

Key Points
-
•
A distinct minor subpopulation of LN CLL cells is activated, interfaces with the immune milieu, and promotes disease progression.
-
•
Clonal evolution primarily occurs in LN and associates with a suppressed T-cell inflammatory response.
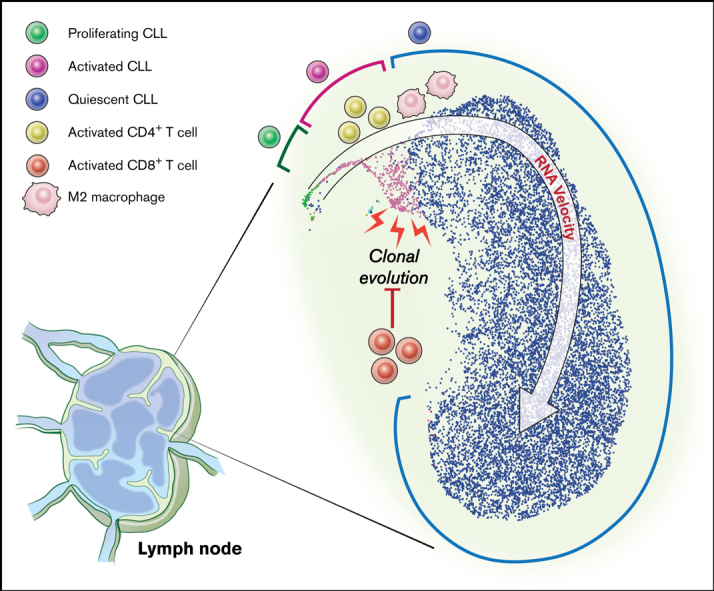
Visual Abstract
Abstract
In chronic lymphocytic leukemia (CLL), B-cell receptor signaling, tumor–microenvironment interactions, and somatic mutations drive disease progression. To better understand the intersection between the microenvironment and molecular events in CLL pathogenesis, we integrated bulk transcriptome profiling of paired peripheral blood (PB) and lymph node (LN) samples from 34 patients. Oncogenic processes were upregulated in LN compared with PB and in immunoglobulin heavy-chain variable (IGHV) region unmutated compared with mutated cases. Single-cell RNA sequencing (scRNA-seq) distinguished 3 major cell states: quiescent, activated, and proliferating. The activated subpopulation comprised only 2.2% to 4.3% of the total tumor bulk in LN samples. RNA velocity analysis found that CLL cell fate in LN is unidirectional, starts in the proliferating state, transitions to the activated state, and ends in the quiescent state. A 10-gene signature derived from activated tumor cells was associated with inferior treatment-free survival (TFS) and positively correlated with the proportion of activated CD4+ memory T cells and M2 macrophages in LN. Whole exome sequencing (WES) of paired PB and LN samples showed subclonal expansion in LN in approximately half of the patients. Since mouse models have implicated activation-induced cytidine deaminase in mutagenesis, we compared AICDA expression between cases with and without clonal evolution but did not find a difference. In contrast, the presence of a T-cell inflamed microenvironment in LN was associated with clonal stability. In summary, a distinct minor tumor subpopulation underlies CLL pathogenesis and drives the clinical outcome. Clonal trajectories are shaped by the LN milieu, where T-cell immunity may contribute to suppressing clonal outgrowth. The clinical study is registered at clinicaltrials.gov as NCT00923507.
Introduction
The compartmentalization of chronic lymphocytic leukemia (CLL) in peripheral blood (PB), bone marrow, and secondary lymphoid tissue has permitted the study of intrinsic and extrinsic drivers of tumorigenesis and progression. We previously showed that the lymph node (LN) tumor microenvironment (TME) activates B-cell receptor (BCR) and nuclear factor κ B signaling and induces the proliferation of CLL cells.1 T–cell-derived CD40L and interleukin-4(IL), and possibly macrophage-derived B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), also promote tumor survival and signaling.2, 3, 4 While receiving support from the TME, CLL cells modulate their surroundings by secreting chemokines, such as CCL3, CCL4, and CCL22, to attract T cells.5 These findings indicate complex, bidirectional crosstalk between CLL cells and their TME.
Beyond these microenvironmental dependencies, a diverse set of putative pathogenic mutations have been identified through large-scale sequencing efforts.6,7 Heterogeneity in the genetic architecture of CLL has been closely linked to disease aggressiveness and its capacity to evolve under the selective pressures of treatment.6,8 Clonal composition furthermore influences the CLL transcriptome in several ways. For example, biased expression of the mutated allele relative to the reference allele has been observed with mutations affecting DNA damage response genes such as ATM and TP53.9 Individual or cooccurrence of putative driver mutations can also result in more global changes in the transcriptome, dysregulating key oncogenic pathways.10,11
Herein, we integrate transcriptome profiling at the bulk and single-cell level with bulk whole-exome sequencing (WES) of paired PB and LN samples to understand the intersection between the TME and molecular events in CLL pathogenesis.
Materials and methods
Details and resources are available in supplemental Methods.
Patient samples
Patient samples were obtained between 12 April 2005 and 26 August 2014 after written informed consent in accordance with the Declaration of Helsinki, applicable federal regulations, and requirements from the National Heart, Lung, and Blood Institute Institutional Review Board. The clinical study is registered at clinicaltrials.gov as NCT00923507.
Sample processing and storage
PB mononuclear cells were isolated by density gradient centrifugation and cryopreserved. LN biopsies were either snap-frozen and stored at −80°C or mechanically disaggregated into single-cell suspensions (SCSs) and cryopreserved. CD19+ CLL cells were purified from PB mononuclear cells or LN SCSs by immunomagnetic positive selection with LS columns and stored as cell pellets at −80°C.
Bulk RNA sequencing and analysis
Total RNA libraries were prepared according to the Illumina TruSeq protocol and sequenced on a HiSeq 2000 (Illumina). Gene set enrichment analysis12 was used to test for enrichment of a curated set of lymphocyte gene expression signatures (supplemental Table 2). CIBERSORT was used for deconvolution of bulk RNA-seq data from LN samples of patients with CLL and normal donors.1
Single-cell RNA sequencing (scRNA-seq) and analysis
Single-cell libraries were created from LN SCSs with the Chromium Single Cell 3′ Reagent Kit V2. Libraries were sequenced on NovaSeq (Illumina) and processed by Cell Ranger Single Cell Software Suite 3.0.1.13 Downstream analyses were performed on Seurat v3.14 For RNA velocity, cell transitions were estimated based on quantification of unspliced and spliced mRNA.15,16
WES and analysis
DNA was extracted from CD19+ CLL cells using DNeasy kits (Qiagen, Germantown, MD). Libraries were prepared using Agilent SureSelect Human All Exon kit and Nextera DNA Library Prep for Enrichment. Pooled libraries were sequenced on Illumina next-generation sequencers. Binary alignment map (BAM) files were generated from the Picard pipeline and then analyzed on the Firehose platform, including quality control, local realignment, mutation calling, small insertion and deletion identification, rearrangement detection, and coverage calculations. ABSOLUTE17 was used to estimate sample purity, ploidy, absolute somatic copy number, and the cancer cell fraction (CCF) of mutations. To distinguish subclonal populations, a Bayesian clustering procedure18,19 was applied to cancer cell fractions (CCFs) across PB and LN samples from each patient.
Immunohistochemical staining
Formalin-fixed, paraffin-embedded LN samples were stained with CD163. Images were captured at a magnification of 10× on an Olympus Bx41 microscope. The number of CD163+ cells was counted in 5 consecutive fields at 40× by a hematopathologist and then averaged to obtain the number of cells per high-power field.
T-cell immunophenotyping
Cryopreserved LN SCSs were thawed and subjected to CD19+ immunomagnetic depletion with LD columns. Cells were preincubated with LIVE/DEAD fixable Aqua Dead Cell Stain then stained with a cocktail of antibodies against CD3, CD4, CD8, CD45RO, CCR7, HLA-DR, CD19, and CD14. Fluorescence minus one controls were used to set gates for positive events. Cells were analyzed on LSRFortessa machine and FlowJo.
Quantification and statistical analysis
Continuous variables were compared between patient subgroups with the 2-sided Student t test. Correlations were computed using Pearson correlation. The Kaplan-Meier method was used to estimate progression-free survival, and differences between patient subgroups were compared using the log-rank test. Statistical analyses were performed in Prism 7 or R.
The bulk and scRNA-seq data have been deposited in the Gene Expression Omnibus database under accession numbers GSE161711 and GSE161610. The WES data have been deposited in the dbGaP database under dbGaP accession phs002297.v1.p1.
Results
Microenvironmental modulation of tumor cells
Prior studies of microarray-based gene expression profiling and in vivo deuterium labeling have revealed the distinct biology of CLL across anatomic compartments.1,20 To deeply investigate the behavior of CLL in the TME, we performed total RNA sequencing (RNA-seq) of purified CD19+ tumor cells from paired PB and LN samples in 29 patients with treatment-naïve CLL or small lymphocytic lymphoma (Figure 1A,B; Table 1). Principal component analysis of bulk transcriptomes separated samples by tissue type (Figure 2A). We identified upregulation of 253 genes and downregulation of 32 genes in purified tumor cells from the LN compared with the PB (fold-change ≥2, false discovery rate [FDR] <0.05) (Figure 2B; supplemental Table 1).
Figure 1.

Patient samples and experimental design. (A) Swimmer plot of each patient from the time of diagnosis to last follow-up or death. Diagnosis of CLL or small lymphocytic lymphoma and timing of sample collection and treatment initiation are shown. Some patients had not received the first treatment at the last follow-up. The types of sequencing performed on PB and LN samples are indicated on the left. (B) Illustration of the types of sequencing performed: (1) bulk RNA-seq (n = 29); (2) WES (n = 14) of CD19+ tumor cells from paired PB and LN samples; (3) scRNA-seq of paired PB and LN samples (n = 5); (4) bulk RNA-seq of unsorted CLL LN samples (n = 34); and (5) bulk RNA-seq of unsorted normal LN samples (n = 4).
Table 1.
Demographic and clinical characteristics and sequencing analysis of patients
| Patient ID | Age | Sex | Dx | IGHV | Cytogenetic abnormalities∗ | Paired LN–PB |
LN |
||
|---|---|---|---|---|---|---|---|---|---|
| RNA-seq (CD19+) | scRNA-seq | WES (CD19+) | RNA-seq | ||||||
| CLL-C01 | 61 | F | CLL | U | Tri 12 | Yes | No | Yes | Yes |
| CLL-C02 | 60 | F | CLL | M | Del 11q | Yes | No | No | Yes |
| CLL-C03 | 60 | M | CLL | M | Del 13q | Yes | Yes | No | Yes |
| CLL-C04 | 51 | M | CLL | U | Del 11q | Yes | No | No | Yes |
| CLL-C05 | 57 | M | CLL | M | Del 11q | Yes | No | Yes | Yes |
| CLL-C06 | 58 | M | CLL | U | None | Yes | No | No | Yes |
| CLL-C07 | 61 | F | CLL | U | None | Yes | No | No | Yes |
| CLL-C08 | 59 | F | CLL | U | Del 11q | Yes | No | Yes | Yes |
| CLL-C11 | 67 | F | CLL | M | Del 17p | Yes | Yes | No | Yes |
| CLL-C13 | 68 | F | CLL | U | Tri 12 | Yes | No | No | Yes |
| CLL-C14 | 55 | F | CLL | U | Del 13q | Yes | No | Yes | Yes |
| CLL-C24 | 52 | M | CLL | U | Del 17p | Yes | Yes | Yes | Yes |
| CLL-C25 | 53 | M | CLL | U | Del 13q | Yes | No | No | Yes |
| CLL-C30 | 43 | M | CLL | U | Del 17p | Yes | No | Yes | Yes |
| CLL-C31 | 48 | M | CLL | U | Del 17p | Yes | No | No | Yes |
| CLL-C32 | 48 | M | CLL | U | Del 13q | Yes | No | Yes | Yes |
| CLL-C33 | 50 | M | CLL | U | Del 11q | Yes | Yes | Yes | Yes |
| CLL-C34 | 49 | F | CLL | M | Del 13q | Yes | No | Yes | Yes |
| CLL-C35 | 60 | F | CLL | U | Del 17p | Yes | No | Yes | Yes |
| CLL-C36 | 57 | F | CLL | U | Del 13q | Yes | No | No | Yes |
| CLL-C37 | 73 | M | CLL | M | Del 11q | Yes | No | No | Yes |
| CLL-C39 | 73 | F | SLL | N/A | Tri 12 | No | No | No | Yes |
| CLL-C41 | 75 | M | CLL | U | Del 11q | Yes | No | Yes | Yes |
| CLL-C42 | 55 | F | SLL | M | Tri 12 | No | No | No | Yes |
| CLL-C43 | 55 | F | CLL | U | Tri 12 | Yes | No | Yes | Yes |
| CLL-C44 | 47 | M | CLL | M | Del 17p | Yes | Yes | No | Yes |
| CLL-C45 | 56 | F | CLL | M | Del 13q | Yes | No | Yes | Yes |
| CLL-C46 | 67 | M | CLL | U | Del 13q | Yes | No | Yes | Yes |
| CLL-C47 | 59 | F | SLL | U | Tri 12 | Yes | No | No | Yes |
| CLL-C49 | 69 | F | CLL | U | Del 17p | Yes | No | No | Yes |
| CLL-C51 | 75 | M | SLL | M | Tri 12 | No | No | No | Yes |
| CLL-C53 | 71 | F | SLL | M | Tri 12 | No | No | No | Yes |
| CLL-C55 | 66 | M | SLL | U | None | No | No | No | Yes |
Dx, diagnosis; IGHV, immunoglobulin heavy-chain gene; M, mutated; sc, single-cell; U, unmutated.
By fluorescent in situ hybridization and according to hierarchal classification (Döhner et al 2000).
Figure 2.

The CLL transcriptome is modulated by the tumor microenvironment. (A) Principal component analysis of bulk RNA-seq in CD19+ tumor cells from paired PB (red) and LN (green) samples (n = 29 pairs). (B) Volcano plot of log2 fold-change (FC) in gene expression between PB and LN bulk RNA-seq vs -log10 false discovery rate (FDR). Differentially expressed genes (dark blue, n = 285) were defined as FC ≥2 and FDR <0.05. (C) Gene signatures enriched (normalized enrichment score ≥|1.6| as shown by the dotted line and FDR <0.05) in either PB or LN bulk RNA-seq. Signatures were categorized as signaling pathways, transcription factor targets, and cellular processes. Signatures comprised of upregulated genes are indicated by an up arrow (↑) while downregulated genes are indicated by a down arrow (↓). (D) Comparison of the indicated signatures between mutated (M; n = 8) and unmutated (U; n = 21) CLL. A signature score is the average expression of genes comprising each signature for a given sample. Box and whiskers show the median, IQR, and 1.5 times IQR of the indicated signature scores across LN samples. IQR, interquartile range.
To evaluate the impact of the TME on the cellular transcriptome, we applied gene set enrichment analysis using a curated list of gene expression signatures of signaling pathways, transcription factor regulation, and cellular processes such as proliferation and metabolism (Figure 2C; supplemental Table 2). Consistent with previously reported gene expression profiling,1,21 we confirmed BCR, downstream NFκB, and NOTCH activation as enriched pathways in the LN. Crosstalk with the TME was supported by signatures of CD40 and cytokine signaling. Proliferation and metabolism signatures were also enriched in the LN. In contrast, PB tumor cells were characterized by 2 signatures: cellular quiescence and the transcription factor Krüppel-like factor 2, which maintains mature B cells in a resting state.22
Somatic hypermutation of the immunoglobulin heavy-chain variable region (IGHV) is associated with cellular energy manifesting as reduced signaling capacity upon in vitro stimulation of the BCR by anti-immunoglobulin–M and better prognosis.23,24 To assess differences between mutated (M-CLL) and unmutated (U-CLL) CLL cases in the TME, we compared the expression of a BCR signature in LN samples. We found overexpression of this BCR signature in U-CLL compared with M-CLL (P = .03; unpaired t test) (Figure 2D). U-CLL cases also overexpressed NFκB, interferon, and T-cell cytokine signatures (P < .05). No signatures were overexpressed in M-CLL cases.
Intratumoral transcriptional heterogeneity within the TME
To further dissect the transcriptional heterogeneity of CLL cells within the TME, we performed a high-throughput, droplet-based single-cell RNA-seq of 5 LN samples. We sequenced a median of 2946 (range, 2605-3377) cells per sample and detected a median of 1171 (range, 916-1374) genes per cell. A total of 15 107 cells were analyzed by Seurat v314 using anchor correspondences across samples to distinguish shared and nonoverlapping cell states (Figure 3A). Three cell lineages (CLL, T cells, and monocytes) were identified (Figure 3B,E). CLL cells were comprised of 3 major subpopulations defined by quiescent, activated, and proliferating cell states (Figure 3B,E). Compared with the major subpopulation of quiescent CLL cells, activated CLL cells, accounting for 2.2% to 4.3% of tumor cells, overexpressed BCR, NOTCH, proliferation, and metabolism signatures (Figure 3F). Proliferating CLL cells, identified by G2/M and S phase markers, comprised 0.4% to 1.0% of tumor cells (supplemental Figure 1A).
Figure 3.

Single-cell transcriptomic analysis reveals intratumoral heterogeneity in LNs. (A) Uniform manifold approximation and projection (UMAP) of integrated scRNA-seq data from 15 107 single cells across 5 LN samples. Each color represents a different LN sample. (B) Clustering of LN single cells into major cell lineages and subpopulations. Each color represents a different cell identity cluster. CLL cells clustered into 3 subpopulations (proliferating, activated, quiescent) based on differences in transcriptional profiles. (C) RNA velocities derived from deterministic modeling projected on a UMAP of LN samples. CLL cells begin in the proliferating state, transition to the activated state, and end in the quiescent state. (D) UMAP of single cells from PB and LN (n = 5 pairs). Cell rendering after integration of PB and LN data is slightly different from that of LN data alone, as expected when computed from combined data. (E) Heatmap of the 5 most differentially expressed genes in each cell identity cluster. The color scale represents the number of reads mapping to the indicated gene per 10 000 reads. The size of the quiescent CLL population has been downscaled to improve the visualization of smaller clusters. Each column representing a quiescent CLL cell is 0.1× the width of columns representing other cells. (F) Significant overlap (hypergeometric test, FDR <0.05) between genes upregulated in activated CLL cells in LN and the indicated gene signatures. (G) Percentage of activated and proliferating CLL cells in LN and PB. Connecting lines indicate paired samples from each patient.
To infer directional dynamics of the 3 CLL cell states, we estimated RNA velocity based on the relative abundance of unspliced pre-mRNA and spliced mRNA in single cells.15,16 For a given gene, unspliced mRNA corresponds to the future state of spliced mRNA. When the balance of unspliced and spliced mRNAs of many genes are analyzed in each single-cell transcriptome, the RNA velocity of these cells can be estimated, which can, in aggregate, reveal the movement from one cell state to the next. In our dataset, the dominant structure comprised of proliferating cells as the root, giving rise to activated cells before entering the final state as quiescent cells (Figure 3C). This program, consistently observed across 3 different types of RNA velocity modeling (supplemental Figure 1B), implies that CLL cell fate in the LN is unidirectional and that most quiescent cells are not reactivated by TME stimuli.
We reasoned the bulk transcriptional profiles of PB and LN samples might be related to the tissue-specific distribution of CLL cell states. Therefore, scRNA-seq was also performed in 5 matched PB samples. A median of 5754 (range, 5071-6248) cells per sample and 29 008 cells in total were projected onto the LN scRNA-seq data to quantify the proportion of tumor cells in each cell state (Figure 3D; supplemental Figure 2A). Proliferating CLL cells were nearly absent in PB, and activated cells were significantly less abundant in PB than LN (Figure 3G). Furthermore, the activated population in PB underexpressed cell cycle genes, CCND2 and DUSP2, and chemokine CCL4 compared with their counterpart in LN (P < .05 for all comparisons) (supplemental Figure 2B).
Using single-cell expression profiles, we derived an activated CLL signature of the 10 most overexpressed genes by mean fold-change (HSP90AB1, ENO1, TUBB, RAN, PRDX1, LDHA, NME1, DNPH1, HSPD1, PKM) among activated cells. We assessed the expression of this signature in bulk RNA-seq of unselected LN samples from 34 patients. These 34 patients included 29 patients who contributed paired LN and PB samples to the tumor transcriptome analysis above and 5 additional patients with small lymphocytic lymphoma (Figure 1A). The activated CLL signature was overexpressed in U-CLL compared with M-CLL (P = .0033), in younger patients <60 years of age compared with older patients (P = .014), and in CLL compared with small lymphocytic lymphoma (P = .049, all unpaired t tests) (Figure 4A). No differences were observed among cytogenetic subgroups (Figure 4B).
Figure 4.

The activated CLL signature is associated with disease aggressiveness. (A,B) Comparison of the activated CLL signature between clinical subgroups. The activated CLL signature was defined as the 10 most differentially expressed genes between activated and quiescent CLL cells in the scRNA-seq dataset. Signature expression was then calculated in bulk RNA-seq data of LN samples. Box and whiskers represent the median, IQR, and 1.5 times IQR. (C-F) Kaplan-Meier plots of TFS in patients with (C) low vs high (below vs equal to or above median) expression of the activated CLL signature, (D) M-CLL vs U-CLL, (E) low vs high signature expression in patients with M-CLL, and (F) low vs high signature expression in patients with U-CLL.
Patients were followed for a median of 62.5 (range, 0.5-155.7) months after their LN biopsy. Consistent with more aggressive disease, higher expression of the activated CLL signature was associated with inferior treatment-free survival (TFS) (hazard ratio [HR], 3.9; 95% confidence interval [CI], 1.7-8.9; P < .0001) (Figure 4C). Median TFS from the time of diagnosis to first treatment or death was 29.0 months among patients with high signature expression and 124.6 months among those with low signature expression. In comparison, we analyzed TFS based on IGHV mutational status, a well-established prognostic marker in CLL.25,26 As expected, TFS of U-CLL was inferior to M-CLL, with a median TFS of 29.2 months vs 137 months (HR, 4.0; 95% CI, 1.9-8.5; P = .0002) (Figure 4D). Within each IGHV subgroup, the activated CLL signature retained independent prognostic information. Median TFS in patients with M-CLL was 76.5 months with high signature expression vs 186.5 months with low signature expression (HR, 3.8; 95% CI, 0.7-20.7; P = .040) (Figure 4E), and in patients with U-CLL was 16.1 months with high signature expression vs 37.4 months with low signature expression (HR, 2.5; 95% CI, 1.0-6.2; P = .025) (Figure 4F).
Interaction between immune cells of the TME and activated CLL
Although cellular interactions within the TME are thought to be critical in the pathogenesis of CLL, understanding the contribution of nonmalignant immune cells has been largely based upon in vitro and murine models. Whereas most studies have focused on specific cell types of interest, we sought to comprehensively assess the immune cell repertoire within the LN TME. The proportion of various cell types can be inferred by deconvolving bulk gene expression profiles.27,28 CIBERSORT27,28 was used to construct a custom gene signature matrix from gene expression profiles of purified CLL cells from LN samples and 20 non-B immune cell types. This signature matrix was then applied to deconvolve bulk RNA-seq data of the 34 unselected CLL LN samples. In parallel, we performed bulk RNA-seq of 4 normal LN samples and deconvolved these data using a validated gene signature matrix of 22 immune cell types, including naïve and memory B cells.28 The median estimated proportion of tumor cells was 77.9% (range, 29.6% to 90.7%). Only 1 sample had <50% tumor content. B cells comprised a median of 34.5% (range, 28.5% to 42.4%) of normal LNs and were predominantly memory B cells. After normalization to tumor or B-cell content, we compared the proportions of the 20 non-B immune cell types between CLL and normal LNs (Figure 5A; supplemental Figure 3). Follicular helper T cells, which provide T-cell help to B cells within germinal centers,29 were expanded while naïve CD4+ T cells and uncommitted macrophages were decreased in CLL LNs compared with normal LNs.
Figure 5.

Activated CLL cells interact with the immune microenvironment. (A) Mean abundance of the indicated cell types as estimated by CIBERSORT deconvolution of bulk RNA-seq data from CLL (n = 34) and normal LN samples (n = 4). There were significantly fewer follicular helper T cells (P < .05) and more CD4+ naïve T cells (P < .0001) and uncommitted macrophages in CLL LNs (P < .01) than normal LNs. (B) Correlation between the activated CLL signature and the abundance of M2 macrophages as estimated by CIBERSORT deconvolution of bulk RNA-seq data (n = 34). (C) Top: heatmap of activated CLL signature expression and CIBERSORT estimated abundance of M2 macrophages. Bottom: triangle and diamond symbols identify LN samples used for immunohistochemical staining of CD163+ M2 macrophages with low and high expression of the activated CLL signature. The average number of CD163+ cells per high-power field (40×) in each sample is provided immediately below. (D) Correlation between the activated CLL signature and the abundance of activated memory CD4+ T cells as estimated by CIBERSORT deconvolution of bulk RNA-seq data (n = 34). (E) Left: gating strategy of HLA-DR+ CD4+ effector memory (TEM CD3+CD19-CD14-CD4+CD8-CD45RO+CCR7-) and central memory (TCM CD3+CD19-CD14-CD4+CD8-CD45RO+CCR7+) cells in a representative flow cytometry dot plot. Right: proportion of CD4+ TEM and TCM cells that are HLA-DR+ in LN samples with low (n = 5) and high (n = 4) expression of the activated CLL signature.
We hypothesized that a subpopulation of tumor cells, specifically that of the activated phenotype, could be interacting with immune cells of the TME. Expression of the activated CLL signature among 34 CLL LNs was correlated against the proportion of 20 non-B immune cell types inferred by deconvolution. The activated CLL signature was significantly correlated with M2 macrophages (Pearson R = 0.44; P = .0087) (Figure 5B) and activated CD4+ memory T cells (Pearson R = 0.53; P = .0013) (Figure 5D). We corroborated these in silico findings with immunohistochemical staining of CD163 for M2 macrophages (Figure 5C) and flow cytometric analysis of activated HLA-DR+ activated CD4+ effector and central memory T cells in a subset of CLL LNs (Figure 5E; supplemental Figure 4). Given the association between the activated CLL signature and IGHV mutation status, we compared the TME immune cell composition between U-CLL and M-CLL. The proportion of the 20 non-B immune cell types was not significantly different based on IGHV mutation status. These data indicate that the TME is abnormal in CLL LNs compared with normal LNs and that an activated subset of CLL cells shapes the immune cell milieu in which they reside.
Permissive TME promotes the expansion of genetic subclones
Genetic lesions have been identified exclusively in either the PB or LN in some patients with CLL.30 We sought to map genotype–phenotype relationships that could explain the compartmentalization of subclones. WES was performed on CD19+ cells of paired PB and LN samples and matched germline DNA from 14 patients. A median of 27 (range, 11-69) somatic single nucleotide variants (sSNVs) and 3 (0-10) insertions and deletions (sIndels) were detected per exome. There were 28 sSNVs and sIndels shared by ≥2 patients (Figure 6A). In addition, all but 1 patient had copy number alterations, most commonly del(11q) and del(13q).
Figure 6.

The immune microenvironment constrains clonal expansion. (A) Distribution of CNAs and nonsilent mutations in WES of paired PB and LN samples in 14 patients. Only CNAs or genes mutated in ≥2 patients are shown. No clonal inframe indels or subclonal nonsense mutations were detected. (B) Top: pie charts of the proportion of patients with genetic compartmentalization of subclones defined as an absolute difference in CCF >0.25 between PB and LN by WES. Bottom: density plots of CCF in PB and LN in patients demonstrating subclonal expansion in each compartment (CLL-C33 and CLL-C43), in LN only (CLL-C24, CLL-C32, CLL-C34, and CLL-C35), and PB only (CLL-C46). The subclone(s) with genetic compartmentalization are highlighted in red in each patient. (C) Comparison of mean FC in gene expression by bulk RNA-seq in LN relative to PB between patients with (shifted) and without (stable) subclonal expansion in LN. Each dot is the FC in the expression of a gene in LN relative to PB averaged across 6 patients in the shifted group (y-axis) and 7 patients in the stable group (x-axis). Colored dots are genes with a significant difference in FC between these 2 groups (Δlog2FC >0.5; FDR <0.05). (D) Heatmap of a T-cell inflammatory signature expression by bulk RNA-seq and hierarchal clustering dendrogram of stable (n = 7) and shifted (n = 6) patients. The top row shows stable patients in white and shifted patients in red or pink. Red and pink colors correspond to patients with expanded subclone(s) in LN only and those with expanded subclone(s) in LN and PB, respectively. CNAs, copy number alterations.
Cancer cell fractions (CCFs) of sSNVs, sIndels, and copy number alterations were inferred from variant allele frequencies using ABSOLUTE,17 then clustered over the 2 anatomic compartments for each patient. Genetic compartmentalization (ΔCCF >0.25; FDR <0.1) was observed in 7 patients (50%), of whom 6 demonstrated subclonal expansion in the LN (Figure 6B). In the remaining 7 patients, no genetic compartmentalization was observed (supplemental Figure 5). To explore factors that contribute to the enrichment of subclones in the TME, we grouped patients based on the presence (shifted group) or absence (stable group) of an expanded subclone in LN. Patient CLL-C46 showed subclonal expansion in PB and was excluded.
Activation-induced cytidine deaminase (AID) is required for somatic hypermutation and class switch recombination but can also give rise to oncogenic mutations.31 In the TCL1 model, AID action has been implicated in clonal evolution.32 Therefore, we assessed AICDA expression in bulk RNA-seq data of purified CLL cells from LN and PB. As expected, LN-resident CLL cells overexpressed AICDA compared with their circulating counterpart (P < .0001) (supplemental Figure 6A). Among LN samples, AICDA expression was significantly higher in U-CLL than in M-CLL (P = .0004) (supplemental Figure 6B). AICDA expression was weakly correlated with the activated CLL signature in bulk RNA-seq (supplemental Figure 6C). However, assessment of AICDA in proliferating, activated, and quiescent single cells could not be performed because AICDA transcripts were detected in only 0.84% of CLL cells in scRNA-seq. Notably, there was no difference in AICDA expression between the shifted and stable groups (supplemental Figure 6C).
Differential gene expression analysis between PB and LN was performed separately in the shifted and stable group. Most differentially expressed genes between PB and LN were shared by both groups of patients. However, each group preferentially overexpressed a small subset of genes in LN relative to PB (log2FC >0.5; FDR <0.05) (Figure 6C). We observed upregulation of cell cycle genes (eg, E2F2, CDC25A, ESPL1) in the shifted group (supplemental Table 3). In the stable group, CD69, a lymphocyte activation marker, and CD83, involved in antigen presentation,33 were upregulated, raising the question of whether immune-mediated control could restrict clonal outgrowth.
To explore this possibility, we used a tumor immune signature comprised of 18 T–cell-associated inflammatory genes, which was developed to predict the clinical outcome with checkpoint blockade in multiple cancer types.34 Unsupervised hierarchal clustering of these signature genes in LN bulk RNA-seq data revealed increased expression in the stable group compared with the shifted group (Figure 6D). Irrespective of the source of mutagenesis, our findings raise the possibility of T–cell-mediated immune surveillance as a factor restricting clonal evolution in the LN.
Discussion
The development and progression of CLL are not isolated to intrinsic properties of leukemia cells but rather involve complex interactions with the TME. In vitro experiments and murine models have identified candidate cellular and soluble components in the local milieu that may be coopted by tumors to support leukemogenesis. In this study, we integrated bulk and single-cell transcriptome and bulk exome sequencing of paired PB and LN samples to define the in situ interactions between CLL and the TME and map clonal trajectories between anatomic compartments.
Our bulk RNA-seq data provided a comprehensive view of signaling pathways and cellular processes that are modulated in leukemic cells by the TME. These findings indicate that multiple support signals from the TME collectively promote growth and survival. Our sample size was sufficient in informing the differences between M-CLL and U-CLL in the TME. Compared with M-CLL, U-CLL demonstrated not only enhanced BCR signaling, consistent with prior studies,23 but also increased engagement with immune cells of the TME, as evidenced by the overexpression of interferon and T-cell cytokine signatures. While both M-CLL and U-CLL exhibit autonomous BCR signaling,35 our data demonstrate that extrinsic factors in the TME augment BCR activation above the basal level established by autonomous signaling in U-CLL more so than in M-CLL.
The gene expression profile provided by bulk RNA-seq is averaged across many cells, each of which has an individual transcriptome. By performing scRNA-seq in a subset of paired PB and LN samples, we found that bulk transcriptome data are, in fact, the aggregate of distinct CLL subpopulations. The transcriptional program associated with LN-resident CLL, that is, increased signaling and proliferation compared with its counterpart in PB, is derived from minor subpopulations comprising <10% of all tumor cells in the TME. By analyzing the RNA velocity of single cells, we found that CLL cells start in the proliferating state, move to the activated state, and end in the quiescent state. The sequential progression of these cell states suggests that most quiescent CLL cells residing in the LN are never reactivated by TME stimuli.
In contrast to the insights gained about tumor heterogeneity, we were only able to identify 3 nontumor immune cell clusters by scRNA-seq of LN samples. Several factors may have contributed to this result. First, adherent populations such as dendritic cells and macrophages are lost during the mechanical dissociation of LN tissue into single cells. Second, the relatively low number of transcripts per cell, which is inherent to scRNA-seq, limits the resolution of closely related immune cell subsets. To overcome these limitations and to analyze more samples in a cost-effective manner, we used in silico transcriptome deconvolution to profile accessory cell types in the LN TME of patients with CLL compared with normal LN. Our data indicate the activated subpopulation shapes the local immune cell composition, specifically of M2 macrophages and activated CD4+ memory T cells, independently confirming conclusions derived from model systems.36, 37, 38 The proportion of activated CLL cells and their capacity to recruit a tumor-supportive TME may underlie the shorter TFS observed in patients with higher expression of the activated CLL signature. Taken together, an identifiable minor tumor subpopulation underlies CLL pathogenesis and drives clinical outcomes.
A prior study detected PB- and LN-specific genetic lesions in 6 of 9 patients,30 but this interpretation overlooked possible differences in the size of subclones common to both PB and LN. In contrast, we included shared and specific mutations to infer and compare the clonal architecture between anatomic compartments. In one-half of patients, the CCF of identical subclones differed by >25% between PB and LN. Although certain mutations could, in theory, confer relative clonal fitness in a given environment, perhaps limited by sample size, we did not identify specific mutations that were preferentially distributed in either PB or LN. Together with the RNA velocity of CLL single cells, the spatial imbalance in genetic composition challenges the lifecycle model of CLL, in which tumor cells transit from a proliferative state in lymphoid tissue to a resting state in the blood and back.39
Our data support differential fitness of subclones within the TME driving genetic compartmentalization. The immune TME is capable of constraining clonal expansion and maintaining tumors in a state of equilibrium.40 In previously treated CLL, nodal relapse without leukemic progression implicates the role of protective niches in clonal evolution.41 Consistent with a permissive TME, subclonal expansion was apparent in the LN in all but 1 case with genetic compartmentalization. Bulk RNA-seq data from the same samples suggested that clonal outgrowth is impeded by an activated immune TME. When immune surveillance is inactivated, subclones with a competitive advantage may expand in response to support signals provided by the TME.
Experimental models have implicated AID in promoting clonal evolution.32 We observed increased AICDA expression in LN compared with PB and in U-CLL compared with M-CLL. However, there was no difference in AICDA expression between cases with or without clonal evolution in situ. Irrespective of the source of mutagenesis, our findings indicate T–cell-mediated immune surveillance as a factor that may restrict clonal evolution in the LN. Cumulative evidence has pointed to the existence of CLL-specific T cells and treatment, such as ibrutinib or lenalidomide, that could restore their cytotoxic activity.42, 43, 44 Therefore, strategies to strengthen host immune surveillance in CLL could represent one avenue to impede the evolution to more aggressive clones. Collectively, our findings underscore the value of an integrated multiomic approach to the evaluation of the LN TME in CLL.
Conflict-of-interest disclosure: C.S. receives research support from Genmab. M.J.B. is an employee at AstraZeneca. S.H.G. is a consultant for Novalgen Ltd and holds patents related to ROR1 therapies; has received honoraria, conference support, and speakers fees from AstraZeneca, AbbVie, and Janssen. N.S.S. is a consultant for and has received speaker fees from AbbVie, Pharmacyclics LLC, an AbbVie company, and Janssen; and is a member of the advisory board for TG Therapeutics, Innocare, BeiGene, KyowaKirin, ADC therapeutics, and Kite. G.G. receives research funds from IBM and Pharmacyclics; and is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, and SignatureAnalyzer-GPU; is a founder, consultant, and holds privately held equity in Scorpion Therapeutics. C.J.W. holds equity in BioNTech, Inc and receives research funding from Pharmacyclics. A.W. receives research support from Pharmacyclics LLC, an AbbVie Company, Acerta LLC, a member of the AstraZeneca Group, Merck, Verastem, Nurix, and Genmab. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the patients, the National Heart, Lung, and Blood Institute (NHLBI) Bioinformatics and Computational Biology Core, the NHLBI Sequencing and Genomics Core, and J. Joseph Melenhorst for his expertise in flow cytometric analysis of T-cell subsets.
This research was supported by the Intramural Research Program of the NHLBI, National Institutes of Health.
Authorship
Contribution: C.S. and A.W. conceptualized the study; C.S., Y.-C.C., A.M.Z., M.J.B., S.P., D.L., D.R., and S.G. provided formal analysis; C.S., M.J.B., N.S.S., and T.D.-H. undertook investigation; C.S. and A.W. provided resources; C.S. curated data; C.S. and A.W. wrote the original manuscript draft; C.S., Y.-C.C., and A.M.Z. performed visualization; G.G., M.P., C.J.W., and A.W. supervised the project; C.J.W. and A.W. acquired funding; and all authors reviewed and edited the manuscript.
Footnotes
The bulk and scRNA-seq data have been deposited in the Gene Expression Omnibus database under accession numbers GSE161711 and GSE161610. The WES data have been deposited in the dbGaP database under dbGaP accession phs002297.v1.p1. For other data sharing, contact the corresponding author: wiestnea@nhlbi.nih.gov.
Contributor Information
Clare Sun, Email: clare.sun@nih.gov.
Adrian Wiestner, Email: wiestnea@nhlbi.nih.gov.
Supplementary Material
References
- 1.Herishanu Y, Pérez-Galán P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–574. doi: 10.1182/blood-2010-05-284984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Granziero L, Ghia P, Circosta P, et al. Survivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood. 2001;97(9):2777–2783. doi: 10.1182/blood.v97.9.2777. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar-Hernandez MM, Blunt MD, Dobson R, et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood. 2016;127(24):3015–3025. doi: 10.1182/blood-2015-11-682906. [DOI] [PubMed] [Google Scholar]
- 4.Nishio M, Endo T, Tsukada N, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood. 2005;106(3):1012–1020. doi: 10.1182/blood-2004-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghia P, Strola G, Granziero L, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32(5):1403–1413. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 6.Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puente XS, Beà S, Valdés-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 8.Landau DA, Sun C, Rosebrock D, et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nat Commun. 2017;8(1):2185. doi: 10.1038/s41467-017-02329-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira PG, Jares P, Rico D, et al. Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res. 2014;24(2):212–226. doi: 10.1101/gr.152132.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin S, Gambe RG, Sun J, et al. A murine model of chronic lymphocytic leukemia based on B cell-restricted expression of Sf3b1 mutation and Atm deletion. Cancer Cell. 2019;35(2):283–296.e5. doi: 10.1016/j.ccell.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Improgo MR, Tesar B, Klitgaard JL, et al. MYD88 L265P mutations identify a prognostic gene expression signature and a pathway for targeted inhibition in CLL. Br J Haematol. 2019;184(6):925–936. doi: 10.1111/bjh.15714. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(1) doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La Manno G, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature. 2018;560(7719):494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38(12):1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 17.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Escobar MD, West M. Bayesian density-estimation and inference using mixtures. J Am Stat Assoc. 1995;90(430):577–588. [Google Scholar]
- 19.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herndon TM, Chen SS, Saba NS, et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia. 2017;31(6):1340–1347. doi: 10.1038/leu.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Onaindia A, Gómez S, Piris-Villaespesa M, et al. Chronic lymphocytic leukemia cells in lymph nodes show frequent NOTCH1 activation. Haematologica. 2015;100(5):e200–e203. doi: 10.3324/haematol.2014.117705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winkelmann R, Sandrock L, Kirberg J, Jäck HM, Schuh W. KLF2--a negative regulator of pre-B cell clonal expansion and B cell activation. PLoS One. 2014;9(5) doi: 10.1371/journal.pone.0097953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007;109(10):4424–4431. doi: 10.1182/blood-2006-11-056648. [DOI] [PubMed] [Google Scholar]
- 24.Muzio M, Apollonio B, Scielzo C, et al. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood. 2008;112(1):188–195. doi: 10.1182/blood-2007-09-111344. [DOI] [PubMed] [Google Scholar]
- 25.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. [PubMed] [Google Scholar]
- 26.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. [PubMed] [Google Scholar]
- 27.Schelker M, Feau S, Du J, et al. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat Commun. 2017;8(1):2032. doi: 10.1038/s41467-017-02289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Giudice I, Marinelli M, Wang J, et al. Inter- and intra-patient clonal and subclonal heterogeneity of chronic lymphocytic leukaemia: evidences from circulating and lymph nodal compartments. Br J Haematol. 2016;172(3):371–383. doi: 10.1111/bjh.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oppezzo P, Navarrete M, Chiorazzi N. AID in chronic lymphocytic leukemia: induction and action during disease progression. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.634383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morande PE, Yan XJ, Sepulveda J, et al. AID overexpression leads to aggressive murine CLL and nonimmunoglobulin mutations that mirror human neoplasms. Blood. 2021;138(3):246–258. doi: 10.1182/blood.2020008654. [DOI] [PubMed] [Google Scholar]
- 33.Kuwano Y, Prazma CM, Yazawa N, et al. CD83 influences cell-surface MHC class II expression on B cells and other antigen-presenting cells. Int Immunol. 2007;19(8):977–992. doi: 10.1093/intimm/dxm067. [DOI] [PubMed] [Google Scholar]
- 34.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dühren-von Minden M, Übelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–312. doi: 10.1038/nature11309. [DOI] [PubMed] [Google Scholar]
- 36.Grioni M, Brevi A, Cattaneo E, et al. CD4+ T cells sustain aggressive chronic lymphocytic leukemia in Eμ-TCL1 mice through a CD40L-independent mechanism. Blood Adv. 2021;5(14):2817–2828. doi: 10.1182/bloodadvances.2020003795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagnara D, Kaufman MS, Calissano C, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. 2011;117(20):5463–5472. doi: 10.1182/blood-2010-12-324210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanna BS, McClanahan F, Yazdanparast H, et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia. 2016;30(3):570–579. doi: 10.1038/leu.2015.305. [DOI] [PubMed] [Google Scholar]
- 39.Calissano C, Damle RN, Marsilio S, et al. Intraclonal complexity in chronic lymphocytic leukemia: fractions enriched in recently born/divided and older/quiescent cells. Mol Med. 2011;17(11-12):1374–1382. doi: 10.2119/molmed.2011.00360. [published correction appears in Mol Med. 2022;28(1):35] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450(7171):903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 41.Kiss R, Alpár D, Gángó A, et al. Spatial clonal evolution leading to ibrutinib resistance and disease progression in chronic lymphocytic leukemia. Haematologica. 2019;104(1):e38–e41. doi: 10.3324/haematol.2018.202085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baptista MJ, Baskar S, Gaglione EM, et al. Select antitumor cytotoxic CD8+ T clonotypes expand in patients with chronic lymphocytic leukemia treated with ibrutinib. Clin Cancer Res. 2021;27(16):4624–4633. doi: 10.1158/1078-0432.CCR-20-4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vardi A, Vlachonikola E, Papazoglou D, et al. T cell dynamics in chronic lymphocytic leukemia under different treatment modalities. Clin Cancer Res. 2020;26(18):4958–4969. doi: 10.1158/1078-0432.CCR-19-3827. [DOI] [PubMed] [Google Scholar]
- 44.Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120(7):1412–1421. doi: 10.1182/blood-2012-02-411678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.