

Abstract
Purpose:
Diffuse gastric cancer (DGC) is an aggressive and frequently lethal subtype of gastric cancer (GC). Because DGC often lacks genomic aberrations that indicate clear candidate therapeutic targets, it has been challenging to develop targeted therapies for this gastric cancer subtype. Our previous study highlighted the contribution of focal adhesion kinase (FAK) in the tumorigenesis of DGC and the potential efficacy of small-molecule FAK inhibitors. However, drug resistance to monotherapy often hinders the efficacy of treatment.
Experimental Design:
We generated a genome-scale library of open reading frames (ORF) in the DGC model of Cdh1−/−RHOAY42C/+ organoids to identify candidate mechanisms of resistance to FAK inhibition. Compensatory activated pathways were also detected following treatment with FAK inhibitors. Candidates were investigated by co-targeting in vitro and in vivo experiments using DGC.
Results:
We found that cyclin-dependent kinase 6 (CDK6) promoted FAK inhibitor resistance in ORF screen. In addition, FAK inhibitor treatment in DGC models led to compensatory MAPK pathway activation. Small molecule CDK4/6 inhibitors or MAPK inhibitors effectively enhanced FAK inhibitor efficacy in vitro and in vivo.
Conclusions:
Our data suggest that FAK inhibitors combined with MAPK inhibitors or CDK4/6 inhibitors warrant further testing in clinical trials for DGC.
Keywords: Diffuse gastric cancer, FAK inhibitor, MAPK inhibitor, CDK4/6 inhibitor, drug combination
Introduction
Gastric cancer (GC) is the fourth leading cause of cancer-related deaths worldwide(1). The Lauren classification, which is based on histology, divides gastric cancer into two main histological types: intestinal gastric cancer (IGC) and diffuse gastric cancer (DGC)(2). DGC is characterized by a highly invasive growth pattern, whereby tumor cells are poorly differentiated and lack cellular adhesion, thus contributing to rapid local invasion and peritoneal metastases(3). According to the genomic/molecular characterization of gastric cancer, DGCs largely fall into the genomically stable (GS) molecular group, which usually lacks conventional hypermutation, harbors less profound chromosomal instability, and has few classic targetable activated oncoproteins(4). The absence of mutations in oncoproteins has slowed the therapeutic development of DGC. Thus, new treatment methods and targets must be developed for patients with DGC.
We previously explored the mechanisms of tumorigenesis of DGC in the context of recently identified recurrent mutations in the RHOA GTPase(5). We demonstrated that engineered murine gastric organoids combining Cdh1 (E-cadherin) loss and RHOA Y42C mutation work together to activate focal adhesion kinase (FAK) and downstream AKT-β-catenin and YAP-TAZ pathways, which transform normal gastric epithelial cells to DGC. We nominated FAK as a potential therapeutic target for DGC(5). FAK has multifaceted functions in cancer, as it is both a non-receptor tyrosine kinase and kinase-independent scaffold. Canonical activation of FAK begins with cell adhesion signaling. When activated by integrins, inactive FAK forms dimers, leading to autophosphorylation of tyrosine 397 (Y397) site(6). SRC family kinases are recruited to phosphorylate the Y576 and Y577 sites of FAK and form the FAK-SRC complex, leading to the full activation of FAK(6,7). Activated FAK regulates multiple biological functions, including the survival, migration, and invasion of cancer cells(7). Autophosphorylation of FAK at the Y397 site is a key step in activation(6,7).
FAK inhibitors have been evaluated in several types of cancer. FAK monotherapy has been tolerated in the clinic but has shown only modest clinical efficacy in cancer treatment(8–10), which inspired us to explore targeted therapeutics in combination with FAK inhibitors in DGC. To this end, we performed a human genome-scale ORF screen and found that CDK6 overexpression promotes FAK inhibitor resistance. We also identified compensatory activation of the MAPK pathway in DGC models when treated with FAK inhibitors. We further demonstrated that FAK inhibitors combined with CDK4/6 or MAPK inhibitors may be promising therapeutic strategies for this lethal malignant tumor.
Materials and methods
Cell lines and organoids culture
HEK293T cells were obtained from (American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin. SNU668 and NUGC4 cells were obtained from the Broad Institute Cell Line Factory and maintained in RPMI-1640 supplemented with 10% fetal bovine serum, penicillin, and streptomycin. All cell lines were maintained in a humidified incubator with 5% CO2 at 37°C. Mycoplasma contamination was queried regularly using a Lonza MycoAlert Mycoplasma Detection Kit.
Cdh1−/−RHOAY42C/+ organoids were generated as previously described(5). Two patient-derived organoids (PDOs) of human DGC were obtained from S. Ryeom (Columbia University) after having been generated from patients with GC under an IRB approved protocol. All patients provided written informed consent, and the studies were conducted in accordance with the Declaration of Helsinki. For passaging, organoids were dissociated using TrypLE Express Enzyme (Gibco) at 37 °C for 6–8 min. Cells (2 × 105) were mixed with 210 μl Matrigel (Corning, 354234) on ice. Seven aliquots of the cell-Matrigel suspension (30 μl per aliquot) were seeded in 6-well plates. To polymerize the Matrigel, plates were incubated at 37 °C and upside down to avoid the attachment of cells to the plate surface. After 3–5 min, plates were returned to the upright orientation. For Cdh1−/−RHOAY42C/+ organoids, 2.5 ml 50% L-WRN conditioned medium(11) (a 1:1 mix of L-WRN conditioned medium and advanced DMEM/F-12 with 20% FBS) were added. The medium for PDOs was described as before(12). Cdh1−/−RHOAY42C/+ organoids were passaged every three days. PDOs were passaged every seven to ten days based upon growth dynamics. Mycoplasma tests (Lonza, Basel, Switzerland) were performed regularly.
Antibodies and drugs
See Supplemental Table S1, S2.
ORFeome Library Titration
Accurate virus volumes to be used in the screen were determined in Cdh1−/RHOAY42C/+ organoids to achieve 30% to 40% infection efficiency. Human ORFeome virus was obtained from the Broad Institute. Cdh1−/−RHOAY42C/+ organoids were dissociated using TrypLE Express enzyme (Gibco). Spinfection was performed in 12well plates containing 1 × 106 or 2 × 106 single cells of Cdh1−/−RHOAY42C/+ per well, with different virus volumes (0, 100, 200, 300, 400, and 500 μl), and a final concentration of 8 μg/mL polybrene with 10 μM Y27632. Cells were spin-infected for 1 h at 600 g, 30 °C, and then incubated at 37 °C for 6–8 h. After infection, the cells were detached using TrypLE and resuspended in matrigel. The cells were then seeded into duplicate wells of 6-well plates. Two days after seeding, the medium was changed, one well as a control, and another well treated with 1 μg/ml puromycin for 2 days. Next, organoids were passaged and selected for another 2 days with 1 μg/ml puromycin. After a total of 4-day puromycin selection, the cells in the control and puromycin-selected groups were counted. The virus volume and cell amount in 12well plates for the spinfection that yielded 30% to 40% infection efficiency, as inferred by survival with puromycin selection, were used for large-scale screening. In this study, 2 × 106 Cdh1−/−RHOAY42C/+ cells and 200 μl virus per well in a 12-well plate were used for experimental infections.
Genome-Scale ORF Screens
The ORFeome barcoded library contains 17,255 barcoded ORFs overexpressing 10,135 distinct human genes with at least 99% nucleotide and protein matches. Screening-scale infections of the ORFeome library were performed with sufficient cells to achieve a representation of at least 1,000 cells per ORF (approximately 2 × 107 surviving cells containing 17,255 ORFs). Infections were performed with the predetermined cell amount and virus volume in 12-well plates using the viral titration described above. After puromycin selection, ETP (early time point) samples were harvested. Next, organoids were divided into drug-treated (2.5 μM defactinib or 2 μM PF-573228) and DMSO-treated arms, with 3 × 107 cells in each arm. Organoids in the DMSO arm were passaged every 3 days. Organoids in the drug-treated arms were passaged every 5–6 days. The indicated drugs or DMSO were added 24 h after the passage. Throughout the screen, we maintained an average representation of 1,000 cells per ORF construct. Samples at the late time point were harvested 20 d after the initiation of treatment.
gDNA was extracted using the phenol-chloroform extraction method. Briefly, the cell pellet was mixed with 5 ml cell lysis (400mM NaCl; 0.2% SDS; 2mM EDTA; 10mM Tris-HCI in ddH2O, pH 8.0). Proteinase K (10mg/ml, 1:100) was added to the mixture and incubated at 55 °C overnight. Next RNase A (10mg/ml, 1:100) was added to the mixture and incubated at 65 °C for 1 h. After digestion, the mixture was aliquoted into gel tubes (phase-lock gel light) with 700 μl cell lysate per tube. An equal volume of phenol/chloroform (Phenol:CHCl:lAA=25:24:1, pH =7.9±0.2, Invitrogen, AM9730) was added to the cell lysate, vortexed vigorously, and incubated for 10 min at room temperature. We centrifuged the gel tube at 12,000–14,000 rpm for 15 min at room temperature and then transferred the supernatant (700 μl) to a 1.5 ml clean Eppendorf tube. Next, 700 μl isopropanol was added to the Eppendorf tube, vortexed vigorously, and centrifuge at 12,000–14,000 rpm for 15 min at room temperature. The supernatant was discarded and the gDNA pellet was washed with 1 ml 70% ethanol, centrifuged at 12,000–14,000 rpm for 5 min at room temperature, the supernatant was removed, and the gDNA pellet was air dried for 15 min. Finally, gDNA was resuspended in 100 μl ultra-pure water. PCR and sequencing were performed at the Broad Institute. The read counts were normalized to reads per million and log2 transformed. The log2 fold change (FC) of each ORF was determined relative to the early time points. Two replicates were performed for the ETP and end-time point samples.
CDK6 overexpression
The lentiviral pHAGE-CDK6 (human) vector (#116725) and empty pHAGE vector (#24526) were obtained from Addgene. The lentiviruses were generated using standard protocols(13). For lentiviral infection of cells grown in a 6 cm dish, SNU668 cells were transduced with 3 ml of viral particle-containing medium with 8 μg/ml polybrene. After incubation for 8 h, the medium was replaced. After 3 days, the infected GFP-positive cells were sorted using a cell sorter (BD FACS Aria II). Protein lysates were collected to confirm plasmid expression by immunoblotting.
Western blot for drug-treated organoids
The organoids were plated and treated with the indicated drugs 24 h later. For organoid collection, Matrigel surrounding the organoids was dissolved using Cell Recovery Solution (Corning) on ice. The released organoids were pelleted and lysed for immunoblotting as previously described(14). The antibodies used are listed in Supplemental Table S1.
Histology and Immunohistochemistry
For H&E staining, organoids were fixed in 10% formalin overnight, during which Matrigel was dissolved. Organoids were pelleted, resuspended in 2% agar, and embedded in paraffin. Standard protocols for sectioning paraffin-embedded tissues and hematoxylin and eosin staining were used. For immunohistochemistry (IHC) staining of xenograft samples, xenograft tumors were excised, fixed with 10% formalin overnight, and embedded in paraffin (FFPE). Unstained sections were stained with the antibodies listed in Supplemental Table S1. A TUNEL staining kit (MERCK, S7101) was used according to the manufacturer’s instructions. The staining was evaluated by an experienced pathologist (Y. Wang).
Flow cytometry for cell cycle analysis
For cell cycle analysis, cells were harvested after treatment with DMSO or inhibitors for 24 or 48h and were then fixed with 70% ethanol at 4 °C for 30 min. Fixed cells were washed with PBS containing 1% FBS and stained with PI/RNase staining solution (Cell Signaling Technologies, 4087). The samples were incubated at room temperature for 30 min in the dark and then analyzed using a cell analyzer (BD LSR Fortessa). The data were analyzed using ModFit LT.
Crystal violet
To detect the effect of the drugs on cell lines, 5 × 104 cells/well NUGC4 or 2 × 104 cells/well SNU668 cells were plated in 6-well plates and treated with the indicated inhibitors or DMSO 24 h after seeding. Fresh media, including DMSO or drugs, were changed every 3–4 days. After 7–10 days of treatment, when the control cells grew confluent, they were stained with 0.1% crystal violet for 5–10 min at room temperature. The plates were then washed with PBS and dried prior to scanning using a scanner (Epson Perfection V600 Photo).
Digital Western Blot (DigiWest)
Cdh1−/−RHOAY42C/+ organoids were plated in a 6-well plate. The next day, the organoids were treated with 2.5 μM defactinib, 2.5 μM PF-573228, or DMSO. After 48 h of treatment, the Matrigel surrounding the organoids was removed using Cell Recovery Solution (Corning) on ice. The organoids were pelleted. All samples were shipped to NMI TT Pharmaservices for further analysis, as previously described(15).
Receptor Tyrosine Kinase Phospho-array
Cdh1−/−RHOAY42C/+ organoids were seeded into a 6-well plate. After 24 h, the organoids were treated with 2.5 μM defactinib, 2.5 μM PF-573228, or DMSO. Organoids were harvested using the Cell Recovery Solution (Corning) after 48 h of treatment. These samples were analyzed using the Proteome Profiler Mouse PhosphoRTK Array Kit (R&D, ARY014) according to the manufacturer’s instructions.
RAS G-LISA assay
RAS-GTP, including HRAS, KRAS, and NRAS, was detected using the Ras GLISA Activation Assay Kit (Cytoskeleton, BK131), according to the manufacturer’s instructions. Briefly, 12.5 μg of whole-cell lysates was added in duplicate in a 96-well plate, and RAS-GTP was bound to an RAS–GTP binding protein linked to each well. Bound active RAS was detected with an RAS (HRAS, KRAS, and NRAS) specific antibody and quantified by measuring the relative absorbance at 490 nM using a TECAN plate reader.
Cell viability assay
Cell viability was measured using the CellTiter-Glo luminescent cell viability assay kit (Promega, G7570) according to the manufacturer’s instructions. Briefly, 1000 cells/well were plated in a 96-well plate. After 24 h, the cells were treated with either the vehicle (DMSO) or the indicated inhibitors. After treatment for the indicated number of days, 50 μL CellTiter-Glo reagent was added to each well. The plates were shaken for 2 min and allowed to rest for 10 min. Luminescence was detected using a Tecan plate reader. In organoids, cell viability was measured using CellTiter-Glo 3D Cell Viability Assay (Promega, G9681). One thousand cells mixed with 5 μl Matrigel were seeded into each well of a 96-well plate, and 100 μl corresponding media was added. After 24 h, organoids were treated with the indicated drugs. After treatment for the indicated days, media in the plate were aspirated, and a mixture of 25 μL CellTiter-Glo 3D reagent and 50 μL 50% L-WRN conditioned media was added to each well. The plate was shaken for 30 min and allowed to rest for 10 min prior to luminescence detection.
The synergy analysis was performed using the zero interaction potency (ZIP) model(16). Briefly, five different concentrations of each drug (0.25, 0.5, 1, 2, and 4 IC50) were administered, either alone or in combination. After the indicated number of days of treatment, cell viability was measured using the CellTiter-Glo assay. The results were analyzed using SynergyFinder(17).
In vivo experiments
All animal experiments were conducted in accordance with the Institutional Animal Care and Use Committee–approved animal protocols at the Dana-Farber Cancer Institute in compliance with NIH guidelines. NSG mice were obtained from Jackson Laboratory. NUGC4 and SNU668 cell lines were detected as pathogen-free and cultured in RPMI-1640 supplemented with 10% FBS. Cells were washed with serum free medium and resuspended in serum-free medium mixed with an equal amount of Matrigel (356231; Corning). Mice were injected with 2*10^6 of NUGC4 or SNU668 cells per injection, with two distinct injections in the flank of each mouse. Tumors were monitored twice weekly using electronic calipers, and tumor volumes were calculated using the formula: volume=0.5 × length × width2. When the tumor volume reached approximately 100 mm3, mice were treated with the indicated drugs. VS4718 was dissolved in 0.5% carboxymethyl cellulose (CMC) with 0.1% Tween 80. VS-6766 was dissolved in 10% (2-Hydroxypropyl)-β-CD (HPCD) in 5% DMSO. Palbociclib was dissolved in HPCD (17%). VS-4718 (50 mg/kg) was administered twice a day. VS-6766 (0.3 mg/kg) and palbociclib (50 mg/kg) were administered once per day. All drugs were administered via oral gavage.
Statistical analysis
Data are presented as mean ± S.D. or S.E.M., as indicated in the figure legends. The number of independent biological experiments for each experiment is noted in the figure legends. Statistical analysis was performed using Prism 9.0 (GraphPad). Comparisons between groups were performed using an unpaired two-tailed Student’s t-test or two-way ANOVA, as appropriate. P<0.05. P-values are denoted by * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001. The sample sizes and animal numbers were determined from pilot laboratory experiments and previously published studies. Animals were excluded from the analysis if they were euthanized for health reasons unrelated to tumor growth. For the in vivo experiments, all mice were randomized before drug treatment.
Data Availability
The data generated in this study are available in the article and its supplementary files. Any additional results can be obtained from the corresponding authors.
Result
Genome-scale ORF screen in Cdh1−/−RHOAY42C/+ organoids identifies CDK6 as driver of FAK inhibitors resistance
To find candidates to target in combination with FAK inhibitors, we first sought to evaluate the potential mechanisms of resistance of DGC to small-molecule FAK inhibitors. We performed a genome-scale open reading frame (ORF) screen and explored the genes or pathways driving FAK inhibitor resistance. In our previous study, we constructed a DGC model of engineered murine Cdh1−/−RHOAY42C/+ organoids containing typical signet-ring cells that recapitulate the phenotype of DGC(5). This prior study revealed the contribution of FAK to the tumorigenesis of Cdh1−/−RHOAY42C/+ organoids(5), making it a good DGC model to explore the mechanism of FAK inhibitor resistance. Given that this model recapitulates the pathological features of the disease, we used Cdh1−/−RHOAY42C/+ organoids to perform this ORF screening despite the technical challenges of screening organoids.
Cdh1−/−RHOAY42C/+ organoids were infected with a pooled lentiviral ORF library containing 17,255 barcoded ORFs, leading to overexpression of 10,135 corresponding human genes. Infected organoids were selected using puromycin and an early time point (ETP) sample was collected. ORF constructs infected Cdh1−/−RHOAY42C/+ organoids were treated in parallel with one of two FAK inhibitors, 2.5 μM defactinib, 2 μM PF-573228, or DMSO for 20 days and then harvested. ORF representation at the ETP and late time point of each treatment arm were evaluated. The log2 fold change (FC) of each ORF was determined relative to ETP (Figure 1A). The upregulated ORFs in the FAK inhibitor treatment arms relative to ETP were considered resistance mediator candidates if they had z-scores (SD from the mean) of log2(FC)≥3 (Figure 1B, C; Table S3).
Figure 1. Genome-scale lentiviral ORFeome library screen identifies drivers of FAK inhibitor resistance in DGC.
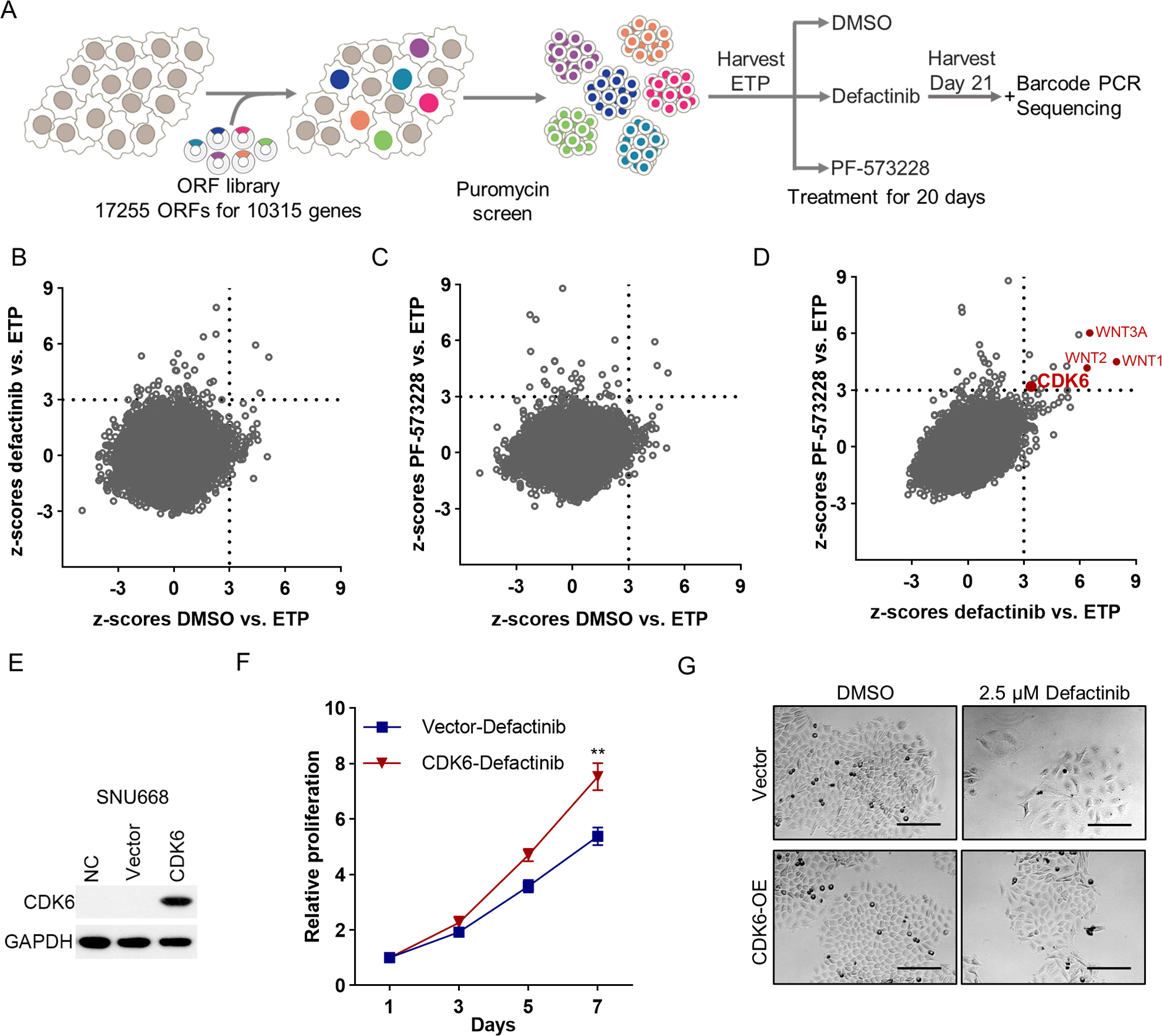
(A) Schematic description of the genome-scale ORFeome library screen. (B, C) Scatter plots presenting the z-scores of average log2(fold change) for defactinib vs. ETP (y-axis) and for DMSO vs. ETP (x-axis) (B); PF-573228 vs. ETP (y-axis) and DMSO vs. ETP (x-axis) (C) in Cdh1−/−RHOAY42C/+ organoids. Z-scores of DMSO vs. ETP < 3 nominate genes not associated with enhanced growth in the DMSO, whereas z-scores of defactinib or PF-573228 vs. ETP ≥ 3 nominate genes associated with resistance to defactinib or PF-573228. Genes with z-scores < 3 for DMSO and ≥ 3 for defactinib or PF-573228 were nominated as candidate genes conferring resistance and classified as significant ORFs. (D) Scatter plots presenting the z-scores of log2(fold change) for defactinib vs. ETP (x-axis) and for PF-573228 vs. ETP (y-axis) in Cdh1−/−RHOAY42C/+ organoids. (E) Immunoblot analysis to validate CDK6 overexpression in SNU668. (F) In vitro proliferation of control or CDK6 overexpressed SNU668 treated with 2.5 μM defactinib for indicated days. The results are the representative of three independent experiments, each done in quadruplicate. Data are mean ± S.D. **P<0.01, two-way ANOVA test. (G) Representative images of control or CDK6 overexpressed SNU668 treated with DMSO or 2.5 μM defactinib. Scale bar, 100 μm.
Eleven candidates emerged from the overlap of resistance to both defactinib and PF-573228 treatment arms. Among these, two genes (WNT9A and GPC6) were removed, as they also had a marked growth-promoting effect in the DMSO arm (Figure S1A). WNT1, WNT3A, and WNT2 were among the nine remaining top hits in the defactinib and PF-573228 arms, indicating the likely biological relevance of Wnt signaling in these tumors. Given the strength of responses to WNT overexpression, we evaluated the combination of Wnt inhibitor (XAV-939) and FAK inhibitor (defactinib) in DGC models. Synergy analysis for the combination was based on the ZIP model(16), with a ZIP synergy score>10 denoting likely synergy. The combination of XAV-939 and defactinib showed synergistic effect in Cdh1−/−RHOAY42C/+ organoids with ZIP score of 26.171 (Figure S1B). XAV-939 also promoted the efficacy of defactinib in one DGC cell line SNU668, however not in a second cell line we evaluated, NUGC4 (Figure S1C).
Notably, CDK6 was one of the top hits in both FAK inhibitor arms, but not in the DMSO arm (Figure 1D), which carries potential translational value as FDA-approved CDK4/6 inhibitors are actively used to treat breast cancer(18). Therefore, we next explored whether CDK4/6 inhibition could be a potential candidate for FAK inhibitor combination in our DGC model. To validate whether CDK6 promotes FAK inhibitor resistance, we overexpressed CDK6 in SNU668, a DGC cell line (Figure 1E). SNU668 cells overexpressing CDK6 were treated with FAK inhibitors (2.5 μM defactinib, 2.5 μM PF-573228, and 1 μM VS-4718). We found that CDK6 promoted resistance to FAK inhibitors (Figure 1F, S1D–E). Furthermore, SNU668 control cells expressing the empty vector demonstrated larger cell morphology upon FAK inhibitor treatment, which was reversed by CDK6 overexpression, suggesting that CDK6 may mitigate FAK inhibitor-induced cell morphology changes (Figure 1G, S1F).
CDK4/6 inhibition sensitizes DGC to FAK inhibition
Given the ability of CDK6 activation to promote resistance to FAK inhibition, we hypothesized that the CDK4/6 inhibitor (palbociclib) may augment the efficacy of FAK inhibitors (defactinib, PF-573228 or VS-4718, with the latter being an additional formulation similar to defactinib). We evaluated this potential in Cdh1−/−RHOAY42C/+ organoids, PDOs and two DGC cell lines, SNU668 and NUGC4. Palbociclib activity was evaluated by assessing the inhibition of Rb phosphorylation(19). We found that 0.5 μM palbociclib was sufficient to attenuate the phosphorylation of Rb (Figure 2A, S2A) and induce G0/G1 arrest in Cdh1−/−RHOAY42C/+ organoids (Figure 2B). Normal gastric organoids maintain hollow and spherical morphology. Our previous research showed that Cdh1 loss and RHOA mutation led to loss of polarity and disorganized organoid growth. Under combined treatment with FAK inhibitors (defactinib, PF-573228, or VS-4718) and palbociclib, we observed shifts in the morphology of Cdh1−/−RHOAY42C/+ organoids back to the normal hollow type (Figure 2C, S2B–C). The number of Cdh1−/RHOAY42C/+ organoids was counted after 48 h of treatment with palbociclib or FAK inhibitor monotherapy or in combination, demonstrating a significant reduction in cell numbers (Figure 2D, S2D–E). Two DGC PDOs, BL62 and DE66, were treated with defactinib and palbociclib (Figure S3A–B), again leading to significant inhibition of cell proliferation of PDO models (Figure S3C–D). The phosphorylation of Rb was inhibited (Figure 2E, S4A–B) and G0/G1 arrest was induced by 0.5 μM palbociclib (Figure 2F) in DGC cell lines, NUGC4 and SNU668. The combination of palbociclib and FAK inhibitors in NUGC4 and SNU668 cell lines, as shown by crystal violet staining, also demonstrated cooperative effects (Figure 2G, S4C–D).
Figure 2. CDK4/6 inhibitor enhanced the efficacy of FAK inhibitors.
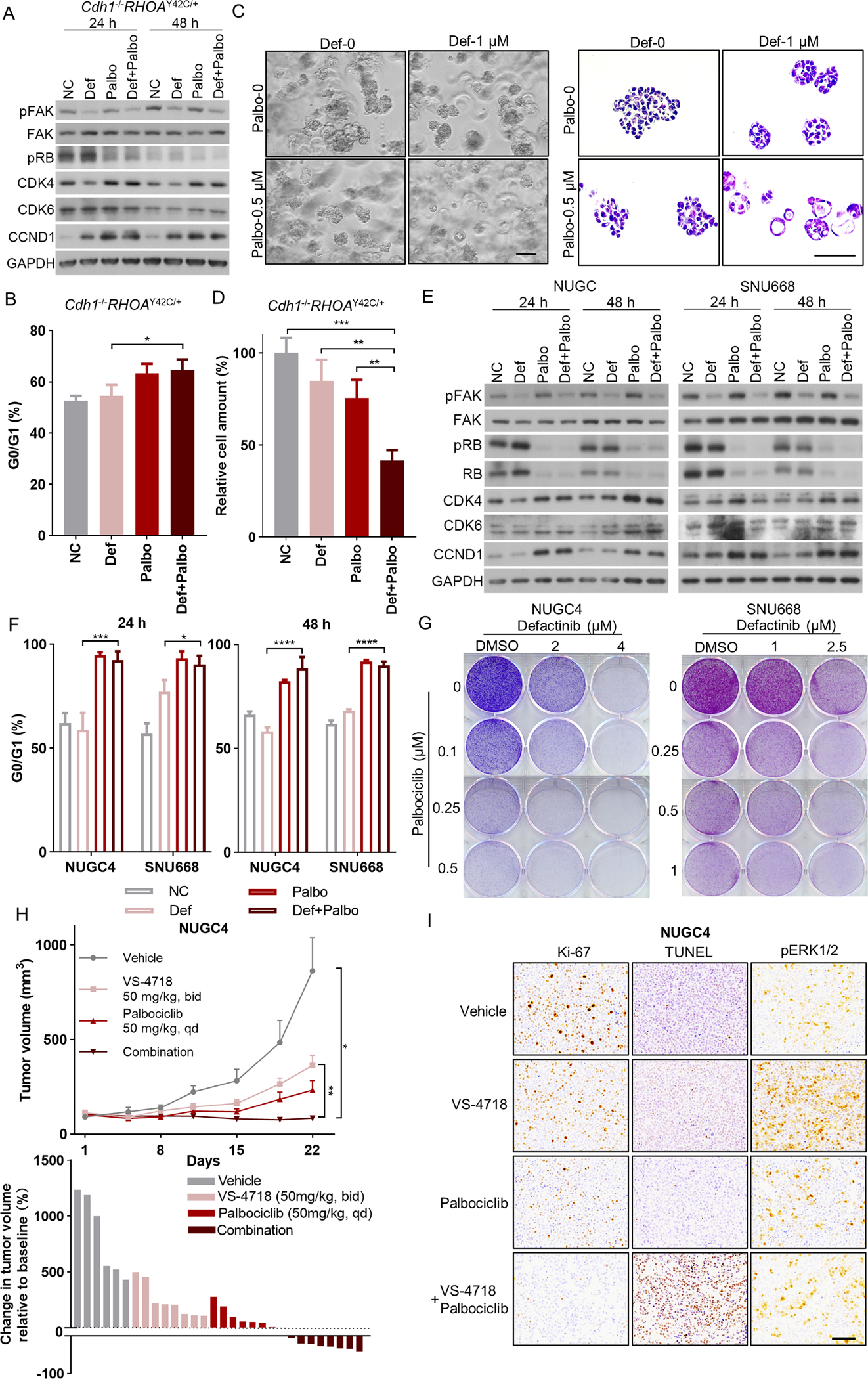
(A) Immunoblot analysis of genes involved in FAK pathway and cell-cycle pathway in Cdh1−/−RHOAY42C/+ organoids treated with 1 μM defactinib (def), 0.5 μM palbociclib (palbo), the combination or with DMSO control. Protein lysates were collected after drug treatment for 24 hours and 48 hours. Immunoblots from one representative experiment (n=2) are shown. (B) The frequency of G0/G1 cells of Cdh1−/−RHOAY42C/+ organoids treated with 24 hours of 1 μM defactinib, 0.5 μM palbociclib, the combination or with DMSO control. Following treatment, the cells were harvested, stained with propidium iodide, and assayed with flow cytometry (n=3). Data are mean ± S.D, *P<0.05, two-way ANOVA test. (C) Representative phase contrast images (left) and HE staining images (right) of Cdh1−/−RHOAY42C/+ organoids treated with 48 hours of 1 μM defactinib, 0.5 μM palbociclib, the combination or with DMSO control. Scale bar, 50 μm. (D) The plots show the relative cell amount of Cdh1−/RHOAY42C/+ organoids treated with 48 hours of 1 μM defactinib, 0.5 μM palbociclib, the combination or with DMSO control (n=3). Cell amount in each group was normalized to that in DMSO control group. Data are mean ± S.D, **P<0.01, ***P<0.001, two-way ANOVA test. (E) Immunoblot analysis of genes involved in FAK pathway and cell-cycle pathway in NUGC4 (left) and SNU668 (right) treated with 2.5 μM defactinib, 0.5 μM palbociclib, the combination or with DMSO control. Protein lysates were collected after drug treatment for 24 hours and 48 hours. Immunoblots from one representative experiment (n=2) are shown. (F) The frequency of G0/G1 cells of NUGC4 and SNU668 treated with 24 hours and 48 hours of 2.5 μM defactinib, 0.5 μM palbociclib, the combination or with DMSO control (n=3). Data are mean ± SD. *P<0.05, ***P<0.001, ****P<0.0001, two-way ANOVA test. (G) Images show representative results of colony formation assays of NUGC4 (left) and SNU668 (right). Cells were cultured in 6-well plates and treated with DMSO, indicated concentration of defactinib or palbociclib alone or together for 7 to 10 days, and then fixed and stained with crystal violet solution. All experiments were repeated at least twice. (H) Top: growth curve for NUGC4 xenograft tumors (n=6–10) treated with vehicle control, VS-4718 (50 mg/kg, bid), palbociclib (50 mg/kg, qd) or the combination. Treatment began on day1. Data are mean ± SEM. *P<0.05, **P<0.01, two-way ANOVA test. Bottom: waterfall plot showing the tumor volume change (at day 22) relative to baseline volume (at day 1). Each bar represents one xenograft tumor. (I) Representative images of Ki-67, TUNEL and pERK1/2 staining of the NUGC4 xenograft tumors. Scale bar = 100 μm.
We next evaluated the FAK inhibitor (VS-4718, given its enhanced in vivo pharmacokinetics relative to defactinib) and CDK4/6 inhibitor (palbociclib) in vivo. NUGC4 or SNU668 cells were injected into the flanks of the NSG mice. When tumors grew to approximately 100 mm3, the mice were treated with vehicle, VS-4718, palbociclib, or a combination of both drugs. During the 3-week treatment, the palbociclib and VS-4178 combination induced regression in the tumor volume of the NUGC4 xenografts (Figure 2H, Figure S5A). Although SNU668 xenograft tumors in the combination group grew slightly during treatment, they were still significantly smaller than those in the monotherapy or vehicle groups (Figure S5B). Tumor tissues from NUGC4 xenografts were obtained when the mice were sacrificed. We then performed immunohistochemistry (IHC) for Ki-67 to evaluate proliferation and TUNEL staining to evaluate apoptosis. We found that Ki-67 staining was significantly weaker and TUNEL staining was significantly stronger in the combination group than in the monotherapy group, indicating that the combination of VS-4718 and palbociclib inhibited proliferation and promoted apoptosis more effectively than monotherapy (Figure 2I, Figure S5C).
FAK inhibitors treatment in DGC models induces the activation of the MAPK pathway
In addition to the potential of CDK4/6 inhibitors in combination with FAK inhibitors, we next explored other candidates that could also improve the efficacy of FAK inhibition. As an alternative approach to identifying candidates for combinatorial therapy, we sought to evaluate the biochemical effects of FAK inhibition to determine how key cancer-relevant pathways were altered by FAK therapy in DGC. For this experiment, we used DigiWest, a high-throughput protein analysis assay(15), to investigate intracellular signaling. Cdh1−/−RHOAY42C/+ organoids were treated with defactinib, PF-573228, or DMSO, and the Digiwest assay was performed to quantify the total and phosphoprotein abundance (Table S4). While we observed attenuation of signaling related to the PI3K pathway, we found that both pERK and the total expression of CDK4 and CCND1 were upregulated, suggesting MAPK activation upon treatment with FAK inhibitors (Figure 3A). Next, we validated MAPK pathway activation by immunoblotting and observed that ERK1/2 compensatory phosphorylation was observed after 24 h of FAK inhibition in organoids (Figure 3B, S6). The DGC cell lines, NUGC4 and SNU668, also showed MAPK activation after FAK inhibitor treatment (Figure 3C–D, S6). Consistent with the in vitro experiment, in the in vivo experiment of NUGC4 xenograft, pERK1/2 staining of tumors in the VS-4718 treatment group was stronger than that in the vehicle group (Figure 2I, Figure S5C).
Figure 3. FAK inhibitors induce MAPK activation in DGC organoids and cell lines.
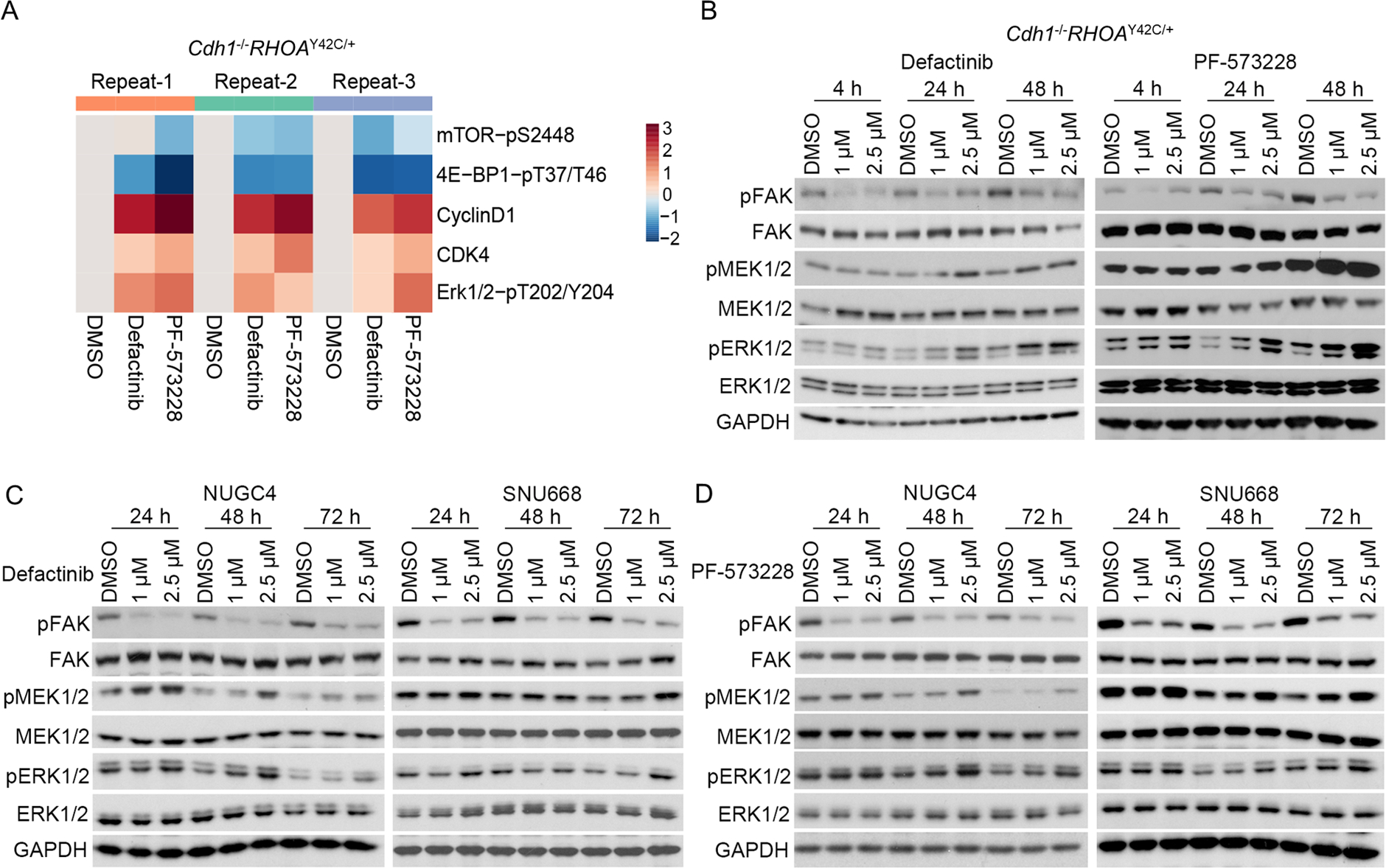
(A) Heat map representation of selected antibodies from Digiwest analysis of Cdh1−/−RHOAY42C/+ organoids treated with DMSO, 2.5 μM defactinib or 2.5 μM PF573228 for 48 hours. Log2fold change of the target signal in treatment group vs DMSO group in each replicate were used in this heatmap. (B, C, D) Immunoblot analysis of Cdh1−/−RHOAY42C/+ organoids (B) and NUGC4, SNU668 cell lines (C, D) treated with defactinib or PF-573228 for indicated time. Immunoblots from one representative experiment (n=2) are shown.
We explored the potential mechanisms of FAK inhibition-induced MAPK pathway activation. Receptor tyrosine kinase (RTK)-RAS signaling is a central regulator of ERK1/2 activation. Therefore, we queried enhanced RTK phosphorylation in Cdh1−/RHOAY42C/+ organoids after treatment with defactinib or PF-573228 and found that pERBB4 was modestly upregulated (Figure S7A). However, the pan-ERBB inhibitor afatinib did not prevent defactinib-induced ERK1/2 activation, suggesting that pERBB4 does not mediate FAK inhibitor-induced ERK1/2 activation (Figure S7B). Next, we evaluated whether RAS-GTP activation could lead to pERK activity in Cdh1−/−RHOAY42C/+ organoids treated with defactinib or PF-573228. We found that compared with the DMSO-treated group, there was no significant change in RAS-GTP in the defactinib treatment group and only a slight upregulation of RAS-GTP in the PF-573228 group (Figure S7C). These data indicate that ERK activation is not mediated by RAS-GTP. The mechanism of ERK1/2 activation promoted by FAK inhibition remains unresolved.
The combination of MAPK inhibitor and FAK inhibitor has a synergistic effect in DGC
Given these data, we explored whether a combination of FAK and MAPK inhibitors could enhance the efficacy of FAK blockade. VS-6766 is a dual RAF/MEK inhibitor (Figure 4A, S8A). We found that FAK inhibitors (defactinib or VS-4718), when combined with VS-6766, significantly changed the morphology of Cdh1−/RHOAY42C/+ organoids, making them smaller and round, thus losing the central filling that accompanies transformation (Figure 4B, S8B). The ZIP score for defactinib+VS-6766 was 11.681, and that for VS-4718+VS-6766 was 23.675, both of which indicated the synergistic effects of the combinations (Figure 4C, S8C).
Figure 4. MAPK inhibitor and FAK inhibitor have synergistic effect in DGC models.
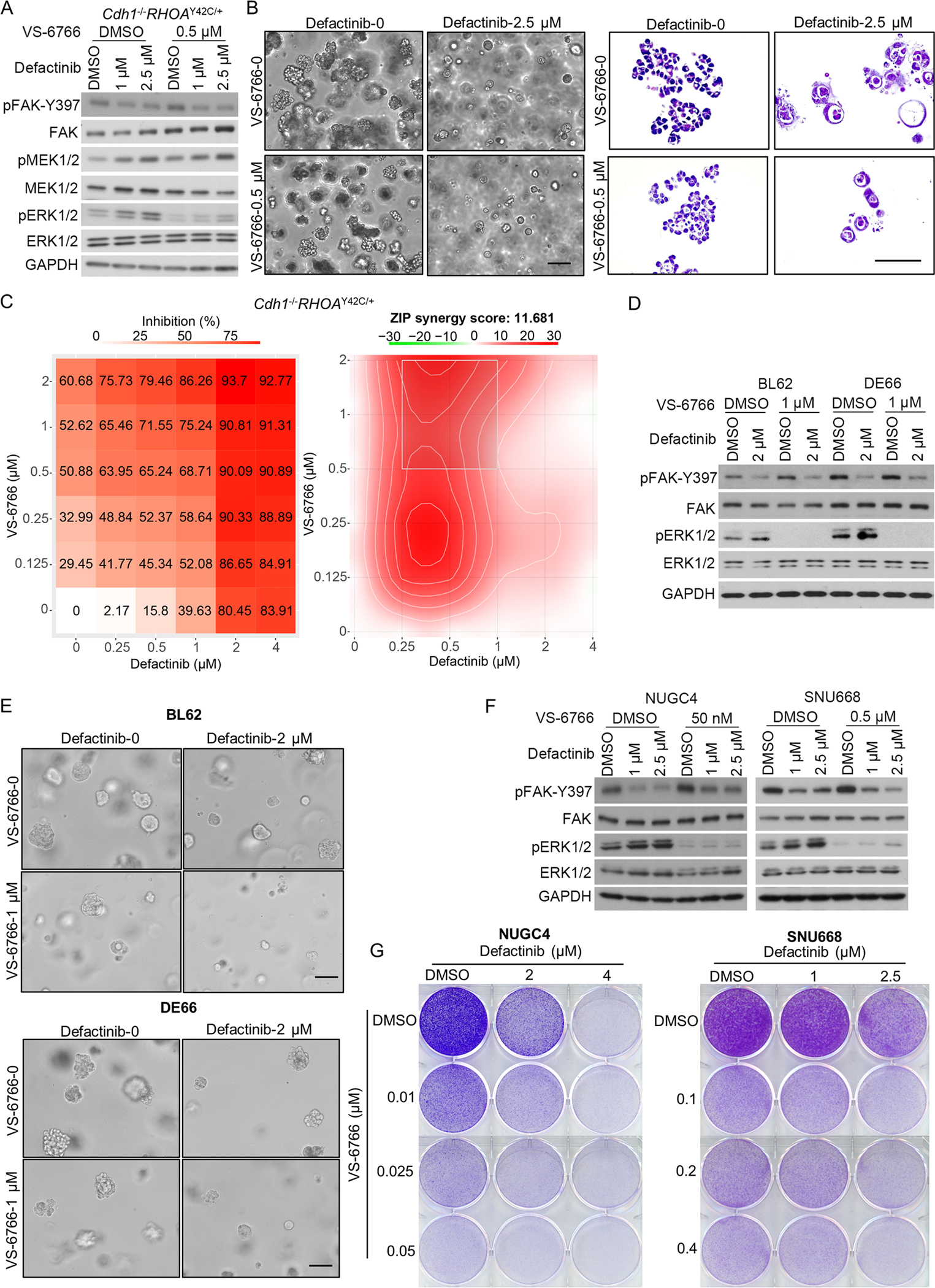
(A) Immunoblot analysis of Cdh1−/−RHOAY42C/+ organoids treated with defactinib, VS-6766, the combination or DMSO control for 24 hours. Immunoblots from one representative experiment (n=2) are shown. (B) Representative phase contrast images (left) and HE staining images (right) of Cdh1−/−RHOAY42C/+ organoids treated for 48 hours with 2.5 μM defactinib, 0.5 μM VS-6766, the combination or with DMSO control. Scale bar, 50 μm. (C) Cdh1−/−RHOAY42C/+ organoids were treated with defactinib (0.25 μM to 4 μM) or VS-6766 (0.125 μM to 2 μM) alone or together for 3 days. Viability in the treatment groups was normalized to DMSO control. The inhibition rate was shown (left). Analysis of synergistic effect in defactinib and VS6766 combination was performed by SynergyFinder using Zero Interaction Potency (ZIP) model. The inhibition rate was used to calculate ZIP synergy score. The box indicates the most synergistic area (right). Representative of two independent experiments were shown. (D) Immunoblot analysis of BL62 (left) and DE66 (right) treated with defactinib, VS-6766, the combination or DMSO control for 5 days. Immunoblots from one representative experiment (n=2) are shown. (E) Representative phase contrast images of BL62 (top) and DE66 (bottom) treated for 5 days with 2 μM defactinib, 1 μM VS-6766, the combination or with DMSO control. Scale bar, 50 μm. (F) Immunoblot analysis of NUGC4 (left) and SNU668 (right) treated with defactinib, VS-6766, the combination or DMSO control for 48 hours. Immunoblots from one representative experiment (n=2) are shown. (G) Images show representative results of colony formation assays of NUGC4 (left) and SNU668 (right). Cells were cultured in 6-well plates and treated with DMSO, indicated concentration of defactinib or VS-6766 alone or together for 7 to 10 days, and then fixed and stained with crystal violet solution. All experiments were repeated for at least twice.
We also tested another potent MEK inhibitor, trametinib, which is currently used to treat BRAFV600E mutant metastatic melanoma and colorectal cancer(20,21). A small dose of 8 nM trametinib was sufficient to inhibit downstream ERK1/2 in Cdh1−/RHOAY42C/+ organoids but caused reactivation of MEK1/2 itself (Figure S8D). We observed similar morphological changes in Cdh1−/−RHOAY42C/+ organoids under defactinib+trametinib combination treatment (Figure S8E), and the synergistic effect was strong, with a ZIP score of 15.978 (Figure S8F).
The combination of defactinib and palbociclib was also evaluated in the two DGC PDOs, (Figure 4D). The organoid numbers of both PDOs in combination groups was significantly smaller than that in monotherapy or control groups (Figure 4E). Synergistic effect was also shown in both BL62 (Figure S9A) and DE66 (Figure S9B), with the ZIP scores of 12.866 and 22.356, respectively.
We next validated the combination of FAK inhibitor (defactinib or VS-4718) with a MAPK inhibitor (VS-6766 or trametinib) in the DGC cell lines NUGC4 and SNU668 (Figure 4F, S10A–B). Using crystal violet staining in NUGC4 and SNU668 cell lines, we observed that VS-6766 enhanced the efficacy of FAK inhibitors defactinib and VS-4718 (Figure 4G, S10C). The combination of trametinib and defactinib also resulted in stronger cell inhibition than monotherapy (Figure S10D).
Next, we tested the VS-4718 and VS-6766 combinations in SNU668 xenografts. We explored the efficacy of the combination of VS-4718 and palbociclib in the SNU668 xenografts (Figure S5B). In parallel to these earlier studies, we also used the VS-4718 and VS-6766 combination, which helped to compare the efficacy of both combination strategies (Figure 5A). We sacrificed one mouse in each treatment group after seven days of treatment for pharmacodynamic and target engagement studies. Tumors were also collected at the end of the treatment. We found that tumor growth in the VS-4718+VS-6766 group was consistently suppressed during the treatment, which was more striking than the reduced growth seen with the palbociclib combination in this model (Figure 5B, S11A). For the proliferation assay, Ki-67 staining in the VS4718+VS-6766 group or VS-4718+ palbociclib group was weaker than that in the control and VS-4718 monotherapy groups (Figure 5C, S11B). In the apoptosis assay, there was no significant difference between the TUNEL staining in the treatment groups, indicating that apoptosis might not contribute to the different growth rates of tumors in these groups (Figure 5C, S11B). Consistent with NUGC4 xenografts (Figure 2I), VS-4718 treatment also upregulated pERK1/2 in SNU668 xenografts (Figure 5C, S11B), but this rebound activation was abrogated by MEK blockade. These data highlight the potential of combination therapy with FAK and MAPK-directed therapy in DGC therapy.
Figure 5. CDK4/6 or MEK inhibition improves FAK inhibition response in DGC xenografts.
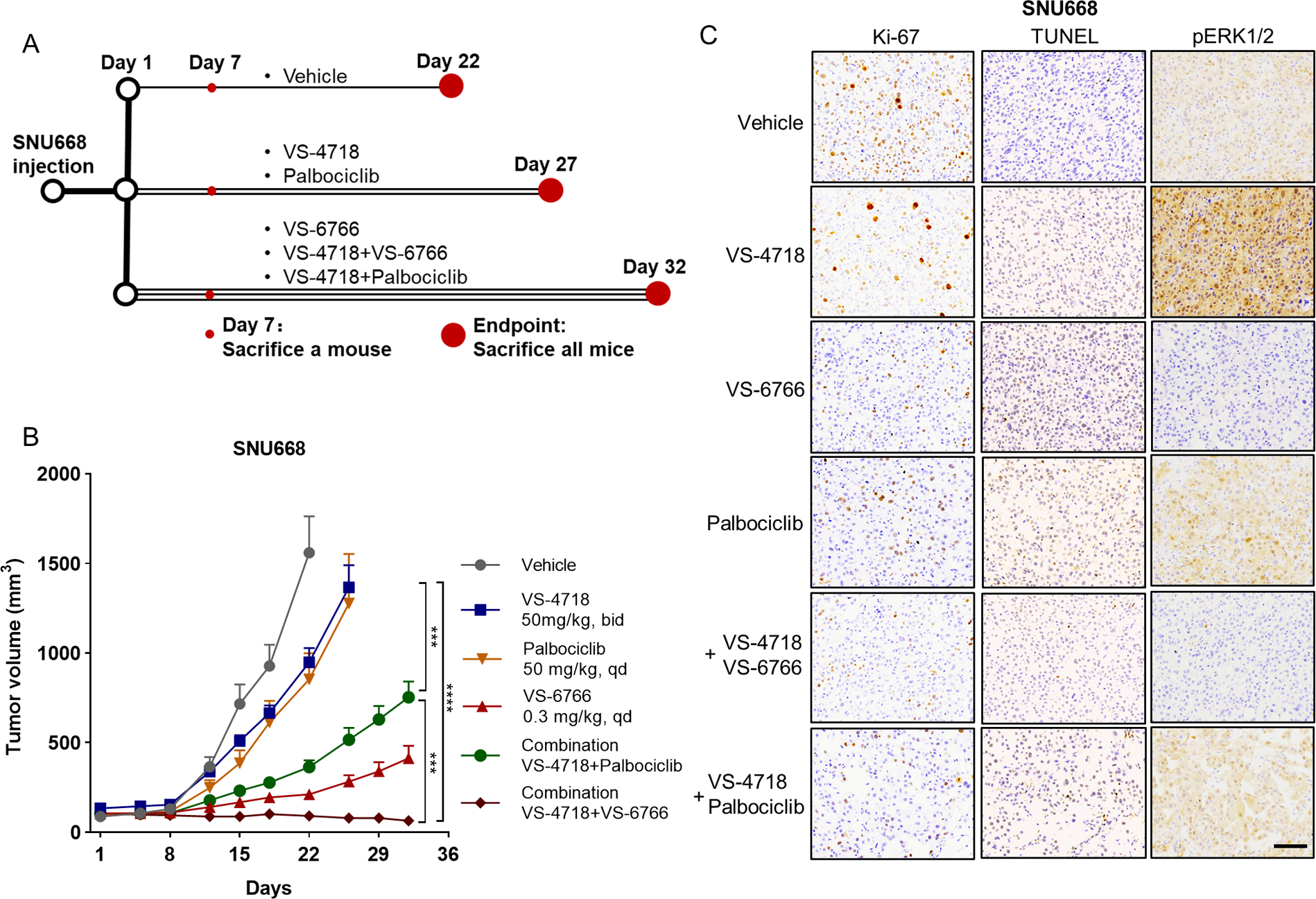
(A) Schematic description of in vivo experiment of SNU668 xenograft. We sacrificed one mouse in each group to obtain the tumor for IHC staining after the treatment of seven days. All mice were sacrificed at the endpoint, and the tumors were also collected. (B) Growth curve for SNU668 xenograft tumors (n=10–12) treated with vehicle control, VS-4718 (50 mg/kg, bid), palbociclib (50 mg/kg, qd), VS-6766 (0.3 mg/kg, qd), VS-4718+Palbociclib combination or VS-4718+VS-6766 combination. Treatment began on day1. Data are mean ± SEM. Statistical analysis was performed using data before day 22, ***P<0.001, ****P<0.0001, two-way ANOVA test. (C) Representative images of Ki-67, TUNEL and pERK1/2 staining of the SNU668 xenograft tumors collected at treatment day 7. Scale bar=100 μm.
Discussion
DGC is characterized by a highly invasive growth pattern, with rapid local invasion and peritoneal metastases. The lack of classic targetable activated oncoproteins such as HER2(4,22) hinders therapeutic development. Thus, patients with DGC usually have a poor prognosis. In our previous study, we found that FAK inhibitors could be potential candidates for the treatment of DGC(5). FAK inhibitors have been developed and evaluated for other tumor types(9,10,23). In a phase 2 study of defactinib monotherapy in KRAS-mutant non-small cell lung cancer (NSCLC), just 28% patients (n=15) met the 12-week PFS endpoint(23). These clinical trials demonstrated that FAK inhibitors are well tolerated, but with only modest clinical efficacy(8–10,23). In our previous study, we also observed that the tumors of Cdh1−/RHOAY42C/+ xenografts grew gradually after FAK inhibitor monotherapy after ten days despite marked growth inhibition relative to negative controls. Therefore, combinatorial strategies are likely to be necessary for FAK inhibition to prove effective in DGC.
In other contexts, the pharmacodynamic effects of FAK inhibitors on tumors have been explored to define potential approaches for overcoming resistance. For example, STAT3 signaling is hyperactivated following prolonged FAK inhibitor treatment, leading to resistance to FAK inhibitor in pancreatic ductal adenocarcinoma (PDAC)(24). Beyond the use of FAK inhibitors as primary targets in cancer, there have been several ongoing efforts to utilize FAK agents as a means to complement other targets, including cytotoxic chemotherapy(25), targeted therapy(26–29), and immunotherapy(30). As FAK activation leads to platinum resistance in high-grade serous ovarian carcinoma(31), the combination of defactinib, paclitaxel, and carboplatin is being evaluated for re-sensitization of carboplatin-resistant ovarian cancer(25). For targeted therapy, treatment with the RAF/MEK inhibitor VS-6766 has been shown to activate FAK in KRAS-mutant tumors(26); thus, the combination of VS-6766 and defactinib is being evaluated for KRAS-mutant ovarian cancer and NSCLC(28,29). FAK inhibition also may increase immune surveillance by overcoming the fibrotic and immunosuppressive tumor microenvironment and renders PDAC models responsive to immunotherapy (30,32), leading to the evaluation of FAK inhibition with immune therapies.
In this study, we sought to define the combinatorial possibilities for FAK inhibition in GC. First, we used a genome-scale ORF screen to select genes or pathways contributing to FAK inhibitor resistance in our organoid model. According to the ORF screening results, WNT1, WNT3A, and WNT2 were genes whose overexpression led to the greatest resistance to treatment with two distinct FAK inhibitors. However, drugs targeting the Wnt pathway are still not mature in clinical settings, although some preclinical outcomes appear promising(33). Thus, we chose to validate CDK6, another top result in our screen, as CDK4/6 inhibitors have been approved for breast cancer and are being evaluated in other tumor types(34). FAK signaling has been reported to mediate CDK4/6-independent CDK2 activation, which drives cell cycle progression under CDK4/6 inhibition and promotes the survival of cells treated with a CDK4/6 inhibitor(35). In intrahepatic cholangiocarcinoma (iCCA), both the FAK and CDK4/6 pathways are highly activated. The combination of FAK and CDK4/6 inhibitors also showed a synergistic effect in iCCA(36). These data inspired us to investigate the effect of FAK and CDK4/6 inhibitor combination in DGC. In our study, the combination of CDK4/6 and FAK inhibitors showed complementary effects in both in vitro and in vivo experimental models.
Our studies on adaptive alterations also led us to evaluate their combination with MAPK inhibition. MAPK and FAK pathways exhibit complex crosstalk and interactions. MAPK blockade promotes FAK activation. In BRAF mutant colorectal cancer, BRAF inhibition activates FAK and the downstream Wnt pathway, leading to resistance to BRAF inhibition(37). In melanoma, the BRAF inhibitor vemurafenib activates the c-Jun/FAK/SRC pathway to induce de-differentiation of melanoma cells, which promotes melanoma cells to tolerate BRAF inhibition(38). Treatment with the RAF/MEK inhibitor VS-6766 also activated FAK in KRAS-mutant cancers(26). Moreover, FAK inhibition can activate the MAPK pathway. In uveal melanoma, FAK is activated by mutant Gαq via the noncanonical TRIO-RhoA pathway(39). FAK inhibitor treatment increases pERK levels, and the combination of FAK and MAPK inhibitors shows a synergistic effect in uveal melanoma(40). In our study, FAK inhibition also activated ERK, and a synergistic effect was observed in the combination of FAK and MAPK inhibitors in DGC.
It is worth noting that the combination of FAK inhibitor defactinib and RAF/MEK inhibitor VS-6766 has been evaluated in phase 1 clinical trials(26,27). The response rate of this combination strategy for KRAS-mutant low-grade serous ovarian cancer was 67% (4/6)(26). Two NSCLC patients with the KRAS G12V mutation (2/2) also showed a partial response in this clinical trial(27). As this combination strategy has achieved promising results, phase 2 clinical trials have been initiated(28,29). Based on our data, this combination strategy for DGC should also be further tested in clinical trials.
In the NUGC4 xenograft model, the combination of VS-4718 and palbociclib significantly inhibited tumor growth. Proliferation was suppressed and apoptosis was elevated according to Ki-67 and TUNEL staining in the combination group. In the SNU668 xenograft model, we used both combination strategies: FAK inhibitor (VS4718) with either RAF/MEK inhibitor (VS-6766) or CDK4/6 inhibitor (palbociclib). We found that SNU668 xenograft tumors showed constant tumor regression under the VS-4718 and VS-6766 combination treatment, but the tumors grew gradually under the VS-4718 and palbociclib combination treatment. The MAPK pathway regulates a wide variety of key cellular processes, including proliferation, differentiation, apoptosis, and stress responses, and is the primary regulator of the cell cycle(41). It is quite reasonable that the combination of MAPK pathway inhibitors may have a better tumor response than that using CDK4/6 inhibitors, as inhibition of the MAPK pathway may have a wider effect than CDK4/6 inhibition alone(42). Nevertheless, there is also the potential for the combination of CDK4/6 and FAK inhibition to have benefits in terms of toxicity and tolerability related to MEK inhibitors. Ultimately, clinical testing will be determined to evaluate these combinations to determine the balance of efficacy and toxicity in DGC.
In conclusion, we identified CDK4/6 and MAPK inhibitors as promising potential FAK inhibitor combinations. We hope that these data will motivate clinical studies on FAK inhibitor combinations in DGC.
Supplementary Material
Translational relevance.
Diffuse gastric cancer (DGC) is a highly lethal subtype of gastric cancer. Because DGC often lacks genomic aberrations that indicate clear candidate therapeutic targets, developing targeted therapies for DGC has been challenging. Our previous study highlighted the contribution of focal adhesion kinase (FAK) in the tumorigenesis of DGC and the potential efficacy of small-molecule FAK inhibitors. However, drug resistance to monotherapies often leads to treatment failure. By employing a genome-scale library of open reading frames (ORF) to identify candidate mechanisms of resistance to FAK inhibition, we found that cyclin-dependent kinase 6 (CDK6) promoted FAK inhibitor resistance. Moreover, we demonstrated that FAK inhibitor treatment in DGC models leads to compensatory MAPK pathway activation. Small molecule CDK4/6 inhibitors or MAPK inhibitors effectively enhance the efficacy of FAK inhibitors. These data suggest that FAK inhibitors combined with MAPK inhibitors or CDK4/6 inhibitors warrant further testing in clinical trials for DGC.
Acknowledgments
We thank Prof. David Root for valuable suggestions regarding the analysis of ORF screen data. Verastem Oncology kindly provided VS-4718 and VS-6766. This work was supported by The V Foundation (to A.J. Bass) and the National Institutes of Health (R01 CA224428, to A.J. Bass).
Footnotes
Conflict of interest: A.J. Bass has received funding from Merck, Novartis, Bayer and Repare and is the advisor to Earli and HelixNano. A.J. Bass and H. Zhang are co-founders and equity holders in Signet Therapeutics. All other authors declare no conflicts.
Reference
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: a cancer journal for clinicians 2021;71(3):209–49 doi 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Lauren P THE TWO HISTOLOGICAL MAIN TYPES OF GASTRIC CARCINOMA: DIFFUSE AND SO-CALLED INTESTINAL-TYPE CARCINOMA. AN ATTEMPT AT A HISTO-CLINICAL CLASSIFICATION. Acta pathologica et microbiologica Scandinavica 1965;64:31–49 doi 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 3.Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nature genetics 2014;46(6):583–7 doi 10.1038/ng.2984. [DOI] [PubMed] [Google Scholar]
- 4.TCGA. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513(7517):202–9 doi 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Schaefer A, Wang Y, Hodge RG, Blake DR, Diehl JN, et al. Gain-of-Function RHOA Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer. Cancer discovery 2020;10(2):288–305 doi 10.1158/2159-8290.cd-19-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acebrón I, Righetto RD, Schoenherr C, de Buhr S, Redondo P, Culley J, et al. Structural basis of Focal Adhesion Kinase activation on lipid membranes. The EMBO journal 2020;39(19):e104743 doi 10.15252/embj.2020104743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer 2014;14(9):598–610 doi 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soria JC, Gan HK, Blagden SP, Plummer R, Arkenau HT, Ranson M, et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2016;27(12):2268–74 doi 10.1093/annonc/mdw427. [DOI] [PubMed] [Google Scholar]
- 9.Jones SF, Siu LL, Bendell JC, Cleary JM, Razak AR, Infante JR, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investigational new drugs 2015;33(5):1100–7 doi 10.1007/s10637-015-0282-y. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu T, Fukuoka K, Takeda M, Iwasa T, Yoshida T, Horobin J, et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer chemotherapy and pharmacology 2016;77(5):997–1003 doi 10.1007/s00280-016-3010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nature protocols 2013;8(12):2471–82 doi 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science (New York, NY) 2018;359(6378):920–6 doi 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nature medicine 2018;24(7):968–77 doi 10.1038/s41591-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou J, Wu Z, Wong G, Pectasides E, Nagaraja A, Stachler M, et al. CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nature communications 2017;8:13897 doi 10.1038/ncomms13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treindl F, Ruprecht B, Beiter Y, Schultz S, Döttinger A, Staebler A, et al. A bead-based western for high-throughput cellular signal transduction analyses. Nature communications 2016;7:12852 doi 10.1038/ncomms12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yadav B, Wennerberg K, Aittokallio T, Tang J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Computational and structural biotechnology journal 2015;13:504–13 doi 10.1016/j.csbj.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic acids research 2020;48(W1):W488–w93 doi 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet (London, England) 2020;395(10226):817–27 doi 10.1016/s0140-6736(20)30165-3. [DOI] [PubMed] [Google Scholar]
- 19.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Molecular cancer therapeutics 2004;3(11):1427–38. [PubMed] [Google Scholar]
- 20.Corcoran RB, Andre T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer discovery 2018;8(4):428–43 doi 10.1158/2159-8290.cd-17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dummer R, Hauschild A, Santinami M, Atkinson V, Mandalà M, Kirkwood JM, et al. Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. The New England journal of medicine 2020;383(12):1139–48 doi 10.1056/NEJMoa2005493. [DOI] [PubMed] [Google Scholar]
- 22.Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature genetics 2014;46(6):573–82 doi 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 23.Gerber DE, Camidge DR, Morgensztern D, Cetnar J, Kelly RJ, Ramalingam SS, et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung cancer (Amsterdam, Netherlands) 2020;139:60–7 doi 10.1016/j.lungcan.2019.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang H, Liu X, Knolhoff BL, Hegde S, Lee KB, Jiang H, et al. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 2020;69(1):122–32 doi 10.1136/gutjnl-2018-317424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osterman CJD, Bean LM, Sulzmaier FJ, Taylor KN, Jiang SA, Tancioni I, et al. Abstract 1991: Vulnerability of platinum-resistant ovarian cancer to FAK inhibition. Cancer research 2018;78(13_Supplement):1991- doi 10.1158/1538-7445.AM2018-1991 %J Cancer Research. [DOI] [Google Scholar]
- 26.Shinde R, Terbuch A, Little M, Caldwell R, Kurup R, Riisnaes R, et al. Abstract CT143: Phase I study of the combination of a RAF-MEK inhibitor CH5126766 and FAK inhibitor defactinib in an intermittent dosing schedule with expansions in KRAS mutant cancers. AACR; 2020. [Google Scholar]
- 27.Krebs MG, Shinde R, Rahman RA, Grochot R, Little M, King J, et al. Abstract CT019: A phase I trial of the combination of the dual RAF-MEK inhibitor VS-6766 and the FAK inhibitor defactinib: Evaluation of efficacy in KRAS mutated NSCLC. Cancer research 2021;81(13_Supplement):CT019–CT doi 10.1158/1538-7445.AM2021-CT019 %J Cancer Research. [DOI] [Google Scholar]
- 28.Banerjee SN, Monk BJ, Van Nieuwenhuysen E, Moore KN, Oaknin A, Fabbro M, et al. ENGOT-ov60/GOG3052/RAMP 201: A phase 2 study of VS-6766 (dual RAF/MEK inhibitor) alone and in combination with defactinib (FAK inhibitor) in recurrent low-grade serous ovarian cancer (LGSOC). Journal of Clinical Oncology 2021;39(15_suppl):TPS5603–TPS doi 10.1200/JCO.2021.39.15_suppl.TPS5603. [DOI] [Google Scholar]
- 29.Capelletto E, Bironzo P, Denis L, Koustenis A, Bungaro M, Novello S. Single agent VS-6766 or VS-6766 plus defactinib in KRAS-mutant non-small-cell lung cancer: the RAMP-202 phase II trial. Future oncology (London, England) 2022. doi 10.2217/fon-2021-1582. [DOI] [PubMed] [Google Scholar]
- 30.Wang-Gillam A, Lockhart AC, Tan BR, Suresh R, Lim K-H, Ratner L, et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer. 2018;36(4_suppl):380- doi 10.1200/JCO.2018.36.4_suppl.380. [DOI] [Google Scholar]
- 31.Diaz Osterman CJ, Ozmadenci D, Kleinschmidt EG, Taylor KN, Barrie AM, Jiang S, et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. eLife 2019;8 doi 10.7554/eLife.47327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature medicine 2016;22(8):851–60 doi 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsons MJ, Tammela T, Dow LE. WNT as a Driver and Dependency in Cancer. Cancer discovery 2021;11(10):2413–29 doi 10.1158/2159-8290.cd-21-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suski JM, Braun M, Strmiska V, Sicinski P. Targeting cell-cycle machinery in cancer. Cancer cell 2021;39(6):759–78 doi 10.1016/j.ccell.2021.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C, Stockwell SR, Elbanna M, Ketteler R, Freeman J, Al-Lazikani B, et al. Signalling involving MET and FAK supports cell division independent of the activity of the cell cycle-regulating CDK4/6 kinases. Oncogene 2019;38(30):5905–20 doi 10.1038/s41388-019-0850-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song X, Xu H, Wang P, Wang J, Affo S, Wang H, et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. Journal of hepatology 2021;75(4):888–99 doi 10.1016/j.jhep.2021.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen G, Gao C, Gao X, Zhang DH, Kuan SF, Burns TF, et al. Wnt/β-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Molecular cancer therapeutics 2018;17(4):806–13 doi 10.1158/1535-7163.mct-17-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fallahi-Sichani M, Becker V, Izar B, Baker GJ, Lin JR, Boswell SA, et al. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Molecular systems biology 2017;13(1):905 doi 10.15252/msb.20166796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer cell 2019;35(3):457–72.e5 doi 10.1016/j.ccell.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paradis JS, Acosta M, Saddawi-Konefka R, Kishore A, Gomes F, Arang N, et al. Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2021;27(11):3190–200 doi 10.1158/1078-0432.ccr-20-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Experimental and therapeutic medicine 2020;19(3):1997–2007 doi 10.3892/etm.2020.8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature reviews Cancer 2004;4(12):937–47 doi 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available in the article and its supplementary files. Any additional results can be obtained from the corresponding authors.