

Abstract
Increasing the speed, specificity, sensitivity, and accessibility of mycobacteria detection tools are important challenges for tuberculosis (TB) research and diagnosis. In this regard, previously reported fluorogenic trehalose analogues have shown potential, but their green-emitting dyes may limit sensitivity and applications in complex settings. Here, we describe a trehalose-based fluorogenic probe featuring a molecular rotor turn-on fluorophore with bright far-red emission (RMR-Tre). RMR-Tre, which exploits the unique biosynthetic enzymes and environment of the mycobacterial outer membrane to achieve fluorescence activation, enables fast, no-wash, low-background fluorescence detection of live mycobacteria. Aided by the red-shifted molecular rotor fluorophore, RMR-Tre exhibited up to a 100-fold enhancement in M. tuberculosis labeling compared to existing fluorogenic trehalose probes. We show that RMR-Tre reports on M. tuberculosis drug resistance in a facile assay, demonstrating its potential as a TB diagnostic tool.
Keywords: Trehalose, fluorescence, molecular rotor, mycobacteria, tuberculosis
Graphical Abstract
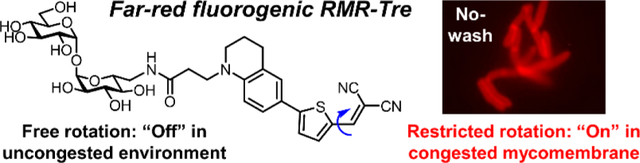
The development of a novel trehalose-based fluorogenic probe that features a molecular rotor turn-on fluorophore with exceptionally bright far-red emission is described (RMR-Tre). RMR-Tre enables rapid, no-wash, low-background fluorescence detection and drug-susceptibility evaluation of live mycobacteria, including Mycobacterium tuberculosis.
Introduction
Mycobacteria and related organisms in the Corynebacterineae suborder have a major impact on human health. The most deadly bacterial pathogen worldwide is Mycobacterium tuberculosis (Mtb), which is estimated to infect a quarter of the globe’s population, every year causing 10 million cases of active tuberculosis (TB) disease and killing 1.5 million people.[1] The emergence and spread of multi- and extensively-drug-resistant strains of Mtb, which are more challenging to diagnose and treat, has further confounded TB control programs.[1] The Corynebacterineae suborder also includes the pathogens that cause leprosy and diphtheria, as well as a group of organisms referred to as non-tuberculous mycobacteria (NTM), some of which are opportunistic pathogens that have clinical manifestations similar to TB.[2] The ability to rapidly and accurately detect mycobacteria in complex samples is critical for researching and diagnosing mycobacterial diseases in these and other contexts. Rapid detection of mycobacteria is often done via microscopy employing either color-based Ziehl-Neelsen or fluorescence-based auramine–rhodamine staining, both of which exploit the hydrophobicity of the mycobacterial outer membrane—also called the mycomembrane—to stain acid-fast bacilli such as mycobacteria.[3] However, both stains have limitations with respect to their specificity, sensitivity, and/or convenience of use, and neither can differentiate live/growing bacteria from dead/non-growing bacteria.[4]
In the past several years, an improved understanding of mycobacterial physiology has converged with advances in chemical biology to open up opportunities for the development of novel mycobacteria-specific detection probes.[5] In this regard, the non-mammalian disaccharide trehalose has proven to be an attractive target, since it is absent from humans but in mycobacteria it is incorporated into the abundant mycomembrane glycolipids trehalose mono- and dimycolate (TMM and TDM) via mycobacteria-specific metabolic pathways.[6] In 2011, the Barry and Davis groups showed that a fluorescein-modified trehalose analogue (FITC-Tre) can metabolically incorporate into trehalose glycolipids in live Mtb through the action of substrate-promiscuous Ag85 mycoloyltransferases.[7] Since then, several groups, including ours, have extended the trehalose probe concept by introducing diverse detection modalities (e.g., fluorophores, “click” chemistry tags, 19F/18F toward PET imaging, photo-cross-linkers) and by modifying the probe scaffold (e.g., trehalose, TMM, TDM) to target different routes of metabolic incorporation.[8]
Recently, there has been a shift toward the development of fluorogenic trehalose-based probes that are designed to emit signal only upon metabolic incorporation into the mycomembrane. Existing fluorogenic trehalose probes include the Bertozzi group’s solvatochromic 4-N,N-dimethylamino-1,8-naphthalimide trehalose (DMN-Tre[8e]) and recently reported hydroxychromone trehalose (H3C-Tre[8l]), which fluoresce post-incorporation due to the hydrophobic environment of the mycomembrane. In addition, FRET-based TMM and TDM analogues (QTF[8f] and FRET-TDM[8h]) developed by Kiessling’s group and our group, respectively, fluoresce upon separation of their attached fluorophore and quencher moieties by mycomembrane-synthesizing or -remodeling enzymes. In principle, such fluorogenic probes should allow specific and sensitive detection of mycobacteria through a one-step, no-wash process. DMN-Tre in particular was shown to embody these features, as it enabled the fast and accurate detection of Mtb in sputum samples from TB patients.[8e] However, a disadvantage is that all of these probes have green-emitting fluorophores, which have liabilities with respect to light penetrance, light scattering, autofluorescence, cellular photo-toxicity, and photo-bleaching.[9] These drawbacks can reduce assay sensitivity and limit applications in complex samples, living systems, and high-throughput screens, all of which could hinder the advancement of trehalose-based probes to preclinical and clinical use. Moreover, although in principle the FRET probe structures can be modified to include red-shifted fluorophores, their trehalose ester-based scaffolds appear to have more limited specificity for mycobacteria,[8h] as further demonstrated herein. To address these issues, here we report the development of the first far-red fluorogenic trehalose probes and demonstrate their ability to specifically and rapidly detect mycobacteria, including in a drug-susceptibility testing scenario.
Results and Discussion
To achieve mycobacteria-specific fluorescence turn-on in the far-red region, here we employed fluorescent molecular rotors, whose fluorescence emission intensity depends on the rotational freedom provided by the surrounding environment.[10] When bond rotation is relatively unrestricted—for example, in a low-steric hindrance environment—fluorescent molecular rotors transition to a twisted intramolecular charge transfer (TICT) state upon photo-excitation, after which energy dissipates predominantly through non-radiative relaxation. By contrast, in a high-steric hindrance environment, rotational freedom and thus transition to the TICT state is suppressed, resulting in energy dissipation through red-shifted fluorescence emission. In 2019, Hsu et al. reported that fluorescent molecular rotors coupled to D-amino acids (RfDAAs) specifically metabolically incorporate into bacterial peptidoglycan, which is a sterically congested environment that led to fluorescence turn-on of the incorporated RfDAAs.[11] Based on this observation, we hypothesized that conjugating a fluorescent molecular rotor to trehalose would enable its targeting to the glycolipid-rich mycobacterial mycomembrane, which has a similarly congested environment that we expected would impede transition to the TICT state and thus trigger fluorescence turn-on after metabolic incorporation (Figure 1A).
Figure 1.
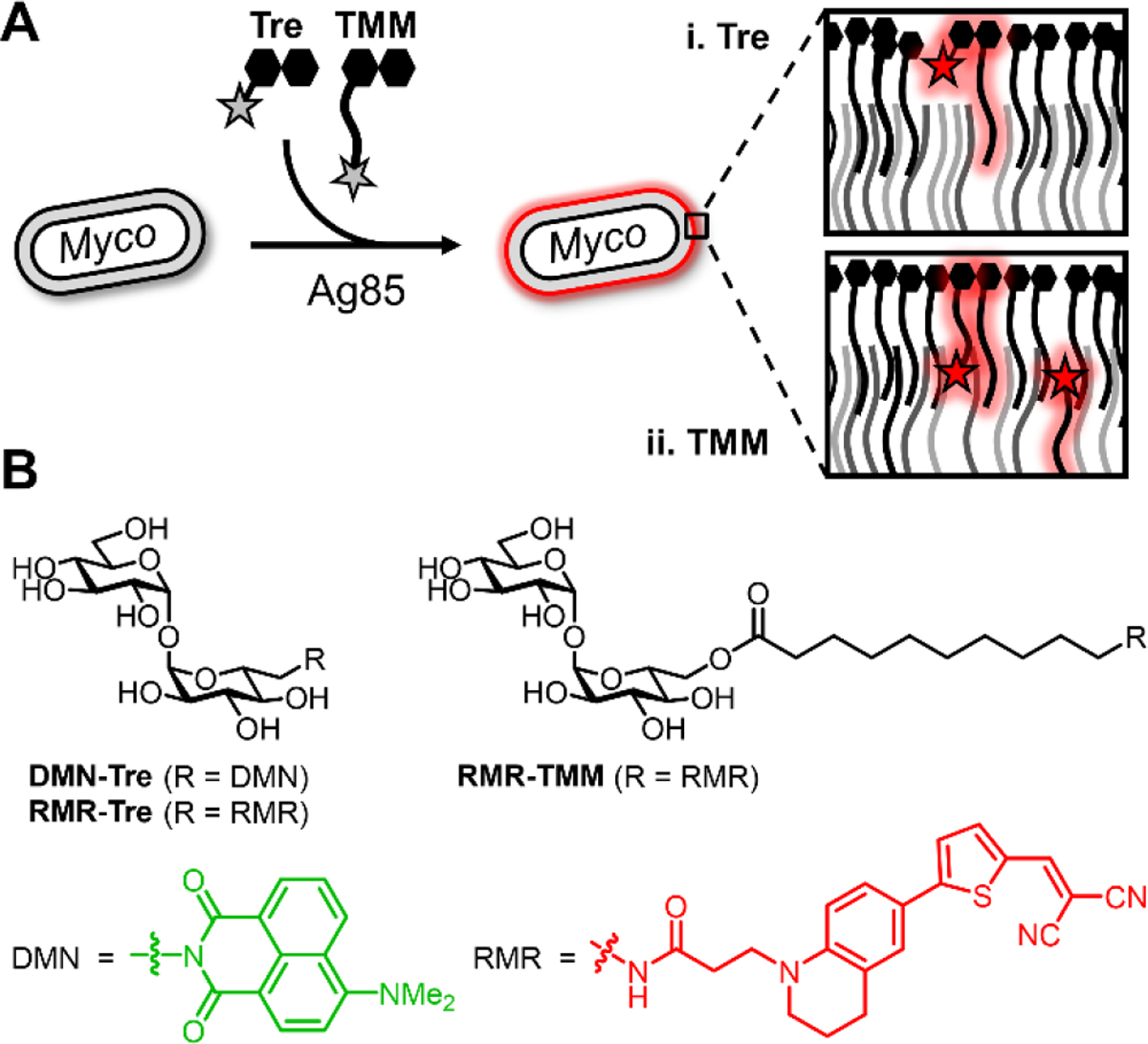
(A) Ag85-mediated incorporation of (i) trehalose- and (ii) TMM-based fluorogenic probes into the hydrophobic, congested mycomembrane of live mycobacteria (see Scheme S1 for incorporation routes and targets). (B) Previously reported solvatochromism-based probe (DMN-Tre) and far-red molecular rotor-based probes (RMR-Tre and RMR-TMM) developed in this work.
The previously reported RfDAAs utilized julolidine-based fluorescent molecular rotors with maximum excitation (max. λEm) and emission (max. λEx) wavelengths ranging from 420–490 nm and 490–660 nm, respectively.[11] Here, we employed a variant with further red-shifted excitation and emission (max. λEx = 536 nm and max. λEm = 684 nm), referred to herein as red molecular rotor (RMR). We designed two probes, trehalose and TMM analogues called RMR-Tre and RMR-TMM (Figure 1B), both of which exploit Ag85 mycoloyltransferase activity for metabolic incorporation into the mycomembrane. Based on our prior work developing trehalose- and TMM-based probes,[8b] RMR-Tre and RMR-TMM were designed to incorporate into different mycomembrane components, potentially leading to complementary fluorogenic properties (Figure 1A and Scheme S1 of the Supporting Information). RMR-Tre was designed to undergo Ag85-catalyzed modification of its free 6-position with a mycoloyl group, thus anchoring the dye into the outer leaflet of the mycomembrane. On the other hand, RMR-TMM was designed to undergo Ag85-catalyzed transfer of its fluorophore-modified 10-carbon acyl chain to mycoloyl acceptors, thus anchoring the dye into both the outer and inner leaflets of the mycomembrane and placing it closer to the interior of the membrane. Overall, we expected that the red-shifted excitation/emission wavelengths and the dual-targeting nature of the RMR trehalose probes—i.e., that their fluorescence activation relies on both mycobacteria-specific metabolism and the distinctive biophysical properties of the mycomembrane—would engender them with ideal properties for mycobacteria-specific fluorescence detection applications.
The syntheses of RMR-Tre and RMR-TMM are shown in Figure 2A. The free RMR carboxylic acid (2) was synthesized from aldehyde 1[11] by condensation with malonitrile, which proceed in 93% yield. Free RMR 2 was then separately conjugated to amino trehalose derivative 3[8d] and amino TMM derivative 4[8g] using peptide coupling conditions. Although several coupling reagents were tested, only N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU) was successful, delivering RMR-Tre and RMR-TMM in 60% and 50% yield, respectively. RMR-Tre and RMR-TMM both had desirable photophysical properties, with each exhibiting red-shifted excitation and emission maxima (max. λEx = 536 nm and max. λEm = 684 nm) compared to the previously reported trehalose probes (Figure 2B and Figure S1 of the Supporting Information). Promisingly, RMR-Tre and RMR-TMM underwent robust fluorescence activation when shifting from water to higher-viscosity glycerol that would more closely resemble the mycomembrane environment (Figure 2B). Interestingly, RMR-TMM showed lower background fluorescence than RMR-Tre in phosphate-buffered saline (PBS), which could be a result of increased rotational freedom of the molecular rotor in RMR-TMM due to the flexible linker separating it from the disaccharide.
Figure 2.
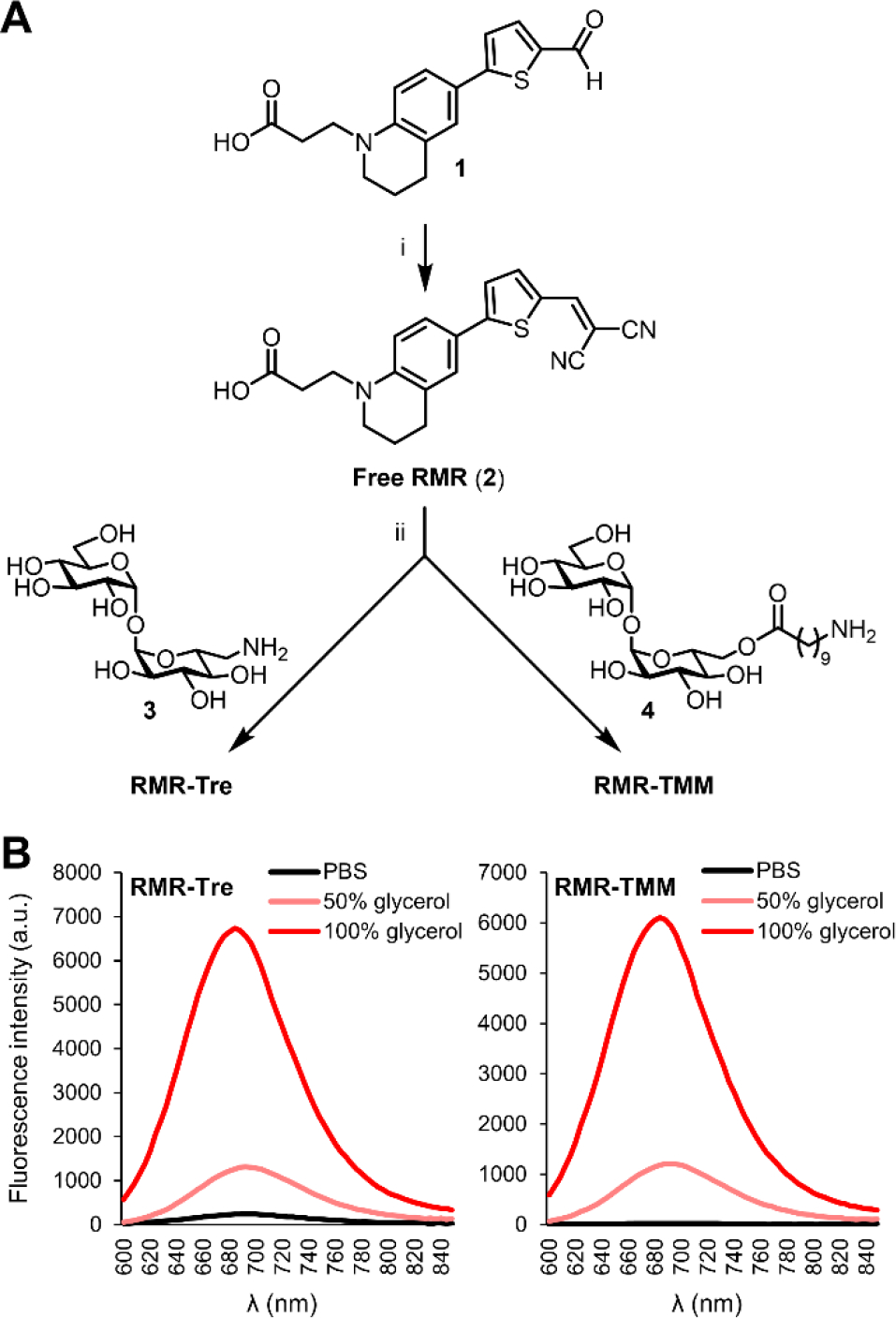
(A) Synthesis of RMR-Tre and RMR-TMM. Reagents and conditions: (i) malonitrile, EtOH, pyridine, 95 °C, 93%; (ii) 3 or 4, TSTU, DMF, DIEA, 60% for RMR-Tre, 71% for RMR-TMM. (B) Fluorescence emission spectra for RMR-Tre and RMR-TMM measured in PBS (pH 7.4) containing 0–100% glycerol (excitation 540 nm).
Evaluation of RMR-Tre and RMR-TMM incorporation into live mycobacteria first focused on the fast-growing, avirulent model organism M. smegmatis. Like all mycobacteria and related species in the Corynebacterineae suborder, M. smegmatis possesses Ag85 mycoloyltransferases capable of inserting trehalose and TMM analogues into the mycomembrane. Furthermore, trehalose probe labeling results in M. smegmatis have generally translated well to other mycobacterial species, including pathogens such as Mtb.[8n] To test for probe incorporation, M. smegmatis was incubated for 4 h in presence of 100 μM of RMR-Tre or RMR-TMM, then the cells were washed, fixed, and analyzed by flow cytometry. Fluorescence fold-change values in probe-treated versus untreated conditions indicated that both RMR probes were incorporated into M. smegmatis with very high efficiency, as RMR-Tre and RMR-TMM treatment led to >2,000- and >1,000-fold increases in fluorescence, respectively (Figure 3A). Treatment with the trehalose-deficient free RMR compound (3) produced negligible fluorescence compared to RMR-Tre and RMR-TMM, consistent with our hypothesis that trehalose conjugation would target the molecular rotor dye to the bacterium (Figure 3A). Free RMR may associate with M. smegmatis non-specifically to a minor extent due to its hydrophobicity, which is likely ameliorated by conjugation to the hydrophilic trehalose moiety.
Figure 3.
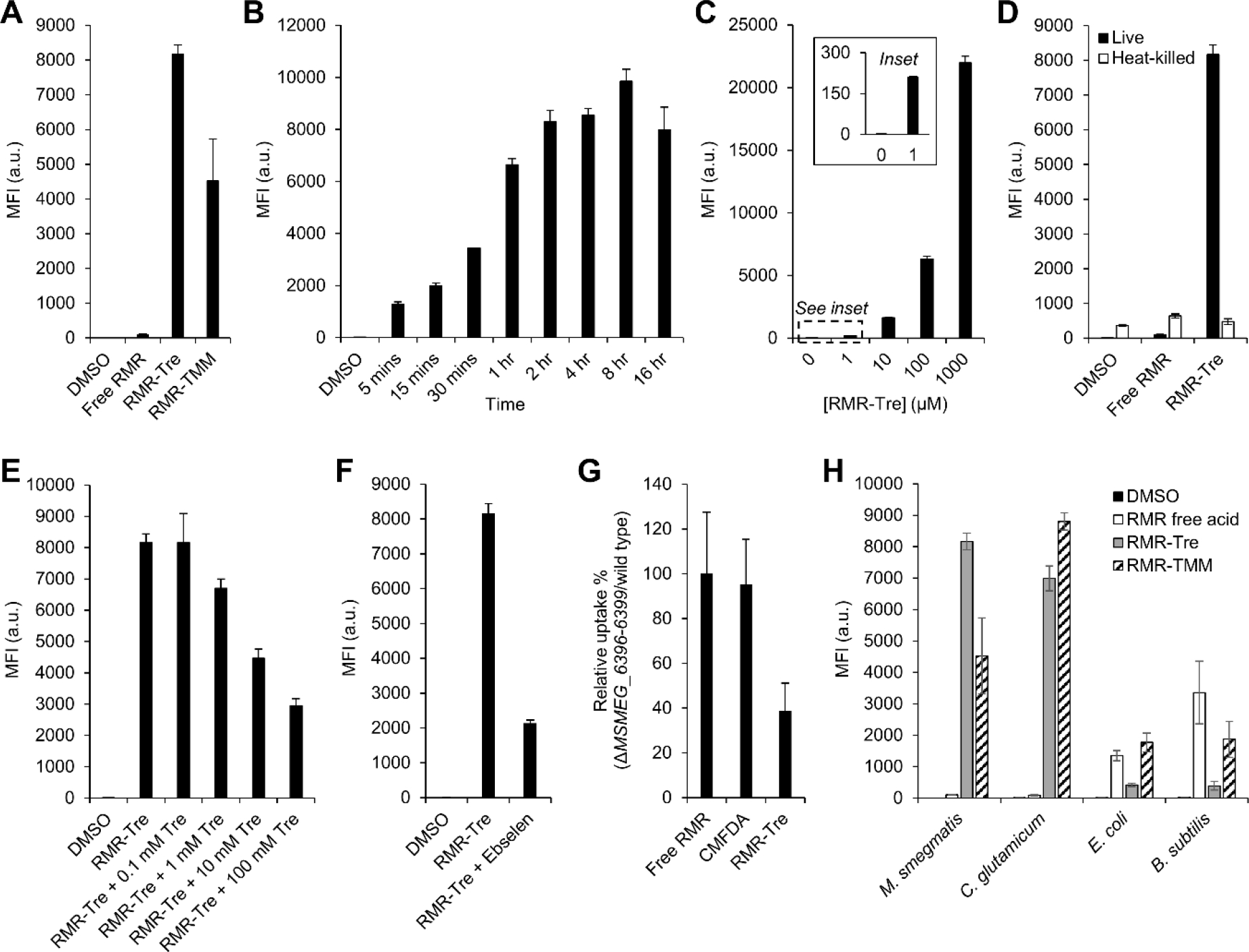
Metabolic incorporation of RMR-Tre into the mycomembrane of live mycobacteria. (A) M. smegmatis was incubated for 4 h in the presence of 100 μM of free RMR, RMR-Tre, or RMR-TMM, or left untreated (DMSO control), then cells were washed, fixed, and analyzed by flow cytometry. (B) M. smegmatis was treated with 100 μM RMR-Tre as in (A) but with varying incubation times. (C) M. smegmatis was treated with RMR-Tre as in (A) but with varying concentrations of probe (inset clearly shows 1 μM concentration results). (D) M. smegmatis was heat-killed or left alone and then treated with 100 μM of free RMR or RMR-Tre as in (A). (E) M. smegmatis was treated with 100 μM RMR-Tre as in (A) with varying concentrations of exogenous native trehalose. (F) M. smegmatis was treated with 100 μM RMR-Tre as in (A) in the presence or absence of Ag85 inhibitor ebselen (100 μg/mL). (G) M. smegmatis wild type or Ag85-deficient mutant ΔMSMEG_6396–6399 were incubated for 30 min in the presence of free RMR (100 μM), CMFDA (5 μM), RMR-Tre (100 μM), or left untreated, then cells were washed, fixed, and analyzed by flow cytometry. Data are expressed as percent relative uptake of each compound in mutant vs. wild-type bacteria to control for permeability effects (see Figure S3 in the Supporting Information for all flow cytometry data and additional discussion). (H) Different bacterial species were treated as in (A). MFI, mean fluorescence intensity in arbitrary units (a.u.). Error bars denote the standard deviation of three replicate experiments.
Although RMR-TMM was incorporated into M. smegmatis, this trehalose ester-based probe also appreciably labeled non-mycobacterial species (discussed below) and thus we focused further characterization efforts on RMR-Tre. RMR-Tre incorporation into M. smegmatis was both time- and concentration-dependent (Figures 3B and 3C). RMR-Tre labeling plateaued after approximately one doubling time (2–4 h), and encouragingly it could be detected with a robust >500-fold fluorescence increase versus untreated cells after an incubation time of only 5 min (Figure 3B). RMR-Tre labeling increased in a dose-dependent manner up to 1 mM, with much lower concentrations of 1 and 10 μM leading to >150 and >1,000-fold fluorescence increases (Figure 3C). Furthermore, bacterial growth was unaffected at RMR-Tre concentrations up to 1 mM (Figure S2 in the Supporting Information). Thus, RMR-Tre provides a means to rapidly label mycobacteria at low reagent concentrations without detectably perturbing normal physiological functions. Undoubtedly, yellow laser excitation (540 nm) and far-red emission (670 nm) contributed to the excellent signal-to-noise values observed for RMR-Tre labeling of M. smegmatis.
Next, we assessed whether RMR-Tre specifically incorporated into live mycobacteria via the targeted trehalose metabolic pathway. Heat-killing of M. smegmatis reduced RMR-Tre labeling to untreated and free RMR-treated background levels, demonstrating that incorporation of RMR-Tre was metabolically driven, while also highlighting the ability of the probe to discriminate between live and dead mycobacteria (Figure 3D). RMR-Tre labeling of M. smegmatis was reduced in a dose-dependent manner by the addition of exogenous native trehalose, which, considered alongside the lack of labeling by the trehalose-deficient free RMR, shows that incorporation is mediated by trehalose metabolism (Figure 3E).
We propose that RMR-Tre incorporates into mycobacteria via extracellular Ag85 mycoloyltransferase activity (Scheme S1 in the Supporting Information). To investigate the labeling route, we first tested RMR-Tre incorporation in the presence of Ag85 inhibitor ebselen,[12] and found that ebselen-treated M. smegmatis exhibited reduced labeling by approximately 75% (Figure 3F). Next, we evaluated RMR-Tre in an Ag85 partial knockout mutant, ΔMSMEG_6396–6399,[8e] which lacks three out of the five predicted Ag85 isoforms encoded by the M. smegmatis genome. Compared to control compounds, including free RMR (3) and 5-chloromethylfluorescein diacetate (CMFDA, a permeability probe[13]), RMR-Tre exhibited a ~60% reduction in ΔMSMEG_6396–6399:wild type fluorescence, consistent with an Ag85-mediated incorporation route (Figure 3G; see also Figure S3 and supplementary discussion of this experiment in the Supporting Information). Although this result does not rule out that some RMR-Tre incorporation may be due to alternative routes, collectively our data and prior literature support our proposed mechanism of incorporation.
We then evaluated the specificity of RMR-Tre and RMR-TMM for mycomembrane-containing bacteria (Figure 3H). RMR-Tre efficiently labeled mycomembrane-containing M. smegmatis and Corynebacterium glutamicum, whereas labeling of mycomembrane-deficient Gram-negative E. coli and Gram-positive B. subtilis was very minor, at a level similar to that of heat-killed M. smegmatis. In contrast, while RMR-TMM efficiently labeled M. smegmatis and C. glutamicum, it also appreciably labeled E. coli and B. subtilis, at an elevated level similar to free RMR. Taken together, our data show that while both RMR-Tre and RMR-TMM efficiently incorporated into live mycobacteria, RMR-Tre exhibited the most promising profile in terms of incorporation efficiency, specificity, and ability to discriminate live from dead bacteria.
Next, we evaluated whether RMR-Tre possessed the critical fluorogenic property desired for mycobacteria detection applications. In prior work, the fluorogenic behavior of DMN-Tre was characterized using fluorescence microscopy,[8e] which is used for TB diagnosis in low-resource settings, so we conducted similar no-wash imaging experiments to assess fluorogenicity of RMR-Tre. We compared RMR-Tre labeling to free RMR, fluorogenic DMN-Tre, and non-fluorogenic 6-FlTre,[8d] a trehalose–fluorescein conjugate. M. smegmatis was incubated in equal concentrations of each compound or left untreated, and cells were directly imaged via fluorescence microscopy without a wash step (Figure 4A). The difference between the non-fluorogenic and fluorogenic probes was striking. Consistent with prior work,[8e] 6-FlTre-treated bacteria were undetectable due to high background, whereas DMN-Tre showed clear labeling of bacteria and good signal above background. The results from RMR-Tre treatment were even more robust, as we observed exceptionally bright fluorescence located at the surface, poles, and septa of the bacteria, consistent with the asymmetric polar growth mode of mycobacteria and with prior imaging experiments employing trehalose probes (Figure 4A).[8n] Notably, the free RMR control compound exhibited low background and did not detectably label M. smegmatis, again underscoring the trehalose-driven fluorescence turn-on capability of RMR-Tre. Furthermore, a no-wash imaging time-course experiment showed that RMR-Tre could label M. smegmatis in as little as 10 min (Figure 4B), demonstrating the possible use of RMR-Tre in rapid detection assays.
Figure 4.
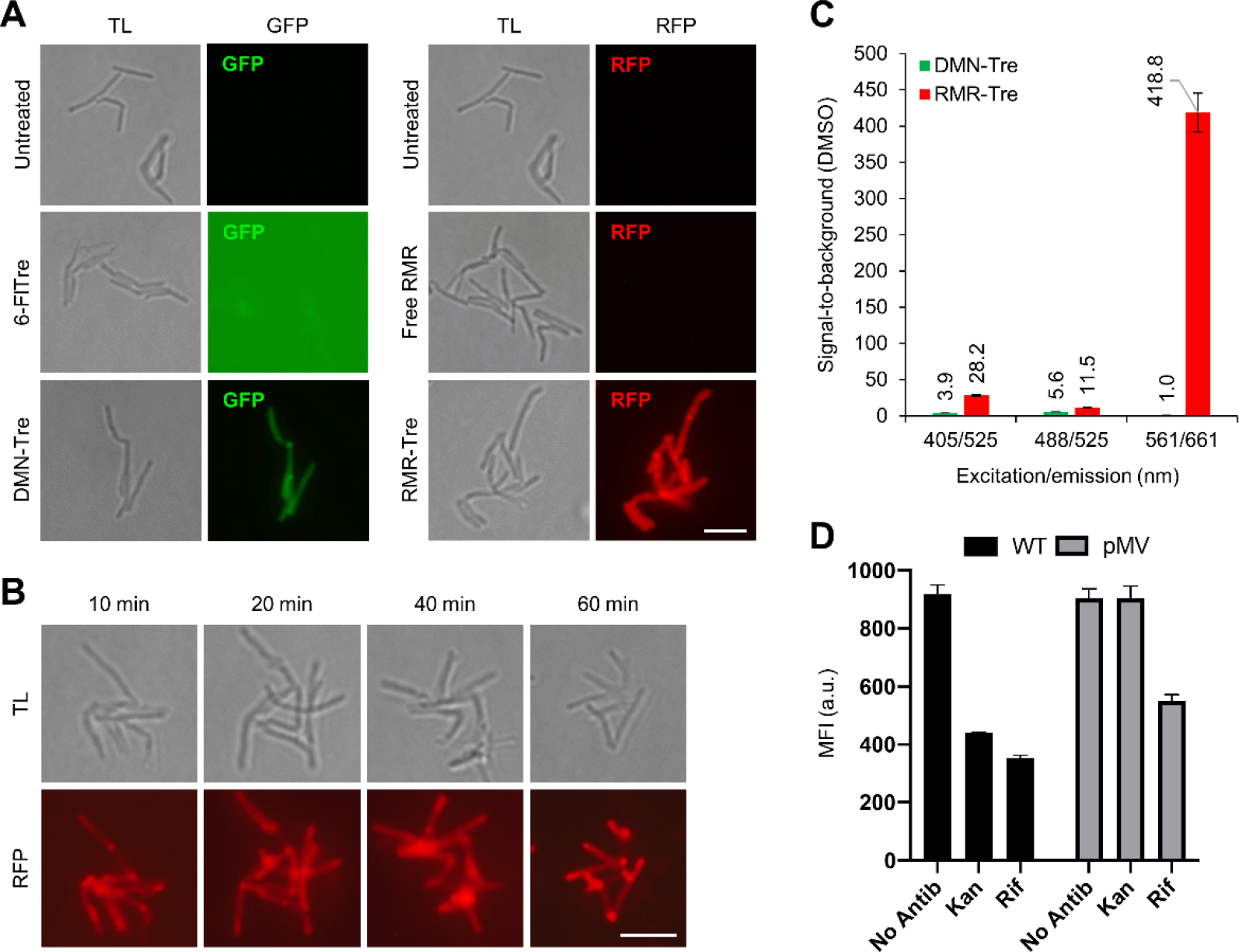
(A) M. smegmatis was incubated for 1 h in the presence of 100 μM of 6-FlTre, DMN-Tre, free RMR, or RMR-TMM, or left untreated, then directly imaged by fluorescence microscopy without washing. Scale bar, 5 μm. GFP, green fluorescent protein channel; RFP, red fluorescent protein channel; TL, transmitted light channel. (B) M. smegmatis was incubated with shaking for the indicated durations in RMR-Tre (100 μM), then directly analyzed by fluorescence microscopy without wash steps. (C) M. tuberculosis H37Rv was incubated for 18 h in the presence of DMN-Tre (100 μM), RMR-Tre (100 μM), or DMSO control, then cells were washed, fixed, and analyzed by spectral flow cytometry (see Figure S4 in the Supporting Information for all spectral flow cytometry data). (D) M. tuberculosis H37Rv mc26206 wild type (WT) or KAN-resistant strain (pMV) were incubated for 24 h in the presence of Rif (1 μg/mL), Kan (2.5 μg/mL), or left untreated, then incubated for an additional 18 h with RMR-Tre (100 μM), fixed without washing, and analyzed by flow cytometry. Error bars denote the standard deviation of three replicate experiments.
Finally, we evaluated the performance of RMR-Tre in the detection of M. tuberculosis and explored the potential of RMR-Tre as a TB diagnostic tool. We compared the labeling efficiencies of RMR-Tre and DMN-Tre in M. tuberculosis H37Rv by spectral flow cytometry, which provided signal-to-background values for both probes across the entire range of excitation and emission wavelengths (Figure 4C shows data for selected excitation and emission wavelengths; Figure S4 in the Supporting Information shows all data). Whereas DMN-Tre exhibited a maximal signal-to-background of ~5 using the green channel (excitation/emission 488/525 nm), RMR-Tre achieved values of >400, nearly 100-fold higher, in the red region (561/661 nm). These results of RMR-Tre labeling in M. tuberculosis further underscore the benefits of the red-shifted molecular rotor fluorophore.
To evaluate the potential of RMR-Tre in TB diagnostics, we investigated whether RMR-Tre could be used as a tool in TB drug-susceptibility testing, which is a critical element of addressing drug-resistant TB but remains hampered by the cost, complexity, slowness, and/or inflexibility of existing technologies in the clinic.[14] RMR-Tre labeling is simple and rapid, and because it specifically labels viable mycobacteria, the probe should be sensitive to drug treatment and thus enable facile detection of drug-resistant M. tuberculosis. Consistently, DMN-Tre was previously shown to detect isoniazid resistance in the model organism M. smegmatis.[8e] Here, we evaluated RMR-Tre labeling in two strains of M. tuberculosis H37Rv mc26206,[15] including a drug-susceptible wild-type strain and a kanamycin (Kan)-resistant strain. Using a no-wash flow cytometry-based labeling experiment, we found that RMR-Tre labeling of wild-type M. tuberculosis was significantly diminished by treatment with both rifampicin (Rif) and Kan, whereas RMR-Tre labeling of the Kan-resistant strain was sensitive to Rif treatment but unresponsive to Kan treatment (Figure 4D and S5). This result establishes proof-of-concept that RMR-Tre can report on M. tuberculosis drug resistance and advances RMR-Tre as a potential tool for TB drug-susceptibility testing.
Conclusion
With mycobacterial diseases such as TB continuing to present a massive burden on global health, new tools are needed to accelerate basic research and improve disease diagnosis and treatment. Among recent efforts to improve the detection of mycobacteria, trehalose-based chemical probes have gained attention due to their speed, simplicity, specificity, and reliance on active metabolic incorporation. The recent integration of fluorogenic turn-on designs has further extended the utility of trehalose probes. However, remaining limitations in probe sensitivity and specificity could impede translational efforts, particularly in the areas of diagnostics and drug development. Here, we developed RMR-Tre, which contains a novel molecular rotor fluorophore that brightly emits signal in the far-red region upon trehalose-guided metabolic integration into the sterically crowded mycomembrane. Our data establish RMR-Tre as having better specificity than trehalose ester-based probes (e.g., RMR-TMM reported herein, FRET-based designs) and enhanced sensitivity compared to existing fluorogenic trehalose probes with green emission. We demonstrate that these attributes may enable applications of RMR-Tre in TB diagnosis and drug-susceptibility testing, and we also anticipate that the probe could have value in imaging and screening studies focused on the mycomembrane, which is an attractive drug target. Finally, with the advantages of molecular rotor-based metabolic probes now established for labeling both bacterial peptidoglycan and mycomembrane, there is interest in the continued development and characterization of this new class of tools.
Supplementary Material
Acknowledgements
This work was supported by NSF CAREER Award 1654408 (B.M.S.), Camille and Henry Dreyfus Foundation Henry Dreyfus Teacher–Scholar Award TH-17–034 (B.M.S.), NIH R35 Award GM136365 (M.S.V.), NIH DP2 Award AI138238 (M.S.S.). NMR instrumentation was supported by NSF MRI Award 2117338 (B.M.S.). We thank Drs. Mallary Greenlee-Wacker, Mireille Kamariza, Bavesh Kana, and Ruby Dewy for helpful discussions, the Indiana University NMR and MS facilities, and the Colorado State University Flow Cytometry Core (RRID: SCR_022000).
References
- [1].World Health Organization, Global Tuberculosis Report 2021. 2021.
- [2].Johansen MD, Herrmann J-L, Kremer L, Nat. Rev. Microbiol. 2020, 18, 392–407. [DOI] [PubMed] [Google Scholar]
- [3].Singhal R, Myneedu VP, Int. J. Mycobacteriol. 2015, 4, 1–6. [DOI] [PubMed] [Google Scholar]
- [4].a) Desikan P, Indian J Med. Res. 2013, 137, 442–444; [PMC free article] [PubMed] [Google Scholar]; b) Alnour TMS, Indian J Tuberc. 2018, 65, 190–194; [DOI] [PubMed] [Google Scholar]; c) Cuevas LE, Al-Sonboli N, Lawson L, Yassin MA, Arbide I, Al-Aghbari N, Bahadur Sherchand J, Al-Absi A, Emenyonu EN, Merid Y, Okobi MI, Onuoha JO, Aschalew M, Aseffa A, Harper G, Anderson de Cuevas RM, Theobald SJ, Nathanson C-M, Joly J, Faragher B, Squire SB, Ramsay A, PLoS Med. 2011, 8, e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Xie H, Mire J, Kong Y, Chang M, Hassounah HA, Thornton CN, Sacchettini JC, Cirillo JD, Rao J, Nat. Chem. 2012, 4, 802–809; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cheng Y, Xie H, Sule P, Hassounah H, Graviss EA, Kong Y, Cirillo JD, Rao J, Angew. Chem. Int. Edit 2014, 53, 9360–9364; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cheng Y, Xie J, Lee KH, Gaur RL, Song A, Dai T, Ren H, Wu J, Sun Z, Banaei N, Akin D, Rao J, Sci. Transl. Med. 2018, 10, eaar4470; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mu R, Kong C, Yu W, Wang H, Ma Y, Li X, Wu J, Somersan-Karakaya S, Li H, Sun Z, Liu G, ACS Infect. Dis. 2019, 5, 949–961. [DOI] [PubMed] [Google Scholar]
- [6].Quémard A, Trends Microbiol. 2016, 24, 725–738. [DOI] [PubMed] [Google Scholar]
- [7].Backus KM, Boshoff HI, Barry CS, Boutureira O, Patel MK, D’Hooge F, Lee SS, Via LE, Tahlan K, Barry CE 3rd, Davis BG, Nat. Chem. Biol. 2011, 7, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Swarts BM, Holsclaw CM, Jewett JC, Alber M, Fox DM, Siegrist MS, Leary JA, Kalscheuer R, Bertozzi CR, J. Am. Chem. Soc. 2012, 134, 16123–16126; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Foley HN, Stewart JA, Kavunja HW, Rundell SR, Swarts BM, Angew. Chem. Int. Edit. 2016, 55, 2053–2057; [DOI] [PubMed] [Google Scholar]; c) Rundell SR, Wagar ZL, Meints LM, Olson CD, O’Neill MK, Piligian BF, Poston AW, Hood RJ, Woodruff PJ, Swarts BM, Org. Biomol. Chem. 2016, 14, 8598–8609; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rodriguez-Rivera FP, Zhou X, Theriot JA, Bertozzi CR, J. Am. Chem. Soc. 2017, 139, 3488–3495; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kamariza M, Shieh P, Ealand CS, Peters JS, Chu B, Rodriguez-Rivera FP, Babu Sait MR, Treuren WV, Martinson N, Kalscheuer R, Kana BD, Bertozzi CR, Sci. Transl. Med. 2018, 10, eaam6310; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Hodges HL, Brown RA, Crooks JA, Weibel DB, Kiessling LL, Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5271–5276; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Fiolek TJ, Banahene N, Kavunja HW, Holmes NJ, Rylski AK, Pohane AA, Siegrist MS, Swarts BM, ChemBioChem 2019, 20, 1282–1291; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Holmes NJ, Kavunja HW, Yang Y, Vannest BD, Ramsey CN, Gepford DM, Banahene N, Poston AW, Piligian BF, Ronning DR, Ojha AK, Swarts BM, ACS Omega 2019, 4, 4348–4359; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Lesur E, Baron A, Dietrich C, Buchotte M, Doisneau G, Urban D, Beau J-M, Bayan N, Vauzeilles B, Guianvarc’h D, Bourdreux Y, Chem. Commun. 2019, 55, 13074–13077; [DOI] [PubMed] [Google Scholar]; j) Zhou X, Rodriguez-Rivera FP, Lim HC, Bell JC, Bernhardt TG, Bertozzi CR, Theriot JA, Nat. Chem. Biol. 2019, 15, 221–231; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Kavunja HW, Biegas KJ, Banahene N, Stewart JA, Piligian BF, Groenevelt JM, Sein CE, Morita YS, Niederweis M, Siegrist MS, Swarts BM, J. Am. Chem. Soc. 2020, 142, 7725–7731; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Kamariza M, Keyser SGL, Utz A, Knapp BD, Ealand C, Ahn G, Cambier CJ, Chen T, Kana B, Huang KC, Bertozzi CR, JACS Au 2021, 1, 1368–1379; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Biegas KJ, Swarts BM, Curr. Opin. Chem. Biol. 2021, 65, 57–65; [DOI] [PubMed] [Google Scholar]; n) Banahene N, Kavunja HW, Swarts BM, Chem. Rev. 2022, 122, 3336–3413; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Dai T, Xie J, Zhu Q, Kamariza M, Jiang K, Bertozzi CR, Rao J, J. Am. Chem. Soc. 2020, 142, 15259–15264; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Li X, Geng P, Hong X, Sun Z, Liu G, Chem. Commun. 2021, 57, 13174–13177. [DOI] [PubMed] [Google Scholar]
- [9].Oliveira E, Bértolo E, Núñez C, Pilla V, Santos HM, Fernández-Lodeiro J, Fernández-Lodeiro A, Djafari J, Capelo JL, Lodeiro C, ChemistryOpen 2018, 7, 9–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a Haidekker MA, Theodorakis EA, J. Biol. Eng. 2010, 4, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sasaki S, Drummen GPC, Konishi G.-i., J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar]
- [11].Hsu YP, Hall E, Booher G, Murphy B, Radkov AD, Yablonowski J, Mulcahey C, Alvarez L, Cava F, Brun YV, Kuru E, VanNieuwenhze MS, Nat. Chem. 2019, 11, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Favrot L, Grzegorzewicz AE, Lajiness DH, Marvin RK, Boucau J, Isailovic D, Jackson M, Ronning DR, Nat. Comm. 2013, 4, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Purdy GE, Niederweis M, Russell DG, Mol. Microbiol. 2009, 73, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gill CM, Dolan L, Piggott LM, McLaughlin AM, Breathe 2022, 18, 210149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sampson SL, Mansfield KG, Carville A, Magee DM, Quitugua T, Howerth EW, Bloom BR, Hondalus MK, Vaccine 2011, 29, 4839–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.