

Abstract

DNA viruses are responsible for many diseases in humans. Current treatments are often limited by toxicity, as in the case of cidofovir (CDV, Vistide), a compound used against cytomegalovirus (CMV) and adenovirus (AdV) infections. CDV is a polar molecule with poor bioavailability, and its overall clinical utility is limited by the high occurrence of acute nephrotoxicity. To circumvent these disadvantages, we designed nine CDV prodrug analogues. The prodrugs modulate the polarity of CDV with a long sulfonyl alkyl chain attached to one of the phosphono oxygens. We added capping groups to the end of the alkyl chain to minimize β-oxidation and focus the metabolism on the phosphoester hydrolysis, thereby tuning the rate of this reaction by altering the alkyl chain length. With these modifications, the prodrugs have excellent aqueous solubility, optimized metabolic stability, increased cellular permeability, and rapid intracellular conversion to the pharmacologically active diphosphate form (CDV-PP). The prodrugs exhibited significantly enhanced antiviral potency against a wide range of DNA viruses in infected human foreskin fibroblasts. Single-dose intravenous and oral pharmacokinetic experiments showed that the compounds maintained plasma and target tissue levels of CDV well above the EC50 for 24 h. These experiments identified a novel lead candidate, NPP-669. NPP-669 demonstrated efficacy against CMV infections in mice and AdV infections in hamsters following oral (p.o.) dosing at a dose of 1 mg/kg BID and 0.1 mg/kg QD, respectively. We further showed that NPP-669 at 30 mg/kg QD did not exhibit histological signs of toxicity in mice or hamsters. These data suggest that NPP-669 is a promising lead candidate for a broad-spectrum antiviral compound.
Keywords: cytomegalovirus, adenovirus, DNA viruses, cidofovir prodrug, antiviral, intracellular metabolism
Introduction
Double-stranded DNA (dsDNA) viruses are a diverse group of infectious pathogens that present a significant risk to human health. Herpes viruses comprise a large family of enveloped dsDNA viruses and are responsible for a wide range of diseases. There are 8 known types of human herpesviruses (HHV1-HHV8), including cytomegalovirus (CMV), herpes simplex (HSV1 and HSV2), varicella zoster virus (VZV), and Epstein–Barr virus (EBV).1 Other dsDNA viruses, including adenoviruses (AdV) and polyomaviruses, are also widespread in the human population and can cause a variety of diseases.2,3 While the host’s immune system generally keeps them in check, these virus infections can become severe and life-threatening in immunocompromised patients.4−8 For the 186,000+ solid organ transplant (SOT) and hematopoietic stem cell transplant (HSCT) recipients annually, CMV is one of the most common and severe infections. CMV infections occur in up to 75% of SOT and HSCT recipients, usually in the first three months after transplantation.9 Furthermore, up to 30% of all high-risk patients develop multiple viral infections, including other herpesviruses besides CMV, AdV, and polyomaviruses, thus, illustrating the need for a broad-spectrum antiviral agent with a better safety profile and better bioavailability than is offered with currently approved products.10
Presently, there are only five FDA-approved drugs for the prevention or treatment of CMV infections: letermovir, ganciclovir and its oral prodrug, valganciclovir, foscarnet, and cidofovir (CDV). Of these five options, only valganciclovir and letermovir can be administered orally; the others require intravenous infusion. Letermovir is only active against CMV and offers no broad-spectrum coverage against other DNA viruses. Furthermore, viral resistance to letermovir is rapidly emerging.11 Similarly, viral resistance against nucleosides such as ganciclovir and valganciclovir is also common due to mutations of the kinase responsible for the first phosphorylation step. However, not all nucleosides are prone to developing this resistance. CDV circumvents this by already containing the initial phosphate group, which is converted intracellularly to its active triphosphate metabolite, CDV-PP.
While CDV does offer broad-spectrum coverage, its cellular uptake is highly inefficient, as reflected by its high EC50 of ∼1 μM against CMV.12 Due to the poor uptake by peripheral tissues, CDV concentrations in the systemic circulation are comparatively high, and the drug is excreted unchanged by the kidneys, where it accumulates and leads to toxicity. Therefore, CDV is administered with probenecid to inhibit the organic anion transporter in the kidneys and prevent the active reabsorption of CDV.13 Additionally, aggressive hydration is recommended to limit the risk of acute renal failure.13 Although CDV-associated neutropenia is commonly reported, it may only be due to high peak CDV concentrations resulting from intravenous (IV) administration, as neutropenia has not been noted with the orally administered CDV prodrug brincidofovir (BCV, see below). Despite its significant side effects, CDV remains the drug of choice for patients with refractory or resistant CMV and coinfections with other DNA viruses.14
The limitations of CDV and other nucleosides are primarily due to their hydrophilic nature, which results in poor transport across lipid bilayer membranes. The prodrug strategies that have been evaluated to date have attempted to mask the negative charge(s) with hydrophobic chemical moieties that are enzymatically cleavable upon intracellular permeation. The chemical strategies that have been explored are acycloxyalkyl,15 alkyloxyalkyl,16S-acylthioethyl (SATE),17 aryl,18 acyloxybenzyl phosphonate ester,19 cyclosaligenyl phosphonate ester,20 phosphonamidate,21,22 and tyrosine phosphonate ester23,24 derivatives. However, these approaches have yet to yield an approved oral therapeutic. The compound that progressed furthest in the pipeline was BCV, which reached Phase II clinical trials.
BCV is a lipid-ester prodrug of CDV that increases oral bioavailability and reduces nephrotoxicity by lowering kidney exposure.25 The lipid moiety allows for increased cellular uptake and improved activity against a broad spectrum of dsDNA viruses.25−30 Although the use of BCV was initially promising, it ultimately failed Phase III clinical trials for CMV and AdV infections. This failure was driven by dose-limiting gastrointestinal (GI) toxicity, which could not be differentiated at the therapeutic dosing regimen from acute graft-versus-host disease. In many cases, the adverse event profile of BCV led to the discontinuation, dose interruption, or dose reduction of BCV.31 Unfortunately, this dose-limiting GI toxicity that has been observed with oral administration of BCV may limit its use to IV administration in adult patients as well.32,33 However, the improved oral bioavailability and efficacy observed with BCV support continued exploration of alternative prodrug moieties with efficacy similar to or better than BCV and a wider therapeutic window following oral administration.
We have successfully developed a platform technology designed to improve the oral absorption and systemic distribution profile of nucleoside phosphonates, specifically CDV and other hydrophilic antiviral drugs, such that they will be effective as oral therapeutic agents. We developed a novel series of CDV prodrug candidates with a highly polar linker to the alkyl chains to improve solubility as well as terminal chain caps to slow or prevent degradation by phosphoesterases and β-oxidation as seen with BCV.34 Three prelead candidates were evaluated in vitro against several DNA viruses and compared to CDV, BCV, and other standard therapeutics. The relative increase in antiviral potency was found to correlate with increases in alkyl chain length that led to elevated intracellular levels of the metabolites CDV and CDV-PP. Based on these studies, compound NPP-669, which exhibits two to three orders of magnitude improved antiviral activity compared to CDV, was selected as a lead candidate. NPP-669 dosed orally was effective in vivo against murine CMV (MCMV) in mice and human AdV in hamsters with reduced kidney toxicity, improved biodistribution compared to CDV, and a wider therapeutic window compared to BCV.12,35,36 These results warrant further development of NPP-669 for use as a broad-spectrum oral antiviral therapeutic.
Experimental Section
Prodrug Design and Synthesis
Prodrug Candidate Physicochemical Property Estimation
The key parameters that influence drug substance solubility as well as systemic and target tissue exposure are pH-adjusted lipophilicity (clog D at pH 7.4, ideally considered to be between 1 and 3), topological polar surface area (TPSA, <125 Å), and molecular weight (MW, ≤500 Da). With this in mind, the physicochemical properties of the prodrugs under consideration were estimated using the ACD/Percepta PhysChem Profiler of the ACD Chemsketch software package (version C30H41 build 87885).
Prodrug Synthesis
Compounds designed to meet the criteria described above were prepared by general convergent synthesis optimized at the University of Michigan. Sulfonyl alkyl chains were originally prepared on a small scale in high yields from commercially available bromoalcohols (Scheme 1, part 1).
Scheme 1. Compounds Prepared by Convergent Synthesis Optimized At SRI International.

The lead candidate, NPP-669 was further scaled to a 30 g batch size at SRI International (under NIAID contract HHSN272201800001I, Scheme 1, part 2). For scale-up, the, sometimes capricious, copper-catalyzed reaction for adding the cyclobutylmethyl group was replaced by a simple Wittig reaction, making all steps amenable to multigram synthesis. Rather than adding the cyclobutylmethyl group via the Grignard-type reaction, the bromoalcohol 2 was converted to the Wittig reagent and reacted with cyclobutyl-aldehyde, and the double bond was reduced with H2/Pd in quantitative yield (Supporting Information S1). Identification and structural confirmation were determined via 1H NMR, 31P NMR, mass spectrometry, LC–MS/MS, and elemental analysis.
Analytical Methods
Tissue Culture Media and Cell Lysate
LC–MS/MS methods were developed on a Shimadzu LCMS 8050 UPLC triple quadrupole mass spectrometer (Shimadzu, Kyoto, Japan) for prodrugs, CDV, and CDV-PP in the tissue culture medium and the cell lysate. For all three analytes, standard curves and QC samples were prepared from blank cell lysates and blank media samples. Samples and standard aliquots were centrifuged at 3000 rpm for 5 min at 4° C. The supernatant was collected and transferred to 96-well plates for analysis using LC–MS/MS. Media samples were mixed by vortexing, diluted 30:70 in methanol (60 μL sample in 140 μL methanol) in a protein precipitation plate, shaken for 5 min using the MicroMix5, and then filtered using the positive pressure manifold for 10 min into a 96-well plate for LC–MS/MS analysis. Sample vials were transferred to an autosampler (4 °C) and 5 μL was injected to measure each component using the following LC–MS/MS conditions. All columns were installed within a column heater compartment and maintained at 60 °C. For the prodrugs, samples were injected into a pentafluoro-phenyl column (2.6 μm, 50 × 2.1 mm) (Phenomenex, Torrance, CA, USA) at a flow rate of 0.7 mL/min. Mobile phases consisted of (A) 6 mM ammonium formate/0.1% formic acid in water and (B) methanol, with an initial composition at 70% B. The composition of mobile phase B increased linearly for 1 min to 95% B, where it was held constant for 1 min. The composition returned to 70% B for the duration of the run (3 min total). For CDV, samples were injected on a C8 reversed-phase column (2.6 μm, 150 × 3 mm) (Phenomenex, Torrance, CA, USA) at a flow rate of 0.5 mL/min. Mobile phases consisted of (A) 6 mM ammonium formate/0.1% formic acid in water and (B) methanol, with an initial composition of 10% B. The composition of mobile phase B increased linearly for 1 min to 90% B, where it was held constant for 1 min. The composition returned to 10% B for the duration of the run (3 min total). For CDV-PP, the same column, mobile phases, and gradient were used, except mobile phase A did not contain formic acid.
The prodrugs and CDV were detected using optimized multiple reaction monitoring (MRM) settings using positive mode electrospray ionization (ESI). CDV-PP was detected using negative ESI. The MRM transitions that were monitored were as follows: NPP-663 (578.5 → 235.10), NPP-666 (606.5 → 235.15), NPP-669 (620.25 → 235.05), BCV (562.30 → 244.15), CDV (280.1 → 112.1), and CDV-PP (439.9 → 269.20). Concentrations of each component were calculated via extrapolation of their analytical signals (peak areas) onto an external calibration curve comprised of serially diluted test compounds in blank cellular lysate. Methods were qualified over a range of 5–2000 nM against a standard set of performance criteria: linearity with r2 ≥ 0.99, accuracy with % RE ≥ 15%, and precision with % CV ≥ 15%.
Plasma and Tissue
Plasma and tissue samples were analyzed under the same LC–MS/MS conditions described for the tissue culture media for prodrug, CDV, and CDV-PP concentrations. Tissue samples were homogenized prior to being subjected to the extraction procedure. NPP-669, CDV, and CDV-PP were extracted from plasma and tissue homogenate by protein precipitation with a 1:1 volumetric equivalent of cold acetonitrile to the sample. The supernatant was collected, filtered (0.22 μm), and analyzed using the qualified LC–MS/MS methods described above.
Metabolic Stability in Rat Hepatocytes
Stability was one of the major factors guiding the design of the novel CDV analogues. To maximize oral absorption, candidates should withstand degradation in the GI tract, yet readily convert intracellularly to release the parent CDV for subsequent conversion to the active CDV-diphosphate metabolite. Analogues were tested in a hepatocyte stability assay at Cyprotex (Watertown, MA) by incubating each compound in duplicate at 1 μM in 0.5 × 106 cells/mL at 37 °C. Samples were collected over 2 h, the reaction was stopped, proteins were precipitated with ice-cold methanol (1:1), and the soluble fraction was analyzed via LC–MS/MS.
Intracellular Conversion to CDV-PP: The Human Foreskin Fibroblast (HFF) Model
Intracellular conversion of prelead candidates in comparison to BCV and CDV was assessed following published methods.37 Briefly, stock solutions of CDV, BCV, and the prodrugs NPP-663, -666, and -669 were prepared in DMSO to a concentration of 200 μM. Each stock solution was diluted in the growth medium (DMEM with 10% FBS + 1× antibiotic-anti fungal + 1× GlutiMAX) to a final concentration of 1 μM (final DMSO concentration 0.5% v/v) and incubated for 24 h in a 37 °C water bath with gentle shaking before use.
Primary human foreskin fibroblast (HFF cells) cells were prepared for subsequent use from human foreskin tissue obtained from the University of Alabama at Birmingham tissue procurement facility, with approval from its institutional review board. The tissue was incubated at 4 °C for 4 h in a cell culture medium consisting of minimum essential medium (MEM) with Earle’s salts supplemented with 10% fetal bovine serum (FBS; HyClone, Inc., Logan, UT) and standard concentrations of l-glutamine, amphotericin B (fungizone), and vancomycin. The tissue was then placed in phosphate-buffered saline (PBS), minced, rinsed to remove the red blood cells, and resuspended in trypsin–EDTA solution. The tissue suspension was incubated at 37 °C and gently agitated to disperse the cells, which were then collected by centrifugation. Cells were resuspended in 4 mL of medium, placed in a 25 cm2 tissue culture flask, and incubated at 37 °C in a humidified CO2 incubator for 24 h. The medium was then replaced with fresh medium, and the cell growth was monitored daily until a confluent cell monolayer was formed. The HFF cells were then expanded through serial passages in a standard growth medium of MEM, with Earle’s salts supplemented with 10% FBS, l-glutamine, penicillin, and gentamicin. The cells were routinely passaged and used for assays at or before passage 10.
HFF cells were revived from cryogenic storage with thawing at 37 °C in a water bath. The cells were then resuspended in growth medium and transferred aseptically to T-75 tissue culture flasks, which were then incubated at 37 °C in a 5% CO2 incubator until confluence was achieved. The confluent cells were trypsinized and passaged at least twice into new T-75 tissue culture flasks before use. Cells were then seeded at a density of 6 × 106 cells per T-75 flask (n = 6) and incubated at 37 °C in a CO2 incubator for 24 h. The growth medium was then removed and replaced with medium (DMEM with 2% FBS + 1× antibiotic-antifungal + 1× GlutiMAX) containing 30 nM of each compound or vehicle in triplicate. Each flask was then incubated at 37 °C with 5% CO2 for 72 h. The cells were examined under a phase contrast microscope every 24 h to monitor for changes in cell morphology. At the end of 72 h, half of the test and control flasks were trypsinized for cell counting. In the remaining flasks, the drug solutions were aspirated, and cells were rinsed twice with 40 mL of chilled saline. Then, 1 mL of a mixture containing chilled methanol and distilled water (70:30) was added to lyse and remove cells. The cell lysates were collected in a sterile vial, vortexed, and stored immediately at −80 °C. The samples were analyzed as described under the Analytical Methods header.
In Vitro Antiviral Potency and Cytotoxicity Assays
Antiviral and cytotoxicity data were obtained in a series of three to five separate experiments to provide an accurate estimate of the antiviral activity and the required statistical data. Every assay included positive and negative control compounds as well as uninfected controls to ensure the integrity of the experiment. The concurrent assessment of cytotoxicity was performed in each assay using the same number of cells at equivalent levels of compound exposure so that accurate selective index (SI) values could be obtained.
Antiviral assays were performed as described against adenovirus (AdV-5), herpesviruses 1 and 2 (HSV-1, HSV-2), human cytomegalovirus (HCMV), murine cytomegalovirus (MCMV), varicella-zoster virus (VZV), and vaccinia.38 The methodology described previously for lymphotrophic herpesviruses, such as Epstein–Barr virus (EBV), human herpes virus-6B (HHV-6B), and human herpes virus 8 (HHV-8), as well as polyomaviruses, such as BK virus and JC virus, was used.39 Briefly, cytopathic effect (CPE) assays using HFF cells (ADV-5, HSV-1,-2, HCMV, VZV, and vaccinia), seeded and incubated for 24 h in 384-well microtiter plates, were done by preparing dilutions of test compounds in a series of 5-fold dilutions in duplicate wells to yield final concentrations that range from 150 to 0.048 μM or from 10 to 0.003 μM. Monolayers were then infected at an appropriate multiplicity of infection (MOI) for the virus used. Assays against MCMV used murine embryo fibroblast (MEF) cells seeded in 96 well plates with drug dilutions performed as mentioned above. Assays against the lymphotrophic viruses (EBV, Akata cell line; HHV-6B, Molt-3 cell line; and HHV-8, and BCBL-1 cell line) and polyomaviruses (BKV, cell line HFF; JCV, and cell line COS7) were performed similarly (in reference to drug ranges and dilutions) but with quadruplicate wells. Infected cells were then incubated further at 37 °C until 100% CPE was observed in the virus control wells. Other cells were incubated for 3 days (EBV) or 7 days (MCMV, HHV-6B, HHV-8, JCV, and BKV), and for these viruses, DNA was extracted and qPCR assays were performed. CPE was determined by the addition of the CellTiter-Glo reagent (Promega, Madison, WI) according to the manufacturer’s suggested protocol. Concentrations of test compounds sufficient to reduce CPE by 50% (EC50) were interpolated from the experimental data. Cytotoxicity was also determined with CellTiter-Glo, and concentrations of the compounds that decreased cell viability by 50% (CC50) were calculated. Selective index (SI) values were calculated as the CC50/EC50 as a measure of specific antiviral activity.
Animals and Ethics Statement
All studies were approved by the Institutional Animal Care and Use Committees at each institution (St. Louis University School of Medicine, University of Cincinnati, and TSRL, Inc.) and were conducted according to federal and institutional regulations. Animals were visually inspected, weighed, and determined to be free of abnormalities and illness upon receipt and at study initiation. Animals were grouped or individually housed as required in wire-bottom cages rested on a plastic pan, with sufficient bedding to cover the wire mesh, or in solid cages with cellulose bedding. Nestlets, and/or cardboard biotunnels were provided for enrichment.
Pharmacokinetics and Biodistribution in Mice
NPP-669, BCV, and CDV were administered to male CFW mice (n = 4) as single equimolar oral doses in 0.5% carboxymethyl-cellulose (CMC) corresponding to 30, 26, and 13 mg/kg respectively. Plasma samples were taken via cardiac puncture at 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose. In addition, target tissue (liver, lung, spleen, kidney, and intestinal) samples were collected at 2, 8, and 24 h. Samples were analyzed by LC–MS/MS for prodrug, CDV, and CDV-PP concentrations as described above. Plasma concentration time profiles were analyzed by standard noncompartmental pharmacokinetic analysis using the WinNonlin, Version 8.0.0.3176 suite of programs. The following parameters were calculated: Tmax, Cmax, AUC, Cl, Vd, and T1/2.
In Vivo Efficacy Models
Mouse Model of CMV (MCMV)
Studies to determine efficacy against MCMV were conducted at Cincinnati Children’s Hospital Medical Center. Groups of female Balb/c mice were (n = 8) inoculated with 1 × 105 pfu of MCMV strain K181 via intraperitoneal (IP) injection and treated with either vehicle (0.5% CMC) or NPP-669 at 30, 10, 3, and 1 mg/kg QD or BID by oral gavage, or IP with 50 mg/kg QD ganciclovir (positive control). Drug treatment started the day before the virus infection and continued for 2 days postinfection. Mice were sacrificed at 3 days postinfection and viral titers were determined from the liver and spleen as previously described.40 Efficacy in male mice will be assessed in the future to assess any potential variability as a function of sex.
Hamster Model of AdV
Studies to determine the efficacy of NPP-669 against AdV were performed at Saint Louis University School of Medicine using previously described methods.41 Briefly, male Syrian hamsters were immunosuppressed using cyclophosphamide (CP) administered IP, starting with an induction dose of 140 mg/kg and then 100 mg/kg for all subsequent injections. The hamsters were sorted into 7 groups (n = 15). Two of the groups were uninfected; these groups served as the vehicle and drug-only (NPP-669 at 1 mg/kg PO QD) controls. The remaining five groups were intravenously inoculated with 3 × 1010 pfu/kg of wild-type human adenovirus type 6 (AdV 6), strain tonsil 99 (ATCC). Three of these infected groups were treated with NPP-669 at 0.1, 0.3, or 1 mg/kg of NPP-669. The remaining two AdV-infected groups were either left untreated or treated with CDV. CDV was administered IP starting with an induction dose of 37 mg/kg, followed by three times weekly at 20 mg/kg. For all drugs, the first dose was given one day before the virus challenge (day −1) and then continued for the duration of the study. Five hamsters were sacrificed to determine the liver virus titer 5 days postinfection. All hamsters were observed and weighed daily. At necropsy, serum and liver were collected. The virus was extracted from the liver and quantified by the 50% tissue culture infectious dose (TCID50) assay in HEK-293 cells. The serum was assayed for liver transaminase levels. Efficacy in female hamsters will be assessed in the future to assess any potential variability as a function of sex.
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software). The log-rank test was used to compare survival, and mixed-effect analysis was used to compare body weight changes. For serum transaminase levels, the variance of samples in all groups was calculated using the Kruskal–Wallis test, and a comparison between groups was performed using the Mann–Whitney U test. p ≤ 0.05 was considered significant.
NPP-669 In Vivo Toxicity
NPP-669 was evaluated in an exploratory toxicology study following a dosing regimen that mimicked the in vivo MCMV efficacy study design reported above. Compounds suspended in 0.5% CMC were administered orally to mice BID at 30, 10, and 1 mg/kg/day for four days. In a follow-up study, NPP-669 was administered once daily for either 4 or 7 days with and without a recovery phase. Animals were observed daily for body weight change and clinical abnormalities. The day after the last dose, the animals were euthanized, blood samples for clinical chemistry and hematology were obtained, and samples of the pancreas, kidney, liver, lung, small intestine, colon, and spleen were processed for histopathology. Separate samples of liver and lung were retained for drug concentration level determination via LC–MS/MS assay.
Results
Prodrug Design, Synthesis, and Theoretical Calculations
CDV exhibits low oral bioavailability and cellular uptake, mediated by the inefficient pathway of endocytosis. This is due to its high hydrophilicity and low lipophilicity, with a calculated log D at pH 7.4 of −7.4. Masking one of the negative charges via phospho-esterification with a long-chain alkyl chain has been shown to improve the cLogD values. The most successful prodrug of CDV to date, developed based on this hypothesis, has been BCV.44 This prodrug is taken up by the cell mostly intact, and CDV is then released through the action of phospholipases.45 Building on the success of BCV to deliver CDV directly into cells, we designed prodrugs that would retain the lipid nature of BCV but overcome its shortcomings. To mitigate the decrease in solubility that the alkyl chain would introduce, we incorporated a sulfonyl group, an isostere of the ether linkage in BCV to increase solubility. The sulfonyl group adds a significant dipole while maintaining the ether bond angle in BCV.46 Because BCV suffers from fatty acid-related β-oxidation metabolism, we added capping groups (t-butyl, cyclobutyl, cyclopropyl, and oxetanes) to the end of the alkyl chain to eliminate β-oxidation and focus the metabolism on the phosphoester hydrolysis. Given the changes made in the alkyl side chain that we introduced, we prepared multiple chain lengths to find the optimum length for our series. Identification and structural confirmation were determined via 1H NMR, 31P NMR, and mass spectrometry. The analytical results for NPP-669 are summarized in the certificates of analysis (Supporting Information S2). The key design parameters of the various CDV analogues are summarized in Figure 1 and illustrate the increased lipophilicity of the analogues, specifically NPP-669 (log P = 2.5), balanced by the ionization potential (log D at pH 7.4 = −1.0) that was achieved.
Figure 1.

Structures of BCV, the novel prodrug platform, and NPP-669, together with the theoretical physicochemical properties of synthesized prodrugs.
Metabolic Stability in Rat Hepatocytes
The metabolic half-life of BCV in hepatocytes was 142 min, while the prodrugs showed a broad range of half-lives, culminating in 202 min for NPP-669 (Table 1). The rank order of hepatocyte stability correlated with the in vivo clearance in the pharmacokinetic (PK) studies discussed further below.
Table 1. Hepatocyte Clearance.
| compound ID | N | clearance (μL/min/million cells) | half-life (min) |
|---|---|---|---|
| BCV | C16 | 9.73 | 142 |
| NPP-662 | C10 | 18.8 | 73.8 |
| NPP-663 | C10 | 22.2 | 62.4 |
| NPP-664 | C12 | 60.6 | 22.9 |
| NPP-666 | C12 | 14.7 | 94.3 |
| NPP-667 | C14 | 154 | 9.01 |
| NPP-669 | C13 | 6.85 | 202 |
| NPP-670 | C12 | 33.3 | 41.6 |
Intracellular Conversion to CDV-PP
In CDV-treated cells, levels of CDV or CDV-PP were below the detection limit of our methods (≤10 ng/mL) in the HFF model (Figure 2). However, NPP-669 and NPP-666 show significantly improved cellular uptake and conversion characteristics compared to BCV and CDV.
Figure 2.
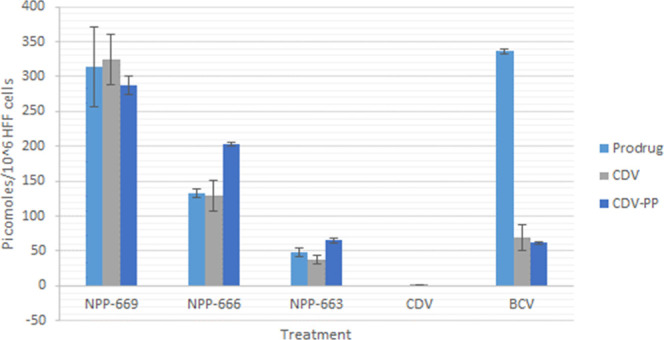
Intracellular prodrug and metabolite levels in HFF cells after incubation with 30 nmol of each drug for 72 h (mean ± SD, n = 3).
While cells treated with BCV had the highest levels of intracellular pro-drug, levels of intracellular CDV and CDV-PP were approximately 5 times greater for NPP-669 treated cells and approximately 3 times greater in NPP-666 treated cells. This indicates that the prodrug candidates convert to CDV and active moiety much more readily than BCV. Interestingly, the chain length affected the cellular uptake. For instance, the uptake of NPP-669 compared to NPP-666 and NPP-663 demonstrated that longer chains increase the uptake of the NPP candidates. Clearly, the low cell permeability of CDV significantly limits its antiviral effectiveness relative to its toxicity to the kidneys. However, the conjugation of CDV to a metabolically cleavable, lipophilic carrier adequately remedies this limitation. It is also clear that the chain length is a critical factor in cell uptake and conversion to CDV and CDV-PP as the one carbon increments in length from C10 to C13 correlate nicely with intracellular penetration. No signs of cell toxicity were observed throughout the 72 h incubation period with any of the test compounds.
In Vitro Antiviral Potency Assays
The prodrug candidates were screened for their antiviral potency at the University of Alabama at Birmingham under the NIAID contract. As shown in Table 2, NPP-669 is more efficacious than antiviral compounds currently in clinical use against adenovirus (AdV-5), herpesviruses (HSV-1 and HSV-2, HCMV, VZV, EBV, HHV-6, and HHV-8) and polyomaviruses (JC and BK virus), while it is on par with BCV. Using published data for comparison, NPP-669 is also significantly more potent than the parent drug CDV against HCMV (mean EC50 < 0.01 μM vs ∼ 1 μM), HSV-1 (EC50 0.02 μM vs 3–5 μM), HSV-2 (EC50 0.14 μM vs 5–6 μM), and VZV (EC50 0.007 μM vs 0.5 μM).42 Similar results were observed against other DNA viruses (Table 2). For HCMV, a secondary screen was conducted with NPP-669, which resulted in an EC90 of <0.0001 and <0.001 μM in standard and resistant isolates, respectively, with a selectivity index of >1000. Further, it was shown that the EC50 of NPP-669 against HADV-C6 and two additional AdV types was similar to that against HAdV-C5 (AdV-5 in Table 2, data not shown). Based on these results, NPP-669 appeared optimal from a pharmacokinetic and in vitro efficacy perspective and was carried forward for evaluation in vivo.
Table 2. In Vitro Potency of the Novel Prodrugs and BCV against Various DNA Viruses (Virus Strain Specified in Parentheses), Compared to a Virus-specific Positive Controla.
| +control | BCV | NPP-662 | NPP-663 | NPP-664 | NPP-666 | NPP-667 | NPP-669 | ||
|---|---|---|---|---|---|---|---|---|---|
| ADV53 | EC50 | 6.64 ± 4.79 | 0.02 ± 0.01 | 10.79 ± 17.45 | 0.01 ± 0.01 | 0.33 ± 0.30 | 0.01 ± 0.01 | 0.24 ± 0.16 | 0.01 ± 0.08 |
| (Adenoid 75) | SI | >23 | 46 | >5 | 83 | >152 | 84 | 180 | 74 |
| HSV11 | EC50 | 0.96 ± 0.19 | 0.03 ± 0.02 | 3.18 ± 3.78 | 0.07 ± 0.04 | 2.38 ± 2.90 | 0.01 ± 0.01 | 1.47 ± 1.73 | 0.02 ± 0.01 |
| (E-377) | SI | >156 | >33 | >16 | 149 | >21 | 1159 | >34 | 469 |
| HSV21 | EC50 | 0.38 ± 0.33 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.29 ± 0.13 | 0.29 ± 0.13 | 0.01 ± 0.01 | 0.29 ± 0.17 | 0.14 ± 0.23 |
| (G) | SI | >395 | >50 | >98 | >3950 | >172 | 498 | >172 | >14 |
| VZV1 | EC50 | 3.24 ± 0.26 | 0.005 ± 0.005 | 1.01 ± 0.22 | 0.02 ± 0.01 | 0.69 ± 0.54 | 0.01 ± 0.01 | 0.03 ± 0.03 | 0.007 ± 0.01 |
| (Ellen) | SI | >46 | 114 | >50 | 25 | 72 | 23 | 1069 | 31 |
| HCMV2 | EC50 | 1.63 ± 0.69 | 0.001 ± 0 | 0.07 ± 0.02 | 0.008 ± 0.01 | 0.017 ± 0.01 | <0.01 ± 0.01 | 0.01 ± 0.01 | <0.01 ± 0.01 |
| (AD169) | SI | >92 | 540 | >714 | 74 | 1821 | >19 | >3000 | >19 |
| *HCMV3 | EC50 | 0.15 | ND | 0.06 | <0.0003 | 0.004 | <0.0003 | 0.003 | <0.0003 |
| (GDGRK17) | SI | >1034 | ND | >17 | >2772 | >250 | >2753 | >333 | >2209 |
| **MCMV2 | EC50 | 8.10 ± 11 | 0.0041 ± 0.0041 | 0.59 ± 0.58 | 0.0015 ± 0.0016 | 0.056 ± 0.064 | 0.00028 ± 0.00003 | 0.36 ± 0.46 | 0.00046 ± 0.00023 |
| (Smith) | SI | >19 | >244 | >2 | >667 | >18 | >3571 | >3 | >2174 |
| EBV3 | EC50 | 10.10 ± 3.44 | 0.09 ± 0.09 | 9.94 ± 13.25 | 14.78 ± 25.52 | 3.81 ± 3.04 | 0.14 ± 0.11 | 5.17 ± 7.32 | 0.08 ± 0.07 |
| (Akata) | SI | >10 | >11 | >5 | >3 | >13 | 241 | >10 | 356 |
| HHV-6B3 | EC50 | 7.09 ± 3.59 | 0.03 ± 0.02 | 24.01 ± 15.25 | 0.09 ± 0.12 | 4.23 ± 5.00 | 0.02 ± 0 | 7.22 ± 9.80 | <0.02 ± 0 |
| (Z29) | SI | 10 | 16 | >2 | 4 | 10 | 4 | >7 | >4 |
| HHV-83 | EC50 | 1.50 ± 0.70 | 0.001 ± 0 | 0.53 ± 0.42 | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.03 ± 0.02 | 0.03 ± 0.01 | 0.01 ± 0.01 |
| (BCBL-1) | SI | >67 | 690 | >94 | 51 | >1667 | 35 | >1667 | 89 |
| **JC virus3 | EC50 | 18.40 ± 16 | 0.35 ± 0.26 | 0.53 ± 0.66 | 0.075 ± 0.007 | 4.2 ± 4.6 | 0.05 ± 0.03 | >25.5 ± 35 | 0.07 ± 0.05 |
| (MAD-4) | SI | >8 | >3 | 48 | 13 | >6 | 17 | 1 | 14 |
| **BK virus3 | EC50 | 5.8 ± 3.9 | 0.005 ± 0.007 | 0.43 ± 0.59 | 0.035 ± 0.02 | 0.38 ± 0.07 | 0.09 ± 0.01 | 0.02 ± 0 | 0.01 ± 0.01 |
| (Gardner) | SI | >26 | 180 | >59 | 34 | 67 | 8 | 1225 | 70 |
| *vaccinia3 | EC50 | 14.6 | ND | ND | ND | ND | 25.75 | ND | 8.95 |
| (Copenhagen) | SI | >10 | ND | ND | ND | ND | >2 | ND | >6 |
Controls: 1acyclovir, 2ganciclovir, and 3CDV. EC50: μM; mean of 3 replicates, unless otherwise stated. *Single replicate and **mean of 2 replicates. ND: not determined.
Pharmacokinetics and Biodistribution in Mice
The PK parameters after IV administration confirmed that distribution volumes increased with the increasing prodrug chain length and that clearance decreased (Table 3), as expected based on the in vitro metabolic half-life (Table 1).
Table 3. PK Parameters of Prodrugs in Mice Following a Single Intravenous (IV) Dose.
| dose (mg/kg) | Cmax (ng/mL) | AUC (ng·h/mL) | T1/2 (h) | Cl (mL/min/kg) | Vd (L/kg) | |
|---|---|---|---|---|---|---|
| BCV | 1 | 491 | 171 | 0.4 | 122 | 3.7 |
| NPP-663 (C10) | 3 | 1155 | 333 | 0.2 | 152 | 2.8 |
| NPP-666 (C12) | 3 | 1693 | 523 | 0.4 | 104 | 4.2 |
| NPP-669 (C13) | 3 | 2617 | 807 | 0.7 | 64 | 4.0 |
NPP-663, NPP-666, and NPP-669 dosed orally to mice showed good oral absorption. Oral dosing of NPP-669 at 30 mg/kg, and of BCV at an equimolar dose rendered plasma levels remaining well above the EC50 (>0.003 μM or 2 ng/mL) over 24 h (Figure 3A). Assessment of target tissue concentrations of NPP-669 compared to BCV showed that BCV was retained at substantially higher levels than NPP-669 in the intestinal tissue (Figure 3B). Importantly, however, conversion to CDV and subsequently to CDV-PP occurred to a substantially greater extent for NPP-669 (Figure 3C) compared to BCV. A similar pattern was observed in liver tissue, where BCV exposure reached significantly higher levels than NPP-669 (Figure 3D), while exposure levels of CDV (and CDV-PP) are substantially higher after NPP-669 administration than after BCV (Figure 3E). These exposure levels are orders of magnitude above the EC50s of the DNA virus strains for which it has shown activity (Table 1). These data are consistent with the cellular conversion data in Figure 2. Furthermore, prodrug exposure to the kidneys is even further reduced over BCV with NPP-669 (Figure 3F). These characteristics underscore the notion that NPP-669 can render higher concentrations of the pharmacologically active species (CDV-PP) to the target tissues of viral replication with significantly prolonged plasma levels that represent the driver for the pharmacodynamic (PD) effect. Exposure to the parent compound is reduced compared to BCV, which is anticipated to result in less toxicity at equal doses and create an adequate safety margin (see further discussion below). These data support the interpretation that NPP-669’s distribution and metabolism characteristics have been significantly optimized to maximize efficacy and minimize toxicity over previous molecular approaches.
Figure 3.

Plasma (A), intestine (B), liver (D), and kidney tissue (F) profiles of NPP-669, BCV, and CDV in mice, with the corresponding CDV levels in the intestine (C) and the liver (E) after equimolar oral (NPP-669, BCV) and IV (CDV) dosing (n = 4, mean ± SD).
In Vivo Efficacy of NPP-669
Mouse Model of CMV
When dosed b.i.d., NPP-669 was effective in suppressing MCMV titers in the liver at all doses tested (Figure 4A shows data for the two lowest doses). As shown, the 3 mg/kg/day dose reduced virus replication in the liver and spleen below the limit of detection and was as effective as ganciclovir (Figure 4B). The lowest dose, 1 mg/kg/day BID, significantly (p < 0.05) reduced viral titers in the liver but was less effective in the spleen. When dosed once a day, NPP-669 significantly suppressed viral replication at 3 mg/kg/day, while at 10 mg/kg/day, QD viral titers were suppressed to below the detection limit (data not shown). A 30 mg/kg/day QD dose was well tolerated with no signs of toxicity.
Figure 4.

Viral titer reduction in the liver (A) and the spleen (B) in a murine model of CMV infection with oral doses of NPP-669 at 1 and 3 mg/kg/day b.i.d. for four days (mean ± SD, n = 8).
Hamster Model of AdV
Once daily treatment of immunosuppressed Syrian hamsters challenged IV with human HAdV-C6 with 0.1, 0.3, and 1 mg/kg of NPP-669 significantly increased survival (Figure 5A) and significantly decreased body weight loss (Figure 5B). Note that a dose of CDV with low-grade toxicity had to be administered to achieve the same level of antiviral efficacy (Figure 5B). The 0.3 and 1 mg/kg QD doses of NPP-669 significantly mitigated the pathogenic effects (liver damage as measured by serum ALT levels, Figure 5C) and inhibited the replication of HAdV-C6 in the liver (Figure 5D). Initial regimens also included cohorts dosed at 3 and 10 mg/kg/day QD, which were well tolerated, with no overt signs of toxicity (data not shown). These are extremely promising efficacy results, which make NPP-669 one of the very few agents that are effective against HAdV infections in vivo. These data support the exceptional efficacy of NPP-669 in the mouse model of CMV and the hamster model of AdV infections.
Figure 5.

NPP-669 inhibits the replication of HAdV-C6 in the liver of immunosuppressed Syrian hamsters and prevents the resulting pathology. (A) Survival. (B) Body weight changes. The symbols represent the group average, and the error bars show the standard error. No group average was calculated after an animal was sacrificed from a given group. (C) Infectious virus load in the liver. (D) Serum alanine transaminase levels. For (C,D), the symbols represent values from individual animals; the horizontal bar shows the group mean, and the error bars depict the standard deviation. Empty symbols for the HAdV-C6 + vehicle and HAdV-C6 + NPP-669 0.1 mg/kg groups signify that the samples were collected from an animal that was sacrificed ahead of schedule for humane reasons. NQ: not quantifiable; ND: not detectable; for statistical analysis, treatment groups were compared to untreated infected groups. *: p < 0.05, **: p < 0.01, and ***: p < 0.001.
In Vivo Toxicity of NPP-669
Uninfected male mice were dosed at 30 mg/kg/day QD for 4 days and at 10 and 3 mg/kg/day QD for either 4 or 7 days, with either a 2 or 3 day reversal phase. The study illustrated that the kidney toxicity associated with CDV has been completely mitigated with NPP-669, as no treatment-related findings were noted in this tissue. Only minimal pathological changes were noted in the GI tract, which were reversible and did not manifest themselves in weight loss or changes in blood chemistry. The no-adverse effect level (NOAEL) dose in uninfected mice was established at 30 mg/kg/day QD.
In the CMV mouse efficacy model, NPP-669 doses of 30 mg/kg/day BID over 4 days did not result in signs of toxicity such as weight loss or inactivity, and since efficacy was achieved with doses as low as 1 mg/kg/day BID, a superior initial therapeutic window of close to 30-fold was established. In contrast, GI toxicity was noted for BCV at daily doses close to those used in the mouse efficacy studies (at 10 mg/kg) and only resolved when treatment frequency was reduced to twice per week. This regimen rendered a NOAEL for BCV of 15 mg/kg, twice weekly, in rats over 13 weeks.36
In the AdV hamster efficacy model, NPP-669 doses of 10 mg/kg/day QD over 15 days did not result in signs of toxicity, and since efficacy was achieved with doses as low as 0.3 mg/kg/day QD, the same therapeutic window seen in the CMV mouse model of close to 30-fold was confirmed. Of note is that to achieve the desired efficacy with CDV, it had to be administered at a toxic dose. This is evidenced by the delayed body weight gain (Figure 5B) and the kidney pathology observed (data not shown) at the day 14 sacrifice.
Discussion
A series of orally bioavailable CDV analogues with excellent solubility, optimized metabolic stability, increased cellular permeability, and rapid intracellular conversion to the pharmacologically active diphosphate (CDV-PP) have been developed. This success utilized the strategy of masking one of the negative charges of CDV via phospho-esterification with a long-chain alkyl moiety that improved lipophilicity. To simultaneously mitigate the decrease in solubility that an alkyl chain would introduce, a sulfonyl group was incorporated to increase solubility. Finally, the addition of capping groups to the end of the alkyl chain was done to further minimize β-oxidation and focus the metabolism on the phosphoester hydrolysis, thereby further optimizing the rate of this reaction. These compound characteristics resulted in (1) significantly enhanced antiviral potency against a wide range of DNA viruses, (2) increased in vitro prodrug stability, (3) decreased systemic clearance in vivo, and (4) a pharmacokinetic profile that maintained plasma and target tissue levels of CDV well above the EC50 for 24 h. Taken together, we identified a novel lead candidate, NPP-669, that has improved drug-like properties, is efficacious against CMV infection in mice and AdV infection in hamsters following oral dosing, and has a favorable initial safety profile compared to CDV and related compounds.
Transplant patients are at high risk for CMV infections.9 Immunocompromised patients infected with CMV experience loss of vision, colitis, esophagitis, hepatitis, encephalitis, and pneumonia.43 Antiviral therapy is the standard practice for treating CMV infections. Of the five FDA-approved CMV drugs, the only one not prone to resistance is CDV. The presence of the initial phosphate group on CDV prevents such resistance. While CDV is efficacious in the treatment of CMV, it has poor cellular uptake leading to high systemic concentrations.12 The accumulation in the kidneys causes severe toxicity. Nevertheless, CDV remains the most commonly prescribed drug for patients with CMV.
To circumvent the toxicity associated with CMV, several chemical strategies have been explored to mask the negative charges. Unfortunately, to date, no method has achieved the desired efficacy and safety. One compound, BCV, reached phase II clinical trials. It was a promising prodrug of CDV as it increased bioavailability, decreased nephrotoxicity, increased cellular uptake, and had activity against other dsDNA viruses.25−33 However, BCV was discontinued as it had dose-limiting GI toxicity. The development of BCV proved that prodrugs of CDV increase the efficacy of the drug. Therefore, alternative prodrug moieties must still be developed with a wider therapeutic window. In the work described here, we masked one of the negative charges on CDV with a long-chain alkyl chain, included a sulfonyl group to increase solubility, and added a capping group to minimize β-oxidation. Several prodrugs were created using these parameters, and the log P, log D, and TPSA were determined. Of these prodrugs, NPP-669 proved to be the most promising. We determined that NPP-669 had a metabolic half-life of 202 min in rat hepatocytes, which was significantly improved compared to BCV. We next determined that the intracellular levels of CDV-PP, the active metabolite of CDV, were 5 times higher for NPP-669 than BCV in the human foreskin fibroblast model. These findings suggest NPP-669 has enhanced conversion to the active moiety compared to BCV.
NPP-669 and the other prodrug candidates were then evaluated for their antiviral properties. In vitro antiviral potency assays were performed to elucidate the antiviral activity of the novel prodrugs against AdV-5, HSV-1, HSV-2, HCMV, MCMV, VZV, EBV, HHV-6B, HHV-8, JC virus, and BK virus. Our study found that NPP-669 is more efficacious than the FDA-approved antiviral compounds for all DNA viruses evaluated and was equally as effective as BCV. We showed that NPP-669 has an EC90 of <0.0001 μM for standard HCMV isolates and an EC90 of <0.001 μM for resistant HCMV isolates (see In Vitro Antiviral Potency Assays section). The findings summarized thus far demonstrated that NPP-669 was the optimal drug candidate for further evaluation in vivo.
Intravenous administration of 3 mg/kg NPP-669 resulted in a Cmax of 2617 ng/mL, an AUC of 807 ng·h/mL, a half-life of 0.7 h, a clearance of 64 mL/min/kg, and a volume of distribution of 4.0 L/kg. Oral administration of 30 mg/kg NPP-669 rendered plasma concentrations above the EC50 for over 24 h, similarly to BCV. Looking deeper into tissue concentrations, BCV was retained in the intestines and liver more than NPP-669; however, CDV and CDV-PP levels were increased for NPP-669 in both the intestine and liver compared to BCV. Interestingly, kidney exposure was reduced with NPP-669 compared to BCV. These findings indicate that NPP-669 increases the concentration of the pharmacologically active metabolite, CDV-PP, in the target tissues and that the exposure of the parent compound is reduced compared to BCV. These in vivo distribution data showed an excellent correlation with the in vitro intracellular conversion data.
To fully evaluate the efficacy and safety of NPP-669, three animal models were utilized. First, a mouse model of CMV demonstrated suppression of viral titers in the liver at doses as low as 1 mg/kg/day and in the spleen at doses as low as 3 mg/kg/day. Doses up to 30 mg/kg/day were well tolerated with no signs of toxicity. Secondly, a hamster model of AdV showed that NPP-669 increased survival, decreased body weight loss, inhibited HAdV-C6 replication in the liver, and significantly decreased liver damage compared to CDV. In this model, doses up to 10 mg/kg/day were well tolerated with no signs of toxicity. For the last model, toxicity was examined in healthy male mice. There was no kidney toxicity with NPP-669 at doses up to 30 mg/kg/day. Furthermore, minimal changes in the GI tract were observed, but these did not cause weight loss or changes to blood chemistry. These data indicate that NPP-669 is effective and safe for the treatment of CMV and AdV.
It should be noted that additional investigations of the PK/PD relationship and dosing regimen are needed to fully characterize and maximize the safety margin and project a safe starting dose for clinical trials. However, based on the abovementioned results, we have shown that NPP-669 has overcome the dose-limiting toxicity associated with both CDV and BCV at a therapeutically relevant dosing regimen.
Conclusions
NPP-669 is a novel prodrug of CDV that has increased lipophilicity, improved oral absorption, reduced nephrotoxicity, increased cellular uptake, and increased activity against several dsDNA viruses compared to CDV. Furthermore, it has reduced GI toxicity compared to another prodrug of CDV, BCV. Our results suggest that NPP-669 is an efficacious antiviral that could be used to treat CMV infections, as well as other dsDNA viral infections, albeit with a wider therapeutic window compared to current agents.
Acknowledgments
We dedicate this paper to John Hilfinger, PhD, who was one of the original inventive drivers of this program, and Mark Prichard, PhD, and William Wold, PhD, two long-time collaborators and friends, who significantly shaped the selection and therapeutic validation of the ultimate lead candidate.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.2c00668.
Author Present Address
¶ Depression Center, University of Michigan, Ann Arbor, MI 48109
Author Present Address
∇ Bioinformatics, University of Michigan Medical School, Ann Arbor, MI 48109.
Author Present Address
○ Pfizer, Inc., Pearl River, NY.
This program was supported by TSRL grants R03AI135760 and Phase I and II SBIR R44AI100401. TSRL has also utilized the nonclinical and preclinical services program offered by the National Institute of Allergy and Infectious Diseases. These services were supported by NIH contracts HHSN272201800001I, HHSN75N93019D00016, HSN272201700017I/HHSN27200003/A05, and HSN272201700041I/75N93021F00004/A56.
The authors declare no competing financial interest.
Author Status
⧫ Deceased.
Supplementary Material
References
- Ho D. Y.; Enriquez K.; Multani A. Herpesvirus Infections Potentiated by Biologics. Infect. Dis. Clin. 2020, 34, 311–339. 10.1016/j.idc.2020.02.006. [DOI] [PubMed] [Google Scholar]
- Wold W. S. M.; Ison M. G.. Adenoviruses. In Fields Virology; Knipe D. M., Howley P. M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2013; pp 1732–1767. [Google Scholar]
- Moens U.; Krumbholz A.; Ehlers B.; Zell R.; Johne R.; Calvignac-Spencer S.; Lauber C. Biology, evolution, and medical importance of polyomaviruses: An update. Infect. Genet. Evol. 2017, 54, 18–38. 10.1016/j.meegid.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Huang Y. T.; Kim S. J.; Lee Y. J.; Burack D.; Nichols P.; Maloy M.; Perales M. A.; Giralt S. A.; Jakubowski A. A.; Papanicolaou G. A. Co-Infections by Double-Stranded DNA Viruses after Ex Vivo T Cell-Depleted, CD34(+) Selected Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1759–1766. 10.1016/j.bbmt.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison M. G.; Hayden R. T.. Adenovirus. Microbiol. Spectr. 2016, 4, 10.1128/microbiolspec.DMIH2-0020-2015 [DOI] [PubMed] [Google Scholar]
- Lion T. Adenovirus persistence, reactivation, and clinical management. FEBS Lett. 2019, 593, 3571–3582. 10.1002/1873-3468.13576. [DOI] [PubMed] [Google Scholar]
- Annaloro C.; Serpenti F.; Saporiti G.; Galassi G.; Cavallaro F.; Grifoni F.; Goldaniga M.; Baldini L.; Onida F. Viral Infections in HSCT: Detection, Monitoring, Clinical Management, and Immunologic Implications. Front. Immunol. 2020, 11, 569381. 10.3389/fimmu.2020.569381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerry Teng C. L.; Wang P. N.; Chen Y. C.; Ko B. S. Cytomegalovirus management after allogeneic hematopoietic stem cell transplantation: A mini-review. J. Microbiol. Immunol. Infect. 2021, 54, 341–348. 10.1016/j.jmii.2021.01.001. [DOI] [PubMed] [Google Scholar]
- Razonable R. R. Cytomegalovirus in Solid Organ Transplant Recipients: Clinical Updates, Challenges and Future Directions. Curr. Pharm. Des. 2020, 26, 3497–3506. 10.2174/1381612826666200531152901. [DOI] [PubMed] [Google Scholar]
- Green M. Introduction: Infections in solid organ transplantation. Am. J. Transplant. 2013, 13, 3–8. 10.1111/ajt.12093. [DOI] [PubMed] [Google Scholar]
- Jung S.; Michel M.; Stamminger T.; Michel D. Fast breakthrough of resistant cytomegalovirus during secondary letermovir prophylaxis in a hematopoietic stem cell transplant recipient. BMC Infect. Dis. 2019, 19, 388. 10.1186/s12879-019-4016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Aziz S. L.; Hartline C. B.; Harden E. A.; Daily S. L.; Prichard M. N.; Kushner N. L.; Beadle J. R.; Wan W. B.; Hostetler K. Y.; Kern E. R. Comparative activities of lipid esters of cidofovir and cyclic cidofovir against replication of herpesviruses in vitro. Antimicrob. Agents Chemother. 2005, 49, 3724–3733. 10.1128/aac.49.9.3724-3733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu V.; de Vieira L. T. V. M.; Zhao P.; Zhang L.; Zheng J. H.; Nordmark A.; Berglund E. G.; Giacomini K. M.; Huang S. M. Towards quantitation of the effects of renal impairment and probenecid inhibition on kidney uptake and efflux transporters, using physiologically based pharmacokinetic modelling and simulations. Clin. Pharmacokinet. 2014, 53, 283–293. 10.1007/s40262-013-0117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Helou G.; Razonable R. R. Safety considerations with current and emerging antiviral therapies for cytomegalovirus infection in transplantation. Expet Opin. Drug Saf. 2019, 18, 1017–1030. 10.1080/14740338.2019.1662787. [DOI] [PubMed] [Google Scholar]
- Starrett J. E. Jr.; Tortolani D. R.; Hitchcock M. J.; Martin J. C.; Mansuri M. M. Synthesis and in vitro evaluation of a phosphonate prodrug: bis(pivaloyloxymethyl) 9-(2-phosphonylmethoxyethyl)adenine. Antiviral Res. 1992, 19, 267–273. 10.1016/0166-3542(92)90084-i. [DOI] [PubMed] [Google Scholar]
- Hostetler K. Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antiviral Res. 2009, 82, A84–A98. 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrottes S.; Egron D.; Lefebvre I.; Gosselin G.; Imbach J. L.; Périgaud C. SATE pronucleotide approaches: an overview. Mini Rev. Med. Chem. 2004, 4, 395–408. 10.2174/1389557043404007. [DOI] [PubMed] [Google Scholar]
- Oliyai R.; Shaw J. P.; Sueoka-Lennen C. M.; Cundy K. C.; Arimilli M. N.; Jones R. J.; Lee W. A. Aryl ester prodrugs of cyclic HPMPC. I: Physicochemical characterization and in vitro biological stability. Pharm. Res. 1999, 16, 1687–1693. 10.1023/a:1018945713623. [DOI] [PubMed] [Google Scholar]
- Jessen H. J.; Schulz T.; Balzarini J.; Meier C. Bioreversible protection of nucleoside diphosphates. Angew. Chem., Int. Ed. Engl. 2008, 47, 8719–8722. 10.1002/anie.200803100. [DOI] [PubMed] [Google Scholar]
- Meier C.; Görbig U.; Müller C.; Balzarini J. cycloSal-PMEA and cycloAmb-PMEA: potentially new phosphonate prodrugs based on the cycloSal-pronucleotide approach. J. Med. Chem. 2005, 48, 8079–8086. 10.1021/jm050641a. [DOI] [PubMed] [Google Scholar]
- Cahard D.; McGuigan C.; Balzarini J. Aryloxy phosphoramidate triesters as pro-tides. Mini Rev. Med. Chem. 2004, 4, 371–81. 10.2174/1389557043403936. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–1791. 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- Krylov I. S.; Kashemirov B. A.; Hilfinger J. M.; McKenna C. E. Evolution of an Amino Acid Based Prodrug Approach: Stay Tuned. Mol. Pharm. 2013, 10, 445–458. 10.1021/mp300663j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharova V. M.; Serpi M.; Krylov I. S.; Peterson L. W.; Breitenbach J. M.; Borysko K. Z.; Drach J. C.; Collins M.; Hilfinger J. M.; Kashemirov B. A.; McKenna C. E. Tyrosine-based 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: synthesis, stability, antiviral activity, and in vivo transport studies. J. Med. Chem. 2011, 54, 5680–5693. 10.1021/jm2001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Spencer J. F.; Dhar D.; Sagartz J. E.; Buller R. M.; Painter G. R.; Wold W. S. M. Hexadecyloxypropyl-cidofovir, CMX001, prevents adenovirus-induced mortality in a permissive, immunosuppressed animal model. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 7293–7297. 10.1073/pnas.0800200105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo J. F.; Morris M. I.; Abbo L. M.; Simkins J.; Saneeymehri S.; Alencar M. C.; Lekakis L. J.; Komanduri K. V. The use of brincidofovir for the treatment of mixed dsDNA viral infection. J. Clin. Virol. 2016, 83, 1–4. 10.1016/j.jcv.2016.07.021. [DOI] [PubMed] [Google Scholar]
- El-Haddad D.; El Chaer F.; Vanichanan J.; Shah D. P.; Ariza-Heredia E. J.; Mulanovich V. E.; Gulbis A. M.; Shpall E. J.; Chemaly R. F. Brincidofovir (CMX-001) for refractory and resistant CMV and HSV infections in immunocompromised cancer patients: A single-center experience. Antiviral Res. 2016, 134, 58–62. 10.1016/j.antiviral.2016.08.024. [DOI] [PubMed] [Google Scholar]
- Florescu D. F.; Keck M. A. Development of CMX001 (Brincidofovir) for the treatment of serious diseases or conditions caused by dsDNA viruses. Expert Rev. Anti Infect. Ther. 2014, 12, 1171–1178. 10.1586/14787210.2014.948847. [DOI] [PubMed] [Google Scholar]
- Florescu D. F.; Pergam S. A.; Neely M. N.; Qiu F.; Johnston C.; Way S.; Sande J.; Lewinsohn D. A.; Guzman-Cottrill J. A.; Graham M. L.; Papanicolaou G.; Kurtzberg J.; Rigdon J.; Painter W.; Mommeja-Marin H.; Lanier R.; Anderson M.; van der Horst C. Safety and efficacy of CMX001 as salvage therapy for severe adenovirus infections in immunocompromised patients. Biol. Blood Marrow Transplant. 2012, 18, 731–738. 10.1016/j.bbmt.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D. C.; Lampert B.; Collins D. J.; Rice T. L.; Painter G. R.; Kern E. R. Efficacy of CMX001 against herpes simplex virus infections in mice and correlations with drug distribution studies. J. Infect. Dis. 2010, 202, 1492–1499. 10.1086/656717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty F. M.; Winston D. J.; Chemaly R. F.; Mullane K. M.; Shore T. B.; Papanicolaou G. A.; Chittick G.; Brundage T. M.; Wilson C.; Morrison M. E.; Foster S. A.; Nichols W. G.; Boeckh M. J. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. 10.1016/j.bbmt.2018.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detweiler C. J.; Mueller S. B.; Sung A. D.; Saullo J. L.; Prasad V. K.; Cardona D. M. Brincidofovir (CMX001) Toxicity Associated With Epithelial Apoptosis and Crypt Drop Out in a Hematopoietic Cell Transplant Patient: Challenges in Distinguishing Drug Toxicity From GVHD. J. Pediatr. Hematol. Oncol. 2018, 40, e364–e368. 10.1097/mph.0000000000001227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter W.; Robertson A.; Trost L. C.; Godkin S.; Lampert B.; Painter G. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrob. Agents Chemother. 2012, 56, 2726–2734. 10.1128/aac.05983-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y.; Yamada J.; Watanabe T.; Suga T.; Takayama H. Participation of the peroxisomal beta-oxidation system in the chain-shortening of PCA16, a metabolite of the cytosine arabinoside prodrug, YNKO1, in rat liver. Biochem. Pharmacol. 1990, 39, 1505–1512. 10.1016/0006-2952(90)90514-l. [DOI] [PubMed] [Google Scholar]
- Kern E. R.; Hartline C.; Harden E.; Keith K.; Rodriguez N.; Beadle J. R.; Hostetler K. Y. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob. Agents Chemother. 2002, 46, 991–995. 10.1128/aac.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier R.; Trost L.; Tippin T.; Lampert B.; Robertson A.; Foster S.; Rose M.; Painter W.; O’Mahony R.; Almond M.; Painter G. Development of CMX001 for the treatment of poxvirus infections. Viruses 2010, 2, 2740–2762. 10.3390/v2122740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cihlar T.; Chen M. S. Identification of enzymes catalyzing two-step phosphorylation of cidofovir and the effect of cytomegalovirus infection on their activities in host cells. Mol. Pharmacol. 1996, 50, 1502–1510. [PubMed] [Google Scholar]
- Hartline C. B.; Keith K. A.; Eagar J.; Harden E. A.; Bowlin T. L.; Prichard M. N. A standardized approach to the evaluation of antivirals against DNA viruses: Orthopox-, adeno-, and herpesviruses. Antiviral Res. 2018, 159, 104–112. 10.1016/j.antiviral.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Keith K. A.; Hartline C. B.; Bowlin T. L.; Prichard M. N. A standardized approach to the evaluation of antivirals against DNA viruses: Polyomaviruses and lymphotropic herpesviruses. Antiviral Res. 2018, 159, 122–129. 10.1016/j.antiviral.2018.09.016. [DOI] [PubMed] [Google Scholar]
- Cardin R. D.; Bravo F. J.; Sewell A. P.; Cummins J.; Flamand L.; Juteau J. M.; Bernstein D. I.; Vaillant A. Amphipathic DNA polymers exhibit antiviral activity against systemic murine Cytomegalovirus infection. Virol. J. 2009, 6, 214. 10.1186/1743-422x-6-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Hussein I. T. M.; Tollefson A. E.; Ying B.; Spencer J. F.; Eagar J.; James S. H.; Prichard M. N.; Wold W. S. M.; Bowlin T. L. Filociclovir Is a Potent In Vitro and In Vivo Inhibitor of Human Adenoviruses. Antimicrob. Agents Chemother. 2020, 64, e01299-20 10.1128/aac.01299-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard M. N.; Williams J. D.; Komazin-Meredith G.; Khan A. R.; Price N. B.; Jefferson G. M.; Harden E. A.; Hartline C. B.; Peet N. P.; Bowlin T. L. Synthesis and antiviral activities of methylenecyclopropane analogs with 6-alkoxy and 6-alkylthio substitutions that exhibit broad-spectrum antiviral activity against human herpesviruses. Antimicrob. Agents Chemother. 2013, 57, 3518–3527. 10.1128/aac.00429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dioverti M. V.; Razonable R. R.. Cytomegalovirus. Microbiol. Spectr. 2016, 4, 10.1128/microbiolspec.DMIH2-0022-2015 [DOI] [PubMed] [Google Scholar]
- Alvarez-Cardona J. J.; Whited L. K.; Chemaly C. R. F. understanding its unique profile and potential role against adenovirus and other viral infections. Future Microbiol. 2020, 15, 389–400. 10.2217/fmb-2019-0288. [DOI] [PubMed] [Google Scholar]
- Heidel K. M.; Dowd C. S. Phosphonate prodrugs: an overview and recent advances. Future Med. Chem. 2019, 11, 1625–1643. 10.4155/fmc-2018-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph F. A. M.; Fuller A. L.; Slawin A. M.; Bühl M.; Alan Aitken A.; Derek Woollins J. D. The X-ray structures of sulfones. J. Chem. Crystallogr. 2010, 40, 253–265. 10.1007/s10870-009-9643-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.