

Abstract
Nitric oxide (NO· ) expression by inducible nitric oxide synthase (iNOS) is an important host defense mechanism against Mycobacterium tuberculosis in mononuclear phagocytes. The objective of this investigation was to examine the role of mitogen-activated protein (MAP) kinase (MAPK) and nuclear factor κB (NF-κB) signaling pathways in the regulation of iNOS and NO· by a mycobacterial cell wall lipoglycan known as mannose-capped lipoarabinomannan (ManLAM). Specific pharmacologic inhibition of the extracellular-signal-regulated kinase (ERK) or NF-κB pathway revealed that both these signaling cascades were required in gamma interferon (IFN-γ)-ManLAM-induced iNOS protein and NO2− expression in mouse macrophages. Transient cotransfection of dominant-negative protein mutants of the c-Jun NH2-terminal kinase (JNK) pathway revealed that the MAP kinase kinase 7 (MKK7)-JNK cascade also mediated IFN-γ–ManLAM induction of iNOS promoter activity whereas MKK4 did not. Overexpression of null mutant IκBα, a potent inhibitor of NF-κB activation, confirmed that the IκBα kinase (IKK)–NF-κB signaling pathway enhanced IFN-γ–ManLAM-induced iNOS promoter activity. By contrast, activated p38mapk inhibited iNOS induction. These results indicate that combined IFN-γ and ManLAM stimulation induced iNOS and NO· expression and that MEK1-ERK, MKK7-JNK, IKK–NF-κB, and p38mapk signaling pathways play important regulatory roles.
In immunocompetent hosts, the innate and adaptive arms of the immune system are relatively efficient in the containment and killing of microbial pathogens. Macrophages have the capacity to produce relatively large quantities of nitric oxide (NO·) and NO·-derived species such as NO2·, NO2−, N2O3, N2O4, S-nitrosothiols, and peroxynitrite (ONOO−). Expression of such reactive nitrogen intermediates from the catalytic action of inducible nitric oxide synthase (iNOS) in response to cytokines or pathogen-derived molecules is essential in the control and elimination of intracellular microorganisms such as Toxoplasma gondii, Leishmania major, Listeria monocytogenes, Mycobacterium leprae, and Mycobacterium tuberculosis (1, 19, 31, 34, 43). iNOS-derived NO· has also been shown to contribute to the host defense against Plasmodium species, Salmonella enterica serovar Typhimurium, Mycoplasma pneumoniae, Chlamydia pneumoniae, and Entamoeba histolytica (54). As a host defense molecule, NO·also inhibits the proliferation of viruses such as ectromelia virus, coxsackie virus B3, and hepatitis B virus (25, 54). Even at very low concentrations, e.g., <100 ppm, NO· is directly toxic to M. tuberculosis (46). In the murine model of tuberculosis (TB), NO· plays an essential role in the killing of M. tuberculosis by mononuclear phagocytes (18, 19, 47). An in vivo example of this is illustrated by the genetically disrupted iNOS mouse strain (iNOS−/−), in which infection with M. tuberculosis is associated with a significantly higher risk of dissemination and mortality than in wild-type C57BL/6 mice (47). NO· is also implicated in mediating apoptosis in infected macrophages, providing an avenue for macrophages that do not produce adequate NO· to chronically harbor M. tuberculosis (52). Furthermore, in mice that express the Bcg/Nramp-1 mycobacterial resistance gene, NO· mediates the resistance to M. tuberculosis (5).
Although the role of NO· in human TB is controversial, there is a growing body of evidence that NO· and related reactive nitrogen species are important in host defense (37, 39, 55, 57, 59, 61, 69). In part, this controversy stemmed from in vitro experiments with human monocyte-derived macrophages or alveolar macrophages that fail to elicit detectable levels of NO· (6). However, this apparent deficiency in human macrophages may be due to experimental limitations such as the lack of iNOS cofactor tetrahydrobiopterin in in vitro human macrophage cultures (8) and the insensitivity of the standard colorimetric assay to detect relatively low but significant levels of nitrite (NO2−), the metabolic product of NO· (37). Despite these technical restrictions, independent laboratories have demonstrated upregulation of active iNOS and NO· in human alveolar macrophages infected with M. tuberculosis (55, 59). Moreover, iNOS inhibition has been shown to enhance intracellular growth of M. tuberculosis in human macrophages (37, 57, 59). In a recent study, it was demonstrated that vitamin D3, known historically to have therapeutic efficacy against TB, suppressed M. tuberculosis growth via induction of iNOS expression and NO· production (61).
The cell walls of mycobacteria contain a variety of molecules that are potentially able to elicit inflammatory and immune responses from host cells. These molecules include complex lipoglycans and lipoproteins. A major component of the cell wall of M. tuberculosis is mannose-capped lipoarabinomannan (ManLAM), an arabinose- and mannose-containing phosphorylated lipoglycan implicated as both a virulence factor and as a stimulus of host defense mechanisms (20). In previous studies, ManLAM was shown to have pleiotropic functions. ManLAM was found to downregulate gamma interferon (IFN-γ)-induced cytocidal and microbicidal capacity of macrophages and to inhibit antigen processing (17, 30, 65). In contrast, ManLAM has also been shown to promote host defense mechanisms by mediating phagocytosis and by inducing interleukin-1β (IL-1β), IL-8, tumor necrosis factor alpha, (TNF-α), and NO· expression (2, 7, 20, 22, 50, 60, 74, 75). ManLAM has also been shown to enhance expression of IL-4, a cytokine thought to play a role in tuberculostasis by inducing the formation of giant cells in granulomatous lesions (23, 33). Thus, we believe that investigations into the signaling mechanisms by which ManLAM regulates iNOS expression and NO· production constitute an important area of research.
Mitogen-activated protein kinases (MAPKs) and their upstream kinases activate a number of transcription factors and signal the induction of a variety of inflammatory genes in response to lipopolysaccharide (LPS) and cytokines (9, 16, 32, 35, 67, 70). MAPKs are serine-threonine kinases that signal the intracellular responses to an array of extracellular stimuli that include mitogens, growth factors, pathogen-derived products, and physical stressors such as hyperosmolality, heat shock, and UV irradiation. There are three principal MAPK family members: (i) p46 and p54 c-Jun–NH2-terminal kinase or stress-activated protein kinase (JNK or SAPK, respectively) with multiple subisoforms, (ii) p38mapk with α, β, γ, and δ isoforms, and (iii) p42 and p44 extracellular-signal-regulated kinase (ERK). MAPKs are activated by specific upstream MAPK kinases (MKKs): (i) MKK4 and -7, also known as JNK kinase 1/2 (JNKK1/2) or SAPK/ERK kinase 1/2 (SEK1/2), activate JNK (26, 44, 64); (ii) MAPK/ERK kinase 1/2 (MEK1/2) activates the ERKs (72); and (iii) MKK3 and -6 activate p38mapk (71). We previously demonstrated that, in NIH3T3 fibroblasts, the MKK4-JNK pathway contributed to the production of IFN-γ–TNF-α-induced iNOS-NO· expression (14, 16). The IκBα kinase (IKK)-nuclear factor κB (NF-κB) signaling pathway also enhances the transcription of an array of inflammatory genes, including the iNOS gene. Thus, the objective of this investigation was to determine the role of the MAPKs and of NF-κB in triggering iNOS and NO· expression in macrophages costimulated with IFN-γ and ManLAM.
Although IFN-γ is a necessary costimulus in iNOS expression, its signaling pathway and the transcriptional elements that control iNOS expression are well characterized (38, 48, 49, 63). The response to IFN-γ has been shown to be localized between positions −913 and −1029 of the 5′ flanking region of the iNOS promoter. This region contains a cluster of motifs characteristic of IFN-γ-responsive genes, including the IFN-γ-activated sequence (GAS) and two IFN-γ-stimulated response elements that bind to transcription factors Stat1α and IRF-1. Because the IFN-γ signaling pathway has been elucidated in regard to iNOS induction, we restricted our study to the MAPK and NF-κB signaling pathways that are activated by ManLAM.
MATERIALS AND METHODS
Materials.
RAW 264.7γNO(−) macrophages were used for all of the studies (51). Unlike the parent RAW 264.7 line (ATCC TIB-71; American Type Culture Collection, Manassas, Va.), RAW 264.7γNO(−) cells do not produce NO· with IFN-γ stimulation alone. ManLAM was produced as previously described (21). To remove any potential LPS contamination, ManLAM preparations were passed through a Detoxi-Gel column using sterile pyrogen-free water, stored in pyrogen-free vials, and reconstituted with sterile pyrogen-free phosphate buffer solution. Evaluation of bacterial endotoxin was done with the amebocyte lysate assay (E. TOXATE kit; Sigma). All plasmids used were isolated by an endotoxin-free plasmid isolation kit (Qiagen, Valencia, Calif.). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Atlanta, Ga.) and routinely tested for LPS contamination; LPS levels were consistently <0.005 ng/ml. Glutathione-Sepharose beads were purchased from Pharmacia (Piscataway, N.J.). Enhanced chemiluminescence assay kits were obtained from Amersham Life Sciences (Arlington Heights, Ill.). Recombinant c-Jun1–79–glutathione S-transferase (GST) was kindly provided by Gary Johnson (University of Colorado Health Sciences Center, Denver, Colo.). The dominant-negative (DN) MKK4 mutant (K116R) in an LNCX expression vector and the null IκBα in a pCMV5 expression vector were gifts from Lynn Heasley (University of Colorado Health Sciences Center). DN-MKK7 (K63R) in a pCMV5 expression vector was generously provided by Hiroshi Itoh (Tokyo Institute of Technology, Tokyo, Japan). Rabbit polyclonal anti-p46 JNK, rabbit polyclonal anti-p42 ERK2, rabbit polyclonal anti-p38mapk (C-20), and mouse monoclonal phospho-specific p46 and p54 JNK antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, Calif.). Mouse IFN-γ was obtained from R & D Systems Inc. (Minneapolis, Minn.). The rabbit anti-iNOS polyclonal antibody was purchased from Alexis Biochemicals (San Diego, Calif.). [γ-32P]ATP (>3,000 Ci/mmol) was purchased from NEN Research Products DuPont (Wilmington, Del.). MEK1 inhibitor PD98059 and the phospho-specific p38mapk antibody were purchased from New England Biolabs (Beverly, Mass.). p38mapk inhibitor SB203580 was purchased from Calbiochem (San Diego, Calif.). BAY 11-7082, an inhibitor of IκBα kinase, was obtained from Biomol Research Laboratories (Plymouth Meeting, Pa.). The full-length iNOS promoter cloned into the pGL2 Basic luciferase reporter gene vector was generously provided by Charles Lowenstein (Johns Hopkins University School of Medicine, Baltimore, Md.) and Robert Scheinman (University of Colorado Health Sciences Center). The phospho-specific ERK rabbit polyclonal antibody and the firefly luciferase reporter assay system were purchased from Promega (Madison, Wis.). The LipofectAMINE reagent used for the transfection experiments was purchased from Gibco BRL (Gaithersburg. Md.). All other reagents were of the highest purity.
Analysis of NO2− accumulation.
The accumulation of NO2− in culture supernatants was quantified using the method described by Ding et al. (27). Briefly, the macrophage monolayers were stimulated with ManLAM (10 μg/ml) plus IFN-γ (10 U/ml) or were coincubated with PD98059 (0.1 to 30 μM) or SB203580 (0.1 to 30 μM) for 18 h. One hundred-microliter aliquots of the culture supernatants were dispensed in duplicate into 96-well plates and were mixed with 100 μl of Greiss reagent composed of 1% (wt/vol) sulfanilamide, 0.1% (wt/vol) naphthylethylenediamine hydrochloride, and 2.5% (vol/vol) H3PO4. A standard curve based on 0.1 to 5.0 nmol of NaNO2 per 100-μl sample was prepared in RPMI growth medium. After incubation at room temperature for 10 min, the absorbances of the wells were quantified at 550 nm in a Biotek Instruments enzyme-linked immunosorbent assay plate reader. The number of cells per well was determined by lysing the cell monolayers in Zapoglobin and quantifying the number of released nuclei with a model ZM Coulter counter. The concentration of NO2− was interpolated from the NaNO2 standard curve, corrected for the volume of the culture supernatant, and normalized to the number of cells per well. Results are presented as nanomoles of NO2− per 106 adherent cells. Each experiment was conducted in triplicate and was conducted a minimum of three times. The results presented are the means ± standard deviations (SD) of three experiments.
Determination of JNK/SAPK activity.
For measurement of JNK activity, the RAW 264.7γNO(−) cell monolayers were lysed at 4°C with 500 μl of ice-cold lysis buffer (50 mM Tris-HCl, pH 8.0, containing 137 mM NaCl, 10% [vol/vol] glycerol, 1% [vol/vol] Nonidet P-40, 1 mM NaF, 10 μg of leupeptin/ml, 10 μg of aprotinin/ml, 2 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride) (36). After the protein content was normalized between samples, JNK/SAPK in each sample of lysate was bound to 15 μl of a 1:1 mixture of slurry of lysis buffer and GST–c-Jun1–79–Sepharose beads and incubated at 4°C for 2 h. The beads were then washed twice with 500 μl of lysis buffer and twice with 500 μl of JNK buffer (20 mM HEPES buffer, pH 7.2, containing 30 mM β-glycerophosphate, 10 mM p-nitrophenylphosphate, 10 mM MgCl2, 0.5 mM dithiothreitol [DTT], and 50 μM Na3VO4). The activity of JNK was detected by phosphorylation of c-Jun–GST in an in vitro kinase assay and was assessed by measuring the incorporation of [γ-32P]ATP (10 μCi/sample) in JNK buffer incubated at 30°C for 30 min. The kinase reactions were then stopped with an equal volume of 2× Laemmli sample buffer containing 20 mM DTT and boiled for 3 min. The proteins present in the supernatants were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) through a 12% polyacrylamide gel and transferred onto nitrocellulose membranes. 32P-labeled c-Jun–GST was detected by autoradiography.
Western blot analysis.
After nucleus-free lysates were normalized for protein content, the samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes as described previously (68). The blots were then washed in Tris-Tween-buffered saline (TTBS; 20 mM Tris-HCl buffer, pH 7.6, containing 137 mM NaCl and 0.05% [vol/vol] Tween 20), blocked overnight with 5% (wt/vol) nonfat dry milk, and probed according to the method described by Towbin et al. (68) with a polyclonal iNOS antibody or with phospho-specific antibodies to p42 and p44 ERK and p38mapk antibodies in 5% (wt/vol) bovine serum albumin dissolved in TTBS. Using a horseradish peroxidase-conjugated secondary antirabbit or antimouse antibody, bound antibodies were detected by enhanced chemiluminescence. To determine equal loading of proteins among samples, the corresponding membranes were probed with rabbit polyclonal p42 and p44 ERK and p38mapk antibodies.
Transient transfection and luciferase assay.
RAW 264.7γNO(−) cells were plated at a density of 106 cells per six-well plate in RPMI 164O containing penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% (vol/vol) heat-inactivated FBS. After 24 h of growth to ∼30 to 40% confluence, the cells were transfected with plasmids using LipofectAMINE as described by the manufacturer's protocol (Gibco BRL) and as previously described (16). Briefly, 0.3 μg of iNOS promoter-luciferase plasmid was combined with 2 μg of DN-MKK4 plasmid, 2 μg of DN-MKK7 plasmid, or 1 μg of null IκBα plasmid, along with 10 μl of LipofectAMINE reagent, and 100 μl of Optimem serum-free medium. To normalize for the amount of DNA transfected, equivalent amounts of LNCX or pCMV5 empty expression vector were cotransfected as controls with the iNOS-luciferase reporter. The LipofectAMINE-DNA mixture was incubated for 30 min at room temperature. Each well was then washed with 2 ml of Optimem serum-free medium and replaced with 1 ml of the LipofectAMINE-DNA mixture. After 5 h of incubation, 1 ml of RPMI 1640 containing 20% (vol/vol) FBS and 1% penicillin–streptomycin–l-glutamine was added to each well. The media were changed 24 h after transfection, and, after an additional 48 h, the cells were stimulated with IFN-γ (10 U/ml) plus ManLAM (10 μg/ml) for 8 h. The cells were then washed with phosphate-buffered saline, lysed in a luciferase lysis buffer, and assayed for luciferase activity according to the manufacturer's instructions (Promega Inc.). The amount of luciferase activity was normalized to protein concentration and reported as the fold increase in activity.
Statistical analysis.
Replicate experiments were independent, and summary results were presented as means ± SD. Differences were considered significant for P values of <0.05. Group means were compared by repeated-measures analysis of variance using Fisher's least significant difference.
RESULTS
IFN-γ synergizes with ManLAM in the induction of iNOS and NO·.
The parental RAW 264.7 macrophage strain expresses NO· in response to IFN-γ stimulation alone, making it difficult to discern the contribution of ManLAM to costimulation; in addition, the use of such cells would pose a conundrum when the hypothesized ManLAM-induced MAPK and NF-κB signaling pathways are studied. To remove this obstacle, we used a subclone of RAW 264.7 cells, designated RAW 264.7γNO(−), that does not respond to IFN-γ alone with respect to iNOS induction (51). However, it is important to emphasize that IFN-γ is still required as a costimulus for induction of NO·.
To determine the ability of ManLAM to induce NO· production, RAW 264.7γNO(−) macrophages were stimulated with IFN-γ (10 U/ml) or ManLAM (10 μg/ml) or were costimulated with IFN-γ (10 U/ml) plus incremental concentrations of ManLAM (0.1 to 10 μg/ml) for 18 h. As can be seen in Fig. 1A, neither IFN-γ (10 U/ml) nor ManLAM (10 μg/ml) alone was capable of inducing NO· expression as measured by the assay for cumulative NO2−, a metabolic product of NO·, in the culture supernatant. However, in cells costimulated with both IFN-γ and ManLAM, there was a significant increase in NO2− accumulation, beginning at a ManLAM concentration between 1 and 5 μg/ml, equivalent to 0.05 to 0.25 μM, respectively. To determine whether the iNOS protein was also being induced, macrophages were treated with IFN-γ (10 U/ml), ManLAM (10 μg/ml), or both stimuli for 18 h followed by cell lysis. After being normalized for protein content, nucleus free lysates were separated by SDS-PAGE and immunoblotted with a rabbit polyclonal anti-iNOS antibody. As shown in Fig. 1B, compared to unstimulated cells (lane 1) or cells stimulated with either ManLAM (lane 2) or IFN-γ (lane 3) alone, treatment with IFN-γ plus ManLAM (lane 4) substantially augmented iNOS protein expression. Because IFN-γ and/or ManLAM can also induce the expression of TNF-α and IL-1β (75), perhaps the expression of iNOS and NO· stimulated by IFN-γ plus ManLAM was secondary to the effects of these cytokines. Thus, we performed neutralization experiments in which IFN-γ and ManLAM were coincubated with an anti-TNF-α antibody (5 μg/ml) or an anti-IL-1β antibody (5 μg/ml) and found that there was no effect of the neutralizing antibodies on NO2− expression (data not shown). Thus, IFN-γ–ManLAM costimulation is capable of inducing NO2− expression independent of TNF-α and IL-1β.
FIG. 1.

IFN-γ synergizes with ManLAM in iNOS and NO2− expression. (A) RAW 264.7γNO(−) cells were stimulated with IFN-γ (10 U/ml), ManLAM (10 μg/ml), or combined IFN-γ (10 U/ml) and ManLAM (0.01 to 10 μg/ml) for 18 h, followed by the Greiss reagent assay for NO2− in the supernatant. Data shown are the means ± SD of three independent experiments. (B) Macrophages were stimulated with IFN-γ (10 U/ml), ManLAM (10 μg/ml), or both for 18 h, and nucleus-free lysates were separated by SDS-PAGE and immunoblotted with a polyclonal iNOS antibody. The immunoblot shown is representative of three independent experiments.
A legitimate concern regarding the effects of ManLAM is the possibility of LPS contamination. We have taken great measures to ensure that ManLAM, fetal calf serum (FCS), transfected plasmids, and other reagents are LPS free by routinely testing for any contamination. In addition, the RAW 264.7γNO(−) cells used do not respond to IFN-γ alone in regard to NO· expression, thus providing an internal control for each weekly experiment. We had previously shown that the peak activation of the MAPKs with LPS stimulation was at ∼1 h (13) whereas peak activation of the MAPKs by ManLAM occurred at ∼30 min. Indirectly, this observation suggests that the effects of ManLAM are not due to LPS contamination. However, we have performed the following experiments to further confirm this (see Fig. 2). First, we stimulated the RAW 264.7γNO(−) cells with 10 U of IFN-γ/ml plus an amount of LPS (0.009 ng/ml) that is contained in 10 μg of ManLAM/ml and found that there was no NO2− produced (data not shown). Second, because binding by LPS to its receptor requires the LPS-binding protein present in serum, we compared the abilities of ManLAM–IFN-γ and LPS–IFN-γ to induce NO2− expression in 10 and 0.1% FCS. As expected, there was substantial reduction of LPS–IFN-γ-induced NO2− production in serum-deprived (0.1% FCS) conditions (data not shown). In contrast, there was abundant expression of NO2− with ManLAM–IFN-γ stimulation in the presence of either 10 or 0.1% FCS (data not shown). This provides further evidence that the effects of ManLAM are not due to LPS.
FIG. 2.

Activation of ERK, p38mapk, and JNK signaling pathways with ManLAM stimulation. (A) RAW 264.7γNO(−) macrophages were stimulated with 5 and 10 μg of ManLAM/ml for 30 min, followed by immunoblotting of nucleus-free whole-cell lysate with phospho-specific antibody to ERK (p-p42 and p-p44). (B) The cells were treated as for panel A, and the separated proteins were immunoblotted with phospho-specific p38mapk antibody (p-p38). Western blots with anti-ERK1/2 (A, p44 and p42) and anti-p38mapk (B, p38) antibodies are also shown. (C) JNK activity was determined by an in vitro kinase assay on nucleus-free lysates using c-Jun–GST as the substrate. Data shown are representative of three independent experiments.
MAPKs are activated by ManLAM.
MAPK signaling molecules are known to mediate the expression of various gene products including iNOS. Although Knutson et al. (41) showed that prolonged pretreatment (16 h) of THP-1 monocytes with ManLAM inhibited activation of p42 and p44 ERK by a second stimulus with phorbol ester, to the best of our knowledge, the ability of ManLAM to directly activate the MAPKs has not been previously reported. Thus, to begin to investigate whether MAPKs play any role in the induction of iNOS-NO· by IFN-γ plus ManLAM, we first determined the effects of ManLAM on MAPK activation. A prerequisite for MAPK activation is the dual phosphorylation of Thr and Tyr residues on a tripeptide motif that is specific for each MAPK (Thr-Glu-Tyr for ERK, Thr-Gly-Tyr for p38mapk, and Thr-Pro-Tyr for JNK). Therefore, to measure ManLAM-induced ERK and p38mapk phosphorylation, RAW 264.7γNO(−) cells were treated with ManLAM (5 and 10 μg/ml) and the separated nucleus-free lysates were immunoblotted with phospho-specific antibodies to ERK or p38mapk. For determination of JNK activation, a solid-phase in vitro kinase assay with c-Jun–GST–Sepharose beads as the substrate was performed on lysates prepared from cells similarly treated (15, 36). As shown in Fig. 2A (top), ManLAM at 5 and 10 μg/ml strongly induced p42 and p44 ERK phosphorylation (p-p42 and p-p44) in comparison to that for unstimulated cells. In contrast, there was little or no detectable p38mapk phosphorylation at 5 μg/ml but a small increase over that for unstimulated cells at 10 μg of ManLAM/ml (Fig. 2B, top). There was equal loading of samples as shown by reprobing the corresponding immunoblots with anti-ERK and anti-p38mapk antibodies, which revealed bands that represent total ERK and p38mapk (Fig. 2A and B, respectively, bottom). An increase in JNK activity was also observed in macrophages stimulated with 5 and 10 μg of ManLAM/ml (Fig. 2C). Peak phosphorylation or activation of all three MAPKs by ManLAM occurred after 30 min of stimulation (data not shown). This time of peak activation was later than that observed with TNF-α stimulation in bone marrow-derived mouse macrophages, where peak activation of the MAPKs occurred after ∼10 min of stimulation (16), and earlier than that seen with LPS stimulation in RAW264.7γNO(−)cells, in which peak activation occurred only after ∼1 h of stimulation (13). Thus, in RAW 264.7γNO(−) cells, ManLAM activates ERK, p38mapk, and JNK in a time- and concentration-dependent fashion. Stimulation of mouse macrophages with IFN-γ alone does not increase any of the MAPK activities over unstimulated levels (16), and there was no additional increase in MAPK activation with IFN-γ–ManLAM stimulation compared to that produced by ManLAM alone (E. D. Chan and D. W. H. Riches, unpublished data).
IFN-γ–ManLAM induction of iNOS and NO· is dependent on the ERK pathway.
To determine the role of the ERK pathway in the regulation of iNOS and NO· expression, we utilized a well-established pharmacologic inhibitor (PD98059) of MEK1, the upstream kinase of p42 and p44 ERK. RAW 264.7γNO(−) macrophages were pretreated with 0.1 to 30 μM PD98059 or with vehicle dimethyl sulfoxide (DMSO) (0.075%) for 1 h, followed by coincubation with 10 U of IFN-γ and 10 μg of ManLAM/ml for 18 h. As can be seen in Fig. 3A, PD98059 strongly inhibited NO2− accumulation in a concentration-dependent fashion, beginning at ∼1 μM PD98059 (P < 0.001). There was approximately an 80% inhibition of NO2− accumulation with 10 μM PD98059 and 100% inhibition with 30 μM. To determine whether this inhibition also occurred at the level of iNOS protein expression, RAW 264.7γNO(−) macrophages were similarly treated with IFN-γ plus ManLAM with and without PD98059 for 18 h and nucleus-free whole-cell lysates were separated by SDS-PAGE and immunoblotted with iNOS polyclonal antibody. As shown in Fig. 3B, inhibition of MEK1-ERK inhibited iNOS protein expression beginning at ∼1 μM PD98059 and was nearly complete at 30 μM. Vehicle DMSO, in an amount equivalent to that contained in 30 μM PD98059 (0.075%), had no effect on either NO2− or iNOS expression (Fig. 3). As previously shown, PD98059 does not inhibit p38mapk or JNK activation (3, 13, 29).
FIG. 3.

Inhibition of MEK1-ERK by PD98059 inhibits iNOS and NO2− expression by IFN-γ plus ManLAM. (A) RAW 264.7γNO(−) macrophages were pretreated with various concentrations of PD98059 (0.1 to 30 μM) or with vehicle DMSO (D) at a concentration of 0.075% for 1 h and then costimulated with IFN-γ (10 U/ml) plus ManLAM (10 μg/ml) for 18 h, followed by the Greiss reagent assay for NO2−. Data shown are the means ± SD of three independent experiments. ∗∗∗, P < 0.001 (versus second bar from left). (B) Macrophages were similarly stimulated with IFN-γ plus ManLAM with or without PD98059 for 18 h, followed by immunoblotting of nucleus-free lysates with iNOS polyclonal antibody. The immunoblot shown is representative of three independent experiments.
IFN-γ–ManLAM induction of iNOS and NO· is inhibited by p38mapk.
To determine the role of p38mapk in the induction of iNOS and NO· by IFN-γ–ManLAM stimulation, RAW 264.7γNO(−) cells were stimulated in the presence of SB203580, a pyridinyl-imidazole derivative that is a specific inhibitor of p38mapk (42). Macrophages were pretreated with SB203580 at 30 μM for 1 h and then costimulated with either IFN-γ (10 U/ml) alone or 10 U of IFN-γ/ml plus 10 μg of ManLAM/ml for an additional 18 h. As demonstrated in Fig. 4A, stimulation with IFN-γ plus ManLAM upregulated NO2− production (third bar from left). Inhibition of p38mapk by SB203580 further augmented IFN-γ–ManLAM-stimulated NO2− accumulation in a consistent manner (right bar; P < 0.001). To determine if SB203580 was inherently capable of inducing NO2− production, macrophages were stimulated with combined 30 μM SB203580 and 10 U of IFN-γ/ml. As can be seen in Fig. 4A (second bar from left), there was no increase in NO2− production over that for unstimulated cells. As shown in Fig. 4B, a similar increase in iNOS protein expression was observed with p38mapk inhibition compared to that for IFN-γ–ManLAM stimulation alone (compare lanes 4 and 3). Lesser but significant augmentations of iNOS and NO2− were observed with 10 and 1 μM SB203580 (data not shown). As was seen with NO2− production, IFN-γ plus SB203580 did not induce iNOS protein expression. These results suggest that the activation of p38mapk may represent a signaling pathway that inhibits IFN-γ–ManLAM-stimulated iNOS-NO· induction. We have also previously showed that SB203580 does not inhibit JNK or ERK activation (13, 42).
FIG. 4.

Inhibition of p38mapk with SB203580 augmented iNOS and NO2− expression stimulated by IFN-γ plus ManLAM. (A) RAW 264.7γNO(−) macrophages were cultured with media alone, IFN-γ (10 U/ml) plus SB203580 (30 μM), IFN-γ (10 U/ml) plus ManLAM (10 μg/ml), or IFN-γ plus ManLAM plus SB203580. The cells were preincubated with SB203580 for 1 h and then cocultured with the indicated stimuli. After 18 h of stimulation, the supernatant was assayed for NO2−. Data shown are the means ± SD of three independent experiments. ∗∗∗, P < 0.001 versus third bar from left. (B) Macrophages were cultured in media alone or with IFN-γ plus SB203580 or were stimulated with IFN-γ plus ManLAM with or without SB203580 for 18 h, followed by immunoblotting of nucleus-free lysate with an iNOS polyclonal antibody. The immunoblot shown is representative of three independent experiments.
DN-MKK7 but not DN-MKK4 inhibits IFN-γ–ManLAM-induced iNOS promoter activity.
JNK is activated after phosphorylation of Thr 185 and Tyr 187 residues by either MKK4 or MKK7. Because a pharmacologic inhibitor of JNK is not currently available, we investigated the role of the JNK pathway in IFN-γ–ManLAM-induced iNOS-NO· expression by determining the effects of DN-MKK4 (K116R) or DN-MKK7 (K63R) on iNOS promoter activity in a reporter assay in which the luciferase gene is cloned downstream of the entire 5′ flanking region of the mouse iNOS gene. These DN-MKKs are able to bind but are unable to phosphorylate their substrate JNK and thus are competitive inhibitors of endogenous MKK4 and MKK7. RAW 264.7γNO(−) cells were cotransfected with 0.3 μg of the iNOS promoter-luciferase (iNOS-luc) plasmid and either 2 μg of the DN-MKK7 plasmid or 2 μg of empty expression vector pCMV5. After transfection and growth for 72 h, the cells were stimulated with 10 U of IFN-γ/ml plus 10 μg of ManLAM/ml for 8 h, followed by measurement of luciferase activity in cell lysates and normalization for protein content. As shown in Fig. 5A, transfection of iNOS-luc and empty vector pCMV5 followed by IFN-γ–ManLAM stimulation resulted in a nearly ninefold induction of luciferase activity compared with that for unstimulated cells (second bar from left versus left bar). Transfection of DN-MKK7 significantly inhibited IFN-γ–ManLAM-induced iNOS promoter activity as measured by the luciferase assay (Fig. 5A, fourth bar from left versus second bar from left; P < 0.01). Similarly, RAW 264.7γNO(−) cells were also cotransfected with the iNOS-luc plasmid and either 2 μg of the DN-MKK4 plasmid or 2 μg of empty vector LNCX, followed by stimulation with IFN-γ and ManLAM. In cells cotransfected with the iNOS-luc plasmid and expression vector LNCX, there was nearly a fourfold induction of iNOS promoter activity with combined IFN-γ–ManLAM stimulation compared to that for unstimulated cells (Fig. 5B, second bar from left versus left bar). In contrast to the inhibitory effect observed with DN-MKK7, transfection of DN-MKK4 had no significant effect on IFN-γ–ManLAM-induced iNOS promoter activation (Fig. 5B, right bar versus second bar from left; P > 0.05). Transfection of both DN-MKK4 and DN-MKK7 was not significantly different than transfection of DN-MKK7 alone (Fig. 5A, right bar versus fourth bar from left; P > 0.05). We also examined the effects of DN-MEK kinase 1 (DN-MEKK1), which we had previously found to inhibit IFN-γ–TNF-α induction of iNOS promoter activity in NIH3T3 cells (16). In contrast to what was found in our previous study, DN-MEKK1 had no inhibitory effect on iNOS promoter activation by IFN-γ plus ManLAM (data not shown).
FIG. 5.
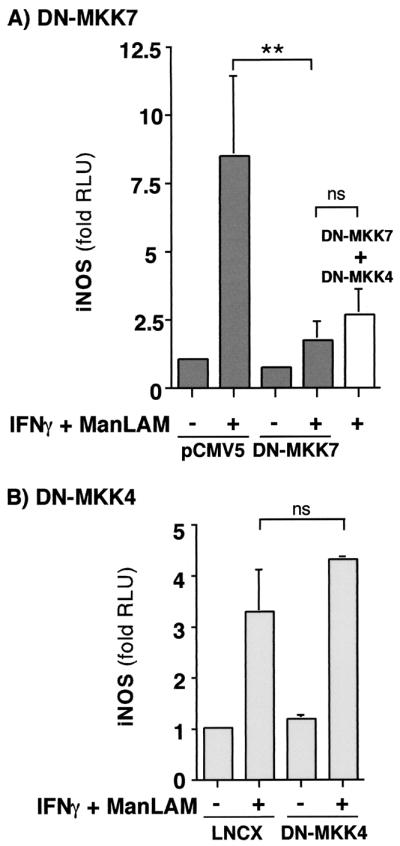
The effects of DN-MKK7 and DN-MKK4 on IFN-γ–ManLAM-induced iNOS-luc activity. (A) RAW 264.7γNO(−) macrophages were cotransfected with 0.3 μg of iNOS-luc plasmid and 2 μg of DN-MKK7 plasmid or 2 μg of pCMV5 empty expression vector, followed by stimulation with IFN-γ (10 U/ml) plus ManLAM (10 μg/ml). After 8 h of stimulation, the cells were lysed and the nucleus-free lysates were then measured for luciferase activity. The results are reported as fold increases in relative light units (fold RLU) and are normalized for protein concentration. Data shown are the means ± SD of three independent experiments. ∗∗, P < 0.01. (B) Macrophages were cotransfected with 0.3 μg of iNOS-luc plasmid and 2 μg of DN-MKK4 plasmid or 2 μg of LNCX empty vector, followed by IFN-γ–ManLAM costimulation. After 8 h of stimulation, the cells were lysed and the nucleus-free lysates were then measured for luciferase activity. The results are normalized for protein concentration. Data shown are the means ± SD of three independent experiments, ns, not significant.
IFN-γ–ManLAM induction of NO2− is dependent on NF-κB activation.
Transcription factor NF-κB has been shown to enhance the expression of a number of proteins that mediate inflammatory responses, including TNF-α, IL-8, E-selection, and iNOS (10, 51, 73). The 5′ flanking region of the iNOS promoter contains two canonical NF-κB-binding sites, both of which are required for maximal induction of iNOS with IFN-γ–LPS stimulation. Brown and Taffet (12) previously showed that ManLAM of M. tuberculosis was capable of activating NF-κB. We have also confirmed these findings by showing that ManLAM was capable of activating a secretory alkaline phosphatase reporter gene that is cloned downstream of four NF-κB-binding cis elements (E. D. Chan and K. R. Morris, unpublished data). Thus, NF-κB is a plausible transcription factor that enhances iNOS induction by ManLAM. To determine the role of NF-κB, we utilized a novel low-molecular-weight compound (BAY 11-7082) shown to specifically inhibit NF-κB activation by inhibiting the phosphorylation and the subsequent degradation of IκBα, the endogenous inhibitor of NF-κB (58). Pierce and coworkers (58) previously demonstrated that BAY 11-7082 did not inhibit ERK, JNK, or p38mapk activation. We pretreated RAW 264.7γNO(−) macrophages with 1 to 10 μM BAY 11-7082 for 1 h as previously described (58) and then costimulated the cells with 10 U of IFN-γ/ml plus 10 μg of ManLAM/ml for 18 h, followed by the Greiss reagent assay for NO2− accumulation. As shown in Fig. 6, inhibition of NF-κB activation significantly inhibited NO2− expression beginning at a concentration between 1 and 5 μM. Vehicle DMSO, used at a concentration equivalent to 10 μM BAY 11-7082 (0.02%), had no effect.
FIG. 6.

The effects of NF-κB inhibition on NO2− expression. Macrophages were pretreated with BAY 11-7082 (1 to 10 μM) or DMSO (0.02%) for 1 h and then costimulated with IFN-γ (10 U/ml) plus ManLAM (10 μg/ml) for 18 h, followed by NO2− assay. Data shown are the means ± SD of three independent experiments.
The effect of mutant (null) IκBα on iNOS promoter activity.
To further confirm that NF-κB enhanced iNOS promoter activity upon IFN-γ–ManLAM stimulation, we determined the effects of a mutant (null) IκBα on the iNOS-luc transient transfection assay. The null IκBα plasmid encodes a mutant IκBα protein in which the serine 32 and 36 phosphorylation sites are deleted. Because null IκBα cannot be phosphorylated by IκBα or -β kinase, it cannot be ubiquitinated and degraded. As a consequence, null IκBα is an even more potent inhibitor of NF-κB than endogenous IκBα by virtue of its ability to remain bound to NF-κB. RAW 264.7γNO(−) cells were cotransfected with 0.3 μg of the iNOS-luc plasmid and either 1 μg of the null IκBα plasmid or 1 μg of empty vector pCMV5, followed by costimulation with 10 U of IFN-γ/ml plus 10 μg of ManLAM/ml. As shown in Fig. 7, in cells cotransfected with the iNOS-luc plasmid and pCMV5, there was an ∼10-fold induction of iNOS promoter activity with IFN-γ–ManLAM stimulation (second bar from left versus left bar). By contrast, in cells transfected with null IκBα plasmid, there was a significant reduction in iNOS promoter activity (right bar versus second bar from left; P < 0.05).
FIG. 7.
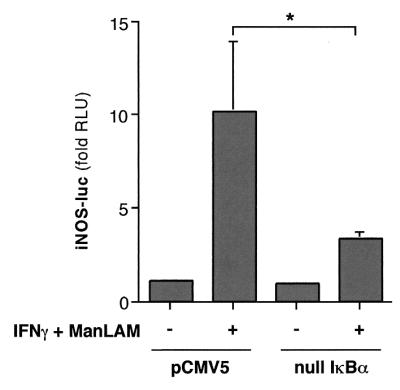
The effects of a mutant (null) IκBα on IFN-γ–ManLAM-induced iNOS promoter activity. RAW 264.7γNO(−) macrophages were cotransfected with 0.3 μg of iNOS-luc plasmid and 1 μg of null IκBα plasmid or 1 μg of pCMV5 vector, followed by stimulation with IFN-γ (10 U/ml) plus ManLAM (10 μg/ml). Nucleus-free lysates were then measured for luciferase activity after 8 h of stimulation. The results are reported as fold increases in relative light units (fold RLU) and are normalized for protein concentration. Data shown are the means ± SD of three independent experiments. ∗, P < 0.05.
DISCUSSION
In this study, we demonstrated that (i) ManLAM, in conjunction with IFN-γ, induces iNOS-NO· expression and (ii) MAPK and NF-κB signaling pathways modulate this induction. Using specific pharmacologic inhibitors, we showed that both the MEK1-ERK and IKK–NF-κB pathways augmented IFN-γ–ManLAM induction of NO2− accumulation by increasing iNOS protein expression in mouse macrophages. We confirmed the important role of NF-κB by showing that overexpression of null IκBα, a potent inhibitor of NF-κB activation, inhibited iNOS-promoter activation by IFN-γ plus ManLAM. Similarly, using DN mutant proteins of the JNK pathway, we showed that IFN-γ–ManLAM induction of iNOS was also dependent on the MKK7-JNK pathway but was independent of MKK4. By contrast, p38mapk inhibited IFN-γ–ManLAM induction of both iNOS and NO· expression by the RAW 264.7γNO(−) cells (Fig. 8).
FIG. 8.

Diagram of the proposed signaling pathways that modulate ManLAM-induced iNOS expression. The required IFN-γ costimulus pathway utilizing transcription factors IRF-1 and Stat1α is also shown.
Utilization of particular signaling pathways in regulating cellular function or inducing gene expression appears to be dependent on, among other factors, the type of stimulus and cell examined. In this regard, augmenting, inhibitory, and neutral roles for the MAPKs have been reported in signaling gene expression, including that for iNOS. For example, both ERK1 and -2 were shown to be necessary for iNOS induction by IL-1β (32) or IL-1β plus IFN-γ (67) or by infection with Leishmania donovani (53). In contrast, ERK played a neutral role in iNOS induction by either TNF-α plus IL-1α in astrocytes (24), IFN-γ plus LPS in glioma cells (56), or IFN-γ plus TNF-α in bone marrow-derived mouse macrophages (16). Furthermore, Lindroos and coworkers (45) showed that ERK2 inhibited IL-1β-mediated induction of α-platelet-derived growth factor receptor expression in rat pulmonary myofibroblasts. We previously demonstrated that the MKK4-JNK pathway was involved in IFN-γ–TNF-α induction of iNOS in NIH 3T3 fibroblasts (16), whereas in the present study this signaling cascade was not involved with IFN-γ–ManLAM stimulation. Instead, we showed that MKK7-JNK regulated IFN-γ–ManLAM induction of iNOS. JNK has also been shown to regulate IL-1β induction of iNOS in an insulin-producing cell line (70). JNK and ERK are able to independently and synergistically activate or increase the expression of a number of transcription factors including c-Fos, c-Jun, NF-κB, activating transcription factor 2, the Ets family, serum response factor, and CREB. Although a possible mechanism by which ERK or JNK contributes to IFN-γ–ManLAM induction of iNOS is via signaling of ManLAM induction of expression of TNF-α or IL-1β, cytokines known to also induce iNOS expression (62), coincubation of IFN-γ and ManLAM with neutralizing antibodies to either TNF-α or IL-1β had no effect on NO2− expression. Moreover, the possibility that the effects of IFN-γ plus ManLAM are secondary to either TNF-α or IL-1β is even less compelling in RAW 264.7γNO(−) macrophages because these cells are poorly responsive to TNF-α and IL-1β (28, 40).
Similar to our findings, Guan and colleagues (35) demonstrated that p38mapk inhibited IL-1β-mediated iNOS induction in mesangial cells, although the precise mechanism for this inhibition is not known. Recently, Alpert and colleagues (4) also found an inhibitory role for p38mapk by showing that the MKK6-p38mapk signaling pathway inhibited TNF-α-induced NF-κB activation. Although the mechanism by which p38mapk inhibits IFN-γ–ManLAM-induced iNOS and NO2− expression appears complex and remains to be elucidated, we have found it not to be due to any inhibition of TNF-α expression by p38mapk. On the contrary, in macrophages treated with SB203580, there was a further inhibition of TNF-α expression, suggesting that p38mapk serves to enhance IFN-γ–ManLAM-stimulated TNF-α protein expression (E. D. Chan and K. R. Morris, unpublished data), consistent with previous work showing that p38mapk may enhance the expression of TNF-α (42).
We and others (12) have found that ManLAM from M. tuberculosis is capable of activating NF-κB. Using a specific inhibitor of IKK, we showed that IFN-γ–ManLAM production of NO2− was also dependent on NF-κB. We have corroborated this finding by showing that transient transfection of null IκBα, which inhibits NF-κB activation by sequestering the latter as a null IκBα–NF-κB binary complex in the cytoplasm, significantly inhibited iNOS promoter activation by IFN-γ–ManLAM. Two canonical NF-κB-binding sites have been identified on the 5′ flanking region of the iNOS promoter, one located at −76 to −85 from the transcriptional start site and the other at −962 to −971. Both sites have been shown to be required for maximal induction of iNOS by LPS. Whether one or both of these cis-acting elements for NF-κB are required by ManLAM remains to be elucidated.
Although we have shown a role for the MAPKs and NF-κB in upregulation of murine iNOS by IFN-γ plus ManLAM, in vivo induction by M. tuberculosis is likely to be considerably more complex. This possibility is due to the fact that an array of other mycobacterial products, such as phospholipase C, lipomannan (LM), dimannosylated phosphatidylinositides (PIM2), mycolyl arabinogalactan-peptidoglycan complex, and lipoproteins contained in the whole organisms also have the potential to induce iNOS expression directly or indirectly. For example, Brightbill and colleagues (11) recently showed that a 19-kDa lipoprotein of M. tuberculosis was also capable of inducing iNOS in RAW 264.7 cells. Barnes et al. (7) also showed that LM and PIM2, simpler derivatives of LAM, were capable of inducing TNF-α, IL-1β, IL-6, IL-8, and IL-10. We have also found that PIM2 induced iNOS and NO2− expression with IFN-γ costimulation (E. D. Chan and K. R. Morris, unpublished data). In addition, Sikora and coworkers (66) demonstrated that, in an autocrine and paracrine fashion, extracellular nucleotides such as ATP released from TB-infected macrophages may also induce iNOS upregulation and NO· production via binding to P2 purinergic receptors present on macrophage cell surfaces.
In summary, we showed that IFN-γ synergized with ManLAM to induce iNOS expression and subsequent NO· production. In addition, the study has shed new light on the signaling pathways utilized by ManLAM in the induction of iNOS and NO·. Our findings support the role of the MEK1-ERK, MKK7-JNK, and IKK-NFκB signaling pathways in ManLAM induction of iNOS and NO· in cells costimulated with IFN-γ and a negative regulatory role for p38mapk. Future directions should examine these signaling pathways in macrophages infected with whole TB organisms, and, if the pathways are found to be significant, a targeted investigation of the role they play in host defense against M. tuberculosis in infected animals should be performed.
ACKNOWLEDGMENTS
Edward D. Chan was supported by Clinical Investigator Development Award 1K08HL03625-01, the Lowerre Foundation for Mycobacteriology Research Award, the Parke-Davis Atorvastatin Research Award, and the Giles F. Filley Memorial Award. John T. Belisle and Patrick J. Brennan were supported by NIH NO1 AI-75320. David W. H. Riches was supported by NIH HL55549 and SCOR HL 56556.
We thank Gary Johnson for the c-Jun–GST construct, Lynn Heasley for the null IκBα, DN-MKK4, and LNCX constructs, Hiroshi Itoh for the DN-MKK7 and pCMV5 constructs, and William Murphy, Charles Lowenstein, and Robert Scheinman for the iNOS-luc constructs and helpful discussions. We are also grateful to Boyd Jacobson, Barry Silverstein, and Nadia de Steckelberg for help with the illustration.
REFERENCES
- 1.Adams L B, Dinauer M C, Morgenstern D E, Krahenbuhl J L. Comparison of the roles of reactive oxygen and nitrogen intermediates in the host response to Mycobacterium tuberculosis using transgenic mice. Tuberc Lung Dis. 1997;78:237–246. doi: 10.1016/s0962-8479(97)90004-6. [DOI] [PubMed] [Google Scholar]
- 2.Adams L B, Fukutomi Y, Krahenbuhl J L. Regulation of murine macrophage effector functions by lipoarabinomannan from mycobacterial strains with different degrees of virulence. Infect Immun. 1993;61:4173–4181. doi: 10.1128/iai.61.10.4173-4181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alessi D R, Cuenda A, Cohen P, Dudley D T, Saltiel A R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 4.Alpert D, Schwenger P, Han J, Vilcek J. Cell stress and MKK6b-mediated p38 MAP kinase activation inhibit tumor necrosis factor-induced IκB phosphorylation and NF-κB activation. J Biol Chem. 1999;274:22176–22183. doi: 10.1074/jbc.274.32.22176. [DOI] [PubMed] [Google Scholar]
- 5.Arias M, Rojas M, Zabaleta J, Rodriguez J I, Paris S C, Barrera L F, Garcia L F. Inhibition of virulent Mycobacterium tuberculosis by Bcg (r) and Bcg (s) macrophages correlates with nitric oxide production. J Infect Dis. 1997;176:1552–1558. doi: 10.1086/514154. [DOI] [PubMed] [Google Scholar]
- 6.Aston C, Rom W N, Talbot A T, Reibman J. Early inhibition of mycobacterial growth by human alveolar macrophages is not due to nitric oxide. Am J Respir Crit Care Med. 1998;157:1943–1950. doi: 10.1164/ajrccm.157.6.9705028. [DOI] [PubMed] [Google Scholar]
- 7.Barnes P F, Chatterjee D, Abrams J S, Lu S, Wang E, Yamamura M, Brennan P J, Modlin R L. Cytokine production induced by Mycobacterium tuberculosis lipoarabinomannan. J Immunol. 1992;149:541–547. [PubMed] [Google Scholar]
- 8.Bertholet S, Tzeng E, Felley-Bosco E, Mauel J. Expression of the inducible NO synthase in human monocytic U937 cells allows high output nitric oxide production. J Leuk Biol. 1999;65:50–58. doi: 10.1002/jlb.65.1.50. [DOI] [PubMed] [Google Scholar]
- 9.Bhat N R, Zhang P, Lee J C, Hogan E L. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackwell T S, Christman J W. The role of nuclear factor-κB in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- 11.Brightbill H D, Libraty D H, Krutzik S R, Yang R-B, Belisle J T, Bleharski J R, Maitland M, Norgard M V, Plevy S E, Smale S T, Brennan P J, Bloom B R, Godowski P J, Modlin R L. Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science. 1999;285:732–735. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 12.Brown M C, Taffet S M. Lipoarabinomannans derived from different strains of Mycobacterium tuberculosis differentially stimulate the activation of NF-κB and KBF1 in murine macrophages. Infect Immun. 1995;63:1960–1968. doi: 10.1128/iai.63.5.1960-1968.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan E D, Riches D W H. IFNγ + LPS-induction of iNOS is modulated by ERK, JNK/SAPK, and p38mapk in a mouse macrophage cell line. Am. J; 2001. . Physiol., in press. [DOI] [PubMed] [Google Scholar]
- 14.Chan E D, Riches D W H. Potential role of the JNK/SAPK signal transduction pathway in the induction of iNOS by TNFα. Biochem Biophys Res Commun. 1998;253:790–796. doi: 10.1006/bbrc.1998.9857. [DOI] [PubMed] [Google Scholar]
- 15.Chan E D, Winston B W, Jarpe M B, Wynes M W, Riches D W H. Preferential activation of the p46 isoform of JNK/SAPK in mouse macrophages by TNFα. Proc Natl Acad Sci USA. 1997;94:13169–13174. doi: 10.1073/pnas.94.24.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan E D, Winston B W, Uh S-T, Wynes M W, Rose D M, Riches D W H. Evaluation of the role of mitogen-activated protein kinases in the expression of inducible nitric oxide synthase by IFNγ and TNFα in mouse macrophages. J Immunol. 1999;162:415–422. [PubMed] [Google Scholar]
- 17.Chan J, Fan X, Hunter S W, Brennan P J, Bloom B R. Lipoarabinomannan, a possible virulence factor involved in persistence of Mycobacterium tuberculosis within macrophages. Infect Immun. 1991;59:1755–1761. doi: 10.1128/iai.59.5.1755-1761.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan J, Tanaka K, Carroll D, Flynn J, Bloom B R. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan J, Xing Y, Magliozzo R S, Bloom B R. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med. 1992;175:1111–1122. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee D, Khoo K-H. Mycobacterial lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology. 1998;8:113–120. doi: 10.1093/glycob/8.2.113. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee D, Lowell K, Rivoire B, McNeil M R, Brennan P J. Lipoarabinomannan of Mycobacterium tuberculosis. J Biol Chem. 1992;267:6234–6239. [PubMed] [Google Scholar]
- 22.Chatterjee D, Roberts A D, Lowell K, Brennan P J, Orme I M. Structural basis of capacity of lipoarabinomannan to induce secretion of tumor necrosis factor. Infect Immun. 1992;60:1249–1253. doi: 10.1128/iai.60.3.1249-1253.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins H L, Schaible U E, Kaufmann S H E. Early IL-4 induction in bone marrow lymphoid precursor cells by mycobacterial lipoarabinomannan. J Immunol. 1998;161:5546–5554. [PubMed] [Google Scholar]
- 24.Da Silva J, Pierrat B, Mary J L, Lesslauer W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J Biol Chem. 1997;272:28373–28380. doi: 10.1074/jbc.272.45.28373. [DOI] [PubMed] [Google Scholar]
- 25.DeGroote M A, Fang F C. NO inhibitions: antimicrobial properties of nitric oxide. Clin Infect Dis. 1995;21(Suppl. 2):S162–S165. doi: 10.1093/clinids/21.supplement_2.s162. [DOI] [PubMed] [Google Scholar]
- 26.Derijard B, Raingeaud J, Barrett T, Wu I-H, Han J, Ulevitch R J, Davis R J. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 27.Ding A H, Nathan C F, Stuehr D J. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- 28.Ding A H, Sanchez E, Srimal S, Nathan C F. Macrophages rapidly internalize their receptors in response to bacterial lipopolysaccharide. J Biol Chem. 1989;264:3924–3929. [PubMed] [Google Scholar]
- 29.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellner J J, Daniel T M. Immunosuppression by mycobacterial arabinomannan. Clin Exp Immunol. 1979;35:250–257. [PMC free article] [PubMed] [Google Scholar]
- 31.Fang F C. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Investig. 1997;100:S43–S50. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finder J D, Litz J L, Blaskovich M A, McGuire T F, Qian Y, Hamilton A D, Davies P, Sebti S M. Inhibition of protein geranylgeranylation causes a superinduction of nitric-oxide synthase-2 by interleukin-1β in vascular smooth muscle cells. J Biol Chem. 1997;272:13484–13488. doi: 10.1074/jbc.272.21.13484. [DOI] [PubMed] [Google Scholar]
- 33.Flesch I E A, Kaufmann S H E. Role of cytokines in tuberculosis. Immunobiology. 1993;189:316–339. doi: 10.1016/S0171-2985(11)80364-5. [DOI] [PubMed] [Google Scholar]
- 34.Green S J, Crawford R M, Hockmeyer J T, Meltzer M S, Nacy C A. Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-γ-stimulated macrophages by induction of TNF-α. J Immunol. 1990;145:4290–4297. [PubMed] [Google Scholar]
- 35.Guan Z, Baier L D, Morrison A R. p38 mitogen-activated protein kinase down-regulates nitric oxide and up-regulates prostaglandin E2 biosynthesis stimulated by interleukin-1β. J Biol Chem. 1997;272:8083–8089. doi: 10.1074/jbc.272.12.8083. [DOI] [PubMed] [Google Scholar]
- 36.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 37.Jagannath C, Actor J K, Hunter R L J. Induction of nitric oxide in human monocytes and monocyte cell lines by Mycobacterium tuberculosis. Nitric Oxide. 1998;2:174–186. doi: 10.1006/niox.1998.9999. [DOI] [PubMed] [Google Scholar]
- 38.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh S I, Kimura T, Green S, Mak T W, Taniguchi T, Vilcek J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 39.Kim H C, Kim J H, Park J W, Suh G Y, Chung M P, Kwon O J, Rhee C H, Han Y C. Difference of nitric oxide production in peripheral blood mononuclear cells and airway epithelial cells between healthy volunteer and patients with tuberculosis. Tuberc Respir Dis. 1997;44(Suppl. 2):72–75. [Google Scholar]
- 40.Kim Y M, Son K. A nitric oxide production bioassay for interferon-gamma. J Immunol Methods. 1996;198:203–209. doi: 10.1016/s0022-1759(96)00162-7. [DOI] [PubMed] [Google Scholar]
- 41.Knutson K L, Hmama Z, Herrera-Velit P, Rochford R, Reiner N E. Lipoarabinomannan of Mycobacterium tuberculosis promotes protein tyrosine dephosphorylation and inhibition of mitogen-activated protein kinase in human mononuclear phagocytes: role of the Src homology 2 containing tyrosine phosphatase 1. J Biol Chem. 1998;273:645–652. doi: 10.1074/jbc.273.1.645. [DOI] [PubMed] [Google Scholar]
- 42.Lee J C, Layton J T, McDonnell P C, Gallagher T F, Kumar S, Green D, McNulty D, Blumenthal M J, Heys J R, Landvatter S W, Strickler J E, McLaughlin M M, Siemens I R, Fisher S M, Livi G P, White J R, Adams J L, Young P R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 43.Liew F Y, Li Y, Millott S. Tumor necrosis factor-α synergizes with IFN-γ in mediating killing of Leishmania major through the induction of nitric oxide. J Immunol. 1990;145:4306–4310. [PubMed] [Google Scholar]
- 44.Lin A, Minden A, Martinetto H, Claret F X, Lange-Carter C, Mercurio F, Johnson G L, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 45.Lindroos P M, Rice A B, Wang Y Z, Bonner J C. Role of nuclear factor-kappa B and mitogen-activated protein kinase signaling pathways in IL-1 beta-mediated induction of alpha-PDGF receptor expression in rat pulmonary myofibroblasts. J Immunol. 1998;161:3464–3468. [PubMed] [Google Scholar]
- 46.Long R, Light B, Talbot J A. Mycobacteriocidal action of exogenous nitric oxide. Antimicrob Agents Chemother. 1999;43:403–405. doi: 10.1128/aac.43.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacMicking J D, North R J, LaCourse R, Mudgett J S, Shah S K, Nathan C F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin E, Nathan C, Xie Q W. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180:977–984. doi: 10.1084/jem.180.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meraz M A, White J M, Sheehan K C F, Bach E A, Rodig S J, Dighe A S, Kaplan D H, Riley J K, Greenlund A C, Campbell D, Carver-Moore K, DuBois R N, Clark R, Aguet M, Schreiber R D. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 50.Moreno C, Taverne J, Mehlert A, Bate C A W, Brealey R J, Meager A, Rook G A W, Playfair J H L. Lipoarabinomannan from Mycobacterium tuberculosis induces the production of tumour necrosis factor from human and murine macrophages. Clin Exp Immunol. 1989;76:240–245. [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy W J, Muroi M, Zhang X, Suzuki T, Russell S W. Both a basal and an enhancer IB element is (sic) required for full induction of the mouse inducible nitric oxide synthase gene. J Endotoxin Res. 1996;3:381–393. [Google Scholar]
- 52.Nabeshima S, Nomoto M, Matsuzaki G, Kishihara K, Taniguchi H, Yoshida S, Nomoto K. T-cell hyporesponsiveness induced by activated macrophages through nitric oxide production in mice infected with Mycobacterium tuberculosis. Infect Immun. 1999;67:3221–3226. doi: 10.1128/iai.67.7.3221-3226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nandan D, Lo R, Reiner N E. Activation of phosphotyrosine phosphatase activity attenuates mitogen-activated protein kinase signaling and inhibits c-FOS and nitric oxide synthase expression in macrophages infected with Leishmania donovani. Infect Immun. 1999;67:4055–4063. doi: 10.1128/iai.67.8.4055-4063.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nathan C, Shiloh M U. Reactive oxygen and nitrogen intermediates in the relationship between hosts and microbial pathogens. Proc Natl Acad Sci USA. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicholson S, da Gloria Bonecini-Almeida M, Lapa e Silva J R, Nathan C, Xie Q-W, Mumford R, Widner J R, Calaycay J, Geng J, Boechat N, Linhares C, Rom W, Ho J L. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J Exp Med. 1996;183:2293–2302. doi: 10.1084/jem.183.5.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishiya T, Uehara T, Edamatsu H, Kaziro Y, Itoh H, Nomura Y. Activation of Stat1 and subsequent transcription of inducible nitric oxide synthase gene in C6 glioma cells is (sic) independent of interferon-γ-induced MAPK activation that is mediated by p21ras. FEBS Lett. 1997;408:33–38. doi: 10.1016/s0014-5793(97)00383-9. [DOI] [PubMed] [Google Scholar]
- 57.Nozaki Y, Hasegawa Y, Ichiyama S, Nakashima I, Shimokata K. Mechanism of nitric oxide-dependent killing of Mycobacterium bovis BCG in human alveolar macrophages. Infect Immun. 1997;65:3644–3647. doi: 10.1128/iai.65.9.3644-3647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pierce J W, Schoenleber R, Jesmok G, Best J, Moore S A, Collins T, Gerritsen M E. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 59.Rich E A, Torres M, Sada E, Finegan C K, Hamilton B D, Toossi Z. Mycobacterium tuberculosis (MTB)-stimulated production of nitric oxide by human alveolar macrophages and relationship of nitric oxide production to growth inhibition of MTB. Tuberc Lung Dis. 1997;78:247–255. doi: 10.1016/s0962-8479(97)90005-8. [DOI] [PubMed] [Google Scholar]
- 60.Riedel D D, Kaufmann S H E. Chemokine secretion by human polymorphonuclear granulocytes after stimulation with Mycobacterium tuberculosis and lipoarabinomannan. Infect Immun. 1997;65:4620–4623. doi: 10.1128/iai.65.11.4620-4623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rockett K A, Brookes R, Udalova I, Vidal V, Hill A V S, Kwiatkowski D. 1,25-Dihydroxyvitamin D3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage-like cell line. Infect Immun. 1998;66:5314–5321. doi: 10.1128/iai.66.11.5314-5321.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rose D M, Winston B W, Chan E D, Riches D W H, Gerwins P, Johnson G L, Henson P M. Fcγ receptor cross-linking activates p42, p38, and JNK/SAPK mitogen-activated protein kinases in murine macrophages. J Immunol. 1997;158:3433–3438. [PubMed] [Google Scholar]
- 63.Salkowski C A, Barber S A, Detore G R, Vogel S N. Differential dysregulation of nitric oxide production in macrophages with targeted disruptions of IFN regulatory factor-1 and -2 genes. J Immunol. 1996;156:3107–3110. [PubMed] [Google Scholar]
- 64.Sanchez I, Hughes R T, Mayer B J, Yee K, Woodgett J R, Avruch J, Kyriakis J M, Zon L I. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 65.Sibley L D, Hunter S W, Brennan P J, Krahenbuhl J L. Mycobacterial lipoarabinomannan inhibits gamma interferon-mediated activation of macrophages. Infect Immun. 1988;56:1232–1236. doi: 10.1128/iai.56.5.1232-1236.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sikora A, Liu J, Brosnan C, Buell G, Chessel I, Bloom B R. Purinergic signaling regulates radical-mediated bacterial killing mechanisms in macrophages through a P2X7-independent mechanism. J Immunol. 1999;163:558–561. [PubMed] [Google Scholar]
- 67.Singh K, Balligand J-L, Fischer T A, Smith T W, Kelly R A. Regulation of cytokine-inducible nitric oxide synthase in cardiac myocytes and microvascular endothelial cells: role of extracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) and Stat 1α. J Biol Chem. 1996;271:1111–1117. doi: 10.1074/jbc.271.2.1111. [DOI] [PubMed] [Google Scholar]
- 68.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang C-H, Liu C-Y, Lin H-C, Yu C-T, Chung K-F, Kuo H-P. Increased exhaled nitric oxide in active pulmonary tuberculosis due to inducible NO synthase upregulation in alveolar macrophages. Eur Respir J. 1998;11:809–815. doi: 10.1183/09031936.98.11040809. [DOI] [PubMed] [Google Scholar]
- 70.Welsh N. Interleukin-1β-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J Biol Chem. 1996;271:8307–8312. doi: 10.1074/jbc.271.14.8307. [DOI] [PubMed] [Google Scholar]
- 71.Winston B W, Chan E D, Johnson G L, Riches D W H. Activation of p38mapk, MKK3, and MKK4 by tumor necrosis factor-alpha in mouse bone marrow-derived macrophages. J Immunol. 1997;159:4491–4497. [PubMed] [Google Scholar]
- 72.Winston B W, Remigio L K, Riches D W H. Preferential involvement of MEK1 in the TNFα induced activation of p42mapk/erk2 in mouse macrophages. J Biol Chem. 1995;270:27391–27394. doi: 10.1074/jbc.270.46.27391. [DOI] [PubMed] [Google Scholar]
- 73.Xie Q, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon γ and bacterial lipopolysaccharide. J Exp Med. 1993;177:1779–1784. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Y, Broser M, Cohen H, Bodkin M, Law K, Reibman J, Rom W N. Enhanced interleukin-8 release and gene expression in macrophages after exposure to Mycobacterium tuberculosis and its components. J Clin Investig. 1995;95:586–592. doi: 10.1172/JCI117702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Doerfler M, Lee T C, Guillemin B, Rom W N. Mechanisms of stimulation of interleukin-1β and tumor necrosis factor-α by Mycobacterium tuberculosis components. J Clin Investig. 1993;91:2076–2083. doi: 10.1172/JCI116430. [DOI] [PMC free article] [PubMed] [Google Scholar]