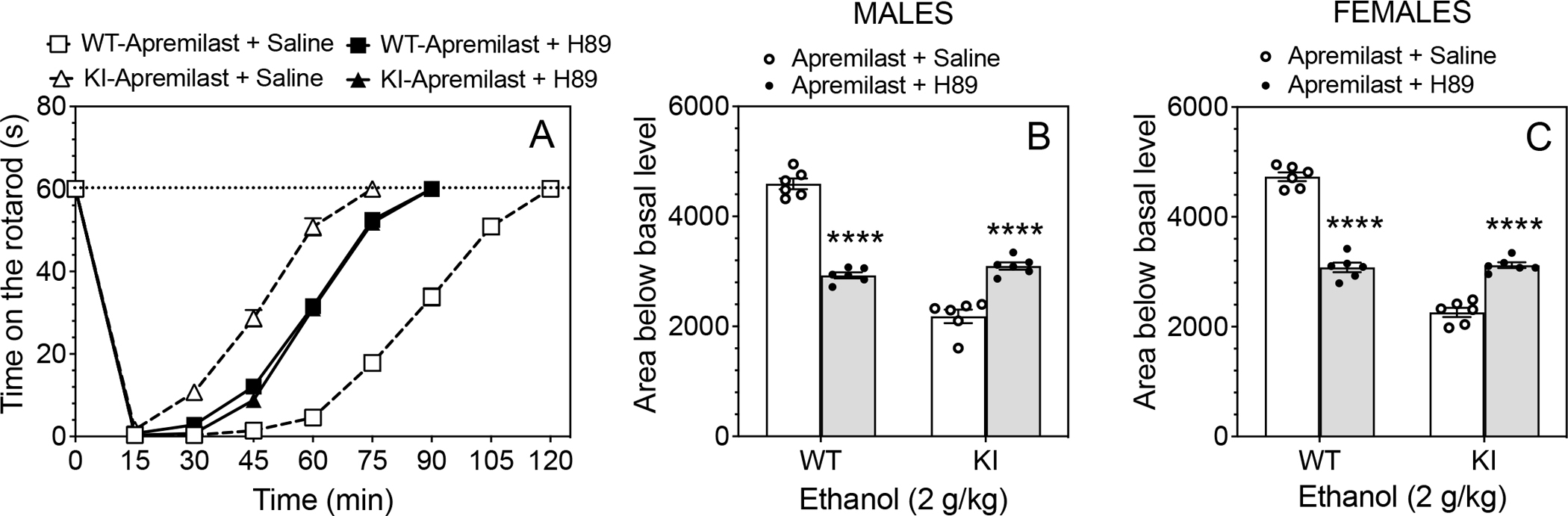
One hour before rotarod testing, mice were given apremilast (20 mg/kg, p.o.) and were then treated with saline (s.c.) or the PKA inhibitor H-89 (10 mg/kg, s.c.) 30 min before injection of ethanol (2 g/kg, i.p.). (A) Time on the rotarod in wild-type (WT) and β3-S408/409A knock-in (KI) mice (n = 12 per group and per genotype, males and females combined). (B) Area below basal level and above the recovery curve in males (n = 6 per group and per genotype). (C) Area below basal level and above the recovery curve in females (n = 6 per group and per genotype). **** p < 0.0001, H-89- vs. saline-treated group of the same genotype (Tukey’s multiple comparisons post-hoc tests).
3.3. PKA sites on β3 subunits are required for apremilast modulation of sedation by ethanol and propofol but not diazepam
We next studied the effects of apremilast (20 mg/kg, p.o.) pretreatment on the sedative-hypnotic effects produced by higher doses of ethanol, propofol, or diazepam (Fig. 3). In both sexes, apremilast prolonged the duration of the LORR induced by ethanol (3.6 g.kg, i.p.) in wild-type mice but had no effect in knock-in mice (Fig. 3A and D). There were significant effects of apremilast treatment (males: F1,31 = 90.04, p < 0.0001; females: F1,26 = 54.42, p < 0.0001), genotype (males: F1,31 = 132.3, p < 0.0001; females: F1,26 = 41.87, p < 0.0001), and treatment × genotype interaction (males: F1,31 = 111.8, p < 0.0001; females: F1,26 = 47.67, p < 0.0001). Apremilast also prolonged the duration of LORR induced by propofol (120 mg/kg, i.p.) in wild-type mice, but it had no effect in mutant mice (Fig. 3B and E). In both sexes, there were main effects of apremilast treatment (males: F1,22 = 123.7, p < 0.0001; females: F1,21 = 8.773, p = 0.0074) and genotype (males: F1,22 = 90.49, p < 0.0001; females: F1,21 = 8.053, p = 0.0099) with a treatment × genotype interaction (males: F1,22 = 83.06, p < 0.0001; females: F1,21 = 15.12, p = 0.0008). In contrast to prolonging the LORR duration due to ethanol and propofol sedation, apremilast shortened the duration of LORR induced by diazepam (50 mg/kg, i.p.) (males: F1,16 = 356.5, p < 0.0001; females: F1,16 = 360.3, p < 0.0001) similarly in both genotypes and sexes (Fig. 3C and F).
Figure 3. Apremilast modulation of ethanol- and propofol-induced, but not diazepam-induced loss of righting, is absent in β3-S408/409A knock-in mice.
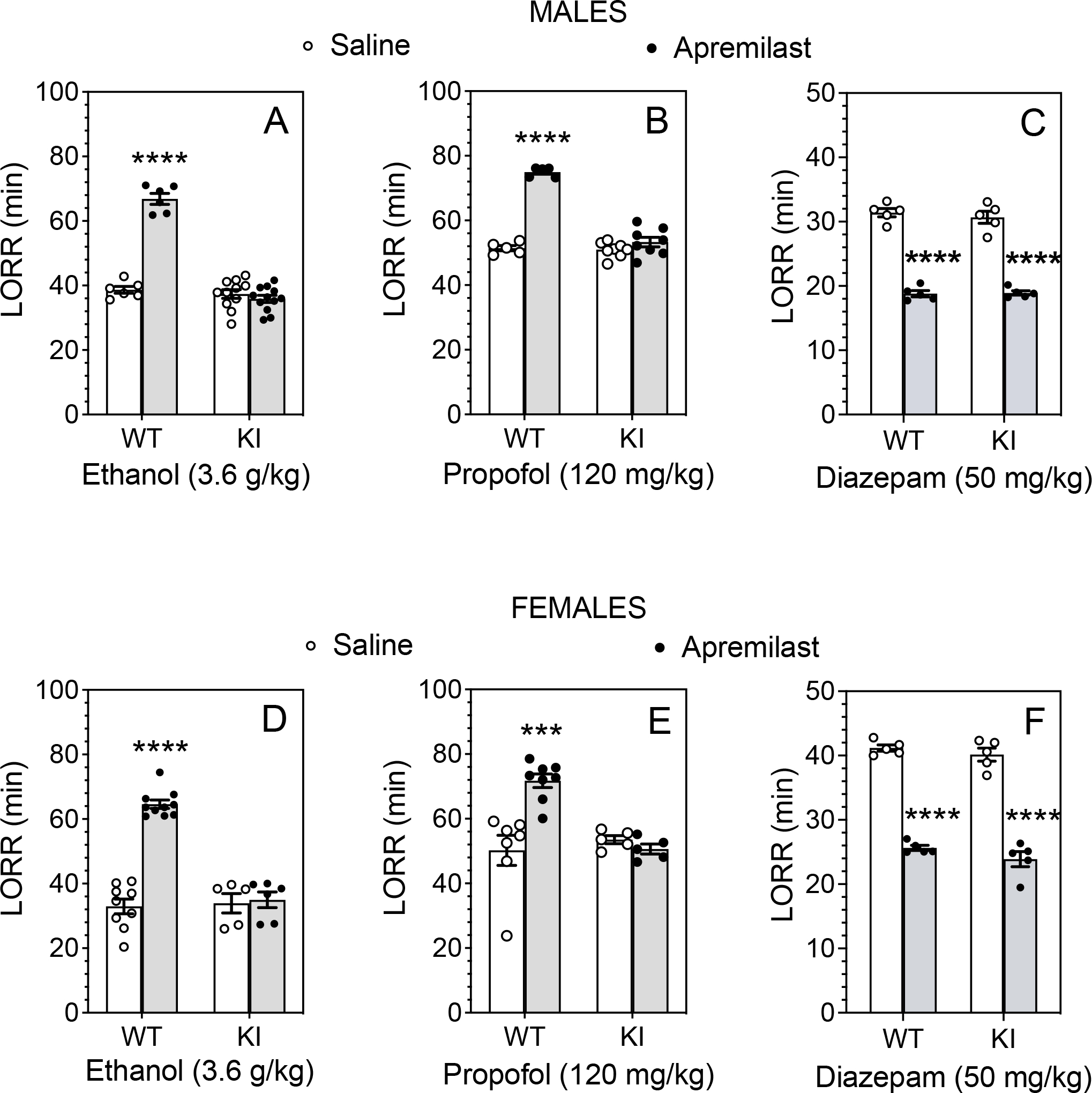
Duration of loss of righting reflex (LORR) in saline (p.o.)- vs. apremilast (20 mg/kg, p.o.)-pretreated male and female β3-S408/409A wild-type (WT) and knock-in (KI) mice after i.p. injections of (A, D) 3.6 g/kg ethanol in males (WT: n = 6 per group; KI: n = 11–12 per group) and females (WT: n = 9–10 per group; KI: n = 5–6 per group), (B, E) 120 mg/kg propofol in males (WT: n = 5 per group; KI: n = 8 per group) and females (WT: n = 7–8 per group; KI: n = 5 per group), or (C, F) 50 mg/kg diazepam in males (n = 5 per group and per genotype) and females (n = 5 per group and per genotype). *** p < 0.001, **** p < 0.0001, apremilast- vs. saline-treated groups of the same genotype (Tukey’s multiple comparisons post-hoc tests).
3.4. Apremilast decreases acute functional tolerance to ethanol ataxia in wild-type but not in β3-S408/409A mice
One possible mechanism by which apremilast might prolong recovery for ethanol intoxication is by enhancing the development of acute functional tolerance (AFT) to ethanol. AFT is behavioral tolerance to ethanol that occurs within an individual test session and can be measured by the time taken to recover from rotarod ataxia. In males (Fig. 4A), apremilast significantly increased the recovery time in wild-type mice but decreased recovery time in β3-S408/409A mice after the first ethanol (1.75 g/kg) injection (F1,30 = 108.3, p < 0.0001, treatment × genotype interaction). These opposite effects between the genotypes are consistent with those found for ethanol-induced ataxia (Fig. 1) after 2 g/kg ethanol. The differences in time of recovery were not due to differences in BECs after the first injection (Fig. 4D). After the second ethanol (2 g/kg) injection, the recovery time was again prolonged in wild-type mice despite a significantly lower BEC2 in this group (Fig. 4E), but not in mutant mice (F1,30 = 105.8, p < 0.0001, treatment × genotype interaction; Fig. 4B). The difference in recovery time between the second and first ethanol injections (Fig. 4C) was increased in wild-type but not in mutant mice treated with apremilast (F1,30 = 55.42, p < 0.0001, treatment × genotype interaction).
Figure 4. Apremilast sensitization to ethanol-induced ataxia is disrupted in male β3-S408/409A knock-in mice.
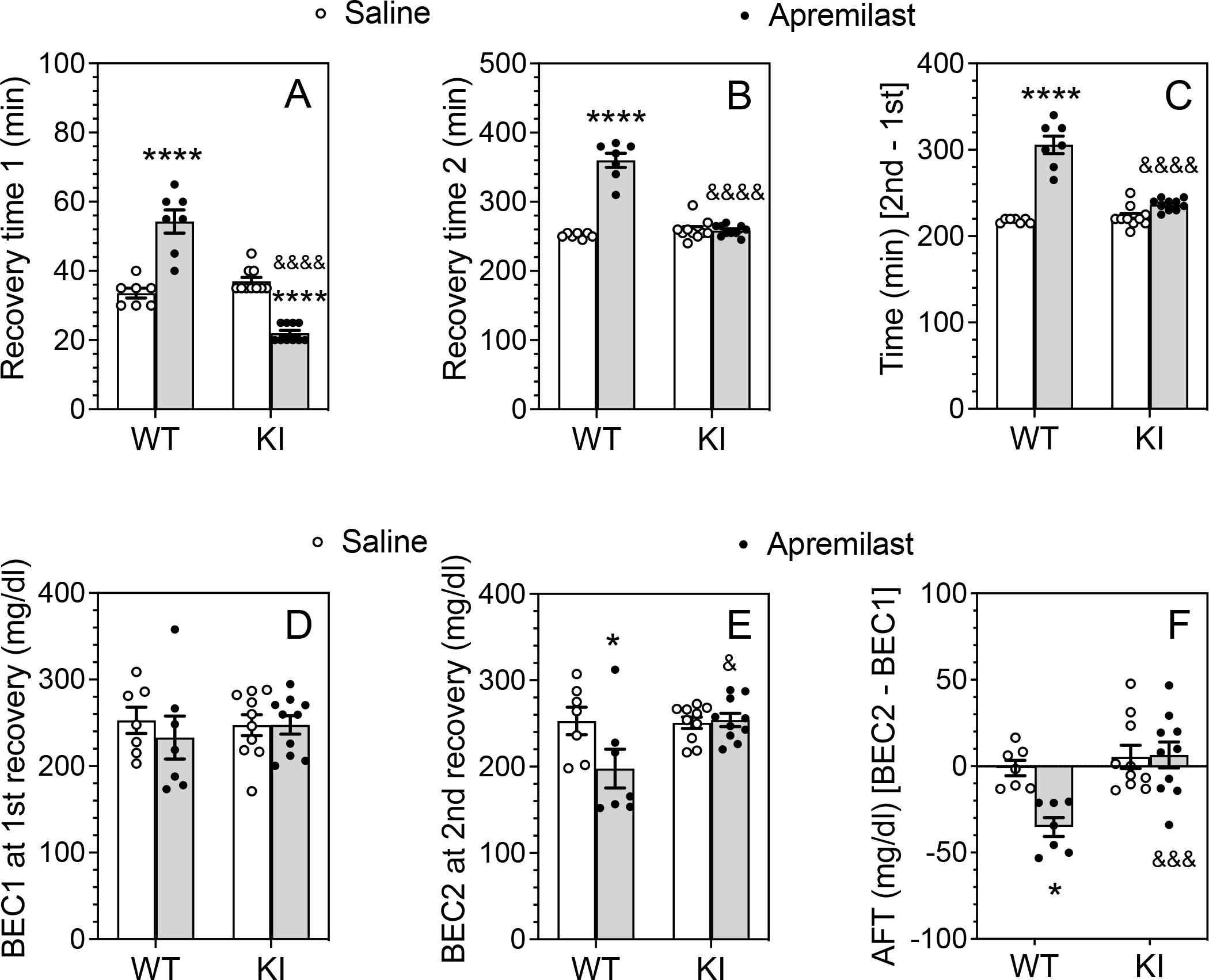
After basal training on the rotarod, male wild-type (WT: n = 7 per group) and β3-S408/409A knock-in (KI: n = 10 per group) mice were pretreated with saline (p.o.) or apremilast (20 mg/kg, p.o) 1h before ethanol injection. (A) Time to regain the ability to remain on the rotarod for 60 s after the first (1.75 g/kg, i.p.) ethanol injection. (B) Time to regain the ability to remain on the rotarod for 60 s after the second (2 g/kg, i.p.) ethanol injection. (C) Difference in recovery time (min) between the second and first ethanol injections. (D) Blood ethanol concentration (BEC1) measured at the time of regaining motor function after the first ethanol injection. (E) BEC2 measured at the time of regaining motor function after the second ethanol injection. (F) Acute functional tolerance (AFT) to ethanol measured as the difference in BECs (mg/dl) after the two ethanol injections (BEC2 - BEC1). * p < 0.05, **** p < 0.0001, apremilast- vs. saline-treated groups of the same genotype; & p < 0.05, &&& p < 0.001, &&&& p < 0.0001, apremilast-treated groups of WT vs. KI mice (Tukey’s multiple comparisons post-hoc tests).
BECs after the first ethanol injection (BEC1 after 1.75 g/kg ethanol) did not differ among the groups (Fig. 4D), but BECs after the second exposure (BEC2 after 2 g/kg ethanol) were decreased only in wild-type mice treated with apremilast (F1,30 = 5.132, p = 0.0309, treatment × genotype interaction; Fig. 4E). Unlike our previous work with B6 male mice (Blednov et al., 2020; Blednov et al., 2018a), the males in the strain used here did not develop AFT in either genotype (p = .8125 for wild-type and p = 0.8203 for knock-in mice treated with saline compared with theoretical median of 0, Wilcoxon tests). There was also no AFT in apremilast-treated knock-in males (p = 0.4316), but in the wild-type, apremilast significantly increased sensitivity to ethanol-induced ataxia (p = 0.0156). Comparison of BEC2-BEC1 values for saline- and apremilast-treated groups also showed that apremilast increased sensitivity to ethanol in wild-type but not in knock-in mice (F1,30 = 6.761, p = 0.0143, treatment × genotype interaction; Fig. 4F).
In female mice, apremilast significantly increased the time to recover from the first ethanol injection in wild-type mice but decreased it in β3-S408/409A mice (F1,42 = 220.5, p < 0.0001, treatment × genotype interaction; Fig. 5A). There were no group differences in BECs at this time (Fig. 5D). After the second ethanol injection (Fig. 5B), apremilast increased the recovery time in wild-type mice but not in β3-S408/409A mice (F1,42 = 198.5, p < 0.0001, treatment × genotype interaction). Upon recovery, BEC2 was reduced in wild-type mice (F1,42 = 9.884, p = 0.0031, genotype) treated with apremilast (F1,42 = 16.95, p = 0.0002, treatment) (Fig. 5E). The difference in recovery time between the second and first ethanol injections (Fig. 5C) was greater in wild-type mice treated with apremilast than in other groups (F1,42 = 107.0, p < 0.0001, treatment × genotype interaction). In addition, AFT (BEC2-BEC1) was decreased only in wild-type mice treated with apremilast (F1,42 = 7.972, p = 0.0072, treatment × genotype interaction; Fig. 5F). In contrast to males, females treated with saline developed AFT (p = 0.0156 for wild-type and p = 0.0068 for knock-in mice compared with a theoretical median of 0, Wilcoxon tests), as did knock-in female mice treated with apremilast (p = 0.0103). In contrast, wild-type female mice treated with apremilast did not develop AFT (p = 0.1484). Collectively, our results in both male and female mice suggest that apremilast prolongs recovery from ataxia in wild-type mice by increasing sensitivity to ethanol in males or by reducing the development of AFT in females, through a process involving PKA-mediated phosphorylation of β3 subunits.
Figure 5. Apremilast reduction in acute functional tolerance to ethanol is disrupted in female β3-S408/409A knock-in mice.
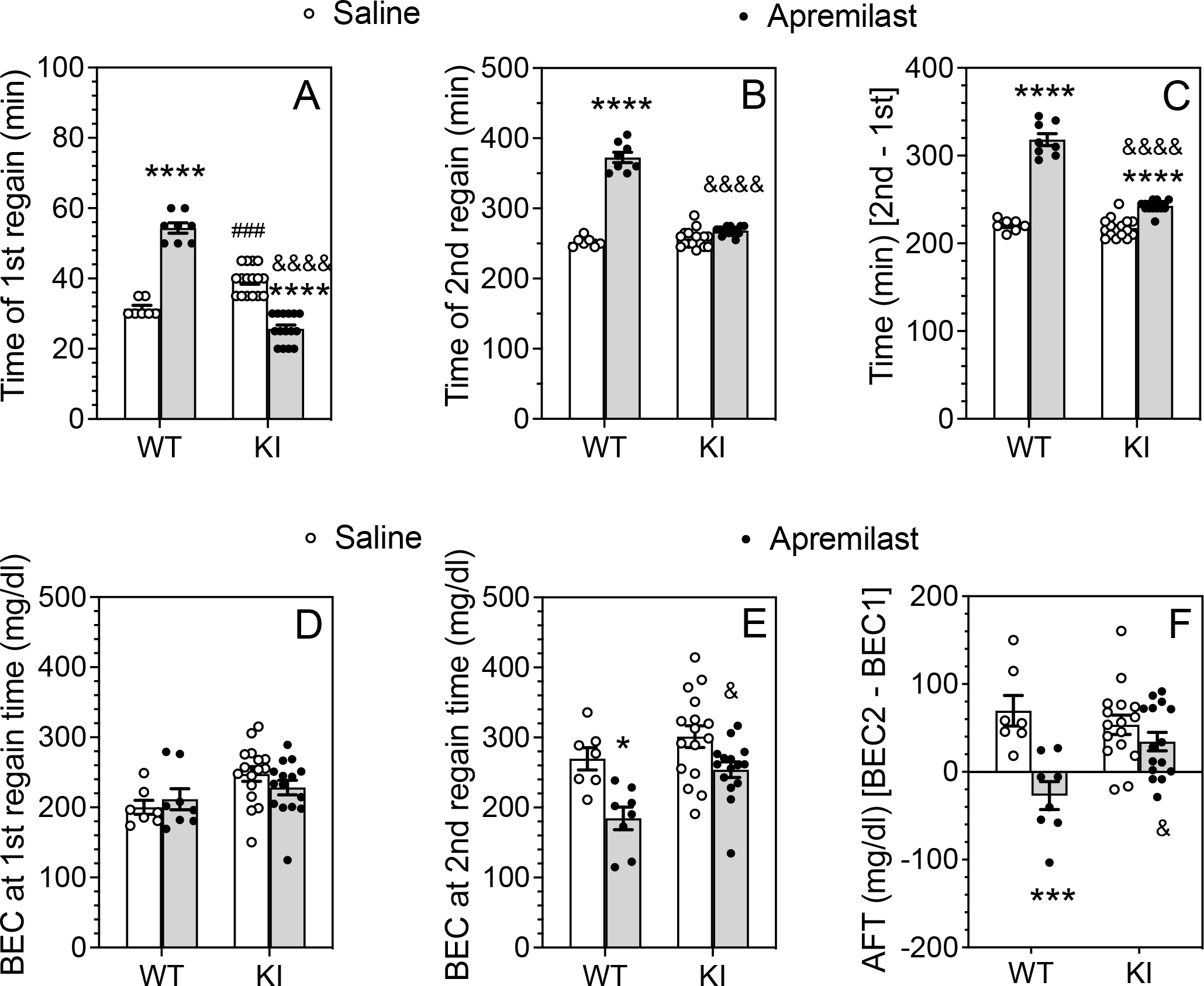
After basal training on the rotarod, female wild-type (WT: n = 7–8 per group) and β3-S408/409A knock-in (KI: n = 15–16 per group) mice were pretreated with saline (p.o.) or apremilast (20 mg/kg, p.o) 1 h before ethanol injection. (A) Time to regain the ability to remain on the rotarod for 60 s after the first (1.75 g/kg) ethanol injection. (B) Time to regain the ability to remain on the rotarod for 60 s after the second (2 g/kg) ethanol injection. (C) Difference in recovery time between the second and first ethanol injections. (D) Blood ethanol concentrations (BECs) measured at the time of regaining motor function after the first ethanol injection. (E) BECs measured at the time of regaining motor function after the second ethanol injection. (F) Acute functional tolerance (AFT) to ethanol measured as the difference in BECs (mg/dl) after the two ethanol injections (BEC2 - BEC1). *** p < 0.001, **** p < 0.0001, apremilast- vs. saline-treated groups of the same genotype. & p < 0.05, &&&& p < 0.0001, apremilast-treated groups of WT vs. KI mice. ### p < 0.001, saline-treated groups of WT (n = 7–8 per group) vs. KI (n = 15–16 per group) mice (Tukey’s multiple comparisons post-hoc tests).
3.5. Voluntary ethanol consumption and preference do not differ between wild-type and β3-S408/409A mice
Because female mice drink more ethanol with higher preference than males and because ethanol concentration and time of exposure also alter consumption, we analyzed the drinking data separately by sex in a 2BC continuous drinking procedure. Wild-type and β3-S408/409A mice were offered increasing concentrations of ethanol (3–15% v/v) with each concentration available for four days. In male (Fig. S1 A–C) and female (Fig. S1 D–F) mice, there were no genotype differences in the amount of ethanol consumed, preference for ethanol, or total fluid intake. Likewise, there were no genotype differences in a 2BC-EOD procedure with 15% (v/v) ethanol in the amount of ethanol consumed, ethanol preference, or total fluid intake in male (Fig. S2 A–C) and female (Fig. S2 D–F) mice.
3.6. Apremilast-mediated inhibition of voluntary ethanol consumption is diminished in β3-S408/409A mice
After undergoing 2BC-EOD drinking, mice from the second experimental group in Fig. S2 were then divided into control and treatment groups to study the effects of apremilast on chronic 2BC-EOD drinking. These data were normalized to the average intake of saline-treated mice of the same genotype. After 2 days of saline injections, basal levels of ethanol intake (g/kg/24h) were similar between genotypes (9.1 ± 0.5 for wild-type and 8.5 ± 0.8 for knock-in male mice; 8.3 ± 0.3 for wild-type and 7.8 ± 0.4 for knock-in female mice). In male mice (Fig. 6A–C), apremilast dose dependently reduced 15% ethanol intake (F5,165 = 34.74, p < 0.0001), and this effect was greater in wild-type than knock-in mice (F1,33 = 5.692, p = 0.0229). Apremilast also dose dependently reduced preference for ethanol (F5,165 = 23.38, p < 0.0001), and the effect was also greater in wild-type mice (F1,33 = 7.596, p = 0.0095). Apremilast slightly decreased total fluid intake in both genotypes (F5,165 = 6.051, p = 0.0002, effect of dose). In female mice (Fig. 6D–F), apremilast dose dependently reduced ethanol intake (F5,180 = 47.22, p < 0.0001) with a greater effect in wild-type mice only at 20 mg/kg apremilast (F5,180 = 3.101, p = 0.0104, dose × genotype interaction). Apremilast also dose dependently reduced ethanol preference (F5,180 = 38.89, p < 0.0001), but there was no dependence on genotype. Apremilast slightly decreased total fluid intake in both genotypes (F5,180 = 5.421, p = 0.0004, effect of dose).
Figure 6. Ability of apremilast to reduce two-bottle choice every-other-day drinking is diminished in β3-S408/409A mice.
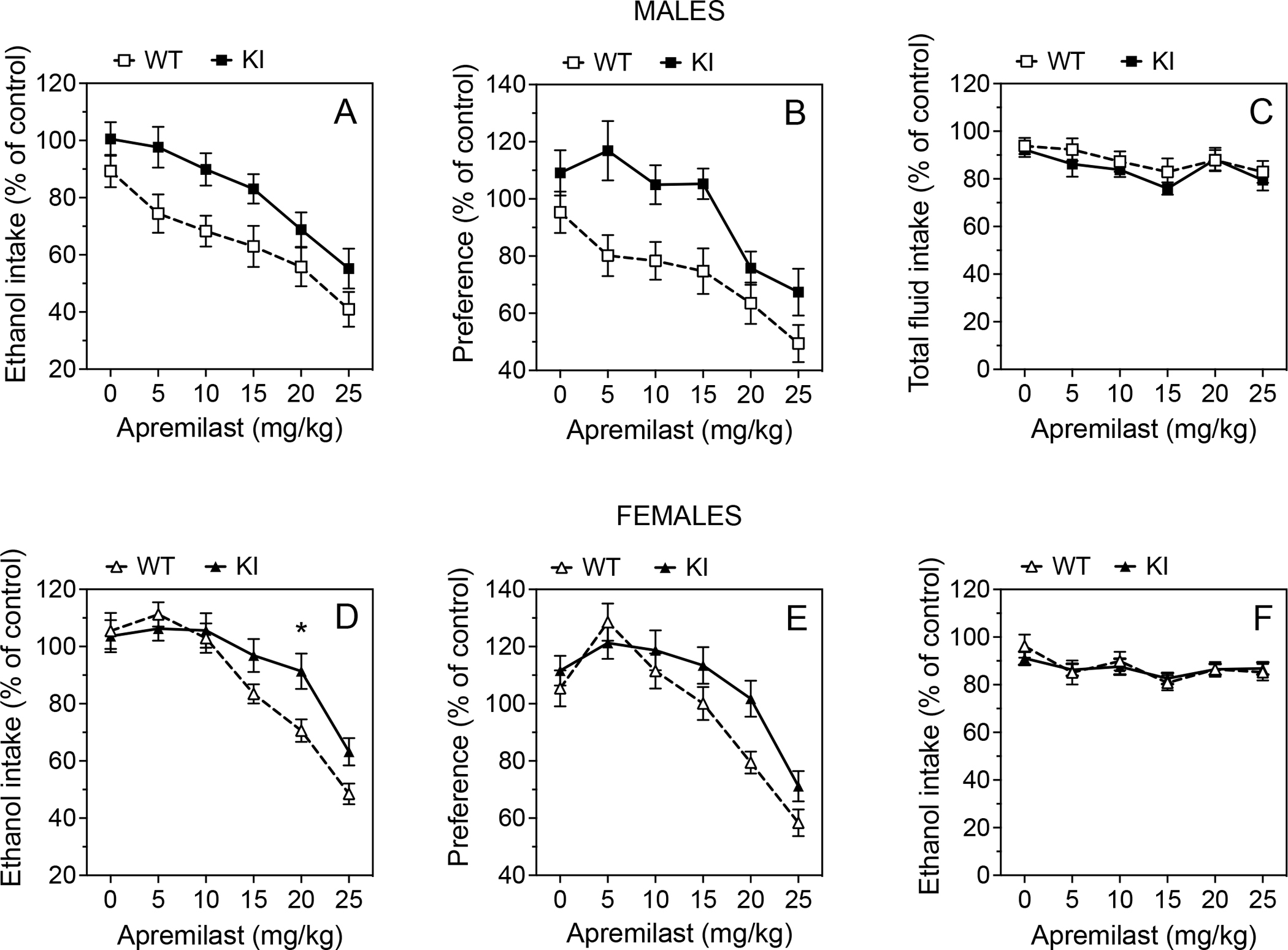
After undergoing 2BC-EOD drinking, mice were divided into matched saline and apremilast treatment groups, and the 2BC-EOD procedure was continued to measure effects of apremilast on the same drinking parameters in the same groups. (A, D) Ethanol (15% v/v) intake (g/kg/24h), (B, E) preference for ethanol, and (C, F) total fluid intake (g/kg/24h) in male (A-C, WT: n = 16 per group; KI: n = 19 per group) and female mice (D-F, WT: n = 16 per group; KI: n = 22 per group). Data were normalized to the average intake of saline-treated mice of the same genotype across the entire study. * p < 0.05, apremilast effect in wild-type (WT) vs. β3-S408/409A knock-in (KI) mice (Tukey’s multiple comparisons post-hoc test).
3.7. Blood ethanol clearance does not differ between wild-type and β3-S408/409A mice
To test the effects of different doses on ethanol clearance from the blood, we examined two ethanol doses (2 g/kg and 4 g/kg) in male (Fig. 7A and B) and female (Fig. 7C and D) mice. The slopes of the regression lines did not differ by dose or genotype in male mice. There was also no genotype difference in female mice, but there was an effect of ethanol dose (F1,26 = 8.891, p = 0.0062) but no dose × genotype interaction. The clearance of 4 g/kg of ethanol in females was slightly faster than the 2 g/kg dose.
Figure 7. Blood ethanol clearance does not differ in wild-type and β3-S408/409A mice.
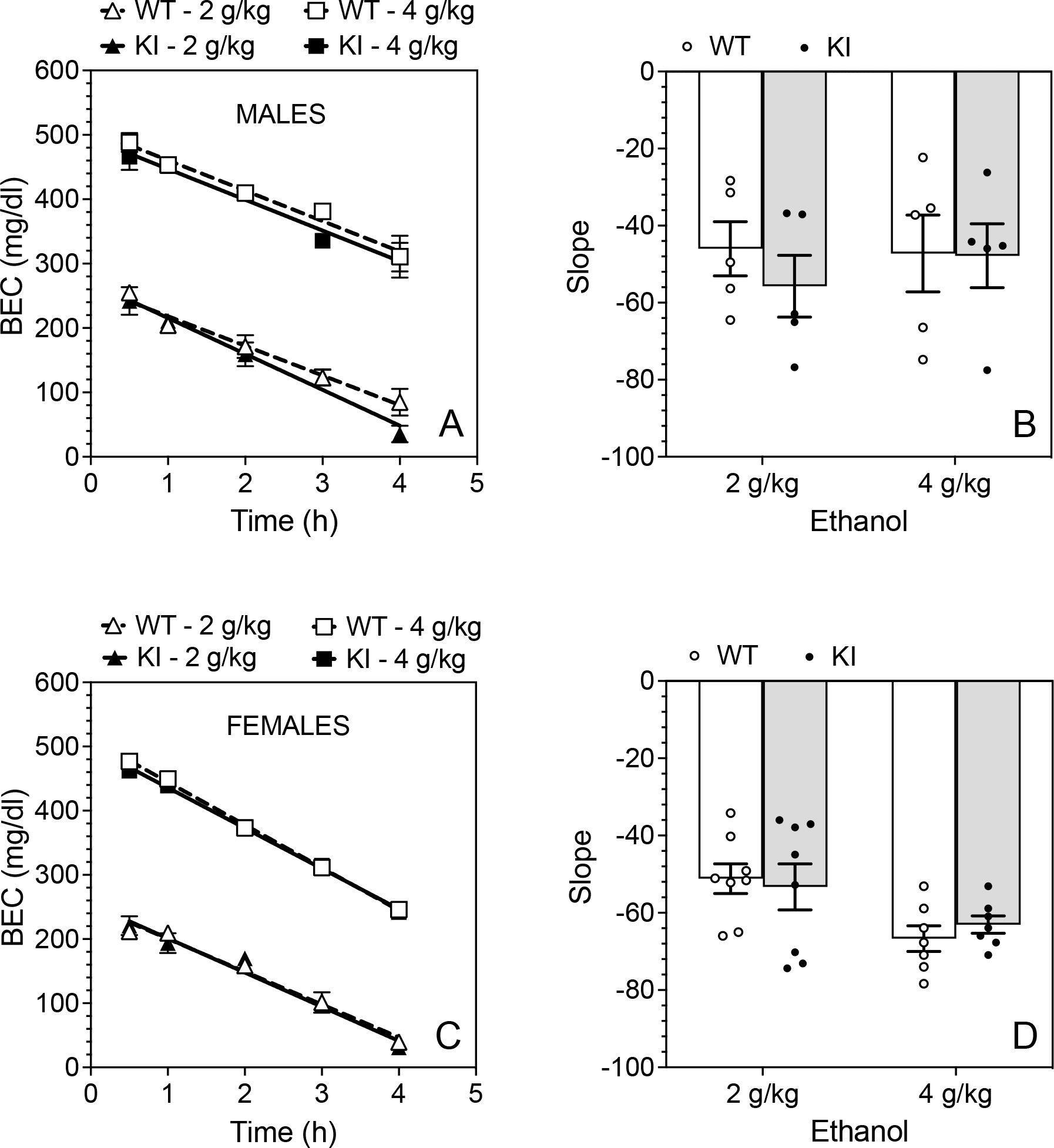
Blood ethanol concentration (BEC) measured over 4 h in (A) male and (C) female wild-type (WT) and β3-S408/409A knock-in (KI) mice after i.p. injection of 2 or 4 g/kg ethanol (n = 5 per group and per genotype for males and n = 7–8 per group and per genotype for females). Slopes of the regression lines in (B) male and (D) female WT and KI mice.
4. DISCUSSION
Our prior studies have shown that apremilast is very effective at reducing alcohol consumption in mice (Blednov et al., 2020; Blednov et al., 2018b), and recent evidence indicates that it is also effective for individuals with AUD (Grigsby et al., 2021). Since AUD has a limited number of pharmacotherapies with limited efficacy, it is important to pursue mechanisms by which apremilast and other PDE4 inhibitors decrease alcohol drinking. Previously, we showed that apremilast profoundly alters the behavioral effects of ethanol and certain GABAA receptor-specific drugs in a PKA-dependent manner in male and female C57BL6/J mice (Blednov et al., 2020). Apremilast produced differential actions depending on the GABAergic drug studied. Based on behavioral pharmacology and functional studies of heterologously expressed GABAA receptors with different phosphorylation states of β1 or β3 subunits, we hypothesized that apremilast’s modulation of ethanol and propofol (but not diazepam) responses was due to PKA phosphorylation of β3-containing GABAA receptors. There are no β3 subunit-selective modulators and no GABAA receptor drugs with sufficient subunit specificity to allow us to directly test this hypothesis. Thus, in the current study, we applied similar pharmacological approaches using β3 knock-in mice with alanine mutations at S408 and S409 to eliminate the sites of PKA phosphorylation (Vien et al., 2015). Our main findings show that the ability of apremilast to increase the ataxic and sedative effects of ethanol is mediated by PKA phosphorylation of the β3 subunit and that this site of action is also important for apremilast’s ability to increase acute sensitivity to ethanol and to decrease ethanol consumption. There were no genotype differences in blood ethanol clearance that could account for these genotype-specific effects of apremilast.
Although we anticipated that the β3-S408/409A mutation would block prolongation of ethanol ataxia by apremilast, we found instead this mutation surprisingly caused apremilast to accelerate recovery. Interestingly, this effect was also due to PKA activation because the PKA inhibitor H-89 prevented it. H-89 can inhibit other kinases (Limbutara et al., 2019; Lochner and Moolman, 2006), but the reversal of PDE4 inhibition by apremilast, which is selective for cAMP and activation of PKA, makes it likely that the predominant effect of H-89 in our study is inhibition of PKA. We propose that apremilast simultaneously induces opposing effects on ethanol-induced ataxia and that prolonged ataxia in wild-type mice (mediated by PKA phosphorylation of β3 subunits) is the predominant mechanism. When PKA-dependent phosphorylation of β3 subunits is blocked by the β3-S408/409A mutation, apremilast-induced PKA phosphorylation of other GABAA receptor subunits is revealed, leading to accelerated recovery from ataxia.
Because apremilast profoundly increases the sensitivity to acute alcohol responses in wild-type animals, we expected it to interfere with the development of AFT to an intoxicating effect of ethanol. As expected, and in agreement with our prior work, apremilast decreased AFT to ethanol-induced ataxia in wild-type (but not knock-in) female mice. Wild-type males behaved differently than B6 males shown in our previous work (Blednov et al., 2020; Blednov et al., 2018a). For example, the current strain of male mice did not develop AFT and drank less ethanol than B6 males normally consume in a continuous two-bottle choice procedure. Since development of AFT in mice depends strongly on the genetic background (Hu et al., 2008), any underlying genetic differences between males and females in the colony generated for this study could account for differences in development of AFT. Apremilast was still able to sensitize wild-type (but not knock-in) male mice to ethanol-induced ataxia. Using β3-S408/409A mice, which were insensitive to apremilast treatment in both males and females, we were able to show that the ability of apremilast to increase sensitivity to ethanol in males or to reduce AFT in females depends on PKA phosphorylation of the β3 subunit.
These responses were accompanied by robust decreases in voluntary alcohol intake in wild-type mice. In the 2BC-EOD procedure, apremilast decreased chronic ethanol consumption and preference in a dose-dependent manner. Compared with wild-type mice, the effect of apremilast was reduced, but not eliminated, in the mutant mice. There was no significant effect of genotype in female mice, suggesting that PKA phosphorylation of β3 subunits by apremilast could have less impact on drinking in females. Overall, our findings demonstrate an important role for β3 phosphorylation in apremilast regulation of chronic drinking (as in acute responses to ethanol), but actions at additional PKA-regulated and alcohol-sensitive receptors likely account for its residual effect in the β3-S408/409A mice. For example, PKA can phosphorylate α1 subunits of glycine receptors (Breitinger et al., 2018) as well as α4 and α7 subunits of nicotinic acetylcholine receptors (McDaid et al., 2016; Miller et al., 2018). The β3 subunit is also implicated in the development of alcohol tolerance and dependence. For example, loss of sensitivity to propofol in a propofol-hyposensitive β3 knock-in mouse model was accompanied by increased tolerance to chronic ethanol and increased severity of ethanol withdrawal, though there was little change in acute ethanol sensitivity (Sanchis-Segura et al., 2007). Also, polymorphisms in genes encoding β3 and β1 subunits, which are also phosphorylated by PKA, have been associated with alcohol dependence in humans (Song et al., 2003).
Further understanding of apremilast’s mechanism of action comes from our study of GABAergic drugs besides ethanol. For example, the prolongation of propofol ataxia and sedation by apremilast was completely blocked in β3-S408/409A mice, indicating that it is dependent on PKA phosphorylation of β3. Although propofol shows no selectivity for β subunits in trimeric receptors (Rudolph and Antkowiak, 2004), PKA activation only phosphorylates β1 and β3, but not β2 subunits (McDonald et al., 1998). By mutating the PKA site on β3 and eliminating apremilast action, we provide strong evidence that β3 phosphorylation is also responsible for apremilast modulation of propofol responses in vivo. As demonstrated in HEK293 cells (McDonald et al., 1998) and in Xenopus oocytes (Blednov et al., 2020), PKA phosphorylation of β3-containing receptors increases inhibitory GABAA receptor-induced currents, which would be expected to increase the effects of propofol and ethanol, as we have consistently observed after apremilast treatment in our previous and current studies. Thus, our results in β3-S408/409A mice support the hypothesis that PKA phosphorylation of β3 subunits is critical for the acute in vivo effects of propofol and ethanol.
In contrast, the faster recovery from ataxia or sedation by diazepam after administration of apremilast does not depend on this mechanism but does depend on PKA activation (Blednov et al., 2020). Based on our results showing that the β1-specific antagonist salicylidene salicylhydrazide can mimic the effect of apremilast on diazepam-induced ataxia (Blednov et al., 2020) and that PKA phosphorylation of β1-containing receptors decreases inhibitory GABAA receptor-induced currents (Blednov et al., 2020; Moss et al., 1992), we speculated that PKA phosphorylation of β1 subunits is a potential mechanism by which apremilast accelerates recovery from diazepam. Current studies using β1-S409A mice are underway to explore this possibility. Our current findings showing that β3 subunits are not involved support this possibility. We also suggest that the behavioral effects mediated by phosphorylation of β1 vs. β3 GABAA receptor subunits are functionally independent.
Recently, evidence indicates that PKA also phosphorylates S359 of the α2 subunit (Nakamura et al., 2020), which decreases the interaction of α2 with gephyrin and collybistin. This reduces the density of synaptic α2-containing GABAA receptor clusters and blocks α2 enrichment at the axon initial segment of cultured cortical and hippocampal neurons, potentially inhibiting GABAA receptor activity. Diazepam acts on GABAA receptors containing α1, α2, α3, or α5 in combination with γ2 subunits (efficacy α3 > α2 > α1 ~ α5) (Sieghart and Savic, 2018). We previously found that α subunits can influence PKA modulation of GABAA receptor function as well as apremilast modulation of GABAergic drugs in vivo (Blednov et al., 2020). As we proposed with β1 subunits, PKA phosphorylation of α2-containing GABAA receptors may be another mechanism by which apremilast decreases neuronal inhibition by diazepam. This may also be important for the faster recovery from ethanol ataxia in β3 mutant mice after apremilast pretreatment, and we know that the α2 subunit plays a role in acute sensitivity to ethanol intoxication since Gabra2 knockout mice show faster recovery from ataxia induced by ethanol or the benzodiazepine flurazepam compared with wild-type littermates (Blednov et al., 2012). We propose that the net in vivo effects of apremilast on ethanol responses may be explained by dominant actions at β3-containing GABAA receptors, while actions at α2- and β1-containing receptors reduce GABAA receptor channel function and limit the ability of apremilast to increase sensitivity to acute alcohol intoxication and reduce alcohol consumption. Apremilast may also modulate propofol responses predominantly by phosphorylation of β3-containing GABAA receptors, while diazepam modulation may occur mainly by phosphorylation of α2- and β1-containing receptors.
To evaluate the differential effects of apremilast, we must also consider that there are 4 isoforms of PDE4 (A-D), each with several splice variants (Richter et al., 2013). All except PDE4C are widely distributed in the brain, especially in the prefrontal cortex, hippocampus, amygdala, and nucleus accumbens, which are regions that regulate rewarding and affective behaviors. The PDE4B gene has become a subject of major interest in the alcohol field because several large-scale human genome-wide association studies found an association between it and alcohol use, although the mechanism behind this association is not yet known (Clarke et al., 2017; Liu et al., 2019; Zhou et al., 2020). Like other PDE4 inhibitors approved for human use, apremilast is nonselective and inhibits multiple isoforms (A1, B1, B2, C1, and D2) (Schafer et al., 2014). The regional distribution and subcellular compartmentalization of each PDE4 isoform suggest that they serve distinct roles in the brain and provide a basis for the separation of therapeutic and adverse effects of PDE4 inhibitors (Fox et al., 2014). For example, Pde4a or Pde4b knockout mice display increased anxiety-like behavior and may have impaired emotional memory (Hansen et al., 2014; Zhang et al., 2008), while Pde4d knockout mice exhibit improved memory (Zhang et al., 2002). Furthermore, selective pharmacological inhibitors of PDE4B (A33) or PDE4D (D159687) produce distinct behavioral effects in mice; A33 has an antidepressant-like profile, whereas D159687 has a pro-cognitive profile (Zhang et al., 2017). Although it is not known if or how different PDE4 isoforms regulate behavioral responses to ethanol, we cannot rule out the possibility that they differentially regulate the phosphorylation of β1 and β3 GABAA receptor subunits. These investigations are currently underway in our lab.
In summary, PKA phosphorylation of β3-containing GABAA receptors by apremilast increases the acute ataxic and sedating effects of ethanol intoxication, decreases AFT to ethanol in females or increases sensitivity to ethanol in males, and decreases ethanol consumption. Our results provide a possible mechanism for how apremilast acts on these three interconnected behaviors. In individuals with normal alcohol metabolism, differences in sensitivity to ataxic and sedative effects of alcohol appear to result mainly from differences in the development of AFT (behavioral tolerance) (Crabbe et al., 2006). In humans, a lower response to the aversive effects of an acute alcohol challenge may contribute to risk of heavy drinking (King et al., 2011). Our work has consistently shown that apremilast-induced increases in sensitivity to the ataxic and sedating effects of an acute alcohol challenge are associated with decreased ethanol consumption, and there is now evidence that apremilast significantly reduces drinking in human subjects with AUD (Grigsby et al., 2021). We propose that apremilast, or other more selective PDE4 inhibitors, may effectively target alcohol drinking behaviors through PKA modulation of GABAergic signaling.
Supplementary Material
Funding:
This work was supported by the National Institutes of Health grants U01 AA013520 to YAB and ROM and U24 AA025479 to ROM.
Footnotes
Declarations of interest: None.
Yuri Blednov: Conceptualization, Validation, Formal Analysis, Investigation, Writing-Original Draft, Writing-Review and Editing, Visualization, Project Administration, Funding Acquisition. Adriana Da Costa: Investigation. Sonia Mason: Investigation. Jody Mayfield: Writing-Original Draft, Writing-Review and Editing. Stephen Moss: Writing-Review and Editing. Robert Messing: Conceptualization, Formal Analysis, Writing-Original Draft, Writing-Review and Editing, Supervision, Funding Acquisition.
REFERENCES
- Blednov YA, Benavidez JM, Black M, Harris RA, 2014. Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front Neurosci 8, 129. 10.3389/fnins.2014.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Borghese CM, Dugan MP, Pradhan S, Thodati TM, Kichili NR, Harris RA, Messing RO, 2020. Apremilast regulates acute effects of ethanol and other GABAergic drugs via protein kinase A-dependent signaling. Neuropharmacology 178, 108220. 10.1016/j.neuropharm.2020.108220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Da Costa AJ, Harris RA, Messing RO, 2018a. Apremilast Alters Behavioral Responses to Ethanol in Mice: II. Increased Sedation, Intoxication, and Reduced Acute Functional Tolerance. Alcohol Clin Exp Res 42, 939–951. 10.1111/acer.13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Da Costa AJ, Tarbox T, Ponomareva O, Messing RO, Harris RA, 2018b. Apremilast Alters Behavioral Responses to Ethanol in Mice: I. Reduced Consumption and Preference. Alcohol Clin Exp Res 42, 926–938. 10.1111/acer.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Mayfield RD, Belknap J, Harris RA, 2012. Behavioral actions of alcohol: phenotypic relations from multivariate analysis of mutant mouse data. Genes Brain Behav 11, 424–435. 10.1111/j.1601-183X.2012.00780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Carlson SL, Morrow AL, 2016. Differential regulation of synaptic and extrasynaptic alpha4 GABA(A) receptor populations by protein kinase A and protein kinase C in cultured cortical neurons. Neuropharmacology 105, 124–132. 10.1016/j.neuropharm.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitinger U, Bahnassawy LM, Janzen D, Roemer V, Becker CM, Villmann C, Breitinger HG, 2018. PKA and PKC modulators affect ion channel function and internalization of recombinant alpha1 and alpha1-beta glycine receptors. Front Mol Neurosci 11, 154. 10.3389/fnmol.2018.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SL, Bohnsack JP, Patel V, Morrow AL, 2016. Regulation of Extrasynaptic GABAA alpha4 Receptors by Ethanol-Induced Protein Kinase A, but Not Protein Kinase C Activation in Cultured Rat Cerebral Cortical Neurons. J Pharmacol Exp Ther 356, 148–156. 10.1124/jpet.115.228056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TK, Adams MJ, Davies G, et al. , 2017. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N=112 117). Mol Psychiatry 22, 1376–1384. 10.1038/mp.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Ponomarev I, Prescott CA, Wahlsten D, 2006. Effects of genetic and procedural variation on measurement of alcohol sensitivity in mouse inbred strains. Behav Genet 36, 536–552. 10.1007/s10519-006-9067-6. [DOI] [PubMed] [Google Scholar]
- Erwin VG, Deitrich RA, 1996. Genetic selection and characterization of mouse lines for acute functional tolerance to ethanol. J Pharmacol Exp Ther 279, 1310–1317. [PubMed] [Google Scholar]
- Fox D 3rd, Burgin AB, Gurney ME, 2014. Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell Signal 26, 657–663. 10.1016/j.cellsig.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KM, Hauser SR, Lasek AW, McClintick J, Ding ZM, McBride WJ, Bell RL, 2015. Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting phosphodiesterase-4 (PDE4). Psychopharmacology (Berl) 232, 2251–2262. 10.1007/s00213-014-3852-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigsby KB, Mangieri RA, Roberts AJ, et al. , 2021. The FDA-approved drug apremilast suppresses alcohol intake: clinical and pre-clinical validation. bioRxiv 2021.05.13.444033. doi: 10.1101/2021.05.13.444033. [DOI] [Google Scholar]
- Hansen RT 3rd, Conti M, Zhang HT, 2014. Mice deficient in phosphodiesterase-4A display anxiogenic-like behavior. Psychopharmacology (Berl) 231, 2941–2954. 10.1007/s00213-014-3480-y. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y, 1984. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry 23, 5036–5041. 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Hu W, Lu T, Chen A, Huang Y, Hansen R, Chandler LJ, Zhang HT, 2011. Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology (Berl) 218, 331–339. 10.1007/s00213-011-2290-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Saba L, Kechris K, Bhave SV, Hoffman PL, Tabakoff B, 2008. Genomic insights into acute alcohol tolerance. J Pharmacol Exp Ther 326, 792–800. 10.1124/jpet.108.137521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AC, de Wit H, McNamara PJ, Cao D, 2011. Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch Gen Psychiatry 68, 389–399. 10.1001/archgenpsychiatry.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Ren Q, Beckley JH, O’Buckley TK, Gigante ED, Santerre JL, Werner DF, Morrow AL, 2012. Ethanol Activation of Protein Kinase A Regulates GABA(A) Receptor Subunit Expression in the Cerebral Cortex and Contributes to Ethanol-Induced Hypnosis. Front Neurosci 6, 44. 10.3389/fnins.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Messing RO, 2008. Protein kinases and addiction. Ann N Y Acad Sci 1141, 22–57. 10.1196/annals.1441.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbutara K, Kelleher A, Yang CR, Raghuram V, Knepper MA, 2019. Phosphorylation Changes in Response to Kinase Inhibitor H89 in PKA-Null Cells. Sci Rep 9, 2814. 10.1038/s41598-019-39116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Jiang Y, Wedow R, et al. , 2019. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 51, 237–244. 10.1038/s41588-018-0307-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hao PD, Yang MF, Sun JY, Mao LL, Fan CD, Zhang ZY, Li DW, Yang XY, Sun BL, Zhang HT, 2017. The phosphodiesterase-4 inhibitor roflumilast decreases ethanol consumption in C57BL/6J mice. Psychopharmacology (Berl) 234, 2409–2419. 10.1007/s00213-017-4631-8. [DOI] [PubMed] [Google Scholar]
- Lochner A, Moolman JA, 2006. The many faces of H89: a review. Cardiovasc Drug Rev 24, 261–274. 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- Logrip ML, 2015. Phosphodiesterase regulation of alcohol drinking in rodents. Alcohol 49, 795–802. 10.1016/j.alcohol.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaid J, Abburi C, Wolfman SL, Gallagher K, McGehee DS, 2016. Ethanol-induced motor impairment mediated by inhibition of alpha7 nicotinic receptors. J Neurosci 36, 7768–7778. 10.1523/JNEUROSCI.0154-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG, 1998. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat Neurosci 1, 23–28. 10.1038/223. [DOI] [PubMed] [Google Scholar]
- Melendez RI, 2011. Intermittent (every-other-day) drinking induces rapid escalation of ethanol intake and preference in adolescent and adult C57BL/6J mice. Alcohol Clin Exp Res 35, 652–658. 10.1111/j.1530-0277.2010.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Wilson RS, Lam TT, Nairn AC, Picciotto MR, 2018. Evaluation of the phosphoproteome of mouse alpha 4/beta 2-containing nicotinic acetylcholine receptors in vitro and in vivo. Proteomes 6. 10.3390/proteomes6040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Smart TG, Blackstone CD, Huganir RL, 1992. Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science 257, 661–665. 10.1126/science.1323140. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Morrow DH, Nathanson AJ, Henley JM, Wilkinson KA, Moss SJ, 2020. Phosphorylation on Ser-359 of the alpha2 subunit in GABA type A receptors down-regulates their density at inhibitory synapses. J Biol Chem 295, 12330–12342. 10.1074/jbc.RA120.014303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton PM, Messing RO, 2006. Intracellular signaling pathways that regulate behavioral responses to ethanol. Pharmacol Ther 109, 227–237. 10.1016/j.pharmthera.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Richter W, Menniti FS, Zhang HT, Conti M, 2013. PDE4 as a target for cognition enhancement. Expert Opin Ther Targets 17, 1011–1027. 10.1517/14728222.2013.818656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwasser AM, Fixaris MC, Crabbe JC, Brooks PC, Ascheid S, 2013. Escalation of intake under intermittent ethanol access in diverse mouse genotypes. Addict Biol 18, 496–507. 10.1111/j.1369-1600.2012.00481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B, 2004. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 5, 709–720. 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Sanchis-Segura C, Cline B, Jurd R, Rudolph U, Spanagel R, 2007. Etomidate and propofol-hyposensitive GABAA receptor beta3(N265M) mice show little changes in acute alcohol sensitivity but enhanced tolerance and withdrawal. Neurosci Lett 416, 275–278. 10.1016/j.neulet.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Schafer PH, Parton A, Capone L, Cedzik D, Brady H, Evans JF, Man HW, Muller GW, Stirling DI, Chopra R, 2014. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal 26, 2016–2029. 10.1016/j.cellsig.2014.05.014. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Savic MM, 2018. International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans. Pharmacol Rev 70, 836–878. 10.1124/pr.117.014449. [DOI] [PubMed] [Google Scholar]
- Song J, Koller DL, Foroud T, et al. , 2003. Association of GABA(A) receptors and alcohol dependence and the effects of genetic imprinting. Am J Med Genet B Neuropsychiatr Genet 117B, 39–45. 10.1002/ajmg.b.10022. [DOI] [PubMed] [Google Scholar]
- Vien TN, Modgil A, Abramian AM, Jurd R, Walker J, Brandon NJ, Terunuma M, Rudolph U, Maguire J, Davies PA, Moss SJ, 2015. Compromising the phosphodependent regulation of the GABAAR beta3 subunit reproduces the core phenotypes of autism spectrum disorders. Proc Natl Acad Sci U S A 112, 14805–14810. 10.1073/pnas.1514657112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen RT, Zhang FF, Zhang HT, 2018. Cyclic nucleotide phosphodiesterases: potential therapeutic targets for alcohol use disorder. psychopharmacol 235, 1793–1805. 10.1007/s00213-018-4895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen RT, Zhang M, Qin WJ, Liu Q, Wang WP, Lawrence AJ, Zhang HT, Liang JH, 2012. The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring Fawn-Hooded rats. Alcohol Clin Exp Res 36, 2157–2167. 10.1111/j.1530-0277.2012.01845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, O’Donnell JM, 2002. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology 27, 587–595. 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, Dlaboga D, Jin SL, Conti M, O’Donnell JM, 2008. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology 33, 1611–1623. 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen L, Beck EM, et al. , 2017. The Discovery of a Novel Phosphodiesterase (PDE) 4B-Preferring Radioligand for Positron Emission Tomography (PET) Imaging. J Med Chem 60, 8538–8551. 10.1021/acs.jmedchem.7b01050. [DOI] [PubMed] [Google Scholar]
- Zhou H, Sealock JM, Sanchez-Roige S, et al. , 2020. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci 23, 809–818. 10.1038/s41593-020-0643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.