

Abstract
Aging is a major risk factor for a number of chronic diseases, including neurodegenerative and cerebrovascular disorders. Aging processes have therefore been discussed as potential targets for the development of novel and broadly effective preventatives or therapeutics for age-related diseases, including those affecting the brain. Mechanisms thought to contribute to aging have been summarized under the term the “hallmarks of aging” and include a loss of proteostasis, mitochondrial dysfunction, altered nutrient sensing, telomere attrition, genomic instability, cellular senescence, stem cell exhaustion, epigenetic alterations and altered intercellular communication. We here examine key claims about the “hallmarks of aging”. Our analysis reveals important weaknesses that preclude strong and definitive conclusions concerning a possible role of these processes in shaping organismal aging rate. Significant ambiguity arises from the overreliance on lifespan as a proxy marker for aging, the use of models with unclear relevance for organismal aging, and the use of study designs that do not allow to properly estimate intervention effects on aging rate. We also discuss future research directions that should be taken to clarify if and to what extent putative aging regulators do in fact interact with aging. These include multidimensional analytical frameworks as well as designs that facilitate the proper assessment of intervention effects on aging rate.
Subject terms: Physiology, Diseases
Introduction
Life expectancy increased from ~50 years in the early 1900s to over 80 years at present [1]. Factors contributing to this development may include advances in medical care as well as the creation of cleaner, safer, and healthier environments for people to live in [1]. Although this represents a great achievement for human societies, the growth in both the size and the proportion of the elderly population also comes with critical challenges. Advanced age is the main risk factor for many common diseases, such as cancers, cardiovascular disorders, and neurodegeneration [1]. Age-related neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and others [2–4], severely compromise the quality of life of affected individuals. Moreover, current demographic developments have substantial socioeconomic implications for health and care systems [5, 6]. Available treatments are symptomatic, despite intensive efforts to develop disease-modifying therapies for these devastating conditions [5, 6].
Among known risk factors for neurodegenerative diseases, the aging process itself has the highest impact [7]. For instance, it has been estimated that the risk for developing AD doubles every 5 years over the age of 65; the risk of death due to AD increases 700-fold between the ages 55 and 85 [8, 9]. Hence, strong mechanistic links between brain aging and neurodegenerative disease have been considered [10] and treatments with putative anti-aging drugs (e.g., rapamycin) have been proposed for clinical trials targeting AD [9]. Thus, studying aging and understanding how exactly aging increases the risk to develop neurodegenerative diseases can provide important clues to inform new strategies for early detection, prevention, and treatment.
The critical outstanding question is: Can aging processes be slowed down? Evidence in nature suggests a positive answer to this fundamental question. For instance, similar pathobiological changes associated with aging develop over very different time scales in different mammalian species [11]. While it may take 70 years for a senile cataract to develop in a human, similar age-related changes develop in horses within 20 years, in dogs within 10 years, and in mice in even only 2 years. Analogous considerations also apply to many other age-related alterations (hair greying, muscle loss, etc.). Although the biology underlying these differences in aging rate are not well understood, these examples demonstrate that similar aging phenomena in comparable tissues can develop over very different absolute time scales. Therefore, there seems to be some plasticity that could be harnessed, in theory, for slowing down the aging process.
Much of what is currently thought to be known about the biological underpinnings of the aging process has been presented in concepts like the “hallmarks of aging” [12–14] which summarize processes claimed to be involved in driving organismal aging phenomena. Here, we carefully examine the evidence presented in favor of such links between these processes and aging. As we will explain in detail below, we identify limitations that are often grounded in the choice of models and/or the way aging is measured. We conclude by outlining experimental designs that are suited to overcome these current limitations and that can be used to address if and to what extent putative aging regulators are in fact involved in regulating organismal aging rate.
The “hallmarks of aging”
The aging field has grown significantly during the 2000s, leading to the testing of many previous hypotheses as well as the emergence of new ideas in the field [15]. In 2013, López-Otín and colleagues published a paper in which they proposed nine hallmarks as the main causes of aging: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [13]. This paper soon became a major reference for researchers in the aging field and beyond, accumulating over 1000 citations per year in recent years [15]. The “hallmarks of aging” also inspired many scientists from other fields, including the field of neurodegeneration, to shape their findings in the form of these nine hallmarks [7, 16]. In fact, researchers from different fields ranging from evolutionary biology [17, 18] to biomedical research [1, 7, 19–23] take these nine hallmarks as a base for their research and tend to think of their findings as being relevant to aging if they can identify compatibility with any of these nine hallmarks. While this clearly shows the importance of aging research for an understanding of different biological and medical issues, a key question here is to what extent these nine hallmarks of aging represent the “causes of aging”. In other words, on which foundational evidence and assumptions have the “hallmarks of aging” been built? To address this question, we have performed a systematic analysis of the papers that were used as supporting evidence for the involvement of each of these hallmarks in the aging process and identified important limitations which need to be discussed and acted upon.
Lifespan —a valid proxy for aging?
Lifespan is often used as a proxy marker for aging. That is, interventions (e.g., genetic manipulations, pharmacological treatments, or other environmental interventions, such as manipulation of dietary factors) found to extend lifespan in model organisms (such as in mice, flies, or worms) are concluded to slow aging because they extend lifespan.
The problem with this assertion is that natural lifespan is often restricted by specific sets of aging-associated pathologies, not by some sort of generalized physiological decline. As a consequence, lifespan-extending interventions are likely to exert their effects on lifespan by targeting whatever pathology is life-limiting in the context of natural aging in that species (hereafter termed lethal age-sensitive phenotypes; lethal ASPs; these might be lethal in isolation or become lethal via combinatorial effects). In species in which lifespan is limited by a very narrow set of pathologies (e.g., specific cancers), treatment-induced longevity effects indicate that this intervention has an effect on the small set of lethal ASPs in this species (Fig. 1A). It is important to note, however, that the observation of such treatment effects has no implications concerning any of the potentially much larger subset of age-dependent changes that do not limit lifespan per se (non-lethal ASPs; such as hair greying, skin aging, sarcopenia, osteoporosis, cognitive decline, etc.; see Fig. 1A).
Fig. 1. Lifespan is not a sufficient readout of aging.
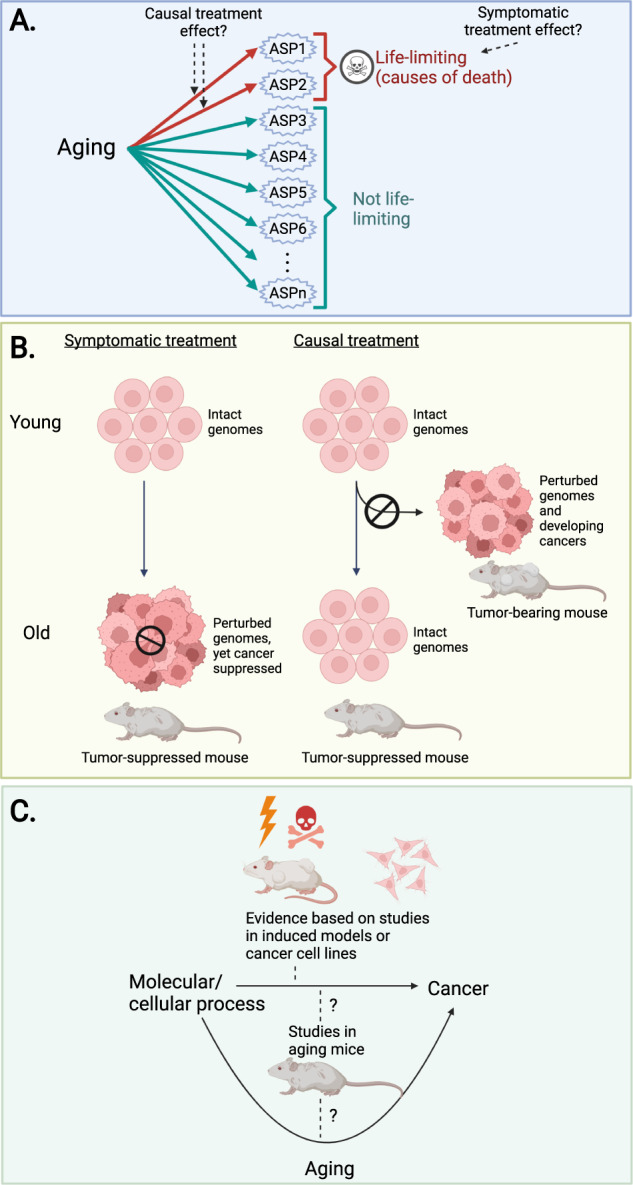
A Only a subset of aging-associated changes (shown here as a collection of age-sensitive phenotypes; ASPs) is life-limiting in the context of natural aging. ASPs could exert their effects on lifespan either individually or via combinatorial effects. Experimental lifespan extension implies that the given intervention interacts with the life-limiting subset of ASPs, but does not speak to the intervention’s ability to influence any of the ASPs that are not life-limiting. In addition, treatments that extend lifespan could be targeting the cause of reduced lifespan in a symptomatic fashion (e.g., inhibition of cancer growth using a cytostatic drug), rather than tackling its underlying causes (e.g., inhibition of mutagenesis to prevent cancer formation in the first place). For further discussion, see the main text. B Symptomatic and causal treatments can lead to the same outcome, but through different mechanisms. The differences between symptomatic and causal treatment are shown here using the age-related pathology “cancer” as an example. Under symptomatic treatment, tumor growth is blocked by non-specifically inhibiting cell proliferation via a cytostatic drug. Importantly, however, the age-related accumulation of genome damage (that underlies cancer predisposition in old age in our example) remains unaffected by this type of approach. Causal treatment prevents the aging-associated accumulation of genome damage, thereby inhibiting cancer by targeting the biology underlying the age-related increase in cancer predisposition. C Treatment-induced anti-cancer effects in aged mice could, in principle, be explained by either anti-aging effects (treatment targets aging-associated changes that predispose for cancer formation in old age, such as genomic instability), by direct anti-cancer effects (i.e., age-independent inhibitory effects on neoplastic diseases, such as a general inhibition of cell division) or by a combination of these two effects. However, direct anti-cancer effects are sufficient to explain any observed anti-cancer effects if a treatment exerts such effects in experimental contexts that do not involve aging, such as experiments performed in young mice (e.g., with chemically induced tumors) or in cell culture models.
Also, note that treatment-induced longevity effects do not necessarily imply that treatment targets the processes causally underlying the aging-associated development of lethal ASPs. While this is one possibility, pro-longevity effects could also be caused by symptomatic treatment effects on lethal ASPs. For instance, a cytostatic drug may extend lifespan by (symptomatically) inhibiting lethal neoplastic disorders; it would do so by generally inhibiting cell proliferation [24] and not by influencing the mechanisms favoring the development of lethal neoplastic disorders in old age (e.g., genomic instability and mutation accumulation in the context of aging) [25–27].
The “hallmarks of aging” paper [13] makes extensive use of data derived from lifespan studies to support claims about roles in the general biology of aging. To make this point clear, we identified all the references cited within [13] that studied genetic variants, dietary factors or pharmacological treatments and showed lifespan extension in the context of natural aging in any one of the different model organisms used (Supplementary Table 1 and Supplementary Table 2). The results are discussed below separately for each species, M. musculus, D. melanogaster, and C. elegans.
Across a range of mouse strains, cancers have been shown to account for ~70 – 90% of natural age-related deaths [28–32]. Hence, given that cancer is the main known life-limited pathology in mice, any pro-longevity intervention in this species is likely exerting its effects via inhibition of carcinogenesis. Again, to examine this point in greater detail, we extracted all genetic, dietary and pharmacological pro-longevity interventions that were cited in [13] and analyzed whether or not each of these interventions has been previously demonstrated to have anti-cancer effects. Consistent with the statements above, these analyses revealed links to cancer inhibition in >80% of these interventions (Table 1) [32–79], indicating that anti-cancer effects could in fact largely explain the lifespan extension induced by these pro-longevity interventions (i.e., there is no need to assume that general aging-associated physiological decline was slowed to explain the pro-longevity effects). Cancer-inhibitory effects were due to a range of mechanisms, including the suppression of de novo cancer formation and the inhibitions of established tumors by reducing cancer growth, promoting apoptosis, and/or inhibiting angiogenesis [32–79].
Table 1.
Established anti-cancer roles of the “hallmarks of aging”.
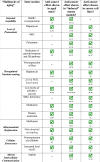 |
The table shows interventions (experimental conditions; examined in mice in the context of natural aging) presented in the “hallmarks of aging” paper [13] as evidence to support a role of each of the given “hallmarks” in aging. For each intervention, the table indicates whether the intervention is associated with known anti-cancer effects or not (in either aged mice and/or cell lines/“induced” mouse models). Note that cancer-inhibitory effects have been established in most cases, indicating that pro-longevity effects of these interventions can be explained by suppressing lethal neoplastic disease in the context of aging.
As discussed above, pro-longevity interventions in mice could, in principle, extend lifespan by targeting the root causes of aging-associated cancers (e.g., by promoting genomic stability / inhibiting mutagenic events). Alternatively, they could inhibit cancers in symptomatic ways, i.e., by tapping into mechanisms unrelated to the ones that mechanistically link aging and cancer formation (this would be the case, for instance, in the scenario where a cytostatic drug inhibits general cell proliferation but leaves genomic instability/mutation accumulation unaffected, or even enhances it due to mutagenic properties that can be associated with this class of drugs [80]). Note that both of these scenarios could cause aged animals to have a lower cancer burden (Fig. 1B). However, it would not be meaningful to consider the animals treated with the symptomatic treatment (the cytostatic drug) to benefit from “slowed aging”. After all, they are as cancer-prone as untreated age-matched controls (or even more cancer-prone considering mutagenic properties of cytostatic drugs) (Fig. 1B) and this would become evident as soon as the symptomatic treatment is stopped. Animals treated with a causal treatment, in contrast, would show a causally reduced cancer risk (due to preserved genomic integrity) (Fig. 1B) that persists even if treatment is terminated.
If a treatment’s anti-cancer effects have been established in the context of naturally aging mice, it is not possible to distinguish between these two scenarios; anti-cancer effects could be due to either symptomatic or causal intervention effects. A distinction between these scenarios is, however, possible in cases in which experimental designs allow the dissociation of direct anti-cancer effects from anti-cancer effects that could be indirectly caused by anti-aging effects. This is possible whenever experiments are carried out in “non-aging” contexts, such as in chemically-induced cancer models (established in young mice) or in cell culture models of cancer (Fig. 1C).
Again, when the manipulation of putative aging regulators (Table 1) suppresses cancers in induced mouse cancer models (in which cancers are induced either chemically, genetically, by radiation, or via xenograft models [81–83]), this strongly implies a direct and symptomatic effect (Fig. 1C). In these mouse models, cancers are generated in young mice and therefore no role of anti-aging effects can be attributed to the anti-cancer effects in these contexts. Similarly, anti-cancer effects in cancer cell lines are also an indication for symptomatic effect (Fig. 1C). Table 1 summarizes the different types of anti-cancer evidence for each of the putative aging regulators considered here. Interestingly, for 100% of these interventions, an anti-cancer role has been shown in “induced” mouse cancer models. For 93% of these interventions, an anti-cancer effect has also been found in cancer cell lines. These observations strongly support the notion of direct and aging-independent anti-cancer effects. As a consequence, given that aging-independent anti-cancer effects are documented for most of the putative aging regulators (Table 1), a straightforward, yet underappreciated mechanistic explanation for much of the pro-longevity effects afforded by these interventions in mice is that they are not induced by “slowing aging” but rather by the direct inhibition of lethal neoplastic disease via aging-independent mechanisms. These considerations point to serious flaws in the sole use of longevity as a proxy marker for aging studies in mice.
While major causes of age-related death in mice have been identified (see above), processes limiting lifespan in Drosophila melanogaster are not that well understood [84, 85]. In flies, the intestinal epithelium constitutes an important barrier against microorganisms and environmental toxins [84]. The structure and function of the intestinal epithelium significantly decline in aging flies, to a point that is thought to become life-limiting [84, 86–88]. One of the well-described pathologies in the aged fly intestine is epithelial dysplasia, driven by intestinal stem cell (ISC) over-proliferation which leads to an increase in intestinal progenitor cells and aberrant differentiation [89–91]. Interestingly, several well-known pro-longevity interventions in flies, such as the mTOR inhibitor rapamycin, caloric restriction, and genetic loss-of-function mutations targeting insulin/insulin-like growth factor signaling (IIS), have been shown to slow down the proliferation rate of ISCs, to delay intestinal dysplasia and to extend lifespan [86, 91–94]. More importantly, several studies have shown that genetic manipulations specifically targeted to ISCs are sufficient to extend lifespan in flies [86, 95–100] (Table 2), consistent with the notion that intestinal dysplasia is a life-limiting pathology in this species. These observations also demonstrate, for flies, that lifespan extension can be induced by eliminating or reducing only one specific life-limiting ASP (without necessitating broader effects on aging). Intriguingly, Drosophila females demonstrate a greater level of intestinal dysplasia compared to males [91] and in line with this, female flies usually also show a greater longevity response to these interventions [101–106].
Table 2.
Examples of ISCs-specific interventions that extend lifespan in Drosophila.
| “Hallmarks of aging” | Intervention | Reference |
|---|---|---|
| Loss of Proteostasis | PERK downregulation | [95] |
| Hsp68 overexpression | [86] | |
| Deregulated Nutrient-Sensing | Inhibition of JNK signaling | [86] |
| Overexpression of Jafrac1 | [86] | |
| Downregulation of lnR | [86] | |
| Downregulation of Dp110 | [86] | |
| Downregulation of Akt | [86] | |
| Downregulation of dMfn | [96] | |
| Activation of Drp1 | [96] | |
| Downregulation of Bsk and BskD | [86] | |
| Mitochondrial Dysfunction | Overexpression of dPGC-1 | [97] |
| Overexpression of Ndi1 | [98] | |
| Altered Intercellular Communication | Activation of Kif1a | [98] |
| PGRP-SC2 overexpression | [99] | |
| Overexpression of Ssk | [100] | |
| Stem Cell Exhaustion | Overexpression of dPGC-1 | [97] |
The table summarizes genetic interventions that have been shown to be sufficient to extend lifespan in D. melanogaster when targeted to intestinal stem cells (ISCs).
It has been shown that specific genetic perturbations in the muscle [107, 108], fat body [101, 109], brain [101, 110] and neurosecretory cells [102, 111, 112] also are sufficient to extend life span in Drosophila melanogaster, however, the underlying mechanism is poorly understood. Note that ISC proliferation is also regulated via extrinsic cues derived from distant tissues, such as muscle, brain, trachea, and fat body [113–116]. In line with this, it has been shown that inhibition of neuronal Hh results in an increase in the number of intestinal progenitor cells as well as defective differentiation, whereas activation of neuronal Hh signaling reduces intestinal progenitor cells, significantly improves intestinal homeostasis [115], and leads to lifespan extension in Drosophila melanogaster [110]. More research is needed to investigate the existence of other possible life-limiting pathologies in other tissues which could be the cause of age-related death in flies.
The roundworm Caenorhabditis elegans is another important model organism for aging research and a large number of genetic and environmental factors have been identified to extend its lifespan. However, what specific life-limiting pathologies naturally play a role in C. elegans remains to be better understood. A longitudinal analysis in C. elegans showed that pharyngeal pumping span (the length of the time interval the pharynx is active) is positively correlated with adult lifespan [117]. Recently, another study revealed that pharyngeal infections and deterioration are among the main life-limiting pathologies in worms [118]. In this study [118], two types of deaths have been reported in adult C. elegans: an early death with a swollen, infected pharynx and a later death with pharyngeal atrophy [118]. Interestingly, additional work analyzing some of the well-known long-lived C. elegans mutants, such as glp-1, eat-2, ced-1, and daf-2 mutant lines, revealed that these interventions change the frequency and/or timing of either form of death, thereby leading to an increase in lifespan [118–120]. Again, these observations in C. elegans are also in line with the notion that the rescue of one specific life-limiting pathology may be sufficient to explain lifespan extension. However, more work is needed to further define life-limiting changes in C. elegans and to explore how those are affected in longevity mutants.
Together, the data and considerations discussed thus far indicate that isolated pro-longevity effects of an intervention, without any further studies, are insufficient to support strong conclusions about any possible broader anti-aging effects this intervention may have.
Choice of models
Shortened lifespan
Conclusions about the biology of aging have also been drawn on the basis of lifespan studies identifying factors that shorten lifespan. As shown in Supplementary Table 1, work focused on shortened lifespan is used to back up claims about aging in [13].
The assertion that lifespan shorting effects can be used to inform the biology of aging is problematic because lifespan can be shortened in many ways entirely unrelated to factors that naturally limit lifespan during aging (lethal ASPs) or are otherwise associated with aging-related phenomena (non-lethal ASPs). There are numerous examples of animal models that live shorter for reasons completely unrelated to aging. For instance, mutations in the TSC1 or TSC2 genes cause tuberous sclerosis complex (TSC) in humans, a neurogenetic condition associated with autism spectrum disorder, epilepsy, and intellectual disability [121, 122]. Neuron-specific conditional Tsc1 and Tsc2 mouse mutants exhibited early premature death due to severe brain pathology [123, 124]. Treatment with an mTORC1 inhibitor rescued brain pathology in the mutant mice and resulted in an increase of lifespan [123]. This is a clear example where a mutation shortens lifespan, yet the cause of death is in no way related to aging and also the mTORC1 inhibitor rescue effects are not related to any aspect of the aging process.
Models that aim to phenocopy aging
Inferences about aging have been also drawn from studies analyzing models that are claimed to phenocopy manifestations of aging but in which the link to aging is either weak or unclear. This includes, for instance, mitochondrial DNA (mtDNA) mutator mice, irradiated mice, mice featuring diet-induced obesity, telomere-dysfunctional mice, mouse models with persistent expression of Wnt1, mouse models with conditional deletion of Tsc1 or mouse models of rare genetic syndromes termed progerias (Werner syndrome (WS), Hutchinson-Gilford syndrome (HGPS)). The latter feature a premature manifestation of some phenotypic changes reminiscent of those observed during normal aging. It remains unclear, however, how these conditions relate to aging. While WS patients, for instance, show a premature manifestation of some phenotypic changes seen in elderly people, such as greying and loss of hair and a development of cataracts, they do not develop prematurely many other consequences of aging, such as cognitive decline, immune dysfunction or cardiovascular disease [125, 126]. In addition, the epithelial and hematopoietic tumors which are very commonly seen during normal aging in humans, are not more common in WS patients [125, 126]. Instead, they show mesenchymal tumors which are rarely seen in normal elderly people [126]. Hence, the relevance of these genetic conditions as models for aging is unclear and it is debated whether they can provide valuable clues about processes involved in aging [126].
As shown in Supplementary Table 1, work focused on models that aim to phenocopy manifestations of aging is used very frequently in [13] to support claims about aging, in particular with regards to some specific “hallmarks”: Approximately 67% of studies cited to support a role for genomic instability in aging were based on such models; also, 63% cited for cellular senescence and 62% cited for stem cell exhaustion refer to studies that were actually not focused on normal aging but instead on models with unclear relevance to aging, such as the ones outlined above.
Cell culture models/Cell-centric view on aging
Organismal aging cannot be simply mimicked in a cell culture dish because it is an emergent property of intact organisms. Hence, examining molecular and cellular hypotheses about aging on the cellular level (cultured cells) does not necessarily yield insights that are relevant to the aging process on the organismal level. If an intervention improves or accelerates cellular function in cultured cells, this observation has, in isolation and without further organismal-level validation in an aging context, no implications regarding organismal aging (Fig. 2A).
Fig. 2. Organismal aging needs to be studied in organismal models.
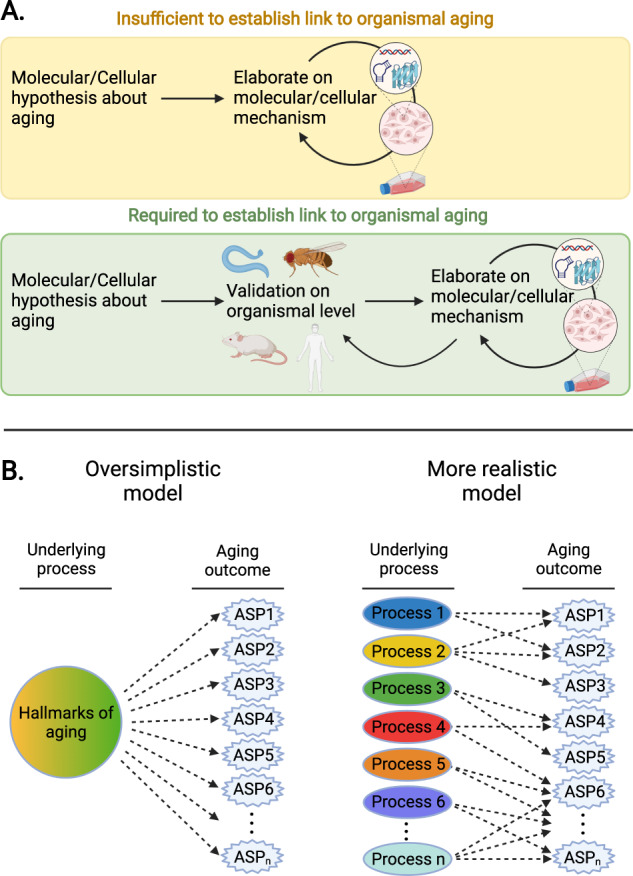
A Examining molecular and cellular hypotheses about aging on the cellular level (cultured cells) is insufficient to truly establish a link to aging. Instead, hypotheses about aging must be validated at the organismal level before potentially additional molecular and cellular studies can be used to elaborate on mechanisms (provided suitable model systems are available). B Aging is a series of outcomes with diverse contributing factors. Rather than a set of outcomes arising from a small set of processes (“hallmarks of aging”), aging outcomes are likely shaped in complex ways by a large number of factors. ASP age-sensitive phenotype.
For instance, reactive oxygen species (ROS) have been hypothesized for a long time to play a role in driving organismal aging. While cells and organisms deficient in ROS defense mechanisms are in fact exquisitely sensitive to oxidative stressors [127–129], only in vivo experiments in the context of natural aging were able to address the question whether and to what extent ROS play a role in organismal aging. However, such studies showed that increased ROS not only does not accelerate aging, but did even extend lifespan in yeast and C. elegans [130–132]. Analyses in mice also showed that genetic manipulations that increase mitochondrial ROS and oxidative damage do not accelerate aging [133, 134] and that manipulations that increase antioxidant defense did not extend longevity [135]. This is a clear example illustrating that cellular damage models require validation in in vivo aging models if they are intended to deliver insights about aging.
Telomere attrition, which is claimed by some to cause organismal aging [13, 136, 137], represents another example of cell-centered models that have been extrapolated to organismal aging despite a lack of evidence on this level of analysis. The relationship between telomere attrition and cellular senescence in vitro has fueled claims that telomere length is a determinant of organismal aging and lifespan [138, 139]. There have been numerous studies on the importance of telomere length on replicative cellular lifespan in human cultured cells [140–143]; however, studies on how telomere attrition could be involved in natural organismal aging are indeed very limited and controversial. Analyses of several different mouse strains revealed no significant correlation between telomere length and longevity in closely related mouse strains and mice with naturally relatively shorter telomere lengths show no significant reduction in lifespan [144]. However, in general, mice have much longer telomeres than humans [145]. As a consequence, mice do not seem to show functionally relevant telomere attrition that takes place during their normal lifespan, indicating that telomere attrition may in fact not underlie aging phenomena observed in these wildtype stocks of mice. Mice engineered to develop short telomeres (Terc-deficient mice) do not show significant adverse effects on many health parameters (lifespan, motor behavior/activity, histological measures, weight gain, etc.) in the first generation. A lifespan-shortening effect becomes only evident after multiple rounds of breeding [137, 146], suggesting that only progressive telomere attrition accumulating across multi-generational cell divisions is capable of eliciting effects on lifespan and health-related outcomes in these mouse models. Such extreme telomere attrition cannot be seen within a normal mouse life [147]. Furthermore, even after multiple generations, these mice feature a pattern and spectrum of pathologies (skin ulceration, infertility, increased frequencies of very specific forms of neoplasias, and frequently lethal gastrointestinal lesions) that looks very different from the organismal changes taking place during normal aging in both mice and humans [126, 137, 146]. Moreover, many studies (with a few exceptions [148, 149]) showed that increasing telomere length promotes carcinogenesis, whereas telomerase-deficiency (leading to telomere attrition) suppresses cancer formation in a number of murine cancer models [150–155]. Studies in other animal models (beyond the mouse), such as in zebrafish (Danio rerio), D. melanogaster and C. elegans, as well as studies in plants (Arabidobsis thaliana) also question the causal involvement of telomeres in aging (see this review for a more detailed discussion [147]). Hence, to date, there is insufficient evidence to support strong conclusions about telomere attrition playing an important role in organismal aging.
Advances in stem cell biology have led some to speculate that brain organoids produced from human pluripotent stem cells (HPSC) could be utilized as a replacement for in-vivo studies. While brain organoids provide an intriguing environment for studying complicated cell-cell interactions, modeling age-related neurodegenerative disorders remains difficult. Brain organoids have a transcriptional profile similar to that of the prenatal brain, and, while they may be suitable to model some features of brain development, their relevance as models for aging-associated change is less clear [156]. Another disadvantage is the lack of complete vascularization, which precludes the modelling of key aspects of brain physiology [157]. Brain organoids also lack the full cellular diversity present in the mammalian brain. Several brain organoids systems, for example, contain astrocytes but no microglial cells [158]. Some others consist of neurons and glial cells but not oligodendrocytes [159]. Furthermore, a wide range of differentiation techniques results in variability in size and structure of brain organoids, hence presenting challenges for reproducibility of research [160, 161]. As a consequence, brain organoids are at present still basic and immature and will require much more additional development.
Altogether, we conclude that links between a molecular/cellular process and aging have to be established in the context of organismal aging to support claims about roles of that process in aging (Fig. 2A). This is because organismal aging is a property emerging in whole intact organisms as time passes by. As such, reductionist systems, like cell culture models, may not necessarily capture processes and features relevant to organismal aging.
Underestimating the complexity of the aging process
Aging is the process that progressively transforms young adult organisms into aged ones with changes across multiple physiological systems [11, 162–165], leading to the emergence of a large number of age-sensitive phenotypes (ASPs), such as loss of bone density, skin thinning, atherosclerotic changes, reduced kidney function, etc. [164]. One of the implicit assumptions in [13] is that putative aging regulators are involved in the development of the majority (if not all) ASPs. However, the complexity of biological systems suggests this is likely an over-simplistic view (Fig. 2B). Recent multi-omics research indicates that aging-associated change take place at varying rates in different organs and systems; also, the underlying biological mechanisms might differ across tissues [166, 167]. It has also been shown that, while some downstream processes are conserved across tissues, transcription factor regulatory networks have limited overlap [166]. In general, if a given intervention improves or accelerates one or a few ASPs in a given organism, this observation cannot be simply extrapolated to other ASPs or organisms. However, such an extrapolation is commonly seen, for instance, when interventions are concluded to slow aging based on the assessment of a small number of ASPs (Supplementary Table 1). As we will discuss in more detail below, considering only a small number of ASPs is insufficient to draw strong conclusions about broader effects on aging.
How can we measure aging: Large-scale assessment of aging outcomes
Aging is a multifactorial process that occurs as adult organisms mature into aged organisms. The results of aging are widespread functional changes across virtually all organ and tissue systems, an increased risk to develop age-related diseases, as well as an elevated mortality risk [126, 162–164]. The process of aging occurs gradually, with changes manifesting across almost all tissue and organ systems and across all levels of biological complexity (i.e., molecular, cellular, tissue and organismal levels of analysis) [164, 168].
Although the mechanistic processes of aging are not well understood and are therefore difficult to quantify, it is straightforward to measure “outcomes” of the aging process (Fig. 3A). In other words, aging is a process with a variety of clearly observable outcomes, such as loss of muscle mass, a loss of bone density, increased numbers of skin wrinkles, hair greying, low-grade tissue inflammation, etc. which can be measured even in the absence of knowledge of their underlying causal processes (Fig. 3A). A subset of parameters may be amenable to longitudinal assessment over time (to derive within-subjects rate of change estimates), others may require cross-sectional study designs comparing young adult vs. aged animals (e.g., all parameters linked to data collection in the context of terminal procedures; these can be used to derive population-level rate of change estimates).
Fig. 3. How to measure aging.
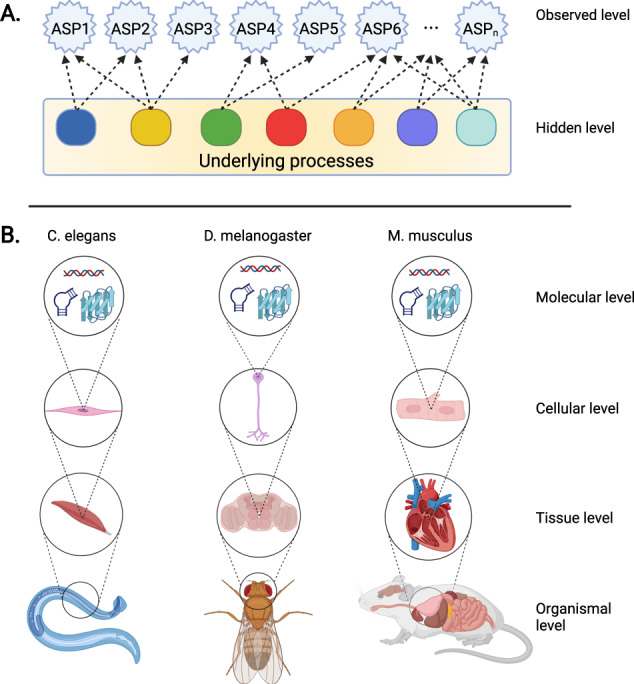
A Aging is a collection of processes with well-characterized and observable phenotypic outcomes (age-sensitive phenotypes; ASPs) but often poorly understood underlying causes. It is likely that a large number of underlying processes drive age-dependent phenotypic changes, each contributing to some of the observed phenotypes. ASPs can be measured directly and can be used as markers to address whether a given intervention (e.g., genetic, dietary or pharmacological) targets the processes underlying their age-dependent change (even if the processes themselves remain poorly defined). B ASPs can be measured in many organisms and across levels of biological complexity (for example, see main text).
An example of this approach are deep-phenotyping studies, which have been used to analyze a wide range of aging-related phenotypic changes across various tissues and organ systems, providing a multi-dimensional view on the phenotypic consequences associated with aging [32, 36, 169–171]. Among hundreds of parameters examined in young and old animals, many differed between the young and old animals, identifying them as ASPs. Once ASPs are identified for any given organism, they can be used collectively as a multi-dimensional representation of aging-associated phenotypic change, which in turn can be used to test putative anti-aging interventions (PAAIs) against. This approach has been successfully used to determine whether PAAIs, such as pharmacological mTOR inhibition using rapamycin, food restriction employing an every-other-day feeding regime or genetically inhibiting growth hormone signaling in dwarf mutant mice, indeed delay aging [32, 36, 169–171].
Remarkable technological advances facilitate an ever-increasing ability to create large-scale phenotypic maps of aging-associated change in model organisms (Fig. 3B), spanning from the molecular to the organismal level. For instance, the assessment of age-dependent phenotypic change in mice can include, but is by no means limited to, an examination of behavioral and neuropsychiatric functions (e.g., learning and memory, attention, sensorimotor gating, motor functions, sensory functions) as well as neuroanatomical (e.g., MRI, histopathology) and neurochemical (e.g., neurotransmitter analyses) measurements, an assessment of endocrine functions (e.g., plasma hormone concentrations, histopathological changes in the thyroid gland, adrenal gland etc.) and metabolism (e.g., body composition changes assessed by NMR, changes in energy metabolism assessed via indirect calorimetry, glucose tolerance measurements, surface and core body temperature measurements, food and water intake, analyses of substrate turnover rates), as well as structural and functional analyses focused on the cardiovascular system (e.g., echocardiography, electrocardiography, blood pressure measurements, histopathological analyses), the respiratory system (e.g., whole body plethysmography, histopathological analyses of lungs and bronchial system), the gastrointestinal tract, liver and pancreas (e.g., histopathological analyses, clinical chemistry, microbiome analysis), the renal system (e.g., assessment of glomerular filtration rate, histopathological analyses, clinical chemistry), skeletal system (e.g., bone densitometry, histopathological analyses), reproductive system (e.g., histopathological analyses, plasma hormone concentrations), immune system (e.g., flow cytometry, antibody measurements, histopathological analyses, immune activation assays), the hematopoietic system (e.g., blood cell counts, histopathological analyses) and the skin (histopathological analyses, transepidermal water loss) [32, 36, 169].
Deep phenotyping approaches are not limited to mammalian model systems. Studies in Drosophila melanogaster, for instance, can also draw from a rich set of aging-associated phenotypic changes, which can be utilized collectively as a proxy to measure aging. This includes, but is not limited to, analyses of molecular alterations (e.g., bulk changes in transcriptome [172–175], proteome [176, 177] and metabolome [178, 179]; single-cell transcriptomic changes [180]); neuromorphological changes (e.g., neurodegeneration [181]), behavioral assessments (e.g., learning and memory [182–184], locomotor activity [185, 186], circadian rhythm and sleep patterns [187, 188]), an assessment of muscle structure and function (e.g., changes in muscle morphology and integrity [189]), analyses of changes in heart function (e.g., assessment of cardiac performance [190, 191]) and gut homeostasis (e.g., histopathological analyses of epithelial dysplasia and barrier function [91]).
A multitude of age-dependent changes can also be observed in the roundworm Caenorhabditis elegans, another popular model organism in aging research. This includes changes at the molecular level (e.g., changes in transcriptome [192–194] and proteome [195, 196]), the subcellular level (e.g., structural and functional changes in mitochondria [197]), and tissue-specific changes, such as those affecting the reproductive system (e.g., rate of reproduction and progeny number [198, 199], deterioration of germline cells and changes in oocyte morphology [200]), muscles (e.g., assessment of muscle structural integrity and sarcomeres structure [201, 202], pharyngeal muscles morphology [203]), neuromuscular functions and the nervous system (e.g., morphology and function of touch receptor neurons [204, 205], neurite sprouting and synapse deterioration [206], locomotion [117, 207, 208], pharyngeal pumping rate [117], learning and memory [209]). For further details, the interested reader is referred to existing review articles, such as [210].
Accounting for aging-independent effects
Interventions that slow, delay or even stop aging must, by definition, interfere with the transformation of a phenotypically young to a phenotypically aged organism. Therefore, studying potential interventions only in aged mice is not sufficient to conclude that a PAAI interferes with the aging process; instead, studies must be designed to examine the PAAI in both young and old animals (either using longitudinal or cross-sectional study designs). This is required to support valid conclusions about the nature of the interactions between a PAAI and aging processes.
Based on the above considerations, designing studies that distinguish between an intervention targeting age-dependent change and a mimicry of such an effect is rather simple. One needs to (1) generate knowledge of lifetime trajectories of ASPs to determine when age-dependent changes in ASPs are first detectable to then (2) design experiments that include young treated reference groups that are subjected to PAAI before age-dependent changes in ASPs become detectable. This allows investigators to dissociate PAAI effects on ASPs from age-dependent changes in these ASPs.
For instance, the number of neurons in the mouse brain could be increased by a specific genetic variant that promotes neurogenetic processes during development of the animal, but has no impact on the rate of neuron loss during aging. Hence, this variant would affect aspects of development without influencing aging-associated change. Although this genetic variant would cause animals to have a larger number of neurons in old age, this observation cannot be interpreted as slowed aging because the rate of age-dependent change remains unaffected [35]. Hence, the true nature of an intervention can only be understood through the examination of the intervention’s effect on both young and old animals, which allows the distinction between aging-independent effects on ASPs (such as in the example above) and a slowing of age-dependent changes in ASPs (Fig. 4). An example of the latter scenario may be a genetic variant that delays or slows aging-associated neuron loss through neuroprotective mechanisms but leaves unaffected neurogenetic processes during development (Fig. 4).
Fig. 4. Mechanisms by which putative anti-aging interventions (PAAIs) could influence age-sensitive phenotypes (ASPs).
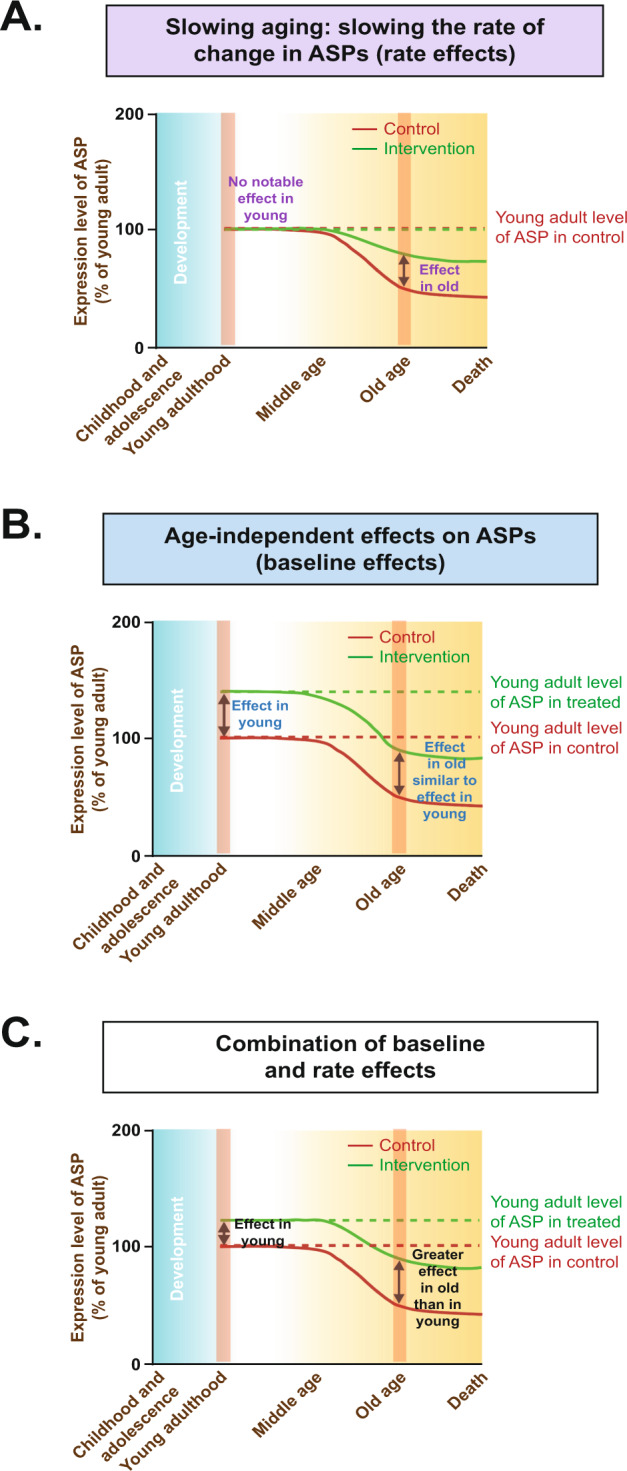
In principle, PAAI effects on ASPs could be attributed to one of the three models: (1) rate model, (2) baseline model, or (3) a combination of rate and baseline models. In the rate model (A), a given anti-aging treatment slows the rate at which an ASP develops but does not have any effects on the ASP prior to the manifestation of age-dependent change in the ASP. This pattern supports the interpretation that age-dependent phenotypic change (aka aging) has been slowed by treatment. In the baseline model (B), a short-term treatment in young animals has similar effects on ASPs as a long-term treatment in aged animals. This pattern indicates age-independent effects unrelated to any influence on the aging process. It is also possible that a treatment influences ASPs in both young and old mice but with larger effects in old mice than in young mice (C). This pattern is more difficult to interpret given that it could be caused by a mixture of age-independent effects and effects on aging rate; alternatively, it could also arise from age-independent effects alone, if treatment duration has an influence on treatment effect size (long-term treatment in old mice resulting in larger effects than short-term treatment in young mice).
This is analogous to the distinction between disease-modifying and symptomatic treatments [211–215]. While both kinds of treatments may have value for the patient, only the former approach targets the underlying cause of the disease, while the latter approach focuses on the presented symptoms. For example, a drug that enhances cognitive function in both Alzheimer’s disease (AD) patients and healthy adults could serve as a symptomatic treatment, but it would not reveal any insights specifically related to the causes of AD. Similarly, a drug that enhances cognitive function in pre-symptomatic AD patients cannot have these effects by targeting the causes underlying cognitive decline (because cognitive decline has not yet emerged in pre-symptomatic patients) and, hence, will not give insight into the pathogenesis of AD. Only a treatment that alters the rate of cognitive decline in AD can be considered a disease-modifying treatment and can be used to better understand the causes underlying AD-associated cognitive decline.
There is currently a shortage of studies with suitable designs (which requires the inclusion of treated and untreated young groups of animals, as outlined above) to allow a judgement of whether or not PAAIs slow the rate of aging. Two unbiased large-scale phenotyping studies including young groups had been published previously, one focusing on the mTOR inhibitor rapamycin and one analyzing effects of a dietary restriction regime on a large set of ASPs [32, 36]. Both inhibition of mTOR signaling and dietary restriction represent important cornerstones in the aging field with many links to the “hallmarks of aging” (e.g., mTOR signaling has well-established links to proteostasis, nutrient sensing, mitochondrial dysfunction, intercellular communication and cellular senescence). Interestingly, in both studies it was observed that intervention effects that are specific to the aged group of mice (supporting the notion of a slowed aging rate) were rather rare. Age-independent effects on ASPs, in contrast, were common, indicating that many intervention effects were unrelated to a slowing of aging rate. For instance, the dietary restriction study mentioned above [32] analyzed 116 ASPs. Strikingly, out of these 116 ASPs only 7 ASPs were influenced by dietary restriction in a way clearly consistent with a slowing of aging rate. 33 ASPs, in contrast, were countered by dietary restriction in both young and old mice, which stresses the importance of controlling for age-independent treatment effects.
Another set of important interventions for the aging field represent those targeting (inhibiting) growth hormone signaling. No large-scale phenotyping study has been published until recently. For growth hormone signaling, we therefore reviewed all available data obtained from studies involving long-lived dwarf and related mouse lines (Ames dwarf [216], Laron dwarf [217], Snell dwarf [218], growth hormone receptor knock-out [219], growth hormone releasing hormone receptor knock-out [218] and Igf1 heterozygous mice [220]) (Supplementary Table 3). Overall, in all papers analyzed, we identified 61 ASPs examined in both young and old groups of mice (Supplementary Table 3). Notably, 30 out of 61 ASPs countered and tested in both young and old mice were clearly influenced in an age-independent fashion (similar effects in young and old). Furthermore, for 9 additional ASPs assessed in both young and old mice, there was a non-significant trend towards similar effects in young and old animals. This is in line with a recently published large-scale phenotyping analysis of Ghrhrlit/lit dwarf mutant mice that found that most ASPs ameliorated by the mutation were influenced in an age-independent fashion with very similar effects in young and old mice [171].
The available data for rapamycin, dietary restriction and growth hormone signaling-related mutants point towards similar conclusions. First, they indicate that only a subset of ASPs countered by rapamycin/dietary restriction/growth hormone signaling-related mutations follows the rate effect model shown in Fig. 4 (indicating slowed aging). Moreover, these analyses show that even for some of the most intensely investigated PAAIs only limited data are available on ASPs and organismal aging. It is therefore far from clear to what extent PAAIs, based on the “hallmarks of aging” have the capability to slow organismal aging rate. More comprehensive studies, based on large-scale approaches and including both young and aged treated animals, are required to further our knowledge regarding possible links between the putative aging regulators and organismal-level aging phenomena.
Conclusion and perspective
To date, key concepts regarding the biology of aging (such as summarized, e.g., in the “hallmarks of aging” [13]) are not sufficiently supported by studies that provide organismal-level aging data. As a consequence, it is not currently clear to what extent any of these putative aging regulators is in fact broadly linked to organismal aging rate. As outlined above, available data, in contrast, suggest that even interventions commonly claimed to “slow aging” in fact have little effect on most age-dependent phenotypic change. Future research can build on unbiased, multidimensional analyses of aging to determine the extent to which molecular regulators with a proposed role in aging do in fact (or do not) influence (aspects of) the aging process.
Aging research essentially deals with a many-to-many mapping problem. There are changes in many age-sensitive phenotypes (collectively representing the aging process, i.e., the transition of a young adult organism to an aged one) that could, in theory, each be influenced by a large set of regulators. Advances in aging research will critically depend on a better definition of this problem. Some important outstanding questions along those lines are:
To what extent do aging-associated phenotypic changes cluster vs. are independent of each other?
Did regulators of age-dependent phenotypic change evolve or do they not exist? It is clear that regulators for some aspects of age-dependent change did not evolve (e.g., neurons lost during aging do not get replaced; no mechanisms exist to repair certain changes of the extracellular matrix), but the answer will differ from phenotype to phenotype.
What are possible regulators of age-dependent phenotypic change (including the putative aging regulators discussed here as well as others)? Again, this needs to be considered on a phenotype-by-phenotype basis.
How complex are aging regulators in biological systems (e.g., how many factors may be required to act in concert to modify change of a phenotype)?
More generally, how many regulators map onto change in how many phenotypes? The extreme ends of the spectrum of possible scenarios are marked by scenarios in which 1) each of a large number of age-dependent phenotypic changes is influenced by a completely different set of regulators and 2) all age-dependent phenotypic change is jointly influenced by a small set of regulators. The middle ground between these extremes is occupied by scenarios in which there are themes of regulators common to subsets of age-dependent phenotypic change.
In conclusion, aging research will benefit from a better definition of how specific regulators map onto age-dependent change, considered on a phenotype-by-phenotype basis. Resolving some of these key questions will shed more light on how tractable (or intractable) the biology of aging is.
Supplementary information
Acknowledgements
Our work was supported by a grant from the Helmholtz Future Topic Program AMPro (Aging and Metabolic Programming). Figures were created with BioRender.com and/or by adapting from BioRender.com templates.
Author contributions
Conceptualization: DE, MK. Manuscript text: DE, MK, DB, KX, KS. Figures: KS, MK, KX, DE. Tables, Supplementary Tables: MK, KS, DE.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41380-022-01680-x.
References
- 1.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22:R741–752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 2.2018 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2018;14:367–429 10.1016/j.jalz.2018.02.001.
- 3.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Prim. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 4.Talbott EO, Malek AM, Lacomis D. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol. 2016;138:225–38. doi: 10.1016/B978-0-12-802973-2.00013-6. [DOI] [PubMed] [Google Scholar]
- 5.Golde TE. The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Mol Neurodegener. 2009;4:8–8. doi: 10.1186/1750-1326-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo X, Castro-Obregón S, Gutiérrez-Becker B, Gutiérrez-Ospina G, Karalis N, Khalil AA et al. Re-thinking the etiological framework of neurodegeneration. Front Neurosci. 2019;13. 10.3389/fnins.2019.00728. [DOI] [PMC free article] [PubMed]
- 7.Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–81. doi: 10.1038/s41582-019-0244-7. [DOI] [PubMed] [Google Scholar]
- 8.Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11:111–28. doi: 10.31887/DCNS.2009.11.2/cqiu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaeberlein M, Galvan V, Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci Transl Med. 2019;11. 10.1126/scitranslmed.aar4289. [DOI] [PMC free article] [PubMed]
- 10.Daniele S, Giacomelli C, Martini C. Brain ageing and neurodegenerative disease: The role of cellular waste management. Biochem Pharmacol. 2018;158:207–16. doi: 10.1016/j.bcp.2018.10.030. [DOI] [PubMed] [Google Scholar]
- 11.Miller RA Biology of Aging and Longevity. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S (eds). Hazzard’s Geriatric Medicine and Gerontology, 6th edition edn. McGraw Hill 2009.
- 12.Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–13. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Chen HZ, Liu DP. The four layers of aging. Cell Syst. 2015;1:180–6. doi: 10.1016/j.cels.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Gems D, de Magalhães JP. The hoverfly and the wasp: A critique of the hallmarks of aging as a paradigm. Ageing Res Rev. 2021;70:101407. doi: 10.1016/j.arr.2021.101407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mattson MP, Arumugam TV. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 2018;27:1176–99. doi: 10.1016/j.cmet.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemoine M. The evolution of the hallmarks of aging. Front Genet. 2021;12:693071. doi: 10.3389/fgene.2021.693071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Rijt S, Molenaars M, McIntyre RL, Janssens GE, Houtkooper RH. Integrating the hallmarks of aging throughout the tree of life: a focus on mitochondrial dysfunction. Front Cell Dev Biol. 2020;8:594416. doi: 10.3389/fcell.2020.594416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aunan JR, Cho WC, Søreide K. The biology of aging and cancer: a brief overview of shared and divergent molecular hallmarks. Aging Dis. 2017;8:628–42. doi: 10.14336/AD.2017.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chmielewski PP. Human ageing as a dynamic, emergent and malleable process: from disease-oriented to health-oriented approaches. Biogerontology. 2020;21:125–30. doi: 10.1007/s10522-019-09839-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guimarães GR, Almeida PP, de Oliveira Santos L, Rodrigues LP, de Carvalho JL, Boroni M, Hallmarks of aging in macrophages: consequences to skin inflammaging. Cells 2021;10. 10.3390/cells10061323. [DOI] [PMC free article] [PubMed]
- 22.Garatachea N, Pareja-Galeano H, Sanchis-Gomar F, Santos-Lozano A, Fiuza-Luces C, Morán M, et al. Exercise attenuates the major hallmarks of aging. Rejuvenation Res. 2015;18:57–89. doi: 10.1089/rej.2014.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rebelo-Marques A, De Sousa Lages A, Andrade R, Ribeiro CF, Mota-Pinto A, Carrilho F, et al. Aging hallmarks: the benefits of physical exercise. Front Endocrinol. 2018;9:258. doi: 10.3389/fendo.2018.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Liu Y, Ma X, Hu H. The influence of cell cycle regulation on chemotherapy. Int J Mol Sci. 2021;22:6923. doi: 10.3390/ijms22136923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Y, Dai W. Genomic instability and cancer. J Carcinog Mutagen. 2014;5:1000165. doi: 10.4172/2157-2518.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 28.Blackwell BN, Bucci TJ, Hart RW, Turturro A. Longevity, body weight, and neoplasia in ad libitum-fed and diet-restricted C57BL6 mice fed NIH-31 open formula diet. Toxicol Pathol. 1995;23:570–82. doi: 10.1177/019262339502300503. [DOI] [PubMed] [Google Scholar]
- 29.Pettan-Brewer C, Treuting PM Practical pathology of aging mice. Pathobiol Aging Age Relat Dis. 2011;1. 10.3402/pba.v1i0.7202. [DOI] [PMC free article] [PubMed]
- 30.Lipman R, Galecki A, Burke DT, Miller RA. Genetic loci that influence cause of death in a heterogeneous mouse stock. J Gerontol A Biol Sci Med Sci. 2004;59:977–83. doi: 10.1093/gerona/59.10.B977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie K, Neff F, Markert A, Rozman J, Aguilar-Pimentel JA, Amarie OV, et al. Every-other-day feeding extends lifespan but fails to delay many symptoms of aging in mice. Nat Commun. 2017;8:155. doi: 10.1038/s41467-017-00178-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol. 2013;15:96–102. doi: 10.1038/ncb2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weaver RL, Limzerwala JF, Naylor RM, Jeganathan KB, Baker DJ, van Deursen JM, BubR1 alterations that reinforce mitotic surveillance act against aneuploidy and cancer. Elife 2016;5. 10.7554/eLife.16620. [DOI] [PMC free article] [PubMed]
- 35.Ehninger D, Neff F, Xie K. Longevity, aging and rapamycin. Cell Mol Life Sci. 2014;71:4325–46. doi: 10.1007/s00018-014-1677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013;123:3272–91. doi: 10.1172/JCI67674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu M, Howes A, Lesperance J, Stallcup WB, Hauser CA, Kadoya K, et al. Antitumor activity of rapamycin in a transgenic mouse model of ErbB2-dependent human breast cancer. Cancer Res. 2005;65:5325–36. doi: 10.1158/0008-5472.CAN-04-4589. [DOI] [PubMed] [Google Scholar]
- 38.Woo Y, Lee HJ, Kim J, Kang SG, Moon S, Han JA, et al. Rapamycin promotes ROS-mediated cell death via functional inhibition of xCT expression in melanoma under γ-irradiation. Front Oncol. 2021;11:665420. doi: 10.3389/fonc.2021.665420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xing D, Orsulic S. A genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proc Natl Acad Sci USA. 2005;102:6936–41. doi: 10.1073/pnas.0502256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amaral CL, Freitas LB, Tamura RE, Tavares MR, Pavan IC, Bajgelman MC, et al. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer. 2016;16:602. doi: 10.1186/s12885-016-2629-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S, Roh HS, Song SS, Shin J, Lee J, Bhang DH, et al. Loss of S6K1 but Not S6K2 in the tumor microenvironment suppresses tumor growth by attenuating tumor angiogenesis. Transl Oncol. 2020;13:100767. doi: 10.1016/j.tranon.2020.100767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai KP, Cheung A, Ho CH, Tam NY, Li JW, Lin X, et al. Transcriptomic analysis reveals the oncogenic role of S6K1 in hepatocellular carcinoma. J Cancer. 2020;11:2645–55. doi: 10.7150/jca.40726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song X, Han X, Yu F, Zhang X, Chen L, Lv C. Polyamine-targeting Gefitinib Prodrug and its near-infrared fluorescent theranostic derivative for monitoring drug delivery and lung cancer therapy. Theranostics. 2018;8:2217–28. doi: 10.7150/thno.24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas TJ, Thomas T, Cellular and animal model studies on the growth inhibitory effects of polyamine analogues on breast cancer. Med Sci. 2018;6. 10.3390/medsci6010024. [DOI] [PMC free article] [PubMed]
- 45.Chen Y, Zhuang H, Chen X, Shi Z, Wang X. Spermidine‑induced growth inhibition and apoptosis via autophagic activation in cervical cancer. Oncol Rep. 2018;39:2845–54.. doi: 10.3892/or.2018.6377. [DOI] [PubMed] [Google Scholar]
- 46.Boguszewski CL, Boguszewski M. Growth hormone’s links to cancer. Endocr Rev. 2019;40:558–74. doi: 10.1210/er.2018-00166. [DOI] [PubMed] [Google Scholar]
- 47.Chhabra Y, Waters MJ, Brooks AJ. Role of the growth hormone-IGF-1 axis in cancer. Expert Rev Endocrinol Metab. 2011;6:71–84. doi: 10.1586/eem.10.73. [DOI] [PubMed] [Google Scholar]
- 48.Takei Y, Saga Y, Mizukami H, Takayama T, Ohwada M, Ozawa K, et al. Overexpression of PTEN in ovarian cancer cells suppresses i.p. dissemination and extends survival in mice. Mol Cancer Ther. 2008;7:704–11. doi: 10.1158/1535-7163.MCT-06-0724. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saga Y, Mizukami H, Suzuki M, Kohno T, Urabe M, Ozawa K, et al. Overexpression of PTEN increases sensitivity to SN-38, an active metabolite of the topoisomerase I inhibitor irinotecan, in ovarian cancer cells. Clin Cancer Res. 2002;8:1248–52. [PubMed] [Google Scholar]
- 51.Li B, Zhang J, Su Y, Hou Y, Wang Z, Zhao L, et al. Overexpression of PTEN may increase the effect of pemetrexed on A549 cells via inhibition of the PI3K/AKT/mTOR pathway and carbohydrate metabolism. Mol Med Rep. 2019;20:3793–801.. doi: 10.3892/mmr.2019.10617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12:2319–30. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 53.Soler A, Serra H, Pearce W, Angulo A, Guillermet-Guibert J, Friedman LS, et al. Inhibition of the p110α isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Med. 2013;210:1937–45. doi: 10.1084/jem.20121571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soler A, Figueiredo AM, Castel P, Martin L, Monelli E, Angulo-Urarte A, et al. Therapeutic benefit of selective inhibition of p110α PI3-Kinase in pancreatic neuroendocrine tumors. Clin Cancer Res. 2016;22:5805–17. doi: 10.1158/1078-0432.CCR-15-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goulielmaki E, Bermudez-Brito M, Andreou M, Tzenaki N, Tzardi M, de Bree E, et al. Pharmacological inactivation of the PI3K p110δ prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018;9:678. doi: 10.1038/s41419-018-0717-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10:8059–67. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 57.Podsypanina K, Lee RT, Politis C, Hennessy I, Crane A, Puc J, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice. Proc Natl Acad Sci USA. 2001;98:10320–5. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srivastava RK, Li C, Khan J, Banerjee NS, Chow LT, Athar M. Combined mTORC1/mTORC2 inhibition blocks growth and induces catastrophic macropinocytosis in cancer cells. Proc Natl Acad Sci USA. 2019;116:24583–92. doi: 10.1073/pnas.1911393116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anisimov VN. Metformin for aging and cancer prevention. Aging. 2010;2:760–74. doi: 10.18632/aging.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang HH, Moro A, Chou CEN, Dawson DW, French S, Schmidt AI, et al. Metformin decreases the incidence of pancreatic ductal adenocarcinoma promoted by diet-induced obesity in the conditional KrasG12D Mouse model. Sci Rep. 2018;8:5899. doi: 10.1038/s41598-018-24337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen K, Qian W, Jiang Z, Cheng L, Li J, Sun L, et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol Cancer. 2017;16:131. doi: 10.1186/s12943-017-0701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aljofan M, Riethmacher D. Anticancer activity of metformin: a systematic review of the literature. Future Sci OA. 2019;5:Fso410. doi: 10.2144/fsoa-2019-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pallavi R, Giorgio M, Pelicci PG. Insights into the beneficial effect of caloric/dietary restriction for a healthy and prolonged life. Front Physiol. 2012;3:318. doi: 10.3389/fphys.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morioka T, Yamazaki S, Yanagihara H, Sunaoshi M, Kaminishi M, Kakinuma S. Calorie restriction suppresses the progression of radiation-induced intestinal tumours in C3B6F1 Apc (Min/+) Mice. Anticancer Res. 2021;41:1365–75. doi: 10.21873/anticanres.14894. [DOI] [PubMed] [Google Scholar]
- 65.Kopeina GS, Senichkin VV, Zhivotovsky B. Caloric restriction—A promising anti-cancer approach: From molecular mechanisms to clinical trials. Biochim Biophys Acta Rev Cancer. 2017;1867:29–41. doi: 10.1016/j.bbcan.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Gates AC, Bernal-Mizrachi C, Chinault SL, Feng C, Schneider JG, Coleman T, et al. Respiratory uncoupling in skeletal muscle delays death and diminishes age-related disease. Cell Metab. 2007;6:497–505. doi: 10.1016/j.cmet.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 67.Shrestha R, Johnson E, Byrne FL. Exploring the therapeutic potential of mitochondrial uncouplers in cancer. Mol Metab. 2021;51:101222. doi: 10.1016/j.molmet.2021.101222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matheu A, Maraver A, Collado M, Garcia-Cao I, Cañamero M, Borras C, et al. Anti-aging activity of the Ink4/Arf locus. Aging Cell. 2009;8:152–61. doi: 10.1111/j.1474-9726.2009.00458.x. [DOI] [PubMed] [Google Scholar]
- 69.Modesitt SC, Ramirez P, Zu Z, Bodurka-Bevers D, Gershenson D, Wolf JK. In vitro and in vivo adenovirus-mediated p53 and p16 tumor suppressor therapy in ovarian cancer. Clin Cancer Res. 2001;7:1765–72. [PubMed] [Google Scholar]
- 70.Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–9. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 71.Zhou X, Singh M, Sanz Santos G, Guerlavais V, Carvajal LA, Aivado M, et al. Pharmacological activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting anti-tumor immunity. Cancer Discov. 2021;11:3090–105. 10.1158/2159-8290.CD-20-1741. [DOI] [PMC free article] [PubMed]
- 72.Ramos H, Soares MIL, Silva J, Raimundo L, Calheiros J, Gomes C, et al. A selective p53 activator and anticancer agent to improve colorectal cancer therapy. Cell Rep. 2021;35:108982. doi: 10.1016/j.celrep.2021.108982. [DOI] [PubMed] [Google Scholar]
- 73.Wang Z, Sun Y. Targeting p53 for novel anticancer therapy. Transl Oncol. 2010;3:1–12. doi: 10.1593/tlo.09250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rohwer N, Kühl AA, Ostermann AI, Hartung NM, Schebb NH, Zopf D, et al. Effects of chronic low-dose aspirin treatment on tumor prevention in three mouse models of intestinal tumorigenesis. Cancer Med. 2020;9:2535–50. doi: 10.1002/cam4.2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guillem-Llobat P, Dovizio M, Bruno A, Ricciotti E, Cufino V, Sacco A, et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget. 2016;7:32462–77. doi: 10.18632/oncotarget.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li XF, Xu BZ, Wang SZ. Aspirin inhibits the proliferation and migration of gastric cancer cells in p53-knockout mice. Oncol Lett. 2016;12:3183–6. doi: 10.3892/ol.2016.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang C, Liu J, Wang Y, Tong J, Wu Y, Liu Y. Aspirin inhibits the proliferation of canine mammary gland tumor cells in vitro and in vivo. Transl Cancer Res. 2017;6:188–97. doi: 10.21037/tcr.2017.01.07. [DOI] [Google Scholar]
- 78.Pathi S, Jutooru I, Chadalapaka G, Nair V, Lee SO, Safe S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS One. 2012;7:e48208. doi: 10.1371/journal.pone.0048208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maity G, De A, Das A, Banerjee S, Sarkar S, Banerjee SK. Aspirin blocks growth of breast tumor cells and tumor-initiating cells and induces reprogramming factors of mesenchymal to epithelial transition. Lab Invest. 2015;95:702–17. doi: 10.1038/labinvest.2015.49. [DOI] [PubMed] [Google Scholar]
- 80.Woods D, Turchi JJ. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther. 2013;14:379–89. doi: 10.4161/cbt.23761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell. 2002;108:135–44. doi: 10.1016/S0092-8674(02)00621-9. [DOI] [PubMed] [Google Scholar]
- 82.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645–58. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 83.Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006;66:3355–8. doi: 10.1158/0008-5472.CAN-05-3827. [DOI] [PubMed] [Google Scholar]
- 84.Rera M, Clark RI, Walker DW. Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci USA. 2012;109:21528–33. doi: 10.1073/pnas.1215849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rera M, Clark RI, Walker DW. Why do old flies die? Aging. 2013;5:586–7. doi: 10.18632/aging.100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010;6:e1001159. doi: 10.1371/journal.pgen.1001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang L, Karpac J, Jasper H. Promoting longevity by maintaining metabolic and proliferative homeostasis. J Exp Biol. 2014;217:109–18. doi: 10.1242/jeb.089920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rera M, Azizi MJ, Walker DW. Organ-specific mediation of lifespan extension: more than a gut feeling? Ageing Res Rev. 2013;12:436–44. doi: 10.1016/j.arr.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Biteau B, Hochmuth CE, Jasper H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila Gut. Cell Stem Cell. 2008;3:442–55. doi: 10.1016/j.stem.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Choi NH, Kim JG, Yang DJ, Kim YS, Yoo MA. Age-related changes in Drosophila midgut are associated with PVF2, a PDGF/VEGF-like growth factor. Aging Cell. 2008;7:318–34. doi: 10.1111/j.1474-9726.2008.00380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Regan JC, Khericha M, Dobson AJ, Bolukbasi E, Rattanavirotkul N, Partridge L. Sex difference in pathology of the ageing gut mediates the greater response of female lifespan to dietary restriction. Elife. 2016;5:e10956. doi: 10.7554/eLife.10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fan X, Liang Q, Lian T, Wu Q, Gaur U, Li D, et al. Rapamycin preserves gut homeostasis during Drosophila aging. Oncotarget. 2015;6:35274–83. doi: 10.18632/oncotarget.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rodriguez-Fernandez IA, Tauc HM, Jasper H. Hallmarks of aging Drosophila intestinal stem cells. Mech Ageing Dev. 2020;190:111285. doi: 10.1016/j.mad.2020.111285. [DOI] [PubMed] [Google Scholar]
- 94.Choi NH, Lucchetta E, Ohlstein B. Nonautonomous regulation of Drosophila midgut stem cell proliferation by the insulin-signaling pathway. Proc Natl Acad Sci USA. 2011;108:18702–7. doi: 10.1073/pnas.1109348108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang L, Ryoo HD, Qi Y, Jasper H. PERK limits drosophila lifespan by promoting intestinal stem cell proliferation in response to ER stress. PLoS Genet. 2015;11:e1005220. doi: 10.1371/journal.pgen.1005220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paredes JC, Welchman DP, Poidevin M, Lemaitre B. Negative regulation by amidase PGRPs shapes the Drosophila antibacterial response and protects the fly from innocuous infection. Immunity. 2011;35:770–9. doi: 10.1016/j.immuni.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 97.Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011;14:623–34. doi: 10.1016/j.cmet.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hu DJ-K, Jasper H. Control of intestinal cell fate by dynamic mitotic spindle repositioning influences epithelial homeostasis and longevity. Cell Rep. 2019;28:2807–23.e2805. doi: 10.1016/j.celrep.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guo L, Karpac J, Tran Susan L, Jasper H. PGRP-SC2 promotes gut immune homeostasis to limit commensal dysbiosis and extend lifespan. Cell. 2014;156:109–22. doi: 10.1016/j.cell.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Salazar AM, Resnik-Docampo M, Ulgherait M, Clark RI, Shirasu-Hiza M, Jones DL, et al. Intestinal Snakeskin limits microbial dysbiosis during aging and promotes longevity. iScience. 2018;9:229–43. doi: 10.1016/j.isci.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–6. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 102.Broughton SJ, Piper MD, Ikeya T, Bass TM, Jacobson J, Driege Y, et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci USA. 2005;102:3105–10. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Libert S, Zwiener J, Chu X, Vanvoorhies W, Roman G, Pletcher SD. Regulation of Drosophila life span by olfaction and food-derived odors. Science. 2007;315:1133–7. doi: 10.1126/science.1136610. [DOI] [PubMed] [Google Scholar]
- 104.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fridell YW, Hoh M, Kréneisz O, Hosier S, Chang C, Scantling D, et al. Increased uncoupling protein (UCP) activity in Drosophila insulin-producing neurons attenuates insulin signaling and extends lifespan. Aging. 2009;1:699–713. doi: 10.18632/aging.100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Magwere T, Chapman T, Partridge L. Sex differences in the effect of dietary restriction on life span and mortality rates in female and male Drosophila melanogaster. J Gerontol A Biol Sci Med Sci. 2004;59:3–9. doi: 10.1093/gerona/59.1.B3. [DOI] [PubMed] [Google Scholar]
- 107.Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–25. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giannakou ME, Goss M, Jünger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 2004;305:361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- 110.Rallis A, Navarro JA, Rass M, Hu A, Birman S, Schneuwly S, et al. Hedgehog signaling modulates glial proteostasis and lifespan. Cell Rep. 2020;30:2627–43.e2625. doi: 10.1016/j.celrep.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 111.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–25. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 112.Broughton SJ, Slack C, Alic N, Metaxakis A, Bass TM, Driege Y, et al. DILP-producing median neurosecretory cells in the Drosophila brain mediate the response of lifespan to nutrition. Aging Cell. 2010;9:336–46. doi: 10.1111/j.1474-9726.2010.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen H, Zheng X, Zheng Y. Age-associated loss of lamin-B leads to systemic inflammation and gut hyperplasia. Cell. 2014;159:829–43. doi: 10.1016/j.cell.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ayyaz A, Li H, Jasper H. Haemocytes control stem cell activity in the Drosophila intestine. Nat Cell Biol. 2015;17:736–48. doi: 10.1038/ncb3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Han H, Pan C, Liu C, Lv X, Yang X, Xiong Y, et al. Gut–neuron interaction via Hh signaling regulates intestinal progenitor cell differentiation in Drosophila. Cell Discov. 2015;1:15006. doi: 10.1038/celldisc.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miguel-Aliaga I, Jasper H, Lemaitre B. Anatomy and physiology of the digestive tract of Drosophila melanogaster. Genetics. 2018;210:357–96. doi: 10.1534/genetics.118.300224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang C, Xiong C, Kornfeld K. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc Natl Acad Sci USA. 2004;101:8084–9. doi: 10.1073/pnas.0400848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao Y, Gilliat AF, Ziehm M, Turmaine M, Wang H, Ezcurra M, et al. Two forms of death in ageing Caenorhabditis elegans. Nat Commun. 2017;8:15458. doi: 10.1038/ncomms15458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao Y, Zhang B, Marcu I, Athar F, Wang H, Galimov ER, et al. Mutation of daf-2 extends lifespan via tissue-specific effectors that suppress distinct life-limiting pathologies. Aging Cell. 2021;20:e13324–e13324. doi: 10.1111/acel.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao Y, Wang H, Poole RJ, Gems D. A fln-2 mutation affects lethal pathology and lifespan in C. elegans. Nat Commun. 2019;10:5087. doi: 10.1038/s41467-019-13062-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sahin M. Targeted treatment trials for tuberous sclerosis and autism: no longer a dream. Curr Opin Neurobiol. 2012;22:895–901. doi: 10.1016/j.conb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Crino PB. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013;125:317–32. doi: 10.1007/s00401-013-1085-x. [DOI] [PubMed] [Google Scholar]
- 123.Crowell B, Lee GH, Nikolaeva I, Dal Pozzo V, D’Arcangelo G, Complex neurological phenotype in mutant mice lacking Tsc2 in excitatory neurons of the developing forebrain(123). eNeuro 2015;2. [DOI] [PMC free article] [PubMed]
- 124.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen L, Oshima J. Werner Syndrome. J Biomed Biotechnol. 2002;2:46–54. doi: 10.1155/S1110724302201011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lombard DB, Miller RA, Pletcher SD, Biology of Aging and Longevity. In: Halter JB, Ouslander JG, Studenski S, High KP, Asthana S, Supiano MA et al. (eds). Hazzard’s Geriatric Medicine and Gerontology, 7e. McGraw-Hill Education: New York, NY, 2017.
- 127.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 128.McDonald RB “Cellular Aging” in Biology of Aging second edition, CRC Press 2019.
- 129.Harman D. The free radical theory of aging: effect of age on serum copper levels. J Gerontol. 1965;20:151–3. doi: 10.1093/geronj/20.2.151. [DOI] [PubMed] [Google Scholar]
- 130.Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22:3236–41. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leão C, et al. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci USA. 2010;107:15123–8. doi: 10.1073/pnas.1004432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 134.Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci. 2009;64:1212–20. doi: 10.1093/gerona/glp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pérez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009;8:73–5. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet. 2009;85:823–32. doi: 10.1016/j.ajhg.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–12. doi: 10.1016/S0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 138.Harley CB, Vaziri H, Counter CM, Allsopp RC. The telomere hypothesis of cellular aging. Exp Gerontol. 1992;27:375–82. doi: 10.1016/0531-5565(92)90068-B. [DOI] [PubMed] [Google Scholar]
- 139.Harley CB. Telomere loss: mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–82. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 140.Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992;89:10114–8. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 142.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 143.Goldstein S. Replicative senescence: the human fibroblast comes of age. Science. 1990;249:1129–33. doi: 10.1126/science.2204114. [DOI] [PubMed] [Google Scholar]
- 144.Hemann MT, Greider CW. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 2000;28:4474–8. doi: 10.1093/nar/28.22.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol. 2013;50:165–74. doi: 10.1053/j.seminhematol.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lee H-W, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–74. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 147.Simons MJ. Questioning causal involvement of telomeres in aging. Ageing Res Rev. 2015;24:191–6. doi: 10.1016/j.arr.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 148.Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012;4:691–704. doi: 10.1002/emmm.201200245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Muñoz-Lorente MA, Cano-Martin AC, Blasco MA. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat Commun. 2019;10:4723. doi: 10.1038/s41467-019-12664-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, et al. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97:515–25. doi: 10.1016/S0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 151.Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–9. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 152.Qi L, Strong MA, Karim BO, Armanios M, Huso DL, Greider CW. Short telomeres and ataxia-telangiectasia mutated deficiency cooperatively increase telomere dysfunction and suppress tumorigenesis. Cancer Res. 2003;63:8188–96. [PubMed] [Google Scholar]
- 153.Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11:461–9. doi: 10.1016/j.ccr.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Artandi SE, Alson S, Tietze MK, Sharpless NE, Ye S, Greenberg RA, et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci USA. 2002;99:8191–6. doi: 10.1073/pnas.112515399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hahn WC, Meyerson M. Telomerase activation, cellular immortalization and cancer. Ann Med. 2001;33:123–9. doi: 10.3109/07853890109002067. [DOI] [PubMed] [Google Scholar]
- 156.Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Bräuninger M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA. 2015;112:15672–7. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huch M, Knoblich JA, Lutolf MP, Martinez-Arias A. The hope and the hype of organoid research. Development. 2017;144:938–41. doi: 10.1242/dev.150201. [DOI] [PubMed] [Google Scholar]
- 158.Yakoub AM. Cerebral organoids exhibit mature neurons and astrocytes and recapitulate electrophysiological activity of the human brain. Neural Regen Res. 2019;14:757–61. doi: 10.4103/1673-5374.249283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gonzalez C, Armijo E, Bravo-Alegria J, Becerra-Calixto A, Mays CE, Soto C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry. 2018;23:2363–74. doi: 10.1038/s41380-018-0229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rambani K, Vukasinovic J, Glezer A, Potter SM. Culturing thick brain slices: an interstitial 3D microperfusion system for enhanced viability. J Neurosci Methods. 2009;180:243–54. doi: 10.1016/j.jneumeth.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Papaspyropoulos A, Tsolaki M, Foroglou N, Pantazaki AA. Modeling and targeting Alzheimer’s disease with organoids. Front Pharm. 2020;11:396. doi: 10.3389/fphar.2020.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rose MR, Evolutionary biology of aging. Oxford University Press, Oxford 1991.
- 163.Rockstein M, Chesky, JA & Sussman, M, Comparative biology and evolution of aging. in Handbook of the biology of aging 3-34 (Van Nostrand Reinhold Company, New York) 1977.
- 164.Aspinall R In: R Aspinall e. Aging of the Organs And Systems. Dordrecht:Springer 2003;3.
- 165.Giaimo S, Traulsen A. The selection force weakens with age because ageing evolves and not vice versa. Nat Commun. 2022;13:686. doi: 10.1038/s41467-022-28254-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schaum N, Lehallier B, Hahn O, Pálovics R, Hosseinzadeh S, Lee SE, et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020;583:596–602. doi: 10.1038/s41586-020-2499-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nie C, Li Y, Li R, Yan Y, Zhang D, Li T, et al. Distinct biological ages of organs and systems identified from a multi-omics study. Cell Rep. 2022;38:110459. doi: 10.1016/j.celrep.2022.110459. [DOI] [PubMed] [Google Scholar]
- 168.Abdulla AR, G.S. The biology of ageing and its clinical implications. Radcliffe Publishing, London 2013.
- 169.Bellantuono I, de Cabo R, Ehninger D, Di Germanio C, Lawrie A, Miller J, et al. A toolbox for the longitudinal assessment of healthspan in aging mice. Nat Protoc. 2020;15:540–74. doi: 10.1038/s41596-019-0256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Freund A. Untangling aging using dynamic, organism-level phenotypic networks. Cell Syst. 2019;8:172–81. doi: 10.1016/j.cels.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 171.Xie K, Fuchs H, Scifo E, Liu D, Aziz A, Aguilar-Pimentel JA et al. Deep Phenotyping and lifetime trajectories reveal limited effects of longevity regulators on the aging process in C57BL/6J Mice. bioRxiv 2022: 2022.2003.2025.485824. [DOI] [PMC free article] [PubMed]
- 172.Hall H, Cooper BR, Qi G, Wijeratne AB, Mosley AL, Weake VM. Quantitative proteomic and metabolomic profiling reveals altered mitochondrial metabolism and folate biosynthesis pathways in the aging drosophila eye. Mol Cell Proteom. 2021;20:100127. doi: 10.1016/j.mcpro.2021.100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bajgiran M, Azlan A, Shamsuddin S, Azzam G, Halim MA. Data on RNA-seq analysis of Drosophila melanogaster during ageing. Data Brief. 2021;38:107413. doi: 10.1016/j.dib.2021.107413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Pacifico R, MacMullen CM, Walkinshaw E, Zhang X, Davis RL. Brain transcriptome changes in the aging Drosophila melanogaster accompany olfactory memory performance deficits. PLoS One. 2018;13:e0209405. doi: 10.1371/journal.pone.0209405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mangleburg CG, Wu T, Yalamanchili HK, Guo C, Hsieh YC, Duong DM, et al. Integrated analysis of the aging brain transcriptome and proteome in tauopathy. Mol Neurodegener. 2020;15:56. doi: 10.1186/s13024-020-00405-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Brown CJ, Kaufman T, Trinidad JC, Clemmer DE. Proteome changes in the aging Drosophila melanogaster head. Int J Mass Spectrom. 2018;425:36–46. doi: 10.1016/j.ijms.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yang L, Cao Y, Zhao J, Fang Y, Liu N, Zhang Y. Multidimensional proteomics identifies declines in protein homeostasis and mitochondria as early signals for normal aging and age-associated disease in Drosophila. Mol Cell Proteom. 2019;18:2078–88. doi: 10.1074/mcp.RA119.001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Laye MJ, Tran V, Jones DP, Kapahi P, Promislow DE. The effects of age and dietary restriction on the tissue-specific metabolome of Drosophila. Aging Cell. 2015;14:797–808. doi: 10.1111/acel.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hoffman JM, Soltow QA, Li S, Sidik A, Jones DP, Promislow DE. Effects of age, sex, and genotype on high-sensitivity metabolomic profiles in the fruit fly, Drosophila melanogaster. Aging Cell. 2014;13:596–604. doi: 10.1111/acel.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Davie K, Janssens J, Koldere D, De Waegeneer M, Pech U, Kreft L, et al. A single cell transcriptome atlas of the aging drosophilia brain. Cell. 2018;174:982–e920. doi: 10.1016/j.cell.2018.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Behnke JA, Ye C, Moberg KH, Zheng JQ. A protocol to detect neurodegeneration in Drosophila melanogaster whole-brain mounts using advanced microscopy. STAR Protoc. 2021;2:100689. doi: 10.1016/j.xpro.2021.100689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tamura T, Chiang AS, Ito N, Liu HP, Horiuchi J, Tully T, et al. Aging specifically impairs amnesiac-dependent memory in Drosophila. Neuron. 2003;40:1003–11. doi: 10.1016/S0896-6273(03)00732-3. [DOI] [PubMed] [Google Scholar]
- 183.Tonoki A, Davis RL. Aging impairs intermediate-term behavioral memory by disrupting the dorsal paired medial neuron memory trace. Proc Natl Acad Sci. 2012;109:6319–24. doi: 10.1073/pnas.1118126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Saitoe M, Saeki S, Hirano Y, Horiuchi J, Age-related memory impairment inDrosophila. In: Dubnau J (ed). Behavioral Genetics of the Fly (Drosophila Melanogaster). Cambridge University Press: Cambridge, 2014, 177-82.
- 185.Jones MA, Grotewiel M. Drosophila as a model for age-related impairment in locomotor and other behaviors. Exp Gerontol. 2011;46:320–5. doi: 10.1016/j.exger.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iliadi KG, Boulianne GL. Age-related behavioral changes in Drosophila. Ann N. Y Acad Sci. 2010;1197:9–18. doi: 10.1111/j.1749-6632.2009.05372.x. [DOI] [PubMed] [Google Scholar]
- 187.Giebultowicz JM, Long DM. Aging and circadian rhythms. Curr Opin Insect Sci. 2015;7:82–86. doi: 10.1016/j.cois.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Brown MK, Chan MT, Zimmerman JE, Pack AI, Jackson NE, Naidoo N. Aging induced endoplasmic reticulum stress alters sleep and sleep homeostasis. Neurobiol Aging. 2014;35:1431–41. doi: 10.1016/j.neurobiolaging.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Miller MS, Lekkas P, Braddock JM, Farman GP, Ballif BA, Irving TC, et al. Aging enhances indirect flight muscle fiber performance yet decreases flight ability in Drosophila. Biophys J. 2008;95:2391–401. doi: 10.1529/biophysj.108.130005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wessells RJ, Bodmer R. Cardiac aging. Semin Cell Dev Biol. 2007;18:111–6. doi: 10.1016/j.semcdb.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 191.Ocorr K, Akasaka T, Bodmer R. Age-related cardiac disease model of Drosophila. Mech Ageing Dev. 2007;128:112–6. doi: 10.1016/j.mad.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tarkhov AE, Alla R, Ayyadevara S, Pyatnitskiy M, Menshikov LI, Shmookler Reis RJ, et al. A universal transcriptomic signature of age reveals the temporal scaling of Caenorhabditis elegans aging trajectories. Sci Rep. 2019;9:7368. doi: 10.1038/s41598-019-43075-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kaletsky R, Murphy CT. Transcriptional Profiling of C. elegans Adult Cells and Tissues with Age. Methods Mol Biol. 2020;2144:177–86. doi: 10.1007/978-1-0716-0592-9_16. [DOI] [PubMed] [Google Scholar]
- 194.Kaletsky R, Yao V, Williams A, Runnels AM, Tadych A, Zhou S, et al. Transcriptome analysis of adult Caenorhabditis elegans cells reveals tissue-specific gene and isoform expression. PLoS Genet. 2018;14:e1007559. doi: 10.1371/journal.pgen.1007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell. 2015;161:919–32. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Narayan V, Ly T, Pourkarimi E, Murillo AB, Gartner A, Lamond AI, et al. Deep proteome analysis identifies age-related processes in C. elegans. Cell Syst. 2016;3:144–59. doi: 10.1016/j.cels.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yasuda K, Ishii T, Suda H, Akatsuka A, Hartman PS, Goto S, et al. Age-related changes of mitochondrial structure and function in Caenorhabditis elegans. Mech Ageing Dev. 2006;127:763–70. doi: 10.1016/j.mad.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 198.Pickett CL, Dietrich N, Chen J, Xiong C, Kornfeld K. Mated progeny production is a biomarker of aging in Caenorhabditis elegans. G3. 2013;3:2219–32. doi: 10.1534/g3.113.008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.McGee MD, Day N, Graham J, Melov S. cep-1/p53-dependent dysplastic pathology of the aging C. elegans gonad. Aging. 2012;4:256–69. doi: 10.18632/aging.100448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Scharf A, Pohl F, Egan BM, Kocsisova Z, Kornfeld K, Reproductive aging in caenorhabditis elegans: from molecules to ecology. Front. Cell Dev Biol. 2021;9. [DOI] [PMC free article] [PubMed]
- 201.Glenn CF, Chow DK, David L, Cooke CA, Gami MS, Iser WB, et al. Behavioral deficits during early stages of aging in Caenorhabditis elegans result from locomotory deficits possibly linked to muscle frailty. J Gerontol A Biol Sci Med Sci. 2004;59:1251–60. doi: 10.1093/gerona/59.12.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–14. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 203.Chow DK, Glenn CF, Johnston JL, Goldberg IG, Wolkow CA. Sarcopenia in the Caenorhabditis elegans pharynx correlates with muscle contraction rate over lifespan. Exp Gerontol. 2006;41:252–60. doi: 10.1016/j.exger.2005.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pan CL, Peng CY, Chen CH, McIntire S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc Natl Acad Sci USA. 2011;108:9274–9. doi: 10.1073/pnas.1011711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tank EM, Rodgers KE, Kenyon C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J Neurosci. 2011;31:9279–88. doi: 10.1523/JNEUROSCI.6606-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Toth ML, Melentijevic I, Shah L, Bhatia A, Lu K, Talwar A, et al. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J Neurosci. 2012;32:8778–90. doi: 10.1523/JNEUROSCI.1494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hahm JH, Kim S, DiLoreto R, Shi C, Lee SJ, Murphy CT, et al. C. elegans maximum velocity correlates with healthspan and is maintained in worms with an insulin receptor mutation. Nat Commun. 2015;6:8919. doi: 10.1038/ncomms9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hsu AL, Feng Z, Hsieh MY, Xu XZ. Identification by machine vision of the rate of motor activity decline as a lifespan predictor in C. elegans. Neurobiol Aging. 2009;30:1498–503. doi: 10.1016/j.neurobiolaging.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kauffman AL, Ashraf JM, Corces-Zimmerman MR, Landis JN, Murphy CT. Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol. 2010;8:e1000372. doi: 10.1371/journal.pbio.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Son HG, Altintas O, Kim EJE, Kwon S, Lee SV. Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell. 2019;18:e12853. doi: 10.1111/acel.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mészáros L, Hoffmann A, Wihan J, Winkler J. Current symptomatic and disease-modifying treatments in multiple system atrophy. Int J Mol Sci. 2020;21:2775. doi: 10.3390/ijms21082775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, et al. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat Rev Drug Disco. 2010;9:560–74. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 213.Cummings J, Fox N. Defining disease-modifying therapy for Alzheimer’s disease. J Prev Alzheimers Dis. 2017;4:109–15. doi: 10.14283/jpad.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Symptomatic vs. Disease-modifying therapies. In: Espay A, Stecher B (eds). Brain Fables: The Hidden History of Neurodegenerative Diseases and a Blueprint to Conquer Them. Cambridge University Press: Cambridge, 2020, 87–94.
- 215.Richardson AM, R. Mechanism of food restriction: change of rate or change of set point. in The potential for nutritional modulation of aging processes(eds Ingram, DK, Baker, GT & Shock, NW) (Food & Nutrition Press) 1992:177–92.
- 216.Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 217.Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc Natl Acad Sci USA. 1997;94:13215–20. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci USA. 2001;98:6736–41. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology. 2000;141:2608–13. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- 220.Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–7. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 221.Dawlaty M, Baker D, Malureanu L, Jeganathan K, van Deursen J. BubR1 overexpression protects against tumorigenesis in mice. Cancer Res. 2008;68:LB-263–LB-263. [Google Scholar]
- 222.Qian Y, Berryman DE, Basu R, List EO, Okada S, Young JA, et al. Mice with gene alterations in the GH and IGF family. Pituitary 2022;25:1–51 10.1007/s11102-021-01191-y. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.