

Abstract
Minimal Residual disease (MRD) in acute myeloid leukemia (AML) is typically measured using multi-parameter flow cytometry (MFC). Detection of leukemia mutations using multi-gene next generation sequencing (NGS) can potentially be used to measure residual disease. We used a targeted 28-gene NGS panel to detect mutations and different-from-normal 10-colour MFC to measure MRD in AML patients before allogeneic hematopoietic stem cell transplant (HCT). Residual disease was defined when any abnormal blast population was detected using MFC and when any leukemia allele was detected with a variant allele frequency (VAF) ≥5% using NGS. We tracked the clearance of leukemia alleles between AML diagnosis and immediately before HCT and found that mutations in DNMT3A, TET2 and JAK2 were less likely to be cleared than NPM1, IDH 1/2 and FLT3-ITD. Despite varying sensitivities, the concordance rate of residual disease detection before HCT using the two assays was 44/62 (71%) evaluable cases. Discordance could be explained by residual mutations in DNMT3A and TET2 that were not detected by MFC and presence of residual leukemia mutations with VAF below the established thresholds for mutation calling. Presence of flow MRD and residual mutations immediately before HCT using the two assays was associated with relapse risk (MFC: HR=4.62 95%CI 1.32–16.09, p=0.016 and NGS: HR=4.35 95%CI 1.63–11.6, p=0.003) and survival (MFC: HR=2.44 95%CI 1–5.97, p=0.05 and NGS: HR=2.1 95%CI 0.97–4.55, p=0.059) after HCT. Residual disease detected concurrently by MFC and NGS conferred the highest relapse risk compared to patients who were either negative by both assays or had discordant status (overall, p=0.008). While MFC is universally applicable, a multi-gene NGS approach to measuring residual disease in AML provides additional information on differential clearance of disease alleles and can assess clonal architecture before transplant.
Keywords: minimal residual disease, flow cytometry, next generation sequencing, Acute myeloid leukemia
Introduction:
Next generation sequencing (NGS) has shown that acute myeloid leukemia (AML) is a genetically heterogeneous disease composed of multiple clones each characterized by combinations of somatic molecular mutations. Sequencing at various time points highlights a specific order in which mutations are acquired. Founder mutations typically occur in genes involved in epigenetic regulation (DNMT3A, TET2, ASXL1). While not enough to cause leukemia on their own, these mutations confer replicative advantage to affected hematopoietic stem cells, a state referred to as clonal hematopoiesis of indeterminate potential (CHIP)1. Secondary genetic events typically arising in genes involved in signal transduction (eg. FLT3, RAS) have the capacity to transform these cells into AML1. Tracking these leukemia alleles can be used as a method of monitoring treatment response.
Studies that track disease specific mutations as markers of AML treatment response have shown differences in the probability of clearing clones characterized by specific mutations2,3. In these studies, mutations in DNMT3A and TET2 had a lower probability of clearance, while NPM1, FLT3 and IDH1/2 mutations were more likely to be cleared. Despite this, detection of any residual mutation following induction chemotherapy was predictive of relapse and survival4. The differential clearance of leukemia alleles results in a potential bias when residual disease is measured by tracking single mutations like NPM15. PCR based assays, which track single alleles are not applicable to all patients given that leukemia mutations vary in incidence and can arise in various regions of affected genes making it difficult to design universally applicable PCR assays. Multi-gene NGS has the potential to overcome the limitations of single-gene PCR techniques. Detection of residual mutations at the time of HCT using multi-gene NGS has never been correlated with outcome in AML patients undergoing HCT.
Multi-parameter flow cytometry (MFC) leverages aberrant antigen expression on leukemic blasts to detect residual disease and has emerged as a powerful predictor of outcome among patients with AML undergoing HCT6,7. A different-from-normal approach may be universally applicable; however, demands analytical and technical expertise7,8. This method is imperfect with approximately 20% of MFC negative patients relapsing and a similar percentage of positive patients remaining in remission after transplant7. Detection of residual disease by MFC has never been correlated with detection of residual mutations by multi-gene NGS.
Methods:
Study cohort:
104 AML patients who underwent allogeneic transplant at our center between 2014 and 2015 were included. Patients were included if NGS was performed at AML diagnosis (n=58), or if NGS (n=83) or MFC (n=77) was performed immediately before HCT. Not all patients had available samples at all time points (table 1). AML was defined according to WHO AML classification and disease status was defined per the international working group criteria9. Relapse was defined as presence of >5% blasts on marrow aspirate. One transplant event per patient was allowed with preference given to events that had residual disease assessment. The median time between the pre-HCT assessment time-point and transplantation was 22 days with no intervening therapy administered between these times. Tandem assessment of residual disease by multi-gene NGS and MFC was performed on the same marrow aspirate in 62 patients who had available samples. Concordant assessment was defined by detection of an abnormal blast population and presence of leukemia mutation(s) with VAF ≥5% (F+M+) or absence of abnormal blasts and absence of mutations (F−M−). Discordant results occurred when one assay was positive and the other was negative (F+M− or F−M+). Disease risk index10, HCT-CI comorbidity score11 and conditioning intensity12,13 were defined according to published guidelines. The institutional review board approved the study and consent was obtained from all participants.
Table 1:
Characteristics of all patients included in the analysis
| Characteristic | N (%) |
|---|---|
| N | 104 |
| Men (%) | 51 (49%) |
| Median age at transplant, years (range) | 58 (21–78) |
| Number of patients with molecular and flow analysis | |
| Molecular analysis at diagnosis | 58 (55.8) |
| Molecular analysis pre-HCT | 83 (79.8) |
| Paired molecular analysis at diagnosis and pre-HCT | 47 (45.2) |
| Flow MRD analysis pre-HCT | 77 (74.0) |
| Paired flow and molecular analysis pre-HCT | 62 (59.6) |
| AML characteristics | |
| Primary AML | 65 (62.5) |
| Secondary AML, prior disease | 39 (37.5) |
| Prior MDS | 21 |
| Prior MPN | 7 |
| Prior CMML | 0 |
| Therapy related AML ^ | 15 |
| CIBMTR Cytogenetics Grouping | |
| Favorable | 2 (1.9) |
| Intermediate | 88 (84.6) |
| Adverse | 14 (13.5) |
| Remission status at time of HCT | |
| CR1 | 59 (56.7) |
| CR2 | 9 (8.7) |
| CR3 | 1 (1.0) |
| CRi | 21 (20.2) |
| Not in CR | 14 (13.5) |
| DRI | |
| Low | 2 (1.9) |
| Intermediate | 71 (68.3) |
| High | 30 (28.8) |
| Very high | 1 (1.0) |
| HCT-CI | |
| <3 | 51 (49) |
| ≥3 | 53 (51) |
| Stem cell source | |
| Peripheral blood stem cells | 73 (70.2) |
| Bone marrow derived stem cells | 12 (11.5) |
| Cord blood | 19 (18.3) |
| CD34+ selection | 29 (27.9) |
| HLA match # | |
| Fully matched | 72 (69.2) |
| Mismatched | 32 (30.8) |
| Conditioning intensity | |
| Ablative | 59 (56.7) |
| Reduced intensity | 45 (43.3) |
| Transplant | |
| First | 95 (91.3) |
| Second | 9 (8.7) |
| Outcomes | |
| Relapse, N (%) | 22 (21.2) |
| Deceased, N (%) | 36 (34.6) |
| Cause of death | |
| Relapse | 13 (12.5) |
| Infection | 10 (9.6) |
| GVHD | 6 (5.8) |
| Toxicity | 2 (1.9) |
| Organ failure | 4 (3.8) |
| Unknown | 1 (0.8) |
2 patients developed therapy related AML after initial diagnosis with therapy related MDS
15 patients did not have available FLT3 mutation testing in the setting of a normal karyotype and thus could not be categorized by the NCCN AML classification.
Number of mismatches at high resolution at HLA class 1 (A, B, C) and class 2 (DR and DQ). All cord blood transplants were mismatched.
Multi-gene NGS
A 28-gene amplicon, capture based NGS assay, designed to identify mutations seen in myeloid neoplasms, was used. Sequenced gene hot-spots are described in the supplementary table 1. Details of this assay and validation have been published14 and are outlined below.
Sample processing:
DNA was extracted from bone marrow aspirates using Qiagen DNAeasy blood kit (Quiagen, Venlo, Netherlands). DNA (500ng) was fragmented using an E-series DNA Sonicator (Covaris, Woburn, MA) and purified using Quiagen MinElute Clean-up (Qiagen). The extracted template DNA and custom primers were merged using the RainDance Thunderstorm instrument to form droplets for amplification (RainDance Technologies, Billerica, MA). Emulsion PCR was performed (RainDance Technologies) using RainDance Droplet Stabilizer and Platinum High Fidelity Taq (Life Technologies; following vendor protocol). Adaptor tails were added to the 5’ ends of the sequence-specific regions of amplicon primers to facilitate secondary PCR. The emulsion was broken and PCR products were recovered using RainDance Droplet Destabilizer (RainDance Technologies) and Qiagen MinElute Clean-up. 10ng of this amplified library was sampled (Agilent Technologies, Santa Clara, CA) for secondary PCR. Custom designed reverse primers (Integrated DNA Technologies, Coralville, IA) with integrated unique Illumina Truseq barcodes and universal forward primers were used for secondary PCR. The product was recovered using Qiagen MinElute Clean-up and 15ng of this library product was sampled using Agilent BioAnalyzer system Agilent. The library pool was quantified using a Qubit dsDNA Broad Range Assay kit, diluted and loaded into a MiSeq 300 cycle reagent kit (Illumina, San Diego, CA) for sequencing. Custom sequencing primers from Integrated DNA Technologies were loaded into the MiSeq cartridge for forward and turn-around read for 150-base pair paired-end sequencing. Sequencing was performed using a Illumina MiSeq v.2 instrument (Illumina) with Illumina MiSeq Library QC sequencing workflow.
Variant Calling:
A Hapmap sample was used as a normal control. Variant calling was performed on target regions that had bidirectional coverage. Variants had to be present on at least 4 reads in each direction. Variants were annotated with Annovar and checked against external databases (COSMIC, Single Nucleotide Polymorphism database and 1000 Genomes). Variant calls were filtered for their annotation as non-synonymous, exonic variants with clear impact on protein function. Variants annotated as ‘synonymous’ or ‘silent’ were removed, along with variants annotated as being located in introns, UTRs, ncRNA or intergenic regions. Coding variants including missense, nonsense, splice-site and frameshift mutations or in-frame indels were catalogued. Know polymorphisms were removed after correlation with the dbSNP database and 1000 genomes. Final mutation calls were reviewed by a molecular pathologist. A minimum threshold variant frequency of 5% was used to call mutations at diagnosis and to identify residual mutations on pre-HCT samples. The catalogued mutations from diagnostic and pre-HCT samples are included in supplementary table 2.
Qualitative testing for FLT3 ITD and TKD were performed separately using a sizing assay with a sensitivity of 5% (performed locally or InVivoscribe).
Flow cytometry MRD
Ten-color multi-parameter flow cytometry (MFC) was performed on marrow aspirates before and after HCT. An abnormal population was identified by visual assessment of populations with antigen expression that was ‘different-from-normal’ as described in prior publications6,7. Antibody panels are included in supplementary table 3. Briefly, up to 1.5 million cells in 100–200 microliters of freshly drawn aspirate sample were stained with three panels, washed with phosphate-buffered saline containing 0.1% bovine albumin and lysed and fixed with ammonium chloride and 0.1% formalin solution. Following the last wash, samples were acquired on FACS-Canto-10 cytometers (BD Biosciences, San Jose CA). The results were analyzed using custom Woodlist software (generous gift of Wood BL, University of Washington). The assay is able to detect abnormal populations down to sensitivity of approximately 1 in 1000 events provided at least 100 000 events are acquired in each of the 3 panels used. MRD detected using MFC was defined by the presence of any abnormal bone marrow blast population meeting the above criteria.
Statistics:
VAFs of patient specific mutations quantified at diagnosis and pre-HCT were compared using paired t-test. Kaplan-Meier methods were used to estimate overall survival, and univariate survival comparisons were evaluated using Cox proportional hazards regression. Cumulative incidence functions were used to estimate the incidence of relapse based on MFC and NGS pre-HCT status, treating death in the absence of relapse as a competing risk. Cause-specific Cox regression was used to compare relapse risk based on pre-HCT characteristics. No adjustments were made for multiple comparisons. All analyses were conducted in R v.3.2.3.
Results:
Patient characteristics
Disease and transplant variables for all included patients are described in Table 1. The majority had primary AML (63%) with intermediate risk DRI (68%) and normal karyotype (44%). In total 21 (21%) relapsed and 36 (35%) died, 13 due to leukemia and 23 due to other transplant related causes. The median follow-up amongst survivors from the time of transplant was 13 months (range 6–26 months).
Mutation clearance prior to HCT
Multi-gene NGS was performed at diagnosis and immediately before HCT in 47 patients, 40 (85%) of whom had somatic mutations allowing measurement of the clearance rate of these leukemia alleles. Allele clearance was defined by a reduction in the mutant gene VAF to <5%. These patients had various induction and consolidation therapies (supplementary table 4); however, 44 (93%) received cytarabine based induction and 21 (45%) received at least one cycle of consolidation. Clearance of all mutations was seen in 22/40 (45%) patients. There were 26 patients with multiple mutations at diagnosis, 5 did not clear any mutation, 7 cleared at least one mutation and 14 cleared all mutations. Mutations were rarely cleared if they occurred in the following genes: JAK2 (0/4, 0%), DNMT3A (3/11, 27%) and TET2 (5/10, 50%) while mutations in the following genes were frequently cleared: NPM1 (8/9, 88%), IDH 1/2 (9/11, 82%) and FLT3 ITD was cleared in 15/20 (75%).
To quantify the absolute change in mutant allele burden we compared the VAF of mutations called at diagnosis with the corresponding mutant VAF measured immediately before HCT (figure 1A). The mean VAF of RUNX1 (0.47 vs 0.13, p=0.012), NPM1 (0.14 vs 0.01, p=0.006), IDH 1/2 (0.25 vs 0.03, p=0.002), TET2 (0.41 vs 0.14, p=0.008) and TP53 (0.32 vs 0.12, p=0.049) were significantly lower pre-HCT, while there was no significant difference in DNMT3A (0.24 vs 0.15, p=0.228) and JAK2 (0.52 vs 0.27, p=0.156) across the two time points.
Figure 1A:
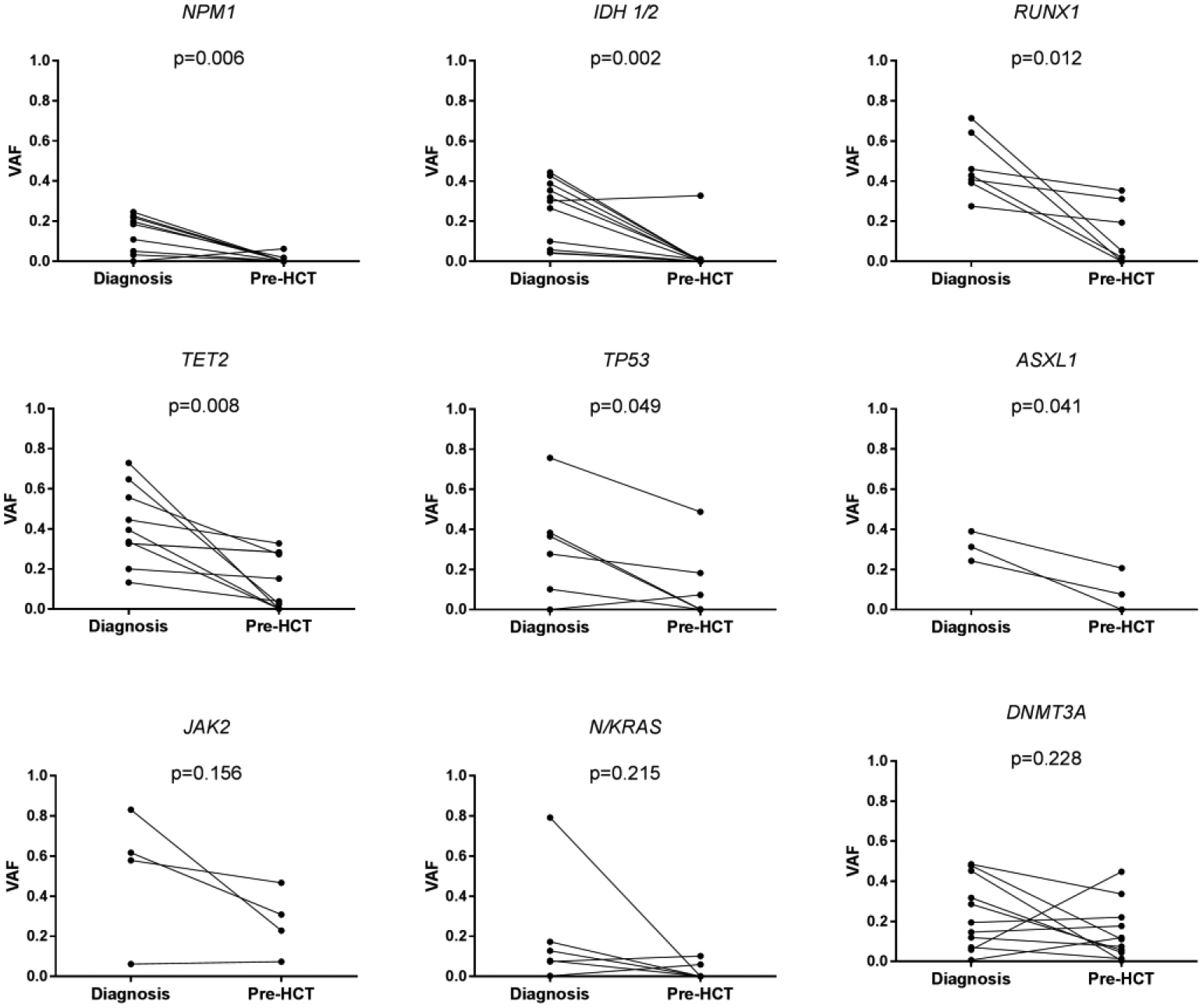
Relative change in mutant gene VAF
Mutant gene VAF was compared between diagnosis and immediately before HCT in patients who had mutations identified at AML diagnosis (n=40). A significant reduction in VAF was noted for NPM1 (n=9), IDH1 and 2 (n=11), RUNX1 (n=7), TET2 (n=10) and TP53 (n=6) while no change in VAF was seen in mutant JAK2 (n=4), N/KRAS (n=6) and DNMT3A (n=11) alleles.
Comparison of flow cytometry and NGS residual disease assessment
Tandem assessment using both MFC and multi-gene NGS on the same marrow aspirate before HCT was performed on 62 patients allowing for direct comparison between the two assays. Concordant results were seen in 44 (71%) cases. Four had no MRD identified by MFC (F−) but had mutations identified with VAF >5% (M+) (ie. F−/M+) and 14 had detectable MFC MRD but no identified mutations (F+/M−) (table 2).
Table 2:
Disease profile of patients with discordant results on tandem assessment immediately before HCT
| Assessment at diagnosis | RD assessment pre HCT | Post HCT | |||||
|---|---|---|---|---|---|---|---|
| Patient | Cytogenetics | Mutations | CR/CRi | MFC MR (%) | Mutations | VAF (%) | Relapse |
| MFC negative and multi gene NGS positive (F−M+) ^ | |||||||
| 1 | 47,XY,+13[21] | DNMT3A p.R882C | CR | 0 | DNMT3A p.R882C | 11.09 | no |
| 2 | 46,XX[20] | NP | CR | 0 | DNMT3A p.R882H | 14.21 | yes |
| 3 | 46,XY,t(X;19), del9(q) |
DNMT3A p.804L TET2 p.M1333fs |
CR | 0 |
DNMT3A p.804L TET2 p.M1333fs |
20.41 13.30 |
no |
| FLT3-ITD# | FLT3-ITD | <5%# | |||||
| 4 | 47,XX,+8[9] | CRi | 0 | SUZ12 p.H6D | 12.31 | no | |
| MFC positive and multi gene NGS negative (F+M−) * | |||||||
| 1 | 46, XX [20] | DNMT3A p.Q402X | CR | 0.35 | DNMT3A p.Q402X | 0.38 | no |
| NRAS p.G12D, | NRAS p.G12D, | 0.00 | |||||
| IDH1 p.R132H | IDH1 p.R132H | 0.00 | |||||
| NPM1 p.L287fs | NPM1 p.L287fs | 0.00 | |||||
| 2 | 46,XX,inv(16) (p13q22 [20] | WT1 p.1H469N | CRi | 0.01 | WT1 p.H469N | 0.00 | no |
| 3 | Complex§ |
NRAS p.Q61K TP53 p.R333fs |
CR | 2.00 |
NRAS p.Q61K TP53 p.R333fs |
0.11 0.22 |
no |
| TP53 p.N210fs | TP53 p.N210fs | 0.10 | |||||
| 4 | 46,XY[20] | IDH1 p.R132H | CRi | 1.20 | IDH1 p.R132H | 0.09 | no |
| TET2 p.P409A | TET2 p.P409A | 3.91 | |||||
| 5 | 46,XY[20] | IDH2 p.R172K | CR | 1.60 | IDH2 p.R172K | 0.15 | no |
| ASXL1 p.T880fs | ASXL1 p.T880fs | 0.00 | |||||
| TET2 p.I873T | TET2 p.I1873T | 0.17 | |||||
| 6 | 46,XX[20] | TET2 p.I1873T | CR | 2.00 | TET2 p.I1873T | 0.19 | no |
| 7 | 46,XY[20] | NPM1 p.L287fs | CR | 1.50 | NPM1 p.L287fs | 0.00 | no |
| FLT3-ITD# | FLT3-ITD | <5%# | |||||
| 8 | 46,XY[20] | IDH p.R132H | CR | 0.28 | IDH p.R132H | 0.46 | no |
NP: not performed, CR: complete remission, CRi: complete remission with incomplete count recovery, VAF: variant allele frequency, MFC multiparameter flow cytometry
All 4 patients in the F−M+ group are presented in the table.
Eight patients in the F+M− group had somatic profiling performed at diagnosis that identified mutations which could be tracked. Of the remaining 6 patients in the F+M− group, 5 did not have somatic mutation profiling performed at AML diagnosis and 1 had no mutations called at the time of diagnosis when assessed using the same NGS assay.
FLT3-ITD assessment was performed using a qualitative stand-alone PCR assay with a sensitivity of 5%, hence VAF is not reported.
43–45,X,-X,+3,del(5)(q?15q?33),−7,?del(13)(q12q14),−15,add(16)(q24),−18,−4mar
Patients in the F−M+ group had residual mutations in either DNMT3A or TET2 with VAF >5%. Eight of fourteen patients in the F+/M− had NGS performed at AML diagnosis that identified at least one somatic mutation, which could be tracked at the time-point prior to HCT. On manual review of pre-HCT samples, 6/8 of these patients had residual disease specific alleles; however, these were present at a VAF below the established thresholds (<5%) for mutation calling (table 2).
The residual disease burden of patients who were in CR immediately before transplant was quantified using tandem assessment by NGS and MFC. Seventeen patients were in hematologic CR or CRi at this time-point but had residual mutations identified using NGS and had MRD assessed by MFC (Figure 1B). The average VAF of residual mutations in DNMT3A (n=9), TET2 (n=7) and JAK2 V617F (n=4) assessed on bulk bone marrow was markedly higher than the percentage of aberrant blasts measured on the same bone marrow aspirates: 19% vs 0.72%; 29% vs 0.64% and 32% vs 0.53%, respectively (figure 1B). This suggests that residual mutations are likely to be present in non-blast cells in patients who are in hematologic CR/CRi.
Figure 1B:
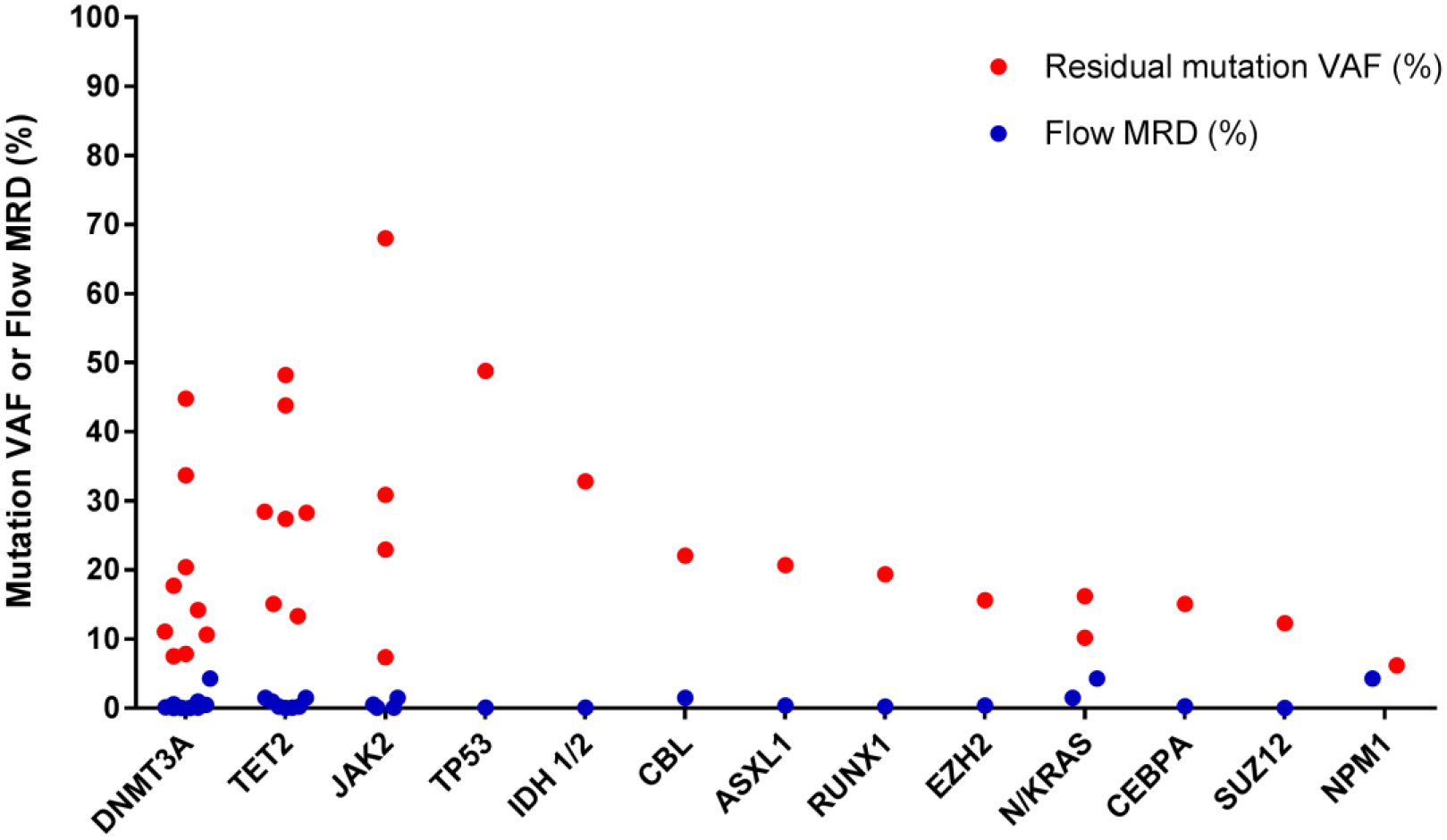
Comparison of residual mutation and flow MRD burden pre HCT
Residual disease in the blast compartment quantified by MFC was matched to the VAF of residual mutations quantified by NGS on the same bone marrow sample for patients who were in hematologic CR/CRi at the time of transplant but who had residual mutations called and had MFC residual disease assessment performed (n=17). The VAF (%) of residual mutations (residual mutations defined by VAF ≥5%) and the percentage of aberrant blasts are plotted on the same Y-axis grouped by leukemia gene. In the vast majority of cases MFC was quantified to be <1% while residual leukemia alleles were quantified ≥5% suggesting that these mutations are present in cells outside of the leukemic blast compartment.
Disease variables associated with transplant outcome
Univariate analysis of factors associated with relapse and survival are presented in table 3. Disease related variables and the HCT-CI score were associated with transplant outcomes, while transplant variables such as donor source, HLA matching, conditioning intensity and GVHD prophylaxis were not.
Table 3:
Univariate analysis of factors associated with overall survival and time-to-relapse after HCT
| Overall Survival | Time-to-Relapse | |||||||
|---|---|---|---|---|---|---|---|---|
| N | Events | HR (95% CI) | p-value | Events | HR (95% CI) | p-value | ||
| Residual disease | ||||||||
| Flow MRD | Negative | 35 | 7 | reference | 0.05 | 3 | reference | 0.016 |
| Positive | 42 | 16 | 2.44 (1–5.97) | 14 | 4.62(1.32–16.09) | |||
| Multi-gene NGS | Negative | 52 | 13 | reference | 0.059 | 6 | reference | 0.003 |
| Positive | 31 | 13 | 2.1 (0.97–4.55) | 12 | 4.35 (1.63–11.6) | |||
| Tandem assessment | F−/M− | 24 | 3 | reference | 0.072 | 2 | reference | 0.008 |
| F−/M+ or F−/M | 18 | 7 | 3.96 (1.02–15.37) | 3 | 2.31 (0.38–13.84) | |||
| F+/M+ | 20 | 8 | 4.53 (1.19–17.18) | 10 | 8.39 (1.83–38.38) | |||
| Blasts >5% | No | 90 | 28 | reference | 0.011 | 16 | reference | 0.007 |
| Yes | 14 | 8 | 2.78 (1.26–6.14) | 6 | 3.64 (1.42–9.33) | |||
| Disease variables | ||||||||
| T-AML | No | 91 | 28 | reference | 0.009 | 18 | reference | 0.165 |
| Yes | 13 | 8 | 2.87 (1.3–6.35) | 4 | 2.16 (0.73–6.41) | |||
| Secondary AML | No | 65 | 18 | reference | 0.076 | 12 | reference | 0.343 |
| Yes | 39 | 18 | 1.81 (0.94–3.48) | 10 | 1.5 (0.65–3.47) | |||
| HCT-CI | <3 | 51 | 12 | reference | 0.006 | 9 | reference | 0.184 |
| ≥3 | 53 | 24 | 2.67 (1.33–5.37) | 13 | 1.78 (0.76–4.17) | |||
| DRI | Low/Intermed | 73 | 21 | reference | 0.013 | 10 | reference | 0.001 |
| High/very high | 31 | 15 | 2.34 (1.2–4.57) | 12 | 3.98 (1.71–9.25) | |||
| Transplant variables | ||||||||
| Stem cell source | CB | 19 | 7 | reference | 0.982 | 1 | reference | 0.315 |
| BM | 12 | 4 | 0.92 (0.27–3.15) | 3 | 4.92(0.51–47.34) | |||
| PB | 73 | 25 | 0.92 (0.4–2.13) | 18 | 4.7 (0.63–35.2) | |||
| HLA matched | Yes | 72 | 23 | reference | 0.337 | 18 | reference | 0.253 |
| No | 32 | 13 | 1.4 (0.71–2.76) | 4 | 0.53 (0.18–1.57) | |||
| CD34+ selection | No | 75 | 25 | reference | 0.86 | 18 | reference | 0.228 |
| Yes | 29 | 11 | 0.94 (0.46–1.91) | 4 | 0.51 (0.17–1.52) | |||
| Conditioning | Ablative | 59 | 18 | reference | 0.237 | 9 | reference | 0.1 |
| RIC | 45 | 18 | 1.48 (0.77–2.86) | 13 | 2.04 (0.87–4.78) | |||
T-AML: therapy related AML, CB: Cord blood, BM: Bone marrow, PB: peripheral blood; RIC: reduced intensity conditioning, DRI: disease risk index, F: flow MRD, M: Residual mutations using NGS, HLA matched: matched sibling or matched unrelated donors assessed at 10 alleles (A, B, C, DR, DQ) and non-matched includes all mismatched related, unrelated and cord blood transplant.
Presence of MFC MRD and residual mutations pre-HCT was associated with relapse and survival. For MFC (n=77), the 18-month relapse incidences for MRD positive (F+) and MRD negative (F−) groups were 37% (95% CI: 21–53%) and 9% (95% CI 2–21%) respectively (p=0.016). For NGS (n=83), the 18-month relapse incidence for those with (M+) and without (M−) residual mutations was 39% (95% CI: 22–56%) and 12% (95% CI: 5–22%) respectively (p= 0.003).
For overall survival, the estimated 18-month OS for F+ and F− was 48% (95% CI: 31–73%) and 73% (95% CI: 57–94%), respectively (p=0.050). For NGS, the estimated 18-month OS for M+ and M− was 54% (95% CI 38–77%) and 69% (95% CI 54–87%), respectively (p=0.059) (figure 2A–D).
Figure 2:
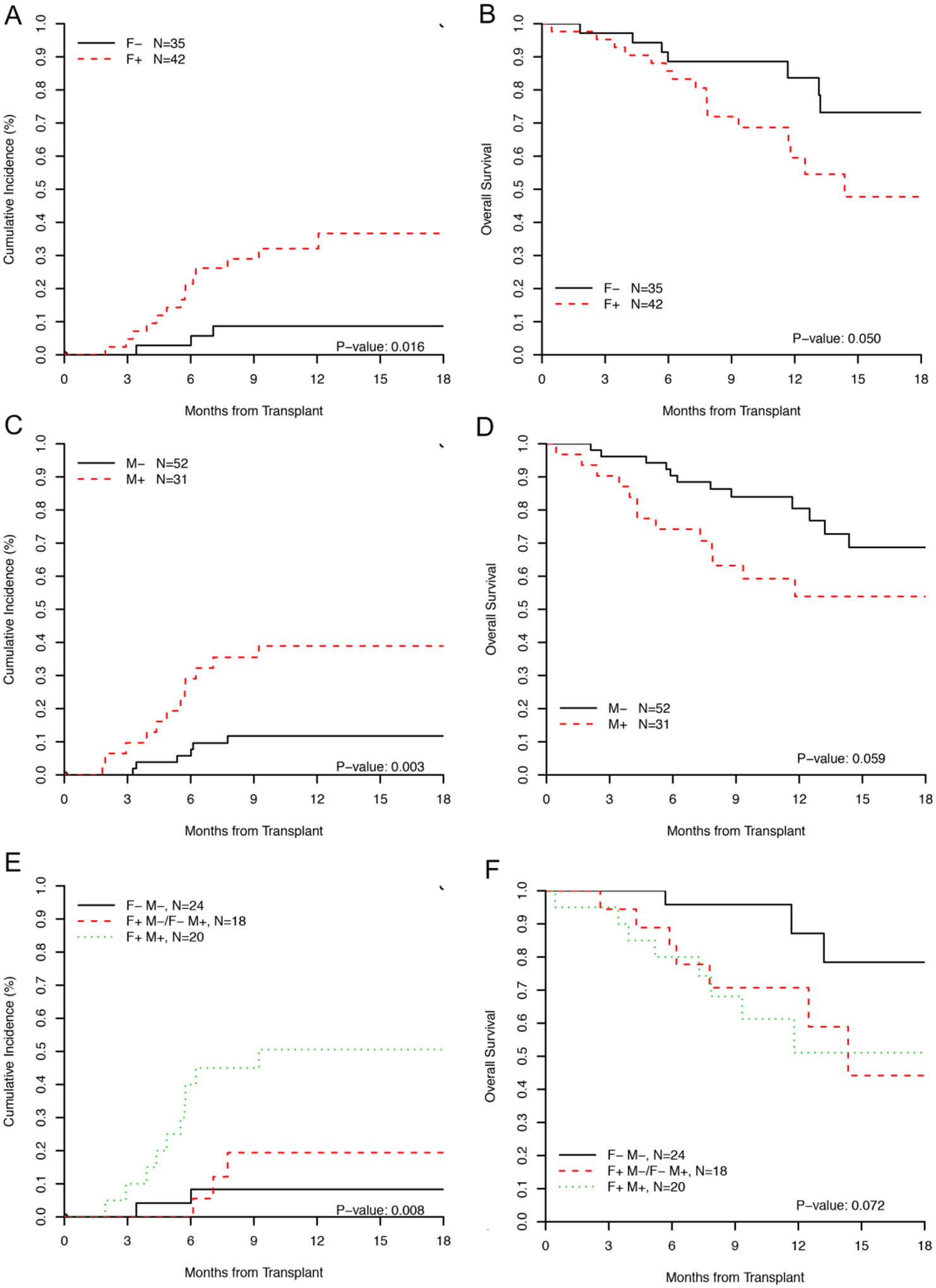
Transplant outcome stratified by residual disease status.
(A) Cumulative incidence of relapse and (B) overall survival by MFC MRD, (C) Cumulative incidence of relapse and (D) overall survival by NGS, (E) Cumulative incidence of relapse and (F) overall survival by tandem assessment using MFC and NGS. In (E) and (F), patients with discordant assessment are grouped together as their outcome tracked with patients in the F−/M− group.
F denotes MFC, M denotes mutation analysis by NGS and status is categorized as +/−
Tandem NGS and MFC assessment predicts relapse and survival
Patients with discordant residual disease assessment appeared to have a low relapse incidence (3/18, 16%) compared to those who were positive by both methods (10/20, 50%). The two methods were combined for those who had tandem assessment (n=62). Patients in the F+/M+ group (n=20) had the highest incidence of relapse with an 18-month estimate of 51% (95% CI: 26–71%) compared to those who were F−/M− (n=24) who had an 18-month estimate of 8% (95% CI: 2–24%) and those with discordant results (F−/M+ or F+/M−, n=18) who had a 18-month estimate of 17% (95% CI 4–43%) (overall p=0.008) (Figure 2E).
Using combined assessment, the estimated 18-month survival of patients in the F+/M+ group was 51% (95% CI 30–86%) for (F+/M+), compared to 78% (95% CI 59%–99%) in the F−/M− group and 44% (95% CI 21–92%) in those with discordant RD assessment (F−/M+ or F+/M−) (overall p=0.072) (Figure 2F).
Discussion:
This analysis is the first to show that a targeted multi-gene NGS panel can be used to detect residual mutations in AML patients immediately before allogeneic transplant. For the first time we compare flow MRD with a multi-gene NGS assay and show that presence of residual mutations as well as atypical blasts are associated with post-transplant relapse and survival. Despite vastly differing sensitivities, the two assays were concordant in 71% of cases; however, the burden of residual disease as measured by residual leukemia alleles was markedly higher than the estimated percentage of aberrant blasts quantified by MFC, suggesting that residual leukemia alleles likely persist in non-blast compartments at the time of remission.
Tracking multiple somatic mutations takes into account both the clonal architecture of AML and the different clearance rates of specific leukemia alleles. Molecular assays that target a single marker include quantitative PCR of core-binding factor fusion transcripts15 or mutant NPM116. Detection of these mutations is associated with outcome; however, assays measuring different markers have different sensitivities in detecting residual leukemia. For instance, measuring residual FLT3-ITD alleles is a poor RD marker as this mutation is typically sub-clonal17. Conversely, early disease mutations arising in DNMT3A may also represent poor disease markers as they often arise in pre-leukemic cells which have unclear malignant potential and have been detected in patients who are in a long-term remission3,5,16,18. Multi-gene NGS considers both founder and sub-clonal alleles. NGS techniques are applicable to almost all patients as they can detect and track mutations irrespective of their genomic location. The targeted 28-gene NGS panel applied here was applicable to 49/58 (84%) patients who had at least one somatic mutation identified at diagnosis. A panel with a more extensive gene set and higher analytical sensitivity is likely to improve accuracy in detecting residual sub-clonal and founder mutations and may further improve the concordance rate with flow cytometry.
The clinical significance of residual DNMT3A and TET2 mutations before HCT is unknown. Patients with DNMT3A and TET2 mutations at diagnosis are highly likely to have these detected at the time of transplant due to the low clearance rate of these alleles with chemotherapy4,5. Such patients often had no MRD detected by flow (F−M+ group) and appeared to have a low relapse incidence. This may represent a state of ‘clonal remission’. The clinical significance of patients who had residual DNMT3A and TET2 alleles and clearance of high risk kinase mutations (FLT3, RAS) requires exploration in a larger cohort.
The high error rate in calling mutations using current NGS methods necessitates setting a high VAF threshold to confidently call mutations. A VAF of 5% was set here based on prior assay validation14. Only one other group has used a multi-gene approach to track mutation clearance in AML and a VAF of 2.5% was arbitrarily chosen in that study4. MFC can generally detect disease at greater depth (1:1000) and would be expected to have a higher sensitivity than our NGS approach (1:20). Despite this difference in sensitivity we found a high concordance between the assays. The issue of low sensitivity with NGS may be overcome if leukemia specific mutations identified at diagnosis are tracked at serial assessment. However, the threshold at which residual alleles are clinically impactful is unknown. Patients who had MRD detected by MFC but had no mutations called at the pre-defined VAF thresholds (F+M−) often had residual alleles below the necessary threshold for mutation calling. The incidence of relapse among these patients was low suggesting that a threshold for residual mutations may exist below which they are unlikely to impact transplant outcomes.
The VAF of residual mutations was markedly higher than the quantified percentage of aberrant blasts when compared immediately before HCT. The high VAF of residual alleles suggests that cells in multiple marrow compartments were clonal even in patients who attained morphologic blast clearance (CR/CRi). This group (F−M+) may represent a state of ‘clonal remission’ where hematopoiesis has reconstituted from progenitors with CHIP-associated mutations but without an aberrant blast population (figure 1b). The clinical significance of this in a pre-transplant setting is unknown. DNMT3A mutations can be detected within both lymphoid and myeloid compartments in patients who remain in long term remission following chemotherapy3. The only allele whose variant fraction was similar to the quantified percentage of blasts was NPM1 (supplementary table 5) suggesting that NPM1 mutations are resident to the blast compartment whereas other alleles are likely to be present in mature myeloid cells and lymphoid cells. This may explain why tracking mutated NPM alleles, has been consistently shown to be a valid disease marker in AML5,19–22. The clinical significance of ‘clonal remission’ (F−M+) before HCT requires further exploration.
Important biases pertaining to this analysis include the small cohort size which limits statistical power to perform multivariate analysis and a short follow-up period such that additional relapses may still occur. Analytical issues exist with the application of amplicon-based NGS to monitor residual mutations. The NGS assay applied here was validated to detect somatic mutations at the time of AML diagnosis, rather then to detect low level residual mutations after chemotherapy hence a relatively high variant threshold of 5% was fixed to call mutations. Germline samples were universally unavailable and as a result it is possible that germline polymorphisms have not been efficiently excluded by post analytical bioinformatics methods. This bias is inherent when sequencing liquid tumors where paired germline tissue is not available to filter out private or non-pathogenic polymorphisms. Finally, the NGS panel was restricted to assess only 28 genes and analysis of a more extensive gene list would be expected to increase the likelihood of detecting residual mutations.
Application of multi-gene NGS in the monitoring of residual disease in AML patients undergoing transplant has several benefits. Although the NGS assay was not designed to monitor disease response, monitoring and tracking mutations was prognostic. Our 28-gene assay allowed broad coverage of known mutation hotspots and detected mutations in 84% of patients at diagnosis. The median number of mutations detected was 2 (range 0–5) per patient, which is lower than expected from whole genome sequencing studies, thus the likelihood to detect residual mutations would be greater if more genes were sequenced23,24. The clonal composition of hematopoiesis before transplantation appears to be associated with HCT outcomes.
Supplementary Material
Highlights:
Multi-gene next generation sequencing can be used to track residual disease in acute myeloid leukemia
Leukemia mutations in DNMT3A, TET2 and JAK2 are less likely to be cleared than NPM1, IDH and FLT3-ITD.
Residual disease measured using multi-gene NGS and 10-colour flow cytometry were associated with relapse and survival after HCT
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Welch JS, Ley TJ, Link DC, et al. : The origin and evolution of mutations in acute myeloid leukemia. Cell 150:264–78, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindsley RC, Mar BG, Mazzola E, et al. : Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 125:1367–76, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ploen GG, Nederby L, Guldberg P, et al. : Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br J Haematol 167:478–86, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Klco JM, Miller CA, Griffith M, et al. : ASsociation between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 314:811–822, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivey A, Hills RK, Simpson MA, et al. : Assessment of Minimal Residual Disease in Standard-Risk AML. New England Journal of Medicine 0:null [DOI] [PubMed] [Google Scholar]
- 6.Walter RB, Gooley TA, Wood BL, et al. : Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J Clin Oncol 29:1190–7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Araki D, Wood BL, Othus M, et al. : Allogeneic Hematopoietic Cell Transplantation for Acute Myeloid Leukemia: Time to Move Toward a Minimal Residual Disease-Based Definition of Complete Remission? J Clin Oncol 34:329–36, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walter RB, Gyurkocza B, Storer BE, et al. : Comparison of minimal residual disease as outcome predictor for AML patients in first complete remission undergoing myeloablative or nonmyeloablative allogeneic hematopoietic cell transplantation. Leukemia 29:137–44, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dohner H, Estey EH, Amadori S, et al. : Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115:453–74, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Armand P, Gibson CJ, Cutler C, et al. : A disease risk index for patients undergoing allogeneic stem cell transplantation. Blood 120:905–13, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorror ML, Maris MB, Storb R, et al. : Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 106:2912–9, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bacigalupo A, Ballen K, Rizzo D, et al. : Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant 15:1628–33, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giralt S, Ballen K, Rizzo D, et al. : Reduced-intensity conditioning regimen workshop: defining the dose spectrum. Report of a workshop convened by the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 15:367–9, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Cheng J, Mitchell TN, et al. : Detection of mutations in myeloid malignancies through paired-sample analysis of microdroplet-PCR deep sequencing data. J Mol Diagn 16:504–18, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin JA, O’Brien MA, Hills RK, et al. : Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML-15 trial. Blood 120:2826–35, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Ivey A, Hills RK, Simpson MA, et al. : Assessment of Minimal Residual Disease in Standard-Risk AML. New England Journal of Medicine 374:422–433, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Palmisano M, Grafone T, Ottaviani E, et al. : NPM1 mutations are more stable than FLT3 mutations during the course of disease in patients with acute myeloid leukemia. Haematologica 92:1268–9, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Debarri H, Lebon D, Roumier C, et al. : IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: a study by the Acute Leukemia French Association. Oncotarget 6:42345–53, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorello P, Cazzaniga G, Alberti F, et al. : Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 20:1103–8, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Papadaki C, Dufour A, Seibl M, et al. : Monitoring minimal residual disease in acute myeloid leukaemia with NPM1 mutations by quantitative PCR: clonal evolution is a limiting factor. Br J Haematol 144:517–23, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Schnittger S, Kern W, Tschulik C, et al. : Minimal residual disease levels assessed by NPM1 mutation-specific RQ-PCR provide important prognostic information in AML. Blood 114:2220–31, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Kronke J, Schlenk RF, Jensen KO, et al. : Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol 29:2709–16, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. New England Journal of Medicine 368:2059–2074, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2059–74, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.