

Abstract
Deregulation of N-myc is a leading cause of malignant brain tumors in children. To target N-myc-driven medulloblastoma, most research has focused on identifying genomic alterations or on the analysis of the medulloblastoma transcriptome. Here, we have broadly characterized the translatome of medulloblastoma and shown that N-myc unexpectedly drives selective translation of transcripts that promote protein homeostasis. Cancer cells are constantly exposed to proteotoxic stress associated with alterations in protein production or folding. It remains poorly understood how cancers cope with proteotoxic stress to promote their growth. Here, our data revealed that N-myc regulates the expression of specific components (~5%) of the protein folding machinery at the translational level through the major cap binding protein, eukaryotic initiation factor eIF4E. Reducing eIF4E levels in mouse models of medulloblastoma blocked tumorigenesis. Importantly, targeting Hsp70, a protein folding chaperone translationally regulated by N-myc, suppressed tumor growth in mouse and human medulloblastoma xenograft models. These findings reveal a previously hidden molecular program that promotes medulloblastoma formation and identify new therapies that may have impact in the clinic.
Introduction
Myc proteins feature prominently in medulloblastoma, the most common malignant pediatric brain tumor. Medulloblastoma exists in four molecular subgroups: WNT and Sonic Hedgehog (SHH), as well as two additional subgroups (Group3 and Group4) that show some overlap (1). SHH and Group 3/4 tumors are characterized by high levels of Myc or N-myc proteins, functional markers that predict poor survival. A highest-risk subgroup of SHH driven-tumors shows amplification of MYCN, rendering tumors resistant to clinical inhibitors that target upstream activators (2–5). Myc proteins remain undruggable and therefore, there is an urgent need to target these tumors, with pathways downstream of N-myc representing potential therapeutic targets.
The cellular-myelocytomatosis (Myc/c-myc) and closely related neuroblastoma-derived myc (N-myc) proteins are basic helix-loop-helix leucine zipper transcription factors that localize to the nucleus and form heterodimers with Myc-associated protein X (Max), binding to DNA at specific “E-box” sequences to drive transcription of targets important for proliferation (6,7). Importantly, Myc is one of the few transcription factors that can control expression of many components of the translation machinery, including translation initiation and elongation factors, tRNA synthetases, nucleolar assembly components and ribosomal proteins (8–15). It has been shown that Myc’s oncogenic capacity relies heavily on its ability to regulate protein synthesis (16). We therefore set out to delineate for the first time the translational landscape of medulloblastoma formation that has historically been solely examined at the level of the transcriptome or genomic analysis.
Here, we harness the power of ribosome profiling to uncover the cancer translatome of medulloblastoma. Interestingly, we identified a specific subset of mRNAs involved in protein folding that were translationally regulated in MYCN-driven medulloblastoma. Moreover, we showed that the major cap-binding protein eIF4E was the key regulator of this translational specificity downstream of N-myc. Reducing eIF4E levels in mouse models for medulloblastoma blocked tumorigenesis. Our data further point to a mechanism by which the translation of specific components of the protein folding machinery is necessary to accommodate increased protein synthesis capacity to prevent proteotoxic stress, a vulnerability for cancer cell survival. In support of this model, we showed that a small molecule inhibitor of Hsp70, the chaperone that guides protein folding, specifically suppressed tumor growth in mouse and human medulloblastoma xenografts. This study unveils a novel translationally controlled pathway that links the protein synthesis and protein folding machineries, enabling cancer cells to augment protein dosage without triggering proteotoxic stress, to maintain cancer cell survival. Thereby, these studies identify a new therapeutic vulnerability for medulloblastoma that may have important implications in the clinic.
Materials and Methods
Cell culture
GTML cells were cultured in complete neurobasal culture media (Gibco) supplemented with B27 (2%), L-Glutamine (1x), EGF (20 μg/L), bFGF (20 μg/L) and Penicillin-streptomycin at 37C. Accutase (Innovate Cell Tech) was used to dissociate the cells. Scramble and eIF4E shRNA expressing plasmids were transiently transfected via Lipofectamine 2000 and cells were harvested 48h post transfection. Cells were regularly tested for mycoplasma contamination by MycoAlert mycoplasma detection kit (Lonza).
Ribosome Profiling
10 million GTML cells were seeded on a poly-ornithine/laminin coated 10 cm plate as triplicates and 24 h post seeding, Rapamycin (5 nM) and Rapalink-1 (50 nM) treatment for 3 hours were performed. Cells were treated with 0.1 mg/ml cycloheximide for 1 min and harvested. Samples were prepared by using ARTseq Ribosome Profiling Kit (Epicentre). For each gene, RNA and ribosome footprint (RFP) abundance were calculated (Supplementary Tables 1 and 2). Translation efficiency is calculated by normalizing the average RFP by the mRNA abundance (Supplementary Table 3). GO term analysis was performed using DAVID. Ribosomal protein and translation factors downregulated upon Rapalink-1 treatment were not taken into consideration in the analysis to highlight the novel targets identified in the experiment.
Western Blotting and Antibodies
The western blot analysis of GTML cells was performed by lysing cell pellets in a RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% Na deoxycholate,1mM EDTA, 1mM DTT) with the addition of PhosSTOP and Complete Mini proteasome inhibitors (Roche). Protein lysates were denatured using a protein sample buffer and separated on an SDS-PAGE gel (BioRad, gradient). The samples were transferred to a nitrocellulose membrane using a wet-transfer system from BioRad. The membrane was then blocked with PBS-Tween-20 (0.1%) containing 5% nonfat dry milk for at least 30 min and incubated with the primary antibody overnight. The membrane was washed three times with PBS-Tween and incubated with the secondary antibodies conjugated to horseradish peroxidase (HRP). Super Signal West Pico Plus Chemiluminescent substrate (Thermo Scientific) was used to develop the membranes on a Chemidoc imaging system (BioRad).
Antibodies used in this study: MYCN (Abcam, ab16898, RRID:AB_443533), p-AKTS473 (Cell Signaling, #4060, RRID:AB_2315049), Total AKT (Cell Signaling, #4685, RRID:AB_2225340), p-4EBP1T37/46 (Cell Signaling, #2855, RRID:AB_560835), Total 4EBP1 (Cell Signaling, #9644, RRID:AB_2097841), p-S6KS240/244 (Cell Signaling, #5364, RRID:AB_10694233), GAPDH (MAB374), P4HB (Bethyl A304–790A), PFDN5 (Bethyl, A304–049A), HSP90AB1 (ProteinTech 11405–1-AP, RRID:AB_2121207), HSPA8 (StressMarq SMC-151, RRID:AB_2120165), ST13 (ProteinTech 26581–1-AP, RRID:AB_2880560), DNAJA2 (ProteinTech 12236–1-AP, RRID:AB_2230709), Actin (Sigma, A5316, RRID:AB_476743), eIF4E (BD, 610270, RRID:AB_397665).
Polysome Profiling and quantitative polymerase chain reaction (qPCR)
10 million GTML cells were seeded on a 10 cm plate. Once they reach 70–80% confluency, the cells were treated for 3 hours with DMSO, Rapamycin and Rapalink-1 (same concentrations indicated in the ribosome profiling). Next, cells were incubated with 100 μg/ml cycloheximide (Sigma) for 3min in the culture. Cells were then washed with PBS containing cycloheximide and resuspended in lysis buffer containing 10mM Tris-HCl (pH 8.0), 150mM NaCl, 1.5mM MgCl2, 0.25% NP-40, 0.1% Triton X-100, 50mM DTT, 100 μg/ml cycloheximide, and 0.5 U/ml Rnasin for 30–45min on ice, vortexing every 10min. Lysates were centrifuged at 10,000rpm at 4 C for 10 min. The supernatants were adjusted by OD260 concentration and loaded onto a 10%–50% sucrose gradient before centrifugation at 38,000 rpm for 2.5 hr at 4C in a Beckman L8–70M ultracentrifuge. Samples were separated on a Biocomp fractionation system to evaluate polysome profiles and collect polysome fractions. 13 fractions were collected for each sample in total and RNA was isolated from each fraction, except the first one. 750ul of Trizol LS (Invitrogen), 0.4ul of 1 mg/ml glycogen (Roche) and 10–20 ng in vitro transcribed Renilla luciferase spike mRNA was added to 250ml of sucrose lysate and incubated for 15min at room temperature. 200ul of chloroform was added to the mix before spinning down the samples for 15 min at 12,000 g at 4C. RNA was precipitated by adding 500ul of isopropanol to the supernatant overnight at 80C. The next day the samples were spun down for 20min at 12,000 g at 4C. The pellet obtained was washed with 70% ethanol and resuspended in dH2O. Any trace DNA was removed with the Invitrogen Turbo DNA-free kit (Invitrogen, AM1907). Single-stranded cDNA was synthesized by using same volume of RNA for each fraction with High-Capacity cDNA Reverse transcription kit (Applied Biosystems, Cat no. 4368814). cDNA samples were diluted 1:10 and 1 μL of template was used in a PowerUP SYBR Green master mix reaction run on an QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems).
35S Metabolic Labeling
GTML cells were incubated in media containing 20 μCi 35S for 1 hour. After washing the cells carefully with D-PBS containing 100 μg/ml cycloheximide, cells were lysed in RIPA buffer and equal amount of total protein were separated out on a 10% SDS-PAGE gel. Once the samples were transferred to a nitrocellulose membrane, the membrane was exposed to autoradiography film for overnight and then developed. 35S methionine/cysteine incorporation was quantified using ImageJ software (RRID:SCR_003070) by normalizing to Actin levels.
Luciferase reporter assay
The 5′UTR of Hspa8 was cloned into the pGL3 luciferase reporter vector (Promega) in which the downstream Firefly luciferase open reading frame (ORF) was replaced with the Renilla luciferase (Rluc) ORF. GTML cells expressing either control or eIF4E shRNAs were seeded at a density of 125k cell/well on a 12-well plate. After 24h, cells were transfected with 500 ng of pGL3–5’UTR-RLuc-Sv40 using Lipofectamine 2000 (Invitrogen). 24h post-transfection, cells were harvested: a fraction was lysed in Trizol for RNA extraction and the rest were lysed in a passive lysis buffer for 20 min. The Rluc activity was measured using the Dual-luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions, using a Glomax microplate luminometer (Promega). RNA extraction was performed using the Zymogen DirectZol kit, following manufacturer’s instructions, and the TurboDNA-free kit (Thermofisher) was used to remove transfected DNA completely. cDNA synthesis was performed by the SuperScript III first-strand synthesis kit (Invitrogen) using specific oligos for RLuc and GAPDH. qPCR analyses were performed as described previously, and Rluc activity was normalized to Rluc mRNA expression.
Edu/DAPI Staining
GTML cells were treated with Rapalink-1 for 24h and stained using Click-iT Plus EdU Alexa Fluor 647 Flow Cytometer Assay kit (Thermofisher, C10634) following manufacturer’s protocol. Newly synthesized DNA is analyzed using the 637 nm laser of the flow cytometer to identify S phase. In parallel, DAPI staining was performed to distinguish G0-G1 and G2 phases, using 405 laser.
IHC
Immunostaining was performed on formalin-fixed paraffin-embedded tumor and brain tissue. In brief, the paraffin blocks were sliced into 5-μM thick sections, deparaffinized with xylene substitute (Sigma) and rehydrated with decreasing concentrations of ethanol in water. For antigen retrieval tissue slides were incubated in 95 °C sodium citrate buffer (pH 6.0) for 20 min in a steamer followed by 20 min of cooling at room temperature. Quenching was performed by incubating the slides in 0.3% hydrogen peroxide (EMD Millipore) for 15 min. Tissue was permeabilized in blocking buffer consisting of 0.4% Triton-X-100/10% normal goat serum/PBS for 30 min at room temperature. Diluted primary antibodies in PBS were applied overnight at 4 °C in a humidified chamber (anti-Hsc70 #ab51052 abcam, 1:50, anti-pan-Akt #4685 cell signaling 1:200). Sections were incubated in biotinylated secondary antibody (goat anti-rabbit IgG or goat anti-mouse, Vector Laboratories) for 60 min at room temperature in a humidified chamber. For signal amplification slides were incubated with Vectastain ABC reagent (Vector Laboratories) for 30 min. Color development was achieved by applying diaminobenzidine tetrahydrochloride (DAB) solution (Vector Laboratories) for two to five minutes, depending on the primary antibody. The duration of DAB incubation was held constant for all of the slides. Sections were counterstained with haematoxylin (Vector Laboratories), dehydrated through ethanol and xylene substitute, and cover-slipped using mounting medium. Stained sections were imaged using a digital microscope (Keyence BZ-X) and BZ-X Keyence Software (RRID: SCR_016979) at 4x and 10x magnification.
In Vivo Studies
All animal experiments were conducted using protocols approved by the University of California, San Francisco’s Institutional Animal Care and Use Committee (IACUC). For GTML xenograft models, 104 cells; for MYCN-NES models, 2×106 cells were subcutaneously injected into NSG mice. Once tumors are palpable, mice were randomized into groups receiving either vehicle or 4 mg/kg JG-231. Both groups were treated every other day for five weeks. Tumor volumes were measured twice every week and mouse body weight were measured every other day for accurate dosing. The formulation to dissolve JG-231 is 10% DMSO, 18% Kolliphor RH 40, 3.6% dextrose, 68.4% HEPES buffer (1M stock, pH 7.3). Sucrose was supplemented to 15% in drinking water. Mice were monitored daily and euthanized when they exhibited neurological deficits or 15% reduction from initial body weight.
Data Availability
All the plasmids used in this manuscript are available upon request. The ribosome profiling data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE215298.
Results
Ribosome profiling reveals preferentially translated mRNAs in MYCN-driven medulloblastoma
To identify the translational landscape of medulloblastoma, we employed unbiased genome wide ribosome profiling in cells originated from our germline genetically engineered mouse model (GEMM) for MYCN-driven medulloblastoma (GLT1-TRE-MYCN/Luciferase-GTML), in which a human MYCN transgene was targeted to murine hindbrain neural progenitor cells (17). We previously showed that MYCN-driven medulloblastoma is characterized by increased mTORC1 activity, a critical regulator of translation control (18). Therefore, we took advantage of two pharmacological approaches to target mTORC1 activity downstream of N-myc. Specifically, medulloblastoma cells were treated for three hours with the ATP-site inhibitor Rapalink-1, which blocks the two translation arms, ribosomal protein S6 kinase (S6K) and eIF4E cap-binding protein downstream of mTORC1; or rapamycin, an allosteric inhibitor that blocks S6K but not eIF4E signaling (Fig. 1A). Western blot analysis confirmed the specificity of the inhibitors (Fig. 1B). Both total mRNA and ribosome protected mRNAs were sequenced. Translational efficiency was measured by the ratio of ribosome footprint density to total RNA density.
Figure 1. Ribosome profiling reveals preferentially translated mRNAs in MYCN-driven medulloblastoma.
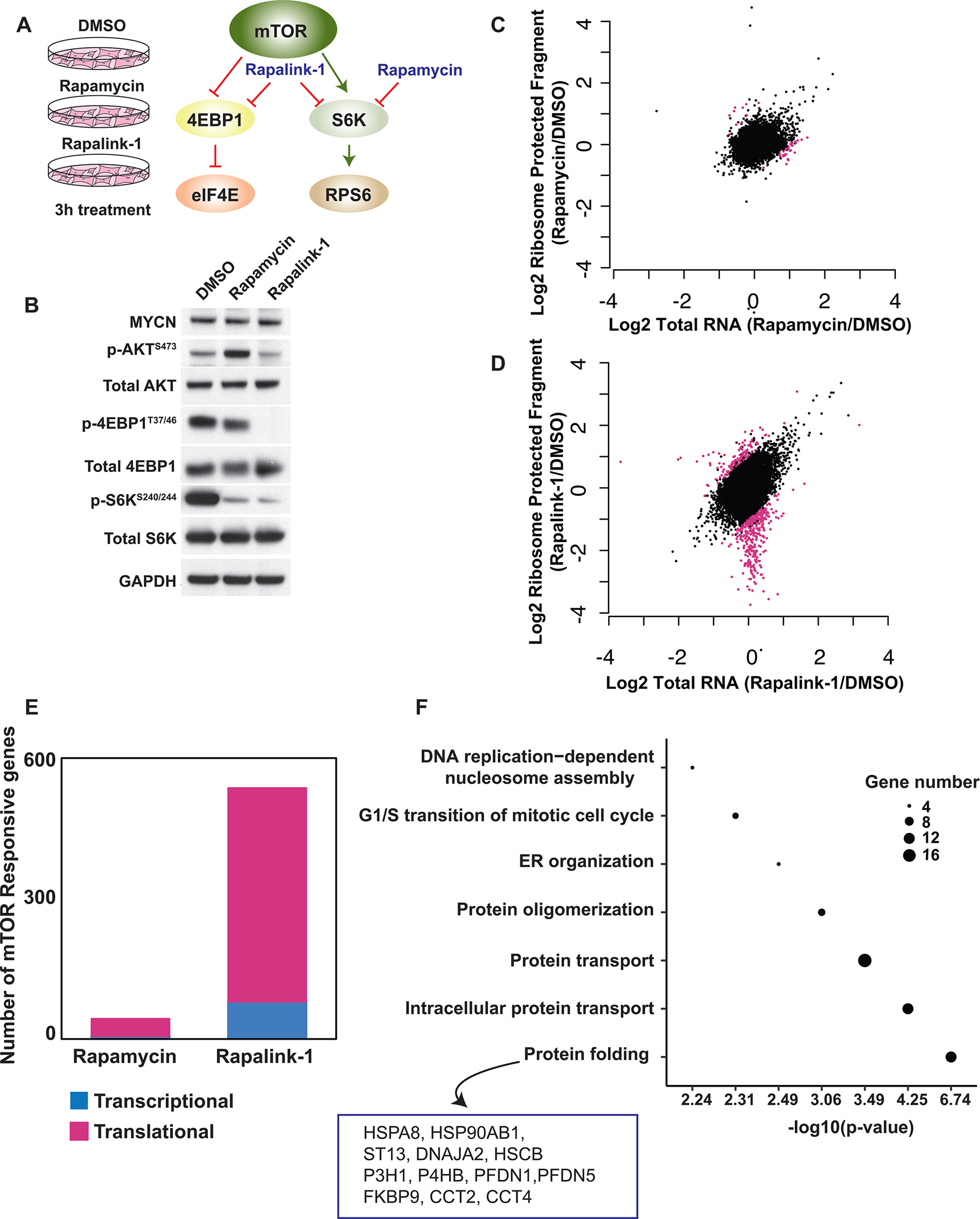
A, The mechanism of action of two mTOR inhibitors, Rapamycin and Rapalink-1, used in the ribosome profiling. B, Western blot analysis showing the specificity of Rapamycin and Rapalink-1 treatment for 3 hours in GTML cells. C-D, Scatter blots comparing total RNA and ribosome protected fragments of each transcript upon 3 hours Rapamycin (C) and Rapalink-1 (D) treatment compared to DMSO treated samples. Pink dots represent translationally regulated mRNAs. E, Bar graph representing number of mTOR responsive genes regulated at the transcriptional (blue) or translational (pink) level upon treatment. F, GO term analysis of translationally regulated targets uncovering specific gene categories. The diameter of each dot represents number of genes in each category. Genes identified in protein folding cluster are listed.
After three hours, rapamycin affected expression of 69 genes at the level of transcription, and 45 at the level of translation (Fig. 1C and E). In contrast, Rapalink-1, while modestly altering expression of 134 genes at the mRNA level, markedly affected translation of a specific network of 650 mRNAs (Fig. 1D and E). Ribosome profiling analysis uncovered 359 target mRNAs which were selectively repressed at the translational level in response to Rapalink-1 (log2 <=−1, (FDR)< 0.01), with limited changes in transcription (Fig. 1E, Supplementary Fig. S1, Supplementary Tables 1–3). As previously described, mTORC1 inhibition affects translation of ribosomal proteins and translation factors (19). In this study, we focused on novel translational targets. Surprisingly, GO term analysis of these select translationally repressed targets showed an enrichment in protein folding, protein transport, protein oligomerization, endoplasmic reticulum organization and DNA replication-dependent nucleosome assembly categories (Fig. 1F). These data characterize a previously unrecognized influence of N-myc on translational specificity through the mTOR pathway.
Protein folding machinery is regulated at the translational level by the mTOR pathway
The translation control of specific components of the folding machinery in Figure 1 was particularly intriguing. We hypothesized that while N-myc increased the protein synthetic capacity of cancer cells, it also specifically activated molecular chaperones that aided protein folding in order to balance protein homeostasis, a crucial hallmark of tumorigenesis.
The protein folding machinery is composed of 262 proteins. Strikingly, only five percent (12 members) of this cluster were regulated at the translational level. The majority of the translationally regulated members were involved in distinct steps of HSP70 and HSP90 machineries (20) that help proteins to fold independently of ribosome-associated chaperones (Fig 2A). To validate the ribosome profiling results, we performed polysome sucrose gradient assays in GTML cells to accurately monitor the translation efficiency of specific mRNAs. Polysome fractionation separates mRNAs that are highly translated (associated with multiple ribosomes or ‘high polysome’) from those that are either poorly translated (associated with few ribosomes or ‘low polysome’) or not translated at all (not associated with actively translating ribosomes or ‘Free/mRNP’) (Fig. 2B).
Figure 2. Protein folding machinery is regulated at the translational level by the mTOR pathway.
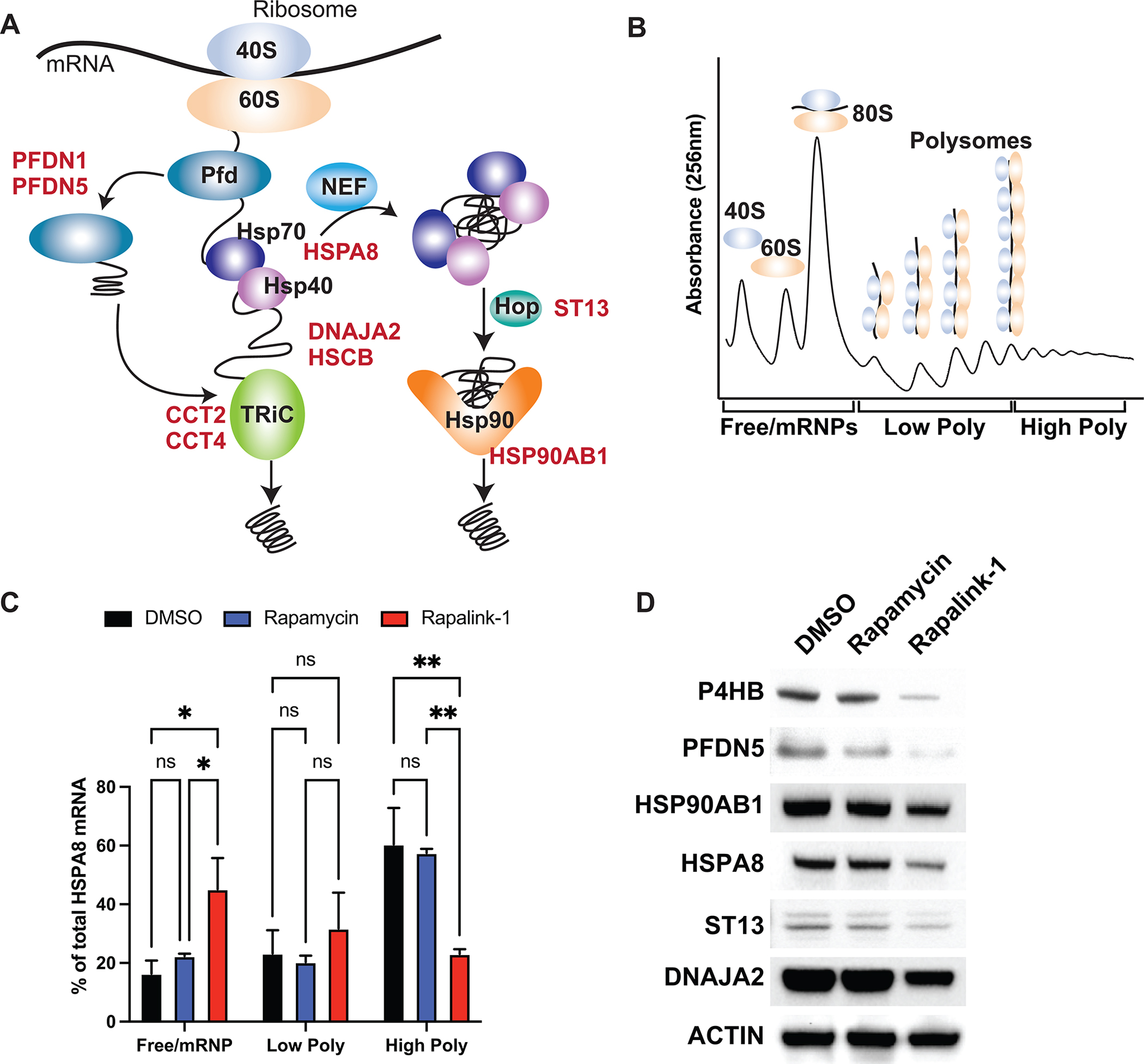
A, Schematic representation of Hsp70 and Hsp90 machineries. B, Cartoon representation of polysome profiling. C, qPCR analysis of HSPA8 mRNA upon Rapalink-1 and Rapamycin treatment, isolated from 10% - 50% sucrose gradient fractions. Pooled fractions corresponding to free ribosomal subunits and messenger ribonucleoprotein (mRNPs) (Free/mRNPs), low-molecular-weight polysomes (Low Poly), and high-molecular-weight polysomes (High Poly) are displayed. A two-way ANOVA test was performed to determine statistical significance (n = 3). D, Western blot analysis of translationally regulated endogenous proteins upon treatment. All values represent the mean + SEM. *p < 0.05, **p < 0.01
The transcripts of molecular chaperones shifted from highly translating polysomes to low polysomes or non-translationally active fractions in Rapalink-1-treated GTML cells, validating their repression at the translational level. We validated translational downregulation of mRNAs involved in protein folding, such as HSPA8 (HSC70, constitutively expressed HSP70 paralog), DNAJA2, PFDN5, P4HB, HSP90AB1 and ST13 (Fig. 2C and Supplementary Fig. S2A and S2B). In agreement with polysome profiling, protein levels of these targets were reduced only in response to Rapalink-1 treatment (Fig. 2D) while mRNA levels did not change (Supplementary Fig. S3A), showing that this specific subset of proteins was translationally regulated in our MYCN-driven GEMM. Importantly, B2M mRNA did not shift significantly upon exposure to Rapalink-1 or rapamycin, highlighting the specificity of the target mRNAs (Supplementary Fig S2B). Although Rapalink-1 treated cells produced a larger 80S peak in polysome profiling (Supplemental Fig. S2A), we did not observe significant changes in global protein synthesis as measured by incorporation of 35S-labeled methionine/cysteine upon 3h drug treatment (Supplemental Fig. S3B). We also treated the cells with Rapalink-1 for 24 hours and examined cell proliferation and survival (Supplemental Fig. S3C–D). Our data showed 20% decrease in cell division and cell proliferation upon Rapalink-1 treatment for longer treatment, suggesting that targeting the pathways regulated by Rapalink-1 can be beneficial to suppress tumor growth.
Inhibition of HSP70-BAG interactions suppresses tumor growth in vivo
Next, we used a pharmacological approach to validate that specific components of the protein folding machinery were required for cell proliferation and growth of tumors in MYCN-driven medulloblastoma. Several compounds have been developed to target two major components of the protein folding machinery, HSP70 and HSP90 (21,22), both of which are translationally regulated. Therefore, we used different compounds targeting these proteins (Fig. 3A), initially monitoring effects on viability by performing Cell-Titer Glo assays on two distinct GTML cell lines derived from two mice (Fig. 3B and C, Supplementary Fig S4A; GTML5 and GTML3).
Figure 3. Inhibition of Hsp70-BAG interactions suppresses tumor growth in vivo.
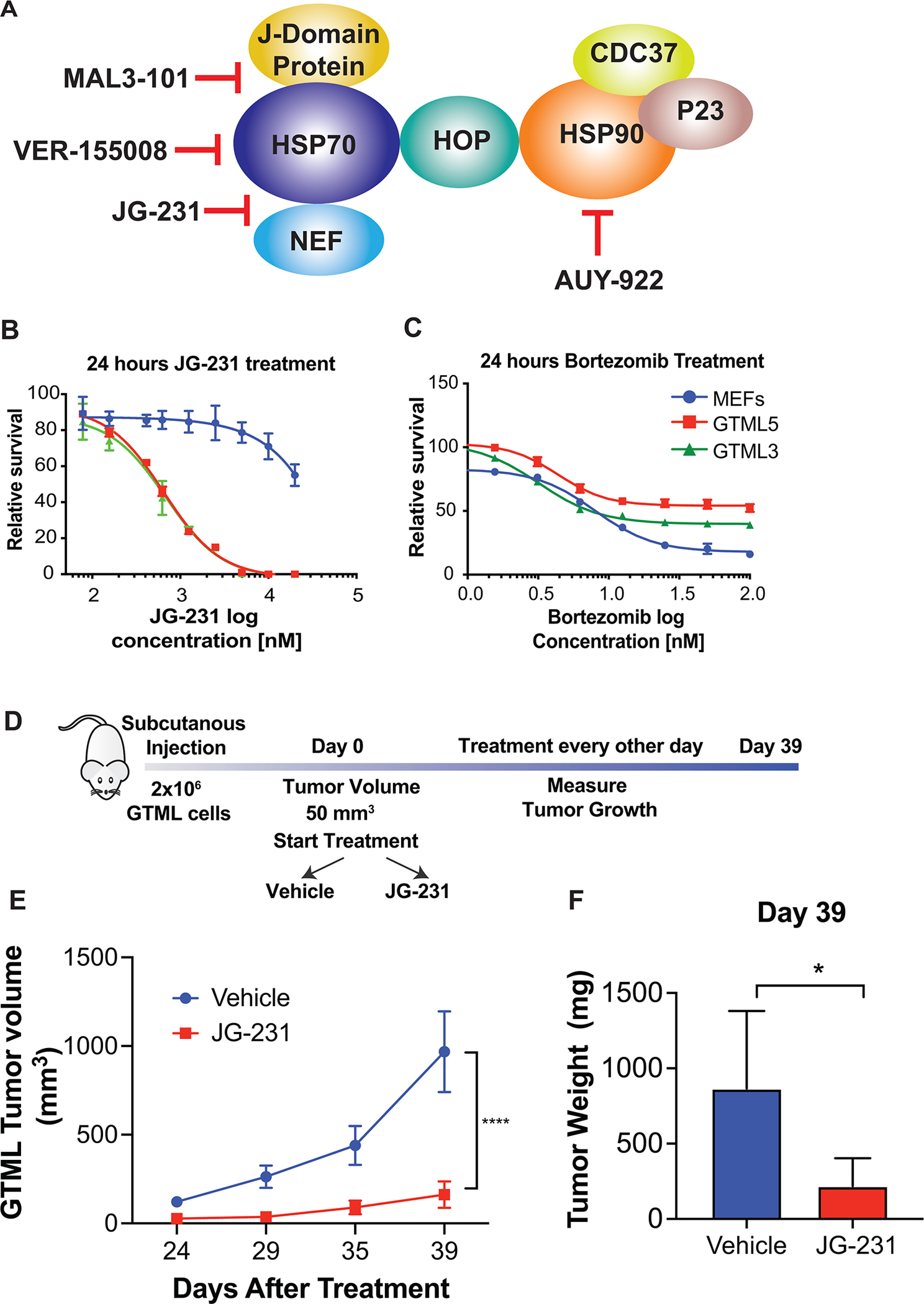
A, Schematic representation of the targets of different compounds used in the study. NEF: Nucleotide Exchange Factor. B-C, Treatment of GTML cell lines derived from two different mice (GTML 5 and GTML 3) and MEFs with JG-231 (B) and Bortezomib (C) for 24 hours. A Cell Titer Glo assay was performed and relative survival for each genotype was plotted normalized to cells treated with DMSO (n > 3). D, Cartoon representing in vivo JG-231 treatment. E, Measurement of tumor volumes over 39 days of JG-231 treatment. A two-way ANOVA test was performed. F, Measurement of tumor weight on Day 39. A multiple t-test was performed.
All values represent the mean + SEM. *p < 0.05, ****p < 0.0001
In this pharmacological approach, we selected a series of compounds with distinct targets and mechanisms-of-action. To target the ATPase activities of HSP70s and HSP90s, we selected NVP-AUY922 and VER-155008. NVP-AUY922 is a potent small molecule ATP-competitive HSP90 inhibitor, which binds to the N-terminal catalytic domain (23). VER-155008 inhibits HSP70 by binding to its N-terminal ATP-binding pocket and preventing allosteric control between nucleotide and substrate binding domains (21). In addition, HSP70 is known to be regulated by over 50 co-chaperones (24). In some systems, targeting the co-chaperones is less toxic than inhibitors of “core” chaperone activity (25), so we considered it important to also test compounds that block interactions with these factors. Specifically, MAL3–101 binds to an allosteric site of HSP70’s ATPase domain and inhibits its catalytic activation by J-domain containing proteins (26,27), such as DNAJA2. Among these co-chaperones, six Bcl-2 associated athanogene (BAG) proteins assist in nucleotide release by HSP70. Tool compounds that block the binding of specific BAGs to HSP70 provide specificity for HSP70 activity, showing anti-cancer activity (28). Therefore, we also tested JG-231, a tool compound that blocks interactions between HSP70 and specific BAG proteins. JG-231 stabilizes the ADP-bound state of the Hsp70s, which “traps” the protein in a high affinity state that is resistant to the activity of BAG proteins (29). Therefore, compared to other inhibitors tested, JG-231 traps Hsp70s in their ADP bound state which inhibits the BAG family proteins, blocking a subset of Hsp70 functions.
Interestingly, among the inhibitors tested, JG-231 was the only one that showed specificity in decreasing viability of MYCN-driven medulloblastoma cells (Fig 3B, Supplementary Fig S4A). JG-231 was >10-fold selective for MYCN-driven tumor cells compared to immortalized mouse embryonic fibroblasts (MEFs). Consistent with the exquisite specificity for translational control of select components of the protein folding machinery downstream of the N-myc:mTORC1 axis, such as HSP70, blockade of the proteasome with bortezomib, which should more broadly affect the general function of molecular chaperones, failed to show efficacy comparing N-myc-driven to control cells (Fig. 3C). To study the effect of JG-231 on global protein synthesis, we performed 35S metabolic labeling. Global levels of protein synthesis in cells treated with JG-231 (200 nM) for 24 hours were not significantly affected (Supplementary Fig S4B). These data uncovered JG-231 as a potential therapeutic compound to suppress tumorigenesis of MYCN-driven medulloblastoma and was remarkably specific among the molecular chaperon inhibitors.
To test whether JG-231 could inhibit N-myc dependent tumor progression in vivo, we established xenografts of GTML cells in NOD scid gamma (NSG) mice. Tumors were implanted subcutaneously, since JG-231 does not cross the blood brain barrier. Animals with established tumors were treated intraperitoneally with vehicle or JG-231 (4 mg/kg) every other day for five weeks (Fig. 3D). This treatment regime did not affect body weight (Supplemental Fig S4C). JG-231 treated animals showed significant reduction in tumor burden measured by tumor volume (Fig. 3E) and tumor weight (Fig. 3F), indicating that JG-231 suppressed tumor growth of MYCN-driven medulloblastoma in vivo. We compared the expression of HSPA8 by immunohistochemistry in mice treated with JG-231 or vehicle and we did not observe any difference (Supplementary Fig S5A). However AKT, a target of HSPA8 (28), was significantly downregulated in the JG-231 treated group, revealing the efficiency of the inhibitor (Supplementary Fig S5A). As expected, the abundance of both HSPA8 and AKT did not change in tumors in which SP6P was wild type, heterozygous or homozygously deleted (Supplementary Fig S5B).
mTOR:eIF4E regulates protein folding in MYCN-driven medulloblastoma
Next, we asked which arm of the mTORC1 pathway is essential for MYCN-driven translation control (Fig. 1A). We hypothesized that the eIF4E arm plays a major role, as we observed translational regulation of specific transcripts in protein folding machinery by Rapalink-1 treatment, which blocks both S6K and eIF4E, but not by rapamycin, which blocks only S6K. To extend our analysis, we generated GTML cell lines expressing either scramble or eIF4E shRNAs and performed polysome profiling. As expected, short-term 48h depletion of eIF4E did not alter global protein synthesis (Supplementary Fig. S6A). HSPA8, HSP90AB1, CCT4, SEC61A1 mRNAs but not B2M mRNA, showed a significant shift from high polysomes to free/mRNP fractions, revealing that eIF4E was important for translation of these select mRNAs (Fig. 4A and Supplementary Fig. S6B). Western blot analysis also showed a clear reduction of Hspa8 at the protein level (Fig. 4B) while mRNA levels did not change (Supplementary Fig. S6C).
Figure 4. eIF4E translationally regulates HSP70 and drives medulloblastoma tumorigenesis.
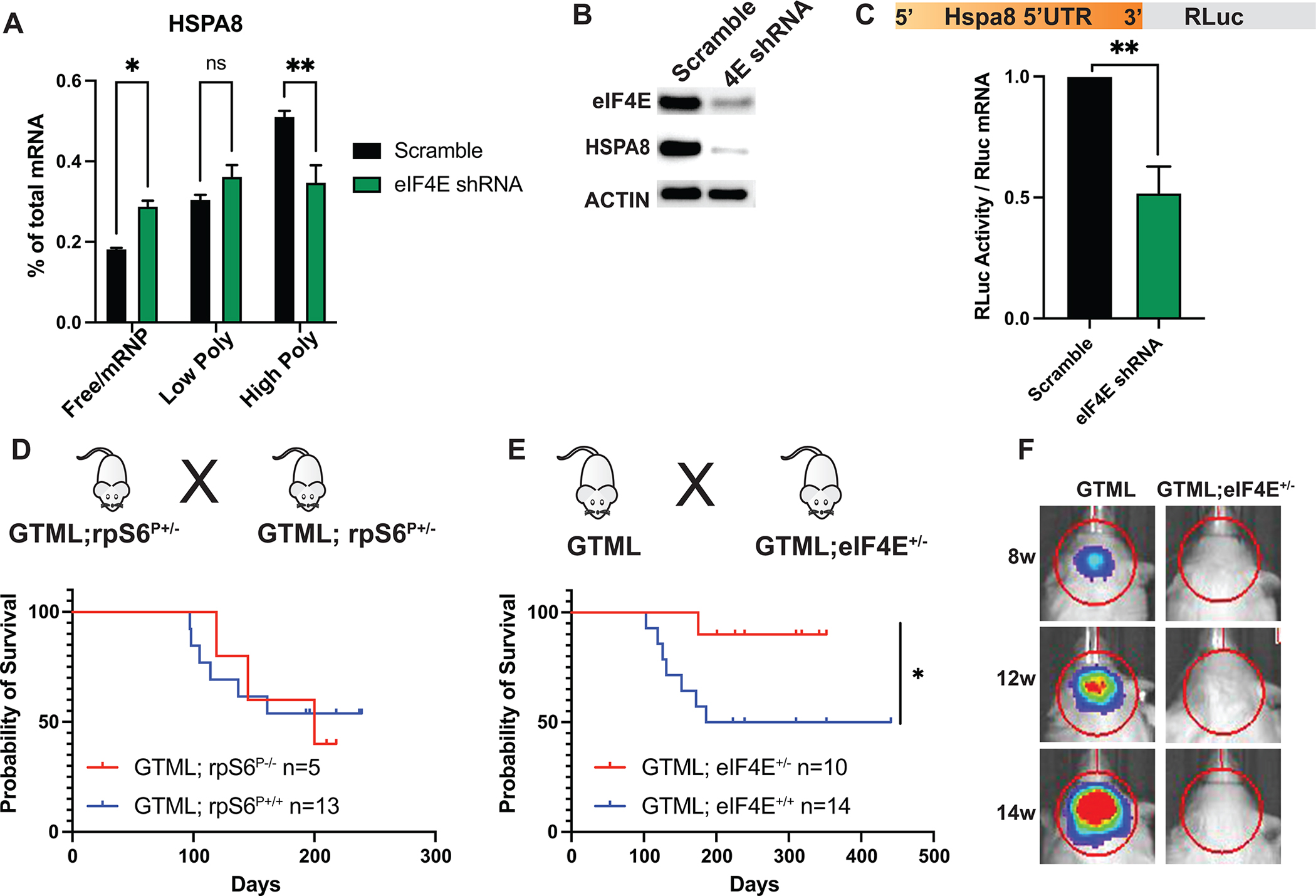
A, qPCR analysis of HSPA8 mRNA in GTML cells expressing Scramble or eIF4E shRNAs, isolated from 10% - 50% sucrose gradient fractions. Fractions are labelled as in Fig. 2c. A two-way ANOVA test was performed to determine statistical significance (n = 3). B, Western blot analysis showing repression in endogenous Hspa8 protein levels.</p>C, Luciferase reporter assay demonstrating that eIF4E is required for 5’ UTR-mediated translation of HSPA8 mRNA. The 5’UTR of HSPA8 mRNA is cloned downstream of Renilla open reading frame and luciferase activity is measured in GTML cells expressing either Scramble or eIF4E shRNAs. Luciferase activity is then normalized to Rluc mRNA levels. A multiple t-test is performed. D-E, Survival plots of GTML;rpS6P−/− (D) and GTML;eIF4E+/− mice (E). n values represent number of animals in each cohort. F, Bioluminescence imaging of GTML and GTML;eIF4E+/− mice at indicated weeks using identical imaging conditions. All values represent the mean + SEM. *p < 0.05, **p < 0.01
The 5’ untranslated regions (5’UTR) of mRNAs, which represent the entry sites for ribosomes, may contain several cis-regulatory elements as well as secondary structures, that are essential for the regulation of translation initiation (30). Therefore, we performed a reporter assay to assess whether the 5’UTR of Hspa8 mRNA was the main driver of eIF4E-regulated translational control in MYCN-driven medulloblastoma. We cloned the 5’UTR of the Hspa8 mRNA upstream of the Renilla luciferase open reading frame and expressed it in scramble and eIF4E shRNA expressing cells. We measured luciferase activity in both conditions and normalized this to mRNA levels to calculate translational efficiency. The Hspa8–5’UTR containing reporter was specifically downregulated in eIF4E depleted cells, revealing that eIF4E was required for the translation of Hspa8 mRNA (Fig. 4C). Therefore, eIF4E is a key determinant of the medulloblastoma translation control program.
The interaction between eIF4E and MYCN drives medulloblastoma tumorigenesis
We showed that eIF4E regulates translation of specific transcripts in MYCN-driven medulloblastoma, which led us to hypothesize that this genetic interaction between MYCN and eIF4E could be essential for tumorigenesis. To address this question, we took advantage of different genetic mouse models. We first focused on one of the key substrates of S6K, the ribosomal protein S6 (rpS6), which has also been shown to be important for translational control. We first generated rpS6 phosphorylation deficient GTML mice (GTML;rpS6P−/−) by crossing GTML with rpS6P−/− mice (31). We did not observe any differences in survival between the wild type and rpS6P−/− cohorts, suggesting that signaling downstream of rpS6 does not contribute to MYCN-driven transformation (Fig. 4D). We next crossed GTML mice to eIF4E heterozygous mice (eIF4E+/−) which express eIF4E at half of normal levels throughout the whole body (32). GTML; eIF4E+/− mice showed significantly decreased tumor growth and increased survival compared with GTML eIF4E+/+ mice (Fig 4E and F), suggesting that cancer cells require eIF4E activity to drive MYCN-dependent tumorigenesis.
JG-231 treatment represses tumor growth in human iPSC-derived medulloblastoma
We next leveraged a second distinct non germline GEMM system where human induced pluripotent stem cells (iPSC)-derived neuroepithelial stem (NES) cells are transformed by overexpression of Flag-tagged MYCN (33). Implanting MYCN-NES cells orthotopically in immunocompromised mice results in generation of tumors aligned with human medulloblastoma (33)). We tested human NES cells expressing either empty vector or MYCN. As a control, we also tested NES cells from patients with Gorlin syndrome, predisposed to medulloblastoma by loss of function mutation PTCH1 [Fig. 5A, (33)]. We aimed to mimic SHH medulloblastoma by using this system: While MYCN-overexpression represents highest risk medulloblastoma, PTCH1-mutant cell line represents low risk SHH. We treated the cells with the HSP70,HSP90 and co-chaperone inhibitors described in Fig. 3A and measured their effects on cell proliferation (Fig. 5B and C, Supplementary Fig. S7A). Again, JG-231 treatment specifically reduced proliferation of N-myc overexpressing NES cells (Fig. 5B) without affecting either control NES cells carrying empty vector or NES cells expressing mutant PTCH1. Consistent with the results obtained in GTML cells, other inhibitors tested did not show efficacy among different NES cell lines (Supplementary Fig. S7A). These data again suggest that HSP70-BAG inhibition by JG-231 reduced tumor growth in a MYCN dependent manner.
Figure 5. JG-231 treatment represses tumor growth in human iPSC-derived medulloblastoma.
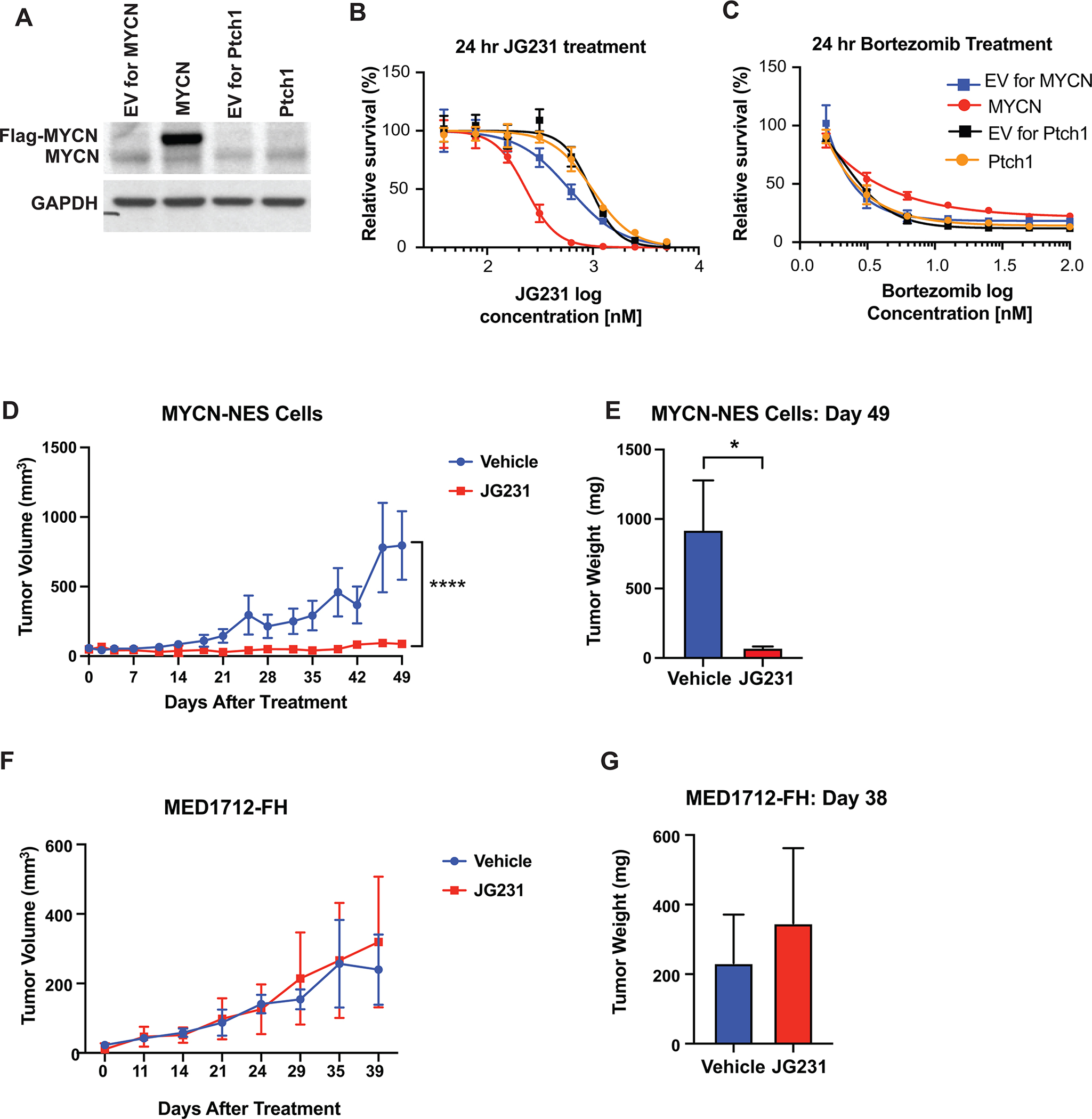
A, Western blot analysis of human iPSC-derived NES cells expressing either empty vector (EV) or Flag-tagged MYCN together with control cells expressing loss of function PTCH1 mutation. B-C, Treatment of NES cells expressing either MYCN or mutant PTCH1 and their corresponding EVs with JG-231 (B) and Bortezomib (C) for 24 hours. A Cell Titer Glo assay was performed and relative survival for each genotype was plotted normalized to cells treated with DMSO (n > 3). D, Measurement of tumor volumes over 49 days of JG-231 treatment. MYCN overexpressing NES cells were orthotopically implanted into immunocompromised mice and treated for 49 days with JG-231. A two-way ANOVA test was performed. E, Measurement of tumor weight on Day 49 of JG-231 treatment, corresponding to (D). A multiple t-test was performed. F, Measurement of tumor volumes over 39 days of JG-231 treatment. MYCN-independent PDX PTCH1 mutant line, MED1712-FH, were grown and treated with JG-231 for 39 days. G, Measurement of tumor weight on Day 49 of JG-231 treatment, corresponding to (F).
Next, we studied the effect of JG-231 on MYCN-dependent tumor growth in vivo. We subcutaneously implanted MYCN-NES cells into immunocompromised mice and treated them with JG-231. Growth of MYCN-dependent tumors was significantly suppressed by JG-231 treatment (Fig. 5D and E), demonstrating MYCN driven human medulloblastoma requires higher activity levels of protein folding machinery and repressing it specifically abolishes tumor growth. As a control, we also tested a MYCN-independent patient-derived xenograft (PDX) PTCH1 mutant line, MED1712-FH. In agreement with our in vitro model, JG-231 treatment did not suppress growth of MED1712-FH, suggesting elevated levels of N-myc are required for the mode of action of the inhibitor (Fig. 5F and G and Supplementary Fig S7B). Therefore, our results revealed that targeting the protein folding machinery specifically in high-risk group medulloblastoma where MYCN is amplified, may have therapeutic benefits.
Discussion
Cancer cells are constantly under proteotoxic stress as a result of heightened protein synthesis, environmental effects, and genetic alterations including point mutations, aneuploidy and gene amplification (34). They overcome these stressors to maintain protein homeostasis via a variety of adaptive mechanisms. Molecular chaperones including heat shock proteins, play a key role in this regulation. Proteotoxic stress is an example of non-oncogene addiction (NOA) where non-oncogenes, such as heat shock proteins, which are not mutated at the genomic level, are required for oncogenic transformation (35–37). NOA genes represent vulnerabilities that can be therapeutically targeted.
In this study, we uncovered previously unknown translation specificity by the N-myc oncogene in medulloblastoma. In this context we found that N-myc driven medulloblastoma is characterized by translational regulation of distinct components (12 of 262 proteins) within the protein folding machinery at least in part through the translation factor eIF4E. Consistent with this specificity within the protein folding machinery, the generalized proteasome inhibitor bortezomib was non-selective comparing N-myc driven with normal cells.
We showed that N-myc increases the dosage of molecular chaperones via translational regulation to prevent proteotoxic stress. While both the HSP70 and HSP90 chaperones were downstream of N-myc, pan-selective inhibitors of these chaperones again did not show selectivity between N-myc driven cells and normal cells. We therefore leveraged our knowledge of this translational specificity in medulloblastoma to design a new therapeutic intervention. Indeed, JG-231, which inhibits the BAG family proteins, blocking a subset of Hsp70 functions, repressed tumor growth of medulloblastoma cells in vitro and in mouse and human xenografts.
This link between protein synthesis and protein folding enables cancer cells to increase protein levels without eliciting any proteotoxic stress, thus serving as an NOA and cancer specific vulnerability. The delicate balance between protein synthesis and protein folding is crucial to maintain the integrity of the proteome. The disruption of such a balance can cause accumulation of misfolded proteins in potentially toxic protein inclusions called ‘aggresomes’. (38,39). In this context, a recent study reported the presence of aggresomes in medulloblastoma tumors (40). Elucidating the role of N-myc driven translation of protein folding machinery and its relationship to the formation of aggresomes in tumorigenesis will be an important future direction.
Cancer cells can adapt to different stress conditions by regulating gene expression and rely on translation control to synthesize proteins urgently needed for this adaptation. Recent studies showed that eIF4E plays a critical role to drive selective translation programs for cancer cells to survive by regulating diverse aspects of cancer such as metabolism (41), immune response (42,43), and mitochondrial stress response (44). Importantly, reducing eIF4E levels to 50% significantly suppresses oncogenic transformation and tumor growth in vivo, without altering global protein synthesis (32), demonstrating that selective eIF4E-dependent translational control represents a new therapeutic target. Our data showing that eIF4E is required for MYCN-driven translational programs opens unexplored avenues for medulloblastoma therapy. It would be informative for future studies to perform a genome-wide analysis of MYCN-driven medulloblastoma after eIF4E knock down to decipher globally the regulation of eIF4E on the translation of specific targets versus the activity of other downstream components of mTOR activation in this process.
SHH driven-tumors (45,46) can be treated with inhibitors of this pathway that target Smo, a key activator of Shh signaling. However, tumors acquire mutations in Smo that block binding of the drug (47), thereby exhibiting resistance to Smo inhibitors. Importantly, MYCN-amplified SHH tumors represent the highest-risk subgroup among SHH tumors (45,48). Our results in MYCN-driven medulloblastoma demonstrate that targeting molecular chaperones, which are translationally regulated, either by translation inhibitors or direct inhibitors, such as JG-231, represents a novel and urgently needed therapeutic intervention.
Supplementary Material
Statement of Significance.
Translatome analysis in medulloblastoma shows that N-myc drives selective translation of transcripts that promote protein homeostasis and that represent new therapeutic vulnerabilities.
Acknowledgments
We thank Dr. Nicholas Ingolia for ribosome profiling data analysis; Dr. Antoine Forget and Dr. Olivier Ayrault, for help with data analysis; Amit J. Sabnis for the reagents.
This study is supported by the following grants: NIH R01NS125668 (W.A.W. and D.R.), R01CA159859 (WAW), R01CA255369 (WAW), R01NS106155 (WAW), the Samuel G. Waxman Cancer Research Foundation (WAW); the Evelyn and Mattie Anderson Chair (WAW); NIH grant R35CA242986 (D.R.), the American Cancer Society (RP-19–181-01-RMC; American Cancer Society Research Professor Award) (D.R.), NIH grant NS059690 (J.E.G.), DoD grant PC180716 (J.E.G.), R35NS122339 (R.W.-R.), R01 CA159859 (R.W.-R.) and R00CA197484 (M.H.), the Wright Trust Foundation and the Saban Research Institute of Children’s Hospital Los Angeles. D.K.-O. was awarded by a EMBO long-term fellowship (ALTF 1005–2015) and a Human Frontier Science Program long-term fellowship (LT000711/2016-L). O.A was awarded by Damon Runyon-Sohn Pediatric Cancer Research Fellowship, Alex’s Lemonade Stand Young Investigator Grant and NIH T32 CA151022–11. Some of the resources were provided by the UCSF Brain Tumor SPORE Biorepository (NIH 5P50 CA097257–18).
Footnotes
Author’s Disclosures
D.R. is a shareholder of eFFECTOR Therapeutics. W.A.W. has ownership interest in StemSynergy Therapeutics.
References
- 1.Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer. 2020;20:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan Q-W, Goldenberg DD, et al. Pleiotropic role for MYCN in medulloblastoma. Gene Dev. 2010;24:1059–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell. 2012;21:601–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Gene Dev. 2002;16:2699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, Alborán IM de, Olson JM, et al. N-myc Is an Essential Downstream Effector of Shh Signaling during both Normal and Neoplastic Cerebellar Growth. Cancer Res. 2006;66:8655–61. [DOI] [PubMed] [Google Scholar]
- 6.Ruggero D The Role of Myc-Induced Protein Synthesis in Cancer. Cancer Res. 2009;69:8839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29:1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuhmacher M, Kohlhuber F, Hölzel M, Kaiser C, Burtscher H, Jarsch M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29:1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 Complex Plays a Prime Role in Regulating N-Myc Phosphorylation and Turnover in Neural Precursors. Dev Cell. 2005;9:327–38. [DOI] [PubMed] [Google Scholar]
- 11.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc National Acad Sci. 2000;97:3260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greasley PJ, Bonnard C, Amati B. Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res. 2000;28:446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, et al. Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 2000;60:5922–8. [PubMed] [Google Scholar]
- 14.Pajic A, Spitkovsky D, Christoph B, Kempkes B, Schuhmacher M, Staege MS, et al. Cell cycle activation by c-myc in a Burkitt lymphoma model cell line. Int J Cancer. 2000;87:787–93. [DOI] [PubMed] [Google Scholar]
- 15.Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: Identification and analysis of c-MYC target genes. Proc National Acad Sci. 2002;99:6274–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan Q-W, Goldenberg DD, et al. Pleiotropic role for MYCN in medulloblastoma. Gene Dev. 2010;24:1059–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Q, Aksoy O, Wong RA, Ilkhanizadeh S, Novotny CJ, Gustafson WC, et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell. 2017;31:424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. [DOI] [PubMed] [Google Scholar]
- 21.Schlecht R, Scholz SR, Dahmen H, Wegener A, Sirrenberg C, Musil D, et al. Functional Analysis of Hsp70 Inhibitors. Plos One. 2013;8:e78443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.John K J B Brian S. The Right Tool for the Job: An Overview of Hsp90 Inhibitors. In: M.L. M, D P, R S-S, editors. HSF1 and Molecular Chaperones in Biology and Cancer [Internet]. Cham: Springer International Publishing; 2020. page 135–46. Available from: 10.1007/978-3-030-40204-4_9 [DOI] [Google Scholar]
- 23.Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, et al. 4,5-Diarylisoxazole Hsp90 Chaperone Inhibitors: Potential Therapeutic Agents for the Treatment of Cancer. J Med Chem. 2008;51:196–218. [DOI] [PubMed] [Google Scholar]
- 24.Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Bio. 2019;20:665–80. [DOI] [PubMed] [Google Scholar]
- 25.Gestwicki JE, Shao H. Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem. 2019;294:2151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, et al. Small Molecule Modulators of Endogenous and Co-chaperone-stimulated Hsp70 ATPase Activity*. J Biol Chem. 2004;279:51131–40. [DOI] [PubMed] [Google Scholar]
- 27.Sabnis AJ, Guerriero CJ, Olivas V, Sayana A, Shue J, Flanagan J, et al. Combined chemical–genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc National Acad Sci. 2016;113:9015–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao H, Li X, Moses MA, Gilbert LA, Kalyanaraman C, Young ZT, et al. Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein–Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem. 2018;61:6163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rinaldi S, Assimon VA, Young ZT, Morra G, Shao H, Taylor IR, et al. A Local Allosteric Network in Heat Shock Protein 70 (Hsp70) Links Inhibitor Binding to Enzyme Activity and Distal Protein–Protein Interactions. Acs Chem Biol. 2018;13:3142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leppek K, Das R, Barna M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Bio. 2018;19:158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1−/−/S6K2−/− Mice Exhibit Perinatal Lethality and Rapamycin-Sensitive 5′-Terminal Oligopyrimidine mRNA Translation and Reveal a Mitogen-Activated Protein Kinase-Dependent S6 Kinase Pathway. Mol Cell Biol. 2004;24:3112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, et al. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell. 2015;162:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang M, Tailor J, Zhen Q, Gillmor AH, Miller ML, Weishaupt H, et al. Engineering Genetic Predisposition in Human Neuroepithelial Stem Cells Recapitulates Medulloblastoma Tumorigenesis. Cell Stem Cell. 2019;25:433–446.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ambrose AJ, Chapman E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J Med Chem. 2021;64:7060–82. [DOI] [PubMed] [Google Scholar]
- 35.Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solimini NL, Luo J, Elledge SJ. Non-Oncogene Addiction and the Stress Phenotype of Cancer Cells. Cell. 2007;130:986–8. [DOI] [PubMed] [Google Scholar]
- 37.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat Shock Factor 1 Is a Powerful Multifaceted Modifier of Carcinogenesis. Cell. 2007;130:1005–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen B, Retzlaff M, Roos T, Frydman J. Cellular Strategies of Protein Quality Control. Csh Perspect Biol. 2011;3:a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–9. [DOI] [PubMed] [Google Scholar]
- 40.Yehia M, Taha H, Salama A, Amer N, Mosaab A, Hassanain O, et al. Association of Aggresomes with Survival Outcomes in Pediatric Medulloblastoma. Sci Rep-uk. 2019;9:12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nature medicine. 2019;25:301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartish M, Tong D, Pan Y, Wallerius M, Liu H, Ristau J, et al. MNK2 governs the macrophage antiinflammatory phenotype. Proc National Acad Sci. 2020;117:27556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuzuoglu-Ozturk D, Hu Z, Rama M, Devericks E, Weiss J, Chiang GG, et al. Revealing molecular pathways for cancer cell fitness through a genetic screen of the cancer translatome. Cell Reports. 2021;35:109321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kool M, Jones DTW, Jäger N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell. 2014;25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skowron P, Farooq H, Cavalli FMG, Morrissy AS, Ly M, Hendrikse LD, et al. The transcriptional landscape of Shh medulloblastoma. Nat Commun. 2021;12:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science. 2009;326:572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tabori U, Baskin B, Shago M, Alon N, Taylor MD, Ray PN, et al. Universal Poor Survival in Children With Medulloblastoma Harboring Somatic TP53 Mutations. J Clin Oncol. 2010;28:1345–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the plasmids used in this manuscript are available upon request. The ribosome profiling data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE215298.