

Abstract
The discovery of the von Hippel-Lindau (VHL) gene marked a milestone in our understanding of clear cell renal cell carcinoma (ccRCC) pathogenesis. VHL inactivation is not only a defining feature of ccRCC, but also the initiating event. Herein, we discuss canonical and noncanonical pVHL functions as well as subsequent breakthroughs shaping our understanding of ccRCC evolution and evolutionary subtypes. Current and evolving strategies to therapeutically exploit effector mechanisms downstream of pVHL are also discussed.
Keywords: AROHIF2, hypoxia-inducible factor, MK-6482, PT2385, PT2977, siRNA, synthetic lethality, tumor evolution
Introduction
Kidney cancer, which is largely made up of renal cell carcinoma (RCC), is among the top ten cancers in the United States, accounting for ~75,000 new cases per year1. RCC is subclassified histologically, and ccRCC comprises ~75% of cases2. ccRCC is resistant to chemotherapy, and until 2005, only one drug was approved by the FDA. However, there has been a surge in the number of drugs available for the treatment of ccRCC, initially with targeted drugs, and most recently, immune checkpoint inhibitors (ICIs). These advances, in particular the development of targeted therapies, were made possible through an improved understanding of ccRCC biology. The first breakthroughs arose from studying a rare inherited disorder, von Hippel-Lindau (VHL) syndrome. These findings propelled decades of research to elucidate the fundamental principles of ccRCC development and set the foundation for novel therapies.
Genetic linkage analysis locates the VHL gene to chromosome 3p
Von Hippel-Lindau syndrome is an autosomal dominant disease with age-dependent penetrance caused by germline mutations in the VHL gene3. This syndrome manifests with multiple malignancies including ccRCCs, hemangioblastomas of the central nervous system (CNS), pheochromocytomas, pancreatic neuroendocrine tumors, as well as renal and pancreatic cysts. Genetic linkage analysis of affected kindreds located the responsible gene to the chromosomal region 3p25-26 in the late 1980s4,5. The VHL gene was identified by positional cloning in 19936, and VHL mutations were subsequently identified in sporadic ccRCC7. Loss of heterozygosity (LOH) and inactivation of the second VHL allele were seen in both sporadic and hereditary cases implicating VHL as a classical two-hit tumor suppressor gene (TSG)8,9. Consistent with VHL functioning as a TSG, reintroduction of VHL into the VHL-deficient ccRCC tumor cell line 786-O decreased tumor formation in mice10. More recent analyses have highlighted the pervasiveness of VHL inactivation in ccRCC. Inactivation of VHL, through intragenic mutation, epigenetic silencing, or large deletion, occurs in up to 91% of sporadic cases11,12. In fact, VHL loss is the earliest event and a truncal finding in ccRCC development. However, contrary to initial suppositions that tumorigenesis is initiated by an intragenic mutation and is followed by 3p loss, recent evidence suggest that loss of 3p is the initiating event. Chromothripsis involving chromosomes 3p and 5q followed by mutation or epigenetic silencing of the second VHL allele is thought to initiate ccRCC development13.
The VHL gene product, pVHL, is a key regulator of the cellular response to hypoxia14. Biochemical studies revealed that pVHL forms a ternary complex with elongins B and C, which link pVHL to cullin-2 (CUL2) and RING-box protein 1 (RBX1)15.This E3 ubiquitin ligase complex binds hypoxia-inducible factor (HIF) α subunits after they have been modified at particular prolyl residues by prolyl hydroxylases (EglN1-3)16, resulting in its ubiquitination and degradation15. Interestingly, elongin C has also been found to be mutated in ccRCC, though to a significantly reduced extent (<5%) and mutations in the corresponding gene (TCEB1) are associated with LOH of chromosome 8, where TCEB1 resides17. In addition, mutations in CUL2 were reported in up to 1% of ccRCCs18. These mutations tend to be mutually exclusive and suggest that the oncogenic properties of the different genes result from disruption of the pVHL complex. While there are differences between ccRCC with mutations in TCEB1 and VHL19,20, overall, inactivation of the pVHL complex may be regarded as a defining signature event in ccRCC.
The paradigmatic substrate of pVHL is HIF-α. The HIF family of transcription factors acts on hypoxia response elements (HREs) distributed throughout the genome controlling the expression of genes implicated, among other processes, in adaptation to hypoxia21. Hypoxia-inducible factor is a heterodimer complex consisting of a labile α subunit and a stable β subunit. Under normoxic conditions, the HIFα subunit is hydroxylated, leading to its recognition and ubiquitination by the pVHL complex22. Loss of VHL impairs HIF regulation, resulting in accumulation of HIFα and constitutive expression of HIF target genes. In humans, there are three HIFα proteins, HIF-1α, HIF-2α, and HIF-3α21. While there is substantial overlap between HIF-1α and HIF-2α target genes, some genes are exclusively regulated by HIF-1 or HIF-223-26. Furthermore, some evidence suggests that HIF-2 is the key driver of ccRCC, whereas HIF-1 may have inhibitory effects27. In pre-clinical models, HIF-2α overexpression is both necessary and sufficient for tumor growth following VHL loss23,28,29. In contrast, overexpression of HIF-1α in cell lines such 786-O, which don’t typically express HIF-1α, inhibits tumor growth in xenograft models30. In addition 14q, which harbors the gene encoding HIF-1α, is lost in some ccRCC, and mutations of the remaining allele are occasionally seen12,31,32. Conversely, genome wide association studies identified a single-nucleotide polymorphism mapping to EPAS1 (which encodes HIF-2α) in association with increased risk of ccRCC33.
The VHL/HIF axis has implications well beyond cancer. Oxygen sensing is a fundamental element of pathophysiology, and plays a critical role in multiple processes. For their discovery and characterization of the cellular mechanism of oxygen sensing, the 2019 Nobel Prize in Physiology or Medicine was awarded to Drs. William G. Kaelin Jr., Sir Peter J. Ratcliffe and Gregg L. Semenza34,35.
Identification of additional tumor suppressor genes on chromosome 3p
Whereas VHL inactivation is nearly universal in ccRCC, it is not sufficient to cause ccRCC. VHL loss has been observed in preneoplastic cysts, and mice with Vhl disruption in the kidneys do not develop ccRCC36–38. This is the case even when both alleles of Vhl are inactivated in the appropriate compartment in the kidney37,39. These data provide unequivocal evidence that VHL loss is not sufficient for ccRCC tumorigenesis, and that additional driver mutations are required.
The additional driver mutations required for ccRCC development remained elusive until the advent of massively parallel sequencing. This technology led to the discovery of three additional ccRCC TSGs located on chromosome 3p. The first to be discovered was the histone-lysine N-methyltransferase SET Domain Containing 2 (SETD2). Targeted sequencing of a ~3,500 gene panel revealed recurring truncating SETD2 mutations40. Subsequent studies found that nonsilent SETD2 mutations occur in ~20% of ccRCC12,17,18. After SETD2, the polybromo 1 (PBRM1) and the BRCA1 associated protein-1 (BAP1) genes were discovered. Varela et al., identified truncating mutations in PBRM1 through whole exome sequencing (WES) of 7 ccRCC41. PBRM1 mutations were subsequently identified in 88 (39.8%) of 221 ccRCCs sequenced, making it the second most common TSG in ccRCC41. A few months later, whole exome sequencing (WES) of paired tumor and patient-derived xenografts (also called tumorgrafts [TGs]) identified BAP1 mutations in ccRCC42. BAP1 targeted sequencing of 176 ccRCC tumors revealed 24 (14%) nonsilent mutations 42. These initial studies were confirmed and expanded upon by sequencing efforts that followed12,17,18. Additional driver mutations and somatic copy number alterations (SCNAs) of some significance include: (i) activating mutations in the mTOR pathway12,17,43, (ii) inactivating mutations in additional chromatin modifying genes12,17,40,44, (iii) loss of TP5312,17,45, and (iv) deletion of chromosomes 14 and 912,17,32,46–48.
SETD2, PBRM1, and BAP1, are all involved in epigenetic regulation of gene expression. SETD2 is a histone methyltransferase for histone 3 lysine 36 (H3K36), and the only known enzyme to trimethylates H3K3649,50. PBRM1 encodes a component of the switching defective/sucrose nonfermenting (SWI/SNF) nucleosome remodeling complex (BAF180). Through its tandem bromo domains, BAF180 recruits the complex to specific nucleosomes characterized by particular histone tail acetylation patterns50. The BAP1 protein is a deubiquitinase that acts on lysine 119 of histone H2A to reverse polycomb-mediated gene repression50,51. Mutations in PBRM1 and BAP1 likely contribute to differences in gene expression we discovered between BAP1- and PBRM1-deficient tumors52.
Interestingly, BAP1, PBRM1, SETD2 are in physical proximity to VHL within a 50 Mb region on chromosome 3p11,32,51. Notably, BAP1 and PBRM1 mutations tend to be mutually exclusive in ccRCC12,42,52. The prevalence of tumors with combined mutations in BAP1 and PBRM1 is lower than expected by chance alone (~1-2% vs. ~5%) given the frequency of PBRM1 (~45%) and BAP1 (~12%) mutations42,52–54. In most tumor types, mutual exclusivity typically indicates that the encoded proteins are in the same pathway (such as for pVHL and elongin C). Once the pathway is deregulated through mutation in one of the genes, there is limited added benefit to mutation of the other. However, inasmuch as ccRCC with loss of BAP1 and PBRM1 differ in grade and gene expression signatures, this is unlikely to be the case in ccRCC42,52. Furthermore, BAP1 mutant tumors are associated with worse RCC-specific survival compared with tumors with loss of PBRM1 52,55–57. The differential association with outcomes between BAP1- and PBRM1-deficient tumors was subsequently extended to metastatic RCC58,59.
These observations provided initial evidence of distinct molecular subgroups within ccRCC. PBRM1 and BAP1 tend to be mutually exclusive and are associated with unique tumor biology, ultimately resulting in divergent clinical outcomes. Multiregional sequencing of human tumors through the TRACERx consortium as well as studies in genetically-engineered mouse models expanded upon these findings.
Distinct evolutionary trajectories in ccRCC
The mouse has traditionally been the animal model of choice in oncology. Remarkably, dysregulation of relevant cancer genes in the corresponding organs often triggers tumor development and tumors reproduce those in humans. However, in contrast to humans, Vhl heterozygous mice were not predisposed to ccRCC36,60. Furthermore, even targeting of two copies of Vhl in the mouse kidney failed to induce tumor development37,61,62. While this may occur when the gene is inactivated in an irrelevant cell type, even when Vhl is disrupted in the appropriate cell type, Vhl alone was not sufficient for tumor development37,61,62. As outlined above, the majority of ccRCCs in humans harbor not only VHL mutations, but also mutations in either PBRM1 (~50%) or BAP1 (~15%), and these mutations tend to be mutually exclusive. These data led us to hypothesize that ccRCC development in the mouse may require combined inactivation of Vhl together with Pbrm1 or Bap1. Indeed, simultaneous targeting of Vhl/Pbrm1 and Vhl/Bap1 in nephron progenitor cells induced ccRCC in the mouse37,39,63,64. Interestingly, as in humans Vhl/ Pbrm1-deficient tumors were of low grade whereas Vhl/Bap1-deficient tumors were of high grade39. In addition, Vhl/Bap1-deficient tumors developed with a shorter latency than Vhl/Pbrm1-deficient tumors. In contrast, Bap1 or Pbrm1 loss by themselves did not induce tumor formation37,39. These data show that: (i) inactivation of VHL, PBRM1 or BAP1 alone is insufficient for ccRCC development; (ii) combined inactivation of VHL with either PBRM1 or BAP1 is necessary; and (iii) PBRM1 and BAP1 are determinants of tumor grade. Interestingly, Vhl/Pbrm1-deficient tumors could be transformed into tumors of high grade by targeting Tuberous Sclerosis Complex 1 (Tsc1)39, which we previously discovered to be mutated in ccRCC43. TSC1 forms a complex with Tuberous Sclerosis Complex 2 (TSC2) and is a critical regulator of mTOR complex 1. A third tumor suppressor gene on chromosome 3p is SETD2, but whether combined inactivation of Vhl and Setd2 is sufficient for ccRCC development remains unknown. In addition, unlike PBRM1 and BAP1, which are generally mutually exclusive and seem to bifurcate tumor development, SETD2 mutations tend to be enriched in PBRM1-deficient tumors54. Parenthetically, whereas in the human VHL, PBRM1 and BAP1 are on the same chromosome, Vhl is on one chromosome, Bap1/Pbrm1 are on another, and Setd2 on a third chromosome in the mouse. This difference implies that whereas in the human a single event (loss of 3p) simultaneously inactivates one copy of all four TSGs, in the mouse, this would require deletions in three different chromosomes. This finding may explain the differential tumor predisposition across the two species.
Additional insight into the biology of ccRCC has been provided by the TRACERx consortium. The TRACERx consortium was established to assess the degree and impact of intratumoral heterogeneity on tumor biology and clinical outcomes, which early studies had revealed may be significant65–67. Turajlic and colleagues performed deep sequencing of 110 oncogenes and TSG across 1,206 ccRCC tumor samples from 101 patients who were prospectively enrolled68,69. This allowed the detection of clonal and subclonal somatic mutations providing an unprecedented view of the molecular diversity within a single tumor. While limited by gene density, the approach also offered a view of SCNAs, and in particular, LOH. After accounting for heterogeneity, the TRACERx consortium estimated the following mutation prevalence: PBRM1 (55%), SETD2 (25%), and BAP1 (19%)69. In keeping with previous analyses52,54, BAP1 and PBRM1 mutations, which were found, in some instances in the same tumor, were typically in spatially distinct regions69.
The analyses of spatially distinct regions within the same tumor enabled an assessment of clonality and inferences about the timing of mutations. Thus, patterns of tumor evolution could be derived. Distinct evolutionary pathways were identified through rule-based clustering69. 3p loss through chromothripsis was the most common initial event in ccRCC development, often occurring decades before clinical presentation13. Consistent with previous results66, after inactivation of the second VHL allele, the most common truncal mutation was in PBRM1. Mutations in BAP1 were sometimes similarly truncal69. As shown previously, the “VHL → BAP1” evolutionary subtype was associated with decreased disease free survival and overall survival42,52,57,68. PBRM1 loss was often followed by other mutations which, when present, were associated with more aggressive disease and increased metastatic potential68,69. Three main branches following PBRM1 loss were: (i) “PBRM1 → SETD2”, (ii) “PBRM1 → mTOR”, and (iii) “PBRM1 → Driver SCNAs”. Another subtype identified was the “multiple clonal drivers” subtype, which contained mutations in two or more of the following genes: BAP1, PBRM1, SETD2, or PTEN. In this group, the clonality of mutations could not be distinguished, indicating they occurred within a relatively short time span of each other. This subtype tended to be the most aggressive, and included the BAP1/PBRM1-deficient tumors which we and others noted to portend a poor prognosis previously42,53,59.
In summary, strong evidence from genetically-engineered mouse models and patient tumors supports the conclusion that BAP1 and PBRM1 mutations represent distinct branches in ccRCC tumor evolution (Figure 1). In the setting of BAP1 mutations, tumors gain aggressive features and as a consequence have high fitness, reducing the need for additional driver mutations. As a result, these tumors have lower intratumoral heterogeneity, higher propensity to metastasize, and reduced overall survival68,69. Conversely, PBRM1-mutant tumors tend to be associated with less aggressive features and are fertile ground for additional driver mutations which enhance tumor fitness. Perhaps the best supported of these are the “PBRM1 → mTOR pathway” and the “PBRM1 → SETD2” trajectories. The former is consistent with the finding that deletion of one allele of Tsc1 in Vhl/Pbrm1 deficient mice reduced tumor latency and increased tumor grade39. The latter is consistent with the observation that mutations in SETD2 are found at increased frequency in PBRM1-deficient tumors suggesting cooperativity54. Furthermore, in the TRACERx cohort, PBRM1 loss preceded SETD2 loss in in all the tumors where mutations in both were found69, consistent with the notion that PBRM1 loss is an earlier event in ccRCC oncogenesis.
Figure 1. Integrated ccRCC development model.
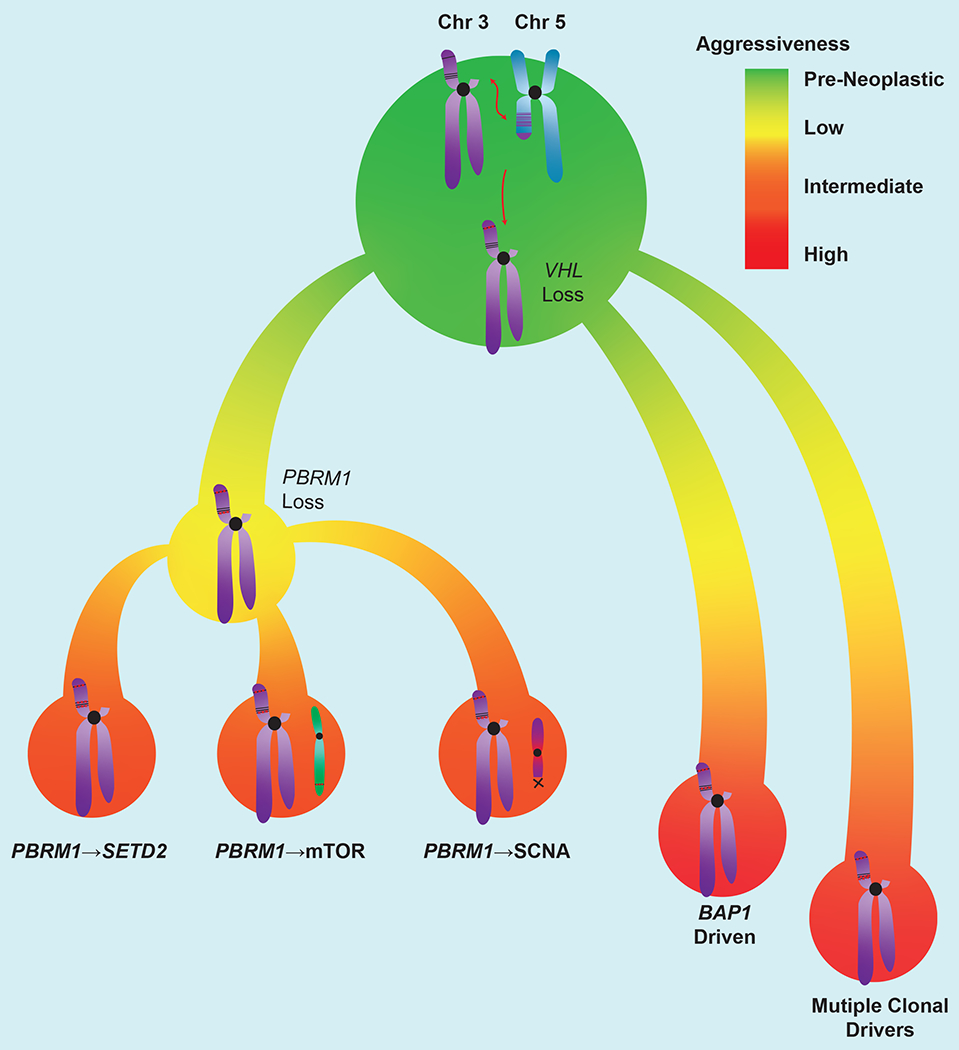
ccRCC development often begins with a chromothripsis event involving chromosomes 3p and 5q, followed by inactivation of the remaining VHL allele. This is followed by loss of BAP1 or PBRM1. Tumors with mutations in VHL and BAP1 are more aggressive than those with VHL and PBRM1 mutations. Further acquisition of driver mutations in PBRM1-deficient tumors increases their aggressiveness. These include SETD2 mutations, activation of the mTOR pathway, or driver SCNAs. Though rare, simultaneous mutantion in BAP1/PBRM1 induce aggressive tumors and represent a distinct molecular subgroup. The trajectories depicted here likely account for ~60% of ccRCC. Remaining to be discovered are cooperative events in VHL-deficient tumors that are wildtype for PBRM1 and BAP1, as well as driving events in VHL wildtype tumors.
Several important questions remain, however. For instance, in the ~40% of tumors that are seemingly wild-type for both PBRM1 and BAP1, what other events cooperate with VHL loss to promote oncogenesis? Also, how do mutations in TCEB1 and CUL2, which are located on chromosome 8 and 10 respectively, lead to ccRCC formation, and what are the specific cooperating events? It is worth noting that in over a third of tumors in the TRACERx cohort, an evolutionary subtype could not be assigned69, suggesting that while the majority of ccRCC tumors may follow the outlined trajectories, there are paths yet to be identified.
Targeting the VHL/HIF axis
Current therapies
An increased understanding of the VHL/HIF axis led to the development of targeted therapies in ccRCC. HIF regulates a number of genes controlling the cellular response to hypoxia, including VEGFA and PDGFB21. Notably, ccRCC appears to be the tumor with the highest levels of VEGFA expression70. The first targeted therapies to receive FDA approval for the treatment of metastatic RCC were sorafenib and sunitinib71,72. These tyrosine kinase inhibitors (TKIs) act primarily on the VEGF receptor (VEGFR1-3), but are also active against the related PDGF receptor (PDGFR)73. The number of TKIs to gain FDA approval in RCC has expanded substantially and today includes pazopanib74, axitinib75, lenvatinib (used in combination with the mTOR complex 1 inhibitor everolimus)76, and cabozantinib77. While all converge on the VEGF pathway, lenvatinib targets the fibroblast growth factor receptor (FGFR), and cabozantinib has activity against the hepatocyte growth factor receptor (encoded by MET) and the tyrosine protein kinase receptor UFO (encoded by AXL)73. As single agents in the frontline setting, TKIs result in objective responses in as many as 35% of patients with advanced ccRCC, with a median progression free survival between 8-11 months78–82. Today, frontline therapy has been replaced with combinations of ICIs. These include combinations of ICIs targeting the PD-1 and CTLA-4 checkpoints or of ICI with TKIs, specifically axitinib73,83–85. These combination therapies result in objective responses rates ranging from 40-60%, and a median progression free survival of 8 – 15 months83–85. In addition, complete responses, which are rare with TKIs and can be durable, are observed in 10% of patients83–85. It is worth noting that HIF also controls the transcription of several genes involved with immune evasion including CD274 (encoding PD-L1), NT5E (encoding CD73), ENTPD1 (encoding CD39)21, which provide one reason as to why ccRCC might be responsive to immunotherapies.
Targeting HIF-2
There are several advantages to targeting the pVHL pathway. First, VHL inactivation is the most common feature of ccRCC and thus, drugs targeting this pathway may be expected to have activity in a large number of patients. Second, VHL mutations are truncal, and thus drugs directed against this pathway may be less affected by intratumoral heterogeneity. Third, as the initiating event in ccRCC tumorigenesis, pVHL loss not only defines the context for future mutations, but likely engenders strong dependencies.
One promising approach is to directly target the HIF-2α/HIF-1β complex, which is immediately downstream of pVHL. Transcription factors such as HIF-2 have historically been considered undruggable as they generally lack pockets suitable for binding small molecules, such as those found in enzymes. The atomic structure of the HIF-2α PAS-B domain, however, revealed a highly structured pocket that could be bound by small molecule inhibitors86,87. This led to the development of the first in-class HIF-2α inhibitor PT2385 by Peloton Therapeutics Inc., a small biotech company in the UT Southwestern Medical Center BioCenter87,88 (the company was acquired by Merck in 2019). PT2385 is a potent and highly selective small molecule inhibitor that specifically dissociates HIF-2α/HIF-1β while leaving HIF-1α/HIF-1β intact88. Preclinical testing of PT2399 (a close analogue of PT2385 and tool compound) demonstrated efficacy in VHL deficient RCC cell lines89 and across an extensive platform of ccRCC tumorgrafts (TG). Overall, PT2399 decreased tumor growth by >80% in 10/22 (45%) independently derived ccRCC TGs, including in sunitinib resistant TGs25. Overall, PT2399 had greater activity than sunitinib; TGs treated with PT2399 (n=96) had a mean tumor volume shrinkage of ~60% vs ~40% in sunitinib treated TGs (n=82, p=0.0126)25. Interestingly, responses to PT2399 in VHL deficient ccRCC cell lines and tumorgrafts were variable, suggesting that VHL loss alone is not sufficient to predict responsiveness25,89. Another possibility could be that pVHL may regulate other substrates, including Zinc finger homeoboxes 2 (ZHX2) and Scm-Like With Four Mbt Domain 1(SFMBT1)90,91. Sensitivity did not appear to be associated with PBRM1 or BAP1 loss either. Overall, sensitive tumors exhibited higher levels of HIF-2α, providing the foundation for future biomarker studies25. In TG analyses, PT2399 did not have activity against non-ccRCC tumors.
PT2385 was the first HIF-2 inhibitor evaluated in humans. In a phase I trial conducted on 51 patients with heavily pretreated metastatic ccRCC, PT2385 demonstrated a favorable safety profile and an efficacy signal; disease control lasting greater than 4 months was seen in 40% (21/51) of patients92. The leading grade 3/4 adverse events were anemia and hypoxia, both of which occurred in 5 (10%) patients (all grade 3). Interestingly, both anemia and hypoxia are on-target adverse events, mediated through suppression of erythropoietin (a HIF-2 regulated gene) and effects on carotid body biology respectively93–95. Fortunately, severe hypoxia was rare (1/51 patients)92. Despite these encouraging results, a few patients who received the recommended phase II dose (800mg BID) had suboptimal circulating drug levels92,96. This was due to extensive hepatic glucuronidation of PT2385 by the UDP-glucuronosyltransferase UGT2B17, which is differentially expressed among individuals96,97. This led to the development of a second-generation inhibitor, PT2779 (MK-6482, after the purchase of Peloton Therapeutics by Merck), which demonstrated improved pharmacokinetics96.
MK-6482 demonstrated a similar safety profile to PT2385 in an open label phase I/II study in advanced ccRCC98,99. In this cohort of heavily pretreated patients, objective response rates were observed in 24% of patients (13/55) and disease control (defined as a best response of stable disease, partial response, or complete response) was achieved in 80% (44/55) of patients. After a median follow up of 13 months, the median duration of response was not reached, with 81% of patients having responses lasting over 6 months99. An ongoing, open-label phase II study evaluating MK-6482 in patients with von Hippel-Lindau associated ccRCC demonstrated similar efficacy in ccRCC lesions, and responses were also observed in other VHL-syndrome associated lesions (CNS and pancreas)100. This has led the Food and Drug Administration to grant MK-6482 breakthrough designation101. An international phase III trial comparing MK-6482 with everolimus in previously treated advanced ccRCC is currently enrolling (NCT04195750)102.
While HIF-2α inhibitors are promising, there is evidence of acquired resistance. Prolonged therapy with PT2399 led to the development of acquired resistance in TG models25. Resistance arose from de novo point mutations which restored HIF-2α/HIF-1β dimerization25. Two mutations were identified, one in HIF-2α (G323E), and another one in HIF-1β (F466L)25. Structural and biochemical analyses of PT2385/HIF-2α/HIF-1β revealed that dissociation of the complex is mediated through allosteric modulation of HIF-2α and disruption of HIF-2α/HIF1β dimerization103. Interestingly, the HIF-2α G323E mutation functions as a gatekeeper mutation, preventing PT2385 binding25,103. HIF-1β F466L, on the other hand, is at the interface of HIF-2α and HIF-1β and is thought to increase binding affinity103. The HIF-2α G323E mutation was subsequently found in two patients who participated in the phase I clinical trial of PT2385104. One potential strategy to overcome resistance mediated by HIF-2α G323E mutation is to develop inhibitors which target different pockets. In fact, comprehensive structural analysis of HIF-2α revealed three additional pockets, potentially suitable for inhibitors103,105.
Another potential strategy to target HIF-2α involves siRNA. While siRNA is an effective tool to inhibit gene expression in-vitro, challenges in the delivery of siRNA have been barriers to its effective use in patients106. Arrowhead Pharmaceuticals has developed an siRNA delivery system (termed dynamic polyconjugates or DPC), which links siRNA with shielding moieties (which improve stability) and targeting ligands specific to RCC. The first generation siRNA exploits Arg/Gly/Asp (RGD) binding to the integrins αvβ3 and αvβ5, which are commonly expressed in ccRCC107. In preclinical models, this DPC-RGD construct was able to inhibit tumor growth in xenografts. A second generation siRNA was developed using the TRIM™ platform, ARO-HIF2, which exhibits increased stability108. A phase Ib trial of this agent in ccRCC has begun enrollment (NCT04169711). A phase Ib trial with a siRNA targeting HIF-1α (RO7070179) in advanced hepatocellular carcinoma was recently reported109. The trial was terminated early due to failure to meet the primary endpoint of HIF-1α mRNA depletion following a single cycle (10mg IV dose), although one (of ten) patient demonstrated a partial response lasting over one year109. In addition to potentially overcoming the HIF-2α resistance mutations highlighted above, the HIF-2α siRNA approach is attractive because it can reduce treatment-related adverse events by specifically targeting ccRCC cells.
Synthetic lethality with VHL loss in cancer
While HIF-2α/HIF-1β is a critical effector downstream of pVHL, evidence suggests that other pathways contribute to tumorigenesis. First, as observed across cell lines, TG and patients, not every tumor with a VHL mutation is sensitive to HIF-2α inhibitors. While pharmacokinetic and other factors may contribute, it is likely that not all VHL-deficient ccRCC tumors are HIF-2α dependent and some tumors, in fact, express low or undetectable levels of HIF-2α25. In addition, pVHL has been shown to regulate other proteins besides the HIF2-α. pVHL substrates other than HIFα may account for different subtypes of VHL syndrome (Type 2A, 2B versus 2C)110–112. Furthermore, Notch and nuclear factor (NF-κB) signaling pathways are constitutively active upon VHL loss in human ccRCC independent of HIF113,114.
By performing an innovative genome-wide in vitro expression screen of ~17,000 proteins, recent studies identified ZHX2 and SFMBT1 as new pVHL substrates90,91. ZHX2 was shown to promote NF-κB activation and SFMBT1 induced the expression of Sphingosine Kinase 1 (SPHK1), both of which acted in a HIF independent manner to promote ccRCC tumorigenesis90,91. N-Myc Downstream-Regulated Gene 3 (NDRG3) was also identified as a potential pVHL substrate, which activates the RAF-ERK1/2 kinase pathway115. Other potential pVHL substrates have been previously described, including EPOR (erythropoietin receptor)116, transcription factor B-Myb117, FLNA (actin cross-linker filamin A)118, CEP68 (centrosomal protein 68)119, CERKL (ceramide kinase like protein)120, hsRPB7 (human RNA polymerase II seventh subunit)121 and EHMT2 (euchromatic histone-lysine methyltransferase 2)122. Further research is needed to elucidate the precise conribution of these potential pVHL substrates and their role in tumorigenesis elicited by VHL loss.
In addition to the canonical role of pVHL as an important component of an E3 ubiquitin ligase, emerging research suggests that pVHL may function as an adaptor in other contexts. For example, Akt1 appears to be hydroxylated on Pro125 and Pro313 which are recognized by pVHL which in turn recruited phosphatase 2A (PP2A) promoting Akt1 dephosphorylation123. A similar mechanism was observed with TANK Binding Kinase 1 (TBK1). TBK1 hydroxylation on Pro48 by EglN1 led to pVHL binding and recruitment of phosphatase PPM1B, ultimately dampening TBK1 activity124. pVHL was also reported to associate with Card9 promoting its phosphorylation by CK2, which led to decreased NF-κB activity113. Finally, pVHL could bind BIM-EL, which inhibited BIM-EL phosphorylation by ERK on Ser69, therefore protecting it from proteasomal degradation125. Thus, the function of pVHL is likely multi-faceted, and remains to be fully characterized ccRCC.
Effector Agnostic Approaches to pVHL Targeting
Thus, arguments can be made to target VHL loss in ccRCC beyond HIF-2. One strategy to exploit loss of function mutations is to leverage “synthetic lethality”, a scenario in which loss of a gene renders a second gene essential126. Several groups have developed high-throughput screening platforms of chemical libraries capable of identifying compounds which exhibit selective killing of VHL-deficient cells. For example, initial reports identified the autophagy modulator STF-62247, and chemical inhibitors of GLUT-1 as selective inhibitors of VHL deficient RCC127,128. Another study revealed that inhibitors of ROCK1 were toxic, another HIF target gene, were toxic to ccRCC with VHL loss129. More recently, a report from the same group showed that inhibition of the Mevalonate pathway had anti-tumor effects in VHL deficient ccRCC cell-lines in a HIF-independent manner130. In a high-throughput screen of ~13,000 small molecules, we identified homoharringtonine (HHT), an FDA-approved drug for chronic myeloid leukemia, as a potential VHL synthetic lethal compound. HHT was further shown to be efficacious in ~30% of tested tumorgrafts131. Finally, TBK1 was also found to be a synthetic lethal partner with VHL loss in ccRCC. Both sgRNA/shRNA mediated depletion and pharmacological inhibition of TBK1 preferentially killed VHL-deficient ccRCC cells124. While synthetic lethal screens of chemical libraries have the advantage of producing hit compounds which can be refined via medicinal chemistry approaches, they target a small fraction of the proteome. Thus, these studies can be complemented by high throughput screens of short hairpin RNA (shRNA) libraries and more recently, small guide RNA (sgRNA)/CRISPR Cas-9.
One initial report utilizing a shRNA library directed against 88 kinases in VHL-deficient RCC cell lines identified CDK6, MET, and MAP2K1 as potential targets132. A follow up effort utilizing an expanded shRNA library targeting ~1,000 genes identified EZH1 as synthetically lethal with VHL133. More recently, both in human and Drosophila, Nicholson and colleagues demonstrated synthetic lethality between CDK4/6 and pVHL134. A genome-wide CRISPR Cas-9 screen identified the selenocysteine biosynthetic pathway as synthetic lethal with VHL135,136. The extent to which these findings are influenced by other mutations in the cell line models is unknown. Synthetic lethality is highly context specific, and future efforts may leverage our greater understanding of ccRCC evolutionary pathways.
Conclusion
Our understanding of ccRCC tumorigenesis has improved substantially since the discovery of the VHL gene. Novel sequencing strategies and insightful experimental designs have refined our understanding of ccRCC biology. New agents targeting the “undruggable” HIF transcription factor have shown promise in the clinic, and strategies to exploit synthetic lethality with VHL loss have been identified. These exciting developments have the potential to build upon a decade which has seen a flurry of new treatments and improved clinical outcomes for ccRCC patients.
Acknowledgements:
J.B receives support from NIH SPORE Grant P50CA196516 and Cancer Prevention & Research Institute of Texas (CPRIT) Grant RP130603. Q.Z is supported by DoD Kidney Cancer Research Program idea development award (W81XWH1910813), the kidney cancer research alliance, CPRIT grant RR190058, American Cancer Society grant RSG-18-059-01-TBE and NCI grant R01CA211732. R.E. receives support from an institutional award provided through the Burroughs Wellcome Fund.
Conflicts of interest:
In addition, J.B. has a patent US Patent No. 15/761,534 issued, a patent US Patent Provisional Application Filed September 23, 2019—US Appl. No. 62/904,268—UTSD 3713 US PZ 1 pending, and a patent US Patent Provisional Application Filed December 19, 2019 pending.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs—part A: renal, penile, and testicular tumours. European urology 2016;70:93–105. [DOI] [PubMed] [Google Scholar]
- 3.Maher ER, Neumann HPH, Richard S. von Hippel–Lindau disease: A clinical and scientific review. European Journal of Human Genetics 2011;19:617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988;332:268–9. [DOI] [PubMed] [Google Scholar]
- 5.Hosoe S, Brauch H, Latif F, et al. Localization of the von Hippel-Lindau disease gene to a small region of chromosome 3. Genomics 1990;8:634–40. [DOI] [PubMed] [Google Scholar]
- 6.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993;260:1317–20. [DOI] [PubMed] [Google Scholar]
- 7.Gnarra JR, Tory K, Weng Y, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994;7:85–90. [DOI] [PubMed] [Google Scholar]
- 8.Zbar B, Brauch H, Talmadge C, Linehan M. Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature 1987;327:721–4. [DOI] [PubMed] [Google Scholar]
- 9.Crossey PA, Foster K, Richards FM, et al. Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Hum Genet 1994;93:53–8. [DOI] [PubMed] [Google Scholar]
- 10.Iliopoulos O, Kibel A, Gray S, Kaelin WG. Tumour suppression by the human von Hippel-Lindau gene product. Nature Medicine 1995;1:822–6. [DOI] [PubMed] [Google Scholar]
- 11.Nickerson ML, Jaeger E, Shi Y, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 2008;14:4726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchell TJ, Turajlic S, Rowan A, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018;173:611–23 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nature reviews Cancer 2015;15:55–64. [DOI] [PubMed] [Google Scholar]
- 15.Kaelin WG Jr, Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2002;2:673–82. [DOI] [PubMed] [Google Scholar]
- 16.Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008;30:393–402. [DOI] [PubMed] [Google Scholar]
- 17.Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 2013;45:860–7. [DOI] [PubMed] [Google Scholar]
- 18.Ricketts CJ, De Cubas AA, Fan H, et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Reports 2018;23:313–26.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hakimi AA, Tickoo SK, Jacobsen A, et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Modern Pathology 2015;28:845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiNatale RG, Gorelick AN, Makarov V, et al. Putative Drivers of Aggressiveness in TCEB1-mutant Renal Cell Carcinoma: An Emerging Entity with Variable Clinical Course. Eur Urol Focus 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends in Cancer 2016;2:758–70. [DOI] [PubMed] [Google Scholar]
- 22.Prabhakar NR, Semenza GL. Oxygen Sensing and Homeostasis. Physiology 2015;30:340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 2005;25:5675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertout JA, Majmundar AJ, Gordan JD, et al. HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proceedings of the National Academy of Sciences of the United States of America 2009;106:14391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016;539:112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du W, Zhang L, Brett-Morris A, et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nature Communications 2017;8:1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaelin WG. Molecular Biology of Kidney Cancer. In: Lara PN, Jonasch E, eds. Kidney Cancer: Principles and Practice. Cham: Springer International Publishing; 2015:31–57. [Google Scholar]
- 28.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002;1:237–46. [DOI] [PubMed] [Google Scholar]
- 29.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 2002;1:247–55. [DOI] [PubMed] [Google Scholar]
- 30.Biswas S, Troy H, Leek R, et al. Effects of HIF-1alpha and HIF2alpha on Growth and Metabolism of Clear-Cell Renal Cell Carcinoma 786–0 Xenografts. J Oncol 2010;2010:757908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen C, Beroukhim R, Schumacher SE, et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov 2011;1:222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore LE, Jaeger E, Nickerson ML, et al. Genomic copy number alterations in clear cell renal carcinoma: associations with case characteristics and mechanisms of VHL gene inactivation. Oncogenesis 2012;1:e14–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purdue MP, Johansson M, Zelenika D, et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nature genetics 2011;43:60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.AB NM. Press Release: The Nobel Prize in Physiology or Medicine 2019. October 7, 2019. Available at: https://www.nobelprize.org/prizes/medicine/2019/press-release/. Accessed September 4, 2020.
- 35.Zhang Q, Yan Q, Yang H, Wei W. Oxygen sensing and adaptability won the 2019 Nobel Prize in Physiology or medicine. Genes Dis 2019;6:328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proceedings of the National Academy of Sciences of the United States of America 2001;98:1583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang SS, Gu YF, Wolff N, et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 2014;111:16538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandriota SJ, Turner KJ, Davies DR, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002;1:459–68. [DOI] [PubMed] [Google Scholar]
- 39.Gu YF, Cohn S, Christie A, et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov 2017;7:900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010;463:360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011;469:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nature Genetics 2012;44:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kucejova B, Pena-Llopis S, Yamasaki T, et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res 2011;9:1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Haaften G, Dalgliesh GL, Davies H, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 2009;41:521–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gad S, Lefèvre SH, Khoo SK, et al. Mutations in BHD and TP53 genes, but not in HNF1beta gene, in a large series of sporadic chromophobe renal cell carcinoma. Br J Cancer 2007;96:336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kovacs G, Frisch S. Clonal chromosome abnormalities in tumor cells from patients with sporadic renal cell carcinomas. Cancer Res 1989;49:651–9. [PubMed] [Google Scholar]
- 47.Cairns P, Tokino K, Eby Y, Sidransky D. Localization of tumor suppressor loci on chromosome 9 in primary human renal cell carcinomas. Cancer Res 1995;55:224–7. [PubMed] [Google Scholar]
- 48.Beroukhim R, Brunet J-P, Di Napoli A, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer research 2009;69:4674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan W, Xie J, Long C, et al. Heterogeneous nuclear ribonucleoprotein L Is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem 2009;284:15701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Cubas AA, Rathmell WK. Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol 2018;15:599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brugarolas J Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol 2014;32:1968–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kapur P, Peña-Llopis S, Christie A, et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol 2013;14:159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joseph RW, Kapur P, Serie DJ, et al. Clear Cell Renal Cell Carcinoma Subtypes Identified by BAP1 and PBRM1 Expression. The Journal of urology 2016;195:180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pena-Llopis S, Christie A, Xie XJ, Brugarolas J. Cooperation and Antagonism among Cancer Genes: The Renal Cancer Paradigm. Cancer research 2013;73:4173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joseph RW, Kapur P, Serie DJ, et al. Loss of BAP1 protein expression is an independent marker of poor prognosis in patients with low-risk clear cell renal cell carcinoma. Cancer 2014;120:1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kapur P, Christie A, Raman JD, et al. BAP1 Immunohistochemistry Predicts Outcomes in a Multi-Institutional Cohort with Clear Cell Renal Cell Carcinoma. The Journal of urology 2014;191:603–10. [DOI] [PubMed] [Google Scholar]
- 57.Hakimi AA, Chen YB, Wren J, et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol 2013;63:848–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carlo MI, Manley B, Patil S, et al. Genomic Alterations and Outcomes with VEGF-Targeted Therapy in Patients with Clear Cell Renal Cell Carcinoma. Kidney Cancer 2017;1:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voss MH, Reising A, Cheng Y, et al. Genomically annotated risk model for advanced renal-cell carcinoma: a retrospective cohort study. Lancet Oncol 2018;19:1688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gnarra JR, Ward JM, Porter FD, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proceedings of the National Academy of Sciences 1997;94:9102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer research 2006;66:2576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frew IJ, Thoma CR, Georgiev S, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. Embo j 2008;27:1747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Espana-Agusti J, Warren A, Chew SK, Adams DJ, Matakidou A. Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat Commun 2017;8:2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nargund AM, Pham CG, Dong Y, et al. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep 2017;18:2893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014;46:225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.consortium TRR. TRACERx Renal: tracking renal cancer evolution through therapy. Nature reviews Urology 2017;14:575–6. [DOI] [PubMed] [Google Scholar]
- 68.Turajlic S, Xu H, Litchfield K, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018;173:581–94 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turajlic S, Xu H, Litchfield K, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018;173:595–610 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science 2017;357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 71.Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. The New England journal of medicine 2007;356:125–34. [DOI] [PubMed] [Google Scholar]
- 72.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. The New England journal of medicine 2007;356:115–24. [DOI] [PubMed] [Google Scholar]
- 73.Choueiri TK, Motzer RJ. Systemic Therapy for Metastatic Renal-Cell Carcinoma. The New England journal of medicine 2017;376:354–66. [DOI] [PubMed] [Google Scholar]
- 74.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8. [DOI] [PubMed] [Google Scholar]
- 75.Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9. [DOI] [PubMed] [Google Scholar]
- 76.Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol 2015;16:1473–82. [DOI] [PubMed] [Google Scholar]
- 77.Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol 2016;17:917–27. [DOI] [PubMed] [Google Scholar]
- 78.Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. New England Journal of Medicine 2013;369:722–31. [DOI] [PubMed] [Google Scholar]
- 80.Sternberg CN, Hawkins RE, Wagstaff J, et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur J Cancer 2013;49:1287–96. [DOI] [PubMed] [Google Scholar]
- 81.Motzer RJ, Hutson TE, McCann L, Deen K, Choueiri TK. Overall survival in renal-cell carcinoma with pazopanib versus sunitinib. N Engl J Med 2014;370:1769–70. [DOI] [PubMed] [Google Scholar]
- 82.Choueiri TK, Hessel C, Halabi S, et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur J Cancer 2018;94:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. New England Journal of Medicine 2018;378:1277–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. New England Journal of Medicine 2019;380:1116–27. [DOI] [PubMed] [Google Scholar]
- 85.Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. New England Journal of Medicine 2019;380:1103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proceedings of the National Academy of Sciences 2009;106:450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scheuermann TH, Li Q, Ma HW, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nature chemical biology 2013;9:271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wallace EM, Rizzi JP, Han G, et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer research 2016;76:5491–500. [DOI] [PubMed] [Google Scholar]
- 89.Cho H, Du X, Rizzi JP, et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016;539:107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang J, Wu T, Simon J, et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018;361:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu X, Simon JM, Xie H, et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Molecular cell 2020;77:1294–306 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Courtney KD, Infante JR, Lam ET, et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol 2018;36:867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith TG, Brooks JT, Balanos GM, et al. Mutation of von Hippel–Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med 2006;3:e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hodson EJ, Nicholls LG, Turner PJ, et al. Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J Physiol 2016;594:1179–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bishop T, Ratcliffe PJ. Genetic basis of oxygen sensing in the carotid body: HIF2α and an isoform switch in cytochrome c oxidase subunit 4. Sci Signal 2020;13. [DOI] [PubMed] [Google Scholar]
- 96.Xu R, Wang K, Rizzi JP, et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. Journal of Medicinal Chemistry 2019;62:6876–93. [DOI] [PubMed] [Google Scholar]
- 97.Bhatt DK, Basit A, Zhang H, et al. Hepatic abundance and activity of androgen-and drug-metabolizing enzyme UGT2B17 are associated with genotype, age, and sex. Drug Metabolism and Disposition 2018;46:888–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jonasch E, Plimack ER, Bauer T, et al. 911PDA first-in-human phase I/II trial of the oral HIF-2a inhibitor PT2977 in patients with advanced RCC. Annals of Oncology 2019;30. [Google Scholar]
- 99.Choueiri TK, Plimack ER, Bauer TM, et al. Phase I/II study of the oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC). Journal of Clinical Oncology 2020;38:611-. [Google Scholar]
- 100.Jonasch E, Donskov F, Iliopoulos O, et al. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. Journal of Clinical Oncology 2020;38:5003-. [Google Scholar]
- 101.Merck & Co. I. FDA Grants Breakthrough Therapy Designation to Merck’s Novel HIF-2α Inhibitor MK-6482 for Treatment of Certain Patients With Von Hippel-Lindau Disease- Associated Renal Cell Carcinoma [news release]. 2020.
- 102.Choueiri TK, Albiges L, Fan L, et al. Phase III study of the hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 versus everolimus in previously treated patients with advanced clear cell renal cell carcinoma (ccRCC). Journal of Clinical Oncology 2020;38:TPS5094–TPS. [Google Scholar]
- 103.Wu D, Su X, Lu J, et al. Bidirectional modulation of HIF-2 activity through chemical ligands. Nature Chemical Biology 2019;15:367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Courtney KD, Ma Y, Diaz de Leon A, et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin Cancer Res 2020;26:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu D, Su X, Potluri N, Kim Y, Rastinejad F. NPAS1-ARNT and NPAS3-ARNT crystal structures implicate the bHLH-PAS family as multi-ligand binding transcription factors. eLife 2016;5:e18790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bobbin ML, Rossi JJ. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu Rev Pharmacol Toxicol 2016;56:103–22. [DOI] [PubMed] [Google Scholar]
- 107.Wong SC, Cheng W, Hamilton H, et al. HIF2α-Targeted RNAi Therapeutic Inhibits Clear Cell Renal Cell Carcinoma. Mol Cancer Ther 2018;17:140–9. [DOI] [PubMed] [Google Scholar]
- 108.Wong SC, Nicholas A, Carlson J, et al. Abstract 4775: Optimizing the potency and dosing design for ARO-HIF2: An RNAi therapeutic for clear cell renal cell carcinoma. Cancer Research 2019;79:4775-. [Google Scholar]
- 109.Wu J, Contratto M, Shanbhogue KP, et al. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1α in advanced hepatocellular carcinoma. World J Clin Oncol 2019;10:149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clifford SC, Cockman ME, Smallwood AC, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Human molecular genetics 2001;10:1029–38. [DOI] [PubMed] [Google Scholar]
- 111.Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG Jr., von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Human molecular genetics 2001;10:1019–27. [DOI] [PubMed] [Google Scholar]
- 112.Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004;103:3924–32. [DOI] [PubMed] [Google Scholar]
- 113.Yang H, Minamishima YA, Yan Q, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell 2007;28:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sjolund J, Johansson M, Manna S, et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest 2008;118:217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee DC, Sohn HA, Park ZY, et al. A lactate-induced response to hypoxia. Cell 2015;161:595–609. [DOI] [PubMed] [Google Scholar]
- 116.Heir P, Srikumar T, Bikopoulos G, et al. Oxygen-dependent Regulation of Erythropoietin Receptor Turnover and Signaling. J Biol Chem 2016;291:7357–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Okumura F, Uematsu K, Byrne SD, et al. Parallel Regulation of von Hippel-Lindau Disease by pVHL-Mediated Degradation of B-Myb and Hypoxia-Inducible Factor alpha. Molecular and cellular biology 2016;36:1803–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Segura I, Lange C, Knevels E, et al. The Oxygen Sensor PHD2 Controls Dendritic Spines and Synapses via Modification of Filamin A. Cell Rep 2016;14:2653–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yin H, Zheng L, Liu W, Zhang D, Li W, Yuan L. Rootletin prevents Cep68 from VHL-mediated proteasomal degradation to maintain centrosome cohesion. Biochim Biophys Acta Mol Cell Res 2017;1864:645–54. [DOI] [PubMed] [Google Scholar]
- 120.Chen J, Liu F, Li H, et al. pVHL interacts with Ceramide kinase like (CERKL) protein and ubiquitinates it for oxygen dependent proteasomal degradation. Cell Signal 2015;27:2314–23. [DOI] [PubMed] [Google Scholar]
- 121.Na X, Duan HO, Messing EM, et al. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel-Lindau protein. Embo Journal 2003;22:4249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Casciello F, Al-Ejeh F, Kelly G, et al. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc Natl Acad Sci U S A 2017;114:7077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo j Chakraborty AA, Liu P, et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016;353:929–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hu L, Xie H, Liu X, et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer discovery 2020;10:460–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li S, Rodriguez J, Li W, et al. EglN3 hydroxylase stabilizes BIM-EL linking VHL type 2C mutations to pheochromocytoma pathogenesis and chemotherapy resistance. Proceedings of the National Academy of Sciences of the United States of America 2019;116:16997–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaelin WG, Jr. Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med 2009;1:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer cell 2008;14:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chan DA, Sutphin PD, Nguyen P, et al. Targeting GLUT1 and the Warburg Effect in Renal Cell Carcinoma by Chemical Synthetic Lethality. Science Translational Medicine 2011;3:94ra70–94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thompson JM, Nguyen QH, Singh M, et al. Rho-associated kinase 1 inhibition is synthetically lethal with von Hippel-Lindau deficiency in clear cell renal cell carcinoma. Oncogene 2017;36:1080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thompson JM, Alvarez A, Singha MK, et al. Targeting the Mevalonate Pathway Suppresses VHL-Deficient CC-RCC through an HIF-Dependent Mechanism. Molecular cancer therapeutics 2018;17:1781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wolff NC, Pavía-Jiménez A, Tcheuyap VT, et al. High-throughput simultaneous screen and counterscreen identifies homoharringtonine as synthetic lethal with von Hippel-Lindau loss in renal cell carcinoma. Oncotarget 2015;6:16951–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bommi-Reddy A, Almeciga I, Sawyer J, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen. Proceedings of the National Academy of Sciences of the United States of America 2008;105:16484–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chakraborty AA, Nakamura E, Qi J, et al. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Science translational medicine 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nicholson HE, Tariq Z, Housden BE, et al. HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Science signaling 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sun N, Petiwala S, Lu C, et al. VHL Synthetic Lethality Signatures Uncovered by Genotype-specific CRISPR-Cas9 Screens. bioRxiv 2019:588707. [DOI] [PubMed] [Google Scholar]
- 136.Sun N, Petiwala S, Lu C, et al. VHL Synthetic Lethality Signatures Uncovered by Genotype-Specific CRISPR-Cas9 Screens. The CRISPR Journal 2019;2:230–45. [DOI] [PubMed] [Google Scholar]