

Abstract
C-reactive protein (CRP) is synthesized in hepatocytes. The serum concentration of CRP increases dramatically during the acute phase response. In human hepatoma Hep3B cells, maximal CRP expression occurs in cells treated with the combination of IL-6 and IL-1β. IL-6 induces transcription of the CRP gene and IL-1β synergistically enhances the effects of IL-6. We investigated the role of IL-6-activated transcription factor STAT3, also known as STAT3α, in inducing CRP expression since we identified four consensus STAT3-binding sites centered at positions −72, −108, −134 and −164 on the CRP promoter. It has been shown previously that STAT3 binds to the site at −108 and induces CRP expression. We found that STAT3 also bound to the other three sites, and several STAT3-containing complexes were formed at each site, suggesting the presence of STAT3 isoforms and additional transcription factors in the complexes. Mutation of the STAT3 sites at −108, −134 or −164 resulted in decreased CRP expression in response to IL-6 and IL-1β treatment, although the synergy between IL-6 and IL-1β was not affected by the mutations. The STAT3 site at −72 could not be investigated employing mutagenesis. We also found that IL-6 activated two isoforms of STAT3 in Hep3B cells: STAT3α which contains both a DNA-binding domain and a transactivation domain and STAT3β which contains only the DNA-binding domain. Taken together, these findings raise the possibility that IL-6 not only induces CRP expression but also regulates the induction of CRP expression by activating STAT3 isoforms and by utilizing all four STAT3 sites.
Keywords: Acute phase protein, Acute phase response, C-reactive protein, STAT3 isoform
1. Introduction
Experiments involving animal models of pneumococcal infection, atherosclerosis and inflammatory arthritis have revealed that C-reactive protein (CRP) performs host defense functions (Jones et al., 2011; Ngwa et al., 2020; Pathak et al., 2020). The mechanisms of CRP gene expression, however, under physiological and pathological conditions, are unknown. CRP is classified as a major, positive, acute phase protein in humans since the serum concentration of CRP may be increased thousand-fold or more during the acute phase response (Gabay and Kushner, 1999; Kushner, 1982). CRP is primarily synthesized by hepatocytes (Ciliberto et al., 1987; Kushner and Feldmann, 1978). The regulation of CRP gene expression in hepatic cells occurs mainly at the transcriptional level (Castell et al., 1990), although post-transcriptional mechanisms have also been shown to contribute to the regulation of CRP expression (Kim et al., 2015). However, the composition of the transcriptional complex formed on the promoter of the CRP gene (Lei et al., 1985; Woo et al., 1985) that induces CRP expression during the acute phase response remains uncharacterized (Bode et al., 2012; Salazar et al., 2014; Zhou et al., 2016).
IL-6 has been found to be the major cytokine that induces CRP expression in hepatocytes and in human hepatoma Hep3B cells (Castell et al., 1990; Ganapathi et al., 1988; Huang et al., 2019; Li et al., 1990; Zhang et al., 1995). STAT3 is one of the transcription factors activated by IL-6 and has been shown to participate in the induction of CRP expression (Akira et al., 1994; May et al., 2003; Voleti and Agrawal, 2006; Zhang et al., 1996; Zhang et al., 2020). C/EBPβ and C/EBPδ, the other two IL-6-activated transcription factors, also participate in the induction of CRP expression (Ramji et al., 1993; Young et al., 2008). IL-1β by itself does not induce CRP expression in Hep3B cells; however, IL-1β synergistically enhances the effects of IL-6 in inducing CRP expression by activating transcription factor NF-κB (Agrawal et al., 2003a; Castell et al., 1990; Ganapathi et al., 1988; Zhang et al., 1995). The first 157 bp region of the CRP promoter is sufficient for the synergy between IL-6 and IL-1β (Agrawal et al., 2003a; Ganapathi et al., 1988; Nishikawa et al., 2008; Toniatti et al., 1990a; Zhang et al., 1995). CRP expression in response to IL-6 alone or in response to the combination of IL-6 and IL-1β, however, was found to be higher when CRP promoters longer than 157 bp were utilized in transactivation assays (Blaschke et al., 2006; Li et al., 1990; Zhang et al., 1996). For example, CRP expression driven by the first 300 bp region of the CRP promoter is five to ten-fold higher than when driven by the first 157 bp region of the promoter in response to IL-6 and in response to the combination of IL-6 and IL-1β (Voleti and Agrawal, 2005). The synergy between IL-6 and IL-1β on 300 bp promoter, however, was not different from the synergy observed on 157 bp promoter (Voleti and Agrawal, 2005).
In addition to STAT3, C/EBPβ, C/EBPδ and classical NF-κB heterodimer p50-p65, the transcription factors HNF-1, HNF-3, C/EBPζ, RBP-Jκ, Oct-1, c-Rel-containing NF-κB heterodimers and p50-p50 NF-κB homodimer have also been shown to participate in transcription of the CRP gene (Agrawal et al., 2001; Agrawal et al., 2003b; Cha-Molstad et al., 2000; Cha-Molstad et al., 2007; Majello et al., 1990; Singh et al., 2007; Toniatti et al., 1990b; Voleti and Agrawal, 2005; Voleti et al., 2012). These transcription factors function either by directly binding to their cognate sites on the promoter or by interacting with other promoter-bound transcription factors, without directly binding to DNA (Agrawal et al., 2003b; Cha-Molstad et al., 2007; Singh et al., 2007; Voleti et al., 2012).
The aim of this study was to investigate the role of IL-6-activated STAT3 in inducing CRP expression in Hep3B cells. There are four isoforms of STAT3 (Caldenhoven et al., 1996; Chakraborty and Twerady, 1998; Hevehan et al., 2002; Schaefer et al., 1997; Schindler et al., 2007; Zhong et al., 1994): STAT3α (or simply STAT3), STAT3β, STAT3γ and STAT3δ. STAT3α (transcriptional activator) is the longest isoform (92 kDa) and contains both a DNA-binding domain and a transactivation domain. STAT3β (transcriptional repressor) is an alternatively spliced isoform (82 kDa) and contains a DNA-binding domain but lacks the transactivation domain, and thus can function as a dominant negative isoform of STAT3α. In addition to being a dominant negative factor, STAT3β can also (indirect activator), by itself, induce the expression of specific target genes by recruiting transcription factors containing a transactivation domain (Schindler et al., 2007). STAT3γ (72 kDa) and STAT3δ (64 kDa) are proteolytically cleaved products of STAT3α and contain a DNA binding domain but lack the transactivation domain. All isoforms of STAT3 can be activated by IL-6 (Levy and Lee, 2002; Maritano et al., 2004; Schaefer et al., 1997). However, these isoforms are not present at the same ratio throughout; their relative amounts have been observed to change in cells depending on the cell growth stage and growth conditions (Hevehan et al., 2002).
The nucleotide sequence of the consensus STAT3-binding site is TT(N)5AA; however, STAT3 has also been shown to bind to TT(N)4AA motifs in the promoter (Akira et al., 1994; Seidel et al., 1995). We identified four consensus STAT3-binding sites within the first 300 bp region of the CRP promoter; the sites are centered at positions −72 (−69 to −76), −108 (−105 to −112), −134 (−130 to −137) and −164 (−160 to −168) (Fig. 1A). In the current study, employing electrophoretic mobility shift assays (EMSA) and luciferase (Luc) transactivation assays, and by using two truncated CRP promoter constructs, −300/+1 and −157/+3 bp regions of the CRP promoter linked to Luc reporter gene, we investigated whether these STAT3 sites bound to IL-6-activated STAT3 and if they were transcriptionally active in controlling IL-6-induced CRP gene expression.
Fig. 1.
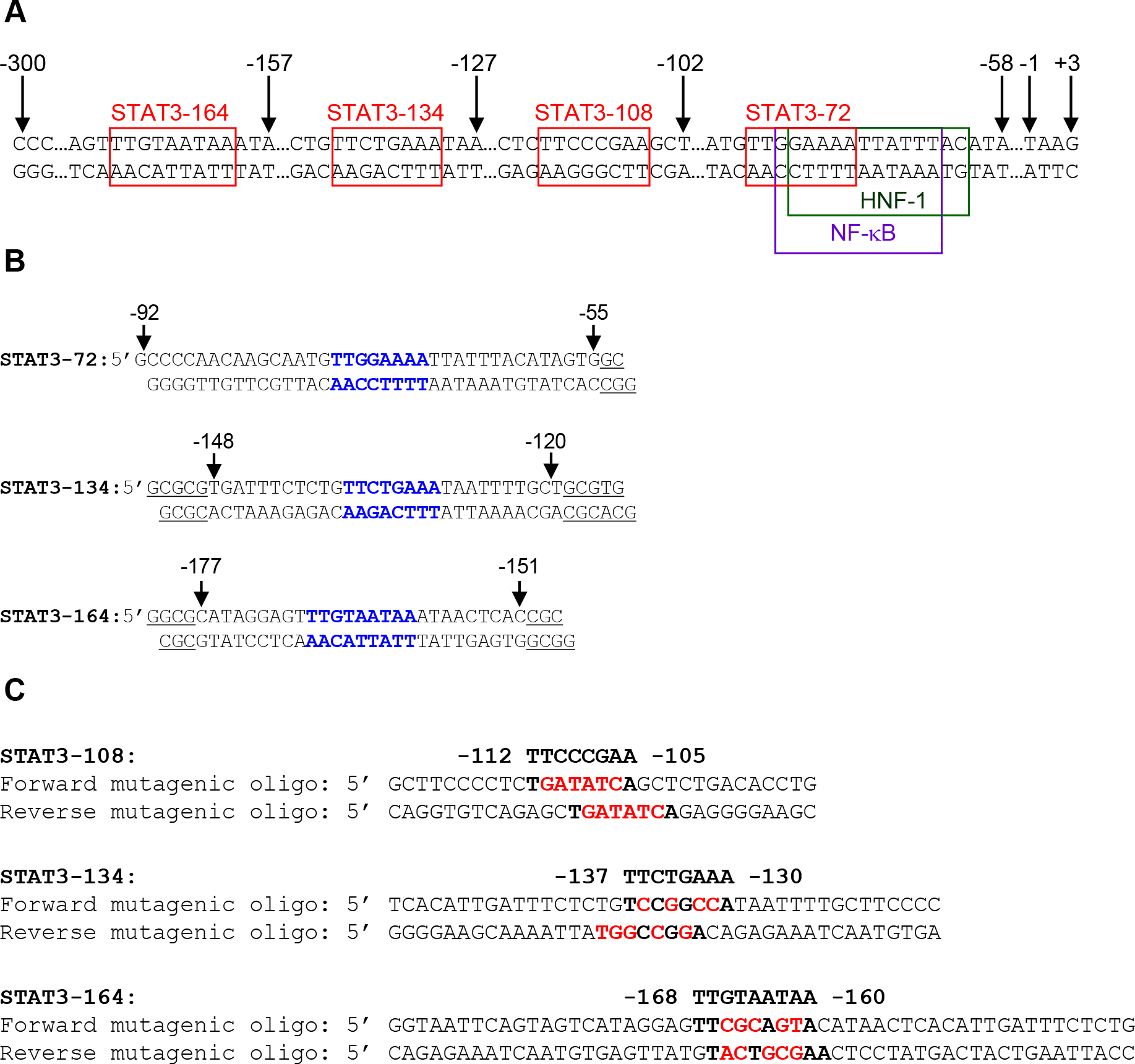
(A) Partial nucleotide sequence of the −300 to +3 region of the CRP gene. The four STAT3-binding sites centered at positions −72 (−69 to −76), −108 (−105 to −112), −134 (−130 to −137) and −164 (−160 to −168) on the promoter are shown in red boxes. The STAT3 site at position −72 overlaps the binding sites for transcription factors HNF-1 and NF-κB. (B) Sequences of the double-stranded oligos derived from the CRP promoter (numbered on the top) and used as probes in EMSA. The STAT3 sites are shown in bold. The underlined nucleotides are not from the CRP promoter; these nucleotides were added to the ends of the oligos according to the instructions from the manufacturer of the radioactive end-labelling kit. (C) Pairs of forward and reverse mutagenic oligo primers used for site-directed mutagenesis of the CRP promoter. The STAT3 sites centered at positions −108, −134 and −164 are shown in bold letters; the mutated bases are shown in red. The primers were designed according to the instructions from the manufacturer of the mutagenesis kit.
2. Materials and methods
2.1. Preparation of nuclear and cytoplasmic extracts
Nuclear and cytoplasmic extracts of Hep3B cells were prepared as described previously (Singh et al., 2007). Hep3B cells (ATCC) were cultured and grown to ~60% confluency and then were subjected to serum starvation overnight. Cells were then treated with IL-6, IL-1β and the combination of IL-6 and IL-1β for 15 min and for 18 h. IL-6 and IL-1β (R&D; catalog numbers 206-IL and 201-LB) were used at concentrations of 10 ng/ml and 1 ng/ml, respectively. Nuclear and cytoplasmic extracts from untreated and cytokine-treated cells were prepared using NE-PER nuclear and cytoplasmic kit (Pierce) according to manufacturer’s instructions.
2.2. EMSA
EMSA was performed as described previously (Voleti et al., 2012). The STAT3 site-containing oligonucleotide (oligo) probes that were used in EMSA are shown in Fig. 1B. Oligos were obtained from Integrated DNA Technologies. Probes were prepared by annealing complementary oligos, followed by labelling with [γ−32P]-ATP (MP Biomedicals, catalog number 3502002) using end labelling with T4 polynucleotide kinase (Promega). Nuclear extracts from Hep3B cells treated with cytokines for 15 min were mixed with the labelled probe and incubated in the gel shift buffer (40 mM KCl, 20 mM HEPES pH 7.9, 1 mM MgCl2, 0.05 mM EGTA, 0.5 mM DTT, 4% Ficoll, and 1 μg of poly dI-dC) for 20 min at room temperature. DNA-protein complexes were resolved in 5% polyacrylamide gels containing 2.5% glycerol and analyzed in a phosphorimager using ImageQuant software (GE Healthcare). To identify STAT3 in the DNA-protein complexes, antibodies (2 μg) to STAT3 (Santa Cruz Biotechnologies, catalog number H-190X) were added to the nuclear extract and incubated on ice for 15 min, prior to addition of the probe.
2.3. Construction of CRP promoter-luc reporter plasmids
The construction of wild-type (WT) CRP promoter constructs, Luc-157 WT (−157 to +3 nucleotides of the CRP gene) and Luc-300 WT (−300 to −1 nucleotides of the CRP gene), has been reported previously (Voleti and Agrawal, 2005; Kleemann et al., 2003). These two WT promoter constructs were used as templates for mutating the STAT3 sites using Quick Change site-directed mutagenesis kit (Stratagene). The STAT3 site centered at position −108 was mutated (mST-108) by substituting −111TCCCGA−106 with −111GATATC−106. The STAT3 site centered at position −134 was mutated (mST-134) by substituting −138TTCTGAAA−131 with −138TCCGGCCA−131. The STAT3 site centered at position −164 was mutated (mST-164) by substituting −169TTGTAATAA−161 with −169TTCGCAGTA−161. The sequences of the mutagenic oligo primers are shown in Fig. 1C. The double mutants with both −108 and −134 STAT3 sites mutated (mST-108-134) were generated by using the single mutants with mutated STAT3-108 site as templates and STAT3-134 oligos as mutagenic primers. The double mutant with both −108 and −164 STAT3 sites mutated (mST-108-164) was generated by using the single mutant with mutated STAT3-108 site as the template and STAT3-164 oligos as mutagenic primers. The double mutant with both −134 and −164 STAT3 sites mutated (mST-134-164) was generated by using the single mutant with mutated STAT3-134 site as the template and STAT3-164 oligos as mutagenic primers. Mutations were verified by nucleotide sequencing, utilizing the services of the Molecular Biology Core Facility of the university. Plasmids were purified using maxiprep plasmid isolation kit (Qiagen).
2.4. Luc transactivation assay
Luc transactivation assays were performed as described previously (Voleti and Agrawal, 2005). Hep3B cells were cultured overnight for transient transfection with the CRP promoter-Luc reporter plasmids and treatment with cytokines in serum-free medium. The confluency of cells at the time of transfection was ~60%. Cells were cultured into 6-well plates and transfection was carried out using FuGENE 6 reagent (Promega). Briefly, per well, 10 μl FuGENE 6 was added to 125 μl RPMI 1640 and incubated for 5 min. Further, 1 μg of the plasmid was added to the RPMI-FuGENE 6 cocktail, incubated for 20 min at room temperature and added to the well. Cytokine treatment was performed 16 h post-transfection. IL-6 and IL-1β were used at concentrations of 10 ng/ml and 1 ng/ml, respectively, and cells were further incubated for 24 h. Luc assays were then performed using Luciferase Assay System (Promega) according to manufacturer’s instructions. WT and all mutant promoter constructs were assayed together.
2.5. Western Blot
Hep3B cytoplasmic extracts were subjected to SDS-PAGE under reducing conditions in a 4–20% gradient polyacrylamide gel. Western blot to visualize STAT3 was performed by using polyclonal antibodies to phosphorylated STAT3 (p-Stat3-Tyr705-R, Santa Cruz Biotechnologies, catalog number sc-7993-R). Pre-stained molecular weight marker was purchased from Bio-Rad.
2.6. Statistical Analysis
Luc activities were measured and plotted as mean ± SEM of three experiments. Unpaired students t-test was used to calculate p-values for the differences in the Luc activities of WT and various mutant promoters.
3. Results
3.1. STAT3 binds to all STAT3 sites on the CRP promoter
EMSA was employed to demonstrate the binding of IL-6-activated STAT3 to its cognate sites on the CRP promoter. The binding of STAT3 to the site at position −108 has already been reported (Zhang et al., 1996). The binding of STAT3 to the sites at positions −72, −134 and −164 are shown in Fig. 2; two images (shorter and longer exposures) of each gel are presented. The results as described below are based on the combined interpretation of both images of each gel.
Fig. 2.

Binding of IL-6-activated STAT3 to CRP promoter. EMSA were performed using the oligos derived from the CRP promoter as probes and nuclear extracts from IL-6-treated and (IL-6 + IL-1β)-treated Hep3B cells as sources of STAT3. Polyclonal anti-STAT3 antibody (α-STAT3) was added to nuclear extracts before the addition of the probe. Transcription factor complexes formed on the probes were visualized using a phosphorimager. Complexes containing STAT3 are indicated by arrows. For each probe, two different exposures of the same gel are shown in left and right panels. Free probes are shown in lanes 1, 6 and 11. A representative of two gels is shown for each probe.
When STAT3-72 oligo was used as the probe, four DNA-protein complexes (I-IV) were formed in response to IL-6 (lane 2). In the presence of antibodies to STAT3, two of these four complexes (II and IV) were abolished and the intensity of the other two complexes (I and III) was decreased (compare lanes 2 and 4) suggesting that all four complexes contained STAT3. In response to the combination of IL-6 and IL-1β, only two DNA-protein complexes (I and III) were formed (lane 3). In the presence of antibodies to STAT3, complex I was abolished and the intensity of complex III was decreased (compare lanes 3 and 5) suggesting the involvement of STAT3 in the formation of both complexes.
When STAT3-134 oligo was used as the probe, three DNA-protein complexes (I-III) were formed in response to IL-6 (lane 7). In the presence of antibodies to STAT3, complex I was abolished while the intensity of the complexes II and III was decreased (compare lanes 7 and 9) suggesting that all three complexes contained STAT3. In response to the combination of IL-6 and IL-1β also, three DNA-protein complexes (I-III) were formed (lane 8). In the presence of antibodies to STAT3, complexes I and II were abolished while the intensity of complex III was decreased (compare lanes 8 and 10) suggesting the involvement of STAT3 in the formation of all three complexes.
When STAT3-164 oligo was used as the probe, three DNA-protein complexes (I-III) were formed in response to IL-6 (lane 12). In the presence of antibodies to STAT3, the intensity of complex I was decreased while complexes II and III were abolished (compare lanes 12 and 14) suggesting that all three complexes contained STAT3. In response to the combination of IL-6 and IL-1β also, three DNA-protein complexes (I-III) were formed (lane 13). In the presence of antibodies to STAT3, the intensity of complex I was decreased while complexes II and III were abolished (compare lanes 13 and 15) suggesting the involvement of STAT3 in the formation of all three complexes.
Overall, these results indicate that all three newly identified consensus STAT3 sites can be occupied by STAT3 to form transcriptional complexes of varying compositions in response to IL-6 and in response to the combination of IL-6 and IL-1β.
3.2. Mutation in each STAT3 site decreases CRP expression
To determine whether all STAT3 sites contribute to CRP expression in response to IL-6 and to maximal CRP expression in response to the combination of IL-6 and IL-1β, we performed Luc transactivation assays. We first performed mutational analysis of the STAT3 sites in the context of 157 bp region of the CRP promoter. This is a region of the promoter that is sufficient for the synergy between IL-6 and IL-1β and contains three STAT3 sites, located at positions −72, −108 and −134. The site at −72 was not mutated at this time due to its overlap with binding sites for other transcription factors. The results of the mutational analysis of the sites at −108 and −134 in the 157 bp promoter context are shown in Fig. 3. As shown, the induction of Luc expression by the promoter in which both sites were mutated (mST-108-134) was significantly lower in response to IL-6 than the WT promoter (red bars). The IL-6 response was also decreased when only the site at −108 was mutated (mST-108) although the decrease was not statistically significant. Mutation of the site at −108, either singly or in mST-108-134, significantly decreased the overall maximal Luc expression in response to the combination of IL-6 and IL-1β compared to WT promoter (blue bars). Compared to WT promoter, there was no significant effect on Luc expression in response to cytokines when the site at −134 alone (mST-134) was mutated.
Fig. 3.
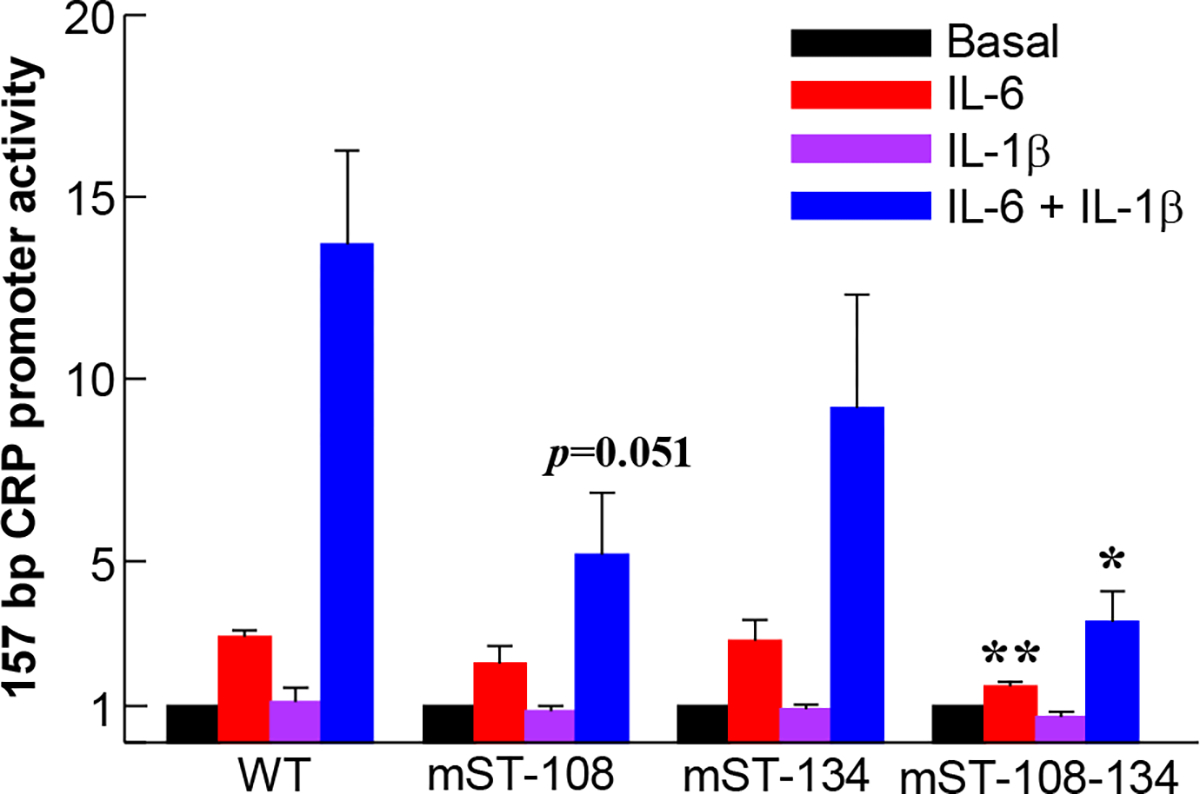
Effects of mutating the STAT3 sites on cytokine-induced promoter (−157/+3) activity. Hep3B cells were transfected with the WT and mutated CRP promoter-Luc constructs, as indicated on the x-axis, and treated with cytokines. CRP promoter activity was measured as transcription of the Luc reporter gene. The basal CRP promoter-Luc activity for each construct is taken as 1 and the activities in response to cytokine treatments are plotted as fold change over basal activity. Data shown are mean ± SEM of three experiments. The p values for the differences in the IL-6 (red bars) and IL-6+IL-1β (blue bars) responses of WT versus mutant promoters are shown as *p < 0.05 and **p < 0.005, unless indicated otherwise.
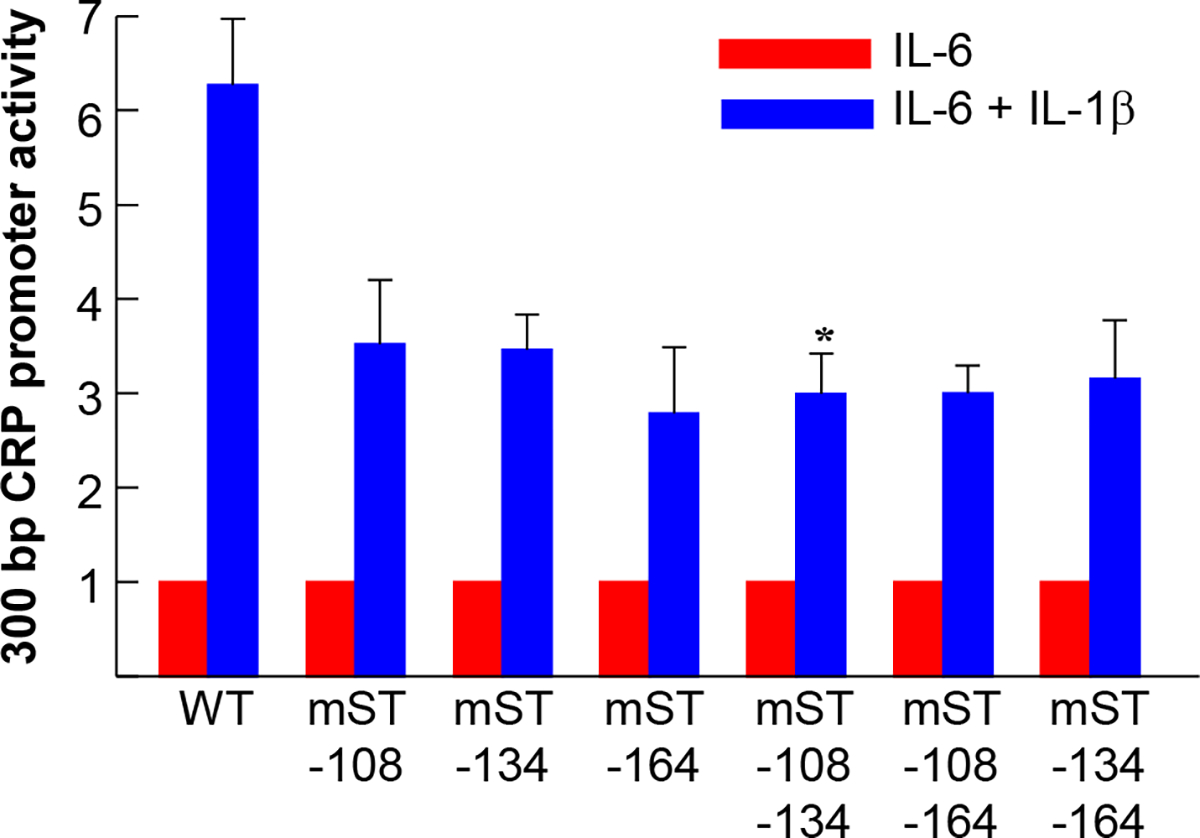
Next, we performed mutational analysis of the STAT3 sites in the context of 300 bp region of the CRP promoter. This region of the promoter contains all four STAT3 sites. The results of the mutational analysis of the sites at −108, −134 and −164 in the 300 bp promoter context are shown in Fig. 4. As shown, compared to WT promoter, the IL-6 response was decreased in all mutant promoters, except mST-134 (red bars), although the decrease was not statistically significant (p > 0.06). The decrease was more pronounced (p = 0.06) when the sites at −108 and −134 (mST-108-134) and the sites at −134 and −164 (mST-134-164) were mutated, compared to other mutants. The overall maximal Luc expression, however, in response to the combination of IL-6 and IL-1β (blue bars), was significantly decreased in all mutant promoters compared to WT promoter.
Fig. 4.
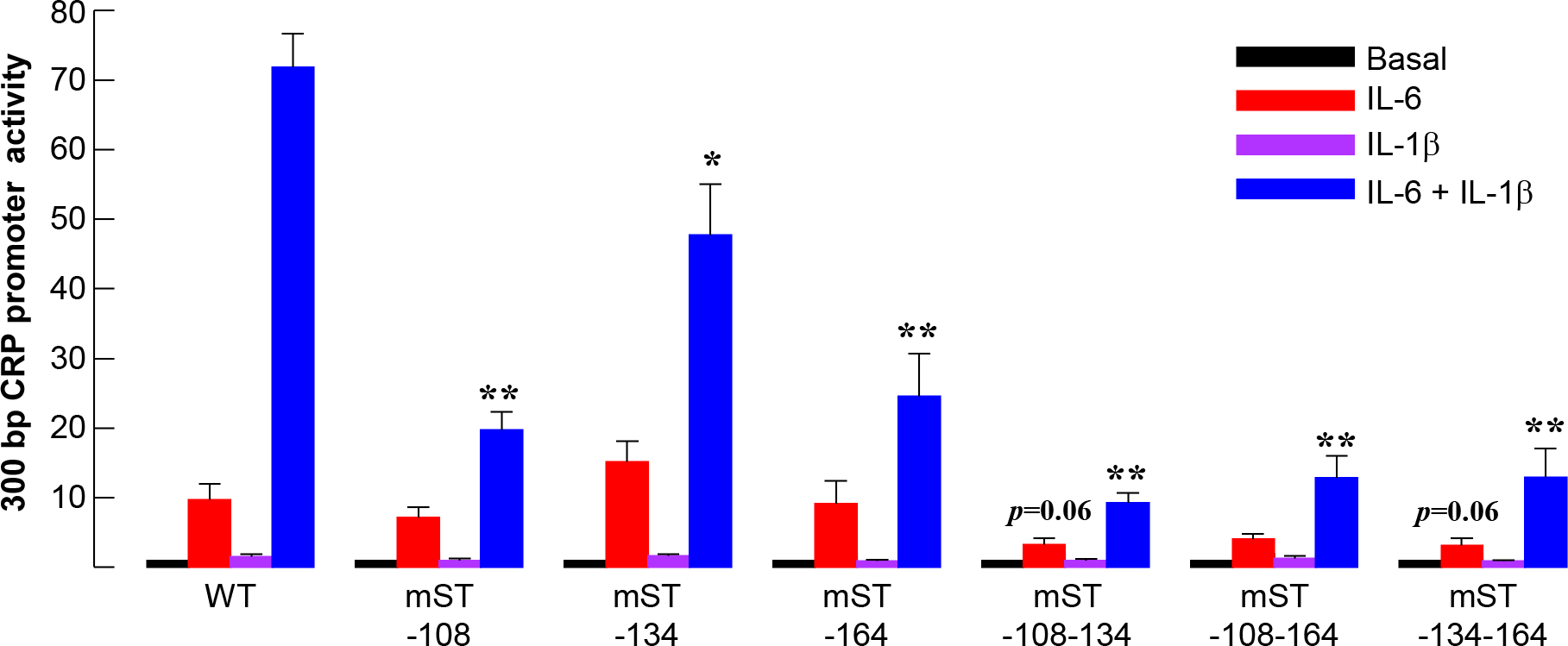
Effects of mutating the STAT3 sites on cytokine-induced promoter (−300/+1) activity. Hep3B cells were transfected with the WT and mutated CRP promoter-Luc constructs, as indicated on the x-axis, and treated with cytokines. CRP promoter activity was measured as transcription of the Luc reporter gene. The basal CRP promoter-Luc activity for each construct is taken as 1 and the activities in response to cytokine treatments are plotted as fold change over basal activity. Data shown are mean ± SEM of three experiments. The p values for the differences in the IL-6 (red bars) and IL-6+IL-1β (blue bars) responses of WT versus mutant promoters are shown as *p = 0.05 and **p < 0.001, unless indicated otherwise.
3.3. Mutations in the STAT3 sites do not affect the synergy between IL-6 and IL-1β
We next investigated whether the decrease in overall maximal CRP expression (Figs. 3 and 4) was due to the statistically insignificant decrease in the IL-6 response or due to the loss of synergy between IL-6 and IL-1β. The effects of mutations on the synergy between IL-6 and IL-1β on CRP promoter was determined by measuring the fold change in the IL-6 response with and without combining IL-1β. The results of the mutational analysis of the sites at −108 and −134 in the 157 bp promoter context are shown in Fig. 5. As shown, the synergy was observed in all single and double promoter mutants. Statistical analysis of the data showed that the differences seen in the synergy between WT and mutant promoters were not significant (p > 0.05). The results of the mutational analysis of the sites at −108, −134 and −164 in the 300 bp promoter context are shown in Fig. 6. As shown, the synergy was observed in all single and double promoter mutants. Statistical analysis of the data showed that the differences seen in the synergy between WT and mutant promoters were not significant (p > 0.05), except for the mutant mST-108-134. Since there was no statistically significant difference in the synergy between any two mutants, the difference seen between WT and mST-108-134 mutant was not obvious.
Fig. 5.
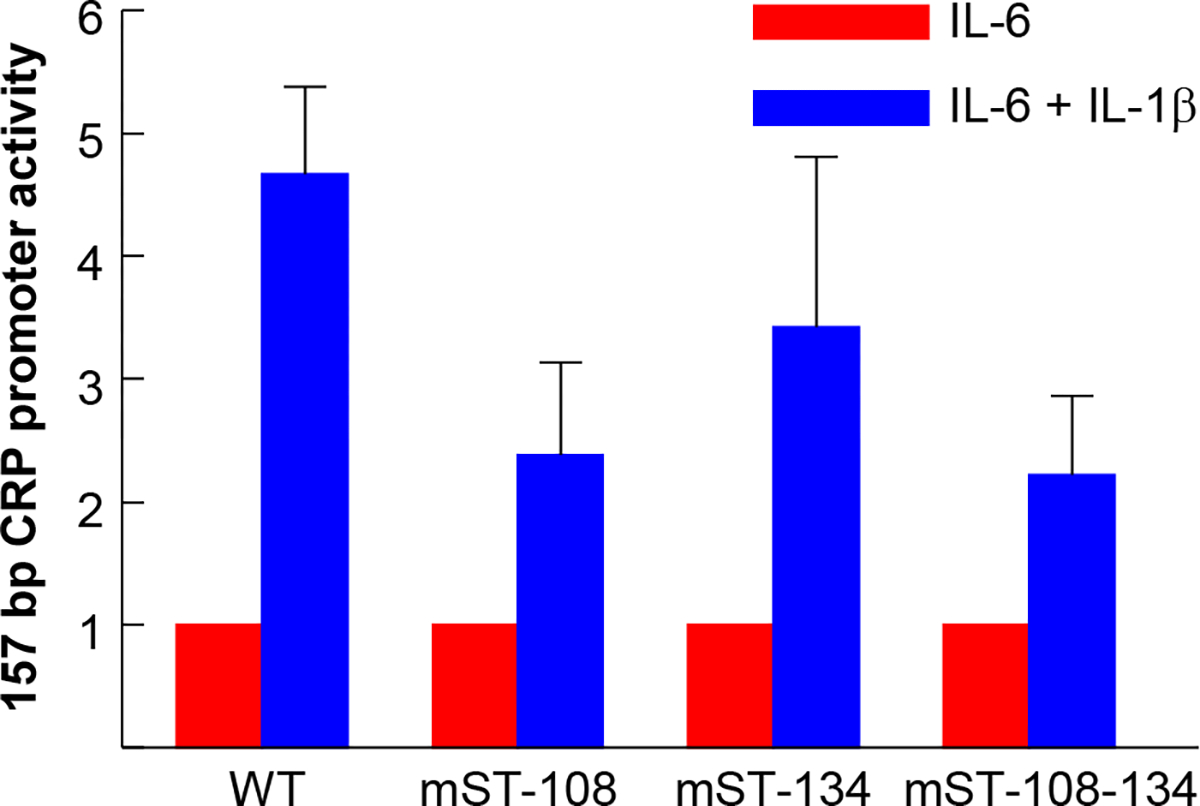
Effects of mutating the STAT3 sites on the synergy between IL-6 and IL-1β in inducing CRP promoter (Luc-157) activity. Hep3B cells were transfected with the WT and mutated CRP promoter-Luc constructs, as indicated on the x-axis, and treated with cytokines. Promoter activity was measured as transcription of the Luc reporter gene. The IL-6-induced CRP promoter-Luc activity for each construct is taken as 1 and the activity in response to the combination of IL-6 and IL-1β treatment is plotted as fold change over IL-6 activity. Data shown are mean ± SEM of three experiments. The differences between the WT and mutant promoters were not statistically significant (p > 0.05).
Fig. 6.
Effects of mutating the STAT3 sites on the synergy between IL-6 and IL-1β in inducing CRP promoter (Luc-300) activity. Hep3B cells were transfected with the WT and mutated CRP promoter-Luc constructs, as indicated on the x-axis, and treated with cytokines. Promoter activity was measured as transcription of the Luc reporter gene. The IL-6-induced CRP promoter-Luc activity for each construct is taken as 1 and the activity in response to the combination of IL-6 and IL-1β treatment is plotted as fold change over IL-6 activity. Data shown are mean ± SEM of three experiments (*p < 0.05).
3.4. IL-6 activates two isoforms of STAT3 in Hep3B cells
Western blot employing antibodies to phosphorylated STAT3 was performed to determine whether IL-6 activated only STAT3α or it also activated other STAT3 isoforms in Hep3B cells. As shown (Fig. 7), untreated Hep3B cells did not contain any phosphorylated STAT3 (lanes 2 and 6), indicating that STAT3 was not constitutively active in Hep3B cells. As expected, IL-1β did not activate STAT3 (lanes 4 and 8). IL-6 activated two isoforms of STAT3 (lanes 3 and 7). The molecular weights of the upper and lower bands were found to be 91.9±0.9 and 84.1±0.8 kDa, respectively, matching with the molecular weights of STAT3α (92 kDa) and STAT3β (82 kDa). Both isoforms were activated within 15 min of IL-6 treatment and persisted in the cell for at least 18 h. Similar results were obtained when the cells were treated with the combination of IL-6 and IL-1β (lanes 5 and 9). The relative amounts of the isoforms were not changed when IL-6 was combined with IL-1β.
Fig. 7.
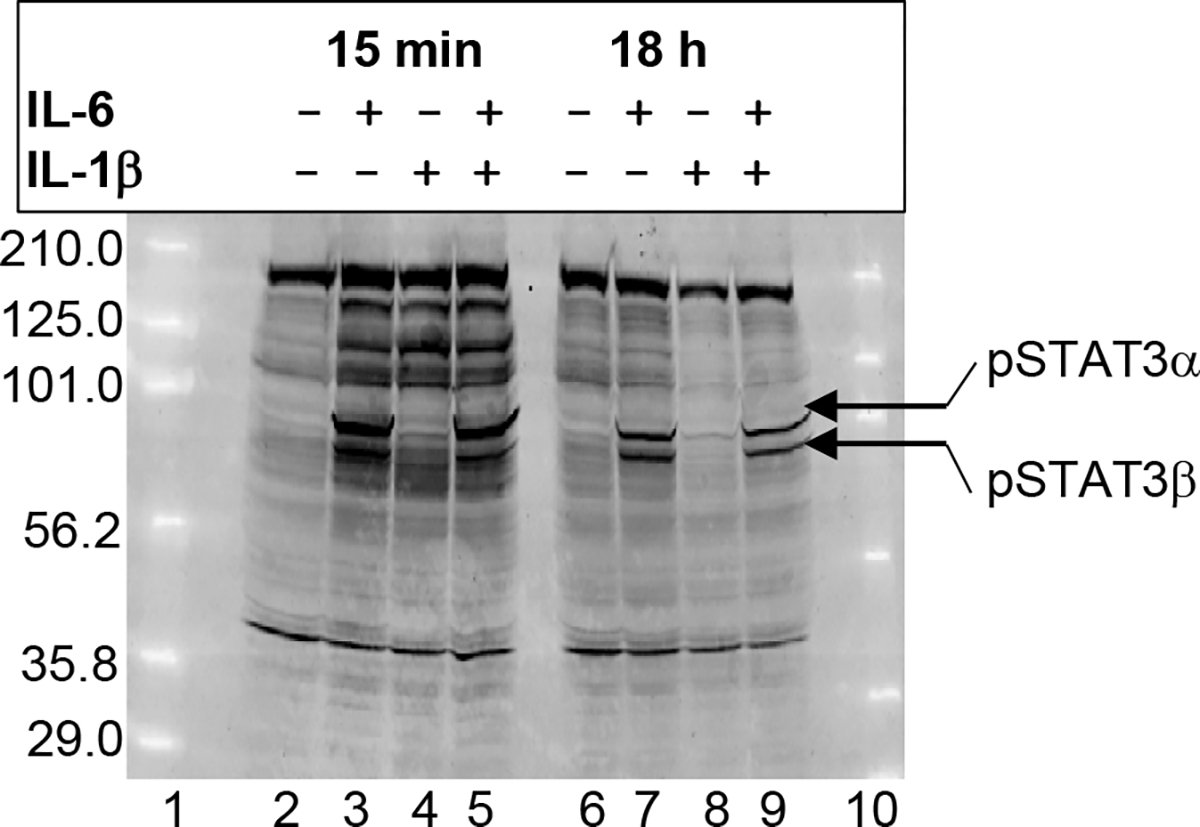
Activation of STAT3 isoforms by IL-6 in Hep3B cells. Cells were treated with cytokines for 15 min and 18 h. Cytoplasmic extracts were subjected to western blot by using polyclonal antibodies to phosphorylated STAT3. Pre-stained marker proteins were used to calculate the molecular weight of STAT3 isoforms. A representative of three blots is shown; however, all three experiments were used to calculate the molecular weight of the STAT3 isoform bands.
4. Discussion
The aim of this study was to investigate the roles of IL-6-activated STAT3 and of the four STAT3-binding sites present on the CRP promoter in the induction of CRP gene expression. Our major findings were: 1. STAT3 bound to its cognate sites centered at positions −72, −134 and −164. 2. Several STAT3-containing complexes were formed at each site. 3. Mutation of the STAT3 site at each position resulted in a decrease in maximal CRP expression in response to the combination of IL-6 and IL-1β. 4. Two isoforms of STAT3, STAT3α and STAT3β, were activated by IL-6 in Hep3B cells.
The presence of two STAT3 isoforms, STAT3α (direct activator) and STAT3β (repressor or indirect activator) in IL-6-treated Hep3B cells and the formation of multiple STAT3-containing complexes at each STAT3 site on the CRP promoter suggest that these complexes contained different isoforms of STAT3. Also, since STAT3β is able to recruit other transcription factors, it is possible that some STAT3 complexes formed on the CRP promoter contain additional transcription factors. It has been shown previously that the transcription factor APE1, which is also IL-6-inducible, complexes with STAT3 on the CRP promoter (Ray et al., 2010). STAT3 has also been shown to form a transcriptional complex with c-Fos and HNF-1α on the CRP promoter (Nishikawa et al., 2008). We hypothesize that STAT3-mediated regulation of induction of CRP expression likely involves the relative amounts of the two STAT3 isoforms in the nuclei (Snyder et al., 2008; Yang et al., 2007).
Our interpretation that STAT3 not only induces but also regulates the induction of CRP expression, is supported by the findings that mutations of all sites, at −108, −134 and at −164, did not significantly affect the IL-6 response, did not significantly decrease the synergy between IL-6 and IL-1β, but did significantly decrease the overall maximal CRP expression in response to the combination of IL-6 and IL-1β. While our work was in progress, the existence of the STAT3 sites at positions −72, −134 and −164 on the CRP promoter had been reported; mutations in none of these sites reduced the IL-6 response (Srivastava et al., 2018). It has been shown previously that the two regions on the CRP promoter, −46 to −94 and −106 to −137, participate in inducing CRP expression and the two regions function independently (Toniatti et al., 1990a). Thus, the STAT3 site at −72 might be more critical for the induction of CRP expression. Our data also suggest that the synergy between IL-6 and IL-1β in inducing CRP expression is most likely through crosstalk between IL-6-activated CEBPβ and IL-1β-activated NF-κB. It is unlikely that the STAT3 site at −72 contributes to synergy since this site overlaps the NF-κB site (Voleti et al., 2012), although an exchange of transcription factors at the STAT3 site at −72 cannot be ruled out.
The role of the STAT3 site centered at −108 had been previously explored (Zhang et al., 1996). Mutational analysis of the site in the context of 123 bp region of the CRP promoter and the data obtained from transactivation assays employing the STAT3 site linked to a TK promoter indicated that the site at −108 was critical for IL-6-induced CRP expression (Zhang et al., 1996). However, in these studies, overexpressed STAT3 was combined with the IL-6 treatment to observe the effects. Overexpressed STAT3 provides only STAT3α while IL-6 activates both STAT3α and STAT3β in Hep3B cells. It is possible that the decrease in CRP expression observed previously (Zhang et al., 1996) by mutating the site was in fact the decrease in overexpressed STAT3-mediated CRP expression. It is also possible that the data presented in this paper would be different if overexpressed STAT3 would have been added to IL-6 treatment. These data further support the hypothesis that the relative ratio of the isoforms of STAT3 is critical for regulating the induction of CRP expression.
One limitation of this study is that the EMSA and western blot were performed employing an anti-STAT3 antibody that recognizes both isoforms of STAT3. Use of the antibodies specific for STAT3α and STAT3β in EMSA and western blots would reveal the complexes containing STAT3α and STAT3β formed on the CRP promoter. We, however, conclude that IL-6 not only induces CRP expression by activating STAT3α but also regulates the induction of CRP expression by activating STAT3β. STAT3 isoforms do so by utilizing all STAT3 sites. The synergy between IL-6 and IL-1β involves crosstalk between IL-6-activated C/EBPβ and IL-1β-activated NF-κB. The occurrence of four STAT3 sites within 100 bp (from −69 to −168) on the CRP promoter, all nearly equally spaced, that is, ~25 bp apart, and located next to a region with overlapping sites for other transcription factors (Singh et al., 2007; Voleti et al., 2012), could be of significance and unique to CRP promoter. It would be interesting to see if such a transcription factor binding sites arrangement is also present on the serum amyloid A (SAA) promoter which is the only other major positive acute phase protein in humans (Gabay and Kushner, 1999; Kushner, 1982; Jensen and Whitehead, 1998) and not present on murine CRP promoter where CRP is not an acute phase protein (Ochrietor et al., 2000; Whitehead et al., 1990).
Acknowledgements
We thank Prem Prakash Singh for performing western blot.
This work was supported by the National Institutes of Health (AR068787 and AI151561; A.A.).
Abbreviations:
- CRP
C-reactive protein
- EMSA
electrophoretic mobility shift assay
- Luc
luciferase
- Oligo
oligonucleotide
- WT
wild-type
Footnotes
Declarations of interest: None
References
- Agrawal A, Cha-Molstad H, Samols D, Kushner I, 2001. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein β (C/EBPβ) and Rel p50. J. Immunol. 166, 2378–2384. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Cha-Molstad H, Samols D, Kushner I, 2003a. Overexpressed nuclear factor-κB can participate in endogenous C-reactive protein induction, and enhances the effects of C/EBPβ and signal transducer and activator of transcription-3. Immunology 108, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A, Samols D, Kushner I, 2003b. Transcription factor c-Rel enhances C-reactive protein expression by facilitating the binding of C/EBPβ to the promoter. Mol. Immunol. 40, 373–380. [DOI] [PubMed] [Google Scholar]
- Akira S, Nishio Y, Inoue M, Wang X-J, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T, 1994. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Takata Y, Caglayan E, Collins A, Tontonoz P, Hsueh WA, Tangirala RK, 2006. A nuclear receptor corepressor-dependent pathway mediates suppression of cytokine-induced C-reactive protein gene expression by liver X receptor. Circ. Res. 99, 88–99. [DOI] [PubMed] [Google Scholar]
- Bode JG, Albrecht U, Häussinger D, Heinrich PC, Schaper F, 2012. Hepatic acute phase proteins - Regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 91, 496–505. [DOI] [PubMed] [Google Scholar]
- Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JAM, Lammers J-WJ, Koenderman L, de Groot RP, 1996. STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 271, 13221–13227. [DOI] [PubMed] [Google Scholar]
- Castell JV, Gomez-Lechon MJ, David M, Fabra R, Trullenque R, Heinrich PC, 1990. Acute-phase response of human hepatocytes: Regulation of acute-phase protein synthesis by interleukin-6. Hepatology 12, 1179–1186. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Agrawal A, Zhang D, Samols D, Kushner I, 2000. The Rel family member p50 mediates cytokine-induced C-reactive protein expression by a novel mechanism. J. Immunol. 165, 4592–4597. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Young DP, Kushner I, Samols D, 2007. The interaction of c-Rel with C/EBPbeta enhances C/EBPbeta binding to the C-reactive protein gene promoter. Mol. Immunol. 44, 2933–2942. [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Tweardy DJ, 1998. Granulocyte colony-stimulating factor activates a 72-kDa isoform of STAT3 in human neutrophils. J. Leukoc. Biol. 64, 675–680. [DOI] [PubMed] [Google Scholar]
- Ciliberto G, Arcone R, Wagner EF, Rüther U, 1987. Inducible and tissue-specific expression of human C-reactive protein in transgenic mice. EMBO J. 6, 4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C, Kushner I, 1999. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340, 448–454. [DOI] [PubMed] [Google Scholar]
- Ganapathi MK, May LT, Schultz D, Brabenec A, Weinstein J, Sehgal PB, Kushner I, 1988. Role of interleukin-6 in regulating synthesis of C-reactive protein and serum amyloid A in human hepatoma cell lines. Biochem. Biophys. Res. Commun. 157, 271–277. [DOI] [PubMed] [Google Scholar]
- Hevehan DL, Miller WM, Papoutsakis ET, 2002. Differential expression and phosphorylation of distinct STAT3 proteins during granulocytic differentiation. Blood 99, 1627–1637. [DOI] [PubMed] [Google Scholar]
- Huang C-F, Chiu S-Y, Huang H-W, Cheng B-H, Pan H-M, Huang W-L, Chang H-H, Liao C-C, Jiang S-T, Su Y-C, 2019. A reporter mouse for non-invasive detection of toll-like receptor ligands induced acute phase responses. Sci. Rep. 9, 19065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LE, Whitehead AS, 1998. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem. J. 334, 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NR, Pegues MA, McCrory MA, Kerr SW, Jiang H, Sellati R, Berger V, Villalona J, Parikh R, McFarland M, Pantages L, Madwed JB, Szalai AJ, 2011. Collagen-induced arthritis is exacerbated in C-reactive protein-deficient mice. Arthrit. Rheum. 63, 2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Noren Hooten N, Dluzen DF, Martindale JL, Gorospe M, Evans MK, 2015. Posttranscriptional regulation of the inflammatory marker C-reactive protein by the RNA-binding protein HuR and microRNA 637. Mol. Cell. Biol. 35, 4212–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HMG, Kooistra T, 2003. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFκB-C/EBP-β complex formation. Blood 101, 545–551. [DOI] [PubMed] [Google Scholar]
- Kushner I, Feldmann G, 1978. Control of the acute phase response: Demonstration of C-reactive protein synthesis and secretion by hepatocytes during acute inflammation in the rabbit. J. Exp. Med. 148, 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner I, 1982. The phenomenon of the acute phase response. Ann. N. Y. Acad. Sci. 389, 39–48. [DOI] [PubMed] [Google Scholar]
- Lei K-J, Liu T, Zon G, Soravia E, Liu T-Y, Goldman ND, 1985. Genomic DNA sequence for human C-reactive protein. J. Biol. Chem. 260, 13377–13383. [PubMed] [Google Scholar]
- Levy DE, Lee CK, 2002. What does Stat3 do? J. Clin. Invest. 109, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S-P, Liu T-Y, Goldman ND, 1990. cis-Acting elements responsible for interleukin-6 inducible C-reactive protein gene expression. J. Biol. Chem. 265, 4136–4142. [PubMed] [Google Scholar]
- Majello B, Arcone R, Toniatti C, Ciliberto G, 1990. Constitutive and IL-6-induced nuclear factors that interact with the human C-reactive protein promoter. EMBO J. 9, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V, 2004. The STAT3 isoforms α and β have unique and specific functions. Nat. Immunol. 5, 401–409. [DOI] [PubMed] [Google Scholar]
- May P, Schniertshauer U, Gerhartz C, Horn F, Heinrich PC, 2003. Signal transducer and activator of transcription STAT3 plays a major role in gp130-mediatd acute phase protein gene activation. Acta Biochim. Pol. 50, 595–601. [PubMed] [Google Scholar]
- Ngwa DN, Singh SK, Gang TB, Agrawal A, 2020. Treatment of pneumococcal infection by using engineered human C-reactive protein in a mouse model. Front. Immunol. 11, 586669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Hagihara K, Serada S, Isobe T, Matsumura A, Song J, Tanaka T, Kawase I, Naka T, Yoshizaki K, 2008. Transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1α is essential for cytokine-driven C-reactive protein gene expression. J. Immunol. 180, 3492–3501. [DOI] [PubMed] [Google Scholar]
- Ochrietor JD, Harrison KA, Zahedi K, Mortensen RF, 2000. Role of STAT3 and C/EBP in cytokine-dependent expression of the mouse serum amyloid P-component (SAP) and C-reactive protein (CRP) genes. Cytokine 12, 888–899. [DOI] [PubMed] [Google Scholar]
- Pathak A, Singh SK, Thewke DP, Agrawal A, 2020. Conformationally altered C-reactive protein capable of binding to atherogenic lipoproteins reduces atherosclerosis. Front. Immunol. 11, 1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramji DP, Vitelli A, Tronche F, Cortese R, Ciliberto G, 1993. The two C/EBP isoforms, IL-6DBP/NF-IL6 and C/EBPδ/NF-IL6β, are induced by IL-6 to promote acute phase gene transcription via different mechanisms. Nucl. Acids Res. 21, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Lee C, Hou T, Bhakat KK, Brasier AR, 2010. Regulation of signal transducer and activator of transcription 3 enhanceosome formation by apurinic/apyrimidinic endonuclease 1 in hepatic acute phase response. Mol. Endocrinol. 24, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar J, Martínez MS, Chávez-Castillo M, Núñez V, Añez R, Torres Y, Toledo A, Chacin M, Silva C, Pacheco E, Rojas J, Bermudez V, 2014. C-reactive protein: An in-depth look into structure, function, and regulation. Int. Scholar. Res. Notices 2014, 653045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer TS, Sanders LK, Park OK, Nathans D, 1997. Functional differences between Stat3α and Stat3β. Mol. Cell. Biol. 17, 5307–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T, 2007. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 282, 20059–20063. [DOI] [PubMed] [Google Scholar]
- Seidel HM, Milocco LH, Lamb P, Darnell JE Jr., Stein RB, Rosen J, 1995. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc. Natl. Acad. Sci. USA 92, 3041–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, Voleti B, Agrawal A, 2007. A novel RBP-Jκ-dependent switch from C/EBPβ to C/EBPζ at the C/EBP binding site on the C-reactive protein promoter. J. Immunol. 178, 7302–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M, Huang XY, Zhang JJ, 2008. Identification of novel direct STAT3 target genes for control of growth and differentiation. J. Biol. Chem. 283, 3791–3798. [DOI] [PubMed] [Google Scholar]
- Srivastava RAK, Cornicelli JA, Markham B, Bisgaier CL, 2018. Gemcabene, a first-in-class lipid-lowering agent in late-stage development, down-regulates acute-phase C-reactive protein via C/EBP-δ-mediated transcriptional mechanism. Mol. Cell. Biochem. 449, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toniatti C, Arcone R, Majello B, Ganter U, Arpaia G, Ciliberto G, 1990a. Regulation of the human C-reactive protein gene, a major marker of inflammation and cancer. Mol. Biol. Med. 7, 199–212. [PubMed] [Google Scholar]
- Toniatti C, Demartis A, Monaci P, Nicosia A, Ciliberto G, 1990b. Synergistic trans-activation of the human C-reactive promoter by transcription factor HNF-1 binding at two distinct sites. EMBO J. 9, 4467–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti B, Agrawal A, 2005. Regulation of basal and induced expression of C-reactive protein through an overlapping element for OCT-1 and NF-κB on the proximal promoter. J. Immunol. 175, 3386–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti B, Agrawal A, 2006. Statins and nitric oxide reduce C-reactive protein production while inflammatory conditions persist. Mol. Immunol. 43, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti B, Hammond DJ Jr., Thirumalai A, Agrawal A, 2012. Oct-1 acts as a transcriptional repressor on the C-reactive protein promoter. Mol. Immunol. 52, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead AS, Zahedi K, Rits M, Mortensen RF, Lelias JM, 1990. Mouse C-reactive protein: Generation of cDNA clones, structural analysis, and induction of mRNA during inflammation. Biochem. J. 266, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P, Korenberg JR, Whitehead AS, 1985. Characterization of genomic and complementary DNA sequence of human C-reactive protein, and comparison with the complementary DNA sequence of serum amyloid P component. J. Biol. Chem. 260, 13384–13388. [PubMed] [Google Scholar]
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR, 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 21, 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DP, Kushner I, Samols D, 2008. Binding of C/EBPβ to the C-reactive protein (CRP) promoter in Hep3B cells is associated with transcription of CRP mRNA. J. Immunol. 181, 2420–2427. [DOI] [PubMed] [Google Scholar]
- Zhang D, Jiang S-L, Rzewnicki D, Samols D, Kushner I, 1995. The effect of interleukin-1 on C-reactive protein expression in Hep3B cells is exerted at the transcriptional level. Biochem. J. 310, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Sun M, Samols D, Kushner I, 1996. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J. Biol. Chem. 271, 9503–9509. [DOI] [PubMed] [Google Scholar]
- Zhang S-C, Wang M-Y, Feng J-R, Chang Y, Ji S-R, Wu Y, 2020. Reversible promoter methylation determines fluctuating expression of acute phase proteins. eLife 9, 51317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JE Jr., 1994. STAT3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Xu M-J, Gao B, 2016. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 13, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]