

Abstract
For over half a century, thousands of tons of triphenylphosphine oxide Ph3P(O) have been produced every year from the chemical industries as a useless chemical waste. Here we disclose efficient transformations of Ph3P(O) with cheap resource-abundant metallic sodium finely dispersed in paraffin oil. Ph3P(O) can be easily and selectively transformed to three reactive organophosphorus intermediates—sodium diphenylphosphinite, sodium 5H-benzo[b]phosphindol-5-olate and sodium benzo[b]phosphindol-5-ide—that efficiently give the corresponding functional organophosphorus compounds in good yields. These functional organophosphorus compounds are difficult to prepare but highly industrially useful compounds. This may allow Ph3P(O) to be used as a precious starting material for highly valuable phosphorus compounds.
Subject terms: Sustainability, Synthetic chemistry methodology
As a byproduct of organic syntheses including the Wittig, Staudinger, and Mitsunobu reactions, triphenylphosphine oxide is often not recycled. Here a transformation of the waste product with metallic sodium to three organophosphorous compounds is presented.
Introduction
Triphenylphosphine oxide Ph3P(O) is a chemically stable compound that is primarily generated, tens of thousands tons a year, as a by-product from the chemical industries, during the preparation of valuable fine chemicals, such as vitamins, pharmaceuticals, agrochemicals etc, and bulk chemicals, such as butanols, using triphenylphosphine PPh3 as an oxophilic reagent or ligand for a metal catalyst (Supplementary Note 1)1–8. A well-known serious problem associated with Ph3P(O) is that thousands of tons of this compound are discarded as useless chemical waste because of its limited utilities. This situation has lasted for more than half a century, and has become a big concern from both industry and academia sides9,10. In order to solve this problem, extensive studies have been carried out world widely. Among them, the reduction of triphenylphosphine oxide Ph3P(O) to its original form triphenylphosphine Ph3P is most studied11. However, either hard conditions or expensive reductants are required in order to break the strong P=O bond. Therefore, a practically operable way that can settle the triphenylphosphine oxide problem has not been found yet12–15.
Herein we disclose a potentially effective solution to this longstanding problem (Fig. 1). By treatment with the cheap, resource-abundant metallic sodium (sodium finely dispersed in paraffin oil with μm-scale sizes; hereafter abbreviated as SD) at 25 °C, triphenylphosphine oxide Ph3P(O), the so far discarded chemical waste (A), can be transformed, easily and selectively, to a variety of organic phosphorus compounds (phosphoryl compounds and phosphines) that are widely used valuable chemicals in the industry16. It is noted that the current process for the production of these organophosphorus compounds is rather dirty, energy-consuming and dangerous since it starts from the highly toxic benzene and phosphorus trichloride (C)17,18. Heavy pollution problems also associate with their preparation because of the poor efficiency. Therefore, the present new finding not only provides a use for waste Ph3P(O) (1) but also may aid the preparation of other useful organophosphorus compounds (2).
Fig. 1. Conversion of waste Ph3P(O) to valuable organic phosphorus compounds.

a Transformation of Ph3P(O) to a variety of organic phosphorus compounds by SD. b A vast amount of Ph3P(O) is produced and discarded as a useless chemical waste. c Costly and dangerous methods for the preparation of organophosphorus compounds. d Selective C-P, C-H and O-P bond cleavage of triphenylphosphine oxide by SD under mild conditions. Condition a: 1 and SD in THF at 25 °C; Condition b: SD/PhCl then 1 at 25 °C, THF; Condition c: SD/PhCl, 1, then SD at 25 °C, THF.
As depicted in Fig. 1d, by cleaving one C-P bond, sodium diphenylphosphinite 2 is generated quantitatively. This intermediate 2 is easily transformed to the corresponding phosphine oxides 2′ (a). On the other hand, by slightly changing the conditions, sodium 5H-benzo[b]phosphindol-5-olate 3 is generated in high yields via one C-P and two C-H bonds cleavage (b). More interestingly, the O-P bond in 3 can be further cleft to sodium benzo[b]phosphindol-5-ide 4 quantitatively (c). Thus, by simply treating with metallic sodium, triphenylphosphine oxide can readily produce three kinds of highly valuable phosphorus compounds.
Results
Reactions of Ph3P(O) with metallic sodium
We serendipitously discovered this rapid reaction of Ph3P(O) with metallic sodium as we added Ph3P(O) to THF that contains trace amount of metallic sodium initially used for its drying. The originally transparent colorless THF solution of Ph3P(O) (Fig. 2a), instantly changed to yellow and then brown (Fig. 2b) at 25 °C. This clear color change is a strong indication that a rapid chemical reaction takes place between Ph3P(O) and sodium. Indeed, this is true. A subsequent experiment surprisingly revealed that, at 25 °C, upon adding sodium to Ph3P(O) dissolved in THF, an exothermic reaction took place rapidly and the starting material Ph3P(O) was completely consumed after 2 h (Fig. 3, equation (1)).
Fig. 2. Reactions of Ph3P(O) with metallic sodium.

a Ph3P(O) dissolved in THF. b Ph3P(O) dissolved in THF in the presence of Na.
Fig. 3. Reaction of triphenylphosphine oxide with sodium.

a Triphenylphosphine oxide 1 rapidly reacted with sodium at 25 °C. Reaction conditions: 0.5 mmol Ph3P(O) was dissolved in 3 mL THF, and 2.5 mmol metallic Na was added at 25 °C. b The reaction mixture was stirred for 1 h, 2 h and overnight and monitored by 31P NMR, respectively.
This easy conversion of Ph3P(O) by sodium was rather unexpected considering that until now literatures all had to conduct this reaction in a highly reducing medium by dissolving sodium in liquid ammonia (the Birch reduction medium) at low temperatures12,13. In addition to the tedious process under the Birch reduction system, that requires difficult handling and operating techniques, yields and selectivity of the desired products were also not satisfactory. A NaH/LiI composite system was recently reported to break down the C-P bond of triarylphosphine oxides. However, NaH is a rather costly reagent. Moreover, a long-time heating (overnight at 60 °C) was required15.
As shown in Fig. 3, metallic sodium (2.5 mmol), cut to small pieces, was added to Ph3P(O) (0.5 mmol) dissolved in THF (3 mL) at 25 °C (equation (1)). The color of the reaction mixture soon changed to brown. After stirring for 2 h, 31P NMR spectroscopy showed that the starting material Ph3P(O) at 32.9 ppm almost disappeared, and three new signals emerged at 91.1 ppm (compound 2) and 102.1 ppm (compound 3) and 6.7 ppm (compound 4) after overnight stirring. In order to identify these compounds, n-BuBr was added to the solution and 2′, 3′, and 4′ were obtained in 70%, 8%, and 21%, respectively, confirming that the new generated phosphorus species are sodium diphenylphosphinite (2), sodium 5H-benzo[b]phosphindol-5-olate (3) and sodium benzo[b]phosphindol-5-ide (4) (Supplementary Figs. 9‒11).
Selective generation of 2 by transformation of Ph3P(O) to Ph2P(O)H and Ph2P(O)R
More excitingly, in addition to the disclosure of the event that Ph3P(O) could readily react with sodium under mild conditions, the reaction conditions for the generation of the three phosphorus species 2, 3, and 4 were tunable, so that these three active phosphorus species could be highly selectively formed, respectively. First, instead of sodium lump, when sodium powder dispersed in paraffin oil (average particle size < 10 μm, here below abbreviated as SD)19 was used, we can produce compound 2 exclusively within a few minutes (ref. 19 Being similar to Na, other alkali metals are expected to react with Ph3P(O) too. Indeed, the reaction of Ph3P(O) with lithium dispersion (metallic lithium finely dispersed in paraffin oil) also proceeded rapidly at 25 °C. However, being different to the reaction with Na, a lot of side products were obtained with Li and the selectivity to Ph2POLi was only ca. 70%.). Thus, 2.5 mmol SD was added to 1.0 mmol Ph3P(O) in 5 mL THF at 25 °C (Fig. 4, equation (2)). After 10 min, the corresponding sodium diphenylphosphinite (2, δ = 91.5 ppm) was produced exclusively and quantitatively! No side products could be detected at all!
Fig. 4. Synthesis of 2.

a Selective transformation to sodium diphenylphosphinite 2. Reaction conditions: sodium (SD) (2.5 mmol) was added to Ph3P(O) (1.0 mmol) dissolved in 5 mL THF at 25 °C and the mixture was stirred for 10 min. b NMR spectra copy of the crude reaction solution.
As to the molar ratios of sodium vs Ph3P(O), we found that more than 2 equivalents of sodium are necessary for the selective complete conversion of Ph3P(O) to Ph2P(ONa). For example, under similar conditions, when one equivalent SD was used, 12% Ph3P(O) remained unchanged, and Ph2P(ONa) and 3 were obtained in 60% and 28% yield, respectively, as determined by 31P NMR spectroscopy. Therefore, the reaction should proceed, being similar to that under the super reducing medium Na/NH312,13, via the cleavage of one C-P bond of Ph3P(O) by two equivalents of sodium generating an equimolar Ph2P(ONa) and PhNa (vide infra) (Fig. 4).
This easy and quantitative conversion of 1 Ph3P(O) to 2 Ph2PONa guaranteed its application as an industrially useful reaction because, now, diphenylphosphine oxide Ph2P(O)H, a widely used but rather expensive industrial chemical, can be easily prepared from the waste chemical Ph3P(O)! Diphenylphosphine oxide Ph2P(O)H20 is widely employed as a versatile starting material for the synthesis of a lot of valuable organophosphorus compounds. This compound is currently industrially produced via the hydrolysis of Ph2PCl. However, Ph2PCl is prepared from a rather inefficient Friedel-Crafts reaction of PCl3 and benzene using AlCl3 that releases a large amount of wastes (more than 3 tones wastes in order to produce one tone product)17,18. As shown in Fig. 5, by simply adding water, the intermediate sodium phosphinite 2 developed above could quantitatively give Ph2P(O)H 2–1 (Fig. 5a). It is noted that Ph2P(O)H is also a starting material for the preparation of 2,4,6-trimethylbenzoyldipenylphosphine oxide (TPO). TPO is a important photoinitiator and thousands of tons of TPO are broadly used in the realm of photopolymerization21. This compound is industrially prepared by two methods: (1) Michaelis–Arbuzov reaction of alkoxyphosphine with acyl chloride22,23 and (2) oxidation of α-hydroxyphosphine oxide generated by the addition of Ph2P(O)H to the aldehyde24 (Fig. 5b). We found that TPO could be conveniently generated directly using Ph2PONa 2 that can eliminate the isolation of Ph2P(O)H and other steps for the synthesis of TPO (Fig. 5b). For example, by adding Ph2PONa 2 to 2,4,6-trimethylbenzoyl chloride (MesC(O)Cl) in THF at 0 °C, the desired product TPO 2–2 was obtained in 56% yield. Beyond its practical utility, it is noted that this reaction is the first example for the preparation of TPO and analogues by the direct nucleophilic substitution reactions with an acylchloride because all of the literature attempts generated side products rather than the desired product TPO21–24.
Fig. 5. Utility of sodium diphenylphosphinite 2.

Reaction conditions: Ph2PONa 2 was prepared from 1.0 mmol Ph3P(O) in 5 mL solvent and 2.5 mmol SD according to the standard procedure. a 2.0 mL saturated aqueous NH4Cl solution was added to Ph2PONa 2 (1.0 mmol, in 1,4-dioxane) at 25 °C. b Ph2PONa 2 (1.0 mmol, in THF) was added to MesC(O)Cl (1.5 mmol) at 0 °C. c An alkyl halide (1.2 mmol) was added to Ph2PONa 2 (1.0 mmol, in THF) at 0 °C. Isolated yield.
Fig. 6. Possible mechanisms.

a The mechanism to 3 using PhNa. b The mechanism to 6 using PhLi (ref. 28).
Finally, Ph2PONa can also efficiently react with an organohalide to give the corresponding phosphine oxide in high yield which is useful in organic synthesis, metal extraction etc. (Fig. 5c)25,26. Although the nucleophilic substitution reaction of Ph2PONa with RBr generating Ph2P(O)R is a known reaction, the high yield of Ph2P(O)R with a slightly excess RBr is surprising considering that an equimolar PhNa is also generated in the reaction mixture but does not interfere in the nucleophilic substitution reaction (vide infra).
Selective generation of 3
Fixing the optimized conditions for the selective generation of 3 was not as easy as 2. The study hardly progressed until we eventually realized that PhNa should be the key for its generation (Fig. 6). Thus, we anticipated that sodium 5H-benzo[b]phosphindol-5-olate 3, would be formally generated by dehydrogenative cyclization. Since PhNa 5 was generated during the reaction of Ph3P(O) with Na, PhNa might act as a base to react with Ph3P(O) to give 3 via cyclization27,28. An early literature reported that the reaction of PhLi with Ph3P(O) in THF under reflux overnight gave 5-phenyl-5H-benzo[b]phosphindole 6 rather than 3 (Fig. 6b)27. Although not fully understood at present, this difference in reactivity between PhLi and PhNa is very interesting. It should be noted that while the current reaction with PhNa took place rapidly at room temperature, the reaction with PhLi required a long-time heating27.
This was indeed the case. When Ph3P(O) (0.45 mmol dissolved in 2 mL THF) was added to PhNa (1.0 mmol prepared from 2.0 mmol SD with 1.1 mmol PhCl) at 25 °C (Table 1, equation (3))20, 5H-benzo[b]phosphindol-5-olate 3 (δ = 101.7 ppm) was generated predominantly (Table 1, run 1). By quenching the organophosphorus species with n-OctBr, 3′b was obtained in 77% yield together with 2–3b generated via the reaction of 2 (11%), respectively, as determined by 31P NMR spectroscopy. Efforts had been devoted to improving the yield and selectivity of 3. Switching the ratio of Ph3P(O) 1 and PhNa to 0.33:1 leaded to lower yield and selectivity of 3 (Table 1, run 2). When a solid PhNa was added to Ph3P(O) dissolved in THF, 3 and 2 were generated in 42% and 38% respectively (Table 1, run 3). Interestingly, by increasing the amount of Ph3P(O) to 0.9 mmol, nearly an equimolar ratio to PhNa, a similar yield of 3 could be obtained (Supplementary Fig. 5), indicating that only one equivalent of PhNa is required for the generation of 3 in the reaction, which not only significantly improved the efficiency of the use of PhNa (Table 1, run 4), but also sustained the proposed mechanism (Fig. 6). The selectivity was not further improved either under a lower or higher temperature (Table 1, runs 5–6).
Table 1.
Selective transformation of Ph3P(O) to sodium 5H-benzo[b]phosphindol-5-olate 3.
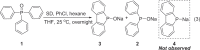 | ||||
|---|---|---|---|---|
| run | Ph3P(O) | PhNa | temperature | Conversion of 1 (yield of 3 and 2) |
| 1 | 0.45 mmol | 1.0 mmol | 25 °C | 88% (77% of 3, 11% of 2) |
| 2 | 0.33 mmol | 1.0 mmol | 25 °C | 85% (41% of 3, 44% of 2) |
| 3 | 0.45 mmol | 1.0 mmol | 25 °C | 80% (42% of 3, 38% of 2) |
| 4 | 0.90 mmol | 1.0 mmol | 25 °C | 100% (82% of 3, 18% of 2) |
| 5 | 0.90 mmol | 1.0 mmol | 0 °C | 42% (39% of 3, 3% of 2) |
| 6 | 0.90 mmol | 1.0 mmol | 40 °C | 86% (64% of 3, 12% of 2) |
Reaction conditions: PhNa was generated in situ by the reaction of SD (2.0 mmol) with PhCl (1.1 mmol) in 2.0 mL hexane at 25 °C for 1 h. A specified amount of Ph3P(O) dissolved in 2.0 mL THF was then added into PhNa at 25 °C and the mixture was stirred for overnight. Yields were estimated from 31P NMR spectroscopy based on 1 used. aSolid PhNa was used
Molecules with dibenzophosphole framework have great potentials as novel optical and electrical materials29. As shown in Fig. 7, old procedures for the synthesis of dibenzophosphole oxides mainly relied on metathesis reaction between dilithiated biphenyl with RPCl2 followed by oxidation (a)30. Recently, a palladium-catalyzed intramolecular arylation of ortho-halodiphenylphosphine (b) and intramolecular dehydrogenative cyclization of secondary hydrophosphine oxides (c) have been developed31,32. All these approaches have to use the toxic phosphine chlorides, and the processes are tedious. As shown in Fig. 7d, the sodium 5H-benzo[b]phosphindol-5-olate 3 derived from Ph3P(O) was easily transformed to the corresponding dibenzophosphole oxides in moderate to high yields. Functional groups like Cl, CN, CF3 are well tolerable. Therefore, an efficient way for the generation of these useful dibenzophosphole oxides by using the chemical waste Ph3P(O) was established.
Fig. 7. Efficient synthesis of dibenzophosphole oxides from the chemical waste Ph3P(O).

Reaction conditions: PhNa was generated in situ by adding PhCl (1.1 mmol) to a suspension of SD (2.0 mmol in 2.0 mL hexane) at 25 °C for 1 h. Ph3P(O) dissolved in THF (2.0 mL) was added into the above PhNa suspension and stirred for overnight. RBr (1.5 mmol) was then added at 0 °C and stirred for 0.5 h. Isolated yield.
Selective generation of 4 through P=O reduction by sodium
A more fascinating phenomenon is that even the trivalent phosphole 4 can be selectively generated starting from Ph3P(O) (Fig. 8). Thus, during the study on further possible reactions with metallic sodium of 2 and 3, we surprisingly found that although no reaction took place with 2, 3 reacted quickly to give 4! Thus, 31P NMR showed that after the addition of SD to 3 at 25 °C for a few minutes, the signal of 3 completely disappeared and a new signal of 4 at 3.0 ppm appeared. As expected, the subsequent addition of n-BuBr to the mixture gave compound 4′ (δ = −13.5 ppm) which was fully characterized by comparing with an authentic sample prepared separately33. This protocol is amenable to use dibromides as electrophiles for the synthesis of bisphosphole 4′b. Since the reaction of sodium benzo[b]phosphindol-5-ide 4 with an alkyl bromide is faster than that with an aromatic bromide, 4′c could be selectively generated. Therefore, by carrying out a one-pot reaction, the so far chemical waste Ph3P(O) could also be easily converted to phosphole 4′ (Fig. 8, equation (5))! This is also a rare example for converting phosphine oxide (P(V)) to phoshine (P(III)) that usually requires highly reactive reducing reagents such as LiAlH4 and hydrosilances, or under harsh conditions, in order to break down the robust P-O bond11–15.
Fig. 8.

a Selective transformation of Ph3P(O) to sodium benzo[b]phosphindol-5-ide 4. b Reaction conditions and monitored by 31P NMR: (a) sodium H-benzo[b]phosphindol-5-olate 3 was generated in situ from Ph3P(O) (0.9 mmol) and PhNa (1.0 mmol); (b) SD (2.5 mmol) was then added and stirred for 2 h; (c) n-BuBr (2.0 mmol) was added to the mixture and stirred for 1 h. c One-pot conversion of Ph3P(O) to phosphle 4′. Isolated yield. a0.4 mmol of BrC4H8Br was added. bThe product was oxidized by H2O2 for easy isolation.
A 10-gram scale reaction
As demonstrated by a lab-scale reaction, the present method is easily applicable to a large-scale preparation of the phosphorus compound (Fig. 9). For example, after treating 10 g of Ph3P(O) 1 with 90 mmol SD (2.5 equiv.) at 25 °C, water was added. The mixture was simply extracted with EtOAc, washed by hexane and passed through a short silica gel column. A spectroscopically pure diphenylphosphine oxide 2–1 was obtained as a white solid (6.93 g, 96% yield).
Fig. 9. 10-g scale reaction.

1 was treated with SD in dioxane followed by quenching with water to give 2–1 in a high yield.
In conclusion, we disclosed that the C-P bond of Ph3P(O) can be efficiently cleft by metallic sodium under normal conditions without using dangerous super reducing media (sodium dissolved in ammonia Na/NH3). Three basic reactive organophosphorus intermediates 2, 3, 4 can be selectively generated from the combination of Ph3P(O) with metallic sodium, giving the corresponding organophosphorus compounds efficiently (Fig. 1). The industrial markets for these organophosphorus compounds derivatives, that are difficult to prepare by other methods, is large enough to consume up all of the Ph3P(O) produced as a chemical waste from the chemical industry. Therefore, Ph3P(O) may serve as a precious chemical stock for highly valuable functional organophosphorus compounds. We believe that this finding can settle the Ph3P(O) problem that has annoyed people for half a century.
Methods
Synthesis of compound 2
Under argon, SD (0.25 mL, 2.5 mmol) was added to Ph3P(O) (278 mg, 1.0 mmol) dissolved in THF (5 mL) at 25 °C with stirring. The initially colourless transparent solution turned to brown soon. After stirring for 10 min, compound 2 was obtained quantitatively. By adding n-BuBr (130 μL, 1.2 mmol) to the above mixture at 0 °C, 2–3a was obtained.
Synthesis of compound 3
PhCl (112 μL, 1.1 mmol) was added dropwise to SD (0.2 mL, 2.0 mmol) suspended in hexane (2.0 mL) at 25 °C under argon. After stirring for 1 h, Ph3P(O) (250 mg, 0.9 mmol) dissolved in THF (2.0 mL) was added and the mixture was stirred at 25 °C for overnight to give compound 3. By quenching the reaction mixture thus generated with n-OctBr (258 μL, 1.5 mmol) at 0 °C afforded 3′b.
Synthesis of compound 4
Under nitrogen, to a solution of compound 3 obtained above was added SD (0.25 mL, 2.5 mmol) at 25 °C. The mixture was stirred for 2 h giving compound 4. By quenching with n-BuBr (215 μL, 2.0 mmol) at 0 °C, 4′ was obtained.
Product derivatizations
Full procedures for transformations of 1 to 2, 3, 4 and their derivatives 2′, 3′, 4′ are available in the Supplementary Methods and Supplementary Figs. 1–6.
NMR spectra
1H,13C and 31P NMR Spectra of all products were provided. See Supplementary Figs. 12–50.
Supplementary information
Acknowledgements
This study was partially supported by a joint-research of National Institute of Advanced Industrial Science and Technology (AIST) with Katayama Chemical Industries Co., Ltd. Yuka Mino and Eiichi Ikawa were acknowledged for their participation. We thank Kobelco Eco-Solutions Co., Ltd. for providing SD samples.
Author contributions
L-B.H. discovered the rapid color change as Na was added to a THF solution of 1 and identified 2. J.Y. isolated 4′a and 4′b. T.H. isolated 2–3a and 2–3b, H.S. and H.F. prepared 3′a and 3′b. J-Q.Z. conducted all the other experiments and prepared the Supplementary Information. L-B.H. oversaw the study and prepared the manuscript with J-Q.Z.
Data availability
The authors declare that all the data supporting the findings of this study are available within the paper and its supplementary information files, and also are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
2/17/2021
A Correction to this paper has been published: 10.1038/s42004-020-0266-5
Supplementary information
Supplementary information is available for this paper at 10.1038/s42004-019-0249-6.
References
- 1.But TYS, Toy PH. Organocatalytic Mitsunobu reactions. J. Am. Chem. Soc. 2006;128:9636–9637. doi: 10.1021/ja063141v. [DOI] [PubMed] [Google Scholar]
- 2.Kohn M, Breinbauer R. The Staudinger Ligation - A Gift to Chemical Biology. Angew. Chem. Int. Ed. 2004;43:3106–3116. doi: 10.1002/anie.200401744. [DOI] [PubMed] [Google Scholar]
- 3.Longwitz L, Werner T. Recent advances in catalytic Wittig-type reactions based on P(III)/P(V) redox cycling. Pure Appl. Chem. 2018;91:95–102. doi: 10.1515/pac-2018-0920. [DOI] [Google Scholar]
- 4.Aroyan CE, Dermenci A, Miller SJ. The Rauhut–Currier reaction: a history and its synthetic application. Tetrahedron. 2009;65:4069–4084. doi: 10.1016/j.tet.2009.02.066. [DOI] [Google Scholar]
- 5.Van Kalkeren HA, Van Delft FL, Rutjes FP. Catalytic Appel reactions. Pure Appl. Chem. 2012;85:817–828. doi: 10.1351/PAC-CON-12-06-13. [DOI] [Google Scholar]
- 6.Meakin P, Jesson JP, Tolman CA. Nature of chlorotris(triphenylphosphine)rhodium in solution and its reaction with hydrogen. J. Am. Chem. Soc. 1972;94:3240–3242. doi: 10.1021/ja00764a061. [DOI] [Google Scholar]
- 7.Ramaotsoa GV, Strydom I, Panayides JL, Riley D. Immobilized tetrakis (triphenylphosphine) palladium (0) for Suzuki–Miyaura coupling reactions under flow conditions. React. Chem. Eng. 2019;4:372–382. doi: 10.1039/C8RE00235E. [DOI] [Google Scholar]
- 8.Evans D, Osborn JA, Wilkinson G. Hydroformylation of alkenes by use of rhodium complex catalyst. J. Am. Chem. Soc. 1968;33:3133–3142. doi: 10.1039/j19680003133. [DOI] [Google Scholar]
- 9.Constable DJC, et al. Key green chemistry research areas – a perspective from pharmaceutical manufacturers. Green. Chem. 2007;9:411–420. doi: 10.1039/B703488C. [DOI] [Google Scholar]
- 10.van Kalkeren HA, Blom AL, Rutjes FPJT, Huijbregts MAJ. On the usefulness of life cycle assessment in early chemical methodology development: the case of organophosphorus-catalyzed Appel and Wittig reactions. Green. Chem. 2013;15:1255–1263. doi: 10.1039/c3gc00053b. [DOI] [Google Scholar]
- 11.Podyacheva E, Kuchuk E, Chusov D. Reduction of phosphine oxides to phosphines. Tetrahedron Lett. 2019;60:575–582. doi: 10.1016/j.tetlet.2018.12.070. [DOI] [Google Scholar]
- 12.Bornancini ER, Rossi RA. Formation and reactions of diorganophosphinite ions in liquid ammonia. Synthesis of triorganophosphine oxides by the SRN1 mechanism. J. Org. Chem. 1990;55:2332–2336. doi: 10.1021/jo00295a019. [DOI] [Google Scholar]
- 13.Stankevič M, Włodarczyk A, Jaklińska M, Parcheta R, Pietrusiewicz KM. Sodium in liquid ammonia–a versatile tool in modifications of arylphosphine oxides. Tetrahedron. 2011;67:8671–8678. doi: 10.1016/j.tet.2011.09.045. [DOI] [Google Scholar]
- 14.Stankevič M, Pisklak J, Włodarczyk K. Aryl group–a leaving group in arylphosphine oxides. Tetrahedron. 2016;72:810–824. doi: 10.1016/j.tet.2015.12.043. [DOI] [Google Scholar]
- 15.Tejo C, et al. Dearylation of arylphosphine oxides using a sodium hydride–iodide composite. Chem. Commun. 2018;54:1782–1785. doi: 10.1039/C8CC00289D. [DOI] [PubMed] [Google Scholar]
- 16.Quin, L. D. A Guide to Organophosphorus Chemistry. (Wiley Interscience, New York, 2000).
- 17.Buchner B, Lockhart LB. Phenyldichlorophosphine. Org. Synth. 1951;31:88. doi: 10.15227/orgsyn.031.0088. [DOI] [Google Scholar]
- 18.Weinberg KG. Synthesis of Arylphosphonous dichlorides, diarylphosphinous chlorides, and 1,6-diphosphatriptycene from elemental phosphorus. J. Org. Chem. 1975;40:24. doi: 10.1021/jo00912a027. [DOI] [Google Scholar]
- 19.Asako S, Nakajima H, Takai K. Organosodium compounds for catalytic cross-coupling. Nat. Catal. 2019;2:297. doi: 10.1038/s41929-019-0250-6. [DOI] [Google Scholar]
- 20.Xu Q, Han L-B. Metal-catalyzed additions of H-P(O) bonds to carbon-carbon unsaturated bonds. J. Organomet. Chem. 2011;696:130–140. doi: 10.1016/j.jorganchem.2010.08.043. [DOI] [Google Scholar]
- 21.Sumiyoshi T, Schnabel W. On the reactivity of phosphonyl radicals towards olefinic compounds. Mukromol. Chem. 1985;186:1811–1823. doi: 10.1002/macp.1985.021860910. [DOI] [Google Scholar]
- 22.Lechtken et al. Photocurable compositions with acylphosphine oxide photoinitiator. US Patent 4710523 (1987).
- 23.Lechtken et al. Acylphosphine oxide compounds. US Patent US4324744 (1982).
- 24.Fischer, M. et al. Preparation of carbonylphosphine oxides. US Patent 5,679,863 (1997).
- 25.Corbridge, D. E. C. Phosphorus: Chemistry, Biochemistry and Technology, 6th edn. (CRC Press, London, 2013)
- 26.Burgard M, Prevost M. The uses of organo phosphorus compounds in liquid-liquid extraction of metal ions. Phosphorus, Sulfur Relat. Elem. 1983;18:319–322. doi: 10.1080/03086648308076030. [DOI] [Google Scholar]
- 27.Ogawa S, Tajiri Y, Furukawa N. Simple preparation of (2, 2′-biphenylylene) phenylphosphine oxide and role of triphenylphosphine oxide as a mediator for formation of biaryl derivatives. Bull. Chem. Soc. Jpn. 1991;64:3182–3184. doi: 10.1246/bcsj.64.3182. [DOI] [Google Scholar]
- 28.Seyferth D, Hughes WB, Heeren JK. Studies in phosphinemethylene chemistry. XI. The reaction of alkyllithium reagents with tetraphenylphosphonium bromide1. J. Am. Chem. Soc. 1965;87:3467–3474. doi: 10.1021/ja01093a033. [DOI] [Google Scholar]
- 29.Matano Y, et al. Fusion of phosphole and 1, 1′‐biacenaphthene: phosphorus (v)‐containing extended π‐systems with high electron affinity and electron mobility. Angew. Chem. Int. Ed. 2011;50:8016–8020. doi: 10.1002/anie.201102782. [DOI] [PubMed] [Google Scholar]
- 30.Chen RF, Fan QL, Zheng C, Huang W. A general strategy for the facile synthesis of 2, 7-dibromo-9-heterofluorenes. Org. Lett. 2006;8:203–205. doi: 10.1021/ol0524499. [DOI] [PubMed] [Google Scholar]
- 31.Cui Y, Fu L, Cao J, Deng Y, Jiang J. Synthesis of dibenzophosphole oxides via palladium‐catalyzed intramolecular direct arylation reactions of ortho‐halodiarylphosphine oxides. Adv. Synth. Catal. 2014;356:1217–1222. doi: 10.1002/adsc.201301081. [DOI] [Google Scholar]
- 32.Kuninobu Y, Yoshida T, Takai K. Palladium-catalyzed synthesis of dibenzophosphole oxides via intramolecular dehydrogenative cyclization. J. Org. Chem. 2011;76:7370–7376. doi: 10.1021/jo201030j. [DOI] [PubMed] [Google Scholar]
- 33.Cornforth J, Cornforth RH, Gray RT. Synthesis of substituted dibenzophospholes. Part 1. Chem. Soc., Perkin Trans. 1982;1:2289–2297. doi: 10.1039/p19820002289. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper and its supplementary information files, and also are available from the corresponding author upon reasonable request.