

Abstract
Cytochrome P450s are important phase I metabolic enzymes located on endoplasmic reticulum (ER) involved in the metabolism of endogenous and exogenous substances. Our previous study showed that a hepatoprotective agent silybin restored CYP3A expression in mouse nonalcoholic fatty liver disease (NAFLD). In this study we investigated how silybin regulated P450s activity during NAFLD. C57BL/6 mice were fed a high-fat-diet (HFD) for 8 weeks to induce NAFLD, and were administered silybin (50, 100 mg ·kg−1 ·d−1, i.g.) in the last 4 weeks. We showed that HFD intake induced hepatic steatosis and ER stress, leading to significant inhibition on the activity of five primary P450s including CYP1A2, CYP2B6, CYP2C19, CYP2D6, and CYP3A in liver microsomes. These changes were dose-dependently reversed by silybin administration. The beneficial effects of silybin were also observed in TG-stimulated HepG2 cells in vitro. To clarify the underlying mechanism, we examined the components involved in the P450 catalytic system, membrane phospholipids and ER membrane fluidity, and found that cytochrome b5 (cyt b5) was significantly downregulated during ER stress, and ER membrane fluidity was also reduced evidenced by DPH polarization and lower polyunsaturated phospholipids levels. The increased ratios of NADP+/NADPH and PC/PE implied Ca2+ release and disruption of cellular Ca2+ homeostasis resulted from mitochondria dysfunction and cytochrome c (cyt c) release. The interaction between cyt c and cyt b5 under ER stress was an important reason for P450s activity inhibition. The effect of silybin throughout the whole course suggested that it regulated P450s activity through its anti-ER stress effect in NAFLD. Our results suggest that ER stress may be crucial for the inhibition of P450s activity in mouse NAFLD and silybin regulates P450s activity by attenuating ER stress.
Keywords: Non-alcoholic fatty liver disease, Silybin, P450s, ER stress, cyt b5, cyt c
Introduction
Nonalcoholic fatty liver disease (NAFLD) has gradually become a global health problem due to increases in high fat and sugar diets. In some industrialized countries, this disease incidence among adults is as high as 30% [1] and has the potential to further develop into liver fibrosis or even hepatocarcinoma. In addition, NAFLD typically causes metabolic alterations in vivo and is closely associated with metabolic syndromes. The hepatic accumulation of lipids subsequently is the hallmark of NAFLD and leads to cellular stress and hepatic injury, which eventually results in chronic liver disease. Abnormal lipid accumulation is associated with perturbed endoplasmic reticulum (ER) proteostasis in hepatocytes and affects the metabolic function as well. One of the most important metabolic enzyme systems is the cytochrome P450s superfamily, members of which participate in both the metabolism of xenobiotics and the biosynthesis of crucial signaling molecules. However, the activity of P450s varies a great deal during liver injury, including NAFLD and NASH [2, 3].
Cytochrome P450 (P450) enzymes are a component of the mixed-function oxidase (MFO) system located in the membrane of the ER and catalyze the oxidative metabolism of 75% of drugs and foreign lipophilic compounds [4]. For example, NADPH-cytochrome P450 reductase (CPR) and cytochrome b5 (cyt b5) are important components that provide electrons required for the monooxygenation reaction [5]. CPR is the redox partner for most human cytochrome P450 enzymes and can bind NADPH to reduce the enzyme by transferring electrons to both the FAD and FMN domains within the protein [6]. Cyt b5 is a small hemoprotein that takes part in many biochemical reactions, including fatty acid desaturation process, fatty acid elongation and cholesterol biosynthesis [7]. This protein is also involved in many microsomal oxidation reactions and monohydroxylation processes by interacting with a variety of cytochrome P450 (CYPs or P450s) and cytochrome c (cyt c) in the mitochondrial outer membrane. The ER membrane is another key factor, since P450s are membrane proteins. The heterogeneity of the membrane affects P450s function by either concentrating or segregating proteins within membrane regions [8]. How ER stress influences P450s activity directly in fatty liver disease is still not clear and this study can provide a better understanding of the underlying mechanisms during ERs in NAFLD.
Silybin is used worldwide as a hepatoprotective agent due to its strong anti-inflammation, anti-lipid peroxidation and anti-hepatic fibrosis effects [9–11]. Previous studies suggested that silybin is a potential inhibitor of multiple drug-metabolizing enzymes (DMEs) in vitro. For example, silybin significantly inhibits UGT activity in a dose-dependent manner in vitro [12–14]. However, little is known regarding its effects on P450s in the in vivo system and the underlying mechanisms. Our previous study revealed that silybin restored CYP3A expression through the SIRT2/NF-κB pathway at the transcriptional level in mouse NAFLD [15]. We also demonstrated that the mechanism associated with the anti-inflammatory effect of silybin is based on inhibiting NLRP3 inflammasome assembly to reduce the release of IL-1β [16], while cytokines are well-defined factors in the transcriptional regulation of P450s [17–19]. However, the underlying mechanisms associated with the direct regulation of P450s activity by silybin under liver disease states have been rarely studied. Considering that silybin is quite effective in preventing ER stress [16], we raise a hypothesis that silybin may regulate liver P450s activity directly through attenuating ER stress during NAFLD.
In the present study, we evaluated how ER stress influences P450s activity in NAFLD. We investigated the variation in P450 components, including CPR, cyt b5, NADPH and NADH, and elucidated how these components influence enzyme activity under ER stress. Furthermore, we provide a model of the underlying mechanism based on ER membrane fluidity and the role of mitochondria-ER interaction in P450s activity regulation.
Materials and methods
Materials
Silybin was purchased from Zeland (Nanjing, China), while tauroursodeoxycholic acid (TUDCA), thapsigargin (TG) and DPH were obtained from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies targeting the following proteins were used in the present study: inositol-requiring transmembrane kinase/endoribonuclease (IRE)-1α (ab37073; RRID: AB_775780), phosphorylated p-IRE-1α (ab48187; RRID: AB_873899) and anti-cytochrome c [7H8.2C12] (ab13575; RRID: AB_300470) (Abcam, Cambridge, MA, USA); and eukaryotic initiation factor 2A (eIF2α) (I45) polyclonal antibody (BS3651; RRID: AB_1663143), eIF2α (phospho-S51) polyclonal antibody (BS4787; RRID: AB_1663144), GAPDH monoclonal antibody (AP0063; RRID: AB_2651132) and cytochrome b5 (G82) polyclonal antibody (BS2410; RRID: AB_1663560). In addition, goat anti-mouse IgG (H + L) DyLight 488 (BS10015; RRID: AB_2819202), donkey anti-rabbit IgG (H + L) DyLight 594 (BS10030; RRID: AB_2819203), and goat anti-rabbit IgG (H + L)-HRP (BS13278; RRID: AB_2773728) (Bioworld Biotechnology, Shanghai, China) were used as secondary antibodies.
Animals and treatment
Male C57BL/6 mice aged 4–6 weeks old were acquired from Changzhou Cavans Experimental Animal Co. Ltd (Changzhou, China). Mice were randomly divided into four groups and housed in a temperature-controlled environment with a 12 h light-dark cycle. All animal protocols were approved by the Animal Ethics Committee of China Pharmaceutical University. Mice were fed a standard chow diet or a high-fat-diet (10% lard, 10% yolk, 1% cholesterol, 0.2% cholate, and 78.8% standard diet) for 8 weeks. Silybin (50 or 100 mg/kg per day) was administered intragastrically for the last 4 weeks of the experiment. All experiments were performed in compliance with the guidelines of the Animal Experimentation Board of China Pharmaceutical University on animal usage.
Cell culture of HepG2 cells
HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific, Madrid, Spain) supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Madrid, Spain), 1% antibiotics (Gibco, Thermo Fisher Scientific, Madrid, Spain) and 1% MEM NEAA (Gibco, Thermo Fisher Scientific, Madrid, Spain) at 37 °C and under a humidified atmosphere with 5% CO2.
Preparation of microsomes from mouse liver and HepG2
Mouse liver microsomes were isolated as follows. Liver tissues were homogenized after the addition of ice-cold PBS (pH 7.4) at a 1:4 ratio (w/v). Then, after the homogenate was centrifuged at 9000 × g for 20 min at 4 °C, the supernatant was carefully transferred to new tubes and centrifuged at 100,000 × g and for 1 h at 4 °C. Subsequently, the supernatant was discarded, and the pelleted liver microsomes were resuspended in a 30% (v/v) glycerol phosphate buffer solution and stored at −80 °C.
ER-enriched HepG2 subcellular fractions were isolated by serial centrifugation at 4 °C. Cell pellets were collected and homogenized in phosphate buffered saline (PBS, pH 7.4) on ice. Subsequently, S9 fractions were obtained after the homogenates being centrifuged at 4 °C at 10,000 × g for 30 min, the supernatants (S9 fractions) were then transferred to a Beckman ultracentrifuge tube and centrifuged at 100,000 × g for 60 min, after which the microsomes at the bottom of the tubes were resuspended in PBS [20]. The protein concentration of the microsomes was determined using a BCA protein assay kit (Beyotime Technology, Shanghai, China), and the microsomes samples were stored at −80 °C until use.
P450s activity assays
To evaluate P450s activity in liver microsomes samples, 75 μL of microsomes (the final concentration was 0.5 mg/mL), 5 μL of substance at different concentrations and 20 μL of NADPH Regeneration System (NRS, 2 mM NADPH, 10 mM G-6-p, 1 U/mL PDH, and 10 mM MgCl2) was mixed and then incubated in a water bath at 37 °C for 5 min. After incubating, the samples were quickly placed on ice and the reaction was stopped by adding 300 μL of methanol containing tamsulosin (100 ng/mL) as an internal standard. Next, the mixtures were vigorously extracted for 5 min and then centrifuged at 18,000 × g for 5 min twice before injection for HPLC–MS/MS analysis. Data were fitted to the Michaelis-Menten model defined by the following equation: V = Vmax × S/(Km + S), where V is the reaction rate, S is the substrate concentration, Vmax is the maximum enzyme velocity and Km is the Michaelis constant, which is equal to the concentration of substrate for half-maximal velocity.
ELISA
Cytochrome b5 (Wuhan EIAab Science Co., Ltd., Wuhan, China), cytochrome P450 reductase (Wuhan Fine Biotech Co., Ltd, Wuhan, China), mitochondrial respiratory chain complex I (Dogesce, Beijing, China) and complex III (Solarbio, Beijing, China) activities were measured with ELISA kits according to the manufacturer’s instructions.
NADPH measurement
Intracellular NADPH levels were detected using a liquid chromatography–tandem mass spectrometry method that was previously established by our laboratory [21].
Measurements of membrane phospholipids
ER lipids were extracted as follows. Briefly, 20 μL of mouse liver microsomes or 40 μL of HepG2 cell microsomes was mixed with 10 μL of phosphatidylcholine (PC, 28:0) as an internal standard. Then, methanol was added to a total volume of 200 μL and mixed, after which 500 μL of methyl tert-butyl ether was added, and the mixture was vigorously extracted for 30 min at room temperature. Then, 125 μL of ultrapure water was added, and the mixture was incubated at room temperature for 10 min before being centrifuged at 14,000 × g for 15 min. Subsequently, a 400 μL aliquot of the upper liquid was transferred to a new tube and evaporated to dryness. Dried lipids were reconstituted in acetonitrile/isopropanol/water (60:35:5, v/v/v) and then centrifuged at 18,000 × g for 5 min at 4 °C twice for mass spectrometry analyses.
Measurements of membrane fluidity
The ER membrane fluidity in mouse liver and HepG2 cells was determined by diphenyl-1,3,5-hexatriene (DPH) according to Gibbons [22]. ER microsomal fractions were resuspended in a KCl-based buffer (150 mM KCl, 10 mM HEPES (pH 7.4) and 2 mM EGTA) to a final concentration of 0.5 mg/mL and then incubated with 10 μM DPH at 45 °C for 30 min. Endpoint fluorescence polarization measurements were performed using a POLARstar Omega plate reader (BMG Labtech, Offenbug, Germany). The temperature was ramped from 27 °C to 45 °C in 2 °C intervals.
Measurements of oxygen consumption
The rates of oxygen consumption in HepG2 cell lines were measured with a Seahorse Bioscience XF96 extracellular flux analyzer (Aglient, Santa Clara, CA, USA) according to the manufacturer’s instructions.
Measurements of cyt c/cyt a
Mitochondria was isolated from cell lines with a mitochondrial extraction kit (Beyotime Technology, Shanghai, China) according to the manufacturer’s instructions. The cyt c/cyt a ratio was measured according to literature [23].
Western blot analysis and immunoprecipitation
Tissue and cell protein lysates were prepared in RIPA buffer, and protein expression was analyzed with 8% SDS-PAGE using the following antibodies: GAPDH (1:10000), IRE1α (1:1000), phosphate-IRE1α (1:1000), eIF2α (1:1000), phosphate-eIF2α (1:1000) and anti-rabbit IgG (1:5000). For immunoprecipitation, the lysates were further diluted with the lysis buffer and then incubated with cyt b5 overnight. Then, the lysates were incubated with 40 μL of protein A/G-Sepharose for an additional 3 h, after which the immunocomplexes were collected by centrifugation. The samples were then washed five times with ice PBS and subsequently eluted in 40 μL of sample buffer. The samples were analyzed by 12% SDS-PAGE using the anti-cytochrome c antibody.
Cytoplasmic and mitochondrial Ca2+
Cells were washed 3 times with PBS after treatment with drugs for 24 h. Before imaging, HepG2 cells were incubated at 37 °C and under an atmosphere with 5% CO2 in 2 μM Fura-2 AM (Beyotime Biotechnology, Shanghai, China) and 2.5 μM Rhod-2 AM (Invitrogen, Carlsbad, CA, USA) for 45 min, after which they were washed with HBSS 3 times. Cytoplasmic and mitochondrial Ca2+ fluorescence was measured using both a Biotek Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek, Vermont, VT, USA) and a fluorescence microscope (Opera phenix, PerkinElmer, Fremont, CA, USA).
Immunofluorescence
Cells were seeded in a confocal petri dish and treated with drugs (1 μM TG, 50 μM SB, and 100 μM TUDCA) for 24 h. Subsequently, the cells were washed 3 times with ice-cold PBS (5 min each time), after which 4% paraformaldehyde was added to each well and the cells were fixed for 20 min at room temperature. The cells were 3 times before the addition of Triton X-100 (Beyotime Biotechnology, Shanghai, China) to permeate cells for 5–10 min. After being washed with PBS, 5% BSA (prepared in PBS) was added to each well for 2 h at room temperature. Then, after being washed with ice-cold PBS, the cells were incubated with the primary antibodies anti‐cyt b5 and anti-cyt c diluted 1:200 at 4 °C overnight. At the end of the incubation, the cells were rinsed with PBST three times to remove unbound antibody. Subsequently, DyLight 488- or DyLight 594-conjugated secondary antibodies diluted 1:500 were added to the cells at room temperature for 2 h, after which the cells were incubated with DAPI for 10 min before being observed by laser confocal microscopy (Olympuc, Tokyo, Japan).
Statistical analysis
The data were presented as the means ± SEM (standard error of the mean) of measurements obtained from five or eight batches of cells or six mice in each group per experiment. The statistical analysis among groups was performed using one-way ANOVA followed by Tukey’s post hoc test. The significance of differences between two groups was determined by Student’s t test (unpaired, two tailed). Values of P < 0.05 were considered statistically significant.
Results
P450s activity varied in HFD-fed mouse liver microsomes
To investigate the P450s activity variation during NAFLD, mice were fed HFD for 8 weeks (Fig. 1a). The morphology of livers from mice subjected to HFD was assessed by H&E and Oil red O staining (Fig. 1b). The hepatic steatosis induced by the HFD was ameliorated by silybin treatment. Then, we evaluated the activity of primary subfamilies of P450s, including CYP1A2, CYP2B6, CYP2C19, CYP2D6, and CYP3A using their specific substrates phenacetin, bupropion, omeprazole, dextromethorphan, midazolam, and testosterone (Fig. 1c–h). The calculated Vmax and CLint values were compared. The means and SD of five calculated parameters were shown, and the values were listed in Table 1. Kinetic analysis results indicated that the activity of all five P450 enzyme subfamilies decreased significantly. Silybin and TUDCA reversed the activity of most enzyme subfamilies to varying degrees.
Fig. 1. HFD induced the suppression of P450s activity and endoplasmic reticulum stress in the mouse liver (n = 5).

Mice were fed a control diet, HFD or silybin for 8 weeks (a). Hematoxylin and eosin (H&E) staining (upper panel) and Oil red O staining (ORO) (lower panel) of mouse livers (b). Analysis of p450s activity, including CYP1A, CYP2B, CYP2C, CYP2D, and CYP3A through microsomes (c–h).
Table 1.
Determination of P450s activity in liver microsomes by HPLC–MS/MS.
| P450 subtype | Control | HFD | Silybin (50 mg/kg) | Silybin (100 mg/kg) | |
|---|---|---|---|---|---|
| CYP1A2 (Acetaminophen) | Vmax (μmol·min-1·g-1 protein) | 0.244 ± 0.006 | 0.191 ± 0.014 | 0.213 ± 0.012 | 0.127 ± 0.007 |
| CLint (mL·min-1·g-1 protein) | 2.071 | 1.048 | 1.694 | 1.164 | |
| CYP2B6 (Hydroxybupropion) | Vmax (μmol·min-1·g-1 protein) | 0.024 ± 0.001 | 0.016 ± 0.001 | 0.032 ± 0.006 | 0.028 ± 0.002 |
| CLint (mL·min-1·g-1 protein) | 0.057 | 0.030 | 0.035 | 0.039 | |
| CYP2C19 (Hydroxyomeprazole) | Vmax (μmol·min-1·g-1 protein) | 0.243 ± 0.179 | 0.113 ± 0.030 | 0.102 ± 0.027 | 0.146 ± 0.012 |
| CLint (mL·min-1·g-1 protein) | 1.873 | 1.463 | 1.688 | 3.628 | |
| CYP2D6 (Dextropan) | Vmax (μmol·min-1·g-1 protein) | 0.111 ± 0.004 | 0.043 ± 0.002 | 0.056 ± 0.004 | 0.038 ± 0.004 |
| CLint (mL·min-1·g-1 protein) | 22.82 | 15.95 | 21.73 | 10.07 | |
| CYP3A (4-hydroxymidazolam) | Vmax (μmol·min-1·g-1 protein) | 0.290 ± 0.049 | 0.267 ± 0.042 | 0.267 ± 0.036 | 0.293 ± 0.077 |
| CLint (mL·min-1·g-1 protein) | 3.756 | 2.935 | 3.470 | 4.741 | |
| CYP3A (6β-hydroxytestosterone) | Vmax (μmol·min-1·g-1 protein) | 0.277 ± 0.077 | 0.147 ± 0.025 | 0.196 ± 0.022 | 0.187 ± 0.063 |
| CLint (mL·min-1·g-1 protein) | 1.271 | 0.569 | 1.146 | 0.820 |
ER stress was involved in P450s activity suppression in NAFLD mice
Because P450s are membrane proteins that are primarily located on the ER, and both silybin and TUDCA are effective at ameliorating ER stress, we hypothesized that ER stress may be an important reason leading to P450s activity decline. Thus, the levels of proteins that sense ER stress were assessed. Consistent with our previous results, HFD enhanced the protein levels of phosphorylated IRE1α (p-IRE1α) and phosphorylated eukaryotic IRF2α (p-eIF2α) along with unchanged total IRE1α and eIF2α expression (Fig. 2a). As for the effect of TG on HepG2, silybin inhibited the activation of IRE1α and IRF2α, lowering the phosphorylation of these protein, similar with the action of positive control TUDCA (Fig. 2b). Then, we studied the effect of TG on P450s activity in HepG2 cells. The rate of metabolite generation for each substrate indicated that most P450s activity was suppressed by TG and ameliorated by medium and low concentrations of silybin and TUDCA (Fig. 2c–h). The results also showed that high concentration of silybin may inhibit the activity of P450s, which was consistent with those of previous studies in vitro [12, 24].
Fig. 2. Effects of endoplasmic reticulum stress on p450s activity.

HepG2 were pretreated with silybin (2, 10, and 50 μM) or TUDCA (100 μM), followed by stimulation with TG (1 μM) for 24 h, and mice were fed an HFD for 8 weeks with oral administration of silybin (50 or 100 mg/kg). IRE1a and eIF2a phosphorylation levels were measured by Western blot (n = 5) (a, b). Analysis of P450s activity, including CYP1A, CYP2B, CYP2C, CYP2D, and CYP3A in HepG2 cells, which was determined by LC-MS/MS (n = 5) (c–h). Statistical significance was determined by two-tailed paired t-test. *P < 0.05; **P < 0.01; ***P < 0.001 vs. model and #P < 0.05; ##P < 0.01; ###P < 0.001 vs. control.
ER stress regulated P450s enzyme system components
To further elucidate how ER stress regulates P450s activity, we studied the effect of ER stress on the primary components of the P450s system. Among the P450s enzyme system components, Cyt b5 was significantly downregulated in the livers of HFD-fed mice and TG-treated HepG2 cells (Fig. 3a). However, the variation trend of CPR was not that obviously (Fig. 3b). In addition, a sharp increase of the ratio of NADP+/NADPH was observed in the livers of HFD-fed mice and TG-treated HepG2 cells, and the ratio decreased after silybin and TUDCA treatment (Fig. 3c). The increased levels of NADP+ can be catalyzed into NAADP, which can induce Ca2+ release from the ER [25–27].
Fig. 3. HFD and endoplasmic reticulum stress induced a decrease in the composition of the P450 catalytic system.

Analysis of cytochrome b5 and CPR levels through ELISA kits (a, b). NADP+ and NADPH levels were determined by LC-QTOF/MS. The values of NADP+/NADPH of other groups were normalized to the control, which was set as 1 (c). n = 6 for animals and n = 5 for cells per group. Data were presented as the means ± SEM. Statistical analysis was performed by two-tailed paired t test. *P < 0.05; **P < 0.01; ***P < 0.001 vs. model and #P < 0.05; ###P < 0.001 vs. control.
ER stress regulated hepatic ER membrane fluidity and phospholipid composition
The ER membrane provides a scaffold for the P450 system proteins that facilitate interactions with their redox partners as well as other P450s such that the composition and fluidity of the ER membrane influences the membrane protein function. Therefore, we assessed whether HFD or TG altered ER membrane fluidity through DPH. We observed that ER membrane fluidity was reduced in the livers of HFD-fed mice and TG-treated HepG2 cells and that DPH polarization was eliminated by increasing temperature (Fig. 4a, b). Then, we determined the phospholipid composition of the ER membrane. Total PC (Fig. 4c), PE (Fig. 4d), PS (Fig. 4e), and PI (Fig. 4f) contents were significantly decreased in the livers of HFD-fed mice and recovered after silybin treatment to different degrees. Similar results were obtained for TG-treated HepG2 cells. PC and PE are most abundant phospholipids on the ER membrane, and a decrease in polyunsaturated phospholipids was observed in the model group, which was closely associated with the membrane fluidity (Fig. 4g, h) [28, 29]. Moreover, alterations in ER fatty acid and lipid composition result in the inhibition of sarco/endoplasmic reticulum calcium ATPase (SERCA) activity and ER stress. The increased ratio of PC/PE (Fig. 4i) suggested that Ca2+ was released through Serca2b under ER stress in both the livers of HFD-fed mice and TG-treated HepG2 cells [30].
Fig. 4. HFD altered the composition of the endoplasmic reticulum membrane through ER stress.

ER membrane fluidity in liver tissues and cells was measured by the inverse correlation of DPH polarization anisotropy, whereby decreased DPH polarization indicates increased membrane fluidity (a, b). Analysis of total phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), polyunsaturated phosphatidylcholine (PUFA PC), and polyunsaturated phosphatidylethanolamine (PUFA PE) in ER fractions from both mouse livers and HepG2 cells treated with TG, with increasing concentrations of silybin and TUDCA (c–h). Ratio of PC and PE in ER fractions from both mouse livers and HepG2 cells (i). Data were presented as the means ± SEM and were analyzed by two-tailed paired t test (n = 5). *P < 0.05; **P < 0.01; ***P < 0.001 vs. model and #P < 0.05; ##P < 0.01; ###P < 0.001 vs. control.
ER stress affected mitochondrial function and cyt c release
Since both the NADP+/NADPH and PC/PE ratios indicate that Ca2+ release and disruption of cellular Ca2+ homeostasis are associated with liver pathological conditions, we quantified the Ca2+ level in the ER and mitochondria in HepG2 cells using Fura-2 AM and Rhod-2 AM, fluorescent indicators of cytoplasmic and mitochondrial Ca2+, respectively. After TG irritation, Ca2+ was released from the ER to the cytoplasm and the mitochondria, while both silybin and TUDCA restrained this process (Fig. 5a, b).
Fig. 5. Effects on the communication between the endoplasmic reticulum and mitochondria induced by ER stress.

Ca2+ in the cytoplasm (Fura-2 AM fluorescence, 20×) and mitochondria (Rhod-2 AM fluorescence, 20×) in cells treated for 24 h with thapsigargin (TG) and silybin or TUDCA (a). Fluorescence intensity of Ca2+ in both cytoplasmic and mitochondria was measured (b). Data were presented as the means ± SEM (n = 8). Analysis of O2 consumption in cells treated as previously described (c). The O2 consumption rates (OCRs) were first measured for 4 × 104 cells of each cell line under basal conditions, after which oligomycin (1 μM), carbonyl cyanide-p-(trifluoromethoxy)-phenylhydrazone (FCCP; 1 μM), rotenone/antimycin A (0.5 μM) were sequentially added at the indicated times to determine different parameters of mitochondrial functions. Graphs present the basal OCR, ATP-linked OCR, proton leak OCR, maximal OCR, and nonmitochondrial OCR for different groups (d). Nonmitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. Basal OCR was determined as OCR before oligomycin minus OCR after rotenone/antimycin A. ATP-linked OCR was determined as OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as basal OCR minus ATP-linked OCR. Maximal OCR was determined as the OCR after FCCP minus nonmitochondrial OCR. Data were presented as the means ± SEM (n = 7). The activities of MRCC I and III were measured using kits. Data were presented as means ± SEM (n = 5) (e, f). Ratios of cyt c/cyt a of mitochondria from mice (n = 6) and cells (n = 5) treated as previously described were measured, and the data were presented as means ± SEM (g, h). The ratios of cyt c/cyt a of mitochondria in cells treated with TG, ALA and CsA were measured (i). Ca2+ levels in the cytoplasm (Fura-2 AM fluorescence, 20×) and mitochondria (Rhod-2 AM fluorescence, 20×) in cells treated for 24 h with digitonin (Dig) and BAPTA-AM (j). Fluorescence intensity of Ca2+ in both cytoplasmic and mitochondria was measured (k). Data are presented as means ± SEM (n = 8). The ratios of cyt c/cyt a of mitochondria in cells treated with digitonin, BAPTA-AM and ALA were measured (l). The activity of CYP3A in cells was measured using midazolam after treatment with Dig and BAPTA-AM (m). The data were presented as the means ± SEM (n = 5). Statistical analysis was performed by two-tailed paired t test. *P < 0.05; **P < 0.01; ***P < 0.001 vs. model, #P < 0.05; ##P < 0.01; ###P < 0.001, ####P < 0.0001 vs. control.
To evaluate the effect of ER stress on mitochondria, we measured the oxygen consumption rate (OCR). As shown in Fig. 5c, d, TG decreased the basal OCR while silybin and TUDCA reversed this decrease. The average basal OCR, maximal OCR, ATP-linked OCR, proton leak OCR, and nonmitochondrial OCR values in the model group also significantly decreased compared to that observed in the other groups. We further measured the activities of mitochondrial respiratory chain complex I (MRCC I) and mitochondrial respiratory chain complex III (MRCC III). As shown in Fig. 5e, f, the activities of MRCC I and III decreased significantly when HepG2 cells were treated with TG and recovered after silybin or TUDCA treatment.
Interestingly, we also observed cyt c release from mitochondria under ER stress (Fig. 5g, h), as evidenced by the decrease in the cyt c/cyt a ratio, which could be prevented by silybin or TUDCA treatment. The opening of the mitochondrial permeability transition pore (mPTP) allows for the release of many substances from mitochondria. The mPTP inhibitors α-linolenic acid (ALA) [31] and cyclosporin A (CsA) [32] could reduce cyt c release (Fig. 5i). In addition, we used digitonin (a detergent that makes the membrane permeable by replacing cholesterol) [33] and BAPTA-AM (a chelator of intracellular Ca2+) [34] to verify the key role of high cytoplasmic Ca2+ levels in cyt c release. As expected, digitonin caused increased levels of cytoplasmic and mitochondrial Ca2+, which could be prevented by BAPTA-AM (Fig. 5j, k). A significant decrease in cyt c release was observed as evidenced by the cyt c/cyt a ratio in mitochondria when calcium overload occurred, as well as a lower CYP3A activity levels after digitonin treatment. Furthermore, this situation was also reversed by BAPTA-AM or ALA (Fig. 5l, m).
Cyt c regulated P450s activity by interacting with cyt b5 under ER stress
We next investigated how mitochondrial dysfunction resulted in feedback on the activity of P450s located on the ER, a process in which the release of cyt c from mitochondria could have a key role. Reducing cyt c release by both ALA and CsA treatment reversed the suppression of CYP3A activity caused by TG (Fig. 6a). Then, we treated cells with ALA and CsA after a high level of calcium stimulation. As expected, ALA and CsA ameliorated the decreased release of cyt c caused by high level of Ca2+ (Fig. 6b), indicating that cyt c release may be caused by calcium overload. After being released into the cytoplasm, cyt c may bind to cyt b5 (Fig. 6c) to disrupt the transfer of electrons, since cyt b5 is an electron transporter participating in electron transfer between the inner and outer mitochondrial membranes and to P450s [35]. The increased interaction between cyt b5 and cyt c after TG and digitonin treatment was also evidenced by the colocalization of these two proteins (Fig. 6d, e).
Fig. 6. Effect of cytochrome c on the P450s activity.

Analysis of CYP3A activity in cells through HPLC–MS/MS after treatment with thapsigargin (TG), digitonin (Dig), ALA, and CsA (a, b). Interaction between cyt b5 and cyt c was determined by immunoprecipitation with an anti-cytochrome c antibody and immunoblotted with antibodies against cyt b5 and GAPDH (c). Representative confocal microscopy images (60×) of nuclei (Blue), cytochrome b5 (cyt b5) (Red) and cytochrome c (cyt c) (Green) in HepG2 cells treated with 1 μM thapsigargin (TG), 50 μM silybin and 100 μM TUDCA (d) as well as 1 μg/mL digitonin, 10 μM BAPTA-AM and 10 μM ALA (e). The data were presented as the means ± SEM (n = 5). Statistical analysis was performed by two-tailed paired t test. **P < 0.01 vs. model, #P < 0.05 ###P < 0.001 vs. control.
Discussion
Activity of metabolic enzymes is the gold standard in enzyme evaluation studies. The results of previous studies have elucidated the transcriptional regulation of cytokines in downregulating the expression of P450s during liver inflammation [17–19]. Our previous study revealed that silybin can lower ER stress and inhibit NLRP3 inflammasome assembly to reduce the release of IL-1β [16] indicating a connection between ER stress and the activity of enzymes located on the ER.
ER stress is involved in many types of liver diseases, including NAFLD, NASH and hepatocellular carcinoma [36]. TG is consistently used as an ER stress inducer which can promote the depletion of Ca2+ in the ER lumen [37]. We assessed the activities of five representative P450 enzymes, and unsurprisingly, their activity was suppressed in HFD-fed mice and in TG-treated HepG2 cells. Therefore, we subsequently evaluated how ER stress affected P450 enzymes activity.
We evaluated the alterations in cytochrome P450 components, since the mechanism of P450 catalysis is a complex cascade of steps involving the interaction with other electron transfer proteins, such as CPR and cytochrome b5 [38]. Generally, NADPH and CPR provide the first electron to P450s, and cyt b5 can substitute NADPH-dependent CPR in this enzymatic reaction by donating the first and second electron in some situations [39]. CPR is the redox partner for most human cytochrome P450 enzymes and binds NADPH, which can reduce the enzyme by transferring electrons to both the FAD and FMN domains within the protein [6]. In addition, cyt b5 is involved in many microsomal oxidation reactions and monohydroxylation processes by interacting with a variety of cytochrome P450 enzymes (CYPs or P450s) as well as cytochrome c (cyt c) in the outer mitochondrial membrane. Our results verified that the interaction between cyt b5 and cyt c under ER stress was involved in the regulation of P450s activity.
Growing evidence has shown that the ratio of NADP+/NADPH is important in physiology and disease. Cells have an endogenously preferred NADP+/NADPH ratio to balance cell growth and biosynthesis of target products [40]. Elevated NADP+ level leads to high levels of NAADP, which is generated from NADP+, inducing Ca2+ release [25–27]. In addition, the heterogeneity of the ER membrane affects P450s function by either concentrating or segregating proteins within membrane regions [8] as well as the activity of calcium channels [41]. Anionic phospholipids have been shown to exert a stimulatory effect on the catalytic activity of CYP1A2 and CYP3A during the course of optimization of reconstituted enzyme activity [42, 43], and some P450s exist in regions where the lipids are more ordered, while others exist in the disordered regions [44].
Moreover, ER membrane phospholipids promote the proper orientation of P450s with their redox partners and generally facilitate substrate binding. Our data suggested that total PC, PE, PI and PS levels were downregulated in the livers of HFD-fed mice and TG-treated HepG2 cells under ER stress. PC is reported to be the major phospholipid constituent of the ER and is also the major component that facilitates P450 interactions with their redox partners [45]. Furthermore, anionic phospholipids, such as PS and PI, can increase the P450s insertion ratio into membranes, which can increase P450s activity [42]. PS is also associated with the expansion of the ER membrane and ER functions [46]. Thus, the decrease of PC, PS and PI in the ER membrane was one of the reasons to reduce enzyme function. The PC/PE ratios in the ER membrane were also upregulated both in vivo and in vitro, which have been associated with increased ER stress due to impaired Serca2b function [47]. SERCA participates in the reuptake of Ca2+ so that the increased level of PC/PE suppresses the uptake of cytoplasmic Ca2+ back to the ER, which can influence the subsequent development.
Further, we studied the interplay between ER and mitochondria. Based on the relationship between ER stress and Ca2+ release, we assessed what occurred in mitochondria after ER stress. When Ca2+ is released from the ER to the cytoplasm, part of the Ca2+ is taken up by mitochondria [48]. If too much Ca2+ is released from the ER, it will cause Ca2+ overload in the mitochondria, contributing to mitochondrial dysfunction [49]. Our results showed that ER stress led to increased Ca2+ levels in both the cytoplasm and mitochondria, which could be suppressed by silybin and TUDCA. As expected, we observed a decrease in the oxygen consumption rate after ER stress, which was alleviated by silybin and TUDCA. Mitochondrial respiratory chain complexes I and III are important elements for electrons to enter the mitochondrial electron transfer chain and for mitochondrial oxidative phosphorylation. Furthermore, the deficient activities of respiratory chain complexes often alter mitochondrial membrane potentials, which is a key indicator of cellular viability [50].
The interaction between the ER and mitochondria has been extensively studied. By checking the variation of mitochondria, we found that TG induced mitochondrial fragmentation (Supplementary Fig. S1), which is consistent with literatures [51, 52]. Silybin and TUDCA could partially restore the morphology of mitochondria and these morphological changes are closely related to ER stress. The interaction may be involved in the regulation of P450s activity in cellular networks. Cyt c, which accepts electrons from cyt b5 [53, 54], was released from mitochondria in HFD-fed mice, while silybin and TUDCA prevented this release. Furthermore, this release of cyt c was also decreased by treatment with the mPTP inhibitors ALA and CsA, indicating that the opening of mPTP participates in cyt c release. Subsequently, we verified high levels of Ca2+ in the cytoplasm caused the release of cyt c through digitonin and BAPTA-AM treatment and observed that it further influenced CYP3A activity. This effect was probably based on the interaction between cyt b5 and cyt c during ER stress, resulting in the disruption of electron transport from cyt b5 to P450s.
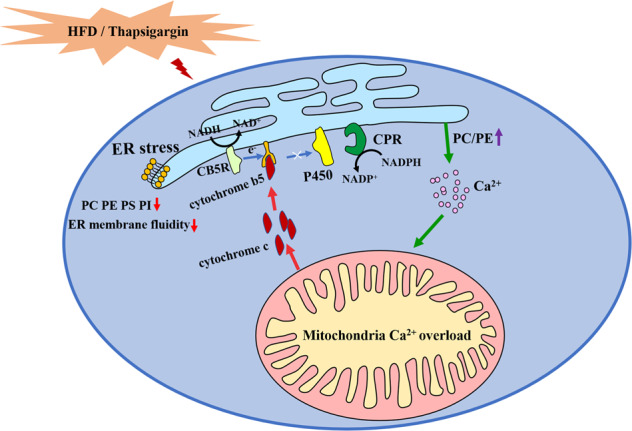
In summary, hepatocyte ER stress induced by HFD resulted in the inhibition of P450s activity due to alterations in membrane phospholipids, ER membrane fluidity and components involved in P450 catalytic system. The interaction between cyt c released from mitochondria and cyt b5 under ER stress indicated that communication between organelles, especially the ER and mitochondria, has an important role in affecting the metabolism of cells and may be a potential target for disease treatment. Silybin restored the P450s activity in NAFLD mice liver based on its regulation of this crosstalk between mitochondria and ER (Fig. 7).
Fig. 7. Proposed mechanism for the suppression P450s activity caused by ER stress.
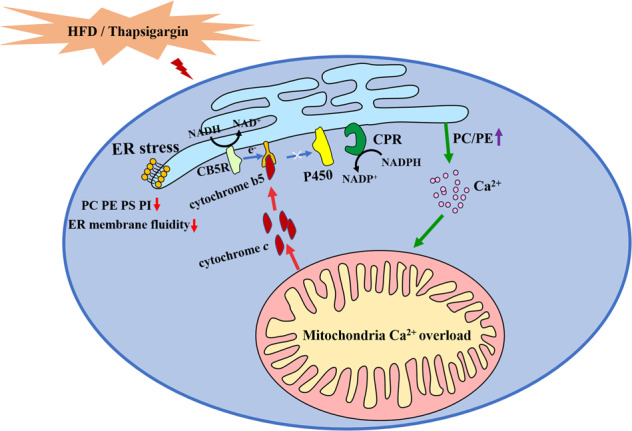
ER stress regulated hepatic ER membrane fluidity and phospholipid composition and cyt c released from mitochondria to interact with cyt b5, which are important reasons in P450s regulation.
Supplementary information
Acknowledgements
This project was supported by the National Natural Science Foundation of China (No. 81872932, 81673679), the Six Talent Peaks Project in Jiangsu Province (SWYY-061), the Sanming Project of Medicine in Shenzhen (SZSM201801060), the Project of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMZZ202001).
Author contributions
YX and JW designed this study. JW, YGL, XLY, RW, and RZ conducted the experiments. JW, YGL, and XLY contributed new reagents or analytic tools. JW, YGL, XLY and RZ performed data analysis. YX, JW, JYA, and GJW wrote or contributed to the writing of the paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Jing Wu, Yun-ge Lou.
Contributor Information
Guang-ji Wang, Email: guangjiwang@hotmail.com.
Yuan Xie, Email: yuanxie@cpu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-022-00924-4.
References
- 1.Pavlides M, Cobbold J. Non-alcoholic fatty liver disease. Medicine. 2019;47:728–33. doi: 10.1016/j.mpmed.2019.08.007. [DOI] [Google Scholar]
- 2.Li H, Canet MJ, Clarke JD, Billheimer D, Xanthakos SA, Lavine JE, et al. Pediatric cytochrome P450 activity alterations in nonalcoholic steatohepatitis. Drug Metab Dispos. 2017;45:1317–25. doi: 10.1124/dmd.117.077644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cobbina E, Akhlaghi F. Non-alcoholic fatty liver disease (NAFLD)—pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab Rev. 2017;49:197–211. doi: 10.1080/03602532.2017.1293683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stiborova M, Indra R, Moserova M, Frei E, Schmeiser HH, Kopka K, et al. NADH: cytochrome b5 reductase and cytochrome b5 can act as sole electron donors to human cytochrome P450 1A1-mediated oxidation and DNA adduct formation by benzo[a]pyrene. Chem Res Toxicol. 2016;29:1325–34. doi: 10.1021/acs.chemrestox.6b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedison TM, Scrutton NS. Tripping the light fantastic in membrane redox biology: linking dynamic structures to function in ER electron transfer chains. FEBS J. 2019;286:2004–17. doi: 10.1111/febs.14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnaba C, Martinez MJ, Taylor E, Barden AO, Brozik JA. Single-protein tracking reveals that NADPH mediates the insertion of cytochrome P450 reductase into a biomimetic of the endoplasmic reticulum. J Am Chem Soc. 2017;139:5420–30. doi: 10.1021/jacs.7b00663. [DOI] [PubMed] [Google Scholar]
- 7.Bhatt MR, Khatri Y, Rodgers RJ, Martin LL. Role of cytochrome b5 in the modulation of the enzymatic activities of cytochrome P450 17alpha-hydroxylase/17,20-lyase (P450 17A1) J Steroid Biochem Mol Biol. 2017;170:2–18. doi: 10.1016/j.jsbmb.2016.02.033. [DOI] [PubMed] [Google Scholar]
- 8.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc. 2013;135:8542–51. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aghazadeh S, Amini R, Yazdanparast R, Ghaffari SH. Anti-apoptotic and anti-inflammatory effects of Silybum marianum in treatment of experimental steatohepatitis. Exp Toxicol Pathol. 2011;63:569–74. doi: 10.1016/j.etp.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Pradhan SC, Girish C. Hepatoprotective herbal drug, silymarin from experimental pharmacology to clinical medicine. Indian J Med Res. 2006;124:491–504. [PubMed] [Google Scholar]
- 11.Feng YY, Yan JY, Xia X, Liang JQ, Li F, Xie TF, et al. Effect and mechanism of total flavonoids of Lichi Semen on CCl4-induced liver fibrosis in rats, and prediction of Q-marker. Zhongguo Zhong Yao Za Zhi. 2020;45:5722–31. doi: 10.19540/j.cnki.cjcmm.20200720.401. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Yan T, Xie Y, Zhao M, Che Y, Zhang J, et al. Mechanism-based inhibitory and peroxisome proliferator-activated receptor alpha-dependent modulating effects of silybin on principal hepatic drug-metabolizing enzymes. Drug Metab Dispos. 2015;43:444–54. doi: 10.1124/dmd.114.061622. [DOI] [PubMed] [Google Scholar]
- 13.Gufford BT, Chen G, Lazarus P, Graf TN, Oberlies NH, Paine MF. Identification of diet-derived constituents as potent inhibitors of intestinal glucuronidation. Drug Metab Dispos. 2014;42:1675–83. doi: 10.1124/dmd.114.059451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu JW, Lin LC, Tsai TH. Drug-drug interactions of silymarin on the perspective of pharmacokinetics. J Ethnopharmacol. 2009;121:185–93. doi: 10.1016/j.jep.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 15.Zhang R, Xu D, Zhang Y, Wang R, Yang N, Lou Y, et al. Silybin restored CYP3A expression through the sirtuin 2/nuclear factor kappa-B pathway in mouse nonalcoholic fatty liver disease. Drug Metab Dispos. 2021;49:770–9. doi: 10.1124/dmd.121.000438. [DOI] [PubMed] [Google Scholar]
- 16.Zhang B, Xu D, She L, Wang Z, Yang N, Sun R, et al. Silybin inhibits NLRP3 inflammasome assembly through the NAD(+)/SIRT2 pathway in mice with nonalcoholic fatty liver disease. FASEB J. 2018;32:757–67. doi: 10.1096/fj.201700602R. [DOI] [PubMed] [Google Scholar]
- 17.Febvre-James M, Bruyere A, Le Vee M, Fardel O. The JAK1/2 inhibitor ruxolitinib reverses interleukin-6-mediated suppression of drug-detoxifying proteins in cultured human hepatocytes. Drug Metab Dispos. 2018;46:131–40. doi: 10.1124/dmd.117.078048. [DOI] [PubMed] [Google Scholar]
- 18.Morgan ET. Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev. 1997;29:1129–88. doi: 10.3109/03602539709002246. [DOI] [PubMed] [Google Scholar]
- 19.Renton KW. Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr Drug Metab. 2004;5:235–43. doi: 10.2174/1389200043335559. [DOI] [PubMed] [Google Scholar]
- 20.Shi J, Wang X, Lyu L, Jiang H, Zhu HJ. Comparison of protein expression between human livers and the hepatic cell lines HepG2, Hep3B, and Huh7 using SWATH and MRM-HR proteomics: focusing on drug-metabolizing enzymes. Drug Metab Pharmacokinet. 2018;33:133–40. doi: 10.1016/j.dmpk.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Cai Q, Zhou F, Liu J, Jin X, Ni P, et al. Impaired pentose phosphate pathway in the development of 3D MCF-7 cells mediated intracellular redox disturbance and multi-cellular resistance without drug induction. Redox Biol. 2018;15:253–65. doi: 10.1016/j.redox.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibbons E, Pickett KR, Streeter MC, Warcup AO, Nelson J, Judd AM, et al. Molecular details of membrane fluidity changes during apoptosis and relationship to phospholipase A(2) activity. Biochim Biophys Acta. 2013;1828:887–95. doi: 10.1016/j.bbamem.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tonshin AA, Saprunova VB, Solodovnikova IM, Bakeeva LE, Yaguzhinsky LS. Functional activity and ultrastructure of mitochondria isolated from myocardial apoptotic tissue. Biochemistry. 2003;68:875–81. doi: 10.1023/a:1025798931614. [DOI] [PubMed] [Google Scholar]
- 24.Jancova P, Anzenbacherova E, Papouskova B, Lemr K, Luzna P, Veinlichova A, et al. Silybin is metabolized by cytochrome P450 2C8 in vitro. Drug Metab Dispos. 2007;35:2035–9. doi: 10.1124/dmd.107.016410. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell KJ, Lai FA, Rutter GA. Ryanodine receptor type I and nicotinic acid adenine dinucleotide phosphate receptors mediate Ca2+ release from insulin-containing vesicles in living pancreatic beta-cells (MIN6) J Biol Chem. 2003;278:11057–64. doi: 10.1074/jbc.M210257200. [DOI] [PubMed] [Google Scholar]
- 26.Lee HC. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu Rev Pharmacol Toxicol. 2001;41:317–45. doi: 10.1146/annurev.pharmtox.41.1.317. [DOI] [PubMed] [Google Scholar]
- 27.Patel S. NAADP-induced Ca2+ release—a new signalling pathway. Biol Cell. 2004;96:19–28. doi: 10.1016/j.biolcel.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Castuma CE, Brenner RR. Effect of fatty acid deficiency on microsomal membrane fluidity and cooperativity of the UDP-glucuronyltransferase. Biochim Biophys Acta. 1983;729:9–16. doi: 10.1016/0005-2736(83)90449-2. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Sheng W, Sun GY, Lee JC. Effects of fatty acid unsaturation numbers on membrane fluidity and alpha-secretase-dependent amyloid precursor protein processing. Neurochem Int. 2011;58:321–9. doi: 10.1016/j.neuint.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–31. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang L, Xu M, Zhang Y, Liu X, Zhang H. The role of mitochondrial permeability transition pore in chronic heart failure rats and potential protective effects of n-3 polyunsaturated fatty acid. J Tongji Univ (Med Sci) 2016;37:18. [Google Scholar]
- 32.Yun B, Lee H, Ghosh M, Cravatt BF, Hsu KL, Bonventre JV, et al. Serine hydrolase inhibitors block necrotic cell death by preventing calcium overload of the mitochondria and permeability transition pore formation. J Biol Chem. 2014;289:1491–504. doi: 10.1074/jbc.M113.497651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilabert-Oriol R, Mergel K, Thakur M, von Mallinckrodt B, Melzig MF, Fuchs H, et al. Real-time analysis of membrane permeabilizing effects of oleanane saponins. Bioorg Med Chem. 2013;21:2387–95. doi: 10.1016/j.bmc.2013.01.061. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Sanderson MJ. Oscillations in ciliary beat frequency and intracellular calcium concentration in rabbit tracheal epithelial cells induced by ATP. J Physiol. 2003;546:733–49. doi: 10.1113/jphysiol.2002.028704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schenkman JB, Jansson I. The many roles of cytochrome b5. Pharmacol Ther. 2003;97:139–52. doi: 10.1016/S0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 36.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes. 2009;58:422–32. doi: 10.2337/db07-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cederbaum AI. Molecular mechanisms of the microsomal mixed function oxidases and biological and pathological implications. Redox Biol. 2015;4:60–73. doi: 10.1016/j.redox.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stiborova M, Indra R, Frei E, Kopeckova K, Schmeiser HH, Eckschlager T, et al. Cytochrome b5 plays a dual role in the reaction cycle of cytochrome P450 3A4 during oxidation of the anticancer drug ellipticine. Monatsh Chem. 2017;148:1983–91. doi: 10.1007/s00706-017-1986-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sung C, Jung E, Choi KY, Bae JH, Kim M, Kim J, et al. The production of omega-hydroxy palmitic acid using fatty acid metabolism and cofactor optimization in Escherichia coli. Appl Microbiol Biotechnol. 2015;99:6667–76. doi: 10.1007/s00253-015-6630-1. [DOI] [PubMed] [Google Scholar]
- 41.Spector AA, Yorek MA. Membrane lipid composition and cellular function. J Lipid Res. 1985;26:1015–35. doi: 10.1016/S0022-2275(20)34276-0. [DOI] [PubMed] [Google Scholar]
- 42.Ahn T, Guengerich FP, Yun CH. Membrane insertion of cytochrome P450 1A2 promoted by anionic phospholipids. Biochemistry. 1998;37:12860–6. doi: 10.1021/bi980804f. [DOI] [PubMed] [Google Scholar]
- 43.Kim KH, Ahn T, Yun CH. Membrane properties induced by anionic phospholipids and phosphatidylethanolamine are critical for the membrane binding and catalytic activity of human cytochrome P450 3A4. Biochemistry. 2003;42:15377–87. doi: 10.1021/bi035280k. [DOI] [PubMed] [Google Scholar]
- 44.Park JW, Reed JR, Brignac-Huber LM, Backes WL. Cytochrome P450 system proteins reside in different regions of the endoplasmic reticulum. Biochem J. 2014;464:241–9. doi: 10.1042/BJ20140787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strobel HW, Lu AY, Heidema J, Coon MJ. Phosphatidylcholine requirement in the enzymatic reduction of hemoprotein P-450 and in fatty acid, hydrocarbon, and drug hydroxylation. J Biol Chem. 1970;245:4851–4. doi: 10.1016/S0021-9258(18)62872-8. [DOI] [PubMed] [Google Scholar]
- 46.Michell RH. Do inositol supplements enhance phosphatidylinositol supply and thus support endoplasmic reticulum function? Br J Nutr. 2018;120:301–16. doi: 10.1017/S0007114518000946. [DOI] [PubMed] [Google Scholar]
- 47.Kirkby B, Roman N, Kobe B, Kellie S, Forwood JK. Functional and structural properties of mammalian acyl-coenzyme A thioesterases. Prog Lipid Res. 2010;49:366–77. doi: 10.1016/j.plipres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Szabadkai G, Simoni AM, Rizzuto R. Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J Biol Chem. 2003;278:15153–61. doi: 10.1074/jbc.M300180200. [DOI] [PubMed] [Google Scholar]
- 49.Boyman L, Karbowski M, Lederer WJ. Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol Med. 2020;26:21–39. doi: 10.1016/j.molmed.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szczepanowska J, Malinska D, Wieckowski MR, Duszynski J. Effect of mtDNA point mutations on cellular bioenergetics. Biochim Biophys Acta. 2012;1817:1740–6. doi: 10.1016/j.bbabio.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 51.Yamashita R, Fujii S, Ushioda R, Nagata K. Ca2+ imbalance caused by ERdj5 deletion affects mitochondrial fragmentation. Sci Rep. 2021;11:20772. doi: 10.1038/s41598-021-99980-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol. 2007;212:498–508. doi: 10.1002/jcp.21051. [DOI] [PubMed] [Google Scholar]
- 53.Huang R, Zhang M, Rwere F, Waskell L, Ramamoorthy A. Kinetic and structural characterization of the interaction between the FMN binding domain of cytochrome P450 reductase and cytochrome c. J Biol Chem. 2015;290:4843–55. doi: 10.1074/jbc.M114.582700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gentry KA, Prade E, Barnaba C, Zhang M, Mahajan M, Im SC, et al. Kinetic and structural characterization of the effects of membrane on the complex of cytochrome b 5 and cytochrome c. Sci Rep. 2017;7:7793. doi: 10.1038/s41598-017-08130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.