

Previous studies have noted that atrophic gastritis is the pathological finding most-correlated with the development of gastric adenocarcinoma [1]. Worldwide, the most common cause of atrophic gastritis is chronic infection with H. pylori. This general loss of acid secreting parietal cells is associated with the development in the corpus of metaplastic lineages including pyloric metaplasia (also known as spasmolytic-polypeptide-expressing metaplasia (SPEM) or pseudopyloric metaplasia) and intestinal metaplasia as direct sequelae of atrophy. The intestinal metaplasia lineages can develop in both the corpus and the antrum. In contrast with H. pylori infection, direct destruction of parietal cells through the production of anti-parietal cell antibodies (most prominently antibodies against the H/K-ATPase) in patients with autoimmune gastritis induces profound atrophy in the corpus, sparing the antrum. While it has been known that autoimmune gastritis is associated with a higher incidence of ECL cell carcinoids in the stomach [2], the risk for adenocarcinoma has been controversial [3, 4, 5]. Many studies have failed to discriminate between cancer arising from H. pylori infection and that emanating from the primary results of autoimmune gastritis. This has led to confusion in how these patients should be followed with endoscopy, especially in younger patients.
In this issue of Gut, Massimo Rugge and colleagues present the results of an important study which clarifies the relationship of autoimmune gastritis with gastric adenocarcinoma versus carcinoid [6]. This study, which followed prospectively 211 patients with autoimmune gastritis, but without H. pylori infection, definitively demonstrates that autoimmune gastritis, on its own, is not a significant precursor for gastric adenocarcinoma. Rather, as reported previously, Rugge, et al. demonstrate that autoimmune gastritis leads to an increased incidence of ECL cell carcinoids in the stomach. These studies provide the most definitive data to date that, in the absence of H. pylori infection, the risk of adenocarcinoma is not significant for autoimmune gastritis patients.
If atrophic gastritis with extensive loss of parietal cells is considered as pre-carcinogenic, why do the H. pylori negative autoimmune gastritis patients fail to demonstrate increases in adenocarcinoma? The answers may lie in the precancerous milieu and its influence on gastric lineages. The present investigation reports that pyloric or pseudopyloric metaplasia was far more commonly observed in autoimmune gastritis patients than intestinal metaplasia. This finding is supported by previous investigations [7, 8]. Since pyloric metaplasia/SPEM is considered a direct response to significant gastric mucosal injury [9], these lineages would be considered predominantly reparative. Increasing evidence suggests that pyloric metaplasia gives rise to intestinal metaplasia in the corpus of the stomach (Figure 1). Glands with intestinal metaplasia can be further sub-classified as either incomplete intestinal metaplasia (containing both intestinal lineages and SPEM lineages) or incomplete intestinal metaplasia (containing absorptive and Paneth cell lineages). Incomplete intestinal metaplasia is considered the lesion with the highest risk for progression to adenocarcinoma [10]. The present study does not report whether the intestinal metaplasia observed in autoimmune gastritis patients was complete or incomplete intestinal metaplasia. Nevertheless, a previous study of 20 H. pylori-negative autoimmune thyroiditis/gastritis patients found only complete intestinal metaplasia in these patients, compared with patients with gastric adenocarcinoma-associated chronic gastritis, who predominantly demonstrated incomplete intestinal metaplasia. Since incomplete intestinal metaplasia is most highly associated with risk for gastric adenocarcinoma [10], it seems more likely that the stomachs of autoimmune gastritis patients are mostly populated with pyloric metaplasia and complete intestinal metaplasia (Figure 1). In rodent studies, treatment for up to a year with the parietal cell-specific toxic drug DMP-777, which causes profound parietal cell loss without inciting a significant immune infiltrate, induced prominent SPEM without the development of intestinal metaplasia or dysplasia [11]. It thus seems that the presence of a pro-carcinogenic immune infiltrate may be needed. In mouse studies, M2-macrophages have been recognized as critical for the promotion of metaplasia and progression towards dysplasia [12, 13]. It is therefore important that, while prominent macrophage infiltrates were observed in the metaplastic mucosa of adenocarcinoma patients with chronic H. pylori-associated gastritis, significantly fewer macrophages were found in the atrophic mucosa of autoimmune gastritis patients without H. pylori [7]. Thus, while autoimmune gastritis patients certainly demonstrate prominent lymphocytic infiltrates, the lack of macrophages may lead to a more benign pattern of metaplasia and abrogate against the increased risk for adenocarcinoma. Why chronic loss of parietal cells from H. pylori infection versus anti-parietal cell antibodies in autoimmune gastritis would elicit different immune responses remains a mystery.
Figure 1: Comparison of lineages changes in H. pylori-associated and autoimmune gastritis-associated atrophic gastritis.
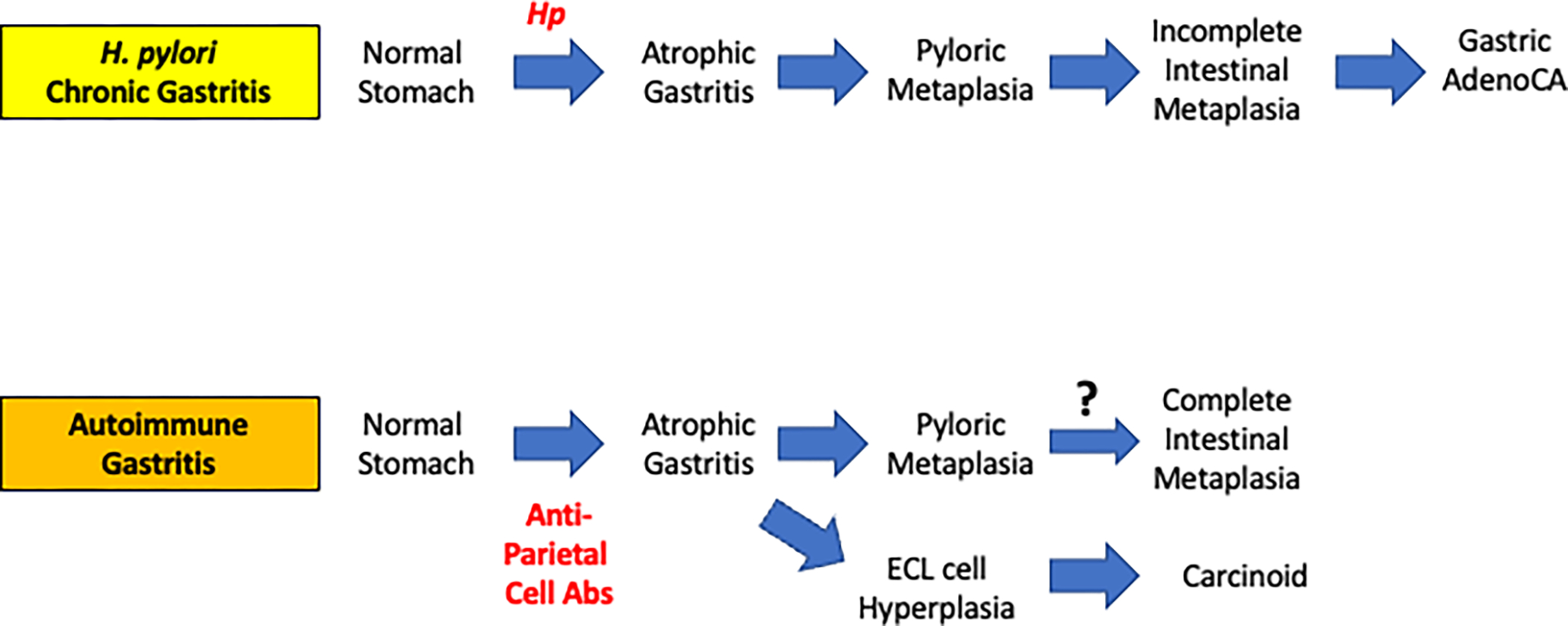
Chronic H. pylori (Hp) infection induces parietal cell loss (atrophic gastritis) followed by induction of pyloric metaplasia. Continued inflammation induces incomplete intestinal metaplasia, which can then develop into gastric adenocarcinoma. In autoimmune gastritis patients, anti-parietal cell antibodies cause atrophic gastritis. Loss of parietal cells leads to subsequent induction of pyloric metaplasia which may progress to complete intestinal metaplasia. Additionally, the atrophic milieu in autoimmune gastritis patients can promote the evolution of ECL cell hyperplasia and eventually carcinoid tumors.
While autoimmune gastritis may not increase risk of gastric adenocarcinoma, it seems likely that autoimmune gastritis coincident with chronic H. pylori infection might provide excess risk for adenocarcinoma. Furthermore, the autoimmune gastritis patients without H. pylori infection demonstrate a continued increased risk for carcinoid tumor development (Figure 1). The results presented by Rugge, et al. demonstrate a prominent progression of atrophy and ECL cell hyperplasia to enteroendocrine cell dysplasia over time. While many have speculated that elevated levels of gastrin may be responsible for carcinoid tumor development from histamine-secreting ECL cells, the exact mechanisms driving this transition remain elusive. Mouse models of autoimmune parietal cell targeted destruction have shown prominent oxyntic atrophy associated with induction of SPEM, but the ability of these models to replicate the ECL cell carcinoids observed in humans remains unclear [14]. Thus, surveillance endoscopy remains important in autoimmune gastritis patients to identify possible progression of ECL cell hyperplasia in atrophic gastric mucosa to frank carcinoid tumors [15]. The study of Rugge, et al. definitively refocuses the care of autoimmune gastritis to eradication of H. pylori when present, and screening endoscopy for progression of lesions towards carcinoid neoplasia.
Acknowledgements:
JRG is supported by grants from a Department of Veterans Affairs Merit Review Award IBX000930, NIH RO1 DK101332 and DOD CA160479.
REFERENCES
- 1.El-Zimaity HMT, Ota H, Graham DY, et al. Patterns of gastric atrophy in intestinal type gastric carcinoma. Cancer 2002;94:1428–36. [DOI] [PubMed] [Google Scholar]
- 2.Borch K, Renvall H, Liedberg G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology 1985;88:638–48. [DOI] [PubMed] [Google Scholar]
- 3.Vannella L, Lahner E, Annibale B. Risk for gastric neoplasias in patients with chronic atrophic gastritis: a critical reappraisal. World J Gastroenterol 2012;18:1279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song M, Camargo MC, Derkach A, et al. Associations between autoimmune conditions and gastric cancer risk among elderly adults in the United States. Am J Gastroenterol 2022;117:486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butt J, Lehtinen M, Öhman H, et al. Association of Helicobacter pylori and autoimmune gastritis with stomach cancer in a cohort of young Finnish women. Gastroenterology 2022;163:305–7. [DOI] [PubMed] [Google Scholar]
- 6.Rugge M, Bricca L, Guzzinati S, et al. Autoimmune gastritis: long-term natural history in naïve Helicobacter pylori -negative patients. Gut 2022;53. doi: 10.1136/gutjnl-2022-327827. [Epub ahead of print: 30 Jun 2022]. [DOI] [PubMed] [Google Scholar]
- 7.Jeong S, Choi E, Petersen CP, et al. Distinct metaplastic and inflammatory phenotypes in autoimmune and adenocarcinoma-associated chronic atrophic gastritis. United European Gastroenterol J 2017;5:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wada Y, Nakajima S, Kushima R, et al. Pyloric, pseudopyloric, and spasmolytic polypeptideexpressing metaplasias in autoimmune gastritis: a case series of 22 Japanese patients. Virchows Arch 2021;479:169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldenring JR, Mills JC, Plasticity C. Cellular plasticity, reprogramming, and regeneration: metaplasia in the stomach and beyond. Gastroenterology 2022;162:415–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao L, Li P, Ye J, et al. Risk of gastric cancer among patients with gastric intestinal metaplasia. Int J Cancer 2018;143:1671–7. [DOI] [PubMed] [Google Scholar]
- 11.Goldenring JR, Ray GS, Coffey RJ, et al. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology 2000;118:1080–93. [DOI] [PubMed] [Google Scholar]
- 12.Petersen CP, Weis VG, Nam KT, et al. Macrophages promote progression of spasmolytic polypeptideexpressing metaplasia after acute loss of parietal cells. Gastroenterology 2014;146:1727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi E, Hendley AM, Bailey JM, et al. Expression of activated Ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology 2016;150:918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bockerstett KA, Osaki LH, Petersen CP, et al. Interleukin-17A promotes parietal cell atrophy by inducing apoptosis. Cell Mol Gastroenterol Hepatol 2018;5:678–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah SC, Piazuelo MB, Kuipers EJ, et al. AGA clinical practice update on the diagnosis and management of atrophic gastritis: expert review. Gastroenterology 2021;161:1325–32. [DOI] [PMC free article] [PubMed] [Google Scholar]