

Abstract
Systematically investigating the scores of genes mutated in cancer and discerning disease drivers from inconsequential bystanders is a prerequisite for Precision Medicine but remains challenging. Here, we developed a somatic CRISPR/Cas9 mutagenesis screen to study 215 recurrent ‘long-tail’ breast cancer genes, which revealed epigenetic regulation as a major tumor suppressive mechanism. We report that components of the BAP1 and the COMPASS-like complexes, including KMT2C/D, KDM6A, BAP1 and ASXL1/2 (“EpiDrivers”), cooperate with PIK3CAH1047R to transform mouse and human breast epithelial cells. Mechanistically, we find that activation of PIK3CAH1047R and concomitant EpiDriver loss triggered an alveolar-like lineage conversion of basal mammary epithelial cells and accelerated formation of luminal-like tumors, suggesting a basal origin for luminal tumors. EpiDrivers mutations are found in ~39% of human breast cancers and ~50% of ductal-carcinoma-in-situ express casein suggesting that lineage infidelity and alveogenic mimicry may significantly contribute to early steps of breast cancer etiology.
Introduction
New genomic technologies hold the promise of revolutionizing cancer therapy by allowing treatment decisions guided by a tumor’s genetic make-up. However, converting genetic discoveries into tangible clinical benefits requires a deeper understanding of the molecular and cellular mechanisms that underlie disease progression (1). In breast cancer, only a few genes such as TP53 and PIK3CA are mutated at high frequencies (~30–50%), while the vast majority are mutated at low frequencies comprising a so called ‘long-tail’ gene distribution (2–4). Whether these long-tail genes functionally contribute to breast cancer progression constitutes a significant knowledge gap. Although mutations in these genes seem to be under positive selection, they are only found in relatively small subsets of patients. It has been proposed that these infrequently mutated genes individually confer a small fitness advantage to cancer cells, but when combined synergize to increase fitness (additive-effects model) (5–7). Alternatively, long-tail genes may work in different ways to produce the same phenotype (phenotypic convergence) and/or affect the same pathway or molecular mechanism (pathway convergence) (8). Recently, we reported the latter mechanism in head and neck cancer, where long-tail genes converge to inactivate NOTCH signaling (9). The biological relevance of long-tail genes in other cancer types remains largely unknown.
Here, we report an in vivo CRISPR/Cas9 screening strategy to identify which long-tail breast cancer genes and associated molecular pathways cooperate with the oncogenic PIK3CAH1047R mutation to accelerate breast cancer progression.
We tested 215 long-tail genes and identified several functionally relevant breast cancer genes, many of which converge on regulating histone modifications and enhancer activity (from here onwards referred to as ‘EpiDrivers’). Single-cell multi-omics profiling of EpiDriver-mutant mammary glands reveals increased cell state plasticity and alveogenic mimicry associated with an aberrant alveolar differentiation program during the early specification of luminal breast cancer. Interestingly, EpiDriver loss in basal cells triggers basal-to-alveolar lineage conversion and accelerated tumor formation. Importantly, EpiDriver mutations are found in ~39% of primary breast tumors, supporting the hypothesis that different genes converge to produce the same cell plasticity that facilitates cancer development.
RESULTS
Direct in vivo CRISPR Gene Editing in the Mouse Mammary Gland
First, we developed a multiplexed CRISPR/Cas9 knock-out approach in the mammary gland of tumor-prone mice. As PIK3CA is the most commonly mutated oncogene in breast cancer, we crossed conditional Lox-Stop-Lox-(LSL)-Pik3caH1047R mice to LSL-Cas9-GFP transgenic mice to generate Pik3caH1047R;Cas9 mice. Intraductal microinjections of a lentivirus that expresses an sgRNA and Cre recombinase (LV-sgRNA-Cre) led to excision of Lox-Stop-Lox cassettes and expression of Cas9, GFP and oncogenic Pik3caH1047R in the mammary epithelium (Fig. 1A). We tested the efficacy of CRISPR/Cas9-mediated mutagenesis by injecting sgRNAs targeting GFP or the heme biosynthesis gene Urod. Knock-out of GFP was detected as a 86±6% reduction in green fluorescence in transduced cells, whereas knock-out of Urod was detected as an accumulation of unprocessed fluorescent porphyrins in 30%±8% of cells (Supplementary Fig. S1A–D) (10). Moreover, Pik3caH1047R;Cas9 mice transduced with an sgRNA targeting Trp53 developed tumors significantly faster than littermate mice transduced with a control sgRNA targeting the permissive Tigre locus (median tumor-free survival of 83 versus 152 days) (Supplementary Fig. S1E). Together, these data demonstrate that this approach recapitulates cooperation between oncogenic Pik3ca and Trp53 loss-of-function (11,12) and can be used to test for genetic interaction between breast cancer genes.
Fig. 1. In vivo CRISPR screen reveals novel epigenetic breast cancer tumors suppressors ‘EpiDrivers’.
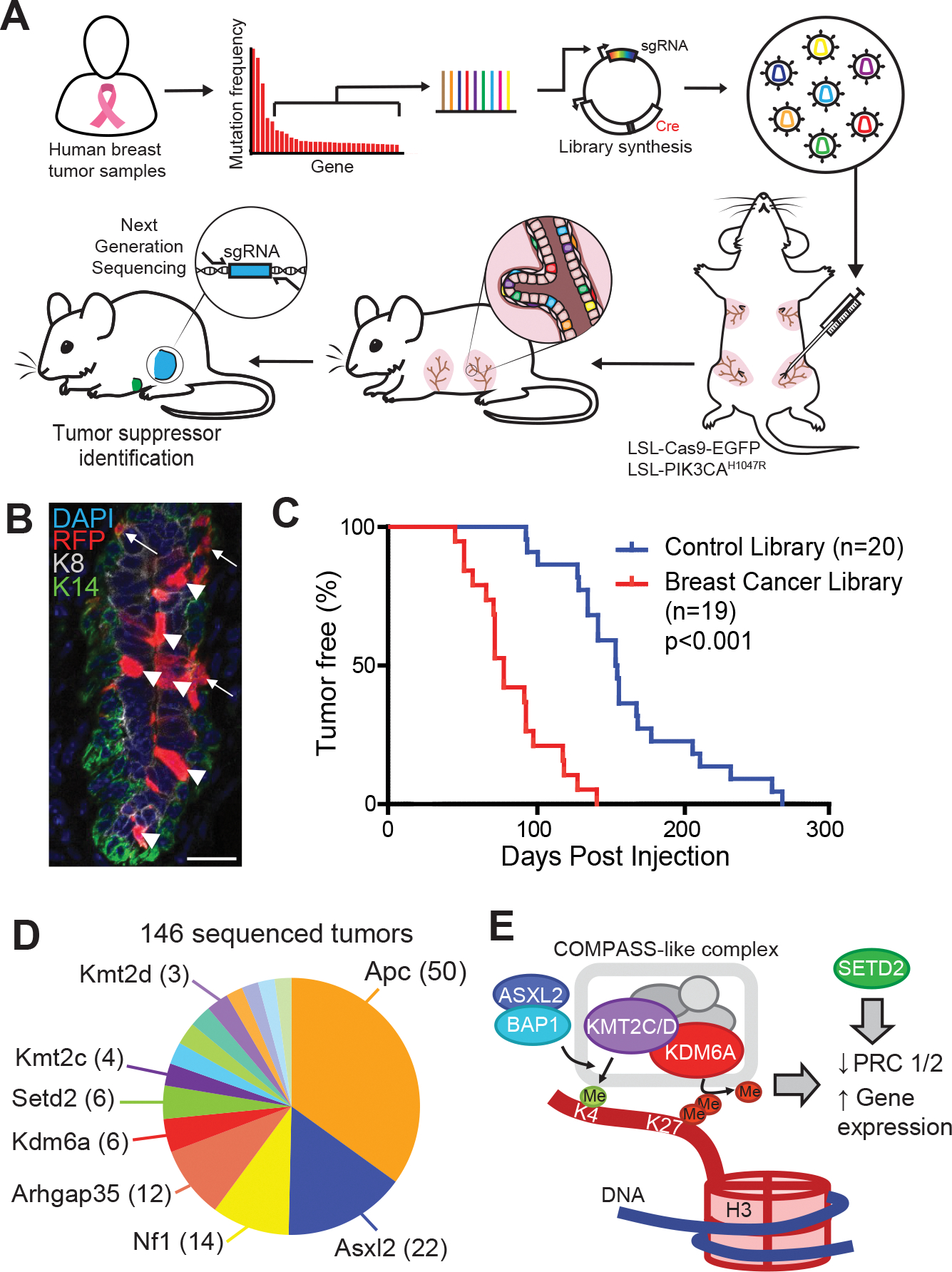
A, Experimental design for in vivo CRISPR screen, showing gene selection from long-tail mutations, intraductal injection of lentiviral libraries and tumor sequencing. B, Mammary epithelium transduced with lentiviral RFP. Arrows denote basal cells and arrow heads denote luminal cells. Scale bar = 25μm. C, Tumor-free survival of Pik3caH1047R;Cas9 mice transduced with a sgRNA library targeting putative breast cancer genes or a control sgRNA library. D, Pie chart showing putative tumor suppressor genes with enriched sgRNAs in tumor DNA (number of tumors are denoted in brackets). E, Schematic of COMPASS-like and ASXL/BAP1 complexes on epigenetic control of gene expression.
CRISPR Screen Identifies Histone Modifiers as Breast Cancer Driver Genes
In breast cancer, 215 long-tail genes show somatic mutations in 2–20% of patients (11,13). To assess disease relevance of these genes in vivo, we established a LV-sgRNA-Cre library targeting the corresponding mouse orthologs (4 sgRNAs/gene; 860 sgRNAs) as well as a library of 420 non-targeting control sgRNAs (Supplementary Table S1). We optimized the parameters for an in vivo CRISPR screen by using a mixture of lentiviruses expressing GFP or RFP to determine the viral titer that transduces the mammary epithelium at clonal density (MOI<1). Higher viral titers were associated with double infections, whereas a 15% overall transduction level minimized double infections while generating sufficient clones to screen (Supplementary Fig. S1F–I). Flow cytometry revealed that the third and fourth mammary gland each contain >3.5×105 epithelial cells, and that EPCAMhi/CD49fmid luminal cells showed a higher infectivity (~30%) compared to EPCAMmid/CD49fhi basal cells (~5%) (Fig. 1B and Supplementary Fig. S1H and S1I). Thus, at a transduction level of 15% and a pool of 860 sgRNAs, each sgRNA was predicted to be introduced into an average of 60 individual cells within a single gland.
To uncover long-tail genes that cooperate with oncogenic PI3K signaling, we introduced the viral libraries into the third and fourth pairs of mammary glands of 19 Pik3caH1047R;Cas9 mice, resulting in an overall coverage of >4,000 clones per sgRNA. Next generation sequencing confirmed efficient lentiviral transduction of all sgRNAs (Supplementary Fig. S2A). Importantly, Pik3caH1047R;Cas9 mice transduced with the long-tail breast cancer sgRNA library developed mammary tumors significantly faster than littermates transduced with the control sgRNA library (74 versus 154 days; p<0.0001) (Fig. 1C). This result was similar to the accelerated tumorigenesis caused by loss of Trp53 (Supplementary Fig. S1E), indicating the existence of strong tumor suppressors within the long-tail of breast cancer-associated genes.
We examined the sgRNA representation in 146 tumors to determine the targets responsible for accelerating mammary tumorigenesis. Most tumors showed strong enrichment for a single or occasionally two sgRNAs (Supplementary Fig. S2B). We prioritized genes that were targeted by ≥2 sgRNAs and knocked-out in multiple tumors, resulting in 29 candidate tumor suppressor genes (Supplementary Table S2). These candidates included well-known tumor suppressors, such as Apc or Nf1, as well as genes with poorly understood function, such as Arhgap35 (14). Intriguingly, several genes encoded histone and DNA modifying enzymes, such as Arid5b, Asxl2, Kdm6a (Utx), Kmt2a (Mll1), Kmt2c (Mll3), and Kmt2d (Mll4), indicating a convergence on epigenetic regulation (Fig. 1D, Supplementary Fig. S2C).
Kdm6a, Kmt2c, Asxl2, Bap1, Setd2 and Apc Suppress Breast Cancer in Mice
KMT2C and KMT2D encode partly redundant histone methyltransferases within the ‘complex of proteins associated with SET1’ (COMPASS)-like complex, which also contains the histone demethylase KDM6A. The KMT2C/D-COMPASS-like complex catalyzes the mono-methylation of lysine 4 as well as demethylation of lysine 27 in histone H3 (H3K4me1/H3K27) at distal enhancers, facilitating recruitment of the CBP/p300 H3K27 histone acetylase (HAT), which ultimately primes enhancers for gene activation (15,16). The KMT2C/D-COMPASS-like complex is recruited to enhancers by the BAP1-ASXL1/2 complex, which facilitates enhancer priming (17,18). In addition, the methyltransferase SETD2 deposits H3K36me3 marks at active enhancers and transcribed gene bodies (19,20). Thus, our top hits converge on regulating enhancer function (Fig. 1E).
We validated each hit by injecting Pik3caH1047R; Cas9 mice individually with one sgRNA from the library, and one newly designed sgRNA targeting Asxl2, Kdm6a, Kmt2c, and Setd2 (termed EpiDrivers) or Trp53 and Apc. We also transduced mice with sgRNAs targeting Asxl1 and Bap1, which were not in the original library. All transduced mice developed multiple highly proliferative breast tumors with much shorter latencies than mice transduced with non-targeting control sgRNAs (sgNT) (Fig. 2A; Supplementary Fig. S2D and S2E). All tested tumors harbored bi-allelic frame-shift mutations in the target genes, and western blot analysis confirmed loss of APC, ASXL2, KDM6A, and p53 expression (Supplementary Fig. S2F–K).
Fig. 2. Validation and transcriptomic profiling of EpiDriver tumors.

A, Tumor-free survival of Pik3caH1047R;Cas9 mice injected with CRISPR lentivirus targeting the indicated gene or non-targeting control sgRNA (sgNT). Two independent sgRNAs/gene were used and data was combined (see Supplementary Fig. S2d for single sgRNA data). B, Tumor-free survival of Pik3caH1047R mice with conditional knockout of Asxl2 or Kdm6a. C, PC plot of all profiled tumor transcriptomes. D and E, METASCAPE analysis showing enriched (D) and depleted (E) pathways in common de-regulated genes in EpiDriver-KO tumors compared to control tumors. (F) K-means clustering of DE ChIP peak regions based on differential signal for H3K27Ac H3K27me3 and H3K4me1 between WT and sgKdm6a cells. Peaks were stratified as promoter proximal or distal based on a minimal distance of >= 2.5kb to an annotated TSS (see Methods).
Histologically, control tumors and Asxl2-, Kmt2c- and Kdm6a-mutant tumors presented mostly as invasive ductal carcinoma usually with glandular and some papillary differentiation. Trp53 and Apc-mutant tumors presented mostly as squamous or basal-like tumors. Detailed analysis by mouse tumor pathologists revealed further glandular, squamous, mixed squamous/glandular (adenomyoepithelioma) or spindle cell differentiation patterns consistent with published reports of Pik3caH1047R-induced mouse mammary tumors (12,21) (Supplementary Fig. S3A–C and Supplementary Table 2). All tumors were estrogen receptor-positive and recapitulated gland morphology with cells marked by basal keratin 14 (K14) or luminal keratin 8 (K8). The Trp53-mutant tumors showed an increased proportion of K14/K8 double positive cells, which were also seen in invasive micro-clusters of EpiDriver-mutant tumors (Supplementary Fig. S3D–H).
Next, we transduced the mammary epithelium of Kdm6afl/fl;Pik3caH1047R/+ and Asxl2fl/fl;Pik3caH1047R/+ mice with lentiviral Cre and observed significantly accelerated tumor formation (68 and 154 days versus 308 days for Pik3caH1047R/+, p<0.002), which not only confirmed our CRISPR/Cas9 results, but also revealed that females with Kdm6afl/+ tumors presented with significantly shorter tumor-free survival (235 days, p=0.001) (Fig. 2B). KDM6A is located on the X-chromosome, but escapes X-inactivation and its expression reflects gene copy number (22,23). Heterozygous Kdm6afl/+ tumor cells still expressed Kdm6a (Supplementary Fig. S4A and S4B), ruling out loss-of-heterozygosity and indicating that Kdm6a functions as haploinsufficient tumor suppressor.
To test whether our hits also function as tumor suppressors in a mouse model of basal-like breast cancer, we transduced the mammary epithelium of Trp53flfl;Rb1flfl;Cas9 mice with LV-sgRNA-Cre targeting Kmt2c or Kdm6a, or sgNT control. Loss of Kmt2c significantly reduced tumor latency (323 versus 436 days; p=0.038) and ablation of Kdm6a resulted in a trend towards reduced tumor latency (348 versus 436 days; p=0.17; Supplementary Fig. S4C), indicating that these EpiDrivers might function as tumor suppressors in several breast cancer subtypes and genetic backgrounds.
EpiDrivers Regulate Genes Involved in EMT, Inflammatory Pathways and Differentiation
Next, we set out to molecularly characterize the EpiDriver knockout tumors. Transcriptional profiling of FACS-isolated Asxl2-, Kdm6a,- Kmt2c-, Setd2-, Trp53- and Apc-mutated Pik3caH1047R tumor cells revealed a wide range of differentially expressed genes compared to control sgNT transduced tumor cells (450–1800 genes; FDR <0.05, fold-change >2, Supplemental Table S3). Principal component (PC) and Pearson’s correlation analyses revealed high concordance between tumors transduced with sgRNAs targeting the same gene (Fig. 2C, Supplementary Fig. S4D). Variance along PC1 and PC2 were driven by Apc and Trp53 loss, respectively. Consistent with their squamous histology, gene set enrichment analysis (GSEA) revealed increased expression of genes linked to keratinization in Apc-mutant tumors, whereas Trp53-mutant tumors showed downregulation of p53-related pathways (Supplementary Fig. S5A and S5B). In addition, intra- and cross-species comparisons revealed that the transcriptome of several Trp53-mutant mammary tumors clustered with basal-like human and mouse breast cancer, while the control and EpiDriver-mutant Pik3caH1047R tumors clustered with human HER2 and/or luminal breast cancers (Supplementary Fig. S5C), further underscoring distinct biology of Apc- and Trp53-mutant tumors.
Compared to Apc- and Trp53-mutant tumors, EpiDriver tumors clustered closely together and closer to control sgNT tumors, indicating that they are transcriptionally less divergent (Fig. 2C). Focusing specifically on EpiDriver-mutant versus control sgNT Pik3caH1047R tumors revealed that EpiDriver inactivation leads to upregulation of ‘epithelial-to-mesenchymal transition (EMT)’, ‘pro-inflammatory interferon-α/γ responses’ and downregulation of cellular metabolism (‘oxidative phosphorylation’ and ‘fatty acid metabolism’) and ‘estrogen responses’ (Supplementary Fig. S5A). Pairwise comparison revealed differences between EpiDriver-mutant transcriptomes, but that overall EpiDriver tumors were more similar to each other than to the control sgNT tumors (3–40 differential pathways in pairwise EpiDriver-mutant comparisons versus 46–111 differential pathways between EpiDriver-mutant and sgNT control tumors) (Supplementary Fig. S6A–G), which is expected for proteins within the same molecular complex. To further elucidate a shared molecular profile, we focused on genes that were commonly dysregulated in all EpiDriver-mutant tumors relative to controls (Supplemental Table S3). Pathway analysis of these 498 ‘commonly dysregulated’ genes revealed enrichment of ‘extracellular matrix organization’ and EMT, and downregulation of ‘epidermis development’ and ‘epithelial cell differentiation’ in EpiDriver-mutant tumors relative to control tumors (Fig. 2D and E; Supplementary Fig. S7A and B).
To identify downstream target genes involved in tumor suppression, we screened 283 genes downregulated in EpiDriver-mutant tumors for their ability to suppress mammary tumor formation in Pik3caH1047R;Cas9 mice (Supplementary Fig. S7C). In this secondary screen, the histone lysine demethylase and nuclear receptor corepressor hairless (Hr), interleukin 4 receptor (Il4ra) and the transcription repressor Bcl6 scored as hits, indicating that these shared downregulated genes function themselves as tumor suppressors (Supplementary Fig. S7D). Of note, Bcl6 also scored in the primary screen and has known function in mammary gland biology and lactation (24,25).
Together these data show that EpiDriver loss leads to significantly accelerated tumor initiation associated with EMT and altered differentiation but does not affect histologic and molecular subtype. By contrast, loss of Apc or Trp53 not only accelerated tumor development, but also caused dramatic transcriptional and histological changes.
Pre-tumorigenic Cells Display Lineage Plasticity and Aberrant Alveogenesis
To elucidate how EpiDriver loss accelerates tumor initiation, we first assessed sphere-forming capacity of Pik3caH1047R-mutant mammary epithelial cells four weeks after EpiDriver mutation. Interestingly, Asxl2-, Kdm6a- or Kmt2c-mutant cells formed significantly more mammospheres that grew to larger diameters compared to LV-sgNT-Cre transduced control mammary epithelial cells (Supplementary Fig. S7E–G), indicating a growth advantage early in tumor formation (26).
Next, we assessed how loss of the COMPASS-like complex affects the histone modification landscape of mammary epithelial tumor cells. We focused on Kdm6a, a core member of the COMPASS-like complex (15,16), and performed ChIP-seq for H3K27me3, H3K27ac, and H3K4me1 and transcriptional profiling on cultured primary Pik3caH1047R mammary tumor cells derived from tumors transduced with either sgKdm6a or control sgNT (Supplementary Fig S8A). We identified differential peaks and clustered them based on the differential ChIP signal for all 3 histone marks at promoter-proximal (TSS +/− 2.5 kb) or previously identified distal enhancer regions. For each of distal and proximal regions, we identified two distinct clusters: cluster 1 displaying increased H3K27me3 and decreased H3K27ac and H3K4me1, indicating repressed regions in KDM6A-mutant cells; and cluster 2 with opposite histone profile, indicating activated regions (Fig. 2F). Indeed, we observed the expected up-/down-regulation of transcription at promoter-proximal regions consistent with the histone profiles (Fig. 2F and Supplementary Fig. S8B and S8C). Gene set-based analysis of differentially expressed genes by RNA-seq again revealed EMT and differentiation as most significant sets upregulated in cultured Kdm6a-mutant mammary tumor cells (Supplementary Fig. S8C–E), consistent with our findings from the EpiDriver-mutant tumors.
Probing deeper into the mechanism of how inactivation of Kdm6a affects transcription and chromatin accessibility at the onset of transformation, we performed parallel single-cell RNA sequencing (scRNA-seq) and single nucleus assay for transposase-accessible chromatin using sequencing (snATAC-seq). First, we analysed scRNA-seq data from FACS-isolated GFP+ LSL-Pik3caH1047R;Kdm6afl/fl; LSL-Cas9-EGFP (Pik3caHR; Kdm6aΚΟ) and LSL-Pik3caH1047R; LSL-Cas9-EGFP (Pik3caHR) and LSL-Cas9-EGFP control mammary epithelial cells two weeks after intraductal Ad-Cre injection. Removing low-quality cells with low read depth (<2,500), high mitochondrial reads (>10%) and/or less than 1000 detected genes resulted in 14,070 high-quality cells composed of 6,160 control, 2,855 Pik3caHR and 5,055 Pik3caHR;Kdm6aΚΟ cells (Supplementary Fig. S9A). Based on canonical markers (27), UMAP clustering revealed the three major epithelial populations corresponding to luminal progenitors (LP; Kit+, Elf5+), hormone-sensing mature luminal (HS-ML; Prlr+, Pr+, Esr1+) and basal cells (Krt5/14+) with distinct subclusters composed of the three genotypes (Fig. 3A and B).
Fig. 3. Single-cell transcriptional profiling reveals alveogenic mimicry.
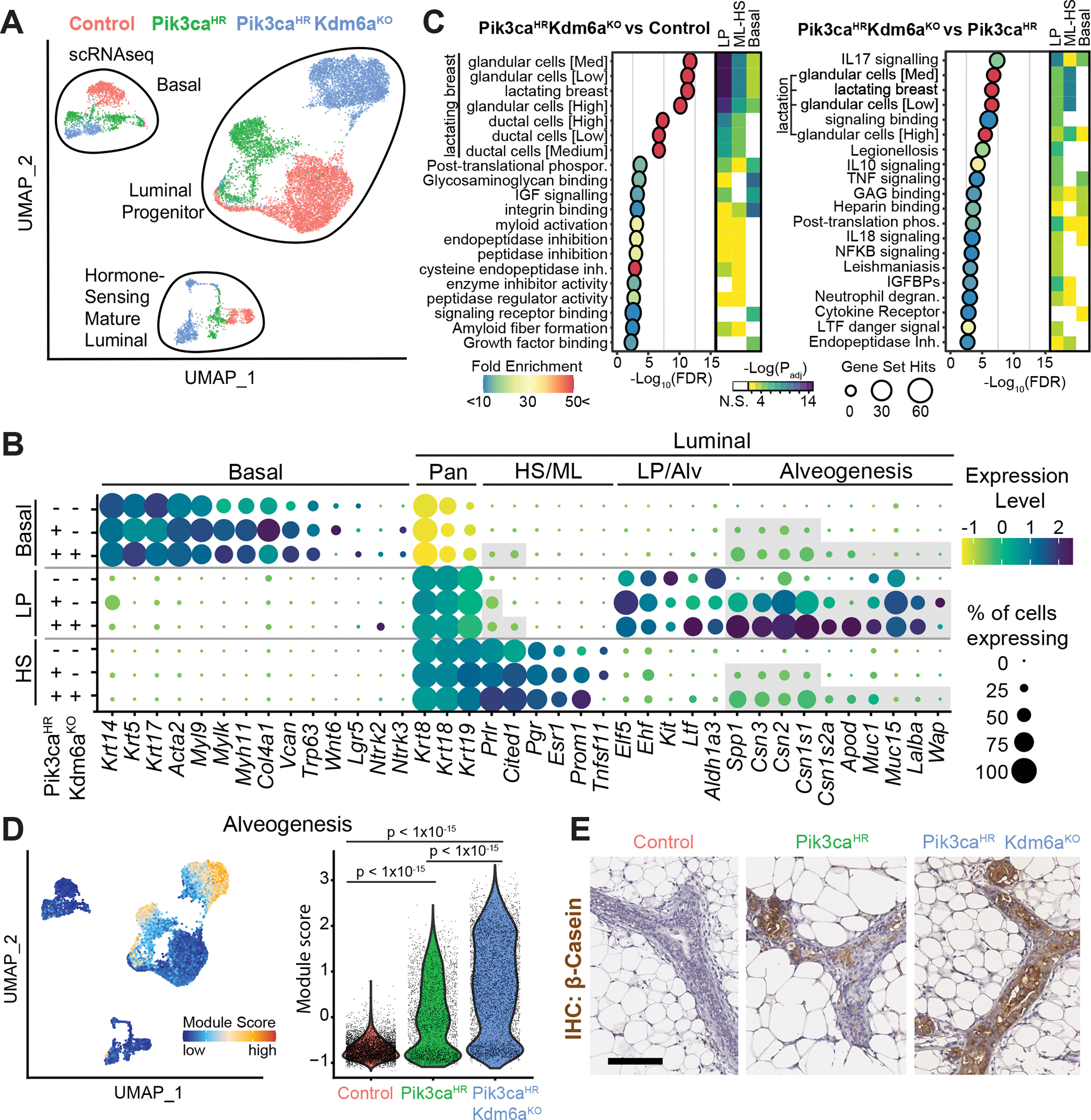
A, UMAP plot showing mammary epithelial cells from control, Pik3caH1047R and Pik3caH1047R;Kdm6afl/fl mutant mice 2 weeks after Ad-Cre injection. B, Dot Blot showing differentially expressed marker genes within the different epithelial lineages stratified by genotypes. C, Pathways differentially enriched in Pik3caH1047R;Kdm6afl/fl versus control and Pik3caH1047R;Kdm6afl/fl versus Pik3caH1047R mammary epithelial LP cells identified using g:Profiler (p <0.05 with Benjamini-Hochberg FDR correction, > 10-fold enrichment). The top 20 enriched pathways are shown. Heat- map depicts how these pathways are altered in the major 3 epithelial lineages. D, UMAP and violin blots showing alveogenesis signature. E, Immunohistochemistry of mammary glands 2 weeks post injection stained with anti-β-Casein. Scale bar is 100 μm.
We performed functional enrichment analysis to reveal the molecular pathways dysregulated upon activation of Pik3caHR and inactivation of Kdm6a within each epithelial lineage. Surprisingly, this analysis revealed ‘lactation’ as the most differentially regulated pathway in Pik3caHR;Kdm6aΚΟ versus control cells. ‘Lactation’ was also upregulated but to a lesser degree in Pik3caHR;Kdm6aΚΟ versus Pik3caHR cells (Fig. 3C). This signature was driven by genes that are typically only expressed upon differentiation of LPs into secretory alveolar cells in a hormone-dependent manner during gestation/lactation, and included caseins (Csn1s1, Csn1s2a, Csn2, and Csn3), milk mucins (Muc1/15), lactose synthase (Lalba), apolipoprotein D (Apod), and milk proteins (Glycam1, Spp1, and Wap) (Fig. 3B). Interestingly, we observed upregulation of these genes in the absence of gestation/parity-induced hormones and not only in LP cells but also in some basal and HS-ML Pik3caHR;Kdm6aΚΟ cells (Fig. 3C and D; Supplementary Fig. S9B). Interestingly, this upregulation of alveogenesis/lactation was associated with a downregulation of genes associated with previously described non-lactation LP cells (28) (Supplementary Fig. S9C). Immunohistochemistry confirmed the increased casein levels in Pik3caHR;Kdm6aΚΟ versus Pik3caHR mammary tissue cells (Fig. 3E). Importantly, genetic ablation of Kmt2c or Asxl2 in Pik3caH1047R-mutant glands also triggered casein expression (Supplementary Fig. S10A and B), indicating a shared phenotype.
Other changes were also evident in Pik3caHR;Kdm6aΚΟ cells. For example, they exhibited upregulation of genes associated with EMT, hypoxia, and involution (Supplementary Fig. S10C and S11A). Pik3caHR and Pik3caHR;Kdm6aΚΟ cells also exhibited higher expression of characteristic HS-ML genes such as Cited1 and prolactin receptor (Prlr) not only in HS-ML cells but also in a subset of LP and/or basal cells (Fig. 3B; Supplementary Fig. S11B). Conversely, basal markers such as Krt14, Lgr5, and Nrtk2 showed aberrant expression in Pik3caHR and/or Pik3caHR;Kdm6aΚΟ LP cells (Supplementary Fig. S11C). Overall, our data reveal reprogramming of transcriptional landscapes, loss of lineage integrity, and induction of alveogenesis in all mammary epithelial lineages upon oncogenic PI3K signaling, and these cancer hallmarks are exacerbated by loss of EpiDrivers.
Chromatin Profiling Confirms Epigenetic Reprogramming and Mimicry of Alveogenesis
In line with the scRNA-seq results and our previous data (29), unsupervised UMAP-clustering of the snATAC-seq data showed that chromatin accessibility clearly separated the three major mammary epithelial lineages (Fig. 4A). While control, Pik3caHR and Pik3caHR;Kdm6aΚΟ cells were intermingled in the HS-ML cluster, indicating that they are indistinguishable with regards to accessible chromatin, they formed distinct sub-clusters in the LP and to a lesser degree in the basal cluster (Fig. 4A). Within the LP clusters there was a modest difference between control and Pik3caH1047R LP cells, large differences were observed between control and Pik3caH1047R;Kdm6aΚΟ, and between Pik3caH1047R and Pik3caH1047R;Kdm6aΚΟ LP cells (Fig. 4B), showing that loss of Kdm6a has a profound effect on chromatin accessibility. In line with KDM6A’s H3K27 demethylase function in COMPASS-like enhancer activation, we found substantially more genomic accessibility in Kdm6a-mutant cells (Fig. 4B).
Fig. 4. Single-cell ATACseq reveals alveogenic mimicry and bridge-like clusters.
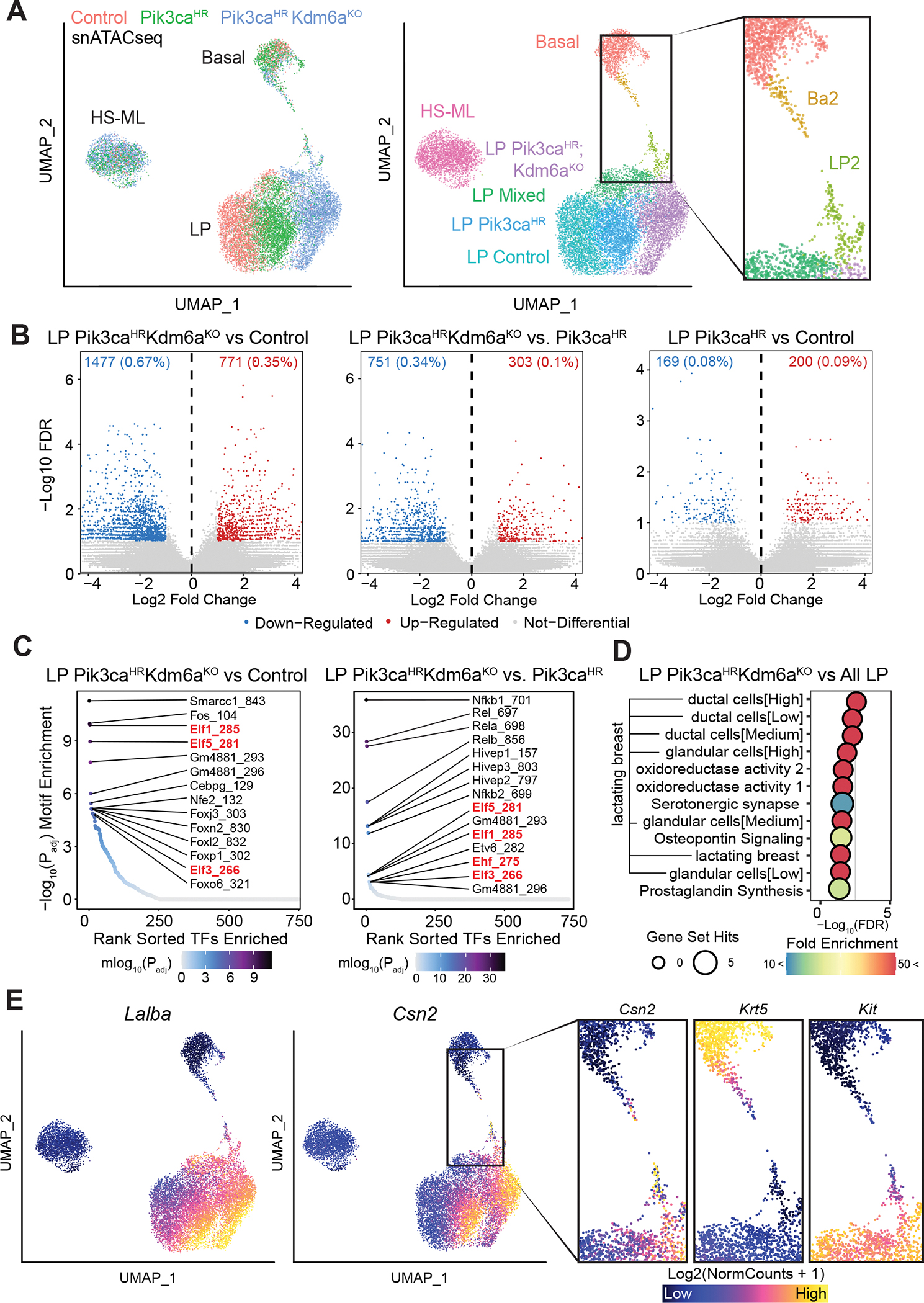
A, Unsupervised UMAP plot of snATACseq profile colored by genotype (left) and identified clusters (middle). Inlet (right) shows BA2 and LP2 clusters. B, Volcano plots showing differentially accessible chromatin peaks between Pik3caH1047R;Kdm6afl/fl and wild-type control or between Pik3caH1047R;Kdm6afl/fl and Pik3caH1047R or between Pik3caH1047R and wild-type control LP cells. C, Enrichment of transcription factor binding sites in differentially accessible chromatin. D, Pathways differentially enriched in Pik3caH1047R;Kdm6afl/fl versus all mammary epithelial LP cells inferred from gene accessibility ArchR Gene Scores. The top 12 enriched pathways are shown as identified using g:Profiler (p <0.05 with Benjamini-Hochberg FDR correction, >10-fold enrichment). E, UMAP plots showing open chromatin associated with alveolar/lactation-associated genes Lalba and Csn2. Inlet (right) shows open chromatin associated with alveolar/lactation gene Csn2, the basal marker gene Krt5 and the LP marker gene Kit in BA2 and LP2 clusters.
We next examined the representation of transcription factor motifs in the differentially accessible genomic regions. The regions with increased accessibility in the Pik3caHR;Kdm6aΚΟ relative to wild-type LP cells were significantly enriched for binding sites of Fos and Smarcc1, followed by the Ets factors Elf1/3/5. Motifs enriched in the Pik3caH1047R;Kdm6aΚΟ relative to the Pik3caH1047 LP cells corresponded to Nfκ-b factors NFκ-B1/2 and Rela/b followed again by core LP regulators Elf1/3/5 and Ehf (Fig. 4C). Similar enrichment profiles were seen from activity inference using chromVAR (30) (Supplementary Fig. S12A and S12B). Consistent with the known function of Elf5 and Ehf in driving alveolar differentiation (27,31), and in line with the scRNA-seq data, gene set-based analysis of accessible loci revealed ‘lactation’ as the most significant set upregulated in Pik3caH1047R;Kdm6aΚΟ LP cells; this association included increased accessibility to multiple alveolar/milk biogenesis-related genes, such as Apod, Csn2/1s1/1s2a, Lalba, Lif, Lipa, and Spp1(Fig. 4D and E; Supplementary Fig. S13A).
Further examination of scATAC-seq results identified a basal-like ‘Ba2’ and a luminal-like ‘LP2’ subcluster enriched in Pik3caH1047R and Kdm6aΚΟ;Pik3caH1047R cells that appear to bridge the basal and LP populations (Fig. 4A). Gene set-based analysis of accessible loci in these subclusters revealed sets associated with ‘chromatin silencing’ (Supplementary Fig. S13A and S13B). In addition, the biological KEGG pathway ‘breast cancer’ was upregulated in the Ba2 versus the basal cluster, with the identification of prominent WNT (Wnt10a, Wnt6, Fzd2, Dvl2, Prickle4, Csnk1g2 and Dlg4) and NOTCH (Dll1 and Jag2) signaling genes (Supplementary Fig. S13A and S13C). In line with this notion, chromVAR analysis showed enrichment of binding sites for transcription factors associated with WNT (Lef1, Tcf7, Tcf7l1, Tcf7l2) and NOTCH signaling (Hes1, Hey1/2, Heyl) in Ba2 cells (Supplementary Fig. S12A). Consistently, we observed upregulation of WNT and NOTCH signaling signatures in Pik3caH1047R and Pik3caHR;Kdm6aΚΟ basal cells in the scRNA-seq dataset (Supplementary Fig. S13D and S13E). Of note, Apc was a major hit in the in vivo CRISPR screen (Fig. 1D), suggesting that elevated WNT signaling is oncogenic in the Pik3caHR model. In addition, WNT and NOTCH signaling are not only known drivers of breast cancer, but also play critical roles in mammary lineage determination (32–34).
Overall, we found that Ba2 cells have reduced chromatin accessibility at basal markers, such as Acta2, Krt5/14, Trp63, and Vim, and increased accessibility of the alveolar genes, such as Csn2, whereas LP2 cells have reduced chromatin accessibility at LP markers, such as Elf5, Ehf, and Kit (Fig. 4E; Supplementary Fig. S14A–C and S15A–C). These data are consistent with the loss of lineage identity observed in the scRNAseq data. Together, our scRNAseq and snATACseq data suggest that Pik3caHR;Kdm6aΚΟ mammary epithelial cells gain lineage plasticity and prior to tumorigenesis reprogram towards the alveolar fate reminiscent of epithelial expansion and differentiation preceding lactation.
To functionally test whether inducing an alveogenic program can indeed accelerate tumorigenesis, we overexpressed ELF5, the key regulator of alveogenesis, in Pik3caHR mammary epithelial cells. Transduction of lentiviruses overexpressing Elf5 (LV-Elf5-Cre) induced faster tumor formation compared to control LV-Ruby-Cre (p<0.05). This is consistent with previous findings of Elf5 overexpression in a PyMT breast cancer mouse model (35,36) (Supplementary Fig. S16A and S16B). In addition, overexpression of ELF5 in Pik3caH1047R mammary epithelial cells triggered casein expression (Supplementary Fig. S16C), reminiscent of the consequences of EpiDriver mutations. Together, these results support a role of alveogenic mimicry in mammary gland tumorigenesis.
The COMPASS-like Complex Inhibits a Tumorigenic Basal-to-Luminal Cell Lineage Conversion
We next determined whether both luminal and basal cells are susceptible to lineage plasticity and contribute to tumor formation using lineage tracing with a basal-specific adenoviral Ad-K5-Cre and luminal-specific Ad-K8-Cre viruses (37) (Supplementary Fig. S17A–E). As previously shown (38,39), expression of oncogenic Pik3caH1047R can lead to lineage plasticity and convert basal and luminal unipotent progenitors into multipotent cells. In line with these reports, induction of Pik3caH1047R in basal cells resulted in a gradual lineage conversion to luminal-like cells, which was dramatically accelerated by Kdm6a or Asxl2 mutation (Fig. 5A–C). In line with a haploinsufficiency tumorigenic effect, heterozygous loss of Kdm6a also significantly accelerated basal-to-luminal lineage conversion (Supplementary Fig. S17F). In contrast, genetic ablation of Kdm6a or Asxl2 did not accelerate lineage conversion from luminal-to-basal cells (Supplementary Fig. S17G).
Fig. 5. Loss of EpiDrivers induces multipotency.
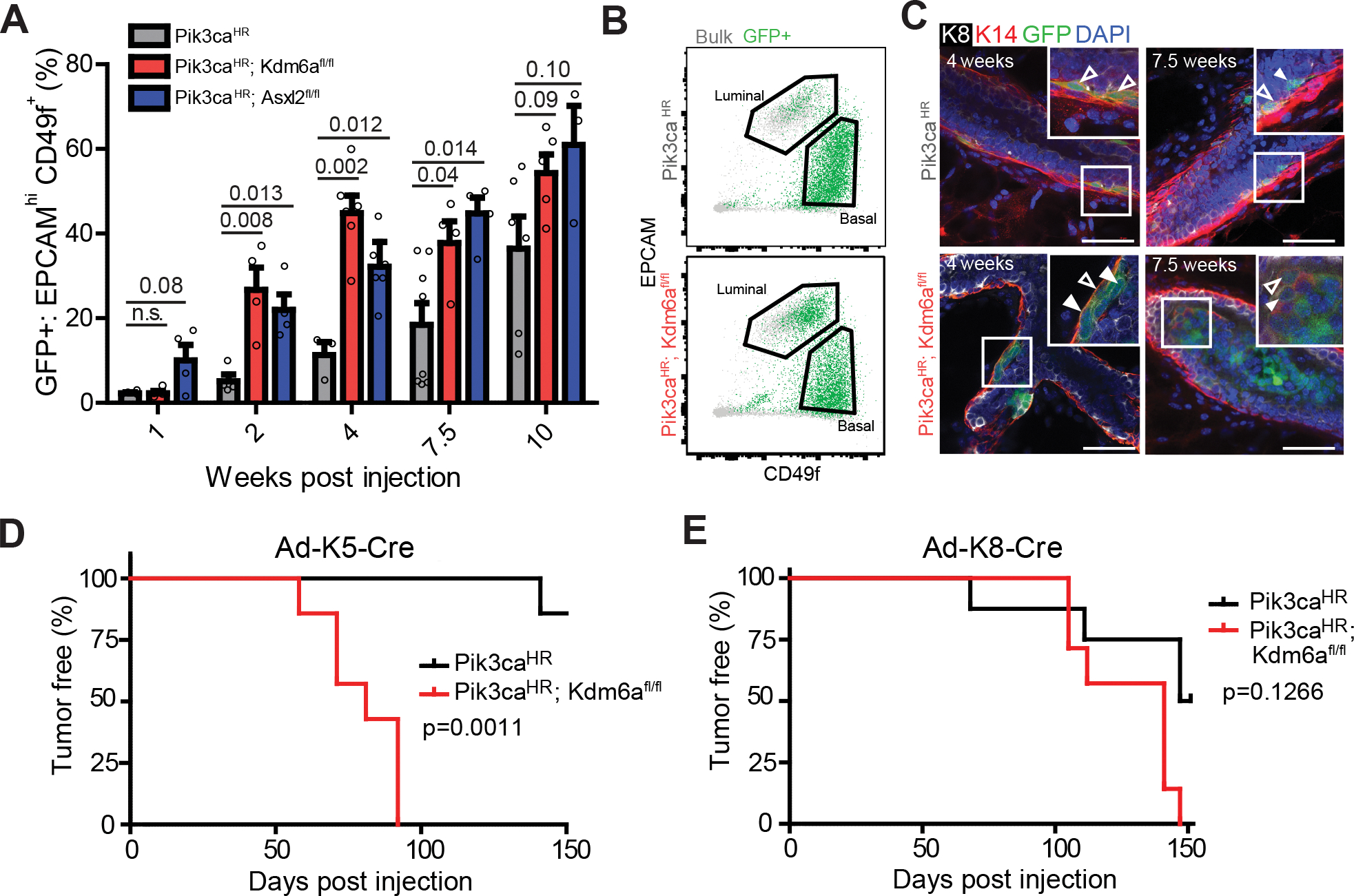
A, Percent of GFP+ EPCAMhigh CD49fmid luminal cells at different time points after Ad-K5-Cre injection into mammary epithelium of mice with the indicated genotype. B, Representative FACS plot at 4 weeks post injection with Ad-K5-Cre. C, Whole-mount image of mammary glands 4 weeks and 7.5 weeks post Ad-K5-Cre injection showing K14+/K8- (empty arrows) as well as K14+/K8+ double-positive and K14-/K8+ GFP+ lineage-traced cells (filled arrows). Scale bar = 50 μm. D and E, Tumor-free survival of Pik3caH1047R;Kdm6afl/fl versus Pik3caH1047R after intraductal injection of Ad-K5-Cre (D) and Ad-K8-Cre (E).
To further characterize this basal-to-luminal lineage conversion, we used a K5-CreERT2 transgenic strain crossed to Pik3caH1047R;Kdm6afl/fl;LSL-Cas9-GFP mice. We used low dose tamoxifen treatment to genetically ablate Kdm6a and concomitantly activate Pik3caH1047R at clonal density in the basal mammary compartment. This approach corroborated our findings and allowed us to quantify converting clones along the epithelial tree. At four weeks after tamoxifen treatment we observed that 50% of GFP+ lineage-traced basal clones have generated K8+ positive luminal-like cells (Supplementary Fig. 17H), demonstrating that this lineage conversion is a frequent event in Pik3caH1047R;Kdm6aKO mammary tissue.
Next, we determined if the cell-of-origin affects the latency and phenotype of tumors arising in Pik3caH1047R;Kdm6afl/fl mice. Loss of Kdm6a in the basal compartment significantly accelerated tumor formation, whereas luminal cell-derived Pik3caHR;Kdm6aΚΟ tumors arose with similar latency as Pik3caHR tumors (Fig. 5D and E). Transcriptome analysis revealed that basal-cell derived tumors clustered with other mouse and human luminal-like tumors (Supplementary Fig. S5C), were indistinguishable from tumors derived upon sgRNA-mediated mutation of Kdm6a, and exhibited K5+, K8+ and K5/K8 double-positive cells and casein+ cells (Supplementary Fig. S17I–K). Together, these results indicate that loss of the COMPASS-like complex in Pik3caH1047R basal cells accelerates their reprogramming into tumor-initiating cells that drive luminal-like breast cancer.
To further characterize the basal-to-luminal-like cell transition, we performed scRNA-seq on control, Pik3caHR or Pik3caHR;Kdm6aΚΟ mammary epithelial cells after two weeks of Ad-K5-Cre lineage-tracing (Fig. 6A–D; Supplementary Fig. S18A). Consistent with the results above, LP-like cells that lost basal markers and gained LP (e.g. Cd14, Elf5, Kit) and alveolar markers (e.g. Apod, Cns3, Wfdc18) emerged from Pik3caHR and Pik3caHR;Kdm6aΚΟ basal cells. We even observed rare Pik3caHR and Pik3caHR;Kdm6aΚΟ cells expressing milk genes, such as Olah and Wap, and HS-ML markers, such as Prlr (Fig. 6E–G; Supplementary Fig. S18B, S18C and S19A–C).
Fig. 6. scRNAseq reveals basal-to-alveolar transdifferentiating at the onset of breast cancer initiation.
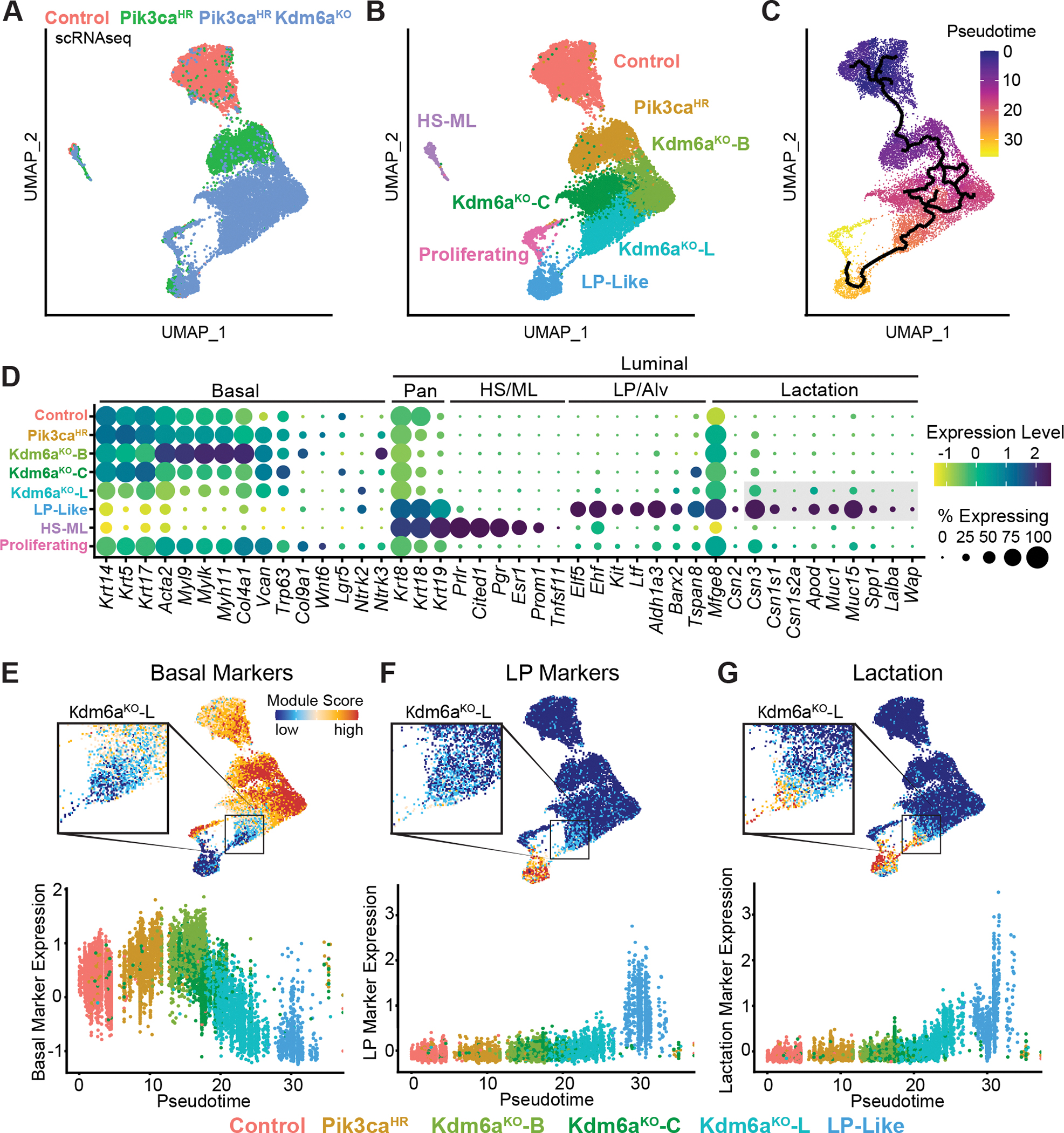
A-C, UMAP plots showing Ad-K5-Cre lineage-traced basal mammary epithelial cells from control, Pik3caH1047R and Pik3caH1047R;Kdm6afl/fl mutant mice 2 weeks post-injection colored by genotype (A), clusters (B) and trajectories inferred by Monocle3 (C). D, Dot plot showing differentially expressed marker genes within the different epithelial clusters. E-G, UMAP and pseudo-time trajectory plots showing basal (E), luminal progenitor (F) and alveolar/lactation (G) marker signatures.
In addition, Pik3caHR;Kdm6aΚΟ basal cells were more heterogenous than wild-type or Pik3caHR cells and comprised three unique subclusters: Kdm6aKO-L, adjacent to the LP-like population, a central cluster (Kdm6aKO-C), and a cluster enriched in basal/myoepithelial markers (Kdm6aKO-B; Acta2, Igfbp2, Myh11, Myl9) (Fig. 6B), further underscoring the notion of increased phenotypic plasticity upon loss of Kdm6a. Importantly, Kdm6aKO-L showed a gradual downregulation of basal markers with concomitant upregulation of alveolar/lactation markers such as Apod, Csn2/3, Muc1/15 or Wfdc18 (Fig. 6E–G; Supplementary Fig. S18B, S19A–C and S20A–C). Kdm6aKO-L was also marked by expression of the EMT master regulators Zeb1 and Zeb2, the latent TGFB binding gene product Ltbp1, as well as Ntrk2 and Socs2 (Supplementary Fig. S20D). Of note, Ntrk2 was previously identified as a basal-to-luminal multipotency breast cancer gene (38) and, together with Ptn, are known drivers of breast cancer (40). Interestingly, this Kdm6aKO-L cluster did not generally express classic luminal progenitor markers (Aldh1a3,Cd14, Elf5, Kit, Lif) (Fig. 6F; Supplementary Fig. S18C). This observation combined with trajectory analysis suggests that Kdm6aΚΟ;Pik3caH1047R basal cells start to gradually activate an aberrant alveolar-like program before acquiring LP characteristics (Fig. 6C–G).
Integrating the Ad-Cre and the Ad-K5-Cre scRNAseq datasets revealed that luminal-like K5-traced Pik3caHR and Pik3caHR;Kdm6aΚΟ cells clustered with LP cells, further supporting the notion of a basal-to-luminal reprogramming. In addition, luminal-like K5-traced Pik3caHR and Pik3caHR;Kdm6aΚΟ cells with high lactation and involution signatures clustered with Pik3caHR and Pik3caHR;Kdm6aΚΟ LP cells, while those without a lactation/involution signature clustered with wild-type LP cells, suggesting functional heterogeneity (Supplementary Fig. S21A–C).
Cells in the proliferating cluster consisted mainly of Pik3caHR;Kdm6aΚΟ with either basal or luminal characteristics (Fig. 6A, B and E–F). This cluster also showed marked elevation of RB1/E2F target genes (Supplementary Fig. S22), reminiscent of RB1 inactivation and E2F activation during pregnancy-induced hyperproliferation in the mammary gland (41). These data further support a role of these proliferating cells and the aberrant alveolar program during tumor initiation.
Human Breast Cancer Shows Frequent EpiDriver Alterations and Signs of Aberrant Alveogenesis
To extend our findings from mouse to humans, we assessed the function of the EpiDrivers in human MCF10A mammary epithelial cells that harbor a PIK3CAH1047R knock-in mutation (42,43). Using CRISPR/Cas9, we generated ASXL2-, KDM6A-, KMT2C-, SETD2-, PTEN- and TP53-mutant cell lines as well as control sgNT cells (Supplementary Fig. S23A–D). Like the parental cells, MCF10A PIK3CAH1047R cells formed polarized and hollow, albeit modestly larger, acini in Matrigel culture (43). In contrast, ASXL2-, KDM6A-, KMT2C-, or PTEN-mutant spheres showed a transformed phenotype with large branching protrusions (Supplementary Fig. S23E and S23F). When grafted orthotopically into the fat pads of immunodeficient (NOD scid gamma, NSG) mice, the KDM6A-, SETD2-, TP53- and PTEN-mutant PIK3CAH1047R cells formed tumors while control sgNT cells did not (Supplementary Fig. S23E). Although the ASXL2- and KMT2C-mutant cells exhibited a transformed phenotype in 3D cultures, they did not efficiently give rise to xenograft tumors in mice. Together, these data indicate that the EpiDrivers ASXL2, KMT2C, KDM6A, and SETD2 suppress transformation of human MCF10A mammary epithelial cells.
Next, we compared our results from mouse Kdm6a-mutant mammary tumor cells to the data obtained from transcriptome and epigenetic profiling of human KDM6A-mutant PIK3CAH1047R MCF10A cells. We used two independent sgKDM6A-knockout and two sgNT control clones (Supplementary Fig. S23G) and performed RNA-seq and ChIP-seq for H3K27me2, H3K27ac, and H3K4me1. As expected, the clones clustered together by genotype for both transcriptional and H3K27me3, H3K27ac and H3K4me1 profiles (Supplementary Fig S23H and I). Clustering of differential promoter-proximal and --distal peaks based on their histone marks again revealed two clusters: cluster 1 displaying increased H3K27me3 and decreased H3K27ac and H3K4me1, indicating repressed regions in KDM6A-mutant cells; and cluster 2 with opposite histone profile, indicating activated regions. Consistent with these histone profiles we observed the expected up-/downregulation of transcription (Supplementary Fig. S23J–L and S24A).
Like mouse Kdm6a-mutant mammary tumor cells, KDM6A-mutant MCF10A cells showed upregulation of gene sets linked to EMT and mammary stem cells, and downregulation of adhesion (Supplementary Fig S24B and S24C). Specifically, we observed upregulation in key mesenchymal markers such as CDH2, VIM, and ZEB1, and downregulation of CDH1 and of a repressor of EMT, GRHL2. KDM6A-mutant cells also showed some signs of aberrant differentiation, including upregulating KRT14, downregulating KRT18, but also gaining expression of lactation-related genes including the prolactin receptor (Supplementary Fig S24D–F). KDM6A-mutant cells also showed upregulation oncogenes (MAFB, ETV1, ROS1, and EPAS1), but downregulation of tumor suppressors (SIRPA, TP63 and PTPRB) (Supplementary Fig. S24D and S24E). Overall, these data indicate that knockout of KDM6A results in coordinated transcriptional and epigenetic alterations that induce EMT and alter differentiation concordant with our findings in mouse Kdm6a-knockout cells.
To test whether the alveogenesis program can also be found in human premalignant breast lesions, we analyzed the transcriptional profiles of 57 ductal carcinoma in situ (DCIS) and 313 invasive breast cancers (44). Remarkably, we found that curated human gene sets corresponding to mammary gland alveogenesis and lactation exhibited significantly higher expression in DCIS compared to invasive breast cancer (Fig. 7A and Supplementary Fig. S25A) and correlated with the signatures of EpiDriver loss derived from the mouse tumor studies (Supplementary Fig. S25B and S25C). To corroborate these findings, we optimized and performed immunohistochemistry for the milk protein casein CSN1S1 on tissue microarrays. Interestingly, 55% of breast atypical hyperplasia, 73% of DCIS, 44% of invasive breast cancer and 47% of breast cancer PDXs exhibited casein staining, while no normal breast or any other cancerous or non-cancerous tissue exhibited casein staining (Fig. 7B and Supplementary Fig. S26A and B). Additional staining of DCIS tumor cores revealed that while Casein staining was generally low in Krt5 single-positive cells, stronger casein staining was observed in both Krt5/Krt8 double-positive cells as well as Krt8 single-positive cells, suggesting that alveogenic mimicry can be observed during basal-to-luminal-like conversion or in intermediate lineage cells (Fig 7C and D, Supplementary Fig. 27A). Analysis of an independent panel of 118 clinically annotated DCIS revealed that 50% of hormone receptor positive (HR+), 56% of HER2+ HR+, 33% of HER2+ HR- and 20% of HER2- HR- DCIS express casein and that HR+ cases showed higher percent of casein positive cells (Supplementary Fig. S27B and S27C). We also found a that casein positive DCIS exhibited more progesterone receptor positive cells, which is in line with progesterone’s role during lobulo-alveogenesis (Supplementary Fig. S27D). Cases with casein staining did not show statistically significant differences with regards to ipsilateral breast cancer recurrence; although trends towards poorer outcome were observed especially in PR+ as well as HER2+ HR+ cases (Supplementary Fig. S27E).
Fig. 7. EpiDrivers function as Tumor Suppressors in Humans.
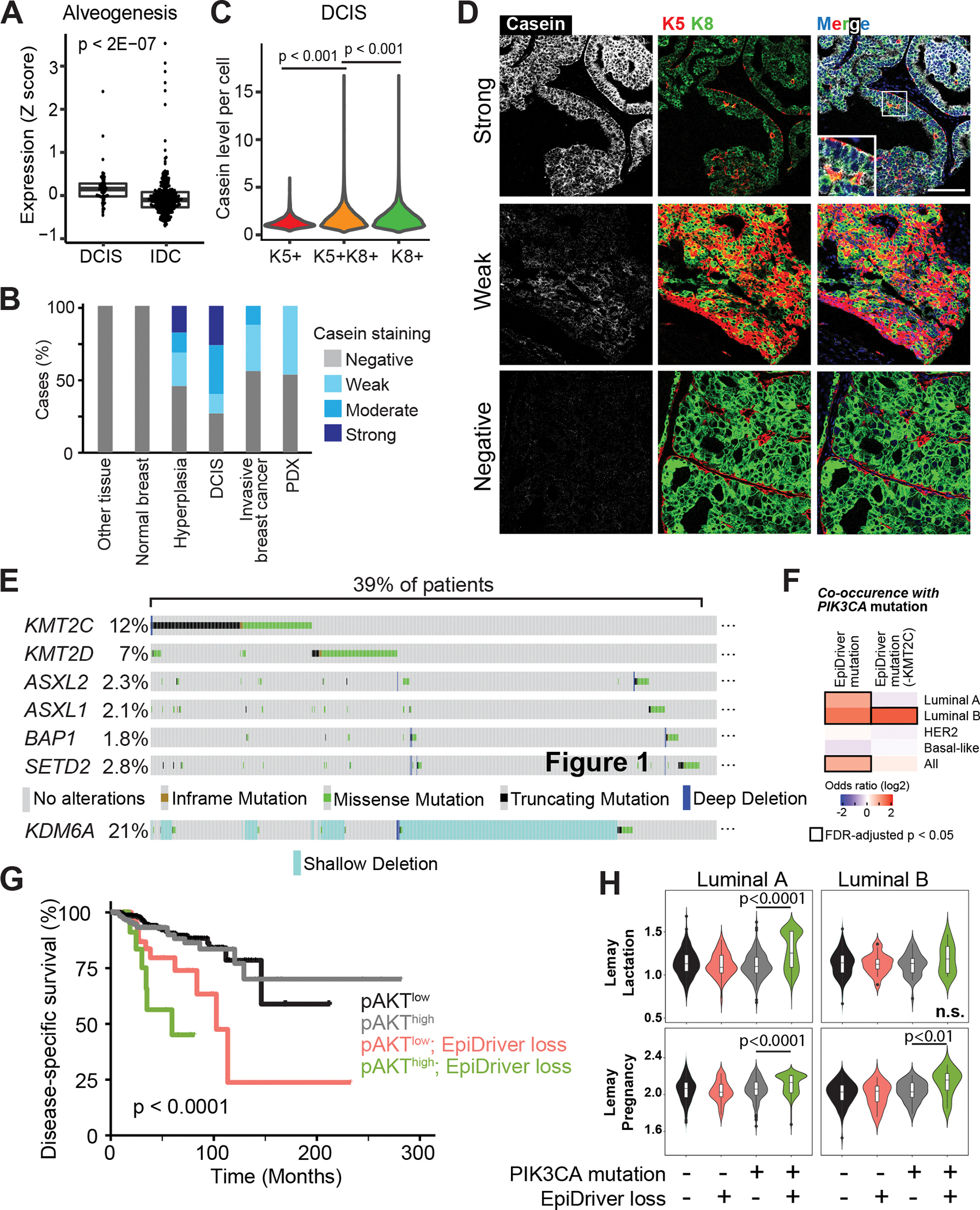
A, Average expression of the ‘Alveogenesis’ gene signature from 57 DCIS and 313 invasive tumors. B, Casein staining level by IHC in each tissue or tumor type. C, Casein staining intensity in individual cells in DCIS tumor cores separated by keratin staining. D, Representative imaging mass cytometry images of DCIS cores stained for casein and Krt5, Krt8 and nuclear stain. Scale bar = 100 μm. E, Prevalence of alterations in EpiDrivers in human breast tumors. Shallow deletion only displayed for KDM6A. F, Co-occurrence analysis of PIK3CA and EpiDriver mutations in the combined breast cancer dataset of TCGA and METABRIC. The results are shown for the complete set of identified EpiDrivers (left), or by excluding KMT2C (right), considering truncating and deleterious missense mutations. The heatmap shows the co-occurrence odds ratios (log2) across breast cancer subtypes, and all tumors considered, and significant (FDR-adjusted p < 0.05) associations are highlighted by black rectangles. g, Disease-specific survival (DSS) of breast cancer patients in the TCGA cohort stratified by phospho-Ser473 AKT (pAKT) and EpiDriver mutations. The long-rank p value is shown. h, Violin plots showing the expression of the Lemay Lactation and Pregnancy signatures in TCGA tumors with concurrent PIK3CA-EpiDriver mutations relative to other groups in luminal A and B breast cancer. The Mann-Whitney test p value is shown. The average value of the group with concurrent PIK3CA-EpiDriver mutations is depicted by a horizontal lilac line.
In human invasive breast cancer, ASXL2, BAP1, KDM6A, KMT2C, KMT2D, and SETD2 are each mutated in 1–12% of breast tumors, as expected for long-tail genes (Fig. 7E; Supplementary Fig. S28A) (11,13). The haploinsufficiency of Kdm6a in mouse mammary tumorigenesis prompted us to also analyze copy number alterations. Interestingly, an additional 19% of patients exhibited shallow deletion indicative of heterozygous KDM6A loss (Fig. 7E, Supplementary Fig. S28A), which coincided with significantly reduced KDM6A expression (Supplementary Fig. S28B). In addition, EpiDriver alterations showed a trend towards mutual exclusivity, and we observed a significant co-occurrence with PIK3CA mutations (Fig. 7F, Supplementary Fig. S28A, C and D, and Supplementary Table S4 and S5). Cases with concurrent PIK3CA and EpiDriver mutations did not show statistically significant differences with regards to overall survival (OS) when compared to cases with only PIK3CA mutation, although we did observe trends towards poorer outcome in luminal A cases (Supplementary Fig. S29). Given that high PI3K signaling can be a consequence of several genetic alterations in cancer, we performed survival analysis of TCGA breast tumors stratified by PI3K signaling defined by means of phospho-Ser473-AKT (45) or a PI3K transcriptional signature (46). Interestingly, concomitant EpiDiver mutations and high PI3K signaling stratified patients with poor survival across subtypes (Fig. 7G) as well as within Luminal A and B breast cancer (Supplementary Fig. S30A–C). Concurrent PIK3CA and EpiDriver mutations also stratified patients with worse outcome in the independent METABRIC dataset across subtypes as well as within HER2+ cases (Supplementary Fig. S31A–B).
Luminal A and/or B tumors with concurrent PIK3CA and EpiDriver mutations were found to be associated with higher expression of gene sets linked to mammary gland alveologenesis and lactation and homologous genes up-regulated in EpiDriver-mutant mouse breast cancers (Fig. 7H, Supplementary Fig. S32A and S32B). GSEA identified hallmarks of EMT and immune system function (interferon-α/γ responses, inflammatory responses, TNFα and TGFβ signaling) and downregulation of cellular metabolism (oxidative phosphorylation and fatty acid metabolism) associated with concurrent PIK3CA and EpiDriver mutations especially in luminal B tumors akin to our mouse model (Supplementary Fig. S32C and D). Together, these data highlight the relevance of the tumor suppressive EpiDriver network and alveogenic mimicry during breast cancer initiation.
Discussion
Large international efforts such as TCGA and ICGC have set out to profile the mutational landscape of many cancers with the goal of cataloguing the genes responsible for tumor initiation and progression. The idea was to identify those genes that are mutated more frequently than expected by random chance and the expectation was that increasing sample size will boost the power to mathematically infer driver mutations (i.e., sensitivity), while weeding out background of random somatic mutations (i.e., specificity). These efforts had considerably expanded the catalogue of cancer genes; however, as these studies advance, it is more evident that the individual contribution of most cancer genes to a given cancer burden is very modest. This observation raises important concerns on how confidently we can identify cancer genes based on their mutation profiles and, most importantly, highlight the fundamental question of which common and/or specific mechanisms endorse carcinogenesis.
Here, we devised and deployed an in vivo CRISPR/Cas9-screening methodology, which allowed us to identify bone-fide cancer drivers in the long-tail of breast cancer genes. Our screen identified several tumor suppressor genes with the top hits converging on epigenetic regulation and mammary epithelial differentiation. Individually, epigenetic regulators are not mutated frequently, but as a group, they are among the most frequently mutated targets in cancer (47–51), indicating that a ‘dysregulated epigenome’ can accelerate tumor development. In particular, we identified several components and auxiliary factors of the COMPASS-like histone methyltransferase complex as potent tumor suppressors and showed that Kdm6a might function in a haploinsufficient manner. Our results show that loss of those EpiDriver accelerates tumor initiation and that the transcriptional profiles of EpiDriver knock-out tumors closely cluster together. However, the results do not rule out the possibility that the individual genes also have distinct functions, perhaps depending on cellular or microenvironmental context. It is noteworthy, however, that, loss of each of the EpiDrivers analyzed triggers a similar alveogenesis program associated with casein expression. This indicates that their loss, at least in part, reflects involvement in shared biological processes that are distinct from, for example, p53 tumor suppressor loss. Importantly, up to 39% of breast cancer patients harbor mutations in the COMPASS-like pathway, highlighting the importance of elucidating the mechanisms by which COMPASS inactivation contributes to breast cancer. In human tumors, EpiDriver genes are deleted or harbor nonsense or missense mutations. Most of the missense mutations are variants with uncertain significance and while many are predicted to be deleterious (Supplementary Table S4), their exact function and effect on cancer etiology remains to be determined. Further studies will also be needed to elucidate potential private functions of these tumor suppressors alone or in combination with a sensitizing oncogene such as Pik3caH1047R.
Components of the COMPASS-like complex were recently implicated as tumor suppressors in leukemia (52), medulloblastoma (53), pancreatic (23) and non-small-cell lung cancer (54) and their loss was associated with substantial enhancer reprogramming and aberrant transcription. We were surprised to find that EpiDriver inactivation did not substantially affect histology or transcriptional profiles of breast tumors. However, it did significantly accelerate tumor initiation, which was coupled with rapid acquisition of phenotypic plasticity. Plasticity plays a central role in development and during tissue regeneration and wound healing (29,55,56). More recently, phenotypic plasticity has also been recognized as a driving forces behind tumor initiation and progression (57–59). For example, elegant lineage-tracing and single cell-profiling experiments have shown that oncogenic signaling can reactivate multipotency within the two epithelial lineages of the mammary gland (38,39,57). Cells that acquire plasticity are thought to gain stem cell features through a process of dedifferentiation (56,60). However, in the system studied here, we did not observe acquisition of fetal mammary stem cell-like transcriptomes as observed in basal-like tumors studies (29,57). Rather, we observed an aberrant differentiation program associated with alveologenesis induced upon PI3K activation and exacerbated by EpiDriver loss. This was most noteworthy in basal cells, which are known to be functionally plastic (61–63). A similar aberrant alveolar differentiation program was recently described in breast cancer models driven by luminal loss of BRCA1 and p53 (27), and upon luminal overexpression of ELF5 and PyMT (35,36). Importantly, we show that overexpression of ELF5 in a Pik3caH1047-mutant background accelerates mammary tumorigenesis. While this indicates that alveogenesis is sufficient to increase tumorigenesis, it still remains to be determined whether alveogenesis in the context of EpiDriver mutations is required for the observed accelerated tumor phenotype.
Together, our data indicate that there are different avenues towards transformation and that the innate but poised program coordinating the proliferative burst during gestation and onset of lactation can be highjacked for rapid expansion at the onset of oncogenic transformation – a phenomenon we term “alveogenic mimicry”. This phenomenon is exacerbated by loss of epigenetic control governed by the COMPASS-like and associated BAP1/ASXL1/2 complexes, and happens not only in the luminal cells, but – given the right combinations of mutations – also in the basal cells. It will be interesting to assess whether other cancers also coerce inherent regenerative or tissue remodeling processes during early transformation.
Another interesting aspect of our study is the potential cell of origin underlying different subtypes of breast cancer. Gene expression studies indicated that mature luminal cells give rise to luminal A/B and HER2 subtypes, while luminal progenitors transform to the basal-like cancers and basal cells give rise to the claudin-low subtype (64–66). Mouse lineage-tracing studies have supported these observations and have shown that certain mutations in specific lineages can indeed give rise to mouse mammary tumors with features similar to different human breast cancer subtypes (38,39,67). Our data now show that, given the right combination of oncogene and cooperating epigenetic alteration, basal cells can also be the cell of origin of luminal tumors. Interestingly, cross-species comparison indicated that Pik3ca-/EpiDriver-mutant mouse tumors share several dysregulated pathways with human luminal B tumors. This supports the idea that the ultimate epigenomic, transcriptomic, and histopathologic characteristics of a tumor depend on the target cell for the initial mutation, the type of mutations, and the collaborating alterations. Clearly, loss of epigenetic regulation needs to be considered as a significant contributor to the loss of lineage integrity that underlie tumor heterogeneity.
Methods
Animals
Animal husbandry, ethical handling of mice and all animal work were carried out according to guidelines approved by Canadian Council on Animal Care and under protocols approved by the Centre for Phenogenomics Animal Care Committee (18–0272H). All mice used in experiments were female. The animals used in this study were R26-LSL-Pik3caH1047R/+ mice (11) [Gt(ROSA)26Sortm1(Pik3ca*H1047R)Egan in a clean FVBN background kindly provided by Egan S, SickKids], R26-LSL-Cas9-GFP [Gt(ROSA)26Sortm1(CAG-xstpx-cas9,-EGFP)Fezh/J #026175 in C57/Bl6 background from Jackson laboratories], LSL-TdTomato [B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, #007908 from Jackson laboratories], Asxl2fl/fl [C57BL/6N-Asxl2tm1c(EUCOMM)Hmgu/Tcp generated by The Canadian Mouse Respiratory] and Kdm6afl/fl [Kdm6atm1.1Kaig] mice kindly provided by Jacob Hanna. Rbfl/fl; Trp53fl/fl; LSL-Cas9-EGFP mice were generated by crossing B6.129;Rb1tm1Brn [#026563 from Jackson laboratories], Trp53tm1Brn [#008462 from Jackson laboratories], and Gt(ROSA)26Sortm1(CAG-xstpx-cas9,-EGFP)Fezh/J mice. CRISPR screens and experiments in the Pik3caH1047R/+; Cas9 cohort were performed in a F1 FVBN/C57Bl6 background. Experiments with Kdm6afl/fl and Asxl2fl/fl were conducted by crossing each strain to LSL-Cas9-EGFP mice resulting in Kdm6afl/fl; LSL-Cas9-EGFP and Asxl2fl/fl; LSL-Cas9-EGFP in a C57Bl6 background. Kdm6afl/fl and Asxl2fl/fl were also crossed to R26-LSL-Pik3caH1047R mice to obtain Kdm6afl/fl; R26-LSL-Pik3caH1047R and Asxl2fl/fl; R26-LSL-Pik3caH1047R mice which were in a mixed FVBN;C57Bl6 background. These mice were then crossed to produce Asxl2fl/fl; R26-LSL-Pik3caH1047R/+; LSL-Cas9-EGFP and Kdm6afl/fl; R26-LSL-Pik3caH1047R/+; LSL-Cas9-EGFP mice, which were of mixed FVBN;C57Bl6 background. NSG mice used for xenograft experiments were NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson laboratories #005557). Genotyping was performed by PCR using genomic DNA prepared from mouse ear punches. For tumor experiments, mice were palpated for tumors weekly by experimenters blinded to experimental group. When total tumor mass per animal exceeded 1000mm3, mice were monitored bi-weekly and scored in accordance to SOP “#AH009 Cancer Endpoints and Tumour Burden Scoring Guidelines”.
Lentiviral constructs and library construction
sgRNAs targeting breast cancer long tail genes were obtained from Hart et al. (68) (4 sgRNAs/gene) and non-targeting sgRNAs were obtained from Sanjana et al. (69), ordered as a pooled oligo chip (CustomArray Inc., USA) and cloned into pLKO sgRNA-Cre plasmid (9) using BsmBI restriction sites. We excluded frequent and known breast cancer tumor suppressor genes such as TP53 or CDH1 from the breast long tail genes library. The non-targeting sgRNAs were designed not to target the mouse genome and served as a negative control. Individual sgRNAs used in this study as well as TIDE primers for evaluating cutting efficiency are listed in Supplemental Table S6. pLKO-mRFP and pLKO-GFP were kindly provided by Elaine Fuchs (RRID:Addgene_26001 and RRID:Addgene_25999). pLEX-306-iCre was cloned from pLEX-306 (RRID:Addgene_41391) by substituting the Puromycin resistance cassette with Cre. ORFs for Ruby fluorescent protein or mouse Elf5 were inserted between the gateway sites. pLKO-mRFP-P2A-Cre was recently described (9) and used for lentiviral injections in Pik3caH1047R;Kdm6afl/fl and Asxl2fl/fl mice.
Virus production and transduction
Large-scale production and concentration of lentivirus were performed as previously described (70–74). Briefly, 293T cells (Invitrogen R700–07, RRID:CVCL_6911) were seeded on a poly-L-lysine coated 15 cm plates and transfected using PEI (polyethyleneimine) method in a non-serum media with lentiviral construct of interest along with lentiviral packaging plasmids psPAX2 (RRID:Addgene_12260) and pPMD2.G (RRID:Addgene_12259). 8 hours post-transfection media was added to the plates supplemented with 10% Fetal bovine serum and 1% Pencillin-Streptomycin antibiotic solution (w/v). 48 hours later, the viral supernatant was collected and filtered through a Stericup-HV PVDF 0.45-μm filter, and then concentrated ~2,000-fold by ultracentrifugation in a MLS-50 rotor (Beckman Coulter). Viral titers were determined by infecting R26-LSL-tdTomato MEFs and FACS based quantification. In vivo viral transduction efficiency was determined by injecting decreasing amounts of a single viral aliquot of known titer, diluted to a constant volume of 8 μl per mammary gland and analyzed by FACS 7 days post infection. Ad5-K5-Cre (VVC-U of Iowa-1174) or Ad5-K8-Cre (VVC-Li-535), or Ad-Cre (VVC-U of Iowa-5) were purchased from the Vector Core at the University of Iowa.
Intraductal injection and viral transduction
Intraductal lentiviral injection has been described. Briefly, to deliver the lentiviral sgRNA library or single sgRNAs targeting gene of interest, a non-invasive injection method was employed which selectively transduces mammary epithelium of female mice. Female mice were injected at >8 and <20 weeks of age, with age at injection matched between groups in all experiments. 8 ul of virus diluted in PBS and visualized with Fast-Green dye was injected into the 3rd and/or 4th mammary glands using pulled glass micropipettes. As previously described (70,72,74), we calculated coverage based on the following parameters: mammary epithelium consist of ~3.5×105 cells; transduction of ~15% results in a minimal double infection rate (~1/10 infected cells); at 15% infectivity every gland has 50,000 infected cells, resulting in 200,000 cells in four glands of a single mouse. To ensure that at least 4000 individual cells were transduced with a given sgRNA, a pool of 860 sgRNAs requires 3.5×106 cells or ~17 animals. To verify the sgRNA abundance and representation in the control and breast long-tail genes libraries, MEFs were transduced with library virus and collected 48h post transfection. For single sgRNA or ORF injection, lentivirus was injected at 1×107 pfu/ml. Ad5-K5-Cre virus was injected at 8×108 pfu/ml and Ad-K8-Cre virus was injected at 3.5×1010 pfu/ml, which infected ~2–20% of basal or luminal cells.
Deep Sequencing: sample preparation, pre-amplification and sequence processing
Genomic DNA from epithelial and tumor cells were isolated with the DNeasy Blood & Tissue Kit (Qiagen). 5μg genomic DNA of each tumor was used as template in a pre-amplification reaction using unique barcoded primer combination for each tumor with 20 cycles and Q5 High-Fidelity DNA Polymerase (NEB). The following primers were used:
FW:5’AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGCTCTTCCGATCTtgtggaaaggacgaaaCACCG-3’
RV:5’CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3’
The underlined bases indicate the Illumina (D501–510 and D701–712) barcode location that were used for multiplexing. PCR products were run on a 2% agarose gel, and a clean ~200bp band was isolated using Zymo Gel DNA Recovery Kit as per manufacturer instructions (Zymoresearch Inc.). Final samples were quantitated then sent for Illumina Next-seq sequencing (1 million reads per tumor) to the sequencing facility at Lunenfeld-Tanenbaum Research Institute (LTRI). Sequenced reads were aligned to sgRNA library using Bowtie version 1.2.2 with options –v 2 and –m 1. sgRNA counts were obtained using MAGeCK count command (75).
Analysis of genome editing efficiency
Tumor cells were live sorted for GFP expression and genomic DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen). For cultured cells, genomic DNA extraction was performed on cells harvested during routine passaging. PCR was performed flanking the regions of sgRNA on genomic DNA from both WT cells and putative knockout cells and was sent for Sanger sequencing. Sequencing files along with chromatograms were uploaded to https://www.deskgen.com/landing/tide.html (76) and genome editing efficiency was estimated. TIDE primers are listed in Supplementary Table S6.
Antibodies
The following primary antibodies were used in this study: rabbit anti-APC (1:200, Santa Cruz sc-896, RRID:AB_2057493), rabbit anti-Kdm6a (1:1000, CST D3Q1I, RRID:AB_2721244), rabbit anti-Asxl2 (1:500, EMD Millipore, ABE1320, RRID:AB_2923141), mouse anti-TP53 (1:1000, CST 1C12, RRID:AB_331743), mouse anti-Pten (1:1000 CST 26H9, RRID:AB_331153), goat anti-Setd2 (1:500 Millipore-Sigma SAB2501940), rabbit anti-Mll3 (1:500 CST D1S1V, RRID:AB_2799442), mouse anti-GAPDH (1:2500 Santa Cruz sc-32233, RRID:AB_627679), rabbit anti-histone H3 (1:1000 CST 4499, RRID:AB_10544537), rabbit anti-Keratin14 (PRB-155P, 1:200 for whole mount, 1:700 for sections, RRID:AB_292096), rat anti-Keratin8 (1:50, TROMA-1, RRID:AB_2891089), mouse anti-ERalpha (R&D Systems, RRID:AB_10890942), APC conjugated anti-CD45, (1:500 rat monoclonal Clone 30 F11, RRID:AB_10376146), APC conjugated anti-CD31 (1:250 rat monoclonal Clone MEC133, Biolegend, RRID:AB_312917), APC conjugated anti-Ter119 (1:250 Biolegend, RRID:AB_313712), PECy7 anti human/mouse CD49f (1:50 clone GoH3, Biolegend, RRID:AB_2561705), APCVio770 mouse anti-CD326 EpCAM (1:50 Miltenyi, RRID:AB_2657525). For casein staining of mouse tissue: HRP conjugated anti-β-casein (1:20 sc-166530HRP H-4). For staining of human tissues: Casein (polyclonal NBP2–55090, Novusbio, 1:5000 dilution, Opal 520, RRID:AB_2923142) and pan-cytokeratin (AE1AE3, Agilent DAKO, Opal 620, RRID:AB_2132885). For IMC: Pr14-conjugated anti-Keratin8–18 (Clone C51, CST-4546BF, RRID:AB_2134843), Nd144-conjugated anti-Keratin5 (Abcam ab214586, RRID:AB_869890), Eu151-conjugated anti-casein (Novus Biologicals NBP2–55090, RRID:AB_2923142). For ChIP-seq: anti-H3K27ac (Active Motif #39133, RRID:AB_2561016), anti-H3K4me1 (EpiCypher #13–0040, RRID:AB_2923143) and anti-H3K27me3 (Millipore #07–449, RRID:AB_310624).
Mammary gland isolation and flow cytometry for lineage tracing and mammosphere assay
Mice were injected with the indicated virus in the #3 or 4 mammary glands with no greater than 2 replicates of a single condition per mouse. Individual mammary glands were harvested digested according to Stemcell Technologies gentle collagenase/hyaluronidase protocol. Briefly glands we digested overnight shaking at 37oC in 250 ul Gentle Collagenase (Stemcell Technologies #07919) in 2.25 ml of complete Basal Epicult media formulated according to manufacture instructions (Epicult Basal Medium Stemcell Technologies #05610, 10% Proliferation Supplement, 5% FBS, 1% Penicillin-Streptomycin, 10 ng/ml EGF, 10 ng/ml bFGF, 0.0004% heparin). Glands were then treated with ammonium chloride and triturated for 2 minutes in pre-warmed trypsin followed by dispase. Cells were stained with CD45, CD31, Ter119, CD49f and EPCAM for luminal and basal cell identification.
Cell culture
Primary mouse tumor cells isolated directly from tumors, which were minced and treated with collagenase for 45 minutes and trypsin for 10 minutes. Single cell suspensions from tumors were sorted to isolate GFP+ cells using fluorescence activated cell sorting (FACS) and were then plated. Primary mouse tumor cells were cultured in DMEM/F12 (1:1) supplemented with MEGS supplement, FBS and Pen-Strep. MCF10A-PIK3CAH1047R cells were purchased from Horizon (Cat# HD 101–011, RRID:CVCL_LD55, acquired in May of 2018) and were cultured as previously described (77) in DMEM/F12 + 5% horse serum, 1% pencillin streptomycin, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 10μg/ml insulin. For sgRNA transfection, cells were cultured in monolayer for growth and transfected with lentiviral CRISPR/Cas9 construct containing puro resistance and sgRNA targeting genes of interest. Cells were tested for cutting efficiency post selection with TIDE analysis and by western blot. All cells were negative for mycoplasma via monthly PCR testing. All cell culture experiments were conducted less than 25 passages after either derivation from tumors (for primary mammary tumor cells) or thaw of the original vial (for MCF10A-PIK3CAH1047R cells). Cell line authentication was not performed after receiving MCF10A-PIK3CAH1047R cells.
Xenograft assay
MCF10A-PIK3CAH1047R cells were infected with the lentiviruses carrying Cas9 and the indicated sgRNAs as well as a puro selection marker. After puro selection and TIDE to determine the more effective guide, cells were used for sphere formation assay or xenograft. For xenograft, 500 000 cells were resuspended in 50 ul PBS, mixed 1:1 with chilled Corning Matrigel (Fisher Scientific, Cat#CB-40234) and injected into each #4 fat pad of NSG mice. Mice were monitored for tumor formation by mammary gland palpation for 6 months. Each fat pad was counted individually.
Sphere formation
For sphere experiments, MCF10A cells were plated on growth-factor-reduced Matrigel (Corning, Fisher Scientific, Cat#CB-40230C) as described previously (77) and imaged by bright field after 10 days of sphere growth. Primary mammospheres were isolated from mouse mammary glands and were plated on Corning® Costar® Ultra-Low Attachment 24-Well Plates (CLS3473–24EA) in serum-free Epicult Basal sphere media (Epicult Basal Medium Stemcell Technologies #05610, 10% Proliferation Supplement, 1% Penicillin-Streptomycin, 10 ng/ml EGF, 10 ng/ml bFGF, 0.0004% heparin, + 2% W21 growth supplement). Mammospheres were counted and imaged 10 days after plating.
Immunofluorescence
Cryosections were fixed with 4% paraformaldehyde for 10 minutes. Following fixation, slides were rinsed 3 times with PBS for 5 minutes. Samples were blocked at room temperature with blocking serum (recipe: 1% BSA, 1% gelatin, 0.25% goat serum 0.25% donkey serum, 0.3% Triton-X 100 in PBS) for 1 hour. For paraffin sections, samples were embedded in paraffin, sectioned, rehydrated and antigen retrieval was performed with Sodium Citrate buffer. Samples were incubated with primary antibody diluted in blocking serum overnight at 4°C followed by 3 washes for 5 minutes in PBS. Secondary antibody was diluted in blocking serum with DAPI and incubated for 1 hour at room temperature in the dark. Following incubation, samples were washed 3 times for 5 minutes in PBS. Coverslips were added on slides using MOWIOL/DABCO based mounting medium and imaged under microscope next day. For quantification, laser power and gain for each channel and antibody combination were set using secondary only control and confirmation with primary positive control and applied to all images.
Casein Staining of breast cancer specimens, tissue imaging and analysis:
Formalin-fixed Paraffin-embedded (FFPE) TMA slides were dried at 60°C for 4 hours. After drying, the slides were placed on the BOND RXm Research Stainer (Leica Biosystems) and deparaffinized with BOND Dewax solution (AR9222, Lecia Biosystems). The multispectral immunofluorescent (mIF) staining process involved serial repetitions of the following for each biomarker: epitope retrieval/stripping with ER1 (citrate buffer pH 6, AR996, Leica Biosystems) or ER2 (Tris-EDTA buffer pH9, AR9640, Leica Biosystems), blocking buffer (AKOYA Biosciences), primary antibody, Opal Polymer HRP secondary antibody (AKOYA Biosciences), Opal Fluorophore (AKOYA Biosciences). All AKOYA reagents used for mIF staining come as a kit (NEL821001KT). Spectral DAPI (AKOYA Biosciences) was applied once slides were removed from the BOND. They were cover slipped using an aqueous method and Diamond antifade mounting medium (Invitrogen ThermoFisher). The duplex mIF panel consisted of the following antibodies: Casein (polyclonal NBP2–55090, Novusbio, 1:5000 dilution, Opal 520) and pan-cytokeratin (AE1AE3, Agilent DAKO, Opal 620).
Slides were imaged on the Vectra® Polaris Automated Quantitative Pathology Imaging System (AKOYA Biosciences). Further analysis of the slides was performed using inForm® Software v2.4.11 (AKOYA Biosciences). Whole TMA spectral unmixing was achieved using the synthetic spectral library supplied within inForm. The operator then created a batch TMA map, which encircles each TMA core as its own individual region of interest (ROI). Next a unique algorithm was created using a machine learning technique, in which the operator selects positive and negative cell examples for each marker. These algorithms were then batch applied across the entire TMA. The operator then conducted a visual review of the phenotyping across all cores to ensure accuracy. Finally, the individual files resulting from batch analysis were consolidated in RStudio using phenoptr reports to determine the percent total Casein per TMA core and this information was aligned with known clinical data.
Mammary gland whole mount
Mammary gland whole mounts were prepared as previously described for visualization of endogenous proteins and fluorescent labelling (78). Briefly, 2 mm3 pieces of mammary gland were fixed for 45 minutes in 4% pfa, followed by a 30-minute wash in WB buffer, 2 hrs in WB1 and an overnight incubation in anti-Keratin8 and anti-Keratin14 antibodies diluted in WB2 buffer. The following day, the pieces underwent 3 × 1hr washes in WB2 buffer prior to overnight incubation in secondary antibody (at 1:200 dilution) with DAPI added at 4°C. Finally, pieces were washed 3 times for 1 hour each and then cleared using FUnGI solution for 2+ hours at room temperature until glands appeared sufficiently cleared, and then were mounted and imaged using confocal microscopy.
RNA-seq and GSEA analyses
Tumors were minced and treated with collagenase for 45 minutes and trypsin for 15min. Single cell suspensions from tumors were sorted to isolate GFP+ cells using fluorescence activated cell sorting (FACS). RNA was extracted from FACS-isolated cells using Quick-RNA Plus Mini Kit (Zymoresearch Inc., #R1057) as per the manufacturer’s instructions. RNA quality was assessed using an Agilent 2100 Bioanalyzer, with all samples passing the quality threshold of RNA integrity number (RIN) score of >7.5. The library was prepared using an Illumina TrueSeq mRNA sample preparation kit at the LTRI sequencing Facility, and complementary DNA was sequenced on an Illumina Nextseq platform. For in vivo mouse tumor samples, sequencing reads were aligned to mouse genome (mm10) using Hisat2 version 2.1.0. For cultured cells, human and mouse RNA-seq datasets were aligned using STAR v2.5.1b (79) to hg38 + GENCODE v27 and mm10 + GENCODE vM4, respectively. Counts were obtained using featureCounts (Subread package version 2.0.0) ) with the settings -s2 and -t gene (80). Differential expression was performed using DESeq2 (81) release 3.8. Gene set enrichment analysis was performed using GSEA version 4.0; utilizing genesets obtained from MSigDB (82). GSEA lists were weighted by −log(p)*sign(FC) for mouse tumors, mouse cells and MCF10A-PIK3CAH1047R cells. For integration with human and existing mouse tumor models, clustering was conducted after normalization and filtering for only intrinsic genes as described previously (83,84). Metascape analysis was performed using default settings (85). g:Profiler (86) was run using the following parameters: version e104_eg51_p15_3922dba; ordered: true; sources: GO:MF, KEGG, REAC, HPA, HP; with all other parameters at default settings. Gene sets are available in Supplementary Table 7.
ChIP-seq sample preparation and sequencing
For ChIP-seq, two biological replicates (separately cultured cell populations) of wild type and Kdm6a-mutant mouse mammary tumor cells, and separate clones of wild type and KDM6A-mutant MCF10A-HR cells were crosslinked with 1% formaldehyde in Solution A (50 mM Hepes–KOH, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) for 10 min at room temperature. Fixation was stopped by addition of glycine at a final concentration of 125 mM. Fixed cells were washed with PBS and lysed using low SDS Chromatin EasyShear Kit (Diagenode #C01020013) following the manufacturer’s instructions. Briefly, cells were resuspended in Lysis Buffer iL1b, incubated for 20 min at 4°C on a rotator, and pelleted by centrifugation at 500 g for 5 min at 4°C. Cells were resuspended in Lysis Buffer iL2 and incubated for 10 min at 4°C while rotating. After centrifugation of 5 min at 500 × g at 4°C, cell pellets were resuspended in iS1b Shearing Buffer (Diagenode #C01020013) supplemented with Protease Inhibitor Cocktail (Roche). Chromatin was shared into 200–500bp fragments with 8 cycles of 30 s sonication and 30 s of pause at 4°C using the Bioruptor Pico sonicator (Diagenode). Chromatin was clarified by centrifugation at 21,000 × g at 4°C for 10 min. An aliquot of 50 ul of shared chromatin from each sample was removed for input DNA extraction. For each ChIP, chromatin lysates from ~6 million cells were combined with 10 ug of anti-H3K27ac (Active Motif #39133), anti-H3K4me1 (EpiCypher #13–0040) or anti-H3K27me3 (Millipore #07–449) antibodies and incubated overnight rotating at 4°C. Chromatin-antibody lysates were then incubated for 4 h with 100 ul of Dynabeads protein G beads (ThermoFisher #10004D) pre-blocked with 0.5 mg/ml BSA while rotating at 4°C. Beads were collected with a magnetic separator (Invitrogen DynaMag-2), washed six times with RIPA buffer (50 mM Hepes–KOH, pH 7.5; 500 Mm LiCl; 1 mM EDTA; 1% NP-40 or Igepal CA-630; 0.7% Na– Deoxycholate) and once with TBS (20 mM Tris–HCl, pH 7.6; 150 mM NaCl), and resuspended in ChIP Elution buffer (50 mM Tris–HCl, pH 8; 10 mM EDTA; 1% SDS). Crosslinking was reversed by incubating the beads at 65°C for 16 h. Cellular proteins and RNA were digested with Proteinase K (Invitrogen #25530049) and RNaseA (Ambion #2271). ChIP and input DNA were purified with phenol:chloroform:isoamyl alcohol (25:24:1) extraction and ethanol precipitation, and used for ChIP-seq library preparation with NEBNext® Ultra™ II DNA Library Prep Kit (NEB #E7645S). In brief, ChIP and input DNA samples were blunt-end repaired and ligated to Illumina sequencing adaptors containing uracil hairpin loop structure and 3′ T overhangs (NEB, #E7337A). Looped adapter sequences were opened by removal of uracil from hairpin structures by adding 3 units of USER enzyme (Uracil-Specific Excision Reagent) (NEB, M5505S) and incubation at 37°C for 15 min. This made DNA accessible for PCR amplification with barcoded primers for Illumina sequencing (NEB #E7335 and #E7500). Agencourt AMPure XP beads (Beckman Coulter) were used to cleanup adaptor-ligated DNA without size selection. PCR amplification was carried out at 98°C for 30 s followed by 9 cycles of 10 s at 98°C and 75 s at 65°C, and a final 5 min extension at 72°C. PCR reactions were cleaned and size selected (200–500bp) with Agencourt AMPure XP beads (Beckman Coulter). Library concentration and size distribution was assessed by Bioanalyzer High Sensitivity DNA chip (Agilent) followed by sequencing on the Illumina NovaSeq 6000 (150 bp paired end reads).
ChIP-seq alignment and peak calling
Human and mouse in fastq format were aligned to their respective genomes (hg38 and mm10) using BWA mem v0.7.8 (87) with default settings and filtered to retain properly paired and uniquely mapping reads with the following command: Samtools view -Shb -q 5 -f 0×2 -F 0×100 -F 0×800. Resultant bam files were processed with picard MarkDuplicates v2.5.0 to remove PCR and optical duplicates. Peak calling was performed with merged replicates and paired input files using MACS v2.1.2 (88) with a q-value cutoff < 0.005 and a fold-enrichment cutoff > 4 for punctate histone modifications (H3K27ac and H3K4me1). A fold-enrichment cutoff=2 and --broad was used for H3K27me3 datasets. A consensus peak set was generated per histone modification by merging peaks sets from WT and KO conditions. Normalized signal tracks (bedgraph/bigwig) were generated during peak calling using the flags --B --SPMR. Fold-change over input tracks were generated using the macs2 bdgcmp utility.
Differential analysis of ChIP-seq regions
Peak level read counts were obtained using bedtools multiBamCov v2.29.2. Differential ChIP enrichment was assessed using DESeq2 v1.34.0 (81). DE peaks were designated as regions passing an FDR-adjusted p-value cutoff of <0.05 (Wald test).
Designation and clustering of promoter-proximal and distal ChIP peaks:
To properly align and cluster ChIP-peaks, we overlapped all peaks with previously published accessible chromatin regions in matched human and mouse cell types (ATAC-seq and snATAC-seq; GSE89013 and (29); respectively). Accessible regions were designated as distal or proximal based on a threshold of <= 2.5kb from the nearest annotated TSS (GENCODE v27 for human, GENCODE vM4 for mouse). Accessible regions overlapping >1 differential ChIP peak were then clustered based on differential ChIP-signal using deepTools v3.6.7 as follows: Differential ChIP-signal was calculated genome-wide using the fold-change over input tracks (described above) with the bigwigCompare utility with pseudocount values of 0.1, 0.01, and 0.05 for H3K27Ac, H3K27me3, and H3K4me1 datasets, respectively. Differential signal was extracted at peak regions using computeMatrix reference-point with the settings -b 6000 -a 6000 -bs 30 –missingDataAsZero –referencePoint center. Clustering was performed using the plotHeatmap utility with –kmeans 2 (number of clusters selected by visual inspection for k=2–4).
Single-cell processing and library preparation
R26-LSL-Pik3caH1047R/+; LSL-Cas9-EGFP (Pik3caHR) and Kdm6afl/fl; R26-LSL-Pik3caH1047R/+; LSL-Cas9-EGFP (Pik3caHR Kdm6aKO) were cohoused for at least 14 days prior to injection to synchronize estrus cycles, with control LSL-GFP-Cas9 mice housed separately due to limitations in mouse numbers per cage. Each mouse was injected with 5×108 pfu/ml Ad-Cre or 8×108 pfu/ml Ad5-Cre in the left and right #4 mammary glands. Two mice per group were harvested except for the Pik3caHRKdm6aKO sample in the K5-Cre experiment, which was preformed on one mouse. Mammary gland digestion was carried out as described in the “Mammary gland isolation” section except two glands were pooled per mouse, and glands were digested in 2x gentle collagenase/hyaluronidase for 2 hours with trituration by P1000 pipette half-way through digestion instead of overnight. Cells were then sorted for GFP+ infected cells and immediately processed for snATACseq or scRNAseq according to 10x Genomics protocol (scRNAseq 3’ kit v.3.1 and snATACseq kit v1.1). Approximately 5000 cells per sample were sequenced with targeted 50 000 reads/cell.
10X single cell RNA-seq data processing
The raw sequencing data from each channel was first aligned in Cell Ranger 4.0.0 using a customized reference based on refdata-gex-mm10–2020-A-R26 to allow quantification of EGFP expression. The EGFP reporter transgene was added to the refdata-gex-mm10–2020-A-R26 reference and rebuilt by running cellranger mkref with default parameters (10x Genomics). To minimize the batch effects from sequencing depth variation, we further used cellranger aggr function to match the depth of mapped reads. The filtered gene-by-cell count matrices from 10x cellranger aggr were further QCed and analyzed in R package Seurat (v3.2.3) (89). Merged library was first processed in Seurat with NormalizeData(normalization.method = “LogNormalize”) function. The normalized data were further linear transformed by ScaleData() function prior to dimension reduction. Principal components analysis (PCA) was performed on the scaled data by only using the most variable 2000 genes (identified using the default “vst” method). Cells were examined in each sample across all clusters to determine the low-quality cell QC threshold that accommodates the variation between cell types. Low-quality cells were removed with the same filtering parameters on the merged object (percent.mt <=10 & nCount_RNA >= 2500 & nCount_RNA <50000 & nFeature_RNA>=1000). Stromal cell contamination from FACS-sorting and doublet clusters were removed to keep only mammary epithelium cells. QCed merged dataset was further integrated using the RunHarmony()function in SeuratWrappers R package to minimize the batch effect between the Ad-Cre batch and K5-Cre batch. Top 30 harmony-PCs were used for subsequent UMAP embedding and neighborhood graph construction of the integrated dataset. To investigate Ad-Cre and K5-Cre separately, the QCed dataset was split into Ad-Cre and K5-Cre subsets and then reprocessed as described above and clusters were labeled with cell types based on marker gene expression and sample/library identity. First 30 PCs in K5-Cre subset and first 40 PCs in Ad-Cre subset were selected as significant PCs for downstream UMAP embedding and neighborhood graph construction in Seurat. Pseudotime analysis was performed using Monocle3 on K5-Cre basal cells. A central point within the WT Control cluster was set as the root node and pseutotime was calculated with automatic partitioning. The ML-HS cluster was portioned separately from the remaining cells and was excluded from visualization. Diffusion mapping was performed on epithelial cells excluding the ML-HS cluster using the destiny package (v3.4.0). The first 3 eigenvectors were used for visualization using the plot3d package.
Cerebro shinyapp of single cell RNA-seq data
Final processed Seurat objects from the harmony integrated dataset, Ad-Cre subset, and K5-Cre subset were further processed using the cerebroApp functions in the cerebroApp R package (v1.3.0) (90). Cerebro processed data was hosted on shinyapps.io server and it is accessible though this link: https://wahl-lab-salk.shinyapps.io/Kdm6aKO_scRNAseq/.
10X single nucleus ATAC-seq data processing.
The raw sequencing data from each mouse was first processed separately in 10x cellranger atac 1.2.0 pipeline using refdata-cellranger-atac-mm10–1.2.0 reference. To minimize the batch effects from sequencing depth variation, we further used the aggr function in cellranger-atac pipeline to match the depth of mapped reads across samples. The post-normalization fragments output from the 10x cellranger-atac aggr pipeline was imported into ArchR (91) and further QCed and analyzed. Arrow files were created with the initial filtering: minTSS=4 and minFrags=1000. Each library was inspected separately to determine the QC filtering thresholds. All samples were further QC filtered with TSSEnrichment > 6 and log10(nFrags) >= 3.4 with the exception of the WT sample, which used a higher threshold log10(nFrags) >= 3.55). The merged samples were first embedded in UMAP by first running latent semantic indexing with 1 iteration with the interativeLSI function. Clusters identify were inferred based on the gene score of marker genes. Clusters of doublets, are marked by shared marker gene expression from two different lineages and higher number of reads per cell on average as previously described (29). Clusters of stromal cell contamination and doublets were removed from subsequent analysis based on marge gene expression and average read-depth distribution as previously described (29). The cleaned mammary epithelial cell dataset was re-processed through a 1-iteration interativeLSI with default parameters. All top 20PCs were used to embed cells in two dimensional UMAP. Clusters were called by using the addClusters(method=‘Seurat’,resolution=1.1,dimsToUse=1:20) function and subsequently labelled with cell types using gene scores of marker genes and sample identity. Pseudo-bulk profile with replicates was generated and reproducible peaks were identified by calling peaks specifically in each clusters or cell types across replicates using the macs2 method. Differentially accessible peaks were identified using the getMarkerFeatures and getMarkers (cutOff = “FDR <= 0.1 & Log2FC >= 1”). TF motif activity was inferred by using the chromVAR TF enrichment deviation z-scores in ArchR (30,91). Heatmaps were generated using the ComplexHeatmap R package using scaled and centered values across cell type groups (92).
TCGA and METABRIC data
Clinical and pathological data, somatic genetic mutations and genomic copy numbers were obtained from the cBioPortal (93). Gene expression (RNA-seq fragments per kilobase of transcript per million mapped reads (FPKM) upper quartile normalized (UQ)) data were obtained from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov). In survival analyses, EpiDriver mutations were defined as somatic gene mutations and/or homozygous genomic deletions of ASXL2, BAP1, KMT2C, KMT2D, KDM6A, and/or SETD2. The TCGA breast cancers were previously scored for PI3K/AKT/mTOR signaling using a transcription-based CMAP signature, in which high values were associated with poor outcome (94). The measures of phospho-Ser473 AKT were downloaded from The Cancer Proteome Atlas (TCPA) (45) and corresponded to level 4 normalized values from assays using reverse-phase protein arrays. The high/low threshold (value = 0) of CMAP and pAKT were confirmed by examining the value distributions in all primary tumors. The Kaplan-Meier curve and log-rank test analyses were performed in R software using the survival and survminer packages. The signature expression scores were derived from the combined expression analysis of the corresponding gene constituents using the single-sample gene set expression analysis (ssGSEA) algorithm (95), calculated with the Gene Set Variation Analysis (GSVA) application (96). Pregnancy, lactation, involution, and alveogenesis gene signatures corresponded to GO Biological Processes terms and to gene sets defined in the study of mouse mammary development (36,97,98). The genes in signature can be found in Supplementary Table 7, MSigDB and in the corresponding referenced papers (36,97,98).
Transcriptomic analyses of ductal carcinoma vs. invasive breast cancer
Gene expression data from 57 DCIS and 313 IDC were obtained and processed as previously described (44). For each gene, standardized gene expression values were calculated by subtracting the mean (across all samples) from the sample’s gene expression value, then dividing by the standard variation. Signature Z-scores were calculated as the mean of standardized gene expression for all genes included in the signature and present in the dataset. The genes in each signature can be found in Supplementary Table 7 or can be found by name on MSigDB.
IMC Staining
Immunofluorescence was used to validate antibodies and metal conjugations were carried out using Maxpar Conjugation Kits (Fluidigm). FFPE slides were baked for 1hr at 60°C, deparaffinized using xylene washes and rehydrated in an ethanol gradient (100%, 95%, 80% and 75%). Heat-induced antigen retrieval was performed using antigen retrieval buffer (Tris-EDTA pH 9.2) at 95°C for 30 minutes. Slides were blocked at room temperature for 1 hr using blocking solution (3%BSA, 5% horse serum, 0.1%Tween in TBS) followed by overnight incubation at 4°C with a panel of metal conjugated antibodies. The following day, slides were washed using TBS and DNA staining was performed using iridium in TBS for 5 minutes at room temperature. Slides were washed three times in TBS and dipped in milliQ before being air dried. Hyperion Imaging System (Fluidigm) was calibrated using a tuning slide and IMC images were acquired at 1um resolution at 200Hz.
IMC Data Analysis Pipeline
Data were preprocessed, segmented, and analyzed using an in-house integrated flexible data analysis pipeline ImcPQ available at https://github.com/JacksonGroupLTRI/ImcPQ. The analysis pipeline is implemented in Python.
Briefly, data were converted to TIFF format and segmented into single cells using the pipeline to classify pixels based on a combination of antibody stains to identify membranes/cytoplasm and nuclei. The stacks were then segmented into single-cell object masks. Single cells were clustered into cell categories based on pre-specified markers and cell phenotypes.
IMC raw data were converted to TIFF format without normalization. ImcPQ pipeline was used for segmentation and to process images to single cell data. Then, based on membranes/cytoplasm and nuclei markers the analysis stacks were generated. First, image layers, or channels, are split into nuclear or cytoplasm/membrane channels and added together to sum all markers that represent nuclei or cytoplasm/membrane. Then Mesmer model (99) were used for segmentation as deep learning method. The resulting single cell mask was used to quantify the expression of each marker of interest and spatial features of each cell. Single-cell marker expressions are summarized by mean pixel values for each channel. The single cell data were normalized and scaled per marker channel. Then data were censored at the 99th percentile to remove outliers.
Clusters of interest Krt5+, Krt8–18+ and double positive population were gated based on the phenotypes. For list of markers used in clustering see. For quantification, the normalized density of marker in gated cell populations is reported.
Statistics and reproducibility
All quantitative data are expressed as the mean ± SE. Significance of the difference between groups was calculated by two-tailed Student’s t-test (with Welch’s correction when variances were significantly different), Wilcoxon Rank-Sum test (when data was not normally distributed) or Log-rank test for survival data using Prism 7 (GraphPad software) unless otherwise specified in figure caption. Where adjustment is indicated and the method is not otherwise specified, p value was adjusted using Bonferroni correction.
Data Availability:
All RNA-seq, scRNAseq, snATACseq and ChIPseq data are available at NCBI Gene Expression Omnibus GEO accession GSE178424. Cerebro processed data is hosted on shinyapps.io server and it is accessible though this link: https://wahl-lab-salk.shinyapps.io/Kdm6aKO_scRNAseq/.
Supplementary Material
Statement of significance.
Infrequently mutated genes comprise most of the mutational burden in breast tumors but are poorly understood. In-vivo CRISPR screening identified functional tumor suppressors that converged on epigenetic regulation. Loss of epigenetic regulators accelerated tumorigenesis and revealed lineage infidelity and aberrant expression of alveogenesis genes as potential early events in tumorigenesis.
Acknowledgements:
We thank all members of our laboratories for helpful comments, with additional thanks to K. Schleicher, and G. Mbamalu for their insight and assistance. We thank H. Melo and D. Durocher for assistance with visualization of g:Profiler data. We also thank The Centre for Phenogenomics, Network Biology Collaborative Centre and Flow Cytometry facility at LTRI as well as the Flow Cytometry Facility at the University of Toronto. Funding: This work was supported by a Career Catalyst Research Grant to D.S. from Susan G. Komen foundation (CCR16377321), a Terry Fox Research Institute Program Projects Grant to J. W. and D.S et al. (TFRI Project #1107) and by Nicol Family Foundation. E.L is a recipient of the Ontario Graduate Scholarship and the Frank Fletcher Memorial Fund, K.N.A. is a recipient of the MBD fellowship and supported by SHSF donation (Mr. Ah Shai), S.K.L. is a Canadian Cancer Society Fellowship recipient (BC-F-16#31919), and L.U.R. is a recipient of the Next Generation of Scientist Scholarship from Cancer Research Society (PIN25558). GMW and ZM are supported by a Cancer Center Core Grant (5 P30CA014195), NIH/National Cancer Institute (R35 CA197687), and the Breast Cancer Research Foundation (BCRF). S.E.E is supported by CIHR. R.P. and M.A.P. was supported by grants from the Carlos III Institute of Health (PI18/01029 and PI21/01306; co-funded by European Regional Development Fund (ERDF), a way to build Europe), Generalitat de Catalunya (SGR 2017–449), and the CERCA Program to IDIBELL.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Mateo J, Steuten L, Aftimos P, Andre F, Davies M, Garralda E, et al. Delivering precision oncology to patients with cancer. Nat Med 2022;28(4):658–65 doi 10.1038/s41591-022-01717-2. [DOI] [PubMed] [Google Scholar]
- 2.Garraway LA, Lander ES. Lessons from the cancer genome. Cell 2013;153(1):17–37 doi 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science 2013;339(6127):1546–58 doi 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017;171(5):1029–41 e21 doi 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castro-Giner F, Ratcliffe P, Tomlinson I. The mini-driver model of polygenic cancer evolution. Nat Rev Cancer 2015;15(11):680–5 doi 10.1038/nrc3999. [DOI] [PubMed] [Google Scholar]
- 6.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A 2010;107(43):18545–50 doi 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar S, Warrell J, Li S, McGillivray PD, Meyerson W, Salichos L, et al. Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell 2020;180(5):915–27 e16 doi 10.1016/j.cell.2020.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173(2):321–37 e10 doi 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loganathan SK, Schleicher K, Malik A, Quevedo R, Langille E, Teng K, et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020;367(6483):1264–9 doi 10.1126/science.aax0902. [DOI] [PubMed] [Google Scholar]
- 10.Ablain J, Durand EM, Yang S, Zhou Y, Zon LI. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell 2015;32(6):756–64 doi 10.1016/j.devcel.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015;163(2):506–19 doi 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams JR, Xu K, Liu JC, Agamez NM, Loch AJ, Wong RG, et al. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Research 2011;71(7):2706–17 doi 10.1158/0008-5472.CAN-10-0738. [DOI] [PubMed] [Google Scholar]
- 13.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479 doi 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505(7484):495–501 doi 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016;352(6290):aad9780 doi 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- 16.Steffen PA, Ringrose L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat Rev Mol Cell Biol 2014;15(5):340–56 doi 10.1038/nrm3789. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med 2018;24(6):758–69 doi 10.1038/s41591-018-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campagne A, Lee MK, Zielinski D, Michaud A, Le Corre S, Dingli F, et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat Commun 2019;10(1):348 doi 10.1038/s41467-018-08255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 2011;21(8):1273–83 doi 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang C, Zhu B. Roles of H3K36-specific histone methyltransferases in transcription: antagonizing silencing and safeguarding transcription fidelity. Biophys Rep 2018;4(4):170–7 doi 10.1007/s41048-018-0063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer DS, Koren S, Leroy C, Brinkhaus H, Muller U, Klebba I, et al. Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R. Oncogenesis 2013;2:e74 doi 10.1038/oncsis.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gazova I, Lengeling A, Summers KM. Lysine demethylases KDM6A and UTY: The X and Y of histone demethylation. Mol Genet Metab 2019;127(1):31–44 doi 10.1016/j.ymgme.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, Tzatsos A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018;33(3):512–26 e8 doi 10.1016/j.ccell.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran TH, Utama FE, Lin J, Yang N, Sjolund AB, Ryder A, et al. Prolactin inhibits BCL6 expression in breast cancer through a Stat5a-dependent mechanism. Cancer Res 2010;70(4):1711–21 doi 10.1158/0008-5472.CAN-09-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Logarajah S, Hunter P, Kraman M, Steele D, Lakhani S, Bobrow L, et al. BCL-6 is expressed in breast cancer and prevents mammary epithelial differentiation. Oncogene 2003;22(36):5572–8 doi 10.1038/sj.onc.1206689. [DOI] [PubMed] [Google Scholar]
- 26.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003;17(10):1253–70 doi 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bach K, Pensa S, Zarocsinceva M, Kania K, Stockis J, Pinaud S, et al. Time-resolved single-cell analysis of Brca1 associated mammary tumourigenesis reveals aberrant differentiation of luminal progenitors. Nat Commun 2021;12(1):1502 doi 10.1038/s41467-021-21783-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pervolarakis N, Nguyen QH, Williams J, Gong Y, Gutierrez G, Sun P, et al. Integrated Single-Cell Transcriptomics and Chromatin Accessibility Analysis Reveals Regulators of Mammary Epithelial Cell Identity. Cell Rep 2020;33(3):108273 doi 10.1016/j.celrep.2020.108273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung CY, Ma Z, Dravis C, Preissl S, Poirion O, Luna G, et al. Single-Cell Chromatin Analysis of Mammary Gland Development Reveals Cell-State Transcriptional Regulators and Lineage Relationships. Cell Rep 2019;29(2):495–510 e6 doi 10.1016/j.celrep.2019.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 2017;14(10):975–8 doi 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oakes SR, Naylor MJ, Asselin-Labat ML, Blazek KD, Gardiner-Garden M, Hilton HN, et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev 2008;22(5):581–6 doi 22/5/581 [pii] 10.1101/gad.1614608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farnie G, Clarke RB. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev 2007;3(2):169–75 doi 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 33.Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 2008;3(4):429–41 doi S1934–5909(08)00399–8 [pii] 10.1016/j.stem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Gu B, Watanabe K, Sun P, Fallahi M, Dai X. Chromatin effector Pygo2 mediates Wnt-notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell Stem Cell 2013;13(1):48–61 doi 10.1016/j.stem.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallego-Ortega D, Ledger A, Roden DL, Law AM, Magenau A, Kikhtyak Z, et al. ELF5 Drives Lung Metastasis in Luminal Breast Cancer through Recruitment of Gr1+ CD11b+ Myeloid-Derived Suppressor Cells. PLoS Biol 2015;13(12):e1002330 doi 10.1371/journal.pbio.1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valdes-Mora F, Salomon R, Gloss BS, Law AMK, Venhuizen J, Castillo L, et al. Single-cell transcriptomics reveals involution mimicry during the specification of the basal breast cancer subtype. Cell Rep 2021;35(2):108945 doi 10.1016/j.celrep.2021.108945. [DOI] [PubMed] [Google Scholar]
- 37.Tao L, van Bragt MP, Laudadio E, Li Z. Lineage tracing of mammary epithelial cells using cell-type-specific cre-expressing adenoviruses. Stem Cell Reports 2014;2(6):770–9 doi 10.1016/j.stemcr.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, Giraddi RR, et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015;525(7567):119–23 doi 10.1038/nature14665. [DOI] [PubMed] [Google Scholar]
- 39.Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015;525(7567):114–8 doi 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- 40.Chang Y, Zuka M, Perez-Pinera P, Astudillo A, Mortimer J, Berenson JR, et al. Secretion of pleiotrophin stimulates breast cancer progression through remodeling of the tumor microenvironment. Proc Natl Acad Sci U S A 2007;104(26):10888–93 doi 10.1073/pnas.0704366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrechek ER, Mori S, Rempel RE, Chang JT, Nevins JR. Patterns of cell signaling pathway activation that characterize mammary development. Development 2008;135(14):2403–13 doi 10.1242/dev.019018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gustin JP, Karakas B, Weiss MB, Abukhdeir AM, Lauring J, Garay JP, et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A 2009;106(8):2835–40 doi 10.1073/pnas.0813351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croessmann S, Wong HY, Zabransky DJ, Chu D, Rosen DM, Cidado J, et al. PIK3CA mutations and TP53 alterations cooperate to increase cancerous phenotypes and tumor heterogeneity. Breast Cancer Res Treat 2017;162(3):451–64 doi 10.1007/s10549-017-4147-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergholtz H, Lien TG, Swanson DM, Frigessi A, Oslo Breast Cancer Research C, Daidone MG, et al. Contrasting DCIS and invasive breast cancer by subtype suggests basal-like DCIS as distinct lesions. NPJ Breast Cancer 2020;6:26 doi 10.1038/s41523-020-0167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, et al. TCPA: a resource for cancer functional proteomics data. Nat Methods 2013;10(11):1046–7 doi 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res 2010;12(3):R40 doi 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valencia AM, Kadoch C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol 2019;21(2):152–61 doi 10.1038/s41556-018-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev 2015;29(3):238–49 doi 10.1101/gad.255182.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 2013;13(7):497–510 doi 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet 2013;14(11):765–80 doi 10.1038/nrg3554. [DOI] [PubMed] [Google Scholar]
- 51.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 2013;45(10):1134–40 doi 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014;25(5):652–65 doi 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol Cell 2018;70(5):825–41 e6 doi 10.1016/j.molcel.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 2020;37(4):599–617 e7 doi 10.1016/j.ccell.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giraddi RR, Chung CY, Heinz RE, Balcioglu O, Novotny M, Trejo CL, et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep 2018;24(6):1653–66 e7 doi 10.1016/j.celrep.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019;24(1):65–78 doi 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dravis C, Chung CY, Lytle NK, Herrera-Valdez J, Luna G, Trejo CL, et al. Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell 2018;34(3):466–82 e6 doi 10.1016/j.ccell.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020;38(2):212–28 e13 doi 10.1016/j.ccell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020;38(2):229–46 e13 doi 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carvalho J Cell Reversal From a Differentiated to a Stem-Like State at Cancer Initiation. Front Oncol 2020;10:541 doi 10.3389/fonc.2020.00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature 2006;439(7079):993–7 doi nature04496 [pii] 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 62.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature 2006;439(7072):84–8 doi 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 63.Centonze A, Lin S, Tika E, Sifrim A, Fioramonti M, Malfait M, et al. Heterotypic cell-cell communication regulates glandular stem cell multipotency. Nature 2020;584(7822):608–13 doi 10.1038/s41586-020-2632-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tharmapalan P, Mahendralingam M, Berman HK, Khokha R. Mammary stem cells and progenitors: targeting the roots of breast cancer for prevention. EMBO J 2019;38(14):e100852 doi 10.15252/embj.2018100852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009;15(8):907–13 doi 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 66.Saeki K, Chang G, Kanaya N, Wu X, Wang J, Bernal L, et al. Mammary cell gene expression atlas links epithelial cell remodeling events to breast carcinogenesis. Commun Biol 2021;4(1):660 doi 10.1038/s42003-021-02201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010;7(3):403–17 doi 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 68.Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015;163(6):1515–26 doi 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 69.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11(8):783–4 doi 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beronja S, Livshits G, Williams S, Fuchs E. Rapid functional dissection of genetic networks via tissue-specific transduction and RNAi in mouse embryos. Nat Med 2010;16(7):821–7 doi nm.2167 [pii] 10.1038/nm.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Endo M, Zoltick PW, Peranteau WH, Radu A, Muvarak N, Ito M, et al. Efficient in vivo targeting of epidermal stem cells by early gestational intraamniotic injection of lentiviral vector driven by the keratin 5 promoter. Mol Ther 2008;16(1):131–7 doi 10.1038/sj.mt.6300332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beronja S, Fuchs E. RNAi-mediated gene function analysis in skin. Methods in molecular biology 2013;961:351–61 doi 10.1007/978-1-62703-227-8_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beronja S, Janki P, Heller E, Lien WH, Keyes BE, Oshimori N, et al. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 2013;501(7466):185–90 doi 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, et al. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science 2014;343(6168):309–13 doi 10.1126/science.1248627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15(12):554 doi 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 2014;42(22):e168 doi 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003;30(3):256–68 doi 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 78.Rios AC, Capaldo BD, Vaillant F, Pal B, van Ineveld R, Dawson CA, et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019;35(4):618–32 e6 doi 10.1016/j.ccell.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 79.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–30 doi 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 81.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu JC, Voisin V, Wang S, Wang DY, Jones RA, Datti A, et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol Med 2014;6(12):1542–60 doi 10.15252/emmm.201404402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J Clin Invest 2016;126(10):3739–57 doi 10.1172/JCI81568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10(1):1523 doi 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019;47(W1):W191–W8 doi 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell 2019;177(7):1888–902 e21 doi 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hillje R, Pelicci PG, Luzi L. Cerebro: interactive visualization of scRNA-seq data. Bioinformatics 2020;36(7):2311–3 doi 10.1093/bioinformatics/btz877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 2021;53(3):403–11 doi 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32(18):2847–9 doi 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 93.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401–4 doi 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH, et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017;31(6):820–32 e3 doi 10.1016/j.ccell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108–12 doi 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lemay DG, Lynn DJ, Martin WF, Neville MC, Casey TM, Rincon G, et al. The bovine lactation genome: insights into the evolution of mammalian milk. Genome Biol 2009;10(4):R43 doi 10.1186/gb-2009-10-4-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lemay DG, Neville MC, Rudolph MC, Pollard KS, German JB. Gene regulatory networks in lactation: identification of global principles using bioinformatics. BMC Syst Biol 2007;1:56 doi 10.1186/1752-0509-1-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Greenwald NF, Miller G, Moen E, Kong A, Kagel A, Dougherty T, et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat Biotechnol 2022;40(4):555–65 doi 10.1038/s41587-021-01094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq, scRNAseq, snATACseq and ChIPseq data are available at NCBI Gene Expression Omnibus GEO accession GSE178424. Cerebro processed data is hosted on shinyapps.io server and it is accessible though this link: https://wahl-lab-salk.shinyapps.io/Kdm6aKO_scRNAseq/.