

Abstract
Aberrant metabolic functions play a crucial role in prostate cancer progression and lethality. Currently, limited knowledge is available on subtype-specific metabolic features and their implications for treatment. We therefore investigated the metabolic determinants of the two major subtypes of castration-resistant prostate cancer (androgen receptor-expressing prostate cancer, ARPC; and aggressive-variant prostate cancer, AVPC). Transcriptomic analyses revealed enrichment of gene sets involved in oxidative phosphorylation (OXPHOS) in ARPC tumor samples compared to AVPC. Unbiased screening of metabolic signaling pathways in PDX models by proteomic analyses further supported an enrichment of OXPHOS in ARPC compared to AVPC, and a skewing toward glycolysis by AVPC. In vitro, ARPC C4–2B cells depended on aerobic respiration, while AVPC PC3 cells relied more heavily on glycolysis, as further confirmed by pharmacological interference using IACS-10759, a clinical-grade inhibitor of OXPHOS. In vivo studies confirmed IACS-10759’s inhibitory effects in subcutaneous and bone-localized C4–2B tumors, and no effect in subcutaneous PC3 tumors. Unexpectedly, IACS-10759 inhibited PC3 tumor growth in bone, indicating microenvironment-induced metabolic reprogramming. These results suggest that castration-resistant ARPC and AVPC exhibit different metabolic dependencies, which can further undergo metabolic reprogramming in bone.
Introduction
Prostate cancer (PCa) initiates as an androgen receptor (AR)-dependent neoplasia that is clinically managed in later stages with androgen deprivation therapy (1). However, through the emergence of various resistance mechanisms, patients eventually advance toward a lethal castration-resistant phenotype, which retains AR-expression and progresses despite low circulating testosterone levels (ARPC) (1). A morphologically heterogenous subtype of advanced castration-resistant PCa has recently emerged, which shares features with neuroendocrine-like/small cell-like PCa, renamed under the comprehensive description of aggressive variant prostate cancer (AVPC) (2). This aggressive PCa subset is distinguished by low/absent AR and circulating prostate-specific antigen (PSA) levels, poor prognosis, no response to castration and second-generation antiandrogens, but sensitivity to platinum-based chemotherapy (2). AVPC is considered a distinct clinical–biologic subset defined by mutations or deletions in the TP53, RB1, and/or PTEN tumor suppressors, which were shown to account for neuroendocrine-like/small cell-like clinical features, resistance to AR inhibition and defined chemotherapy sensitivity (3). Notably, while small-cell/neuroendocrine prostate carcinoma is rarely diagnosed de novo (<2% of PCa cases (4)), an increasing proportion of castration-resistant PCa patients (20–25%) progress towards this phenotype, likely as a mechanism of therapy resistance to second-generation anti-androgen treatments (2,5). Advanced ARPC and AVPC are often associated with metastatic dissemination, primarily to bone, representing a major site for distant growth in 84% of metastatic patients (2,6). While the localized disease is manageable, with an almost 100% survival rate at five years after diagnosis, the survival rate of metastatic PCa drops to 30% (1).
The deregulation of cellular energetics and the emergence of aberrant metabolic programs is a crucial hallmark of cancer progression (7). Such metabolic rewiring is meant to support high replication rates in a nutrient-limited environment, balancing sufficient ATP production, biosynthetic needs, and redox homeostasis (7). Pro-cancer metabolic rewiring is achieved differently across tumor types and disease stages (8). Typically, cells rely on citrate oxidation as a key step in the Krebs cycle for aerobic respiration. Healthy peripheral prostatic epithelium, instead, hinders this step to enable citrate accumulation and secretion into the seminal vesicle fluid (9). This truncated Krebs cycle occurs through zinc-mediated inhibition of the enzyme m-aconitase that converts citrate to isocitrate (9). During the initial phases of prostatic adenocarcinoma development, decreased expression of zinc transporters depletes intracellular zinc, leading to a restoration of the Krebs cycle and oxidative phosphorylation (OXPHOS) in PCa cells (9). When progressing into the later stages of the disease, the classic Warburg effect plays an increased role. While the initial phases of metabolic reprogramming in situ have been elucidated, the role of this rewiring in the late phases of ARPC and AVPC progression has been poorly characterized (9,10).
In this work, we sought to elucidate the metabolic determinants of ARPC and AVPC by integrating transcriptomics and proteomics with functional in vitro and in vivo studies to test the importance of OXPHOS across PCa states.
Materials and methods
Animal studies
Animal studies were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center and Fred Hutchinson Research Cancer Center and performed according to the institutional guidelines for animal care and handling. For in vivo testing, 8-week-old athymic nude or non-obese diabetic/severe combined immunodeficiency (NOD-SCID) male mice were purchased from the Department of Experimental Radiation Oncology, MD. Anderson Cancer Center. Mice were housed with a maximum of 5 animals per cage in a state-of-the-art, air-conditioned, specific-pathogen–free animal facility and all procedures were performed in accordance with the NIH Policy on Humane Care and Use of Laboratory Animals. Surgical procedures were performed with mice under general anesthesia (isoflurane), and analgesia was provided at the end of each procedure (buprenorphine, 0.05 mg/kg, one dose immediately before the start of the surgery, a subsequent dose postoperatively within 24 hours). Tumor-bearing animals were observed daily and examined by a veterinarian 5 days/week for signs of morbidity (e.g. matted fur, weight loss, limited ambulation, and respiratory difficulty). In case of discomfort, the animals were euthanized consistently with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. PDXs were generated and passaged as previously described (11).
Cell culture and Reagents
Luciferase-expressing PC3 human prostate cancer cells (RRID:CVCL_0035) were provided by Dr. Gary Gallick, UT MD Anderson Cancer Center. Cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Sigma) and 1% penicillin and streptomycin (both 100 μg/ml, Sigma). Luciferase-expressing C4–2B human PCa cells (RRID:CVCL_4784) were provided by Dr. Timothy Thompson, UT MD Anderson Cancer Center. Cells were cultured in RPMI (Invitrogen) supplemented with 10% fetal bovine serum (Sigma), 1% penicillin and streptomycin (both 100 μg/ml, Sigma) and 1% Hepes. The absence of mycoplasma was tested regularly (once every two months). Cell cultures were monitored for doubling times and morphology, and passaged less than 20 times. The identity of tumor cell lines was verified by Short Tandem Repeat DNA profiling (Characterized Cell Line Core Facility, MD Anderson).
Reverse-phase protein array (RPPA) construction, processing, probing and analysis
Protein microarrays were printed and processed as previously described (12,13). More details are provided in the supplementary materials and methods.
Mitochondrial activity and glycolysis measurements studies
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) experiments were performed using the XF-96 Analyzer apparatus from Seahorse Bioscience according to manufacturer’s instructions. Cells were plated (20,000 cells/well; n=8 wells per group) in Seahorse 96-well plates precoated with Cell-Tak. After 24 hours, IACS-10759 was administered to the cells at different concentrations (0, 0.3, 3, and 30 nM). After additional 24 hours, the medium was replaced with reconstituted Seahorse XF base DMEM medium supplemented with 10 mM glucose, 2 mM glutamine and 1 mM sodium pyruvate adjusted to pH ~7.4 and warmed up to 37°C in a CO2-free incubator. For the Mitochondrial Stress test (Seahorse Agilent), oligomycin, trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), and a mixture of antimycin and rotenone were injected to final concentrations of 1.25 μM, 0.625 μM, and 0.625 μM, respectively. For the Glycolytic Rate assay (Seahorse Agilent), a mixture of rotenone/antimycin and 2-deoxyglucose were injected to final concentrations of 5 μM and 50 mM, respectively. OCR and ECAR were normalized to live-cell area as determined by analysis of cell nuclei stained with 25 μg/ml propidium iodide and 50 μg/ml Hoechst at the end of the experiments. OCR and ECAR values were used to compute basal respiration, maximal respiration, spare respiratory capacity, ATP production, basal glycolysis, and compensatory glycolysis. Data were analyzed using Wave software and calculations were performed using ExcelMacro Report Generator provided by Seahorse Biosciences.
Metabolomic analysis of glucose-derived metabolites in 2D cultured PC3 and C42B cells using mass spectrometry
PC3 and C4–2B cells were cultivated in a 24-well plate (n=3 wells per group, 42,000 cells/well). After 24 hours, cells were treated with 200 nM IACS-10759 and 10 μM antimycin. After additional 24 hours, cell culture medium was collected, centrifuged at 1200 rpm for 5 minutes and analyzed by mass spectrometry, as previously reported (14). More details are provided in the supplementary materials and methods.
JC-1 mitochondrial staining
PC3 and C4–2B cells were plated in a 96-well plate and cultured overnight, then cell medium was removed and 50 μl/well of a 1:5 growth factor reduced Matrigel solution were added and let polymerize for 45 minutes at 37°C 5% CO2. Cells were treated with 0 or 10 μM antimycin for 1 hour, then incubated in a JC-1 solution (10 μg/ml in complete medium) for 1 hour at 37°C 5% CO2, washed with PBS and imaged with a multiphoton microscope using a 32X objective. Detection of the monomeric and polymeric probe was achieved by spectral separation of green fluorescence (525/50, 920 nm), and red fluorescence (620/60, 920 nm). Image analysis was performed with FIJI software: the background was removed from each channel, and GFP and RFP intensities (mean grey value) were measured in the area delimited by cell perimeter; RFP/GFP ratio was then calculated for each cell.
Growth and vitality assessment of cells treated with IACS-10759
Luciferase-expressing PC3 and C4–2B cells were cultivated in a 96-well plate (n=8 wells/group, 1,000 cells/well) and treated after 24 hours with IACS-10759 (0–10 μM, 1:10 dilutions). The growth was measured over time on live cells by bioluminescence signal after addition of luciferin solution (150 μg/ml), or on 100% ice-cold ethanol-fixed cells by 570 nm absorbance after crystal violet staining (2.5 μg/ml in 20% methanol), using an automatic plate reader (PerkinElmer, EnVision 2104 multilabel plate reader). Experimental data were used to develop nonlinear regression three parametric inhibition curves using Prism GraphPad software. Inhibitory concentration 50 (IC-50) was computed and used to compare PC3 and C4–2B response to IACS-10759 treatment. Cell viability was measured by staining cells for 15 minutes with 25 μg/ml propidium iodide and 50 μg/ml Hoechst and by subsequent imaging using an EVOS FL Cell Imaging System (AMG) equipped with 10X objective. Data were analyzed through FIJI software: images were thresholded and the area occupied by stained cells was computed. Then the percentage of dead cells was obtained dividing the area occupied by PI stained cells by the total area covered by cells (PI stained cells + Hoechst stained cells).
PC3 or C4–2B cells were plated in a 96-well plate (n=4/group; 1000 cells/well). After 1 day, cells were incubated with 0.4–5 g/L glucose in 18 or 1% O2. After 1 day, cells were treated with IACS-10759 (0–10 μM or 0–3 μM, 1:10 dilutions, for PC3 or C4–2B cells, respectively). 3 days-post treatment, cells were fixed in 100% ice-cold ethanol and stained with crystal violet. Each well was automatically acquired at the EVOS microscope (1 image/well, 4x objective, equal to ~75% of the total area). The resulting image was thresholded in FIJI and the area occupied by stained cells was measured.
IACS-10759 treatment, in vivo
Male athymic nude mice were implanted with luciferase-expressing PC3; NOD-SCID mice were implanted with luciferase-expressing C4–2B cells. Subcutaneous tumors were generated by implanting 5×106 luciferase-expressing PC3 or 10×106 luciferase-expressing C4–2B cells (n=5 mice/group) in 30% Matrigel in 150 μl total volume. Bone tumors were generated by implanting 2.5×105 cells/tibia luciferase-expressing PC3 or 5×105 cells/tibia luciferase-expressing C4–2B cells (n=12 mice/group). Tumors were randomized the day of the treatment. Mice were treated by oral gavage for 5 days/week with 10 mg/kg IACS-10759 in 0.5% methyl cellulose for 17 days. Dosing schedule was adjusted according to mouse overall health status, monitored daily, e.g. suspended for 1 day if the mouse appeared severely lethargic. Tumor growth was monitored by macroscopic bioluminescence imaging using an IVIS 200 imaging system (Perkin Elmer, Waltham, MA).
Transcriptomic analysis
The data analyzed in this study were obtained from GSE35988 and dbGap: phs000915.v1.p1.
Statistical analysis
For statistical analyses, Student’s t-test was used for paired samples with Gaussian distribution. For independent samples, irrespective of distribution, the one-way ANOVA test was used. For all multiple analyses, Tukey’s HSD post hoc correction was performed. GraphPad Prism 9.2.0 software was used for statistical analysis.
Data availability
The data generated from this study are available within the article and its Supplementary Data.
Results
Metabolic programs are differentially enriched in ARPC and AVPC
To identify the metabolic status of ARPC and AVPC in patient-derived samples, we applied previously defined AR and NE score gene sets (15) to primary tumors in the Grasso dataset (16), obtaining 24 ARPC and 9 AVPC samples, and performed Gene Set Enrichment Analysis (GSEA (17)) on metabolic-related sets of genes (Fig. 1A). This analysis revealed significant enrichment of gene sets involved in OXPHOS, mitochondrial complex I biogenesis, mitochondrial protein import, mitochondrial translation, and citric acid cycle in ARPC tumor samples compared to AVPC (Fig. 1B), suggesting that distinct metabolic pathways underlie ARPC and AVPC subtypes. To evaluate whether this transcriptional outcome extended to the protein level, we applied functional proteomic analysis to castration-resistant prostatic carcinoma patient-derived xenografts (PDXs) of the LuCap series (Fig. 1C)(11,18). The ARPC CR sublines were established from tumors that grew after castration and still express AR at significantly higher levels compared to AVPC sublines (Fig. 1D). We analyzed 13 different PDXs (9 ARPC, 4 AVPC; 3 tumors/PDX, for a total of 39 tumors) using a reverse-phase protein phase array (RPPA; Fig. 1C). The Uniform Manifold Approximation and Projection (UMAP) of the proteomic dataset showed ARPC and AVPC subtype tumors fell into distinct clusters (Fig. 1E). Unbiased screening of metabolic signaling pathways in PDX models using RPPA showed significantly increased levels of proteins involved in OXPHOS in ARPC and significantly increased expression of glycolysis-related enzymes in AVPC (Fig. 1F). Overall, these unbiased analyses suggest defined alterations in metabolic pathways associated with subtype-specific progression at the transcriptional and protein levels.
Fig. 1. Transcriptomic and proteomic analysis of ARPC and AVPC samples.
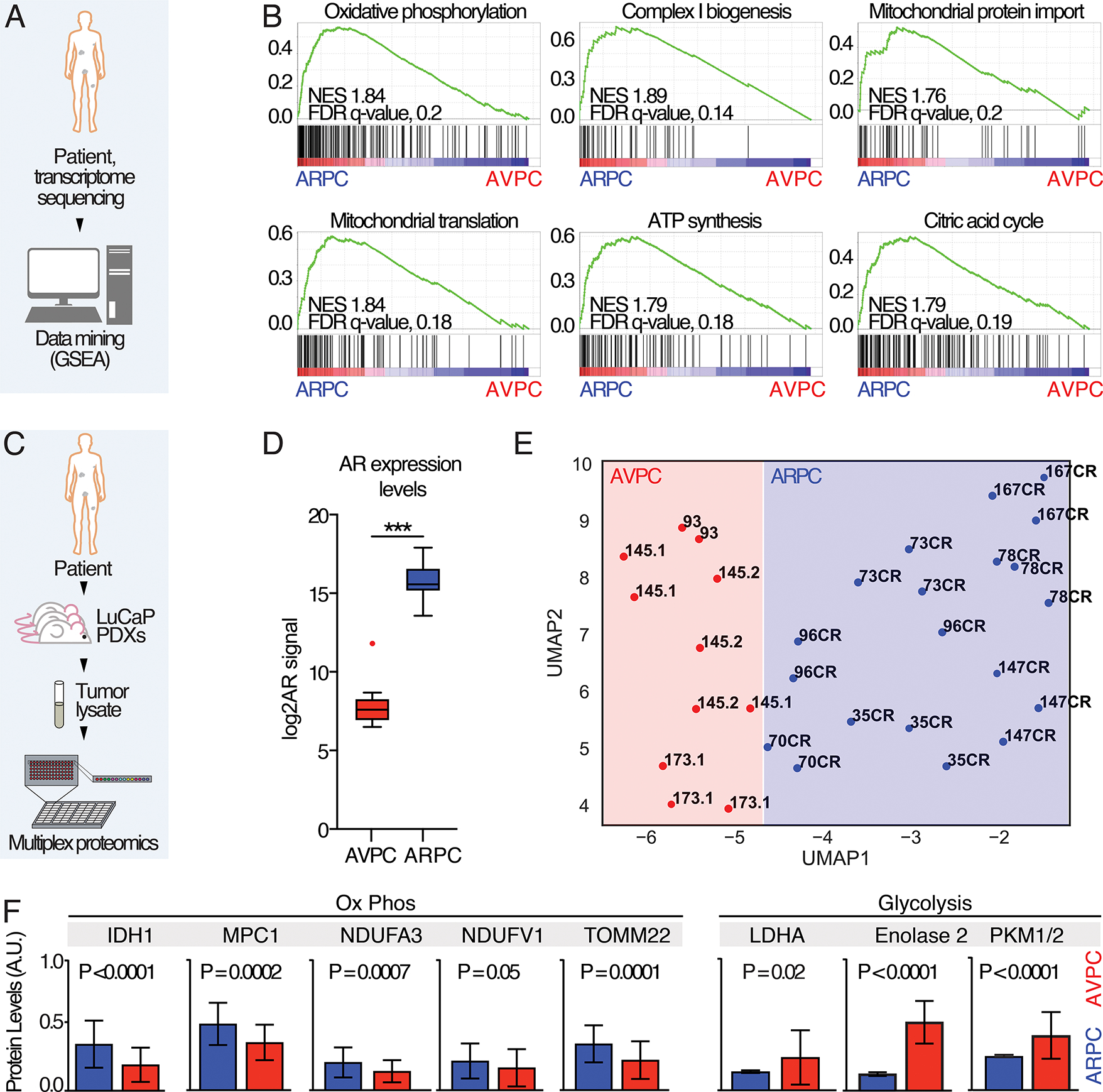
A) Schematic representation of the experiment: data from (16) were segregated in ARPC and AVPC based on AR and NE scores and GSEA analysis performed. B) GSEA enrichment plots; Normalized enrichment scores (NES) and false discovery rate (FDR)-q values are shown. C) Schematic representation of the RPPA study using PDX samples. D) Androgen expression levels in ARPC and AVPC. P value was estimated through Mann Whitney test, (***) P < 0.001. E) A UMAP shows clustering within ARPC and AVPC LuCaP PDX models. CR, Castration-resistant. F) Representative plots showing 8 different proteomic measurements in APRC and AVPC tumor lysate samples. P values are reported.
Metabolic characterization of C4–2B and PC3 cells in 2D culture
Despite being a valuable model for in vivo therapeutic responses, the application of PDXs for metabolic studies in vitro is limited by their inability to survive in conventional cell culture. Therefore, to further characterize the metabolic status of ARPC and AVPC, we used C4–2B and PC3, two cell lines that recapitulate key features of each subtype. C4–2B is an adenocarcinoma cell line derived from LNCaP bone metastatic lesions generated in castrated mice that retains both AR and PSA protein expression. PC3 cells are negative for AR and PSA expression, and display a homozygous mutation in TP53 and a homozygous loss of PTEN (19,20), thus exhibiting the molecular signature of AVPC (3). To characterize C4–2B and PC3 metabolic profiles and determine whether they recapitulated the metabolic features identified in patient-derived and PDX samples, we performed mass spectrometry-based metabolomics, Seahorse analysis, and evaluated the cells’ mitochondrial membrane potential (MMP).
For metabolomics, C4–2B cells had significantly higher levels of the Krebs cycle intermediates malate and succinate, suggesting a greater reliance on OXPHOS. Conversely, PC3 cells had higher baseline lactate levels, and glycolytic index, suggesting that PC3 cells are more glycolytic than C4–2B cells (Fig. 2A, B). As orthogonal measures of metabolism, we then exploited Seahorse technology to test both the mitochondrial activity and the glycolytic capacity of C4–2B and PC3 cells. Seahorse Mito Stress tests (Fig. 2C; Fig. S1A) revealed higher maximal respiration and spare respiratory capacity in C4–2B cells, suggesting a greater reliance on oxidative processes for energy production and no differences in the basal respiration. Consistently, the basal OCR/basal ECAR ratio, which indicates cell preferences for oxidative or glycolytic energetic processes, was higher for C4–2B cells than PC3 cells. Conversely, Seahorse Glycolytic Rate assays (Fig. 2D; Fig. S1B) revealed elevated basal glycolysis in PC3 cells but no significant difference in compensatory glycolysis. Accordingly, PC3 cells displayed higher glucose transporter 1 (GLUT-1) levels (Fig. 2E), suggesting a more substantial reliance on glycolysis. Furthermore, administration of 2-deoxy-D-glucose (2-DG), a glucose analog which inhibits glycolysis through competitive binding of glucose hexokinase, revealed significantly higher sensitivity by PC3 cells (Fig. 2F). Next, we investigated the presence of functional mitochondria in PC3 and C4–2B cells using JC-1 to monitor MMP by fluorescence microscopy (Fig. 2G). Both cell lines showed monomeric and polymeric JC-1 accumulation, indicative of functional mitochondria polarization, which was impaired by administration of antimycin A, a complex III inhibitor (Fig. 2H). C4–2B cells however, exhibited a significantly higher red/green fluorescence ratio (Fig. 2H), reflecting a higher mitochondrial potential. Overall, these results indicate increased OXPHOS in C4–2B cells.
Fig. 2. Metabolic characterization of PCa cell lines.
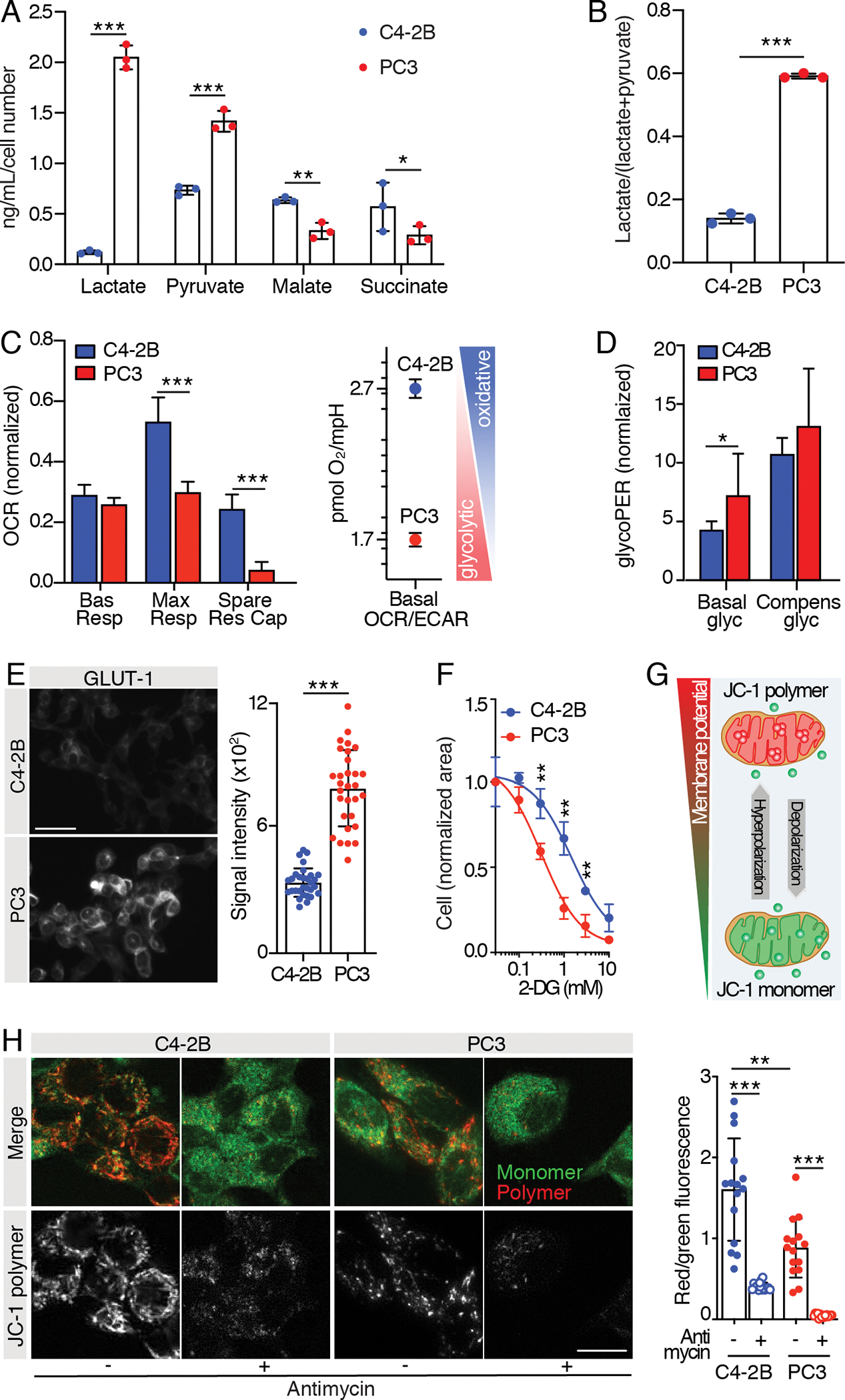
A) Mass spectrometry analysis of glucose-derived metabolites (lactate, pyruvate, malate and succinate) and B) glycolytic index “lactate/(lactate + pyruvate)” in C4–2B and PC3 24 hours-conditioned extracellular medium; n=3 wells/group, metabolite concentration values were normalized over cell-free culture medium metabolite-content and total cell number. C, D) Mitochondrial function parameters from Seahorse Mito Stress test (C) and Seahorse Glycolytic Rate assay (D) on C4–2B and PC3 cells; the experiments were performed 2 times, 1 representative experiment is shown, n=8 wells/group. Oxygen Consumption Rate (OCR) and glycolytic Proton Efflux Rate (glycoPER) values were normalized on the live cell area/well. E) Representative images of C4–2B and PC3 cells immunostained for glucose transporter 1 (GLUT-1) (scale bar: 50 μm); signal quantification is shown from 30 cells (10 cells/independent experiment); statistical significance was calculated on the average of the three experiments. F) Dose-response tumor cell growth curves in the presence of 2-DG; the experiments were performed 2 times, 1 representative experiment is shown, n=3 wells/group. G) Cartoon showing JC-1 probe potential-dependent accumulation and aggregation in functional mitochondria. H) Representative images of C2–2B and PC3 cells stained with JC-1 probe (scale bar: 20 μm); JC-1 polymer/monomer (red/green) fluorescence ratio is shown; n= 15 cells/group.
All the values are presented as mean ± SD; p-values were estimated through unpaired Student’s t test (A-F) or one-way ANOVA, followed by Tukey’s HSD post-hoc test (H): (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (no *) not significant, as indicated.
Inhibition of aerobic respiration in C4–2B and PC3 cells, in cell culture
Our results suggest that ARPC and AVPC cells are metabolically skewed towards glucose aerobic pathways or glycolysis, respectively. To test the dependency of C4–2B and PC3 cells on aerobic respiration, we performed interference experiments with IACS-10759, a clinical-grade, highly potent and selective small-molecule that inhibits complex I within the mitochondrial electron transport chain by preventing its binding with ubiquinone, thus severely impairing OXPHOS (21). IACS-10759 was dramatically more potent (~1500 fold) in C4–2B cells (IC-50, 0.312 nM) compared to PC3 cells (IC-50, 516.5 nM), suggesting that PC3 growth was largely independent of aerobic respiration (Fig. 3A). To monitor potential cytotoxic effects induced by IACS-10759, we quantified the number of PI+ cells at day 1 and 6 post-treatment at doses that inhibited cell growth (30 nM and 10 μM for C4–2B and PC3, respectively, as reported in Fig. 3A). We identified no significant difference in the percentage of PI+ dead cells between treated and control samples for both PCa cell subtype (Fig. 3B), indicating that IACS-10759 works as a cytostatic rather than cytotoxic agent in these cells, as previously reported for other cancer types (22).
Fig. 3. C4–2B and PC3 cells treatment with IACS-10759 in vitro.
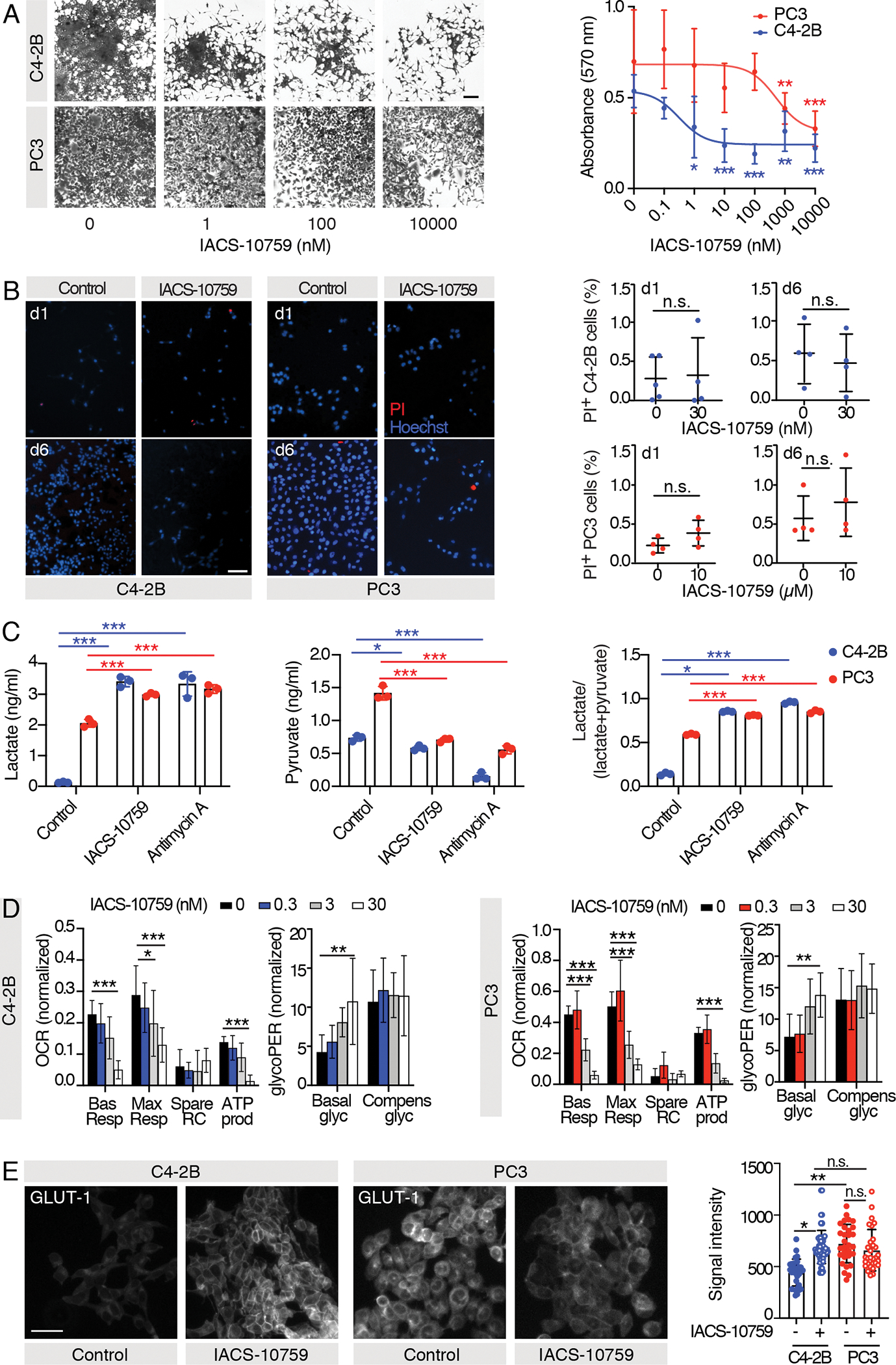
A) Representative images of crystal violet-stained C4–2B and PC3 cells cultured in 2D and treated with different concentrations of IACS-10759 and dose-response curves; the experiments were performed 2 times, 1 representative experiment is shown, n=8 wells/group; the P values shown refer to analyses performed against control-treated samples. B) Representative images of Propidium Iodide (PI)- and Hoechst-stained C4–2B and PC3 cells after 1 or 6 days of treatment with IACS-10759 and graphs showing the percentage of dead (PI+) cells; the experiments were performed 2 times, 1 representative experiment is shown, n=4–5 wells/group. C) Mass spectrometry analysis of glucose-derived metabolites (lactate, pyruvate) and glycolytic index “lactate/(lactate + pyruvate)” in 24 hours-conditioned extracellular medium of C4–2B and PC3 C4–2B cells treated with 200 nM IACS-10759 and 10 μM Antimycin-A; n=3 wells/group, metabolite concentration values were normalized over cell-free culture medium metabolite-content and total cell number. D) Mitochondrial function parameters from Seahorse Mito Stress test and Seahorse Glycolytic Rate assay on C4–2B and PC3 cells cultured in 2D and treated with different IACS-10759 concentrations; the experiments were performed 2 times, 1 representative experiment is shown, n=8 wells/group. E) Representative images of C4–2B and PC3 cells treated with 30 nM or 10 μM IACS-10759 and immunostained for glucose transporter 1 (GLUT-1) (scale bar: 50 μm); signal quantification is shown from 30 cells (10 cells/independent experiment); statistical significance was calculated on the average of the three experiments.
All the values are presented as mean ± SD; p-values were estimated through unpaired Student’s t test (B) or one-way ANOVA, followed by Tukey’s HSD post-hoc test (A,C-E): (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (n.s.) not significant, as indicated.
As glycolysis is negatively controlled by OXPHOS (Pasteur effect) (7), we expected that pharmacological inhibition of OXPHOS would lead to a compensatory upregulation of glycolysis to cope with the energetic stress. Accordingly, mass spectrometry showed decreased levels of pyruvate upon treatment with IACS-10759 and Antimycin A (used as positive control), while both lactate and glycolytic index increased (p < 0.0001) in both cell lines (Fig. 3C). Then, Seahorse Mito Stress tests and Glycolytic Rate assays (Fig. 3D; Fig. S2) were performed on PC3 and C4–2B cells treated for 24 hours with different doses of IACS-10759 (0–30 nM). We observed a dose dependent decrease in basal and maximal OCR and ATP produced by mitochondria in cells treated with IACS-10759. Notably, IACS-10759 efficiently inhibited PC3 cell aerobic metabolism, despite having no impact on cell proliferation (Fig. 3A), suggesting that this compound is biologically active but that PC3 cells are not dependent on OXPHOS for growth in cell culture conditions. We also observed a dose dependent increase in basal glycolysis following IACS-10759 treatment, but compensatory glycolysis remained near constant, indicating saturation. These data suggest the induction of a glycolytic switch by IACS-10759. Accordingly, C4–2B cells treated with 30 nM IACS-10759 for 24 hours showed increased expression of GLUT-1 compared to controls, while both control and IACS-10759-treated (10 μM) PC3 cells expressed comparable levels of this transporter (Fig. 3E), suggesting that maximum expression levels were achieved prior to complex I inhibition. Altogether, these results indicate that IACS-10759 rewired the metabolic demand, which was paralleled by an increased rate of glycolysis.
Inhibition of aerobic respiration in C4–2B and PC3 cells, in vivo
While in vitro studies of metabolism have been extremely informative over the years, emerging data have highlighted the importance of studying metabolism in vivo where the tumor microenvironment can have profound effects on cancer cell metabolism (23). To test the relative importance of OXPHOS in diverse models of ARPC and AVPC in vivo, we generated subcutaneous and intra-osseous prostate tumors with luciferase-expressing PCa cells in mice. We prioritized the use of PC3 and C4–2B cells lines, which were thoroughly characterized in vitro, and facilitated the longitudinal monitoring of therapeutic response in a non-invasive manner. Luciferase activity consumes ATP, which binds to the carboxy-terminal of a luciferin molecule as the first step of a series of reactions that lead to photon release (Fig. S3A), a process potentially affected by IACS-10759 administration. To rule out any bias, crystal violet staining (independent of cell metabolic activity) and bioluminescence analysis were carried out in parallel on luciferase-expressing C4–2B and PC3 cells treated with IACS-10759. Cell growth was assessed by means of bioluminescence imaging (BLI) and crystal violet staining at day 5 after treatment administration, showing no significant differences (Fig. S3B, C). This experiment validated the use of bioluminescence monitoring for our in vivo detection of tumor growth. Luciferase-expressing C4–2B and PC3 cells were implanted into NOD-SCID or athymic nude mice, respectively, and allowed to grow for 15 (PC3) or 30 (C4–2B) days subcutaneously or for 5 days in the tibia (Fig. 4A, D). Tumor burden was evaluated through BLI, mice were randomized based on the average BLI signal and treated with 0 or 10 mg/kg IACS-10759, 5 days/week for two weeks. Consistent with our in vitro results, C4–2B tumor growth was impaired by IACS-10759 in both the subcutaneous and intratibial models (Fig. 4B, C). Also consistent with our 2D culture results, IACS-10759 treatment had negligible effects on subcutaneous PC3 tumor growth. Surprisingly however, PC3 tumors grown in bone were sensitized to IACS-10759 (Fig. 4E, F). This response was also visible following a qualitative macroscopic observation of isolated bones, where the bone marrows of control-treated tibiae appeared to be largely replaced by tumor cells, while the bone marrow cavity of IACS-10759-treated bones was only partially colonized (Fig. S4). The observation of decreased glycolysis in AVPC samples from bone relative to other metastatic sites is supported by clinical data as interrogation of transcriptomic data from the Stand Up To Cancer (SU2C)/Prostate Cancer Foundation (PCF) database, which contains RNA-seq data from cohorts of bone as well as soft tissue biopsies (Fig. 4G) (24). These clinical correlates suggest that the organ-site specific differences in response an OXPHOS inhibitor and thus metabolism, may extend beyond PC3 cells to additional AVPCs. Overall, these data confirmed IACS-10759 efficacy in C4–2B cells in vivo but importantly revealed a new therapeutic window for AVPC PC3 cells implanted in bone.
Fig. 4. Macroscopic therapy response of PCa subcutaneous and intratibial tumor to IACS-10759 monitored by bioluminescence detection.
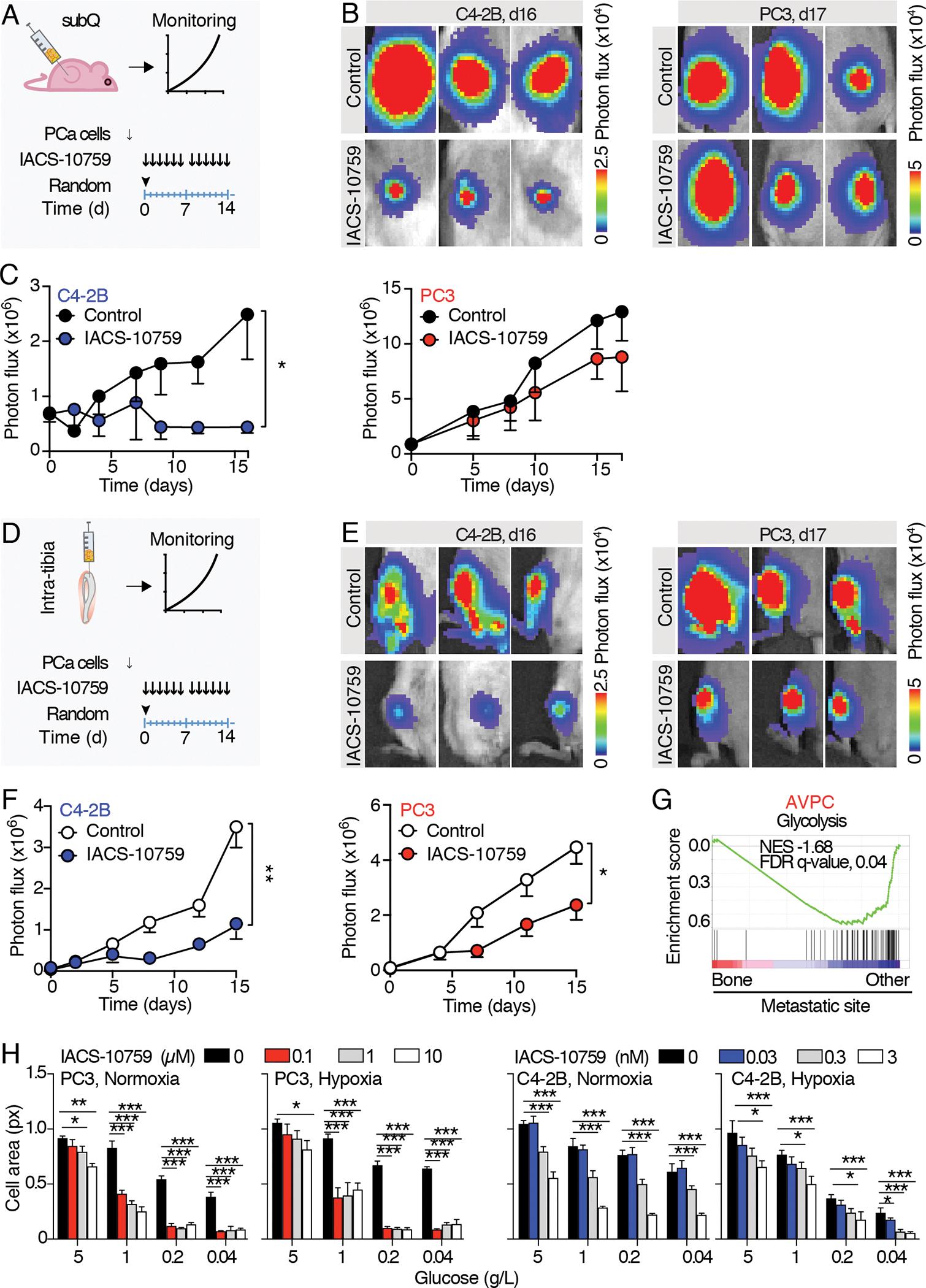
A) Schematic representation of the experimental schedule for subcutaneous tumors. 15 to 30 days after PC3/C4–2B tumor cells injection, mice were randomized and treated with IACS-10759 (10 mg/Kg in 0.5% methyl cellulose by oral gavage following a 5 days/week schedule). B) Representative images of subcutaneous tumor-derived bioluminescence detected by IVIS-200. C) Longitudinal monitoring of subcutaneous tumor growth; n=5 tumors per group. D) Schematic representation of the experimental schedule for intratibial tumors. 5 days after PC3/C4–2B tumor cells injection, mice were randomized and treated with IACS-10759 (10 mg/kg in 0.5% methyl cellulose by oral gavage following a 5 days/week schedule). E) Representative images of intratibial tumor-derived bioluminescence monitored by IVIS-200. F) Longitudinal monitoring of intratibial tumor growth; n=12 tibiae per group. Mean ± SD, p-values were estimated through unpaired Student’s T-test: (*) p < 0.05, (**) p < 0.01, as indicated. subQ = subcutaneous; random = randomization time; d = days. G) GSEA enrichment analysis performed on (24); Normalized enrichment scores (NES) and false discovery rate (FDR)-q values are shown. H) Tumor cell growth monitored by cell area of C4–2B and PC3 cells cultured in 2D in normoxic (18%) or hypoxic (1%) conditions in combination with different glucose concentrations (0.04–5 g/L) and treated with different IACS-10759 concentrations (0–10 μM, PC3; 0–3 nM, C4–2B); the experiments were performed 2 times, 1 representative experiment is shown, n=4 wells/group.
All the values are presented as mean ± SD; p-values were estimated through unpaired Student’s T-test (C, F) or one-way ANOVA, followed by Tukey’s HSD post-hoc test (H): (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (no *) not significant, as indicated.
In an attempt to recapitulate conditions mimicking the bone metastatic microenvironment such as hypoxia (where oxygen tension ranges from <1% – 6%), and hypoglycemia (25,26), we performed in vitro dose-responses with IACS-10759 (0–10 μM) under oxygen- and glucose-deprived conditions. PC3 and C4–2B cells were incubated in normoxia or hypoxia (18% or 1 % O2) in 5, 1, 0.2 or 0.04 g/L of glucose. IACS-10759 was added at 0, 0.1, 1, 10 μM or 0, 0.03, 0.3 or 3 nM, for PC3 or C4–2B cells, respectively, spanning 3 logarithms based on the cell-line specific IC-50. In PC3 cells, standard cell culture conditions (5 g/L glucose) prevented IACS-10759 from inhibiting cell growth at lower doses, as shown above, regardless of O2 status. However, decreasing levels of glucose sensitized cells to IACS-10759, an effect that was only partially enhanced under hypoxic conditions (Fig. 4H). Glucose reduction no or minimally sensitized C4–2B cells to IACS-10759, suggesting that maximum inhibition levels were already achieved (Fig. 4H).
These results suggest that PC3 cell metabolic vulnerabilities can be tuned differently based on local conditions and that a glucose-low environment can cause a metabolic shift away from dependence on glycolysis and towards increased OXPHOS.
Discussion
Although recent studies have provided some insight into PCa metabolism, a thorough characterization of the metabolic status of PCa subtypes has been lacking (9,10). This work showed that castration-resistant ARPC and AVPC rely on distinct metabolic functions, as monitored in patient-derived samples at the transcriptomic and proteomic levels, and in representative PCa cells models. Aerobic respiration is a driver of ARPC, both in vitro and in vivo, while AVPCs prefer glycolysis over respiration, but can reprogram their metabolism based on the site of growth. We hypothesized that pharmacological interference might provide a tool for monitoring the metabolic dependencies of PCa subtypes. To do this, we used IACS-10759, a potent complex I inhibitor that showed striking in vivo preclinical activity in multiple, preclinical tumor models, including leukemia, lung, brain, (21,27,28) and more recently, enzalutamide-resistant prostate cancer cells, in vitro (29,30).
AR drives key functions in both healthy and transformed prostate cells, including the regulation of glucose metabolism (10,31,32). Prostatic tissue has a unique metabolism that supports the secretion of citrate into the gland lumen. Secretion of citrate is mediated by the accumulation of zinc which inhibits its further metabolism by aconitase in the Krebs cycle. Conversely, PCa cell transformation drives a healthy and metabolically inefficient cell into a more energy efficient malignant counterpart, which re-reacquire the ability to complete the Krebs cycle. At this stage, AR expression persists. Then, disease progression toward an AR-indifferent disease results in a further transition from an OXPHOS-driven to a glycolytic-driven phenotype (33). Accordingly, ARPC tumors have been shown to express the mitochondrial pyruvate carrier under AR-mediate transcriptional regulation, which enables mitochondrial metabolism. AR-indifferent models, instead, lack mitochondrial pyruvate carrier expression (34). Our results are in line with this evidence, with ARPC cells being relatively skewed towards OXPHOS, whereas the more aggressive AVPC relies more on glycolysis.
Despite reliably reducing oxygen consumption rate in both cell lines, IACS-10759 induced dose-dependent cytostatic effects in ARPC C4–2B but not in AVPC PC3 cells in culture. Accordingly, when administered in vivo, C4–2B tumor growth was inhibited by IACS-10759 both in subcutaneous and intratibial models, while PC3 subcutaneous tumors were, as anticipated, not impacted. Interestingly, IACS-10759 impaired the growth of PC3 bone lesions, suggesting that the tumor microenvironment reprogrammed PC3 cell metabolism. A cross-species analysis comparing a novel genetically engineered mouse model that spontaneously develops bone metastases to two clinical cohorts of men with localized disease or bone metastases also reported that one of the top pathways increased in bone metastases was OXPHOS (35). Consistently, enrichment of OXPHOS signature has been recently identified in patient-derived bone samples compared to primary tumors (36). These correlative data, combined with our own functional data, support the idea that the bone forces cancer cells to shift their metabolism towards OXPHOS to survive this unique microenvironment. Some highly proliferating cancer cells generate energy through glycolysis even when oxygen levels are adequate and they have functional mitochondria, a phenomenon known as the Crabtree effect (37,38). This reversible and glucose-induced suppression of aerobic respiration can enhance the growth of tumor cells, allowing them to adapt their metabolism to heterogeneous microenvironments (37). Furthermore, it enables resistance to agents that target mitochondrial function, which is reverted in the presence of low glucose (38). PC3 cells followed this trend, and, in line with this evidence, were vulnerable to IACS-10759 at reduced glucose levels (Fig. 4H). A similar outcome was identified in chronic lymphocytic leukemia cells, which were sensitized to IACS-10759 in low-glucose culture conditions (39). However, both tumor intrinsic and extrinsic determinants of metabolic reprogramming by PCa cells in bone warrant further investigation, including the potential role of resident stromal cells and specific gene mutations that may also contribute to this environmental-dependent metabolic rewiring.
We detected significant metabolic differences across a cohort of patient-derived data at the transcriptomic level and of LuCaP PDXs at the proteomic level. Then, we performed a thorough characterization of PC3 and C4–2B models both at baseline and after IACS-10759 treatment, in vitro, followed by in vivo monitoring. To more extensively characterize the metabolic status of PCa subtypes in vivo, and their response to metabolic interference, we envision future studies testing different ARPC and AVPC PDXs that are derived from both primary and metastatic sites. While the LuCaP series was mostly generated from primary tumors and soft tissue-derived metastasis (11), different bone-derived PDX models have recently been described (40). Comparison of response to metabolic inhibition by bone-derived PDXs implanted subcutaneously or in bones, if possible, could further define a persistent bone-induced metabolic reprogramming of tumor cells vs local microenvironmental modulation.
IACS-10759 has advanced into the clinic for leukemia and advanced-stage solid tumors (including 3 castration-resistant PCa patients). As a drawback, some patients displayed severe neurological side effects (41). Interestingly, a patient with heavily pretreated castration-resistant PCa achieved partial response with resolution of cancer-related pain. We observed that IACS-10759 has an IC-50 of 0.312 nM in C4–2B cells, ~10 times more effective than AML cells (21), which may explain the promising clinical results achieved in the castration-resistant PCa patient (41). The remarkable response to IACS-10759 by both C4–2B and PC3 bone lesions support further clinical investigation in castration-resistant PCa patients with bone metastasis. This disease currently has limited options available for its treatment and remains lethal for patients, but a unique therapeutic window may exist for OXPHOS inhibitors. Interestingly, enzalutamide-resistant AR+ PCa cells rely more on mitochondrial metabolism as compared to enzalutamide-sensitive AR+ PCa cells by switching from glycolysis to OXPHOS, and show increased sensitivity to agents that inhibit the electron transport chain, including IACS-10759 (29,30). This evidence further supports the important role that OXPHOS plays in helping ARPCs cope with various environmental stresses and provides further rationale for testing inhibitors of central carbon metabolism in CRPC patients.
In conclusion, our data highlight subtype and site-specific-induced metabolic vulnerabilities in PCa, which can be further explored to develop targeted treatment options.
Supplementary Material
Implications:
These vulnerabilities may be exploited with mechanistically novel treatments, such as those targeting OXPHOS alone or possibly in combination with existing therapies. In addition, our findings underscore the impact of the tumor microenvironment in reprogramming prostate cancer metabolism.
Acknowledgments
Funding: The National Cancer Institute, Prostate Cancer SPORE (P50CA140388–09; E.D., D.E.F, C.J.L.); the DH Koch Center for Applied Research of Genitourinary Cancers (E.D., C.J.L.); the National Institutes of Health (P30CA016672; C.J.L.); the establishment and characterization of the LuCaP PDX models was supported by the PNW Prostate Cancer SPORE P50CA097186 and P01CA163227 (E.C, T.S.G).
We would like to thank the patients who generously donated tissue that made the generation of these models possible and the GU Cancer lab personnel and the UW Comparative Medicine Animal Caregivers for assistance with the LuCaP xenograft work.
Footnotes
Conflict of interest statement: the authors declare no potential conflicts of interest.
References
- 1.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res 2014;20:2846–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin Cancer Res 2016;22:1520–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helpap B, Kollermann J, Oehler U. Neuroendocrine differentiation in prostatic carcinomas: histogenesis, biology, clinical relevance, and future therapeutical perspectives. Urol Int 1999;62:133–8 [DOI] [PubMed] [Google Scholar]
- 5.Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N Engl J Med 2019;381:121–31 [DOI] [PubMed] [Google Scholar]
- 6.Gandaglia G, Abdollah F, Schiffmann J, Trudeau V, Shariat SF, Kim SP, et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014;74:210–6 [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 2017;14:11–31 [DOI] [PubMed] [Google Scholar]
- 8.Kelly RS, Sinnott JA, Rider JR, Ebot EM, Gerke T, Bowden M, et al. The role of tumor metabolism as a driver of prostate cancer progression and lethal disease: results from a nested case-control study. Cancer Metab 2016;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin C, Salzillo TC, Bader DA, Wilkenfeld SR, Awad D, Pulliam TL, et al. Prostate Cancer Energetics and Biosynthesis. Adv Exp Med Biol 2019;1210:185–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bader DA, McGuire SE. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nature Reviews Urology 2020;17:214–31 [DOI] [PubMed] [Google Scholar]
- 11.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017;77:654–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bello T, Paindelli C, Diaz-Gomez LA, Melchiorri A, Mikos AG, Nelson PS, et al. Computational modeling identifies multitargeted kinase inhibitors as effective therapies for metastatic, castration-resistant prostate cancer. Proceedings of the National Academy of Sciences 2021;118:e2103623118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue C, Corey E, Gujral TS. Proteomic and Transcriptomic Profiling Reveals Mitochondrial Oxidative Phosphorylation as Therapeutic Vulnerability in Androgen Receptor Pathway Active Prostate Tumors. Cancers (Basel) 2022;14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zacharias N, Lee J, Ramachandran S, Shanmugavelandy S, McHenry J, Dutta P, et al. Androgen Receptor Signaling in Castration-Resistant Prostate Cancer Alters Hyperpolarized Pyruvate to Lactate Conversion and Lactate Levels In Vivo. Molecular Imaging and Biology 2019;21:86–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017;32:474–89.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487:239–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007;23:3251–3 [DOI] [PubMed] [Google Scholar]
- 18.Corey E, Brown LG, Quinn JE, Poot M, Roudier MP, Higano CS, et al. Zoledronic acid exhibits inhibitory effects on osteoblastic and osteolytic metastases of prostate cancer. Clin Cancer Res 2003;9:295–306 [PubMed] [Google Scholar]
- 19.Seim I, Jeffery PL, Thomas PB, Nelson CC, Chopin LK. Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3 (Bethesda) 2017;7:1731–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai S, Sun Y, Squires JM, Zhang H, Oh WK, Liang CZ, et al. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011;71:1668–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036–46 [DOI] [PubMed] [Google Scholar]
- 22.Vangapandu HV, Alston B, Morse J, Ayres ML, Wierda WG, Keating MJ, et al. Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells. Oncotarget 2018;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayers JR, Vander Heiden MG. Famine versus feast: understanding the metabolism of tumors in vivo. Trends in Biochemical Sciences 2015;40:130–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014;508:269–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiedemann K, Hussein O, Komarova SV. Role of Altered Metabolic Microenvironment in Osteolytic Metastasis. Front Cell Dev Biol 2020;8:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nature Medicine 2018;24:1047–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu X, Han L, Busquets J, Collins M, Lodi A, Marszalek JR, et al. The Combined Treatment With the FLT3-Inhibitor AC220 and the Complex I Inhibitor IACS-010759 Synergistically Depletes Wt- and FLT3-Mutated Acute Myeloid Leukemia Cells. Front Oncol 2021;11:686765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu HS, Wilganowski N, Robertson S, Reuben JM, Cohen EN, Zurita A, et al. Prostate cancer cells survive anti-androgen and mitochondrial metabolic inhibitors by modulating glycolysis and mitochondrial metabolic activities. The Prostate 2021;81:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowell PD, Giafaglione JM, Jones AE, Nunley NM, Hashimoto T, Delcourt AML, et al. Androgen receptor inhibition induces metabolic reprogramming and increased reliance on oxidative mitochondrial metabolism in prostate cancer. bioRxiv 2022:2022.05.31.494200 [Google Scholar]
- 31.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. Embo j 2011;30:2719–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giunchi F, Fiorentino M, Loda M. The Metabolic Landscape of Prostate Cancer. European Urology Oncology 2019;2:28–36 [DOI] [PubMed] [Google Scholar]
- 33.Spratt DE, Gavane S, Tarlinton L, Fareedy SB, Doran MG, Zelefsky MJ, et al. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate 2014;74:1153–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bader DA, Hartig SM, Putluri V, Foley C, Hamilton MP, Smith EA, et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nature Metabolism 2019;1:70–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arriaga JM, Panja S, Alshalalfa M, Zhao J, Zou M, Giacobbe A, et al. A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance. Nature Cancer 2020;1:1082–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitburn J, Rao SR, Morris EV, Tabata S, Hirayama A, Soga T, et al. Metabolic profiling of prostate cancer in skeletal microenvironments identifies G6PD as a key mediator of growth and survival. Science Advances 2022;8:eabf9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2011;1807:568–76 [DOI] [PubMed] [Google Scholar]
- 38.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree Effect: Replacing Media Glucose with Galactose Increases Susceptibility of HepG2 Cells to Mitochondrial Toxicants. Toxicological Sciences 2007;97:539–47 [DOI] [PubMed] [Google Scholar]
- 39.Vangapandu HV, Alston B, Morse J, Ayres ML, Wierda WG, Keating MJ, et al. Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells. Oncotarget 2018;9:24980–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palanisamy N, Yang J, Shepherd PDA, Li-Ning-Tapia EM, Labanca E, Manyam G, et al. The MD Anderson prostate cancer patient-derived xenograft series (MDA PCa PDX) captures the molecular landscape of prostate cancer and facilitates marker-driven therapy development. Clin Cancer Res 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yap TA, Ahnert JR, Piha-Paul SA, Fu S, Janku F, Karp DD, et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. Journal of Clinical Oncology 2019;37:3014–3014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated from this study are available within the article and its Supplementary Data.