

Abstract
Advances in long-read sequencing technologies and genome assembly methods have enabled the recent completion of the first Telomere-to-Telomere (T2T) human genome assembly, which resolves complex segmental duplications and large tandem repeats, including centromeric satellite arrays in a complete hydatidiform mole (CHM13). Though derived from highly accurate sequences, evaluation revealed evidence of small errors and structural misassemblies in the initial T2T draft assembly. To correct these errors, we designed a novel repeat-aware polishing strategy that made accurate assembly corrections in large repeats without overcorrection, ultimately fixing 51% of the existing errors and improving the assembly QV from 70.2 to 73.9 measured from PacBio HiFi and Illumina k-mers. By comparing our results to standard automated polishing tools, we outline common polishing errors and offer practical suggestions for genome projects with limited resources. We also show how sequencing biases in both HiFi and Oxford Nanopore Technologies reads cause signature assembly errors that can be corrected with a diverse panel of sequencing technologies.
Introduction
Genome assembly is a foundational practice of quantitative biological research with increasing utility. By representing the genomic sequence of a sample of interest, genome assemblies enable researchers to annotate important features, quantify functional data, and discover/genotype genetic variants in a population1–6. Modern draft eukaryotic genome assembly graphs are typically built from a subset of four Whole Genome Shotgun (WGS) sequencing data types: Illumina short reads7,8, Oxford Nanopore Technologies (ONT) long reads9, PacBio Continuous Long Reads (CLR), and PacBio High-Fidelity (HiFi) long reads9,10, all of which have been extensively described7–10. However, we note that even the high-accuracy technologies produce sequencing data with some noise caused by platform-specific technical biases that require careful validation and polishing11,12,1,10,13.
Current genome assembly software attempts to reconstruct an individual or mosaic haplotype sequence from a subset of the above WGS data types. Some assemblers do not attempt to correct sequencing errors14, while others attempt to remove errors at various stages of the assembly process15–19. Regardless, technology-specific sequencing errors usually lead to distinct assembly errors13,20. Additionally, suboptimal assembly of specific loci often causes small and large errors in draft assemblies21,22. Here, we define “polishing” as the process of removing these errors from draft genome assemblies. Most polishing tools use an approach that is similar to sequence-based genetic variant discovery. Specifically, reads from the same individual are aligned to a draft assembly, and putative “variant”-like sequence edits are identified22,23. For diploid genomes, heterozygous “alternate” alleles are interpreted as genuine heterozygous variants, while homozygous alternate alleles are interpreted as assembly errors to be corrected. Some polishing tools, such as Quiver/Arrow, Nanopolish, Medaka, DeepVariant, and PEPPER leverage specialized models and prior knowledge to correct errors caused by technology-specific bias24–28. Others, such as Racon29, use generic methods to correct assembly errors with a subset of sequencing technologies29–31. These generic tools can utilize multiple data types to synergistically overcome technology-specific assembly errors.
The Telomere-to-Telomere (T2T) consortium recently convened an international workshop to assemble the first-ever complete sequence of a human genome. Because heterozygosity can complicate assembly algorithms, the consortium chose to assemble the highly homozygous genome of a complete hydatidiform mole cell line (CHM13hTERT; abbr. CHM13). Primarily using HiFi reads and supplemented with ONT reads, the consortium built a highly accurate and complete draft assembly (CHM13v0.9) that resolved all repeats with the exception of the rDNAs1. CHM13v0.9 contained about 1 error in every 10.5 Mb (Q70.22), and while this was highly accurate by traditional standards, we, as part of the consortium, sought to correct all lingering errors and omissions, including those within repeats, in this first truly complete assembly of a human genome.
Alignment-based validation and polishing commonly underperform within genomic repeats where alignments are ambiguous and inaccurate. For example, this challenge was identified while validating the first complete centromere and satellite repeats of the Chromosome X, requiring a customized conservative marker-assisted alignment32. To address this challenge, specialized repeat-aware alignment methods were recently developed, such as Winnowmap233,34 and TandemMapper35. However, to the best of our knowledge, no studies have utilized such methods to reliably validate and polish an entire genome assembly, including the most notoriously repetitive regions.
Here, we describe techniques developed to carefully evaluate the accuracy and completeness of a complete human genome assembly using multiple complementary WGS data types. Our evaluation of the initial draft CHM13 assembly discovered a number of assembly errors, therefore we created a custom polishing pipeline that was robust to genomic repeats and technology-specific biases. By applying this polishing pipeline to CHM13v0.9, we made 1,457 corrections, replacing a total of 12,234,603 bp of sequence with 10,152,653 bp of sequence, ultimately leading to the landmark CHM13v1.1 assembly representing the first complete human genome ever assembled. Our edits increased the estimated quality value to Q73.94 while mitigating haplotype switches. Further, we extended the truncated p-arm of chromosome 18 to encompass the complete telomere, and polished all telomeres with a new specialized PEPPER-DeepVariant model. Our careful evaluation of CHM13v1.1 confirmed that polishing did not overcorrect repeats (including rDNAs) nor did it cause false-positive edits causing invalid coding sequence reading frames. Additionally, we identified a comprehensive list of putatively heterozygous loci in the CHM13 cell line, as well as sporadic loci where read alignments still indicated exceptionally low coverage. Finally, we uncovered common mistakes made by standard automated polishing pipelines and provide best practices for other genome assembly projects.
Results
Initial evaluation of CHM13v0.9
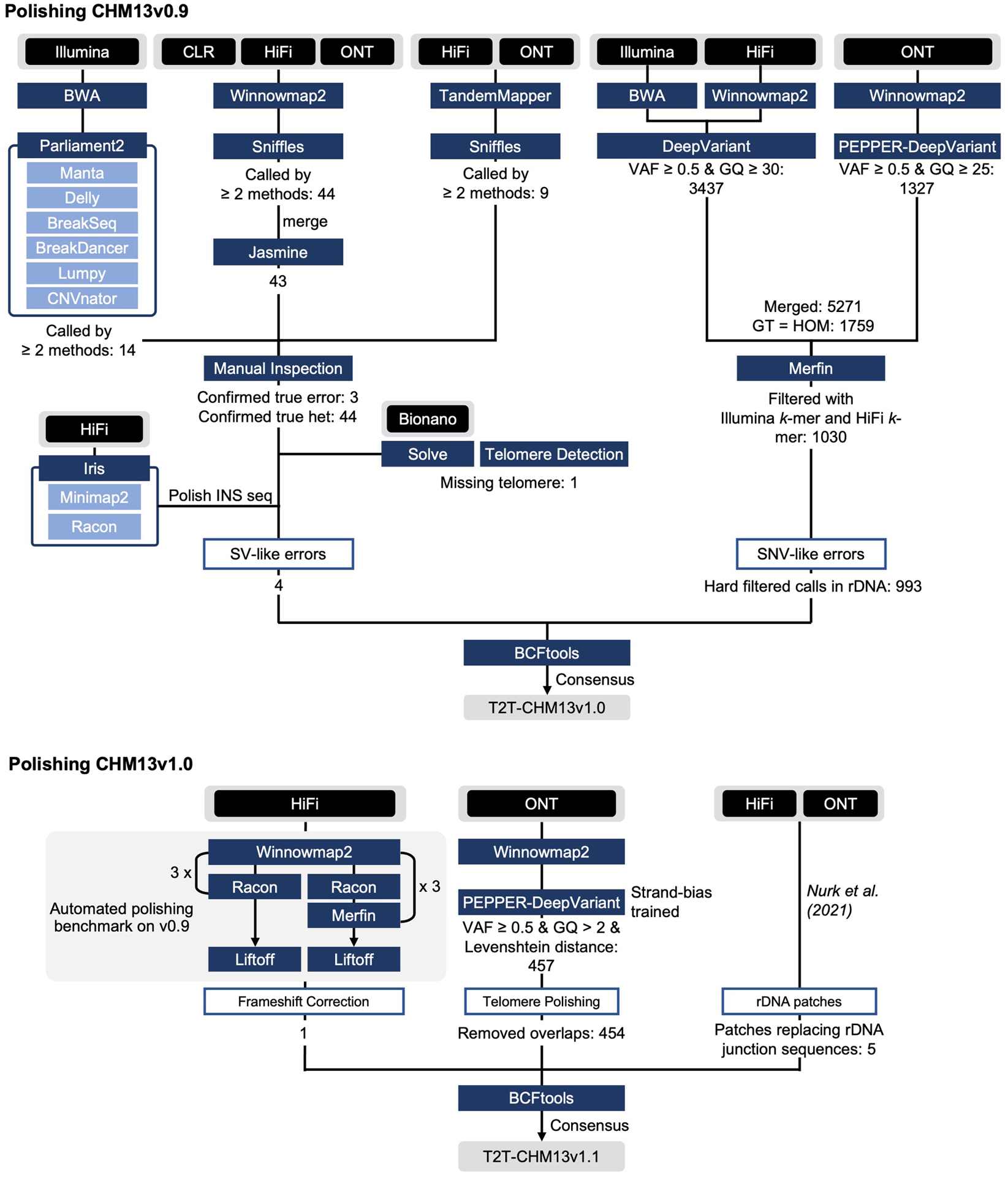
The T2T Consortium has collected a comprehensive and diverse set of publicly available WGS sequencing and genomic map data (Illumina PCR-free, PacBio HiFi, PacBio CLR, ONT, and Bionano optical maps) for the nearly-completely homozygous CHM13 cell line (https://github.com/marbl/CHM13). As part of the consortium, we drew upon these sequencing data to generate a custom pipeline (Fig. 1) to evaluate, identify and correct lingering errors in CHM13v0.9.
Figure 1 |. An overview of the evaluation and polishing strategy developed to achieve a complete human genome assembly.
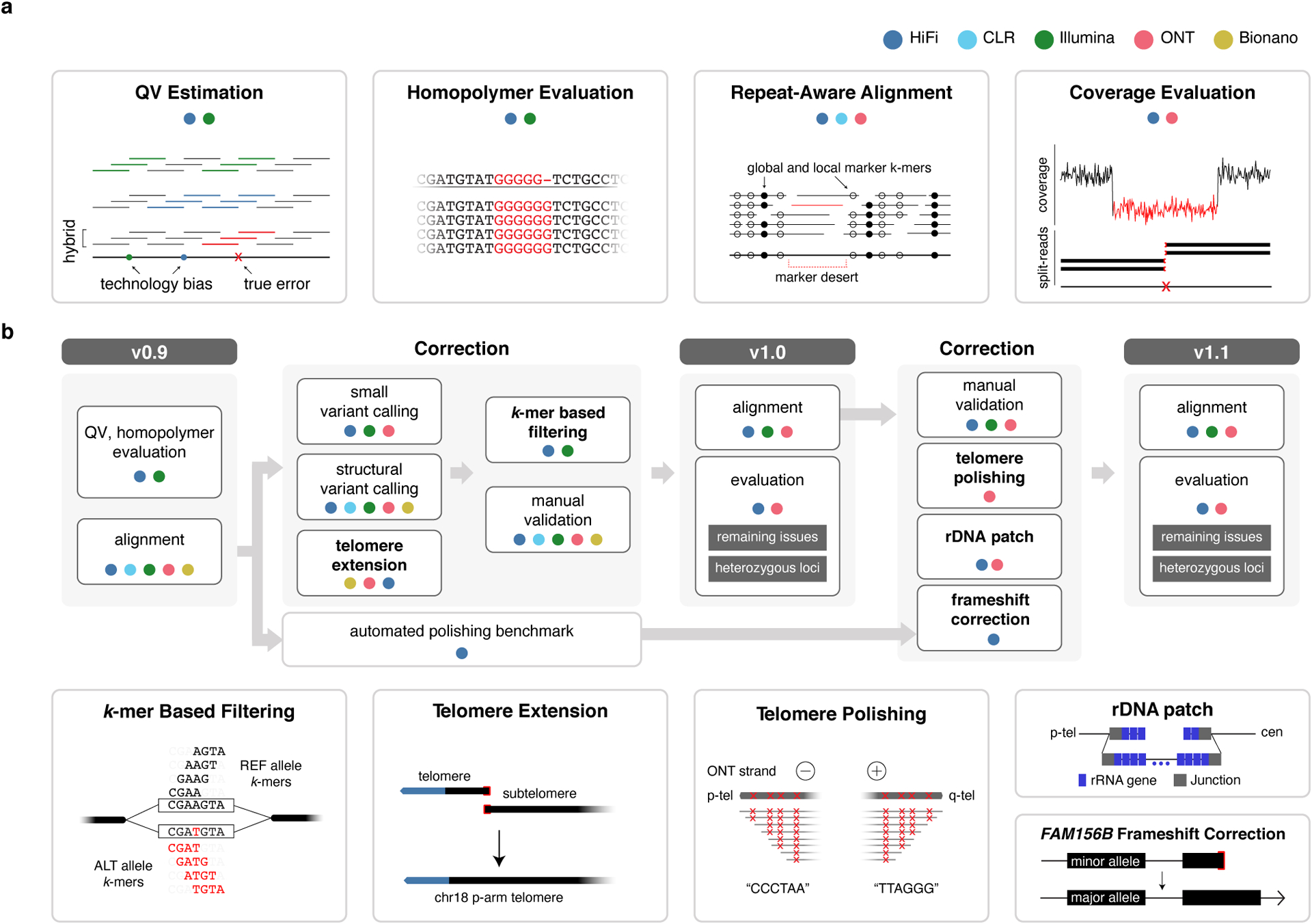
a, The evaluation strategies used to assess genome assembly accuracy before (CHM13v0.9) and after (CHM13v1.0 and CHM13v1.1) polishing. b, The “do no harm” polishing strategy developed and implemented to generate CHM13v1.0 and CHM13v1.1.
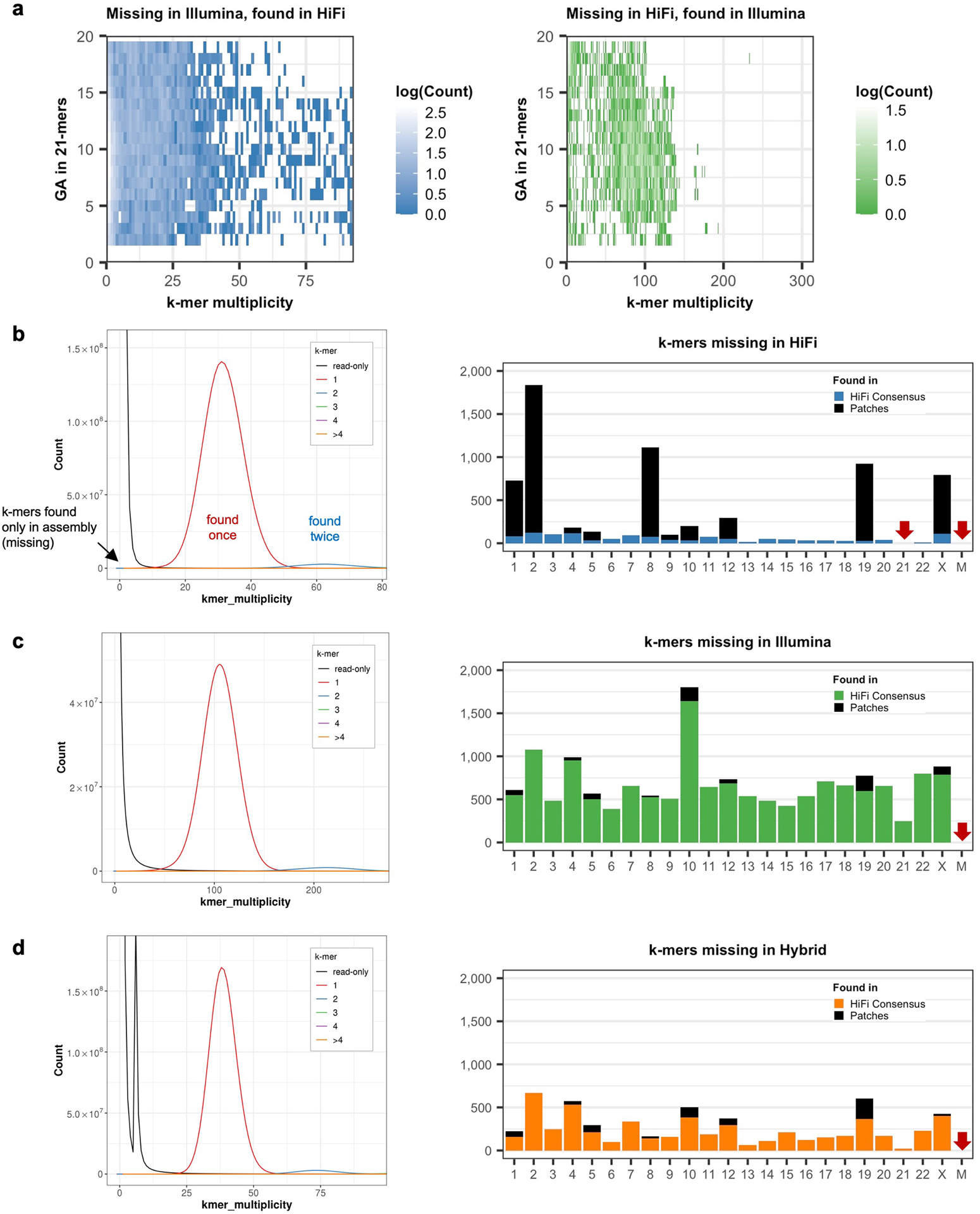
We first derived k-mer-based quality estimations (k = 21 bp) of CHM13v0.9 using Merqury36 using both Illumina and HiFi reads. The k-mer size was chosen to limit the collision rate to 0.5% given the estimated genome size of 3.05 Gbp of CHM1337. While estimating the Illumina reads QV, we found 15,723 k-mers present in the assembly and not the reads (erroneous k-mers), leading to an estimated base quality of Q66.09. Using HiFi reads, we found 6,881 error k-mers (Q69.68) (Fig. 2a). To test how technical sequencing bias may have influenced this QV estimation, we examined the k-mer multiplicity and sequence content of assembly k-mers absent from one technology but present in the other. Here, our results indicated that k-mers missing from Illumina reads were present with expected frequency in HiFi and were enriched for G/C bases. Conversely, k-mers missing in HiFi were present with higher frequency in Illumina reads with A/T base enrichment (Fig. 2b). However, we identified no particular enrichment pattern in the number of GA or CTs within the k-mers, possibly due to the short k-mer size chosen (Extended Data Fig. 1a). Most of the k-mers absent from HiFi reads were located in patches derived from a previous ONT-based assembly (CHM13v0.7), which were included to overcome regions of HiFi coverage dropout1 (Extended Data Fig. 1b–c). These findings highlighted that platform-specific sequencing biases were underestimating the QV when measured from a single sequencing platform. To overcome this, we created a hybrid k-mer database that combined these platforms to be used for QV estimation (Extended Data Fig. 1d). Unlike the default QV estimation in Merqury, we removed low frequency k-mers to avoid overestimated QVs caused by excessive noise accumulated from both platforms. We estimated base level accuracy as Q70.22 with 6,073 missing k-mers (Table 1). We note that this estimate does not account for the rarer case of k-mers present in the reads but misplaced or falsely duplicated in the assembly.
Figure 2 |. Sequencing biases in PacBio HiFi and Illumina reads.
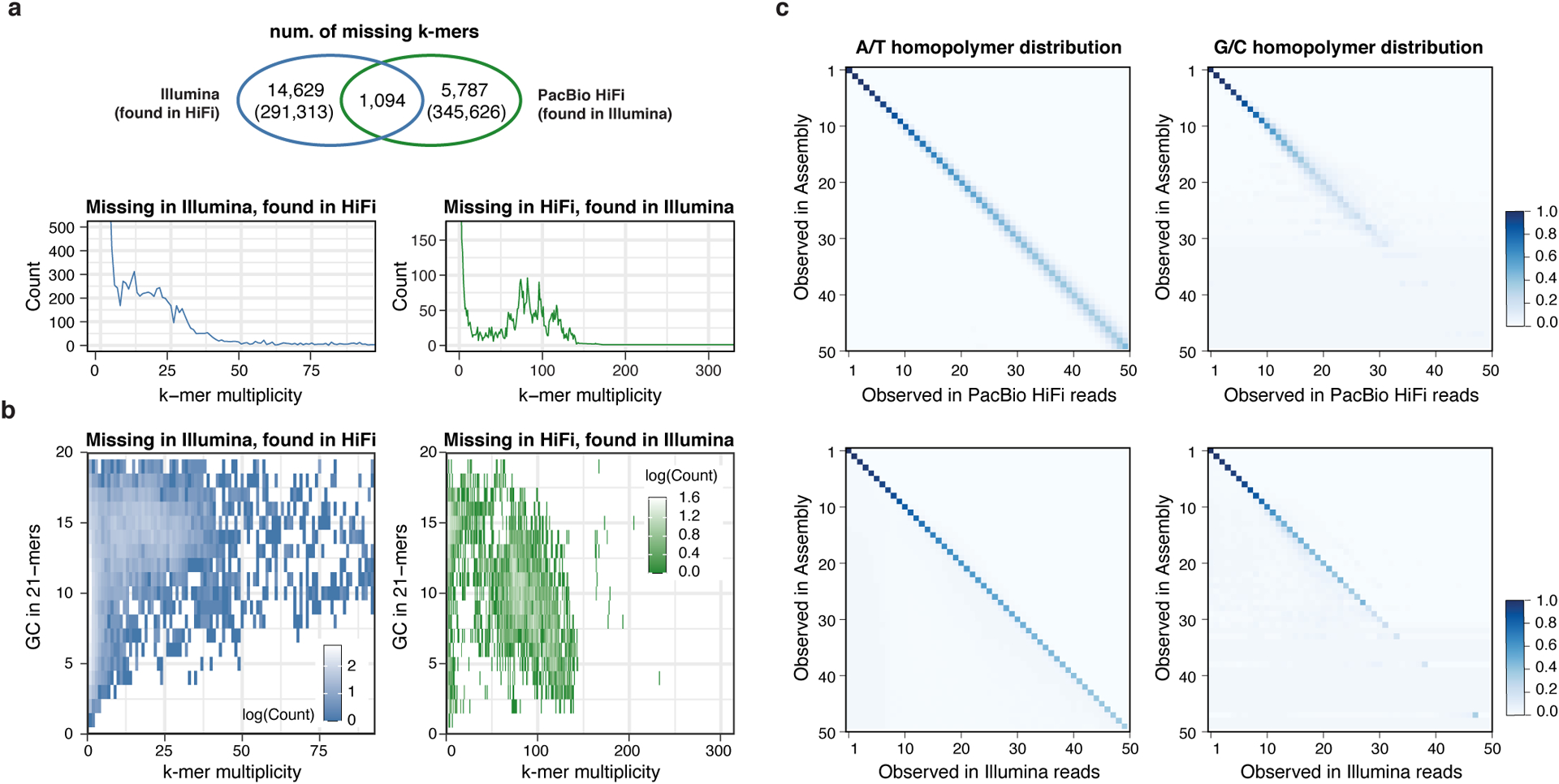
a, Venn Diagram of the “missing” k-mers found in the assembly but not in the HiFi reads (green) or Illumina reads (blue). Except for the 1,094 k-mers that were absent from both HiFi and Illumina reads, error k-mers were found in the other sequencing platform with expected frequency, matching the average sequencing coverage (lower panels). b, Missing k-mers from a with its GC contents, colored by the frequency observed. Low frequency erroneous k-mers did not have a clear GC bias. k-mers found only in HiFi had a higher GC percentage, while higher frequency k-mers tend to have more AT rich sequences in Illumina. c, Homopolymer length distribution observed in the assembly and in HiFi reads (upper) or Illumina reads (lower) aligned to that position. Longer homopolymers in the consensus are associated with length variability in HiFi reads especially in the GC homopolymers. The majority of the Illumina reads were concordant with the consensus.
Table 1. K-mer based consensus quality evaluation.
From each sequencing dataset and assembly versions, 21-mers were collected and compared with Merqury36.
| PacBio HiFi | Illumina | Hybrid | |
|---|---|---|---|
| QV | |||
| v0.9 | 69.68 | 66.09 | 70.22 |
| v1.0 | 69.88 | 67.28 | 72.62 |
| v1.1 | 69.80 | 67.86 | 73.94 |
| K-mers found only in assembly (Error k-mers) | |||
| v0.9 | 6,881 | 15,723 | 6,073 |
| v1.0 | 6,581 | 11,961 | 3,496 |
| v1.1 | 6,724 | 10,497 | 2,591 |
| K-mers found in both assembly and reads | |||
| v0.9 | 3,045,438,411 | 3,045,438,411 | 3,045,438,411 |
| v1.0 | 3,045,440,942 | 3,045,440,942 | 3,045,440,942 |
Despite the high accuracy of CHM13v0.9 (Q70.22), we expected to find consensus sequence errors related to the systematic presence of homopolymer- or repeat-specific issues in HiFi reads9,38. To detect these, we generated self-alignments by aligning CHM13 reads to CHM13v0.9 for each WGS sequencing technology. Though each data type required technology-specific alignment methods (Methods), we highlight our use of Winnowmap2 that enabled robust alignment of long-reads to both repetitive and non-repetitive regions of CHM13v0.933,34. To understand the homopolymer length differences between the assembly and the reads, we derived a confusion matrix from Illumina read alignments showing discordant representation of long homopolymers between the Illumina reads and the assembly (Fig. 2c). Altogether, the QV and homopolymer analysis suggested that CHM13v0.9 required polishing to maximize accuracy of a complete human genome.
Identification and correction of assembly errors
To address assembly flaws identified during evaluation, we aimed to establish a customized polishing pipeline that would avoid false positive polishing edits (especially in repeats) and maintain local haplotype consistency (Fig. 1b) (Extended Data Fig. 2). We identified and corrected small errors (<=50bp) using several small variant calling tools from self-alignments of Illumina, HiFi, and ONT reads to CHM13v0.9. To call both single-nucleotide polymorphisms (SNPs) and small insertions and deletions (INDELs), we applied a hybrid mode of DeepVariant26 that exploited both HiFi and Illumina read alignments39. Simultaneously, we used PEPPER-DeepVariant27 to generate additional SNP calls with ONT reads as it can yield high-quality SNP variants in difficult regions of the genome27,39 (See Supplementary Fig. 1 in ref. 27 for more details). We rigorously filtered all calls using Genotype Quality (GQ < 30 for the hybrid calls and GQ < 25 for ONT SNP calls) and Variant Allele Frequency (VAF < 0.5) to exclude any low-frequency false-positive calls (Extended Data Fig. 2). We chose VAF < 0.5 to avoid including heterozygous variants and the GQ threshold was chosen based on the previously reported calibration plot of DeepVariant that shows that calls that have quality above 25 or 30 are highly unlikely to result in false positives27,26. We then filtered all of the suggested alternate corrections with Merfin40, a tool concurrently developed by members of the T2T consortium, to avoid introducing error k-mers (Fig. 1b, Fig. 3c). Finally, we ignored variants near the distal or proximal rDNA junctions on the short arms of the acrocentric chromosomes to avoid homogenizing the alleles from the un-assembled rDNAs. After merging all variant calls, we identified 993 small variants (<=50bp) that represented potential assembly errors and heterozygous sites. From these 993 assembly edits, about two-thirds were homopolymer corrections (512) or low-complexity micro-satellite repeats composed of 2 distinct bases in homopolymer-compressed space (hereby noted as “2-mer”) consistent with prior observations of HiFi sequence errors or bias16. Across all 617 loci, we evaluated the edit distribution using both Illumina and HiFi reads and found that the majority of Illumina reads supported the longer homopolymer or 2-mer repeat lengths compared to HiFi reads, thereby uncovering systemic biases in both homopolymer and 2-mer length in HiFi reads16 that caused the propagation of these errors into the consensus assembly sequence (Fig. 3d).
Figure 3 |. Errors corrected after polishing.
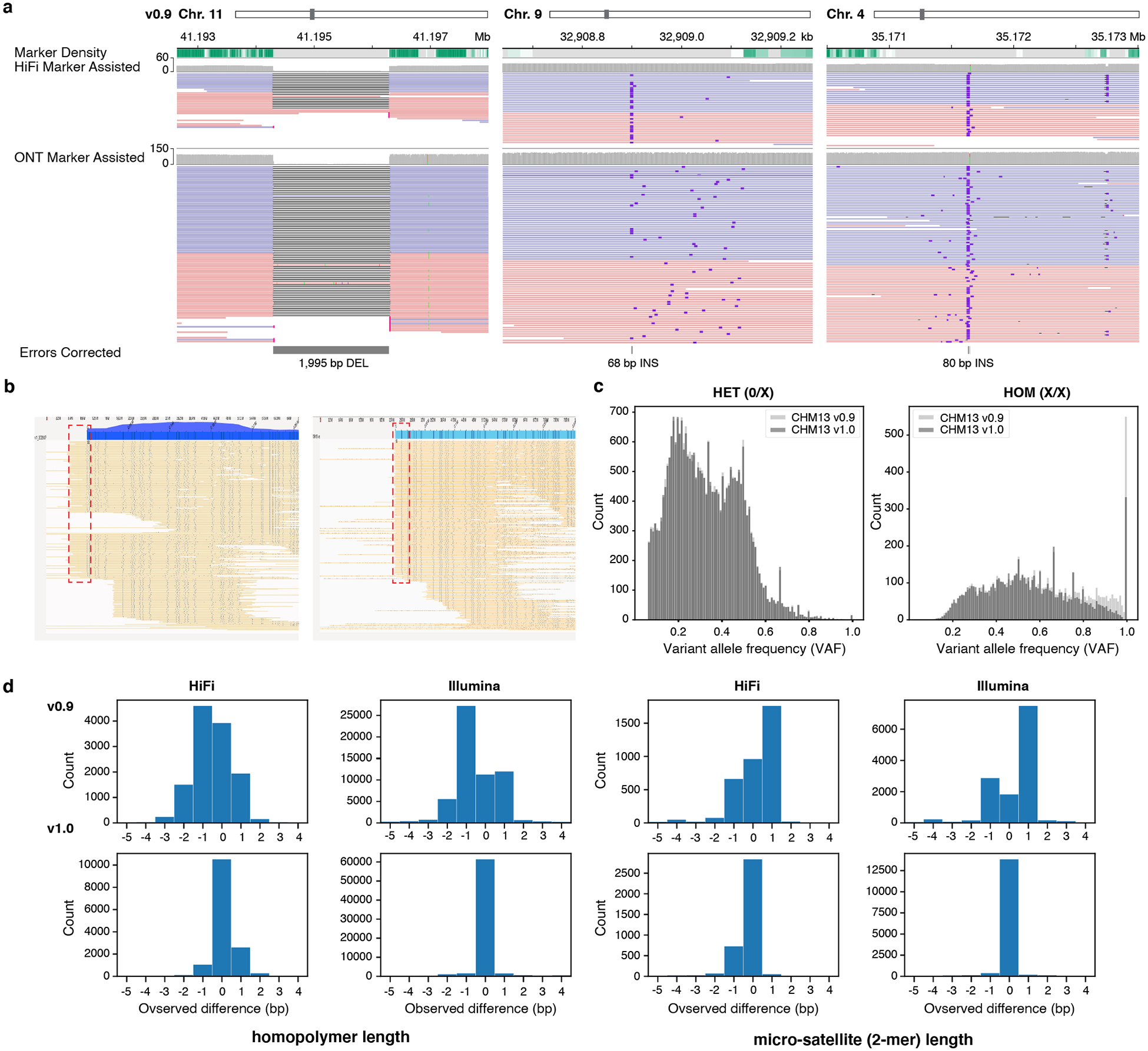
a, Three corrected SV-like errors. b, Bionano optical maps indicating the missing telomeric sequence on Chr. 18 p-arm (left) with a higher than average mapping coverage. This excessive coverage was removed after adding the missing telomeric sequence (right) and most of the Bionano molecules end at the end of the sequence. c, Variant allele frequency (VAF) of each variant called by DeepVariant hybrid (HiFi + Illumina) mode, before and after polishing. Most of the high frequency variants (errors) are removed after polishing, which were called ‘Homozygous’ variants. d, Total number of reads in each observed length difference (bp) between the assembly and the aligned reads at each edit position. Positive numbers indicate more bases are found in the reads, while negative numbers indicate fewer bases in the reads. Both the homopolymer and micro-satellite (2-mers in homopolymer compressed space) length difference became 0 after polishing.
We used Parliament241 and Sniffles42 to identify medium-sized (>50bp) assembly errors and heterozygous structural variants (SVs). Parliament2 runs 6 structural variant callers41 using short-read data, while the Sniffles detects structural variants using one of the long-read technologies (HiFi, ONT, and CLR). To improve specificity, we only considered Parliament2 calls supported by at least two SV callers and Sniffles calls supported by at least two long-read technologies. Similar to small variant detection, we excluded SVs called in the partial rDNA arrays and the HSat3 satellite repeat on chromosome 9. This pipeline identified a relatively small number of SV calls (66, see Extended Data Fig. 2) that we were able to manually curate via genome browsing. In total, we corrected three medium-sized assembly errors (replacing 1,998 bp of CHM13v0.9 sequence with 151 bp of new sequence) and we identified 44 heterozygous SVs (Fig. 3a and Extended Data Fig. 3b). We also identified a missing telomere sequence on the p-arm of chromosome 18 — a potential result of the string graph simplification process and confirmed through Bionano mapping (Fig. 1b and 3b). To correct this omission, we used the CHM13v0.9 graph to identify a set of HiFi reads expected to cover this locus1 and found ONT reads that mapped to the corresponding subtelomere and contained telomeric repeats. We used the ONT reads to derive a consensus chromosome 18 extension that was subsequently polished with the associated HiFi reads. After patching this telomere extension, we used Bionano alignments to confirm the accuracy of this locus (Fig. 3b). Altogether, the small and medium-sized variant calls along with the chromosome 18 telomere patch were combined into two distinct VCF files: a polishing edits file (homozygous ALT variants and the telomere patch) and a file for heterozygous variants (all other variants). We created the polished CHM13v1.0 assembly by incorporating these edits into the CHM13v0.9 with bcftools43.
We ensured polishing accuracy by extensive manual validation through visual inspection of the repeat-aware alignments, error k-mers, marker k-mers, and marker-assisted alignments. Here, we define “marker” k-mers as k-mers that occur only once in the assembly and in the expected single-copy coverage range of the read k-mer database and are highly likely to represent unique regions of the assembly (Extended Data Fig. 3b–d)44. To generate marker-assisted alignments, we filtered Winnowmap233 alignments to exclude any alignments that did not span marker k-mers (https://github.com/arangrhie/T2T-Polish/tree/master/marker_assisted). Our findings supported that most genomic loci contained a deep coverage of marker k-mers to facilitate marker-assisted alignment, except for a few highly repetitive regions (11.3 Mb in total) that lacked markers (termed “marker deserts”) (Fig. 1a and Extended Data Fig. 3c–d). In parallel, we used TandemMapper35 to detect structural errors in all centromeric regions, including identified marker deserts. TandemMapper35 used locally unique markers for the detection of marker order and orientation discrepancies between the assembly and associated long reads. We manually validated all large polishing edits and heterozygous SVs, and many small loci were validated ad hoc.
Validation of CHM13v1.0
Given the high completeness and accuracy standards of the T2T consortium, and knowing that polishing may introduce additional errors40, we took extra precautions to validate polishing edits and to ensure that edits did not degrade the quality of CHM13v0.9. First, we repeated self-alignment variant calling methods on CHM13v1.0, confirming that all edits made were correct (Fig. 3a). Through Bionano optical map alignments, we validated the structural accuracy of the chromosome 18 telomere patch and confirmed that all 46 telomeres were represented in CHM13v1.0 (Fig. 3b). Notably, our polishing led to a marked improvement in the distribution of GQ and VAF of small variant calls (Fig. 3c and Extended Data Fig. 4a). Our approach also increased the base level consensus accuracy from Q70.22 in CHM13v0.9 to Q72.62 in CHM13v1.0. Further, we found that error k-mers were uniformly distributed along each chromosome, suggesting that remaining errors were not clustered within certain genomic regions (Extended Data Fig. 4b–c). Upon re-evaluation of the homopolymers and 2-mers, we noted most of the biases we found in CHM13v0.9 from HiFi reads had been accurately removed, achieving an improved concordance with Illumina reads (Fig. 3d). Polishing did not induce invalid open reading frames (ORFs) in CHM13v0.9 transcripts with valid ORFs, and polishing corrected 16 invalid CHM13v0.9 ORFs (Supplementary Table 1).
Overall, we made a total of 112 polishing edits (impacting 267 bp) in centromeric regions32, with 15 (35 bp) of these edits occurring specifically in centromeric alpha-satellite higher-order repeat arrays. We made 134 edits (4,975 bp) in non-satellite segmental duplications45. Moreover, the polishing edits were neither enriched nor depleted in satellite repeats and segmental duplications (p=0.85, permutation test), suggesting that non-masked repeats were not over- or under-corrected compared to the rest of the genome (Extended Data Fig. 4c). Finally, through extensive manual inspection, we confirmed the reliability of the alignments for the three SV associated edits incorporated into CHM13v1.0 (Extended Data Fig. 5), and these efforts uncovered some heterozygous loci in the centromeres. These regions are under active investigation by the T2T consortium to both ensure their structure and understand their evolution32.
As an additional validation, we investigated potential rare or false collapses as well as rare or false duplications in CHM13v1.0. Here, based on k-mer estimates from both GRCh38 and CHM13v1.0 and from Illumina reads for 268 Simon’s Genome Diversity Project (SGDP) samples, we identified regions in CHM13v1.0 with a lower or higher copy number than both GRCh38 and 99% of the SGDP samples2. We found six regions of rare collapses in CHM13v1.0 that were not in GRCh38 (covering 205 kb, four from one single segmental duplication family). Both our HiFi read depth and Illumina k-mer-based copy number estimates suggest these six regions are likely rare copy number variants in CHM13 (e.g.,CHM13v1.0 has only a single copy of the 72 kb tandem duplication in GRCh38, Fig. 4a). Additionally, we found that CHM13v1.0 had 33x fewer false or rare collapses than GRCh38 (~185 loci covering 6.84 Mbp)6. We identified five regions (160 kb) with rare duplications in CHM13v1.0. This included a single 142 kb region that appeared to be a true, rare tandem duplication based on HiFi read depth and Illumina k-mer-based copy number estimates (Fig. 4b). Two of the smaller regions appeared to be true, rare tandem duplications, and two other small regions were identified during polishing as heterozygous or mosaic deletions, revealing potential tandem duplications arising during cell line division or immortalization. In summary, we found 7.5x fewer rare or falsely duplicated bases in CHM13v1.0 relative to the 12 likely falsely-duplicated regions affecting 1.2 Mb and 74 genes in GRCh386, including the medically relevant genes: CBS, CRYAA, and KCNE146.
Figure 4 |. Examples of the largest CHM13 regions with a copy number in the reference that differs from GRCh38 and most individuals.
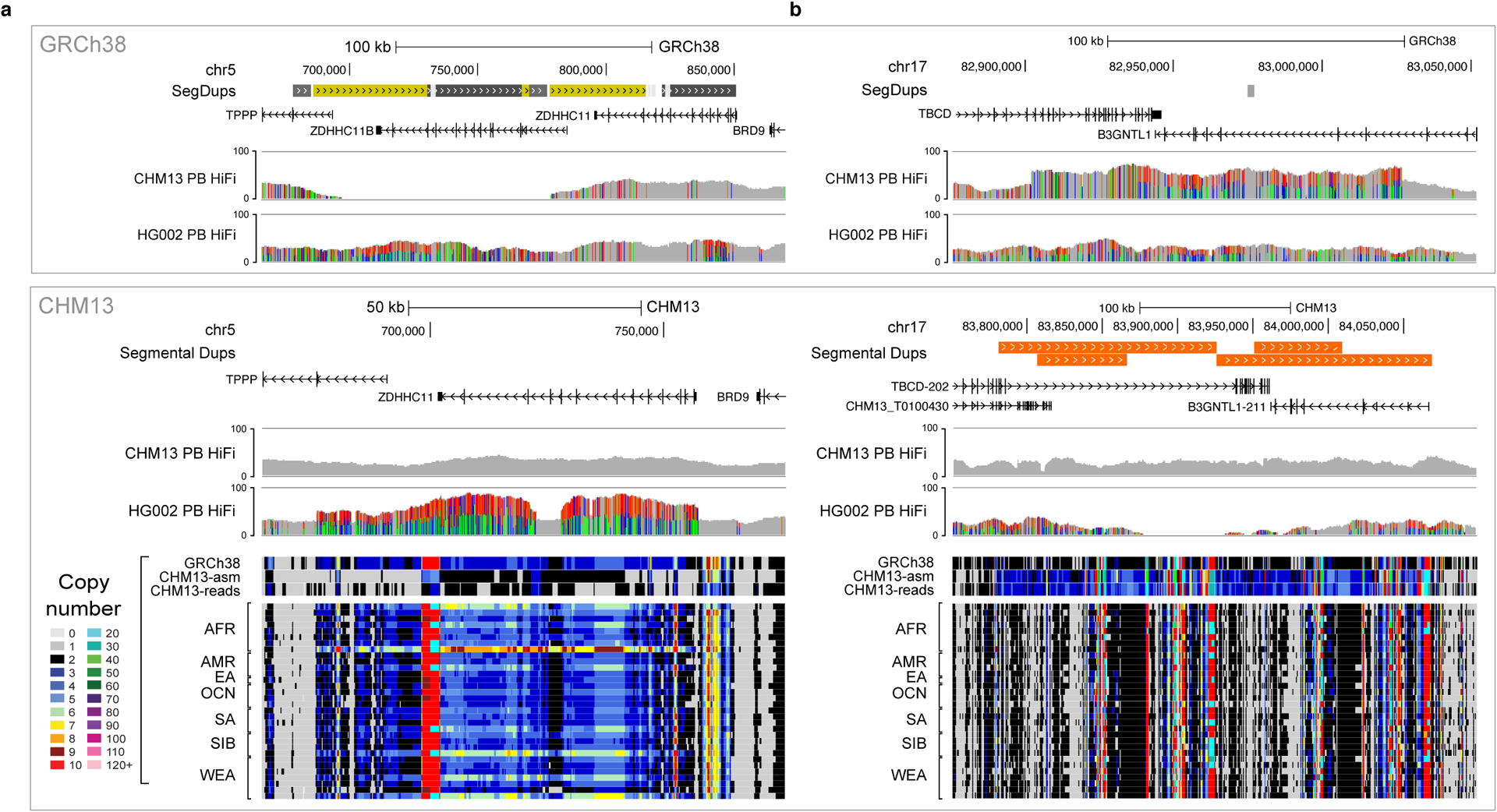
a, One of the two largest examples of rare collapses in CHM13, where one copy of a common 72 kb tandem duplication is absent in CHM13. b, The largest rare duplication in CHM13, a 142 kb tandem duplication of sequence in GRCh38 that is rare in the population. CHM13 and HG002 PacBio HiFi coverage tracks are displayed for both references, GRCh38 (top) and CHM13v1.0 (bottom), to demonstrate that CHM13 reads support the CHM13 copy-number but HG002 reads are consistent with the GRCh38 copy-number. Read-depth copy-number estimates in CHM13 are shown at the bottom for ‘k-merized’ versions of GRCh38 and CHM13v1.0 references, CHM13 Illumina reads, and Illumina reads from a diverse subset (n=34) of SGDP individuals.
Toward a completely polished sequence of a human genome
While evaluating CHM13v1.0, the T2T consortium successfully completed the construction of the rDNA models and their surrounding sequences on the p-arms of the five acrocentric chromosomes1. In parallel, we determined that all telomeric sequences remained unpolished. Specifically, in canonical [TTAGGG]n repeats, we found both HiFi read coverage dropouts and ONT strand bias impeded high quality variant calling (Extended Data Fig. 6a). For ONT, we observed only negative strands on the p-arm and only positive on the q-arm across all telomeric repeats at chromosomal ends; we suspect the ONT ultra-long transposon-based library preparation prevents reads from starting at chromosome ends, causing reads to only read into the telomere44,47. We tailored our PEPPER-based polishing approach and performed targeted telomere polishing to remove these errors remaining in telomeric sequences (Methods). Finally, automated polishing (described below), indicated that the FAM156B gene was heterozygous in CHM13v0.9 and CHM13v1.0 represented the rare minor allele (encoding a premature stop codon) at this locus. We replaced this minor allele with the other CHM13 allele encoding a full-length protein sequence. Overall, we made 454 telomere edits, producing longer stretches of maximum perfect matches to the canonical k-mer at each position across these telomeres compared to CHM13v1.0 (Extended Data Fig. 6b). Combined with the parallel completion of the five rDNA arrays, our final round of polishing led to an improved QV of Q73.94 for CHM13v1.1.
Again, to ensure updates did not compromise the high accuracy of the assembly and to identify any remaining issues, we carried out an additional round of SV detection and manual curation using HiFi and ONT with an updated Winnowmap2 alignment (Online Methods and Extended Data Fig. 7), classifying seven loci as remaining issues in CHM13v1.1 (Supplementary Table 2). We excluded CLR because the lower base accuracy compared to HiFi and ONT and shorter read length compared to ONT were adding no information. Bionano was also excluded as the molecules were lacking coverage in centromeric regions (Extended Data Fig. 8) and did not detect any structural issues beyond the missing telomere and a few heterozygous structural variants already identified by HiFi and ONT. Two loci located in the rDNA sequences appear to be a potential discrepancy between the model consensus sequence and actual reads or an artifact of mapping or sequencing bias. Lower consensus quality is indicated at two other loci, one detected with read alignments that were both low in coverage and identity, and one of which contained error k-mers detected by the hybrid dataset. One locus consisted of multiple insertions (<1kb) with breakpoints detected in low-complexity sequences associated with heterozygous variants and indicated a possible collapsed repeat (Extended Data Fig. 9) and an additional two loci joined and created an artificial chimeric haplotype (Extended Data Fig. 10). Additionally, we found 218 low coverage loci using HiFi (Supplementary Table 3), with 81.2% associated with GA-rich (78.0%) regions. The remaining 41 loci had signatures of lower consensus quality and alignment identity, and 30 had error k-mers detected from the hybrid k-mer dataset. In contrast, we detected one low-coverage locus using ONT that overlapped the GA-rich model rDNA sequence. We associated most remaining loci, totalling only 544.8 kb or <0.02% of assembled sequence, with lower consensus quality in regions lacking unique markers. Overall, we found 394 heterozygous regions, including regions with clusters of heterozygous variants (https://github.com/mrvollger/nucfreq), totalling 317 sites (~1.1 Mb).
We manually curated, both the breakpoints and alternate sequences associated with 47 heterozygous SVs, including sites previously inspected (CHM13v1.0) for SV-like error detection. We then investigated HiFi read alignment clippings and confirmed an association with clipping to both true heterozygous variant and spurious low frequency alignments. Additionally, we detected a further heterozygous inversion that went previously undetected.
A comparison to automated assembly polishing
To demonstrate the efficacy of the customized DeepVariant-based approach, we compared our semi-automated polishing approach used to create CHM13v1.0 (Q72.62) to a popular state-of-the-art automated polishing tool, Racon29,45. We iteratively polished CHM13v0.9 (three rounds) using Racon with PacBio HiFi alignments. While the QV improved from Q70.22 to Q70.48 after the first round of Racon polishing, it degraded with the subsequent second (Q70.26) and third (Q70.15) rounds, ultimately diminishing assembly accuracy as a result of overcorrection. We also found that Racon incorporated 7,268 alternate alleles from heterozygous variants identified by DeepVariant, thus potentially causing undesirable haplotype-switching in originally haplotype-consistent blocks. To examine how Racon polished large, highly similar repetitive elements, we counted the number of corrections in non-overlapping 1 Mb windows of the CHM13v0.9 assembly and measured local polishing rates. Unlike CHM13v1.0, Racon polishing showed a clear right-tail in the distribution of polishing rates, indicating the presence of polishing “hotspots”, defined here as loci with >60 corrections/Mb (Fig. 5a). The proximal and distal junctions of the rDNA units (masked from CHM13v1.0 polishing) were prevalent among these loci, a finding that reinforced the importance of masking known collapsed but resolved loci to avoid overcorrection. We also found non-rDNA loci that were preferentially polished by Racon, including satellite repeats such as the highly repetitive HSat3 region in chromosome 9. Finally, CHM13v1.0 made two corrections, recovering two protein-coding transcript’s open reading frames (ORFs), but Racon did not make these corrections (Supplementary Table 1). While CHM13v1.0 did not induce invalid ORFs in any transcripts, Racon made 10 corrections that caused invalid ORFs in 22 transcripts (from nine genes) (Fig. 5b). Most of these corrections occurred at homopolymer repeats, consistent with our previous findings that homopolymer bias in HiFi reads could lead to false expansion or contraction of homopolymers during polishing.
Figure 5 |. Errors made by automated polishing.
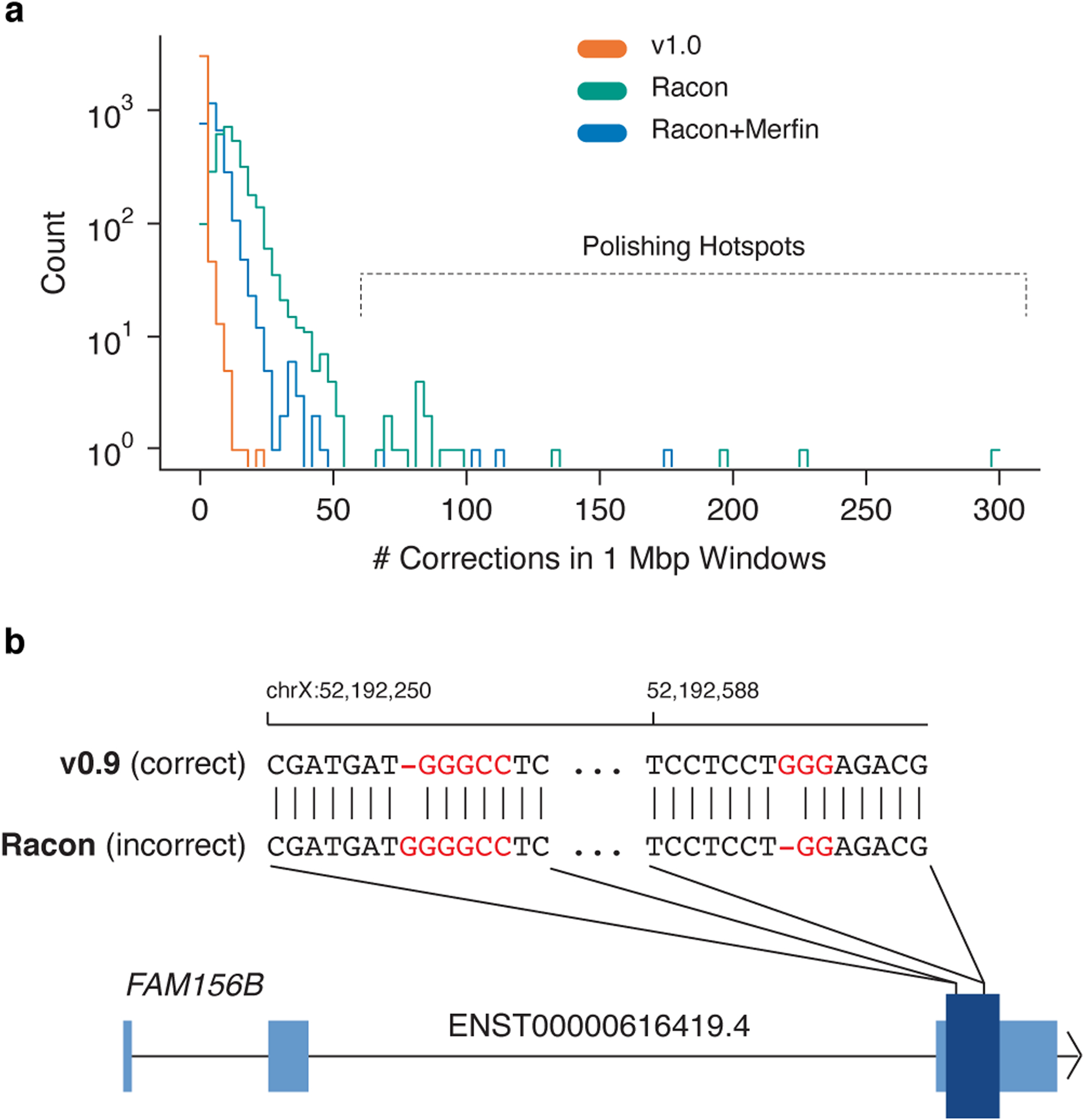
a, The distribution of the number of polishing edits made in non-overlapping 1 Mb windows of the CHM13v0.9 assembly. b, Two Racon polishing edits causing false frameshift errors in the FAM156B gene. Light blue indicates UTR and dark blue indicates the single coding sequence exon. Highlighted sequence indicates GC-rich homopolymers.
To overcome these relative shortcomings of Racon polishing, we tested polishing the CHM13v0.9 assembly with three iterative rounds of Racon followed by filtering with Merfin (Racon+Merfin). After each round of polishing, Merfin removed proposed Racon edits that incorporated false assembly k-mers. As expected, the Racon+Merfin assembly QV monotonically increased from Q70.22 to Q77.34, Q77.99 and Q78.12. However, Racon+Merfin still incorporated 2,274 alternate alleles from heterozygous variants and polishing hotspots were still evident, suggesting that some repeats were overcorrected (Fig. 5a). These overcorrections are not reflected in the QV measurements as k-mers from true heterozygous variants are considered ‘valid’ sequences. Merfin mitigated the 10 ORF-invalidating Racon corrections, however, Merfin also failed to correct the two reading frame corrections made in CHM13v1.0 but not Racon (Supplementary Table 1). Overall, when considering only automated polishing, we suggest that Racon and Merfin can be used together as a highly effective strategy for building reference assemblies with minimum false positive corrections. However, we would like to emphasize that a custom polishing pipeline with manual interventions is still required for preserving haplotype consistency and avoiding repeat overcorrection.
Discussion
The CHM13v0.9 human genome assembly represented a landmark achievement for the genomics community by representing previously unresolved repeats in a locally haplotype consistent assembly. Though it was imperative to validate and correct this draft assembly, successful polishing faced three major obstacles. First, while repeats are challenging to polish in any draft assembly, the CHM13v0.9 assembly represented hundreds of megabases of exceptionally large and complex repeats genome-wide, which could potentially induce false positive (overcorrection) or false negative polishing corrections. Secondly, though the CHM13 genome is mostly homozygous, we identified non-negligible levels of interspersed heterozygous variation. Therefore, it was essential to distinguish between heterozygous variants and polishing edits in order to maintain the original haplotype consistency. Finally, our evaluation of CHM13v0.9 discovered how homopolymer and coverage bias in HiFi reads caused assembly errors genome-wide. This analysis also revealed that standard methods for measuring QV can be influenced by technology-specific biases.
These obstacles necessitated a custom and contextualized polishing and evaluation model that capitalized on the wealth of available data to exploit the advantages of each sequencing platform. It also required the use of specialized aligners, hard masking, and manual intervention to avoid false polishing corrections within repeats. This polishing approach called for just 1,457 corrections including: p-arm of chromosome 18; 454 telomere corrections; 1 large deletion; 2 large insertions; 993 SNPs; 113 small insertions and 880 small deletions. Although the final CHM13v1.1 is highly accurate (Q73.9), we identified 225 loci that were recalcitrant to validation, and we have documented these loci along with 394 heterozygous loci (317 merged loci) (https://github.com/marbl/CHM13-issues/).
The high accuracy of CHM13v1.1 showcases the effectiveness of our informed selection and implementation of appropriate repeat-aware aligners33,35, k-mer evaluation and filtration tools, and highly accurate and sensitive variant callers27,40 whilst also highlighting the utility of capitalizing on the synergistic nature of multiple sequencing technology platforms. The minimal number of corrections implemented by our approach and uniform coverage (99.86%) exemplifies the high accuracy of the initial graph construction, with sequencing biases being associated with the remaining coverage fluctuations (223 regions were regions of HiFi dropouts, 77.5% found in GA/TC-rich, and AT-rich satellite sequences such as HSat2/3 and HSat1, were associated with in HiFi coverage increases and ONT coverage depletion, respectively)1.
In many respects, the T2T CHM13 genome assembly initiative is not representative of typical assembly projects. The success of the CHM13v1.0 assembly was enabled by the low level of heterozygosity of the CHM13 genome, advancements in sequencing technologies, a combination of sequencing technologies (HiFi, ONT, and Illumina), customized assembly algorithms, and a large dedicated team of scientists, yielding results currently not possible with limited resources and automated algorithms16,17. However, despite the unique and semi-automated nature of our polishing and evaluation endeavor, recent trends in DNA sequencing and genome assembly algorithms suggest that CHM13v1.1 is just a preview of an imminent wave of high-quality T2T reference genomes in other species48–50. It is therefore critical that the lessons outlined here be incorporated into the next generation of automated bioinformatics tools29,33,35,40. For immediate projects, combining data types, using phased reads with repeat-ware alignments, and carefully filtering polishing edits can improve automated polishing accuracy.
METHODS
Evaluating homopolymer concordance
By analyzing the homopolymer length agreement, we assessed sequencing platform-specific biases between reads and the assembly using both Illumina and HiFi reads through the runLengthMatrix submodule of Margin (https://github.com/UCSC-nanopore-cgl/margin). Here, we used Margin to convert the assembly sequence to a run-length encoded (RLE) sequence. For example, the sequence ACTTG became (ACTG, {1,1,2,1}) where ACTG represented the encoded sequence, and {1,1,2,1} represented the run-length for each nucleotide base. While encoding the sequence to run-length, Margin created a map of positions in the assembly to the RLE position. Using the position map, Margin converted the raw sequence alignment to run-length alignment by iterating through the matches between the read and the assembly and keeping track of the previous match in RLE space. This way, Margin created a matrix where each row represents a run-length of a nucleotide base observed in the reads, and each column represents the run-length observed at the corresponding position in the assembly where the read mapped.
Identifying potential polishing edits and heterozygous variants
To find potential polishing edits and heterozygous variants, we aligned a variety of public CHM13 WGS sequencing reads to CHM13v0.9 (https://github.com/marbl/CHM13). We refer to these alignments as “self-alignments” as both the query reads and reference assembly represent the CHM13 genome. Further, we aligned Illumina reads with BWA-MEM (v0.7.15)51 and removed PCR duplicate-like redundancies using `biobambam2 bamsormadup` (v2.0.87)52 with default parameters. Pacific Biosciences Continuous Long Read (CLR) and Circular Consensus Sequencing (CCS/HiFi), and Oxford Nanopore (ONT) reads were aligned using Winnowmap2 (v1.1).
We used both Illumina and HiFi read alignments to call SNPs and indels with the “hybrid” model of DeepVariant (v1.0) but only ONT alignments were used to call SNPs using PEPPER-DeepVariant (v1.0)27. To exclude potentially spurious variant calls, we removed variants with low allele fraction support or low genotype quality (VAF<=0.5, GQ<=30 for Illumina/HiFi, and GQ<=25 for ONT). We then combined Illumina/HiFi hybrid and ONT variant calls using a custom script (https://github.com/kishwarshafin/T2T_polishing_scripts/blob/master/polishing_merge_script/vcf_merge_t2t.py). Finally, we filtered small polishing edits using Merfin40 to ensure all retained edits did not introduce any false 21-mers that were absent from the Illumina or HiFi reads.
Our approach implemented structural variant (SV) inference tools to detect medium-sized polishing edits and structural heterozygosity. For short-read-based SV calling, we used Illumina alignments as input to Parliament241 (v0.1.11) using default settings. For long-read SV calling, we relied on HiFi, CLR, and ONT alignments to call SVs with Sniffles42 (v1.0.12, -s 3 -d 500 -n −1) and we removed all SVs with less than 30% of reads supporting the ALT allele. After this, we generated and refined insertion and deletion sequences with Iris (v1.0.3, using Minimap253 and Racon29 for aligning and polishing, respectively)(https://github.com/mkirsche/Iris). Our approach yielded three independent technology-specific call sets that we merged using Jasmine (v1.0.2, max_dist=500 min_seq_id=0.3 spec_reads=3 --output_genotypes)54. Through manual inspection in IGV (v2.6), we validated all long-read variant calls longer than 30 bp supported by at least two technologies and all short-read SV calls55.
Our approach combined small and structural variant calls into two distinct VCF files: one for potential polishing edits (homozygous ALT alleles) and one for putative heterozygous variants (heterozygous ALT alleles) and we excluded all edits within known problematic loci - prone to producing false variant calls (rDNA gaps as well as the large HSat3 region on chromosome 9). To generate the CHM13v1.0, we applied `bcftools consensus` (v1.10.2–140-gc40d090) to incorporate the suggested polishing edits into CHM13v0.956 and repeated same previously detailed methods with respect to CHM13v1.0 to ensure that no additional polishing edits were apparent and to call heterozygous loci.
Patching the chromosome 18 p-arm telomere
As a result of the string graph simplification process, we found a telomere missing from the graph representing the p-arm of chromosome 18. Bionano molecules and CMAPs44 were mapped to CHM13v0.9 and CHM13v1.0 using Bionano Solve v3.6 and and all scaffolds were manually inspected end to end to search for assembly errors. No issues were detected except this missing telomere. We identified five ONT reads associated with these telomeric sequences using the telomere pipeline developed by the VGP (https://github.com/VGP/vgp-assembly). Using these reads we ran Medaka (v1.0.3)28 to generate a consensus sequence and manually patched it into the assembly (https://github.com/malonge/PatchPolish]. We obtained seven matching HiFi reads, not in the assembly graph and confirmed to have telomeric repeats, and used Racon (v1.6.0)29 to further polish. In total, we added 4,862 bp of telomere sequence to the start of chromosome 18.
Evaluating polishing accuracy
We repeated self-alignment variant calling methods on CHM13v1.0 and confirmed that no additional polishing errors were apparent. In addition to the self-alignments used for polishing and heterozygous variant calling, we derived marker-assisted alignments from previously created HiFi, CLR, and ONT Winnowmap2 alignments34. For marker-assisted alignment production, we removed Winnowmap2 alignments that did not span “marker” k-mers. We define marker k-mers as any 21-mer present once in CHM13v1.0 and between 42 and 133 times in the Illumina reads44 and filtered reads using technology specific length thresholds with HiFi having a 10kbp, CLR a 1kbp and ONT a 25kbp threshold. Our approach relied on both CHM13v1.0 self-alignments and marker-assisted alignments for manual inspection.
We also assessed the genome assembly using Merqury v1.3 QV estimations based on 21-mer databases we created for both Illumina PCR-free and HiFi reads36. Following this, we derived a “hybrid” Merqury k-mer database using Meryl v1.3 by combining Illumina and HiFi k-mers that occurred over 1 times, and adjusting the k-mer frequency to match the k-mer frequency at 35x in the diploid (2-copy) peak by increasing k-mer frequency in HiFi reads by 4 and dividing the Illumina k-mer frequency by 3 and combined the k-mer databases by taking the union and setting frequency to the maximum observed in the two datatypes.
To identify regions with rare collapses or rare duplications in CHM13v1.0, we compared copy number estimates of CHM13v1.0 to copy number estimates of 268 human genomes (Simons Genome Diversity Project; SGDP) using short reads45. We averaged these copy number estimates for each genome across 1 kbp windows and flagged a potential false or rare duplication if the copy number in CHM13v1.0 was greater than the copy number in 99% of the other genomes and GRCh38. Moreover, we flagged a potential false or rare collapse if the copy number in CHM13v1.0 was less than the copy number in 99% of the other genomes and GRCh38 and assigned all flagged regions a value of 1 and unflagged regions a value of 0. We included GRCh38 in this analysis to help remove rare technical artifacts where the assembly-based k-mer copy number estimate is systematically different from the Illumina read-based k-mer estimate. To filter the flagged regions, we used a median filter approach with a window size of 3 kbp where the binary value of each 1 kbp region was replaced with the median value of the complete window. Finally, we merged all adjacent flagged regions and reported the start and end coordinates with respect to CHM13v1.0, and we curated and removed flagged regions if they overlapped LINEs as SGDP copy number estimates are less reliable in these high copy number repeats.
Polishing enrichment or depletion within repeats
We performed a permutation test to check if our polishing pipeline suggested significantly more or fewer polishing edits within repeats compared to the rest of the genome. We established two distinct samples of genomic intervals. For the first, we randomly sampled 20,000 100 kbp windows from the genome and removed any windows that intersected repeats. For the second, we randomly sampled 20,000 100 kbp windows and removed any windows that intersected non-repeats. By measuring the number of polishing edits in each 100 kbp window, we established two different random distributions of polishing rates: one within and one without repeats. We utilised SciPy (v1.7.0) stats.ttest_ind using 10,000 permutations to derive our p-value57. SciPy is available in Python package v3.5.
Telomere polishing
We employed a targeted polishing of telomeres by retraining PEPPER (v0.4)27 on HG002 chr20 with all forward strand reads removed to correct for the original model’s dependence on having reads from both strands. Using this retrained model, we generated a set of candidate variants in the telomere regions and the coverage depth was calculated using samtools58 depth (v1.9). Finally, we implemented a custom script (https://github.com/kishwarshafin/T2T_polishing_scripts/blob/master/telomere_variants/generate_telomere_edits.py) that took these candidate variants and calculated the Levenshtein distance between the canonical telomere k-mer and the sequence we derived after the candidate variant had been applied. We selected only those variants as true telomere edits if the candidate had a minimum allele frequency of 0.5, a minimum genotype quality of 2 and reduced the Levenshtein distance to the canonical telomere k-mer when compared to the existing sequence. Further, we trimmed the consensus sequence where ONT read depth support was lower than 5.
Coverage supports and excessive clippings
We employed SV detection to identify regions with low coverage support, excessive read clippings, and enriched secondary alleles, and to further ensure that accuracy was not compromised but also to identify and document outstanding issues with CHM13v1.1 (Fig. 1a). On inspection of both Winnowmap233 and Minimap253 read clippings, artificial alignment breaks were highlighted that caused clipping and coverage drops in regions with highly identical satellite sequences. Notably, we did not identify these breaks in alignments from TandemMapper35, a more conservative aligner specifically designed for alignment in satellite repeats. On further inspection of clipped reads, we found the chaining algorithm of Winnowmap2 handled lower confidence alignment blocks incorrectly, and so we updated accordingly (v2.0 to v2.01) for all future evaluations of both CHM13v1.0 and CHM13v1.1 (Extended Data Fig. 7).
Comparison to automated polishing approaches
To evaluate our newly proposed approach to polishing, we compared it to the off-the-shelf tools available for HiFi reads. We performed three rounds of iterative polishing using the Racon consensus tool with each iteration including the following steps. (1) Alignment of input HiFi reads to the input target sequences using Winnowmap 1.11 (https://github.com/marbl/Winnowmap/releases/tag/v1.11; options: “--MD -W bad_mers.txt -ax map-pb”) as used for polishing CHM13v0.9. We used CHM13v0.9 (unpolished) as the first iteration target, while every following iteration used the polished output of the previous stage as the input target. (2) We filtered secondary alignments and alignments with excessive clipping using the “falconc bam-filter-clipped” tool (available in the “pbipa” Bioconda package; options: “falconc bam-filter-clipped -t -F 0×104”). By default, maximum clipping on either left or right side of an alignment is set to 100bp, but this was applied only if the alignment was located at least 25bp from the target sequence end (to prevent clipping due to contig ends which could otherwise cause false alignment filtering). (3) Finally, we used Racon (https://github.com/isovic/racon, branch “liftover”, commit: 73e4311) to polish the target sequences using these filtered alignments. For the purposes of this work, we extended the “master” branch of Racon to include two custom features: BED selection of regions for polishing and logging all changes introduced to the input draft assembly to produce the final polished output (in VCF, PAF or SAM format). We then ran Racon with default options with the exception of two new logging options: “-L out_prefix -S” implemented to store the liftover information between the input and output sequences. We used Liftoff (v1.6.0, -chroms -copies -exclude_partial -polish) using gencode v35 to annotate each of the polished assemblies59,60.
Extended Data
Extended Data Fig. 1 |. Sequencing biases observed in missing k-mers.
a, missing k-mers with its GA composition. b-d, v0.9 assembly and k-mer copy number spectrum from HiFi, Illumina, and hybrid k-mer sets (left) and per-chromosome missing (likely error) k-mer counts from the HiFi derived consensus or patches (right). Most missing k-mers in HiFi overlapped sequences from patched regions. No missing k-mer was found on chromosomes indicated with red arrows.
Extended Data Fig. 2 |. Error detection and polishing pipeline.
A detailed overview of the polishing pipeline along with the number of errors identified and polished at each step. Additionally, data type and polishing tools utilized are highlighted. Illumina, 100X PCR-free library Illumina reads; HiFi, 35x PacBio HiFi reads; ONT, 120x Oxford Nanopore reads.
Extended Data Fig. 3 |. Number of SV-like errors and globally unique single copy k-mers used for marker assisted alignment.
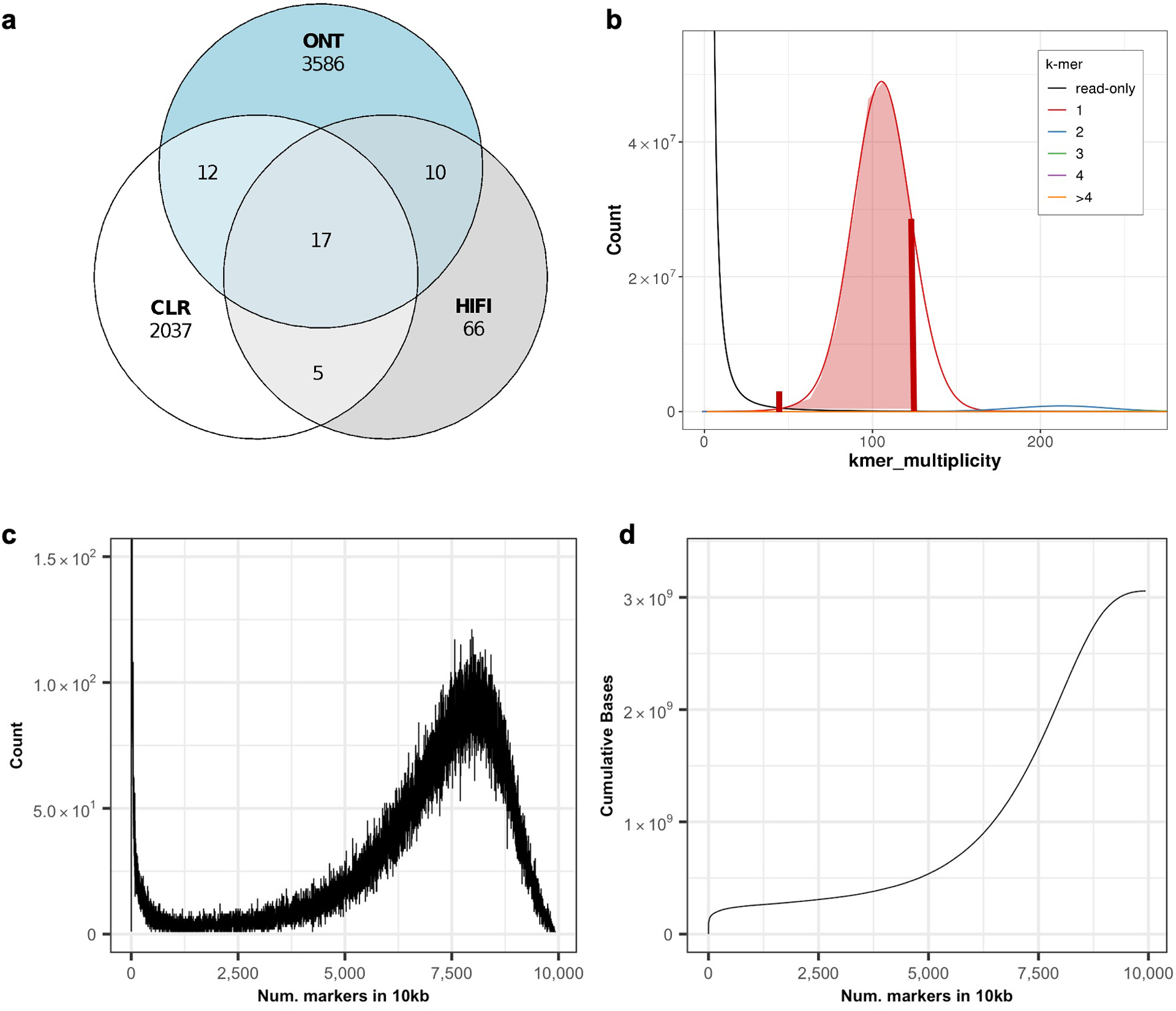
a. Number of SV-like errors called from long-read platforms. b. Range of k-mer counts defined as ‘single-copy’ markers from Illumina reads and in the assembly. The cutoffs were chosen to minimize inclusion of low-frequency erroneous k-mers and 2-copy k-mers. c. Number of markers in every 10 kb window. d. Cumulative number of bases covered by the number of markers in each 10 kb window.
Extended Data Fig. 4 |. Post-polishing evaluation.
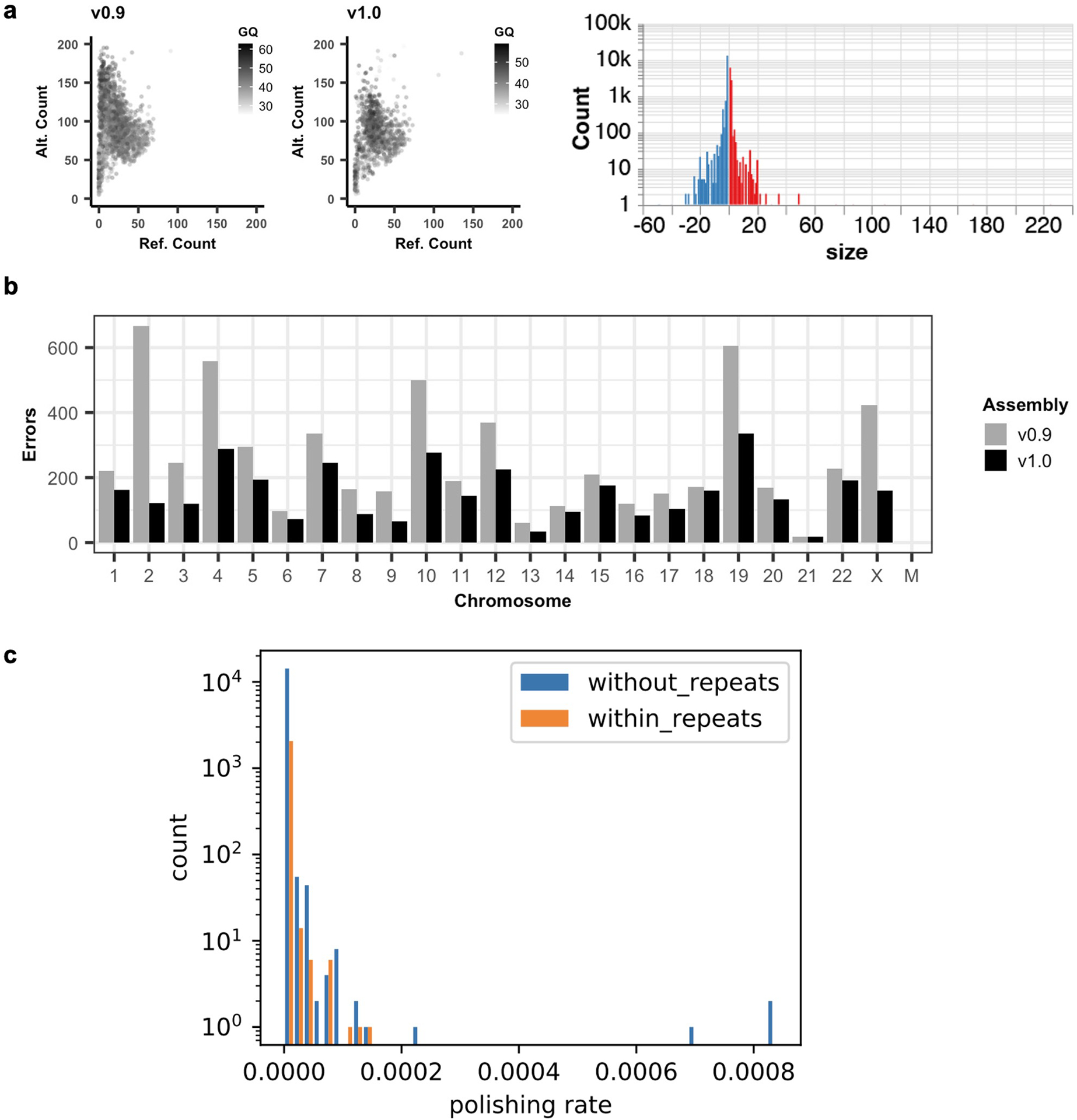
a. Left, genotype quality and number of reads supporting the reference and alternate alleles from the combined Illumina-hifi hybrid and ONT homozygous variant calls, with AF > 0.5. Right, balanced insertion (red) and deletion (blue) length distribution from the Illumina-HiFi hybrid DeepVariant heterozygous calls in CHM13v1.0. b. Number of errors detected in each chromosome, before and after polishing. c. Polishing inside and outside of repeats. The distribution of CHM13v0.9 polishing rates within and without repeats.
Extended Data Fig. 5 |. Three SV-like errors corrected.
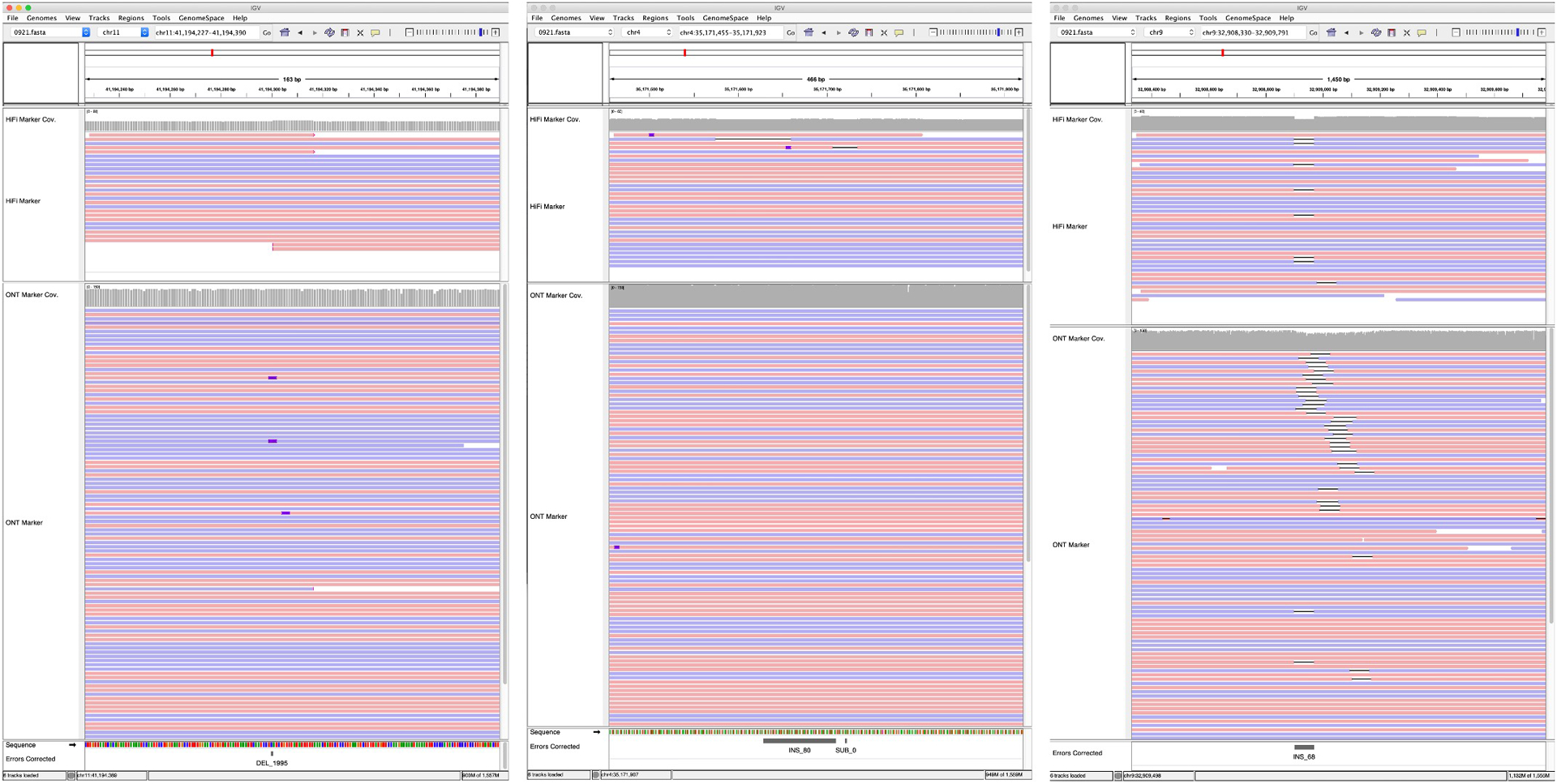
HiFi and ONT marker assisted alignments, post correction of the 3 large SV-like edits visualized with IGV. HiFi coverage track is shown in data range up to 60, ONT up to 150. Clipped reads are flagged for >100bp. INDELs smaller than 10 bp are not shown. Reads are colored by strands; positive in red and negative in blue.
Extended Data Fig. 6 |. Telomere polishing.
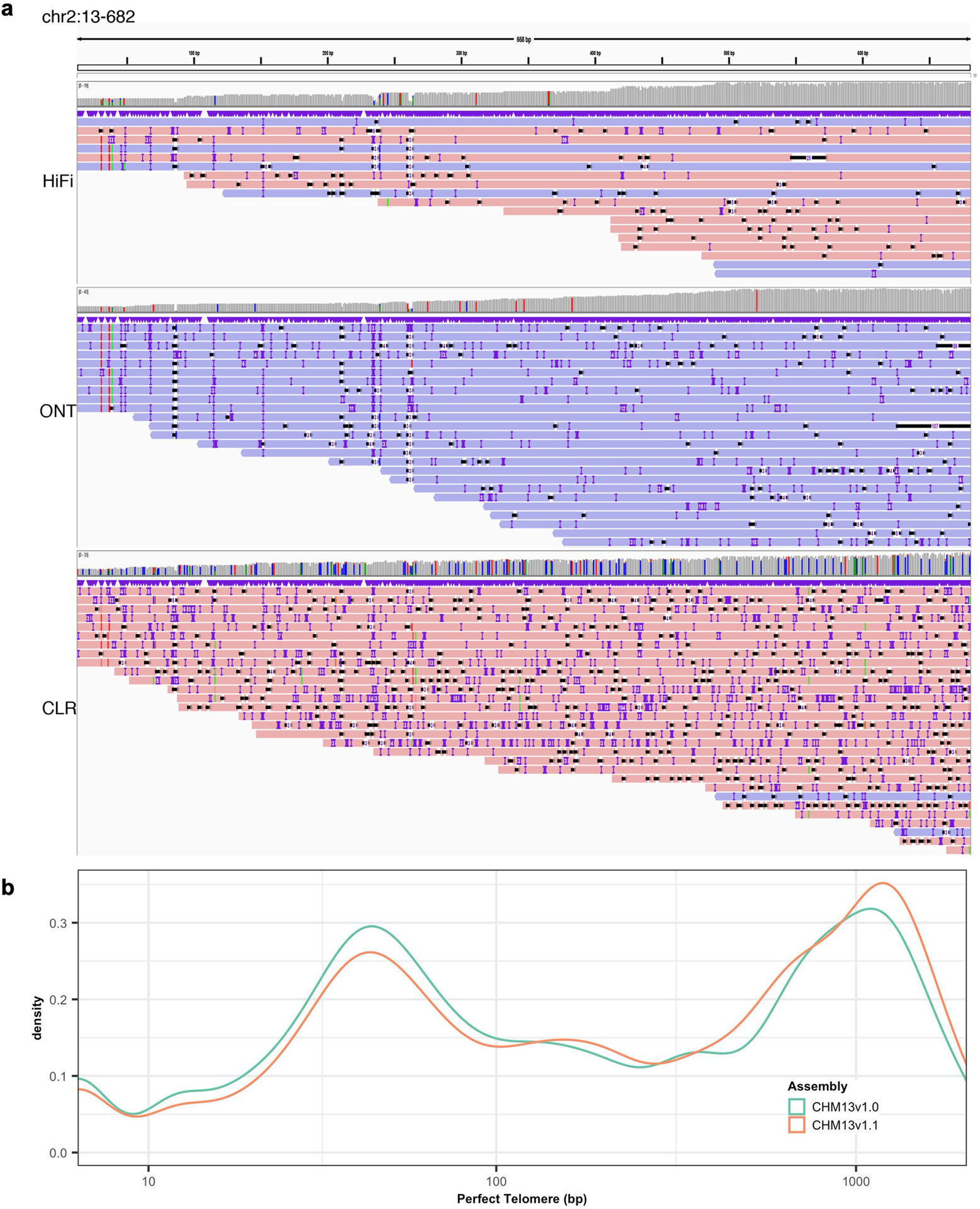
a. An illustration of Chr. 2 telomere sequence reads from HiFi, ONT and CLR platform. b. Distribution of maximum perfect match to the canonical k-mer observed at each position in the telomere before (CHM13v1.0) and after (CHM13v1.1) polishing the telomeres.
Extended Data Fig. 7 |. Mapping biases found and corrected.
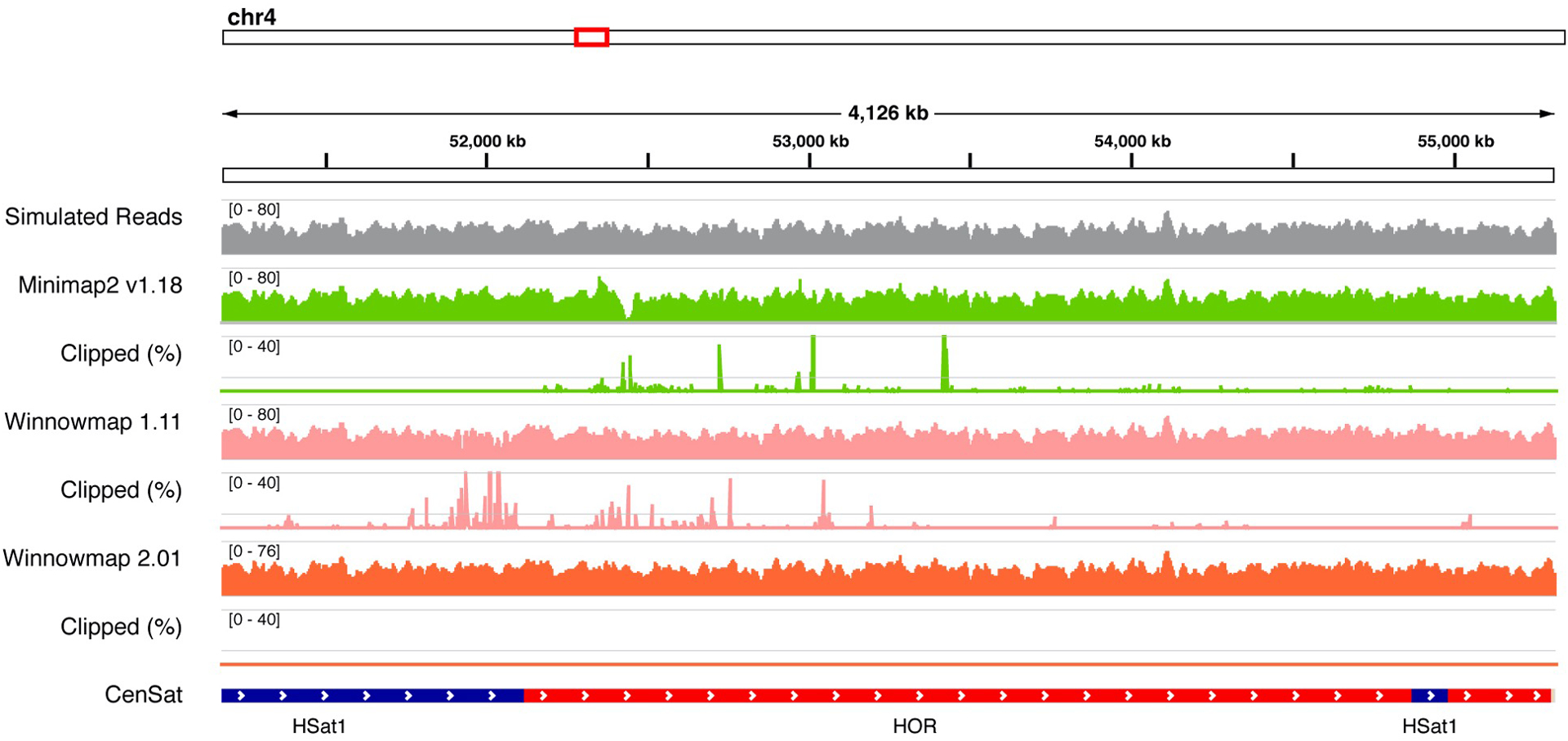
On simulated HiFi reads, we found excessive clippings in highly identical satellite repeats in Minimap and Winnowmap by the time of evaluation. We have addressed this issue in Winnowmap 2.01+. Clipped (%) indicates the percentage of reads clipped in every 1024 bp window, shown in 0~40% range with a midline of 10%.
Extended Data Fig. 8 |. HiFi, CLR, ONT read coverage, alignment identity, and read length from Winnowmap2 v2.01 alignments and Bionano DLE-1 molecule coverage from Bionano Solve.
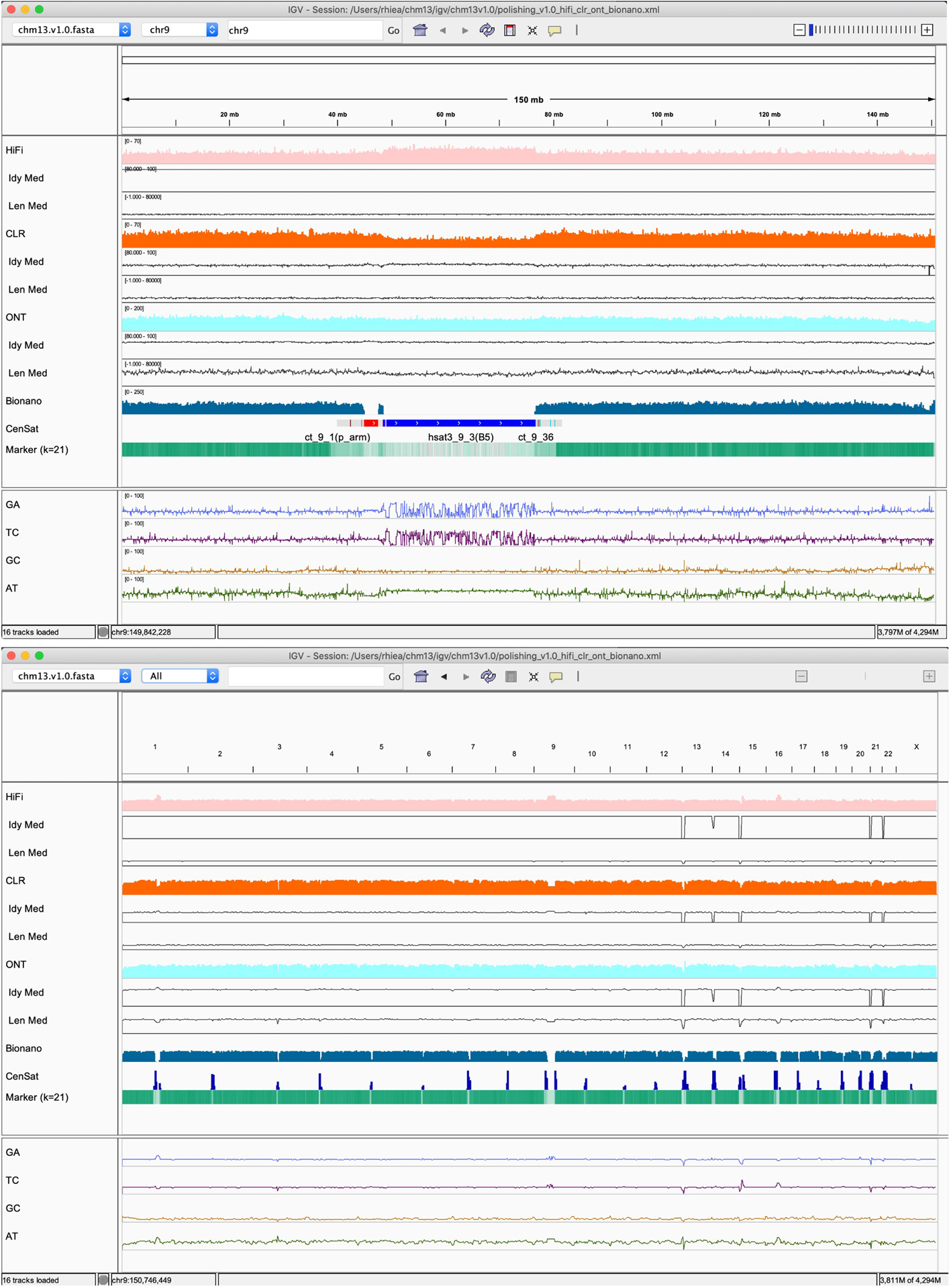
Upper panel shows a zoomed in region of Chromosome 9, while the upper panel shows the whole-genome alignment view. HiFi, CLR, ONT, and Bionano coverage are shown up to 70x, 70x, 200x, and 250x, respectively. Median read identity in every 1024 bp is shown in 80–100% range. Median read length in every 1024 bp is shown in 0–100kb range. Read identity was the worst in CLR, and between HiFi and ONT. Bionano molecules were lacking coverage in most of the centromeric repeats.
Extended Data Fig. 9 |. Collapsed simple tandem repeat.
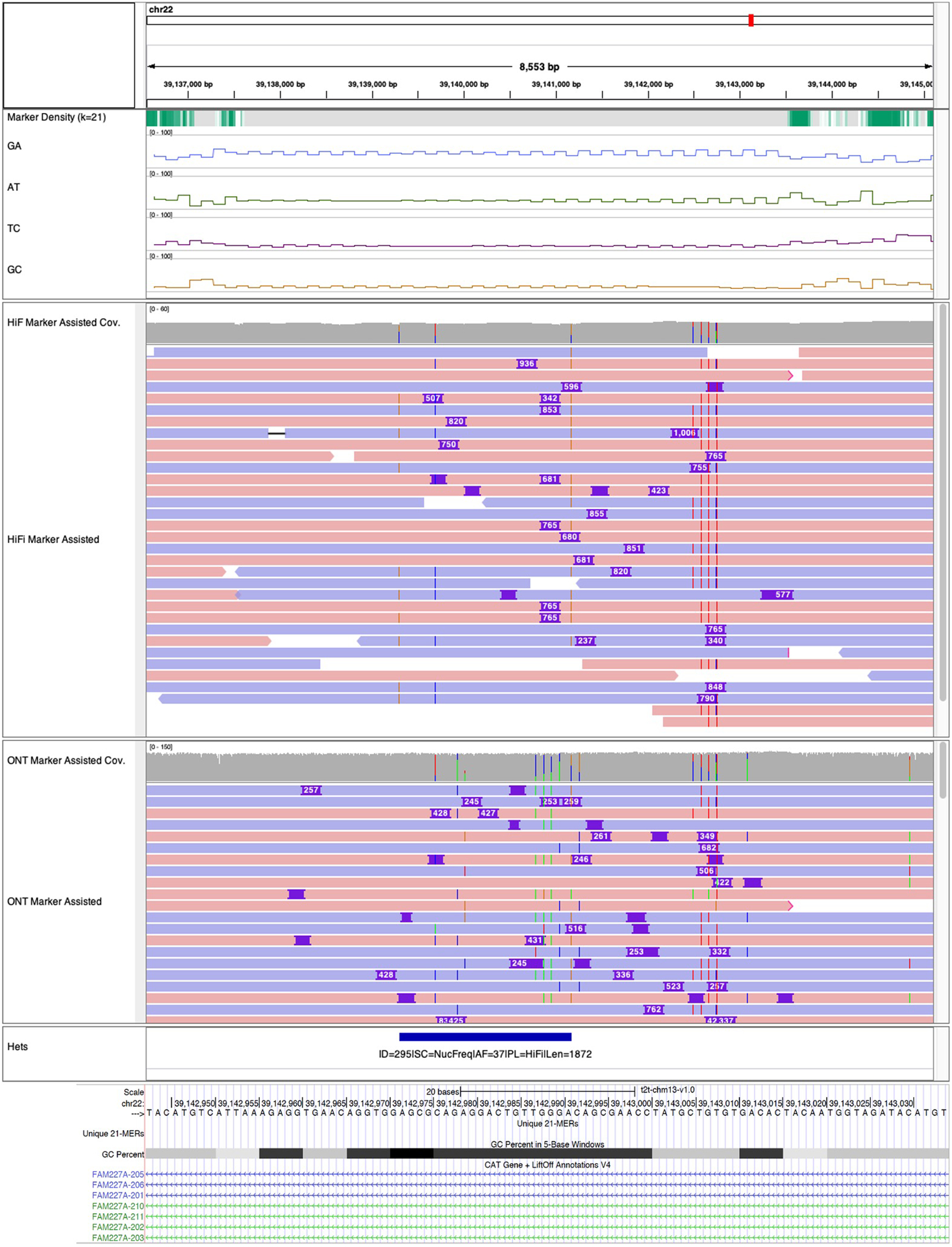
The collapse in the Intronic sequences of gene FAM227A was undetected, due to the variable insertion breakpoints and insertion length in the HiFi and ONT alignments. The panels above the alignments show marker density and percent microsatellites (GA / AT / TC / GC) in each 64 bp window, which indicates this region is highly repetitive with GA enriched sequences, which later alternates with AT enriched sequences.
Extended Data Fig. 10 |. Chimeric junction of two haplotypes.
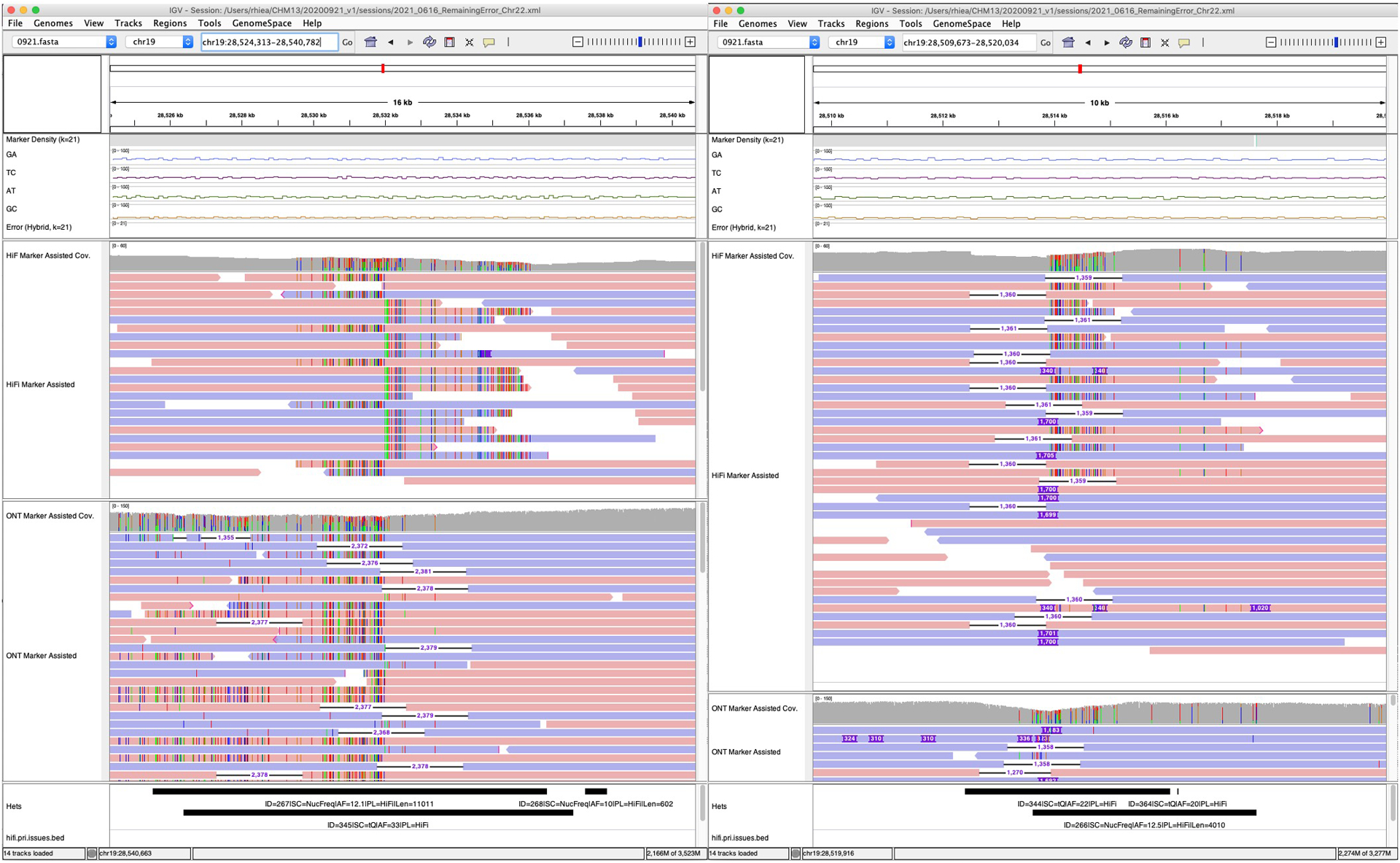
In the shown above regions, both HiFi and ONT reads indicate that the consensus has a chimeric junction of the two haplotypes.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Intramural Research Program of the National Human Genome Research Institute (NHGRI), National Institutes of Health (NIH) (AMM, CJ, SK, AMP, AR); National Science Foundation: DBI-1350041 and IOS-1732253 (MA); NIH/NHGRI R01HG010485, U41HG010972, U01HG010961, U24HG011853, OT2OD026682 (KS, BP); HHMI (GF); Wellcome WT206194 (KH, JMW); NIGMS F32 GM134558 (GAL); NIH/NHGRI R01 1R01HG011274-01, NIH/NHGRI R21 1R21HG010548–01, and NIH/NHGRI U01 1U01HG010971 (KM); St. Petersburg State University grant ID PURE73023672 (AM); NIH/NHGRI R01HG006677 (AS); Fulbright Fellowship (DCS); Intramural funding at the National Institute of Standards and Technology (JZ). This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). Certain commercial equipment, instruments, or materials are identified to specify adequately experimental conditions or reported results. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the equipment, instruments, or materials identified are necessarily the best available for the purpose.
COMPETING INTERESTS STATEMENTS
Ivan Sović is an employee of PacBio. Arkarachai Fungtammasan is an employee of DNAnexus. Sergey Koren has received travel funds to speak at symposia organized by Oxford Nanopore. The remaining authors declare no competing interests.
Footnotes
CODE AVAILABILITY
To facilitate usability of our evaluation and polishing strategy, we made the up-to-date version of tools that have been used within our workflows openly available on https://github.com/arangrhie/T2T-Polish. Exact codes used for polishing CHM13v0.9 and CHM13v1.0 are available on https://github.com/marbl/CHM13-issues. Both GitHub repositories are available through a public domain, and have been deposited to Zenodo61,62. Custom scripts used for merging small variants, and generating telomere edits are available at https://github.com/kishwarshafin/T2T_polishing_scripts and deposited to Zenodo63 under a MIT license.
DATA AVAILABILITY
All data types and assemblies are available on https://github.com/marbl/CHM13 and under NCBI BioProject PRJNA559484 with the Assembly GenBank accession GCA_009914755. Polishing edits, catalogued remaining issues and known heterozygous regions are available on https://github.com/marbl/CHM13-issues. All the data in the two GitHub repositories are directly downloadable from https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=T2T/CHM13/ with no restrictions. The retrained PEPPER model used for telomere polishing is available to download at https://storage.cloud.google.com/pepper-deepvariant-public/pepper_models/PEPPER_HP_R941_ONT_V4_T2T.pkl. Source data for generating plots in this manuscript are available on https://github.com/arangrhie/T2T-Polish/tree/master/paper/2022_Mc_Cartney.
REFERENCES
- 1.Nurk S, Koren S, Rhie A, Rautiainen M, et al. The complete sequence of a human genome. Science (2022). doi: 10.1126/science.abj6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollger MR, et al. Segmental duplications and their variation in a complete human genome. Science (2022). doi: 10.1126/science.abj6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gershman A, et al. Epigenetic Patterns in a Complete Human Genome. Science (2022). doi: 10.1126/science.abj5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ebert P et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hufford MB et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science (2021) 10.1126/science.abg5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aganezov S, Yan SM, Soto DC, Kirsche M, Zarate S, et al. A complete reference genome improves analysis of human genetic variation. Science (2022). doi: 10.1126/science.abl3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Dijk EL, Auger H, Jaszczyszyn Y & Thermes C Ten years of next-generation sequencing technology. Trends Genet. 30, 418–426 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Metzker ML Sequencing technologies - the next generation. Nat. Rev. Genet 11, 31–46 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Logsdon GA, Vollger MR & Eichler EE Long-read human genome sequencing and its applications. Nat. Rev. Genet 21, 597–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baran N, Lapidot A & Manor H Formation of DNA triplexes accounts for arrests of DNA synthesis at d(TC)n and d(GA)n tracts. Proceedings of the National Academy of Sciences vol. 88 507–511 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guiblet WM et al. Long-read sequencing technology indicates genome-wide effects of non-B DNA on polymerization speed and error rate. Genome Res. 28, 1767–1778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y-C, Liu T, Yu C-H, Chiang T-Y & Hwang C-C Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS One 8, e62856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolmogorov M, Yuan J, Lin Y & Pevzner PA Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol 37, 540–546 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Nurk S et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng H, Concepcion GT, Feng X, Zhang H & Li H Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods (2021) doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimin AV et al. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27, 787–792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson JT et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watson M Mind the gaps – ignoring errors in long read assemblies critically affects protein prediction. doi: 10.1101/285049. [DOI] [PubMed] [Google Scholar]
- 21.Salzberg SL et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 22, 557–567 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhie A et al. Towards complete and error-free genome assemblies of all vertebrate species. Cold Spring Harbor Laboratory 2020.05.22.110833 (2020) doi: 10.1101/2020.05.22.110833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimin AV & Salzberg SL The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol 16, e1007981 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GenomicConsensus. (Github).
- 25.Loman NJ, Quick J & Simpson JT A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Poplin R et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol 36, 983–987 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Shafin K et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat Methods 18, 1322–1332 (2021). 10.1038/s41592-021-01299-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oxford Nanopore Technologies. https://github.com/nanoporetech/medaka. medaka: Sequence correction provided by ONT Research. https://github.com/nanoporetech/medaka.
- 29.Vaser R, Sović I, Nagarajan N & Šikić M Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H, Jain C & Aluru S A comprehensive evaluation of long read error correction methods. BMC Genomics 21, 889 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu S, Wang A & Au KF A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 20, 26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altemose N, et al. Complete genomic and epigenetic maps of human centromeres. Science (2022). doi: 10.1126/science.abl4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain C et al. Weighted minimizer sampling improves long read mapping. Bioinformatics 36, i111–i118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain C, et al. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat Methods (2022). doi: 10.1038/s41592-022-01457-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mikheenko A, Bzikadze AV, Gurevich A, Miga KH & Pevzner PA TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36, i75–i83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhie A, Walenz BP, Koren S & Phillippy AM Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fofanov Y et al. How independent are the appearances of n-mers in different genomes? Bioinformatics 20, 2421–2428 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Lang D et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. GigaScience vol. 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olson ND et al. precisionFDA Truth Challenge V2: Calling variants from short- and long-reads in difficult-to-map regions. Cell Genomics (2022) doi: 10.1016/j.xgen.2022.100129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Formenti G, Rhie A, et al. Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation. Nat Methods (2022) doi: 10.1038/s41592-022-01445-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zarate S et al. Parliament2: Accurate structural variant calling at scale. Gigascience 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miga KH et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vollger MR, et al. Segmental duplications and their variation in a complete human genome. Science (2022). doi: 10.1126/science.abj6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner J et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat Biotechnol. 2022. May;40(5):672–680. doi: 10.1038/s41587-021-01158-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naish M, Alonge M, Wlodzimierz P & Tock AJ The genetic and epigenetic landscape of the Arabidopsis centromeres. Science (2021) doi: 10.1126/science.abi7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J et al. Gapless assembly of maize chromosomes using long-read technologies. Genome Biol. 21, 121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du H et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun 8, 15324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN] (2013). [Google Scholar]
- 52.Tischler G & Leonard S biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol. Med 9, 13 (2014). [Google Scholar]
- 53.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirsche M et al. Jasmine: Population-scale structural variant comparison and analysis. bioRxiv 2021.05.27.445886 (2021) doi: 10.1101/2021.05.27.445886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorvaldsdóttir H, Robinson JT & Mesirov JP Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Virtanen P et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H, Twelve years of SAMtools and BCFtools, GigaScience (2021) 10(2) giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shumate A & Salzberg SL Liftoff: accurate mapping of gene annotations. Bioinformatics (2020) doi: 10.1093/bioinformatics/btaa1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rhie A, Formenti G, Shafin K, Fungtammasan A, & Jain C arangrhie/T2T-Polish: v1.0 (Zenodo, 2021). 10.5281/zenodo.5649017 [DOI] [Google Scholar]
- 62.Rhie A and Phillippy A marbl/CHM13-issues: v1.1. (Zenodo, 2021) 10.5281/zenodo.5648989 [DOI] [Google Scholar]
- 63.Shafin K kishwarshafin/T2T_polishing_scripts: v0.1 release for zenodo. (Zenodo, 2021). 10.5281/zenodo.6127865 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data types and assemblies are available on https://github.com/marbl/CHM13 and under NCBI BioProject PRJNA559484 with the Assembly GenBank accession GCA_009914755. Polishing edits, catalogued remaining issues and known heterozygous regions are available on https://github.com/marbl/CHM13-issues. All the data in the two GitHub repositories are directly downloadable from https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=T2T/CHM13/ with no restrictions. The retrained PEPPER model used for telomere polishing is available to download at https://storage.cloud.google.com/pepper-deepvariant-public/pepper_models/PEPPER_HP_R941_ONT_V4_T2T.pkl. Source data for generating plots in this manuscript are available on https://github.com/arangrhie/T2T-Polish/tree/master/paper/2022_Mc_Cartney.