

Abstract
The BA.2 sub-lineage of the Omicron (B.1.1.529) severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant rapidly supplanted the original BA.1 sub-lineage in early 2022. Both lineages threatened the efficacy of vaccine-elicited antibodies and acquired increased binding to several mammalian ACE2 receptors. Cryoelectron microscopy (cryo-EM) analysis of the BA.2 spike (S) glycoprotein in complex with mouse ACE2 (mACE2) identifies BA.1- and BA.2-mutated residues Q493R, N501Y, and Y505H as complementing non-conserved residues between human and mouse ACE2, rationalizing the enhanced S protein-mACE2 interaction for Omicron variants. Cryo-EM structures of the BA.2 S-human ACE2 complex and of the extensively mutated BA.2 amino-terminal domain (NTD) reveal a dramatic reorganization of the highly antigenic N1 loop into a β-strand, providing an explanation for decreased binding of the BA.2 S protein to antibodies isolated from BA.1-convalescent patients. Our analysis reveals structural mechanisms underlying the antigenic drift in the rapidly evolving Omicron variant landscape.
Keywords: SARS-CoV-2, COVID-19, Omicron, cryoelectron microscopy
Graphical abstract
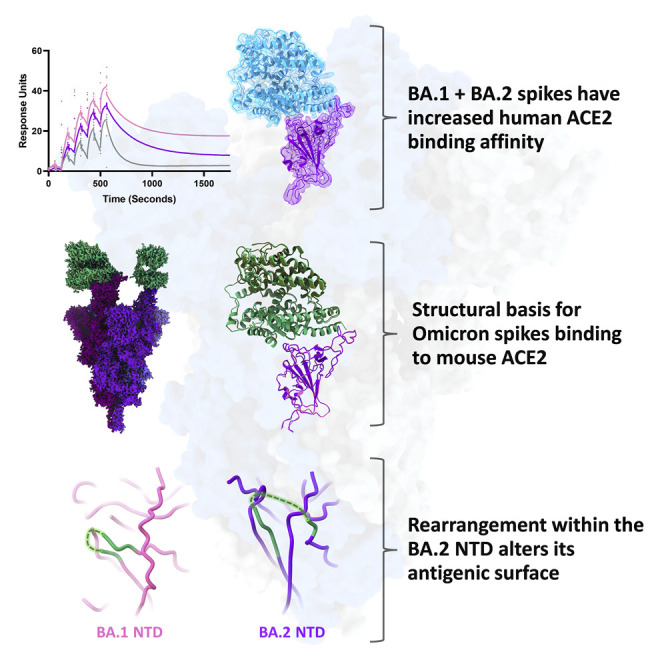
Saville et al. employ biochemical and structural techniques to compare the Omicron BA.1/BA.2 spike proteins and report increased human ACE2 affinity for both variants. Residues mutated in both variants contact non-conserved residues between human and mouse ACE2. Structural rearrangements within the BA.2 NTD rationalize its escape from BA.1-convalescent antibodies.
Introduction
The extensively mutated Omicron variant (B.1.1.529) is threatening the efficacy of vaccines and antibody therapeutics after replacing the Delta variant (B.1.617.2) as the globally dominant severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lineage. Initially, five sub-lineages of Omicron were identified as BA.1, BA.2, BA.3, BA.4, and BA.5. Lineages BA.1 and BA.1.1—a further sub-lineage of BA.1 that differs only by an additional R346K mutation in the spike (S) protein—comprised the vast majority of Omicron infections in late 2021 (Figure 1A).1 In early 2022, however, BA.2 infections began increasing and displacing the BA.1 lineages, with BA.2 constituting the majority of SARS-CoV-2 infections by March. The S protein mutational profile of sub-lineage BA.3 resembles the BA.1 mutational profile most closely, yet this variant never exceeded 1% of global SARS-CoV-2 infections. The BA.4 and BA.5 sub-lineages share identical S protein mutations, which differ from the BA.2 S protein by only a few mutations (Δ69/70, L452R, and F486V), with these lineages combining to comprise less than 20% of global infections by the end of May 2022 (Figure 1A). BA.2 shares many S protein mutations with BA.1 with the exception of a number of unique mutations in the receptor-binding domain (RBD) and amino-terminal domain (NTD) (Figure 1B). Given the functional importance of the RBD and NTD in receptor engagement and their susceptibility to vaccine-elicited neutralizing antibodies,2 , 3 we sought to understand the molecular consequences of BA.2 S protein mutations within these two domains.
Figure 1.

Global prevalence and S protein mutations of the Omicron sub-lineages
(A) Global prevalence of Omicron sub-lineages BA.1, BA.1.1, and BA.2 from November 2021 to March 2022. Only Omicron sub-lineages surpassing 1% global frequency are displayed. Sequence data were downloaded from the Global Initiative on Sharing All Influenza Data (GISAID) and graphed as weekly prevalence.1
(B) SARS-CoV-2 spike (S) protein amino acid sequence boxplots for the wild-type (D614G), BA.1/BA.1.1, and BA.2 Omicron sub-lineages. The BA.1.1 lineage is identical to BA.1 with the exception of an additional R346K mutation. NTD, N-terminal domain; RBD, receptor-binding domain; RBM, receptor-binding motif.
(C) Cryo-EM-derived atomic model of the BA.2 S glycoprotein. Each S protein protomer is colored in different shades of blue. The locations of modeled amino acid mutations are shown as spheres on one protomer. BA.1-, BA.1.1-, and BA.2-specific mutations are colored in magenta, light magenta, and purple, respectively. Shared mutations across BA.1, BA.1.1, and BA.2 sub-lineages are colored in gray.
Results
Cryoelectron microscopy (cryo-EM) structural analysis of the BA.2 S protein ectodomain reveals preserved overall architecture compared with the wild-type S, BA.1 S, and other previously emerged SARS-CoV-2 variant S structures (Figures 1C and S1).4 , 5 , 6 , 7 , 8 , 9 Wild type (WT) refers to the Wuhan-Hu-1 isolate with the addition of the D614G mutation (lineage B.1). Similar to our observations for the BA.1 S trimer,9 the RBD of a single protomer within the BA.2 trimer is well resolved in the “up” position, with poor densities for the other 2 RBDs demonstrating flexibility of these regions relative to the rest of the trimer. This suggests a dynamic nature of the BA.2 RBD and is consistent with recent structural analyses: Stalls et al. found 3-RBD-down, 1-RBD-up, and 1.5-RBD-up conformations in their cryo-EM analysis of the BA.2 ectodomain,10 while Zhang et al. found 3-RBD-down, 1-RBD-up, and 1-RBD conformations in an intermediate up conformation for the BA.2 full-length S protein.11
The unique BA.1 G496S mutation, which is the sole differentiating mutation between BA.1 and BA.2 within the receptor-binding motif (the portion of the S protein that directly interacts with ACE2), was recently shown to add a new hydrogen bonding interaction with human ACE2 (hACE2) residue K353.9 We measured the affinity of the BA.2 S protein ectodomain or RBD binding to hACE2 using three different surface plasmon resonance approaches: (1) measurement of binding of dimeric hACE2 to immobilized WT, BA.1, and BA.2 RBDs (Figure 2 A); (2) measurement of binding of WT, BA.1, and BA.2 RBDs to immobilized dimeric hACE2 (Figure 2B); and (3) measurement of binding of WT, BA.1, and BA.2 S protein ectodomains to immobilized dimeric hACE2. Collectively, these measurements reveal increased binding affinity of the BA.2 S protein to hACE2 relative to WT and is comparable to the increased binding affinity observed for BA.1.
Figure 2.

Binding affinity and cryo-EM structure of the Omicron BA.2 S protein-human ACE2 complex
(A) Surface plasmon resonance experiments measuring dimeric human ACE2 (hACE2) binding to immobilized wild-type (WT), BA.1, and BA.2 RBDs, performed in technical triplicates. Summary data are shown at the top with representative surface plasmon resonance (SPR)-binding curves (colored solid line), and fitted 1:1 binding models (black dashed line) are shown on bottom.
(B) As in (A) but measuring WT, BA.1, and BA.2 RBDs binding to immobilized dimeric hACE2, performed in at least technical quadruplicates.
(C) As in (A) but measuring WT, BA.1, and BA.2 ectodomains binding to immobilized dimeric hACE2, performed in at least technical duplicates. The WT and BA.1 data in (C) were previously reported.9 Pairwise statistical significance test was performed using a one-way ANOVA test (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001, ns, not significant).
(D) Focus-refined cryo-EM density map and fitted atomic model of the BA.2 RBD in complex with hACE2 at 2.8 Å.
(E) Aligned atomic models of hACE2 bound to BA.1 and BA.2 RBDs. The BA.1 RBD (PDB: 7T9L) and complexed hACE2 atomic models are shown in magenta and dark blue, respectively. The BA.2 RBD and complexed hACE2 atomic models are shown in purple and light blue, respectively.
(F) Atomic model of the BA.1 S protein-hACE2 complex, focused on residue S496. The hydrogen bonding interaction between BA.1 S protein residue S496 and hACE2 residue K353 is indicated by an orange dashed line.
(G) As in (F) but for the BA.2 S protein-hACE2 complex, focused on residue G496.
To visualize the structural impacts of BA.2 RBD mutations on hACE2 binding, we performed cryo-EM analysis of the BA.2 S protein in complex with hACE2 (Figures 2D and S1). Structural alignment of BA.1 and BA.2 RBD-hACE2 complexes demonstrates a high degree of structural similarity across both the RBDs and hACE2 molecules (0.97 Å root-mean-square deviation; Figure 2E). Seven mutations distinguish the BA.1 and BA.2 RBDs—BA.1 contains S371L, G446S, and G496S mutations and BA.2 contains S371F, T376A, D405N, and R408S mutations—yet only the G496S mutation makes differential interactions with hACE2 residues between these two sub-lineages (Figures 2F and 2G). The S496G revertant mutation present in the BA.2 receptor-binding motif (RBM) no longer makes a hydrogen bonding interaction that is present in the BA.1 RBD-hACE2 complex. However, the loss of this hydrogen bonding interaction in the BA.2 variant S does not impart a measurable difference in overall hACE2 binding (Figures 2A–2C); therefore, we conclude that similar to BA.1, the numerous mutations within the BA.2 S protein enable enhanced hACE2 affinity relative to the WT S protein.
The BA.1 and BA.2 S proteins uniquely exhibit a significant increase in binding affinity for mouse ACE2 (mACE2) compared with previous SARS-CoV-2 lineages.12 , 13 Additionally, increased cell entry—relative to WT—was observed in authentic BA.1 virus infecting cells overexpressing mACE2.14 Mouse-adapted SARS-CoV-2, generated by the serial passaging of the virus in mice, reproducibly (across multiple studies) selected for mutations at positions Q493 and Q498,15 , 16 which are mutated sites in the BA.1 and BA.2 Omicron sub-lineages. To provide a structural basis for the increased mACE2 affinity exhibited by both the BA.1 and BA.2 Omicron variants, we solved cryo-EM structures of their S proteins in complex with the ectodomain of mACE2 (Figures 3A–3C and S1). mACE2 is observed to be similarly positioned in its binding of the BA.1 and BA.2 RBDs compared with hACE2 (Figures 3A–3C and S2). Focused refinement of the BA.1- and BA.2-mACE2 regions was possible, resulting in 2.8 and 2.7 Å local reconstructions, respectively, and allowing for side-chain placement at the RBD-mACE2 interfaces. As shown for the hACE2 structures, the BA.1- and BA.2-mACE2 focused refinements exhibit a high degree of structural similarity relative to one another (0.87 Å root-mean-square deviation), with identical side-chain interactions made at the RBD-mACE2 interface. In the BA.1 and BA.2 RBD-hACE2-bound structures, the sole differentiating mutation at this interface is the BA.1-specific G496S mutation, which makes a hydrogen bonding interaction with hACE2 residue K353 (Figures 2F and 2G). In mACE2, position 353 is a histidine residue that is not positioned to hydrogen bond with S496 (>4.8 Å distance) in BA.1 (Figure S2), thus the BA.1-specific G496S mutation does not distinguish the BA.1 and BA.2 variants at the mACE2 interface as it did in hACE2. Our analysis proceeds with the higher-resolution BA.2-mACE2 focus-refined structure, with direct parallels possible for the BA.1-mACE2 structure.
Figure 3.

Cryo-EM structure of the Omicron BA.2 S protein-mouse ACE2 complex
(A) Cryo-EM density map of BA.2 S protein in complex with mouse ACE2 at 2.5 Å. Mouse ACE2 is shown in green, and protomers of the BA.2 S protein are shown in shades of purple.
(B) Focus-refined cryo-EM density map and fitted atomic model of the BA.2 RBD-mouse ACE2 (mACE2) complex at 2.7 Å.
(C) Aligned atomic models of the BA.1 and BA.2 RBD-mACE2 complexes. The BA.1 RBD and complexed hACE2 atomic models are shown in magenta and dark green, respectively. The BA.2 RBD and complexed hACE2 atomic models are shown in purple and light green, respectively.
(D) Atomic model of the BA.2 RBD-mACE2 complex, focused on residues Y501 and H505.
(E) As in (D) but focused on residue R493.
(F) Atomic model of the WT RBD-hACE2, focused on residues N501 and Y505.
(G) As in (H) but focused on residue Q493.
(H) Atomic model of mACE2 from the perspective of a binding RBD. Black labels are mACE2 residues, and gray labels denote the interacting residues in a bound RBD. Gold labels denote the interacting residues in a bound RBD that are mutated in the BA.1 and BA.2 Omicron sub-lineages.
mACE2 has a high degree of overall amino acid sequence homology with hACE2 (82% identity), with lower sequence homology (73% identity) at the RBD-interaction interface comprising ACE2 amino acid residues 18–46, 78–91, 324–358, and 392–394.17 There are two sites within the Omicron RBD-mACE2 interface that differ relative to the Omicron RBD-hACE2 interface. The first site (Figure 3D) involves residue H353 in mACE2 (K353 in hACE2) forming π-π stacking and hydrogen bonding interactions with Omicron-mutated RBD residues Y501 and H505, respectively. Both of these interactions are not possible in the WT RBD, which harbors N501 and Y505 residues at these positions (Figure 3F). Site two (Figure 3E) involves Omicron-mutated residue R493 forming a hydrogen bonding interaction with residue N31 of mACE2 (K31 in hACE2). Across these two sites, we see that non-conserved ACE2 residues between hACE2 and mACE2 (H353, N31, Q34) are complemented by mutated residues in the Omicron variants, providing us with the conclusion that the mutations Q493R, N501Y, and Y505H engage non-conserved ACE2 residues unique to mACE2, rationalizing the enhanced binding of mACE2 by the Omicron variant S proteins (Figure 3H).
Given that the vast majority of mutational differences between the BA.1 and BA.2 S proteins are within the immunogenic NTD and RBD regions, we sought to probe the antigenic differences between these proteins. We first assessed antibody binding of WT, BA.1, and BA.2 S proteins via ELISA using a small panel of RBD- and NTD-directed antibodies.18 , 19 , 20 , 21 , 22 , 23 BA.1 and BA.2 S proteins exhibited similarly decreased binding by all antibodies compared with the WT S protein, with a pronounced decrease in BA.2 S binding by S309 compared with BA.1. S309 is the precursor to the clinical monoclonal antibody sotrovimab, which is the only FDA-approved monoclonal antibody that retained neutralization capability for the BA.1 variant.12 , 20 , 24 Both BA.1 and BA.2 share N440K and G339D mutations, which are within the S309 epitope, while the sub-lineages are differentiated by S371L (BA.1) and S371F (BA.2) mutations in an alpha helix close to the N343 glycan, an important feature of the S309 epitope (Figure S2).20 A recent study by Iketani et al. showed that the S371F mutation alone was sufficient to decrease the neutralization potency of S309 by 20-fold, suggesting that this distal mutation may disrupt the S309-binding epitope via allosteric mutational mechanisms.25 Superposition of WT, BA.1, and BA.2 RBDs reveals no prominent structural changes that could account for this finding, although a shift in the antigenic surface of these domains is evident upon mapping BA.1- and BA.2-specific mutations (Figures 4C and 4D). There is a distinct shift from the location of two BA.1-specific RBD mutations to three BA.2-specific RBD mutations, which are located within the footprints of different patient-derived antibodies.
Figure 4.

Antigenic shift of the BA.2 S protein
(A) Percentage of binding of monoclonal antibodies against the BA.1 and BA.2 S proteins relative to WT as assessed by ELISA, performed in technical triplicates.
(B) Antibody epitopes with the side chains of contacted residues within the RBD or NTD shown and colored. BA.1- and BA.2-mutated residues are labeled within the antibody epitopes in magenta and purple, respectively, with shared mutations labeled in gray.
(C) Alignment of WT, BA.1, and BA.2 RBDs, with shared, BA.1-specific, and BA.2-specific mutations labeled in gray, magenta, and purple, respectively.
(D) Alignment of select patient-derived RBD-directed antibodies on the RBD. CB6, PDB: 7C01; REGN10933/REGN10987, PDB: 6XDG; CV2-75, PDB: 7M31; CR3022, PDB: 6YLA.
(E) Side-by side comparison of WT (PDB: 7KRS), BA.1 (PDB: 7TNW), and BA.2 NTDs with a focused view on the structural rearrangement of the 67–79 loop and N1 antigenic loop.
(F) Alignment of deposited patient-derived NTD-directed antibody atomic models with PDB IDs listed in Table S2. The labels of antibodies that make intermolecular contacts with the N1 and/or 67–79 loop are colored in orange and/or underlined in green, respectively. The NTD is shown in gray with its N1 loop highlighted in orange and the 67–79 loop in green.
(G) Schematic and BA.2/BA.1 antibody-binding ratio for domain-enriched (ectodomain, NTD, or RBD) BA.1-convalescent polyclonal sera. Serum was pooled from 18 BA.1-convalescent patients (16 breakthrough cases and 2 infections in non-vaccinated patients) prior to incubation with either BA.1 ectodomain, NTD, or RBD to enrich domain-specific BA.1-convalescent antibodies. The samples were washed prior to quantification of IgG binding by ELISA and plotting of the BA.2/BA.1 ratio of domain-specific antibody binding. Data are derived from serum from 18 pooled BA.1-convalescent patients, and the ELISA assays were performed in technical duplicates.
To assess structural differences within the flexible NTD region, we performed focused refinement on the NTD of the BA.2 S protein and were able to resolve a structure at 2.9 Å (Figures S1 and S3). In contrast to the RBD, inspection of the WT, BA.1, and BA.2 NTDs reveals a structural reordering of the immunodominant N1 loop within the BA.2 NTD (Figure 4E). While the N1 region is adjacent to the loop formed by residues 67–79 within the WT and BA.1 NTDs, the BA.2 N1 region is located between both ends of the 67–79 loop as a beta strand within an anti-parallel beta sheet, resembling a “threading” of the BA.2 N1 region within the 67–79 loop. Given the inability of proline to contribute favorable hydrogen bonding contacts within beta sheets,26 the structural rearrangement and ordering of the BA.2 N1 region is likely driven by the loss of two proline residues due to the BA.2-specific deletion of residues 24–26. This structural threading of the N1 loop is a distinguishing feature of the BA.2 variant and is significant given the inclusion of the N1 loop within the “NTD neutralization supersite”—designated for the propensity of patient-derived neutralizing antibodies to bind this location.3 , 27 Figure 4F shows a comparison of PDB-deposited structures of patient-derived NTD binding antibodies aligned to a single NTD. From this alignment, one can see that a significant portion of NTD-binding antibodies contact the N1 and 67–79 loops, which are rearranged in the BA.2 NTD and rationalizes the reported antibody escape.25 , 28 Further, this rearrangement likely perturbs the NTD glycan shield, with glycosylated amino acid residue N74 significantly rearranged in the BA.2 NTD (Figure S3).29 Additionally, the BA.2 S protein lacks the N17-linked glycan present in both the WT and BA.1 NTDs due to the T19I mutation, which destroys the N-linked glycosylation consensus sequence (N-X-S/T/C, where X is any amino acid except proline). Therefore, rearrangement and the loss of glycans within the BA.2 NTD likely further contribute to its antibody escape.
Despite detecting a large decrease in 4A8 binding against BA.2 relative to WT (Figure 4A), we were able to complex the BA.2 S protein with 4A8 under conditions with high antibody excess and solve a cryo-EM structure of the complex, yielding a slightly higher-resolution (2.5 Å) focus-refined structure of the BA.2 NTD (Figures S1 and S3). This structure also shows the threading of the N1 loop through the 67–79 loop and confirms that this BA.2 NTD rearrangement is present when the NTD is bound to an antibody.
We hypothesized that the mutational changes in the antigenic surface of the RBD along with the structural reordering of the NTD represent an antigenic drift between the BA.1 and BA.2 S proteins. To test this, we purified BA.1 S ectodomain-, RBD-, and NTD-specific polyclonal antibodies from patients with a history of BA.1 infection and assessed the ability of the immunoglobulin G (IgG) component of these preparations to bind the BA.2 S ectodomain via ELISA (Figures 4G and S4). The IgG component of all three polyclonal antibody preparations bound the BA.2 S ectodomain to a lower extent than BA.1. Interestingly, the disparity between BA.2 and BA.1 S ectodomain binding was greater for both RBD-specific antibodies (∼40% decrease in BA.2 S binding) and NTD-specific antibodies (∼60% decrease in BA.2 S binding) relative to whole S ectodomain-specific antibodies (∼20% decrease in BA.2 S binding). This result suggests that antibodies targeting the BA.1 RBD and NTD are particularly sensitive to mutations within the BA.2 S protein, demonstrating an antigenic drift between BA.1 and BA.2 lineages that is driven by S protein RBD and NTD mutations.
Discussion
Herein, we have conducted a structural comparison of the spike proteins from the original Omicron BA.1 sub-lineage and the BA.2 lineage that replaced it. We provide a structural basis for the acquired ability of BA.1 and BA.2 S proteins to engage mACE2, finding Omicron mutations at the ACE2-binding site to complement non-conserved residues between hACE2 and mACE2. As for BA.1, BA.2 exhibits dramatic escape from monoclonal antibodies through direct mutational effects within the RBD and allosteric mutational effects within the BA.2 NTD, for which we determined a structural basis. Finally, our discovery that BA.1 convalescent polyclonal sera exhibits decreased binding for the BA.2 ectodomain, RBD, and NTD relative to BA.1 highlights the antigenic difference between the BA.1 and BA.2 S proteins. Our overarching finding is that the BA.1 and BA.2 S proteins do not differ greatly with regards to ACE2 binding (human or mouse) yet are distinguished by the arrangement of their NTDs, with implications for the evasion of convalescent serum antibodies. It is likely that mutations within proteins other than the S protein that underlie processes such as transmission and replication also rationalize the emergence and global dominance of the BA.2 sub-lineage.
The Omicron BA.1 and BA.2 sub-lineages share mutations with previously circulating SARS-CoV-2 variants, such as Alpha (B.1.1.7) and Delta (B.1.617.2)—the only other variants to surpass 50% global prevalence.1 We previously showed that SARS-CoV-2 S protein mutations are highly modular, with a given mutation in one variant imparting similar effects on hACE2 binding and antibody evasion in another variant S.30 For example, the N501Y mutation within the RBM of the Alpha and BA.1/BA.2 S proteins contributes additional interactions with hACE2, likely explaining—in part—their enhanced hACE2 binding affinity. While the Alpha and Delta variants emerged prior to population-level vaccination, the BA.1/BA.2 variants arose since widespread vaccination had been achieved. This widespread global vaccination may have shifted viral fitness pressure toward variants that exhibit significant evasion of pre-existing immunity.31 , 32 The Alpha and Delta variant S proteins were primarily characterized as enabling enhanced cell infectivity compared with the WT S,7 , 33 , 34 while the BA.1 and BA.2 S proteins exhibit preserved hACE2 binding efficiency and dramatically altered antigenic surfaces, enabling them to significantly escape vaccine- and infection-elicited immunity. Therefore, the enhanced immune evasion achieved by the Omicron sub-lineage S proteins, in combination with mutational effects in non-S viral proteins, likely rationalizes their rapid and sustained global prevalence.
Omicron spillover into non-human animals has been reported in white-tailed deer and cats.35 , 36 , 37 The acquired ability of SARS-CoV-2 variants to access animal reservoirs allows them to sample different evolutionary pressures in these organisms, with potential impacts on “spillback” transmission, which has recently been reported for deer-to-human transmission.36 Our identification of Q493R, N501Y, and Y505H Omicron mutations as complementing the non-conserved residues N31 and H353 (between hACE2 and mACE2) may be extended to the ACE2 sequences of other animals to enable predictions of their susceptibility to SARS-CoV-2 infection. Interestingly, mACE2 is distinguished from human, cat, ferret, deer, and bat ACE2 by non-conserved N31 and H353 residues (Figure S5), and the fact that these residues make intermolecular contacts with Omicron-specific RBD mutations provides a structural rationale for the acquisition of BA.1 and BA.2 sub-lineages binding to mACE2.
The restructuring of the N1 antigenic loop within the BA.2 NTD rationalizes, in part, the antigenic drift reported between the BA.1 and BA.2 Omicron sub-lineages. While BA.1 and BA.2 appear to be similarly immune evasive,25 , 26 these variants are distinguished by different antigenic properties in their S proteins. Our finding of decreased BA.1 ectodomain-, NTD-, and RBD-directed antibodies to the BA.2 S protein ectodomain provides a domain-specific assessment of this antigenic drift. While the rearrangement of the BA.2 NTD represents the largest structural distinction between the BA.1 and BA.2 S proteins, it is likely that the differential mutations within the BA.1 and BA.2 RBDs have the greatest influence on this Omicron sub-lineage antigenic drift, given the immunodominance of the RBD in the humoral immune response.3
Limitations of the study
Our analyses make use of timeric HexaPro-stabilized S protein ectodomains, which differ from native S proteins by six stabilizing proline mutations in the S2 domain and the transmembrane domain replaced with a trimerization motif.38 We have focused the present study on characterizing the BA.1 and BA.2 S glycoproteins; however, there exist distinguishing mutations outside of the S protein open reading frame that may influence different aspects of viral fitness (replication kinetics and tropism, for example). Future studies will provide further insight into these aspects of viral fitness to garner a better understanding of the evolution and global prevalence of SARS-CoV-2.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| VH-FC ab8 | (Li et al.)18 | N/A |
| Fab S309 | (Pinto et al.)20 | N/A |
| Fab S2M11 | (Tortorici et al.)19 | N/A |
| Fab 4-8 | (Liu et al.)21 | N/A |
| Fab 4A8 | (Chi et al.)22 | N/A |
| Fab DH1052 | (Li et al.)23 | N/A |
| Goat anti-human IgG | Jackson ImmunoReserach | Cat. # 109-035-088; RRID: AB_2337584 |
| Mouse anti-his tag antibody | abcam | Cat# ab18184; RRID: AB_444306 |
| Chemicals, peptides, and recombinant proteins | ||
| Ace2 (18-615) | New England Biolabs | Cat. # 73775S |
| BA.1 RBD | Sino Biological | Cat# 40592-V08H121 |
| Critical commercial assays | ||
| Pierce 1-Step Ultra Substrate Solution | ThermoFisher | Cat. # 34028 |
| Deposited data | ||
| S(BA.2) | This paper | Global refinement: EMDB 27523, PDB: 8DM1 Focus refinement: EMDB 27524, PDB: 8DM2 |
| S(BA.2)+hACE2 | This paper | Global refinement: EMDB 27527, PDB: 8DM5 Focus refinement: EMDB 27528, PDB: 8DM6 |
| S(BA.2)+mACE2 | This paper | Global refinement: EMDB 27529, PDB: 8DM7 Focus refinement: EMDB 27530, PDB: 8DM8 |
| S(BA.1)+mACE2 | This paper | Global refinement: EMDB 27531, PDB: 8DM9 Focus refinement: EMDB 27532, PDB: 8DMA |
| S(BA.2)+4A8 | This paper | Global refinement: EMDB 27525, PDB: 8DM3 Focus refinement: EMDB 27526, PDB: 8DM4 |
| Experimental models: Cell lines | ||
| Expi293F | ThermoFisher | Cat# A14527 |
| Recombinant DNA | ||
| pcDNA3.1 HexaPro BA.1 | Mannar et al.9 | N/A |
| pcDNA3.1 BA.1 NTD | This paper | N/A |
| pcDNA3.1 HexaPro BA.2 | This paper | N/A |
| pcDNA3.1 BA.2 NTD | This paper | N/A |
| pcDNA3.1 BA.2 RBD | This paper | N/A |
| pcDNA3.1 hACE2 (1-615) | Mannar et al.9 | N/A |
| pcDNA3.1 mACE2 (1-615) | This paper | N/A |
| pCSCG hACE2-FC | Yang et al.39 | Addgene: 164222 |
| pcDNA3.1 Fab S309 Light Chain | Mannar et al.30 | N/A |
| pcDNA3.1 Fab S309 Heavy Chain | Mannar et al.30 | N/A |
| pcDNA3.1 Fab S2M11 Light Chain | Mannar et al.30 | N/A |
| pcDNA3.1 Fab S2M11 Heavy Chain | Mannar et al.30 | N/A |
| pcDNA3.1 Fab 4-8 Light Chain | Saville et al.40 | N/A |
| pcDNA3.1 Fab 4-8 Heavy Chain | Saville et al.40 | N/A |
| pcDNA3.1 Fab 4A8 Light Chain | Saville et al.40 | N/A |
| pcDNA3.1 Fab 4A8 Heavy Chain | Saville et al.40 | N/A |
| pcDNA3.1 Fab DH1052 Light Chain | This paper | N/A |
| pcDNA3.1 Fab DH1052 Heavy Chain | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad 7.0 | https://www.graphpad.com/scientific-software/prism/ |
| EPU automated acquisition | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home/electron-microscopy/products/software-em-3d-vis/epu-software.html |
| UCSF Chimera | Pettersen et al.41 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al.42 | https://www.cgl.ucsf.edu/chimerax/ |
| RELION 3.1 | Scheres43 | https://github.com/3dem/relion/releases/tag/3.1.0 |
| cryoSPARC live (v3.0.1) | Punjani et al.44 | https://cryosparc.com/live |
| COOT | Emsley et al.45 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Liebschner et al.46 | https://phenix-online.org/ |
| MolProbity | Williams et al.47 | http://molprobity.biochem.duke.edu/ |
| PyMOL | v.2.2 Schrodinger, LLC | https://pymol.org/2/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sriram Subramaniam (sriram.subramaniam@ubc.ca).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell lines
Expi293F cells (ThermoFisher, Cat# A14527) were grown in suspension culture using Expi293 Expression Medium (ThermoFisher, Cat# A1435102) at 37°C, 8% CO2 with agitation at 130 rpm.
Method details
Ethics statement
Patient derived sera samples were collected according to the CARE COVID Study (http://www.bccdc.ca/health-professionals/clinical-resources/covid-19-care/covid-19-serology-care-covid-study) with ethics approval from the UBC Clinical Research Ethics Board.
Expression and purification of Omicron recombinant spike protein constructs
The production of the SARS-CoV-2 wild-type (D614G) and BA.1 HexaPro S proteins and human ACE2 (residues 1-615) were described previously.9 , 30 , 48 The BA.1 RBD was obtained from Sino Biological (Cat# 40592-V08H121).
The BA.2 Omicron HexaPro S protein gene was synthesized and inserted into pcDNA3.1 (GeneArt Gene Synthesis, Thermo Fisher Scientific). The mouse ACE2 gene was ordered from Addgene (Plasmid #158087) and amino acid residues 1-615 were amplified and inserted into pcDNA3.1 with a C-terminal HRV 3C site, 8x histidine tag and twin strep tag.
Expi293F cells were transiently transfected at a density of 3 × 106 cells/mL using linear polyethylenimine (Polysciences Cat# 23966-1). The media was supplemented 24 hours after transfection with 2.2 mM valproic acid, and expression was carried out for 3 days at 37°C, 8% CO2. The supernatant was harvested by centrifugation and filtered through a 0.22-μm filter prior to loading onto a 5 mL HisTrap excel column (Cytiva). The column was washed for 20 CVs with wash buffer (20 mM Tris pH 8.0, 500 mM NaCl), 5 CVs of wash buffer supplemented with 20 mM imidazole, and the protein eluted with elution buffer (20 mM Tris pH 8.0, 500 mM NaCl, 500 mM imidazole). Elution fractions containing the protein were pooled and concentrated (Amicon Ultra 100 kDa cut off, Millipore Sigma) for gel filtration. Gel filtration was conducted using a Superose 6 10/300 GL column (Cytiva) pre-equilibrated with GF buffer (20 mM Tris pH 8.0, 150 mM NaCl). Peak fractions corresponding to soluble protein were pooled and concentrated (BA.2 spike protein: Amicon Ultra 100 kDa cut off, Millipore Sigma; mouse ACE2 protein: Amicon Ultra 30 kDa cut off, Millipore Sigma). Protein samples were flash-frozen in liquid nitrogen and stored at −80°C.
Surface plasmon resonance (SPR)
A Biacore T200 instrument was used for all SPR experiments. All experiments were performed at 25°C, using 10 mM HEPES, 150 mM NaCl, 3 mM EDTA and 0.05% v/v Surfactant P20 as the SPR running buffer. The surface was regenerated using 10 mM glycine pH 1 for all experiments which strips all proteins from the SPR chip surface. Fresh protein was immobilized onto the SPR chip at the beginning of each experimental run. Reference-subtracted curves were fitted to a 1:1 binding model using Biacore evaluation software. Specific details pertaining to each experimental setup are described below.
ACE2-FC as ligand, RBD as analyte
Human ACE2 attached to a human FC tag (ACE2-FC) was immobilized using the series S protein A chip in SPR running buffer. Increasing concentrations of RBD protein constructs (6.25 nM, 31.25 nM, 62.5 nM, 125 nM, 250 nM) were flowed over the surface for single cycle kinetic experiments.
RBD as ligand, ACE2-FC as analyte
CM5 chips were functionalized with anti-his tag antibody (abcam Cat# ab18184) using an amine coupling kit (Cytiva) and used to capture his tagged RBD constructs. Increasing concentrations of ACE2-FC (2.5 nM, 25 nM, 50 nM, 100 nM, 200 nM) were flowed over the surface for single cycle kinetic experiments.
ACE2-FC as ligand, spike protein ectodomain as analyte
Human ACE2 attached to a human FC tag (ACE2-FC) was immobilized using the series S protein A chip in running buffer. Increasing concentrations (6.25 nM, 31.25 nM, 62.5 nM, 125 nM, 250 nM) of BA.2 spike protein trimers were flowed over the surface for single cycle kinetic experiments and compared to previously published data9 for WT and BA.1 spike ectodomains obtained in an identical assay.
Antibody production
VH-FC ab8, Fab S309, Fab S2M11, Fab 4-8, Fab 4A8, and Fab DH1052 were produced as previously described.30 , 40 Briefly, plasmids encoding the light and heavy chains for each Fab were used to co-transfect Expi293 cells at a density of 3 × 106 cells/mL using linear polyethylenimine (Polysciences Cat# 23966-1). The heavy chains were designed to incorporate a C-terminal 6x histidine tag. 24 hours following transfection, the media was supplemented with 2.2 mM valproic acid, and expression was continued for 3–5 days at 37°C, 8% CO2. The supernatant was harvested by centrifugation and filtered through a 0.22-μm filter prior to loading onto a 5 mL HisTrap excel column (Cytiva). The column was washed for 20 CVs with wash buffer (20 mM Tris pH 8.0, 500 mM NaCl) and 5 CVs of wash buffer supplemented with 20 mM imidazole. The protein was eluted with elution buffer (20 mM Tris pH 8.0, 500 mM NaCl, 500 mM imidazole). Elution fractions containing the protein were pooled and concentrated (Amicon Ultra 10 kDa cut off, Millipore Sigma) prior to gel filtration. Gel filtration was conducted using a Superose 6 10/300 GL column (Cytiva) pre-equilibrated with GF buffer (20 mM Tris pH 8.0, 150 mM NaCl). Peak fractions corresponding to soluble protein were pooled and concentrated to 8–20 mg/mL (Amicon Ultra 10 kDa cut off, Millipore Sigma). Fabs were stored at 4°C until use.
Monoclonal antibody enzyme linked immunosorbent assay
For ELISA, 50 μL of wild-type (D614G), BA.1, or BA.2 SARS-CoV-2 S proteins were coated onto 96-well MaxiSorp plates at 2 μg/mL in TBS overnight at 4°C. All washing steps were performed three times with TBS +0.05% Tween 20 (TBS-T). After washing, wells were incubated with blocking buffer (TBS-T + 2% casein) for 1 hour at room temperature. After washing, wells were incubated with dilutions of primary antibodies in TBS-T + 0.5% casein buffer for 1 hour at room temperature. After washing, wells were incubated with goat anti-human IgG (Jackson ImmunoResearch) at a 1:8,000 dilution in TBS-T + 2% casein buffer for 1 hour at room temperature. After washing, the substrate solution (Pierce 1-Step) was used for color development according to the manufacturer’s specifications. Optical density at 450 nm was read on a Varioskan Lux plate reader (Thermo Fisher Scientific).
Polyclonal antibody purification and ELISA
BA.1 spike ectodomain, RBD, NTD, and irrelevant antibody functionalized resin were prepared using Pierce NHS-Activated Agarose Slurry according to manufacturer’s instructions. Pooled sera from BA.1 convalescent patients was diluted by a factor of two in PBS, split into four batches, and incubated with each resin at 4°C overnight. After washing three times with PBS, antigen specific polyclonal antibody preparations were eluted from resin using 100 mM Glycine pH 2.5 and immediately neutralized using 1 M Tris pH 8 buffer. Polyclonal preparations were concentrated using 10 kDa cut off spin columns (Amicon).
For ELISA, 100 μL of either BA.1 or BA.2 spike protein ectodomain in PBS were coated on 96-well MaxiSorp plates at 2 μg/mL in PBS overnight. All washing steps were performed three times with PBS +0.05% Tween 20 (PBS-T). After washing, wells were incubated with blocking buffer (PBS-T + 1% casein) for 1 h at room temperature. Then polyclonal antibody preparations were serially diluted in PBS-T + 0.5% casein and incubated in wells for 1 h at room temperature. After washing, wells were incubated with goat anti-human IgG (Jackson ImmunoResearch) at a 1:5,000 dilution in PBS-T + 1% casein buffer for 1 h at room temperature. After washing, the substrate solution (Pierce 1-Step) was used for color development according to the manufacturer’s specifications. Optical density at 450 nm was read on a Varioskan Lux plate reader (Thermo Fisher Scientific). An identical ELISA was performed using anti-his tag antibody (abcam Cat# ab18184) as a spike protein loading control. Area under the curve (AUC) was calculated for each experiment and normalized to the anti-his tag ELISA control for data analysis.
Electron microscopy sample preparation and data collection
For cryo-EM, 2.25 mg/mL S protein, S protein-ACE2 complex (1:2.3 S protein trimer: ACE2 molar ratio), and S protein-4A8 complex (1:6.4 S protein trimer: 4A8 Fab) samples were vitrified on Quantifoil R1.2/1.3 Cu mesh 200 holey carbon grids using a Vitrobot Mark IV (ThermoFisher Scientific) plunge freezing device. Grids were first glow discharged for 15 seconds using a Pelco easiGlow glow discharge unit (Ted Pella) and then 1.8 μL of protein suspension was applied to the surface of the grid at a temperature of 10°C and a humidity level of >98%. Grids were then blotted (12 seconds, blot force −10) and plunge frozen into liquid ethane. S protein-ACE2 complex grids were imaged using a 300 kV Titan Krios G4 transmission electron microscope (Thermo Fisher Scientific) equipped with a Falcon4 direct electron detector in electron event registration (EER) mode. Movies were collected at 155,000x magnification (calibrated pixel size of 0.5 Å per physical pixel) over a defocus range of −0.5 μm to −2 μm with a total dose of 40 e−/Å2 using EPU automated acquisition software (ThermoFisher Scientific). Grids containing the BA.2 Omicron S protein alone were imaged using a 200 kV Glacios transmission electron microscope (ThermoFisher Scientific) equipped with a Falcon4 camera operated in EER mode. Movies were collected using EPU at 190,000x magnification (physical pixel size 0.7 Å) over a defocus range of −0.5 μm to −2 μm and a total accumulated dose of 40 e−/Å2.
Image processing
The detailed data processing workflow is illustrated in Figure S1. All data processing was carried out in cryoSPARC. Motion correction in patch mode (EER upsampling factor 1, EER number of fractions 40), CTF estimation in patch mode, blob particle picking, and particle extraction (box size 400 Å) were performed in real time in cryoSPARC live. Multiple rounds of 3D and/or 2D classification were used to clean the particle stack. The final homogeneous refinement was performed with per particle CTF estimation and aberration correction. For the complexes of spike protein ectodomain and human/mouse ACE2, local refinement was performed with a soft mask covering a single RBD and its bound ACE2. For apo spike protein and complexes of spike protein with 4A8, to better resolve the N-terminal domain or the interface between the N-terminal domain and 4A8, particles were symmetry expanded after another round of global refinement with C3 symmetry, and then refined with a soft mask covering a single N-terminal domain or a single N-terminal domain and its bound 4A8.
Model building and refinement
For models of spike protein ectodomain alone, the SARS-CoV-2 HexaPro S trimer (PDB ID 7MJG) was used as an initial model and docked into the map. Then, mutation and manual adjustment were performed in COOT, followed by iterative rounds of real-space refinement in COOT and Phenix. For models of spike-ACE2/4A8 complex, the subcomplexes RBD-ACE2 or NTD-4A8 were built and refined against local refinement maps. The resulting models were then docked into global refinement maps together with the other individual domains of the spike protein. Models were validated using MolProbity. Figures were prepared using UCSF Chimera, UCSF ChimeraX, and PyMOL (Schrodinger, LLC).
Quantification and statistical analysis
Quantification and statistical analysis information can be found in the figure legends associated with the data. GraphPad Prism 7.0 was used to perform the one-way ANOVA tests presented in Figures 2A–2C. Normal population distribution, similar variance, and sample independence were assumptions made during this ANOVA analysis.
Acknowledgments
We thank the study participants and The BC Center for Disease Control Public Health Laboratory along with Drs. Inna Sekirov and Ana Citlali Marquez for providing BA.1 convalescent sera and patient demographic information. This work was supported by awards to S.S. from a Canada Excellence Research Chairs, Government of Canada Award; the VGH Foundation; Genome BC, Canada; and the Tai Hung Fai Charitable Foundation. J.W.S. is supported by a CIHR Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award (CGS-D) and a University of British Columbia President’s Academic Excellence Initiative PhD Award. D.M. is supported by a CIHR Vanier Canada Graduate Scholarship. S.C. is supported by a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship Master’s Award (CGS-M).
Author contributions
J.W.S. and D.M. carried out expression and purification of the S proteins and antibodies. D.M. and J.W.S. performed the surface plasmon resonance (SPR)-binding assays. J.W.S., D.M., S.C., and F.V. performed the antibody-binding experiments. A.M.B. and K.S.T. carried out the experimental components of cryo-EM and electron microscopy including specimen preparation and data collection. X.Z. carried out all computational aspects of image processing and structure determination. J.W.S., D.M., and X.Z. interpreted and analyzed the cryo-EM structures. J.W.S., D.M., and S.S. prepared the manuscript with input from all authors.
Declarations of interests
S.S. is the founder and CEO of Gandeeva Therapeutics, a drug discovery company based in Vancouver.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111964.
Supplemental information
Data and code availability
-
•
Cryo-EM reconstructions and atomic models generated during this study are available at the PDB and EMBD databases (https://www.rcsb.org; http://emsearch.rutgers.edu), with PDB and EMBD identifiers listed in the key resources table and Table S1.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Starr T.N., Czudnochowski N., Liu Z., Zatta F., Park Y.-J., Addetia A., Pinto D., Beltramello M., Hernandez P., Greaney A.J., et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature. 2021;597:97–102. doi: 10.1038/s41586-021-03807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCallum M., De Marco A., Lempp F.A., Tortorici M.A., Pinto D., Walls A.C., Beltramello M., Chen A., Liu Z., Zatta F., et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell. 2021;184:2332–2347.e16. doi: 10.1016/j.cell.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J., Cai Y., Xiao T., Lu J., Peng H., Sterling S.M., Walsh R.M., Rits-Volloch S., Zhu H., Woosley A.N., et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science. 2021;372:525–530. doi: 10.1126/science.abf2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J., Xiao T., Cai Y., Lavine C.L., Peng H., Zhu H., Anand K., Tong P., Gautam A., Mayer M.L., et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science. 2021;374:1353–1360. doi: 10.1126/science.abl9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gobeil S.M.-C., Janowska K., McDowell S., Mansouri K., Parks R., Stalls V., Kopp M.F., Manne K., Li D., Wiehe K., et al. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science. 2021;373:eabi6226. doi: 10.1126/science.abi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannar D., Saville J.W., Zhu X., Srivastava S.S., Berezuk A.M., Tuttle K.S., Marquez A.C., Sekirov I., Subramaniam S. SARS-CoV-2 Omicron variant: antibody evasion and cryo-EM structure of spike protein - ACE2 complex. Science. 2022;375:760–764. doi: 10.1126/science.abn7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stalls V., Lindenberger J., Gobeil S.M.C., Henderson R., Parks R., Barr M., Deyton M., Martin M., Janowska K., Huang X., et al. Cryo-EM structures of SARS-CoV-2 Omicron BA.2 spike. Cell Rep. 2022;39:111009. doi: 10.1016/j.celrep.2022.111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Tang W., Gao H., Lavine C.L., Shi W., Peng H., Zhu H., Anand K., Kosikova M., Kwon H.J., et al. Cold Spring Harbor Laboratory; 2022. Structural and Functional Characteristics of SARS-CoV-2 Omicron Subvariant BA.2 Spike. [DOI] [PubMed] [Google Scholar]
- 12.Cameroni E., Bowen J.E., Rosen L.E., Saliba C., Zepeda S.K., Culap K., Pinto D., Vanblargan L.A., De Marco A., Di Iulio J., et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2022;602:664–670. doi: 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y., Wu C., Cao X., Gu C., Liu H., Jiang M., Wang X., Yuan Q., Wu K., Liu J., et al. Structural and biochemical mechanism for increased infectivity and immune evasion of Omicron BA.2 variant compared to BA.1 and their possible mouse origins. Cell Res. 2022;32:609–620. doi: 10.1038/s41422-022-00672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann M., Krüger N., Schulz S., Cossmann A., Rocha C., Kempf A., Nehlmeier I., Graichen L., Moldenhauer A.-S., Winkler M.S., et al. The Omicron variant is highly resistant against antibody-mediated neutralization: implications for control of the COVID-19 pandemic. Cell. 2022;185:447–456.e11. doi: 10.1016/j.cell.2021.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang K., Zhang Y., Hui X., Zhao Y., Gong W., Wang T., Zhang S., Yang Y., Deng F., Zhang Q., et al. Q493K and Q498H substitutions in Spike promote adaptation of SARS-CoV-2 in mice. EBioMedicine. 2021;67:103381. doi: 10.1016/j.ebiom.2021.103381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gawish R., Starkl P., Pimenov L., Hladik A., Lakovits K., Oberndorfer F., Cronin S.J., Ohradanova-Repic A., Wirnsberger G., Agerer B., et al. ACE2 is the critical in vivo receptor for SARS-CoV-2 in a novel COVID-19 mouse model with TNF- and IFNγ-driven immunopathology. Elife. 2022;11:e74623. doi: 10.7554/elife.74623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conceicao C., Thakur N., Human S., Kelly J.T., Logan L., Bialy D., Bhat S., Stevenson-Leggett P., Zagrajek A.K., Hollinghurst P., et al. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 2020;18:e3001016. doi: 10.1371/journal.pbio.3001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W., Schäfer A., Kulkarni S.S., Liu X., Martinez D.R., Chen C., Sun Z., Leist S.R., Drelich A., Zhang L., et al. High potency of a bivalent human VH domain in SARS-CoV-2 animal models. Cell. 2020;183:429–441.e16. doi: 10.1016/j.cell.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tortorici M.A., Beltramello M., Lempp F.A., Pinto D., Dang H.V., Rosen L.E., McCallum M., Bowen J., Minola A., Jaconi S., et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science. 2020;370:950–957. doi: 10.1126/science.abe3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinto D., Park Y.-J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 21.Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q., Luo Y., Chan J.F.W., Sahi V., Figueroa A., et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584:450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- 22.Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y., et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369:650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li D., Edwards R.J., Manne K., Martinez D.R., Schäfer A., Alam S.M., Wiehe K., Lu X., Parks R., Sutherland L.L., et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell. 2021;184:4203–4219.e32. doi: 10.1016/j.cell.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanblargan L.A., Errico J.M., Halfmann P.J., Zost S.J., Crowe J.E., Purcell L.A., Kawaoka Y., Corti D., Fremont D.H., Diamond M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022;28:490–495. doi: 10.1038/s41591-021-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iketani S., Liu L., Guo Y., Liu L., Chan J.F.W., Huang Y., Wang M., Luo Y., Yu J., Chu H., et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature. 2022;604:553–556. doi: 10.1038/s41586-022-04594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisk J.D., Powell D.R., Gellman S.H. Control of hairpin formation via proline configuration in parallel β-sheet model systems. J. Am. Chem. Soc. 2000;122:5443–5447. doi: 10.1021/ja9929483. [DOI] [Google Scholar]
- 27.Lok S.-M. An NTD supersite of attack. Cell Host Microbe. 2021;29:744–746. doi: 10.1016/j.chom.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ai J., Wang X., He X., Zhao X., Zhang Y., Jiang Y., Li M., Cui Y., Chen Y., Qiao R., et al. Antibody evasion of SARS-CoV-2 Omicron BA.1, BA.1.1, BA.2, and BA.3 sub-lineages. Cell Host Microbe. 2022;30:1077–1083.e4. doi: 10.1016/j.chom.2022.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannar D., Saville J.W., Zhu X., Srivastava S.S., Berezuk A.M., Zhou S., Tuttle K.S., Kim A., Li W., Dimitrov D.S., Subramaniam S. Structural analysis of receptor binding domain mutations in SARS-CoV-2 variants of concern that modulate ACE2 and antibody binding. Cell Rep. 2021;37:110156. doi: 10.1016/j.celrep.2021.110156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konishi T. Mutations in SARS-CoV-2 are on the increase against the acquired immunity. PLoS One. 2022;17:e0271305. doi: 10.1371/journal.pone.0271305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., COVID-19 Genomics UK COG-UK Consortium. Peacock S.J., Robertson D.L. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu X., Mannar D., Srivastava S.S., Berezuk A.M., Demers J.P., Saville J.W., Leopold K., Li W., Dimitrov D.S., Tuttle K.S., et al. Cryo-electron microscopy structures of the N501Y SARS-CoV-2 spike protein in complex with ACE2 and 2 potent neutralizing antibodies. PLoS Biol. 2021;19:e3001237. doi: 10.1371/journal.pbio.3001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayarri-Olmos R., Johnsen L.B., Idorn M., Reinert L.S., Rosbjerg A., Vang S., Hansen C.B., Helgstrand C., Bjelke J.R., Bak-Thomsen T., et al. The alpha/B.1.1.7 SARS-CoV-2 variant exhibits significantly higher affinity for ACE-2 and requires lower inoculation doses to cause disease in K18-hACE2 mice. Elife. 2021;10:e70002. doi: 10.7554/elife.70002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandegrift K.J., Yon M., Surendran-Nair M., Gontu A., Amirthalingam S., Nissly R.H., Levine N., Stuber T., Denicola A.J., Boulanger J.R., et al. Cold Spring Harbor Laboratory; 2022. Detection of SARS-CoV-2 Omicron Variant (B.1.1.529) Infection of White-Tailed Deer. [Google Scholar]
- 36.Pickering B., Lung O., Maguire F., Kruczkiewicz P., Kotwa J.D., Buchanan T., Gagnier M., Guthrie J.L., Jardine C.M., Marchand-Austin A., et al. Cold Spring Harbor Laboratory; 2022. Highly Divergent White-Tailed Deer SARS-CoV-2 with Potential Deer-To-Human Transmission. [Google Scholar]
- 37.Sánchez-Morales L., Sánchez-Vizcaíno J.M., Pérez-Sancho M., Domínguez L., Barroso-Arévalo S. Cold Spring Harbor Laboratory; 2022. The Omicron (B.1.1.529) SARS-CoV-2 Variant of Concern Also Affects Companion Animals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh C.-L., Goldsmith J.A., Schaub J.M., Divenere A.M., Kuo H.-C., Javanmardi K., Le K.C., Wrapp D., Lee A.G., Liu Y., et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science. 2020;369:1501–1505. doi: 10.1126/science.abd0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Q., Hughes T.A., Kelkar A., Yu X., Cheng K., Park S., Huang W.-C., Lovell J.F., Neelamegham S. Inhibition of SARS-CoV-2 viral entry upon blocking N- and O-glycan elaboration. Elife. 2020;9:e61552. doi: 10.7554/elife.61552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saville J.W., Mannar D., Zhu X., Srivastava S.S., Berezuk A.M., Demers J.-P., Zhou S., Tuttle K.S., Sekirov I., Kim A., et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat. Commun. 2022;13:742. doi: 10.1038/s41467-022-28324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheres S.H.W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development ofCoot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liebschner D., Afonine P.V., Baker M.L., Bunkóczi G., Chen V.B., Croll T.I., Hintze B., Hung L.-W., Jain S., McCoy A.J., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019;75:861–877. doi: 10.1107/s2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mannar D., Saville J.W., Sun Z., Zhu X., Marti M.M., Srivastava S.S., Berezuk A.M., Zhou S., Tuttle K.S., Sobolewski M.D., et al. SARS-CoV-2 variants of concern: spike protein mutational analysis and epitope for broad neutralization. Nat. Commun. 2022;13:4696. doi: 10.1038/s41467-022-32262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Cryo-EM reconstructions and atomic models generated during this study are available at the PDB and EMBD databases (https://www.rcsb.org; http://emsearch.rutgers.edu), with PDB and EMBD identifiers listed in the key resources table and Table S1.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.