

Abstract
Aging is the major risk factor for Alzheimer’s disease (AD). Mitochondrial dysfunction and neuronal network hyperexcitability are two age-related alterations implicated in AD pathogenesis. We found that levels of the mitochondrial protein deacetylase sirtuin-3 (SIRT3) are significantly reduced, and consequently mitochondria protein acetylation is increased in brain cells during aging. SIRT3-deficient mice exhibit robust mitochondrial protein hyperacetylation and reduced mitochondrial mass during aging. Moreover, SIRT3-deficient mice exhibit epileptiform and burst-firing electroencephalogram activity indicating neuronal network hyperexcitability. Both aging and SIRT3 deficiency result in increased sensitivity to kainic acid-induced seizures. Exposure of cultured cerebral cortical neurons to amyloid β-peptide (Aβ) results in a reduction in SIRT3 levels and SIRT3-deficient neurons exhibit heightened sensitivity to Aβ toxicity. Finally, SIRT3 haploinsufficiency in middle-aged App/Ps1 double mutant transgenic mice results in a significant increase in Aβ load compared with App/Ps1 double mutant mice with normal SIRT3 levels. Collectively, our findings suggest that SIRT3 plays an important role in protecting neurons against Aβ pathology and excitotoxicity.
Keywords: Amyloid plaques, Electroencephalogram, Epileptic seizures, Excitotoxicity, Protein deacetylase, SOD2
Introduction
A diagnosis of Alzheimer’s disease (AD) is based on progressive cognitive decline and the accumulation of extra-cellular amyloid β-peptide (Aβ) plaques and intracellular neurofibrillary tangles comprised of hyperphosphorylated Tau in the brain. Aging is the major risk factor for AD. Alterations that occur in the brain during aging that may contribute to AD include oxidative stress, impaired cellular energy metabolism, DNA damage, impaired autophagy, impaired adaptive cellular stress responses, and aberrant neuronal network activity (Mattson & Arumugam, 2018). Analyses of postmortem brain tissue from AD patients and mouse models have provided evidence for oxidative damage to mitochondrial DNA, compromised electron transport chain function and ATP depletion, and impaired mitophagy (Kerr et al., 2017). Positron emission tomography studies have consistently revealed impaired glucose utilization by brain cells in patients with mild cognitive impairment (MCI) or AD compared to age-matched neurologically normal controls (Cunnane et al., 2020; Kapogiannis & Avgerinos, 2020). Analyses of brain tissue samples from AD patients and animal models have documented the accumulation of damaged dysfunctional mitochondria in vulnerable neurons (Kerr et al., 2017).
Throughout the brain, the core neuronal circuits consist of excitatory neurons that deploy the neurotransmitter glutamate and inhibitory neurons that deploy GABA. Approximately 90 percent of neurons in the brain are glutamatergic. Accumulating evidence suggests that an excitatory imbalance occurs very early in AD and contributes to the degeneration of synapses and the death of neurons. Epileptic seizures are common in AD patients, and subclinical epileptiform activity occurs very early in the disease process and is associated with cognitive decline (Vossel et al., 2016, 2017). Data from studies of cultured neurons and transgenic mice that develop Aβ plaques have shown Aβ aggregation on neuronal membranes where the Aβ impairs the function of ion-motive ATPases and glucose transporters, rendering neurons vulnerable to cellular calcium overload, and excitotoxicity (Keller et al., 1997; Mark et al., 1995a, b, 1997a, b; Mattson et al., 1992). Studies of animal models of AD have demonstrated the efficacy of interventions that prevent neuronal network hyperexcitability in attenuating Aβ and Tau pathologies and cognitive decline (Hijazi et al., 2020; Hunsberger et al., 2015; Mark et al., 1995b; Zhang et al., 2014).
Impaired mitochondrial function can render neurons vulnerable to excitotoxicity, while excessive activation of glutamatergic synapses causes oxidative damage to mitochondria (Plotegher et al., 2021). Moreover, conditions that impair mitochondrial function or cause neuronal hyperexcitability accelerate the production and accumulation of Aβ (Cunnane et al., 2020). The roles of specific mitochondrial proteins in protecting against Aβ and neuronal network hyperexcitability in AD are largely unknown. The mitochondrial protein deacetylase Sirtuin-3 (SIRT3) removes acetyl groups from lysine residues in several hundred mitochondrial proteins (Carrico et al., 2018). These SIRT3 substrates include several enzymes involved in oxidative phosphorylation or fatty acid metabolism, including the antioxidant enzyme superoxide dismutase 2 (SOD2), acetyl coenzyme A synthetase 2, pyruvate dehydrogenase, and cyclophilin D. Studies of cultured neurons and SIRT3-deficient mice have shown that SIRT3 protects neurons against excitotoxicity and oxidative stress (Cheng et al., 2016; Gleave et al., 2017; Shi et al., 2017). Electrophysiological recordings from neurons in brain tissue slices from wild type and SIRT3-deficient mice provided evidence that SIRT3 enhances GABAergic neurotransmission thereby suppressing the activity of glutamatergic neurons (Mark et al., 1995b). The results of a recent study showed that SIRT3 haploinsufficiency exacerbates loss of GABAergic neurons and neuronal network hyperexcitability in a mouse model of AD (Cheng et al., 2020).
Here we show that SIRT3 levels decline, and protein acetylation increases in brain cells during aging. We provide evidence that SIRT3 suppresses Aβ accumulation in a mouse model of AD and protects neurons against the neuronal network hyperexcitability and degeneration otherwise caused by Aβ. These findings suggest that SIRT3 plays a critical role in AD pathogenesis. Interventions that bolster SIRT3 expression or activity may provide protection against AD.
Materials and Methods
Animals
Double mutant transgenic mice (App/Ps1 mice) expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9) under the control of a prion protein promoter were purchased from the Jackson Laboratories. Breeding pairs of Sirt3 knockout mice used to establish an in-house colony were a generous gift from David Gius (Northwestern University, Evanston, IL). Both mouse strains were on a congenic C57BL/6J background. Methods for genotyping have been described previously (datasheet on AppPs1 mouse strain 005864, Jackson Laboratories; and Cheng et al., 2016). Sirt3-haploinsufficient App/Ps1 mice (Sirt3± App/Ps1) were generated by cross-breeding Sirt3± and AppPs1 mice as described previously (Cheng et al., 2020). All animal procedures were approved by the Animal Care and Use Committee of the National Institute on Aging Intramural Research Program. Mice were provided a standard diet (Teklad Global 18% Protein Rodent Diet, Envigo, Indianapolis, IN, USA). They were maintained on a 12 h light/dark cycle at 20–22 °C. All mice used in this study were male.
Primary Neuronal Culture and Cell Death Quantification
Primary cultures of WT, Sirt3± and Sirt3−/− cerebral cortical mouse neurons were established from embryonic day 15 mouse embryos. WT embryos were obtained by breeding WT male and female mice, Sirt3± embryos were obtained by breeding WT males with Sirt3−/− females, and Sirt3−/− mice were obtained by mating Sirt3−/− males with Sirt3−/− females. Cultures were established and maintained using methods described previously (Cheng et al., 2016). Briefly, pregnant mice were euthanized, embryo brains were removed, and the cerebral hemispheres were removed in sterile ‘Hank’s Balanced Saline Solution. The brain tissues were incubated in 0.25% trypsin EDTA for 15 min at 37 °C, and then transferred to MEM+ (Minimal Essential Medium supplemented with 10% fetal Clone III bovine serum (HyClone, Logan, UT, USA) and were dissociated by trituration using a fire-polished Pasteur pipet. The dissociated cells were seeded into polyethyleneimine-coated plastic culture dishes or glass coverslips at a density of 60,000 cells/cm2. After a 4-h incubation in MEM+ to allow for cell attachment, the medium was replaced with Neurobasal medium containing B27 supplements, 2 mM l-glutamine, antibiotic and antimycotic, and 1 mM HEPES (Thermo Fisher Scientific, Waltham, MA, USA).
Aβ1-42 was purchased from Bachem (Torrance, CA, USA). Aβ1-42 oligomers were freshly prepared before treatment by dissolving the peptide in distilled water at a concentration of 200 μM. This stock solution was incubated at 37 °C for 24 h to promote peptide self-aggregation. After treatment for designated time periods, primary cultured neurons were stained with DNA-binding dye Hoechst 33342 by directly adding to the culture medium for 30 min to 1 h. Each well (or plate) was observed and images of three or four randomly selected fields were acquired using EVOS FL auto imaging system (Thermo fisher Scientific, Waltham, MA, USA). The numbers of dead neurons with bright condensed nuclei, and live neurons with intact neuronal morphology and faint diffuse nuclear staining in each microscope field were counted.
Analysis of Mitochondrial DNA
For analysis of mitochondrial DNA (mtDNA) was isolated from cortical tissues using a mitochondrial DNA isolation kit (Biovision, Milpitas, CA, USA). Briefly, fresh cortical tissues were washed thoroughly in PBS and then transferred into 1 ml of cytosol extraction buffer. After homogenization using a Dounce homogenizer, the homogenates were separated into cytosol (supernatant) and mitochondrial fractions (pellet) by centrifugation. The mitochondrial pellets were lysed and incubated at 50 °C in 30 μl of mitochondrial lysis buffer containing protein degradation enzymes. mtDNA was isolated by ethanol precipitation. An aliquot of homogenate was reserved for protein quantification and mtDNA content was normalized to the protein concentration.
Immunohistochemistry
Mice were anesthetized and perfused transcardially with PBS followed by 4% paraformaldehyde in PBS (pH 7.4). Brains were postfixed for 2 days and then transferred to a solution of 30% sucrose in PBS for cryopreservation. After 3–5 days in cryopreservation solution, cryostat sections were cut in the coronal plane at a thickness of 30 μm. Coronal sections containing the hippocampus were collected on Super-frost Plus slides (VWR International, Radnor, PA, USA). For Aβ immunostaining, brain sections were permeabilized and pre-incubated with blocking solution (0.2% Triton ×−100, 10% normal goat serum) in PBS for 30 min and then incubated overnight with primary antibody (6E10, Biolegend, San Diego, CA, USA) diluted in the same blocking solution at 4 °C. The sections were then washed with PBS and incubated with FITC goat anti-mouse IgG secondary antibodies diluted in the same blocking solution for 2 h at room temperature. Then the sections were counterstained with propidium iodide (PI) or DAPI (0.02% and 1% RNase in PBS) for 10 min; they were then washed with PBS and mounted on microscope slides by using antifade medium (Vector Laboratories, Burlingame, CA, USA).
Confocal Imaging and Imaging Analysis
Quantitative analysis of Aβ accumulation was performed to determine the Aβ load, which is the percentage of the neuropil in an anatomical area occupied by the Aβ-immunoreactivity. All the images were acquired using an Olympus Multiphoton Laser Scanning Microscope with 10× (NA, 0.4) or 4× (NA, 0.16) objectives. All images were acquired using identical confocal system parameters. To quantify the Aβ load the confocal images were converted to an 8-bit gray scale and a threshold was determined to include all immunoreactive deposits in the area using Image J NIH software. Aβ loads were quantified in a cortical segment through the entire dorsal to ventral extent of the cortex and in dentate gyrus regions of hippocampus. Determinations were made in three adjacent sections and the average was used as the Aβ load for each mouse.
Immunoprecipitation and Immunoblot Analysis
Mice were sacrificed by cervical dislocation and the cerebral cortex was removed and placed in a tube on dry ice. The tissues were stored at − 80 °C before processing for immunoblot analysis. For immunoblot analysis, tissues were lysed in RIPA buffer containing protease and phosphatase inhibitors (Millipore Sigma). Lysates were sonicated and centrifuged at 14,000×g for 5 min at 4 °C. A final protein concentration of each sample was determined using a protein assay kit (Bio-Rad) with BSA as the standard. Thirty micrograms of protein/lane were resolved in a 4–10% polyacrylamide gradient gel (Invitrogen, Waltham, MA, USA) and then transferred electrophoretically to a nitrocellulose membrane (Invitrogen, Waltham, MA, USA). Nonspecific binding sites were blocked in by incubation in a solution containing 5% milk for 2 h at room temperature. Then the membranes were incubated overnight in primary antibody followed by incubation in secondary antibody for 1.5 h at room temperature. The reaction products in the membranes were visualized using an enhanced chemiluminescence Western Blot Detection Kit (Kindle Bioscience, LLC). Primary antibodies recognized SIRT3 (1:1000), acetyl-lysine (1:500, Cell Signaling Technology, Danvers, MA, USA), SOD2 (1:1000, Sigma-Aldrich, St. Louis, MO, USA), cytochrome c oxidase subunit 1 (COX1) (1:500, Molecular Probes, Eugene, OR, USA), VDAC (1:1000, Abcam, Cambridge, UK), or actin (1:1000, Cell Signaling Technology, Danvers, MA, USA).
For immunoprecipitation analysis cortical tissues were solubilized in lysis buffer (150 mM NaCl, 20 mM Tris, 1.0% Triton ×−100, 1% deoxycholate, and 5 mM EDTA; pH 7.2) with proteinase inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). Equal amounts of protein extracts (500 μg) were pre-cleaned by incubating with 50 μl of protein A/G agarose (Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at 4 °C on a rotating shaker and were then centrifuged to precipitate the beads. Supernatants (pre-cleaned protein lysates) were collected and incubated with 1.5 μg of antibodies against SOD2 and 50 μl protein A/G agarose beads for 4 h at 4 °C on a shaker. Equal amounts of supernatants were incubated with 1.5 μg normal rabbit IgG and 50 μl protein A/G agarose beads for 4 h at 4 °C on a shaker as a negative control. Protein A/G beads were pelleted by centrifugation and washed four times with 1 ml of lysis buffer, the final pellets were eluted in protein loading buffer, and samples were vortexed and heated at 70 °C for 10 min. Beads were precipitated by centrifugation, and supernatants were collected for immunoblot analysis using the acetyl-lysine antibody. Images of blots were analyzed using Image J software.
Kainic Acid Administration and Seizure Scoring
Kainic acid (KA; Abcam, Cambridge, UK) was prepared as a stock solution in PBS at a concentration of 20 mg/ml. Mice were injected intraperitoneally at a single dose of 20 mg/kg (10 μl stock solution per gram of body weight). Seizure severity was scored using a modified version of the Racine scale (Cheng et al., 2020; Hamilton et al., 2018). The scale ranged from normal behavior with a score of 0 to death with a score of 6, and with the following intermediate scores: 1, hypoactivity, including crawling, fixed gaze, and/or a hunched postures, with occasional wet-dog shakes; 2, unilateral forelimb clonus, head nodding, frequent wet-dog shakes; 3, mild generalized clonus, rearing, bilateral forelimb clonus, loss of upright posture; 4, severe generalized clonus, falling, and/or uncontrolled running and jumping; 5, status epilepticus with severe loss of balance and tonic limb extension; and 6, seizures culminating in death. Behaviors were scored every 10 min for a period of 2 h after KA administration by a trained experimenter blinded as to the genotype of the mice. For each mouse the scores at each time point were averaged, and the scores for mice of each genotype and age were grouped (8–10 mice per group).
Telemetric EEG Recording
A commercially available telemetry system (Data Sciences International, St. Paul, MN, USA) was used to record the EEG, core body temperature, and general activity (movement in the home cage) of the mice. For electrode implantation mice were anesthetized with isoflurane and a radio transmitter (ETA-F10) was surgically implanted intraperitoneally. The transmitter electrodes were passed subcutaneously to the base of the skull. Two burr holes were drilled through the skull at the following coordinates relative to bregma: the positive recording electrode (parietal cortex: AP, − 2.0 mm; L, 1.8 mm) and the reference electrode (cerebellum: AP, − 6.2 mm; L, 2.0 mm). The electrodes were inserted into the holes above the dura and were secured with a tissue adhesive (Loctite 454, Henkel) and dental cement. Body temperature was maintained using a heating pad. Immediately after the surgical procedures mice received extended-release buprenorphine (0.5 mg/kg; ZooPharm, Laramie, WY, USA). Warm sterile saline (1 ml/mouse) mixed with antibiotics Enrosite (enrofloxacin, 2.5 mg/kg; Norbrook Laboratory) was injected immediately after implantation and twice daily during the next 3 days. The mice were allowed to recover for 7–10 days after the surgical procedure and then EEG activity was recorded continuously for 2 days or 1 week with the mice in their home cages. EEG data were collected using Dataquest ART (DSI, version 4.36) and analyzed offline using NeuroScore software (DSI; version 3.2.0). The EEG signal from artifact-free epochs was subjected to fast Fourier transformation by the algorithm embedded in the Neuroscore software with normalization of spectral calculation. The seizure-like spikes were identified using the spike detector tool in NeuroScore (DSI, 3.2.0), which can determine individual spikes and spike trains (defined as at least four spikes within a 200-ms time period). A seizure-like spike was defined using the analytic criteria of the minimum value of 100 μV and having a peak amplitude 5× greater than the average baseline value. The occurrence of spikes was assessed in each of the 10 s epochs and summed in a defined time duration as indicated.
Statistics
All data are presented as mean ± SEM. The power analysis used to determine the sample size was based on numbers of animals used in experiments that measured the same endpoints in previously published studies (Cheng et al., 2016, 2020). Statistical analyses were performed using Prism 7.0 software. The data were analyzed by unpaired Student’s t test or one-way or two-way ANOVA, followed by Tukey post hoc tests. A p value < 0.05 was considered statistically significant.
Results
Evidence that SIRT3 Level is critical for Maintaining Mitochondrial Integrity in the Cerebral Cortex During Aging
To determine whether SIRT3 levels are altered in aging brain, we performed immunoblot analysis of SIRT3 protein levels in cortical tissues collected from 8, 12, 16, and 20-month-old mice. We found that SIRT3 levels in 16 and 20-month-old mice were significantly reduced by 20 percent and 30 percent, respectively, as compared to 8-month-old mice (Fig. 1A, B). We next performed immunoblot analysis of cortical tissue from wild type (WT) and SIRT3-deficient (Sirt3−/−) mice of different ages using an antibody that recognizes acetyl-lysine residues. Numerous proteins exhibited increased acetylation in Sirt3−/− mice compared to WT mice of all ages examined (Fig. 1C). Wild-type mice at 20 months of age exhibited a significant increase in protein acetylation levels compared with younger, 8 and 12-month-old mice; these increases were associated with a significant reduction of SIRT3 levels in old compared to younger mice (Fig. 1A, D). An age-related increase in protein acetylation was also evident in Sirt3−/− mice leading to hyperacetylation in old Sirt3−/− mice (Fig. 1D). We next determined the acetylation status of SOD2 by immunoprecipitating SOD2 from cortical tissue samples from young and old WT and Sirt3−/− mice, and then probing the immunoblot with the acetyl-lysine antibody. As expected, SOD2 was hyperacetylated in Sirt3−/− mice compared to WT mice. SOD2 was hyperacetylated in an age-dependent manner in both WT and Sirt3−/− mice (Fig. 1E, F).
Fig. 1.
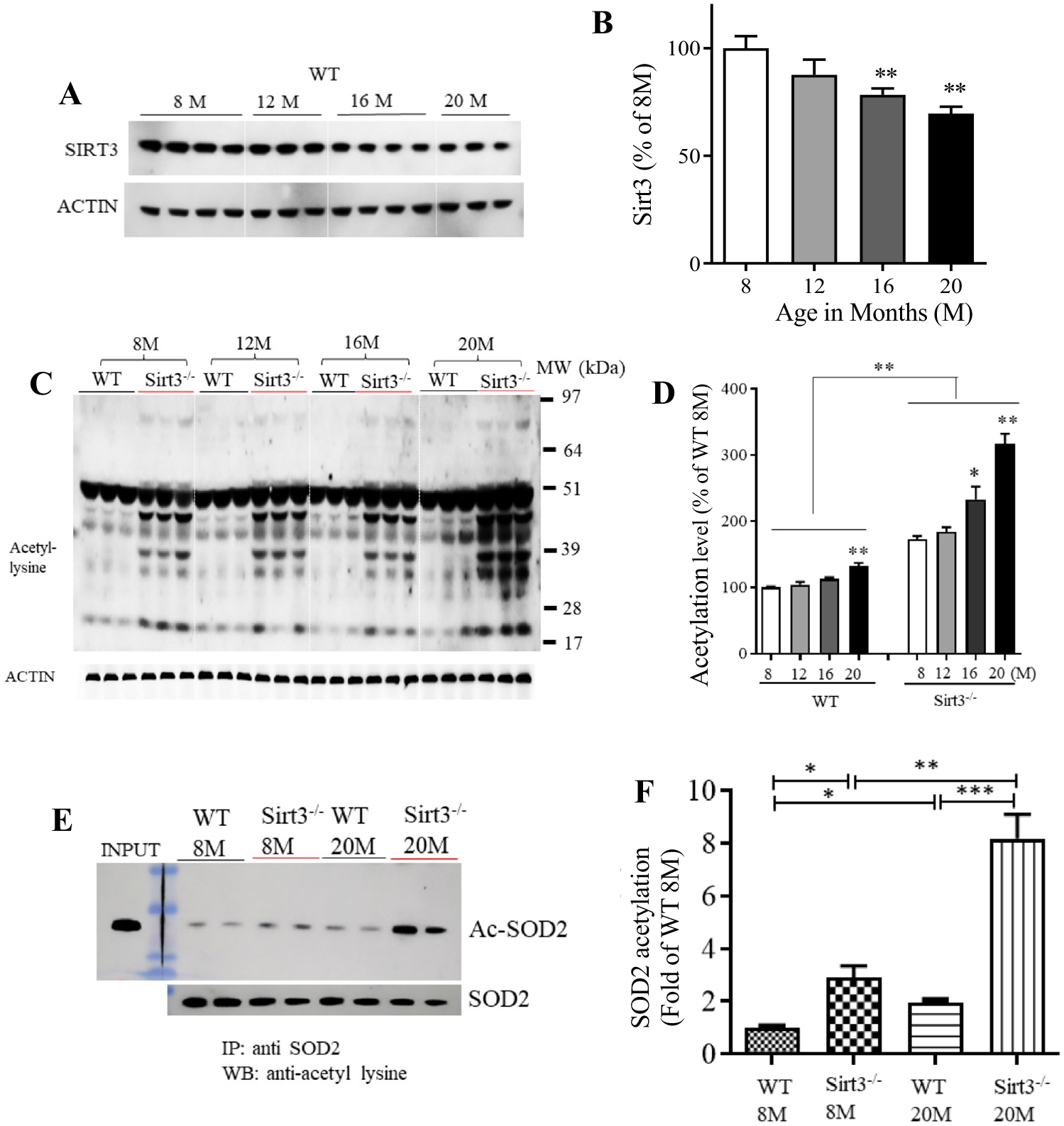
SIRT3 levels decrease and mitochondrial protein acetylation increases in the cerebral cortex during aging. A Immunoblot analysis of SIRT3 protein in cortical tissue from wild type (WT; Sirt3+/+) mice at different ages (8, 12, 16 and 20 month). Actin was used as a loading control. B SIRT3 levels were normalized to actin protein levels and expressed as a percentage of the value for 8-month-old mice. Values are mean ± SEM (n = 7 mice). **p < 0.01 (One-way ANOVA with Tukey post hoc test). C Immunoblot analysis of acetylated proteins detected using an acetyl-l-lysine antibody in protein samples prepared from cortical tissues of Sirt3+/+ and Sirt3−/− mice of 8, 12, 16 and 20 months (M) of age. Actin was used as a loading control. D Total protein acetylation levels (densitometric analysis of immunoreactivity in the entire lane) were normalized to actin protein levels and expressed as a percentage of the value for Sirt3+/+ 8-month-old mice. Values are mean ± SEM (n = 6 mice). *p < 0.05; **p < 0.01 (one-way ANOVA with Tukey post hoc test). E Proteins in lysates of cortical tissues from young (8 months) and old (20 months) Sirt3+/+ and Sirt3−/− mice were immunoprecipitated with an SOD2 or antibody and subjected to immunoblot analysis with antibodies to acetyl-lysine (Ac-SOD2). The blots were re-probed with SOD2 antibody to control for total SOD2 level. Input refers to the direct immunoblotting of tissue lysate using an antibody against SOD2. F Acetylated SOD2 level normalized to the SOD2 level and expressed as percentage of the value for 8-month-old Sirt3+/+ mice. Values are mean ± SEM (n = 6 mice). *p < 0.05; **p < 0.01 (one-way ANOVA with Tukey post hoc test)
Previous studies have provided evidence that mitochondrial function declines in neurons during normal aging (Toescu et al., 2000). We next determined whether age-related alterations in mitochondria are affected by SIRT3. Immunoblots were performed on cortical tissue samples from WT and Sirt3−/− mice of different ages using antibodies against the mitochondrial proteins cytochrome c oxidase subunit 1 (COX1) and voltage-dependent anion channel (VDAC). Levels of these two mitochondrial proteins did not change significantly during aging in WT mice, although there was a trend toward reduced COX1 levels (Fig. 2A, B). In contrast, Sirt3−/− mice exhibited significant age-related reductions in VDAC and COX1 protein levels in the cerebral cortex (Fig. 2C, D). To determine whether mitochondrial mass was affected by SIRT3 deficiency, we isolated mitochondria from cerebral cortex tissue of 20-month-old WT and Sirt3−/− mice and extracted the mitochondrial DNA (mtDNA). The mtDNA content of the cortical tissues was significantly decreased in aged Sirt3−/− mice by approximately 30 percent compared to WT mice (Fig. 2E, F). We further found that the reduction of mitochondrial mass is apparently not due to the suppression of mitochondrial biogenesis because the mRNA levels of several master mitochondrial biogenesis regulators (Cheng et al., 2012), peroxisome proliferator-activated receptor gamma co-activator (Pgc-1α), nuclear respiratory factor 1 (Nrf1) and mitochondrial transcription factor A (Tfam), were not altered in the cerebral cortex of Sirt3−/− mice compared to that of WT mice (Fig. S1).
Fig. 2.
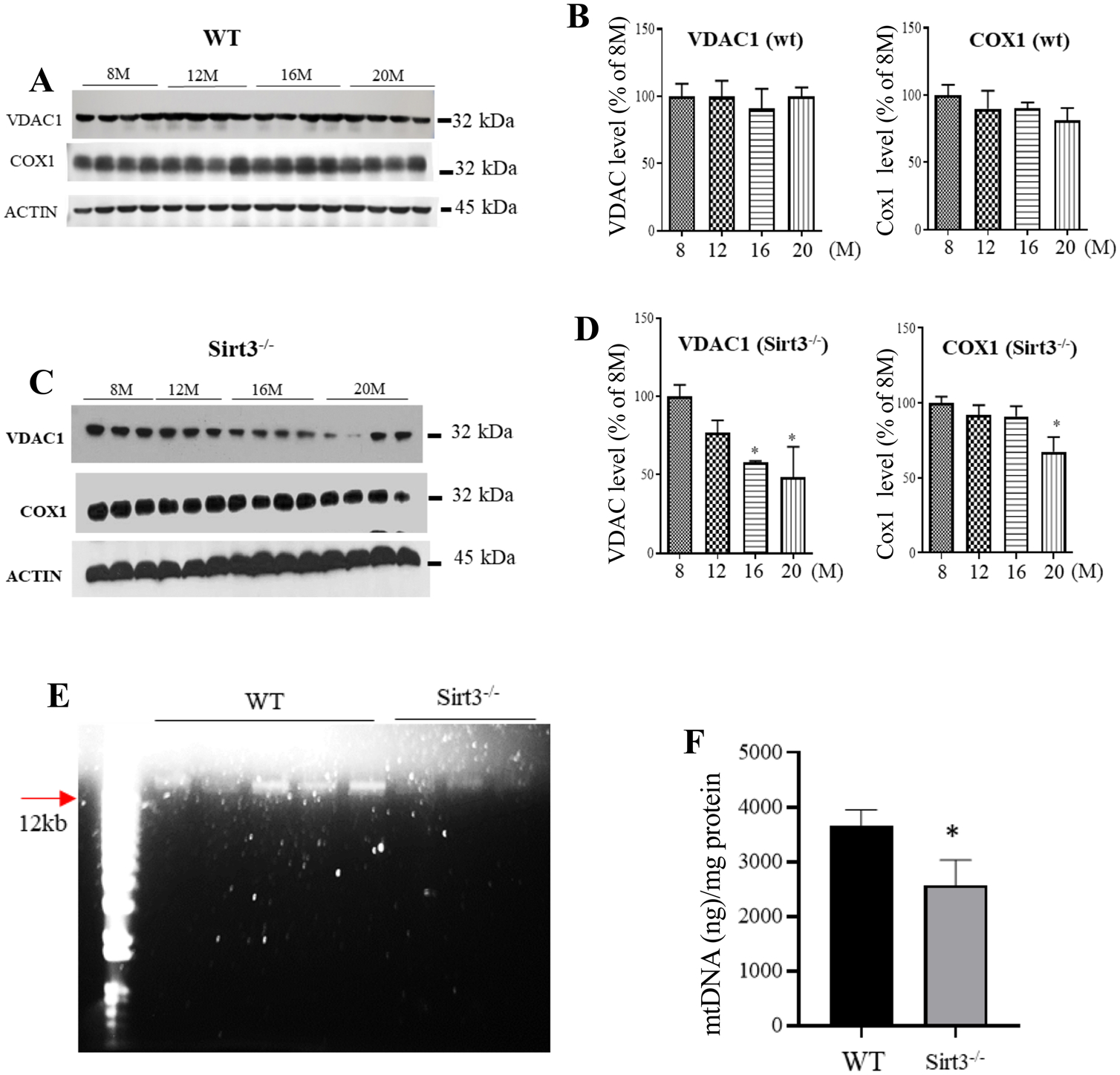
Levels of mitochondrial COX1 and VDAC, and DNA (mtDNA) are significantly reduced in the cerebral cortex of Sirt3−/− mice but not WT mice during aging. A, C Immunoblots of COX1 and VDAC proteins in cortical tissue of WT (A) and Sirt3−/− (C) mice at different ages (8, 12, 16 and 20 months). Actin was used as a loading control. B, D COX1 and VDAC levels (densitometric analysis) were normalized to actin protein levels and expressed as a percentage of the value for 8-month-old WT mice (B) or Sirt3−/− mice (D). Values are mean ± SEM (n = 6 mice). **p < 0.01 (One-way ANOVA with Tukey post hoc test). E, F A representative gel showing mtDNA (left) and results of measurement of the amount of mtDNA normalized to total protein concentration in cortical tissue from 20-month-old WT and Sirt3−/− mice. Values are mean ± SEM (n = 3–5 mice). *p < 0.05 (Student’s t test)
SIRT3 Plays a Critical Role in Constraining Neuronal Network Excitability
We previously reported that Sirt3 haploinsufficiency potentiates neuronal network hyperexcitability and seizures in an Alzheimer’s disease mouse model (Cheng et al., 2020). To determine whether SIRT3 is important for regulating neuronal network activity in normal mice, we implanted surface electrodes in the parietal cortex of 12-month-old WT, Sirt3±, and Sirt3−/− mice, and recorded EEG activity telemetrically with the mice in their home cages. We found that all Sirt3−/− mice exhibited frequent epileptiform spikes (sharp-wave discharges ≥ five-fold the amplitude of the basal voltage) and occasional burst-firing (Fig. 3A). In contrast, sharp-wave discharges were rarely seen, and no burst-firing was evident in WT and Sirt3 ± mice. We quantified total high-amplitude spikes and burst firings over a 24 h period, and the data are shown in Fig. 3B and C, respectively. We next studied whether SIRT3 levels influenced the susceptibility of mice to seizures induced by the glutamate receptor agonist kainic acid (KA). WT and Sirt3−/− of two different ages (8 and 20 months) were administered KA intraperitoneally (20 mg/kg) and were scored for behavioral manifestations of seizures every 10 min during the ensuing 120 min. Seizure severity in 8-month-old WT mice never exceeded a score of 1. In older mice (20-month-old), both WT and Sirt3± mice exhibited a significant increase in seizure score (Fig. 3D–F). For 20-month-old Sirt3−/− mice, the seizure score reached the maximum of 5, with the animals exhibiting generalized clonus and status epilepticus (Fig. 3F). There was a non-significant trend (p < 0.07) toward increased seizure scores in 20-month-old Sirt3−/− mice compared to 20-month-old WT mice (Fig. 3G). Altogether, the EEG and seizure score data show that SIRT3 is important in maintaining normal neuronal network activity and can protect against hyperexcitability during aging.
Fig. 3.
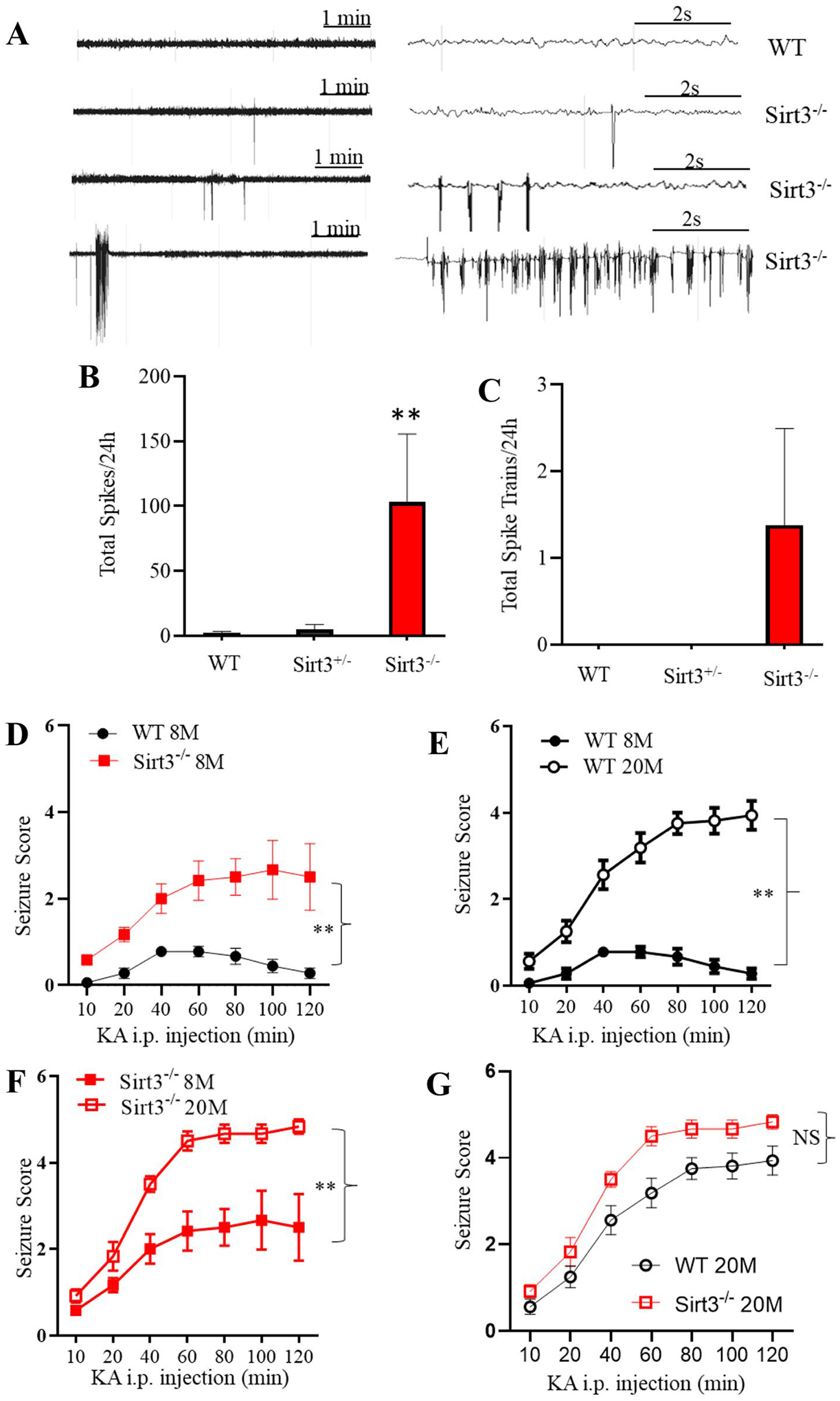
Evidence that SIRT3 is critical for maintaining normal neuronal network activity during aging. A EEG recordings showing examples of high-voltage spikes and bursts of epileptiform activity in 16-month-old WT and Sirt3−/− mice. B, C Quantitative analysis of spikes (B) and spike trains/burst (C) during a 24-h recording period in WT and Sirt3−/− mice. Values are mean ± SEM (n = 8 mice). **p < 0.01 (one-way ANOVA with Tukey post hoc test). D–G Both SIRT3 deficiency and aging sensitize mice to seizures and death induced by kainic acid (KA). WT and Sirt3−/− mice at 8 and 20 months of age were administered KA (20 mg/kg, intraperitoneally), and seizure scores were recorded at 10-min intervals during a 120-min period. The average values for every two consecutive 10-min intervals were plotted. Values are mean ± SEM (n = 6–9 mice/group). **p < 0.01 (two-way ANOVA followed by Tukey post hoc test)
Evidence that SIRT3 Protects Cerebral Cortical Neurons Against Aβ-Induced Apoptosis
We first treated primary cultured mouse cerebral cortical neurons with increasing concentrations of aggregating Aβ for 24 h, and then measured relative levels of SIRT3 by immunoblot analysis. Exposure of the neurons to Aβ resulted in a concentration-dependent reduction in SIRT3 levels (Fig. 4A, B). We next established cerebral cortical cell cultures from WT, Sirt3±, and Sirt3−/− mice, and performed experiments to determine whether SIRT3 affects the vulnerability of neurons to apoptosis induced by exposure to Aβ. Cultured neurons were exposed to 0, 1 μM, 5 μM, or 8 μM Aβ for 24 h and were then stained with the fluorescent DNA-binding dye Hoechst 33,342. The percentages of neurons exhibiting condensed and/or fragmented nuclear DNA were quantified We found that significantly more neurons lacking SIRT3 were killed by Aβ compared to WT and Sirt3± neurons (Fig. 4C, D). These data suggest that SIRT3 plays a protective role against Aβ neurotoxicity, as SIRT3-deficient neurons were significantly more vulnerable to apoptosis than WT neurons.
Fig. 4.
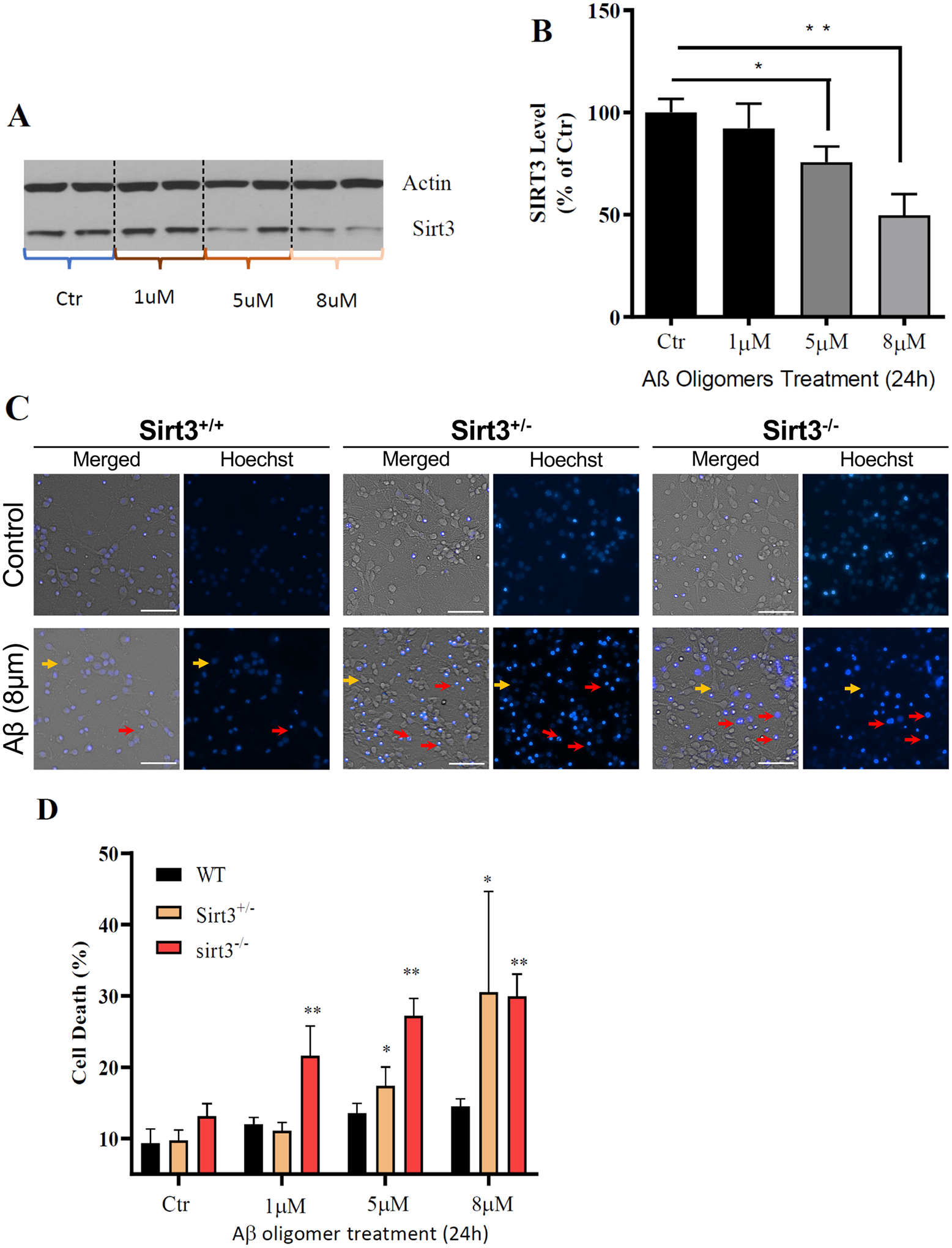
SIRT3 deficiency renders cultured cerebral cortical neurons vulnerable to Aβ toxicity. A Immunoblot analysis of SIRT3 protein levels in primary mouse cerebral cortical cultures (from WT mice) that had been treated for 24 h with vehicle or Aβ 1–42 at indicated concentrations. Actin was used as a loading control. B SIRT3 levels were normalized to actin protein levels and expressed as a percentage of the value for vehicle control group. Values are mean ± SEM (n = 4–6 cultures from two separate experiments). **p < 0.01 (one-way ANOVA with Tukey post hoc test). C Representative merged phase-contrast and Hoechst dye fluorescence (blue) images of cultured cortical neurons from Sirt3+/+, Sirt3± and Sirt3−/− mice that had been exposed for 24 h to Aβ. Viable neurons (examples indicated by the yellow arrows) exhibit faint/no diffuse nuclear DNA-associated (Hoechst) fluorescence and intact neurites, whereas dying/dead neurons (examples indicated by the red arrows) exhibit intense punctate Hoechst fluorescence as a result of nuclear chromatin condensation, and fragmented neurites. Scale bar = 50 μm. D Results of quantitative analysis of neuronal death. Values are mean ± SEM of cell counts performed on cultures established from 3 mice. *p < 0.05, **p < 0.01 (two-way ANOVA test with Tukey post hoc test)
Genetic Reduction of SIRT3 Levels Accelerates Aβ Plaque Accumulation in App/Ps1 Double Mutant Transgenic Mice
Transgenic mice expressing the human App Swedish and presenilin 1 delta E9 mutations under the control of a prion promoter (App/Ps1 mice) exhibit robust accumulation of Aβ plaques (Borchelt et al., 1996). We first used monoclonal antibody 6E10, which selectively recognizes human but not rodent Aβ, for immunoblot analysis of protein samples from the cerebral cortex of WT and App/Ps1 mutant mice of two different ages. No immunoreactivity was evident in blots from WT mice (Fig. S2). Only full-length APP was evident in 4-month-old App/Ps1 mutant mice, while full-length APP, Aβ monomer, and two higher-molecular-weight bands presumed to be Aβ aggregates were present in blots from 12-month-old App/Ps1 mutant mice (Fig. S2).
To determine whether SIRT3 influences the development of Aβ plaque pathology, we quantified Aβ plaque loads in the cerebral cortex and hippocampus of App/Ps1 mutant mice and App/Ps1 mutant mice with a genetic reduction in SIRT3 levels (Sirt3± App/Ps1). Coronal brain sections from 8 to 12-month-old mice were stained using immunofluorescence methods with antibody 6E10 and counterstaining with either DAPI or propidium iodide to label cell nuclei. Representative confocal images showing staining in the hippocampus and cerebral cortex are shown (Fig. 5A, B). Quantitative analysis showed that Aβ plaque load was significantly higher in both the cerebral cortex and hippocampus of 12-month-old Sirt3± App/Ps1 mutant mice compared to App/Ps1 mice (Fig. 5C).
Fig. 5.
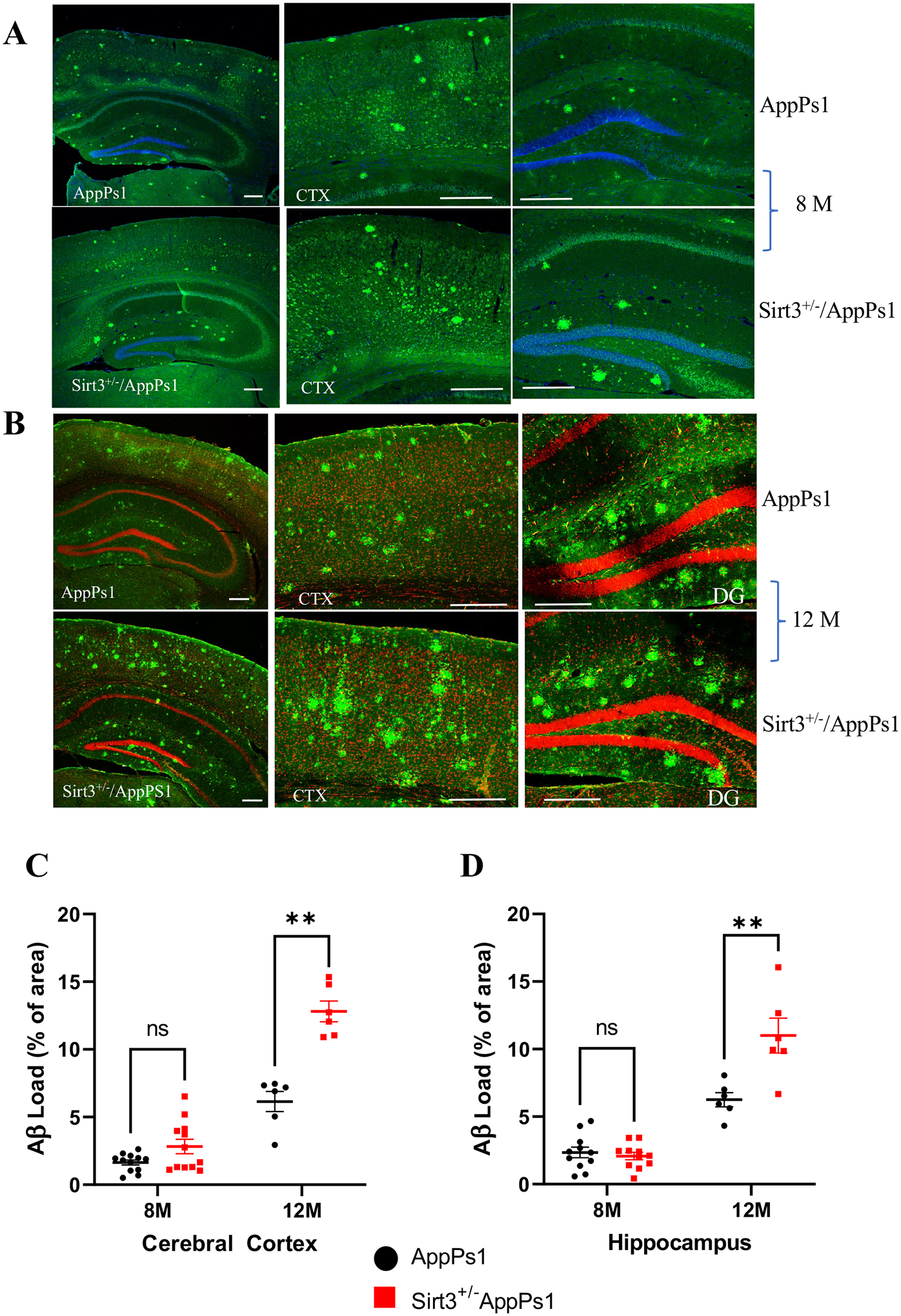
SIRT3 haploinsufficiency exacerbates Aβ accumulation in the cerebral cortex and hippocampus of AppPs1 mutant mice. A, B Confocal images showing Aβ immunoreactivity in the cerebral cortex and hippocampal regions of App/Ps1 and App/Ps1 Sirt3± mice at 8 months (A) and 12 months (B) of age, respectively. Coronal brain slices were stained for with Aβ antibody 6E10 (green) and cell nuclei were counterstained with propidium iodide (red) or DAPI (blue). C, D Quantitative analysis of Aβ load in matching cortical (C) and hippocampal (D) regions. Values are mean ± SEM (n = 6–12 brain slices analyzed from 3 to 6 mice). **p < 0.01 (two-way ANOVA followed by Tukey post hoc tests)
Discussion
While considerable evidence suggests that mitochondrial function is compromised in AD, the roles of specific mitochondrial proteins in the disease process are unknown. The protein deacetylase SIRT3 is increasingly recognized as a regulator of mitochondrial proteins and involved in a range of processes, including mitochondrial energy metabolism, free radical removal, and membrane channel function (Baeza et al., 2016; Carrico et al., 2018; Cheng et al., 2016). We found that SIRT3 levels decrease and protein acetylation levels increase in the cerebral cortex of mice during aging. Mice lacking SIRT3 exhibit hyperacetylation of numerous proteins including SOD2, and reduced markers of mitochondrial mass and function during aging. Previous studies have shown that SIRT3 expression declines and mitochondrial protein acetylation increases in several peripheral tissues during normal aging (Fu et al., 2016). Data from studies of animal models suggest that such age-related decrements in SIRT3-mediated protein deacetylation play roles in several common disorders of aging, including hypertension, osteoarthritis, and fatty liver disease (Dikalova et al., 2020; Fu et al., 2016; Kendrick et al., 2011). Therefore, a reduction in SIRT3 expression may be a common feature of aging that contributes to functional decline and susceptibility to disease.
The consequences of age-related mitochondrial protein hyperacetylation for neuronal network function and vulnerability to disease have not been established. Our EEG data show that mice lacking SIRT3 exhibit abnormal spike discharges and burst-firing. This neuronal network hyperexcitability is similar to what has been reported to be associated with cognitive decline in people with mild cognitive impairment and AD (Xu et al., 2021). The first evidence that Aβ contributes to neuronal network hyperexcitability came from experiments showing that Aβ renders cultured human and rodent cerebral cortical neurons vulnerable to glutamate excitotoxicity (Koh et al., 1990; Mattson et al., 1992). Subsequent studies demonstrated that as Aβ aggregates, it generates reactive oxygen species that cause membrane lipid peroxidation (Hensley et al., 1994; Mark et al., 1995a). The lipid peroxidation product 4-hydroxynonenal has been shown to impair the function of neuronal membrane ion-motive ATPases (sodium and calcium pumps) and glucose transporters (Keller et al., 1997; Mark et al., 1995a, 1997b). As a consequence, Aβ impairs the ability of neurons to restore ion gradients when stimulated by glutamate resulting in excessive accumulation of intracellular Ca2+, damage to dendrites, and cell death (Mattson et al., 1992). Additional evidence that Aβ accumulation contributes to neuronal network hyperexcitability in AD comes from studies of mouse models (Kazim et al., 2021; Mattson, 2020; Palop & Mucke, 2010).
We found that SIRT3 deficiency greatly increases the vulnerability of mice to seizures induced by kainic acid and increases the vulnerability of cultured cerebral cortical neurons to Aβ toxicity. Previous findings suggest that SIRT3 deficiency results in reduced SOD2 activity, increased oxidative damage, and depletion of ATP (Cheng et al., 2016). Oxidative stress and reduced ATP levels result in impaired function of the plasma membrane Na+ pump and may thereby promote membrane depolarization.
Increasing evidence suggests that inhibitory GABAergic interneurons may be particularly vulnerable to mitochondrial dysfunction and degeneration in AD (for review see Mattson, 2020). Fast-spiking interneurons that express parvalbumin are vulnerable to excitotoxicity because of their high numbers of mitochondria and high energy demand. A recent study (Cheng et al., 2020) showed that whereas fast-spiking GABAergic interneurons do not degenerate in App/Ps1 mice, they do degenerate in App/Ps1 Sirt3± mice. Moreover, feeding App/Ps1 Sirt3± mice a ketone ester to boost SIRT3 expression and their energy level prevented seizures. These observations and those of the present study collectively support the notion that SIRT3 plays a major role in preserving mitochondrial function in highly active neurons and is therefore critical for maintaining optimal neural network excitability.
Tyagi et al. (2020) recently reported that SIRT3 homozygous knockout mice exhibit systemic insulin resistance. They generated APP/PS1 mutant mice lacking SIRT3 and found that those mice exhibit increased cerebral Aβ accumulation and neuroinflammation. They suggested that the exacerbation of cerebral Aβ pathology in SIRT3-deficient mice resulted from peripheral insulin resistance. Previous studies have compared insulin sensitivity in WT and SIRT3 homozygous mice (Hirschey et al., 2011; Jing et al., 2011). In the present study we found that APP/PS1 with an approximately 50% reduction in SIRT3 levels exhibit increased cerebral Aβ pathology and seizure susceptibility. We did not evaluate peripheral glucose regulation in the present study and so do not know whether SIRT3 haploinsufficient mice exhibit insulin resistance.
We found that SIRT3 haploinsufficiency accelerates Aβ plaque accumulation in the cerebral cortex and hippocampus of App/Ps1 mutant transgenic mice. Previous findings suggest that aberrant neuronal hyperexcitability increases the production and deposition of neurotoxic Aβ (reviewed in Mattson, 2020; Ovsepian & O’Leary, 2016). Accordingly, treatment of App/Ps1 mutant transgenic mice with anticonvulsant drugs lessens the accumulation of Aβ plaques (Zhang et al., 2014). Our findings therefore suggest a scenario in which age-related decreases in SIRT3 promotes neuronal hyperexcitability which, in turn, accelerates Aβ accumulation. However, the increased accumulation of Aβ resulting from reduced SIRT3 levels could also result from reduced clearance of Aβ. Further studies will be required to elucidate if and how mitochondrial SIRT3 affects Aβ accumulation.
The present findings suggest that mitochondrial SIRT3 plays an important role in protecting against neuronal network hyperexcitability and Aβ accumulation. These findings further support the view that interventions that increase SIRT3 expression and/or deacetylase activity might protect neuronal networks against hyperexcitability and degeneration in AD. Consistent with this possibility, we previously reported that running wheel exercise increases SIRT3 protein levels in cerebral cortical cells in mice and protects neurons against excitotoxicity (Cheng et al., 2016). It has also been reported that SIRT3 mediates the enhancement of GABAergic inhibitory neuron activity in the hippocampus in response to intermittent fasting (Mark et al., 1995b). The latter study also showed that intermittent fasting ameliorates deficits in hippocampal synaptic plasticity in APP mutant knock-in mice in a SIRT3-dependent manner. Other potential interventions that target hyperexcitability and have been reported to ameliorate deficits in transgenic mouse models of AD include a ketone ester (Kishiwaya et al., 2013; Cheng et al., 2020) and anticonvulsant drugs (Mark et al., 1997a; Zhang et al., 2014). One study reported that a low dose of the drug levetiracetam improved cognition in elderly patients with mild cognitive impairment (Bakker et al., 2012), providing support for further trials of anticonvulsants in people with AD.
Supplementary Material
Acknowledgements
We thank Dr. David Gius (NCI) for providing breeding pairs of Sirt3−/− mice. This work was supported by the Intramural Research Program of the National Institute on Aging, NIH.
Funding
This research was supported by the Intramural Research Program of the National Institute on Aging of the National Institutes of Health.
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s12017-022-08713-2.
Conflict of interest None of the authors have any conflicts of interest to disclose.
Data Availability
Data are available from the National Institute on Aging Intramural Research Program.
References
- Baeza J, Smallegan MJ, & Denu J (2016). M. Mechanisms and dynamics of protein acetylation in mitochondria. Trends in Biochemical Sciences, 41, 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, & Gallagher M (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron, 74, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, & Sisodia SS (1996). Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron, 17, 1005–1013. [DOI] [PubMed] [Google Scholar]
- Carrico C, Meyer JG, He W, Gibson BW, & Verdin E (2018). The mitochondrial acylome emerges: Proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metabolism, 27, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wang J, Ghena N, Zhao Q, Perone I, King TM, Veech RL, Gorospe M, Wan R, & Mattson MP (2020). SIRT3 haploinsufficiency aggravates loss of GABAergic interneurons and neuronal network hyperexcitability in an Alzheimer’s disease model. Journal of Neuroscience, 40, 694–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, & Mattson MP (2012). Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nature Communications, 3, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, Bohr VA, & Mattson MP (2016). Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metabolism, 23, 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, Eckert A, Harvey J, Jeggo R, Jhamandas JH, Kann O, la Cour CM, Martin WF, Mithieux G, Moreira PI, … Millan MJ (2020). Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nature Reviews Drug Discovery, 19, 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalova AE, Pandey A, Xiao L, Arslanbaeva L, Sidorova T, Lopez MG, Billings FT, Verdin E, Auwerx J, Harrison DG, & Dikalov SI (2020). Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circulation Research, 126, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR, Hakim M, Pan Y, Verdin E, & Griffin TM (2016). Aging promotes sirtuin 3-dependent cartilage superoxide dismutase 2 acetylation and osteoarthritis. Arthritis & Rhematology, 68, 1887–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave JA, Arathoon LR, Trinh D, Lizal KE, Giguère N, Barber JHM, Najarali Z, Khan MH, Thiele SL, Semmen MS, Koprich JB, Brotchie JM, Eubanks JH, Trudeau LE, & Nash JE (2017). Sirtuin 3 rescues neurons through the stabilisation of mitochondrial biogenetics in the virally-expressing mutant α-synuclein rat model of parkinsonism. Neurobiology of Diseases, 106, 133–146. [DOI] [PubMed] [Google Scholar]
- Hamilton KA, Wang Y, Raefsky SM, Berkowitz S, Spangler R, Suire CN, Camandola S, Lipsky RH, & Mattson MP (2018). Mice lacking the transcriptional regulator Bhlhe40 have enhanced neuronal excitability and impaired synaptic plasticity in the hippocampus. PLoS One, 13(5), e0196223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, & Butterfield DA (1994). A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proceedings of the National Academy of Sciences USA, 91, 3270–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi S, Heistek TS, Scheltens P, Neumann U, Shimshek DR, Mansvelder HD, Smit AB, & van Kesteren RE (2020). Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Molecular Psychiatry, 25, 3380–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stančáková A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV Jr., … Verdin E (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular Cell, 44, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger HC, Weitzner DS, Rudy CC, Hickman JE, Libell EM, Speer RR, Gerhardt GA, & Reed MN (2015). Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. Journal of Neurochemistry, 135, 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, & Kahn CR (2011). Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proceedings of the National Academy of Sciences USA, 108, 14608–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, & Avgerinos KI (2020). Brain glucose and ketone utilization in brain aging and neurodegenerative diseases. International Review of Neurobiology, 154, 79–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, Okun E, Clarke K, Mattson MP, & Veech RL (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiology of Aging, 34, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazim SF, Seo JH, Bianchi R, Larson CS, Sharma A, Wong RKS, Gorbachev KY, & Pereira AC (2021). Neuronal network excitability in Alzheimer’s disease: The puzzle of similar versus divergent roles of amyloid beta and tau. eNeuro. 10.1523/ENEURO.0418-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, & Mattson MP (1997). Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. Journal of Neurochemistry, 69, 273–284. [DOI] [PubMed] [Google Scholar]
- Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J, Gius D, Sack MN, Jing E, Kahn CR, Friedman JE, & Jonscher KR (2011). Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. The Biochemical Journal, 433, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, & Fang EF (2017). Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends in Neurosciences, 40, 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Yang LL, & Cotman CW (1990). Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Research, 533, 315–320. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Ashford JW, Goodman Y, & Mattson MP (1995a). Anticonvulsants attenuate amyloid beta-peptide neurotoxicity, Ca2+ deregulation, and cytoskeletal pathology. Neurobiology of Aging, 16, 187–198. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Hensley K, Butterfield DA, & Mattson MP (1995b). Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. Journal of Neuroscience, 15, 6239–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, & Mattson MP (1997a). A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. Journal of Neurochemistry, 68, 255–264. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, & Mattson MP (1997b). Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. The Journal of Neuroscience, 17, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2020). Involvement of GABAergic interneuron dysfunction and neuronal network hyperexcitability in Alzheimer’s disease: Amelioration by metabolic switching. International Review of Neurobiology, 154, 191–205. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, & Rydel RE (1993). Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron, 10, 243–254. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, & O’Leary VB (2016). Neuronal activity and amyloid plaque pathology: An update. Journal of Alzheimer’s Disease, 49, 13–19. [DOI] [PubMed] [Google Scholar]
- Palop JJ, & Mucke L (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural net- works. Nature Neuroscience, 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Filadi R, Pizzo P, & Duchen MR (2021). Excitotoxicity revisited: Mitochondria on the verge of a nervous breakdown. Trends in Neurosciences, 44, 342–351. [DOI] [PubMed] [Google Scholar]
- Shi H, Deng HX, Gius D, Schumacker PT, Surmeier DJ, & Ma YC (2017). Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Human Molecular Genetics, 26, 1915–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, & Remy S (2014). Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron, 84, 1023–1033. [DOI] [PubMed] [Google Scholar]
- Tyagi A, Mirita C, Taher N, Shah I, Moeller E, Tyagi A, Chong T, & Pugazhenthi S (2020). Metabolic syndrome exacerbates amyloid pathology in a comorbid Alzheimer’s mouse model. Biochimica et Biophysica Acta Molecular Basis of Disease, 1866, 165849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, Karydas AM, Coppola G, Roberson ED, Miller BL, Garcia PA, Kirsch HE, Mucke L, & Nagarajan SS (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Annals of Neurology, 80, 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, & Miller BL (2017). Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurology, 16, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lavrencic L, Radford K, Booth A, Yoshimura S, Anstey KJ, Anderson CS, & Peters R (2021). Systematic review of coexistent epileptic seizures and Alzheimer’s disease: Incidence and prevalence. Journal of the American Geriatrics Society, 69, 2011–2020. [DOI] [PubMed] [Google Scholar]
- Yao J, Sun B, Institoris A, Zhan X, Guo W, Song Z, Liu Y, Hiess F, Boyce AKJ, Ni M, Wang R, Ter Keurs H, Back TG, Fill M, Thompson RJ, Turner RW, Gordon GR, & Chen SRW (2020). Limiting RyR2 open time prevents Alzheimer’s disease-related neuronal hyperactivity and memory loss but not beta-amyloid accumulation. Cell Reports, 32, 108169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MY, Zheng CY, Zou MM, Zhu JW, Zhang Y, Wang J, Liu CF, Li QF, Xiao ZC, Li S, Ma QH, & Xu RX (2014). Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiology of Aging, 35, 2713–2725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the National Institute on Aging Intramural Research Program.