

Summary
The mammalian immune system employs various pattern recognition receptors (PRRs) to recognize invaders and host damage and transmits this information to downstream immunometabolic signaling outcomes. Laccase domain-containing 1 (LACC1) protein is an enzyme highly expressed in inflammatory macrophages and serves a central regulatory role in multiple inflammatory diseases, such as in inflammatory bowel diseases (IBDs), arthritis, and clearance of microbial infection1-4. However, the biochemical roles required for LACC1 functions remain largely undefined. Here, we elucidated a shared biochemical function of LACC1 in mice and humans, converting L-citrulline (L-Cit) to L-ornithine (L-Orn) and isocyanic acid (HNCO) and serving as a bridge between proinflammatory nitric oxide synthase (NOS2) and polyamine immunometabolism. We validated the genetic and mechanistic connections among NOS2, LACC1, and ornithine decarboxylase 1 (ODC1) in mouse models and bone marrow derived macrophages (BMDMs) infected by Salmonella enterica Typhimurium. Strikingly, LACC1 phenotypes required upstream NOS2 and downstream ODC1, and Lacc1−/− chemical complementation with its product L-Orn could significantly restore wildtype activities. Our findings illuminate a previously unidentified pathway in inflammatory macrophages, explain why its deficiency may contribute to human inflammatory diseases, and suggest that L-Orn could serve as a nutraceutical to ameliorate LACC1-associated immunological dysfunctions such as arthritis or IBD.
The innate immune system is the first line of defense against infection and other injuries. It is activated by PRRs that broadly distinguish between foreign and endogenous host molecules, initiating inflammatory responses and recruiting macrophages as well as other immune cells to the site of injury4. In addition to a variety of essential proteins, a few small molecule families such as reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive carbonyl species (RCS) are indispensable for immune function. ROS can both promote and prevent cell death, cancer, aging and inflammation in a context-dependent manner5,6. RNS have been recognized to play a crucial role in the physiologic regulation of many cell types, such as smooth muscle cells, cardiomyocytes, platelets, and nervous and juxtaglomerular cells7. As the major source of RNS in macrophages, inducible NOS2 was originally described as an enzyme that is expressed in classically activated macrophages, generates nitric oxide (NO) from the amino acid L-arginine (L-Arg)8, and thereby contributes to the control of replication or killing of intracellular microbial pathogens9. The other product of NOS2, L-Cit, can be used as a substrate to regenerate L-Arg via argininosuccinate synthetase 1 (ASS1) and argininosuccinate lyase (ASL)10. However, the fate of NOS2 products remain only partially defined.
The laccase domain-containing 1 protein (LACC1) serves a central regulatory role in a variety of inflammatory diseases and is expressed predominantly in myeloid cells1,2. LACC1, also known as C13orf31 or FAMIN, is robustly activated by lipopolysaccharide (LPS) or poly-I:C in mouse bone marrow derived macrophages (BMDMs)2,3. Consistent with LACC1 activity in inflammatory macrophages, its expression is induced by macrophage colony-stimulating factor (M-CSF) in an AKT-mTOR-dependent manner during human monocyte-macrophage differentiation11. Frame-shift mutations and single-nucleotide polymorphisms K38E, I254 and C284R in LACC1 identified in genome-wide association studies (GWAS) are correlated with early-onset Crohn’s disease (CD), ankylosing spondylitis, systemic juvenile idiopathic arthritis (JIA), and a high-risk state for leprosy (Mycobacterium leprae infections)12-17. Mouse models deficient in LACC1 showed exacerbated arthritis, psoriasis, T cell transfer colitis (Rag2−/− background), and intestinal bacterial infection (Citrobacter rodentium and Salmonella enterica serovar Typhimurium)2,3. Mouse LACC1 (mLACC1) deficient animals exhibited elevated proinflammatory cytokines and worsened intestinal bacterial clearance, supporting both anti-inflammatory and antibacterial functions2,3. Yet how such an enzyme could mediate such broad effects was unclear. Recently, human LACC1 (hLACC1) was shown to interact with a family of autophagy-associated proteins, including autophagy inducers RACK1 and AMPK, and Lacc1 deficiency reduced autophagic flux in primary human macrophages, defining a novel form of genetically inherited JIA associated with impaired autophagy in macrophages11. This is consistent with enhanced expression of autophagy proteins in wildtype (WT) relative to Lacc1−/− mouse BMDMs and partial restoration of LACC1-mediated bacterial clearance in the presence of the autophagy inducer rapamycin3. While LACC1 was reported to participate in several purine nucleotide metabolic reactions in mouse BMDMs18, these biochemical activities do not explain the autophagy phenotype. Overall, these findings support LACC1 as an important regulator of macrophage immunometabolic function19, but the major catalytic roles required for LACC1 function in inflammatory macrophages remained unknown.
LACC1 is an isocyanic acid synthase
To establish the biochemical function of LACC1, we first conducted in vitro biochemical assays using isolated recombinant mLACC1. Mouse and human LACC1 amino acid sequences predict a multi-copper polyphenol oxidoreductase laccase-like domain (pfam02578). To first establish the metal composition of the enzyme, we analyzed isolated mLACC1 versus vector control using inductively coupled plasma mass spectrometry (ICP-MS) (Extended Data Fig. 1a-b). However, specific enrichment of Zn (0.94 metal:enzyme ratio) was observed, whereas there was no enrichment of Cu or other transition metals, suggesting that LACC1 is rather a Zn-dependent metalloenzyme. To establish a physiologically-relevant substrate mixture for LACC1, we generated a Lacc1−/− mouse model and extracted the metabolites from its isolated inflammatory BMDM cells (Fig. 1a). We reasoned that the LACC1 substrates would accumulate in these cells. The metabolite extract was incubated with mLACC1 (1 h; 37°C) in phosphate buffered saline supplemented with Zn2+ and then analyzed by ultra-performance liquid chromatography quadrupole time-of-flight mass spectrometry (UPLC-QTOF-MS). The metabolomics data were processed and presented as a volcano plot showing fold change (FC) versus false discovery rate (FDR) values. Intriguingly, L-Orn was the most upregulated metabolite among significant hits (FDR <0.05) under these conditions (positive ion mode, Fig. 1b; negative ion mode, Extended Data Fig. 1d)18. L-Orn is a product of L-Arg metabolism, which is the major metabolism classification between classical and alternatively activated macrophages. In inflammatory macrophages, L-Arg is metabolized to NO and L-Cit via NOS2. By contrast, in IL-4 stimulated anti-inflammatory macrophages, L-Arg is metabolized to L-Orn and urea via arginase20,21.
Fig. 1.

LACC1 is an endogenous isocyanic acid synthase, converting L-Cit to L-Orn and HNCO. a, Pipeline used for in vitro enzymatic evaluation of mLACC1. b, Volcano plot showing the fold change (FC) (substrate with mLACC1 versus substrate with heat inactivated mLACC1) and false discovery rate (FDR) values in positive ion mode. c, Abundance of L-Orn in enzymatic assays validating the substrate of mLACC1. d, Summary of kinetic parameters of mLACC1, hLACC1, hLACC1 K38E, hLACC1 I254V, and hLACC1 C284R. e, Abundance of HNCO in enzymatic assays, as assessed by the 2-aminobenzoate-HNCO carbamoylation assay. f, Direct detection of isocyanic acid (HNCO) through 13C-NMR spectroscopy. g, Major biochemical transformation mediated by LACC1. The mean and SEM (error bars) are derived from three biological replicates (n = 3). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ***P<0.001; nd, not detectable.
To validate the enzymatic transformation observed, we performed assays using L-Arg, L-argininosuccinate (L-ArgSuc), or L-Cit and found that L-Cit was the only viable substrate (Fig. 1c). The stereoconfiguration of product L-Orn was confirmed via Marfey’s analysis (Extended Data Fig. 1i). To assess the catalytic efficiencies and roles of putative hLACC1 disease polymorphisms, we also conducted enzyme kinetic analysis of isolated WT mLACC1 and hLACC1 and the polymorphic human enzyme variants: K38E, I254V and C284R. Michaelis-Menten kinetics were established using varying L-Cit substrate concentrations and measuring L-Orn product concentrations over time by liquid chromatography triple quadrupole mass spectrometry (LC-QQQ-MS) (pH 7.4, Extended Data Fig. 2a-f; pH rate profiling, Extended Data Fig. 1e). mLACC1 and hLACC1 showed comparable binding (Km) and catalytic turnover (kcat) values (Fig. 1d), supporting a conserved biochemical activity between mice and humans. Disease associated human polymorphisms I254V and C284R exhibited a significant reduction in turnover (kcat) and overall catalytic efficiency (kcat/Km), providing a catalytic mechanistic contribution to their associated immunopathologies (see below). However, disease correlative mutant K38E showed improved catalytic efficiency and binding (Km), which would accelerate the timing of the response in inflammatory macrophages (i.e., LACC1 response would occur sooner with accumulating NOS2-derived L-Cit in this variant versus wildtype). Finally, consistent with our metal analysis, presence of metal- (ethylenediaminetetraacetic acid, EDTA) or Zn2+-specific (N,N,N′,N′-tetrakis (2-pyridylmethyl) ethane-1,2-diamine, TPEN) chelators in the reaction buffer significantly inhibited LACC1 activity (Extended Data Fig. 1f).
While we established that LACC1 cleaves L-Cit to L-Orn, the second fragment of the reaction could not be identified under the conditions of the studies. To directly characterize the unidentified fragment, we analyzed the mLACC1 reaction with 6-13C1-labelled L-Cit in D2O by nuclear magnetic resonance (NMR) spectroscopy (Fig. 1f). Strikingly, we established that LACC1 is a novel HNCO synthase, converting L-Cit to L-Orn and the RCS-like molecule HNCO (Fig. 1g), which was confirmed using a standard of its conjugate base, sodium cyanate (−OCN) (Fig. 1f). Only three carbon peaks were observed for this fragment, which correspond to different protonation states of the product. To detect HNCO, a carbamoylation reagent associated with uraemic syndrome22, by MS, we conducted 2-aminobenzoate-HNCO carbamoylation assays and measured the product by LC-QQQ-MS23. End-point HNCO production under saturating substrate conditions was consistent with our L-Orn kinetic measurements, including a significant reduction of activity in the human I254V and C284R mutants (Fig. 1e). Strikingly, these data suggest that LACC1 has a previously unrecognized role in L-Arg metabolism in inflammatory macrophages, bridging NOS2-derived L-Cit formation with L-Orn, a precursor in polyamine signaling, and HNCO, an RCS-like cytotoxin.
LACC1 protects from bacterial infection
To probe the molecular mechanism of LACC1 in inflammatory BMDMs and mice, we first cultured BMDMs from WT and Lacc1−/− mice and stimulated them with LPS and interferon-γ (IFNγ) for 16 hours. Key proinflammatory cytokines such as TNFα, IL6 and IL12b were upregulated in Lacc1−/− BMDMs (Fig. 2a, Extended Data Fig. 3i-k), further supporting the anti-inflammatory function of LACC1. To test the function of LACC1 products on cytokine regulation, we conducted chemical complementation studies using exogenous L-Orn and HNCO in Lacc1−/− BMDMs. Importantly, supplementation with L-Orn, but not HNCO, could significantly complement Lacc1 cytokine suppression (Fig. 2a, Extended Data Fig. 3i-k). To validate whether L-Orn could complement Lacc1 antibacterial deficiency in vivo, we performed Salmonella Typhimurium infection in WT and Lacc1−/− mice with or without L-Orn in the drinking water. In the absence of L-Orn, the body weight loss of Lacc1−/− mice was significantly higher than WT littermates (Fig. 2b). This phenotype was accompanied with worsened survival and severe bacterial burden in the feces, caecum, spleen and liver of Lacc1−/− mice compared to controls (Fig. 2c-g). These data are consistent with LACC1’s antibacterial role3,24. Strikingly, 1% L-Orn in the drinking water significantly complemented Lacc1 deficiency in vivo (Fig. 2b-g). Indeed, this oral administration of L-Orn significantly improved the effects of weight loss, survival, and bacterial burden. In the absence of S. Typhimurium infection, 1% L-Orn in the drinking water did not perturb the body weight of WT or Lacc1−/− mice, supporting its immunological role during infection (Extended Data Fig. 4). To further validate whether the function of LACC1 depends on its enzymatic activity, we constructed Lacc1C284R/C284R mice and subjected them to the same S. Typhimurium infection experiment. Lacc1C284R/C284R mice showed an intermediate phenotype with exacerbated bacterial infection and more severe bacterial burden (Extended Data Fig. 3a-e), which was consistent with the Lacc1−/− mouse studies and the C284R mutant biochemical studies. Moreover, inflammatory BMDMs from the Lacc1C284R/C284R mice showed an intermediate elevation of TNFα, IL6, and IL12b upon LPS and IFNγ stimulation (Extended Data Fig. 3f-h). These data suggest that the evaluated anti-inflammatory and antibacterial functions of LACC1 are primarily mediated by L-Orn and that the human disease associated mutation reduces catalytic efficiency and confers a deficient antibacterial response in vivo.
Fig. 2.

LACC1 inhibits proinflammatory cytokine signaling and protects from S. Typhimurium infection. a, ELISA measurements of TNFα, IL6 and IL12b secreted by BMDMs in the presence or absence of immunostimulant (LPS + IFNγ), exogenously supplied LACC1 product L-Orn (1, 5 mM), and/or the conjugate base of LACC1 product HNCO, NaOCN (100 μM, 1 mM). b, Body weight of each mouse was monitored daily after S. Typhimurium infection with or without 1% L-Orn administration, and the body weight loss was depicted as the percentage (%) compared to the initial body weight (n = 10). c, Mouse survival curves (n = 10). d, Colony-Forming Units (CFUs) in fecal pellets were measured at day 4 after infection. e, f, g, CFUs in caecum, spleen and liver, respectively, were measured at day 6 after infection. The mean and SEM (error bars) are derived from three (Fig. 2a) or ten (Fig. 2b-g) biological replicates (n = 3 or n = 10). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ns, not significant.
LACC1 contributes to polyamine synthesis
In macrophages, L-Orn is decarboxylated by ornithine decarboxylase 1 (ODC1) to produce putrescine for polyamine synthesis25. Odc1 expression is upregulated during bacterial infections26,27. During polyamine synthesis, L-methionine (L-Met) is converted into S-adenosyl-L-methionine (AdoMet or SAM), which is then decarboxylated by AdoMet decarboxylase (AdoMetDC) to produce decarboxylated AdoMet (dcAdoMet or dcSAM). dcSAM is then used as an aminopropyl group donor by spermidine synthase (SRM) to convert putrescine into spermidine and spermine synthase (SMS) to convert spermidine into spermine25 (Extended Data Fig. 7a). To examine the impact of the enzymatic function of LACC1 on polyamine synthesis in BMDMs, we first cultured BMDMs from WT and Lacc1−/− mice supplemented with 1,2,3,4,5-13C5 labeled L-Cit during differentiation. Indeed, we observed significantly reduced 13C-L-Orn, 13C-putrescine (13C-Put), 13C-spermidine (13C-Spd), and 13C-spermine (13C-Spm) in inflammatory BMDMs from Lacc1−/− mice than those from WT mice (Fig. 3a). Additionally, there was a consistent inverse relationship with dcSAM production, but its enhancement in Lacc1−/− BMDMs was not statistically significant (Extended Data Fig. 5a-b). Because L-Cit can also be recycled to L-Arg via ASS1 and ASL10 and ARG2 can directly convert L-Arg to L-Orn in IL-4 activated macrophages, we performed additional 13C-L-Arg, 13C-L-Cit, and 13C-L-ArgSuc isotope feeding experiments in the presence or absence of a NOS2 inhibitor, 1400W, or an ARG2 inhibitor, CB-1158 (Extended Data Fig. 5c-e). While our data support that NOS2 is the major source of L-Cit, which can indeed be recycled to L-Arg, in inflammatory macrophages as anticipated, inactivation of LACC1 significantly reduces 13C-L-Arg-mediated polyamine labeling regardless of inhibitor and consistent with our hypothesis. No significant LACC1 effects were observed in the 13C-L-ArgSuc feeding studies. These data support LACC1 as a contributor to L-Cit-mediated polyamine synthesis in inflammatory macrophages (Fig. 3b).
Fig. 3.

LACC1 bridges arginine metabolism with polyamine synthesis in inflammatory macrophages. a, LC-QQQ-MS intensity of 13C-Orn, 13C-Put, 13C-Spd and 13C-Spm in inflammatory BMDMs from WT and Lacc1−/− mice. b, Key metabolism pathways in inflammatory BMDMs, highlighting LACC1’s novel and central role in L-Arg and polyamine metabolism. The mean and SEM (error bars) are derived from three biological replicates (n = 3). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01.
Because L-Cit is derived from NOS2 in inflammatory macrophages8, we proposed that LACC1 function would be dependent upon NOS2 as the latter provides the substrate for the former (Fig. 4a). To test this hypothesis, we bred Nos2−/− mice with Lacc1−/− mice to generate Nos2-Lacc1 double knockout mice and measured their performance in vivo relative to controls in the S. Typhimurium infection model. We observed that NOS2 protected mice from S. Typhimurium infection based on body weight loss, survival, and bacterial burden in feces and invaded organs as anticipated (Fig. 4b-g), consistent with the diminished killing of S. Typhimurium by macrophages from Nos2−/− mice28. However, the antibacterial function of LACC1 was abolished in Nos2 deficient mice relative to the double knockout mice (Fig. 4b-g). Consistent with our biochemical model and BMDM results, these mouse model data suggest that the observed LACC1 function is dependent on NOS2.
Fig. 4.

NOS2 is indispensable for the antibacterial function of LACC1. a, Illustration of the NOS2-LACC1-ODC1 signaling axis. b, Body weight of each mouse was monitored daily after S. Typhimurium infection (n = 10, P = 0.0414). c, Mouse survival curves (n = 10). d, CFUs in fecal pellets were measured at day 4 after infection. e, f, g, CFUs in caecum, spleen and liver, respectively, were measured at day 6 after infection. h, i, j, ELISA measurements of TNFα, IL6 and IL12b, respectively, secreted by BMDMs stimulated by LPS and IFNγ with or without 1 mM DFMO treatment. k, CFUs of intracellular S. Typhimurium in inflammatory BMDMs. l, LDH assay in S. Typhimurium infected inflammatory BMDMs. The mean and SEM (error bars) are derived from three (Fig. 4h-l) or ten (Fig. 4b-g) biological replicates (n = 3 or n = 10). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ns, not significant.
To test whether the L-Orn-polyamine axis is necessary for the anti-inflammatory and antibacterial function of LACC1, we blocked the enzymatic activity of ODC1 using the irreversible ODC1 inhibitor α-difluoromethylornithine (DFMO) in BMDMs (Fig. 4a). In contrast to cells treated with solvent vehicle, the secreted proinflammatory cytokines TNFα, IL6, and IL12b in DFMO treated BMDMs remained unchanged between WT and Lacc1−/− BMDMs following LPS and IFNγ stimulation, supporting that LACC1 may suppress proinflammatory cytokine production via the downstream polyamine pathway (Fig. 4h-j). We also infected DFMO treated inflammatory BMDMs with S. Typhimurium. Although the normalized intracellular bacterial CFUs/macrophage remained unchanged with DFMO treatment (Fig. 4k), the lactate dehydrogenase (LDH) activity assay showed that irreversible inhibition of ODC1 phenocopied exacerbated S. Typhimurium induced cell death from Lacc1−/− inflammatory BMDMs (Fig. 4l). This phenotype was not complemented using exogenous putrescine treatment, possibly due to dose related toxicity limiting the concentration of exogenous polyamine that could be employed (Extended Data Fig. 6e). These data suggest that intracellular LACC1-mediated polyamine synthesis protects macrophages from S. Typhimurium induced cell death, thereby enhancing antibacterial function. Thus, we propose a model in which LACC1 contributes to anti-inflammatory and antibacterial functions through the L-Orn-polyamine immunometabolism signaling axis (Extended Data Fig. 7a). The RCS-like cytotoxin and carbamoylation reagent HNCO29 likely contributes as an antimicrobial and signaling molecule, but our evaluation of activities with exogenous −OCN supplementation could be masked by the overall effects of cellular ROS, RNS, and RCS in these inflammatory macrophage phenotypes.
Taken together, our study could explain how loss of Lacc1 function in inflammatory macrophages leads to both augmented inflammatory signaling and cell death, which is consistent with both the immunopathologies and antibacterial roles associated with LACC1 in mouse models and in human diseases. This proposed inflammation equilibrium of LACC1 is analogous to other immunometabolism pathways in macrophages, such as the proinflammatory succinate- and downstream anti-inflammatory and antimicrobial itaconate signaling axis that is the result of a rewired tricarboxylic acid (TCA) cycle in inflammatory macrophages30-33. While other enzymatic activities of LACC1 may contribute,18,34 our results support a molecular mechanism in which LACC1 is a central immunometabolic regulator in L-Arg macrophage metabolism, and they may reveal the missing mechanistic connection between proinflammatory NOS2 signaling and subsequent anti-inflammatory polyamine-mediated cytokine suppression35,36, putative antioxidant activity37, and autophagy stimulation (Extended Data Fig. 7b)38. Indeed, polyamines such as spermidine have been reported to: 1) have a role in decreasing oxidative damage, although the mechanism is unclear39; 2) stimulate autophagy in mammalian cells38; and 3) alleviate experimental autoimmune encephalomyelitis through inducing inhibitory macrophages40,41. In anti-inflammatory macrophages, polyamine biosynthesis modulates mitochondrial metabolism through eIF5A hypusination, maintaining the TCA cycle and the electron transport chain (ETC) integrity42. In addition to supporting polyamine metabolism, LACC1 serves as an unprecedented endogenous mammalian HNCO synthase, an activity observed for antimicrobial myeloperoxidase only with specific exogenous environmental metabolites from high-fat diet and chronic exposure to cyanide, mimicking exposure to pollution and smoking43. While we cannot rule out other activities of LACC1-mediated L-Orn and HNCO production in cells, our molecular studies support an alternative immunometabolic proposal for LACC1 in autophagy regulation through its contribution to polyamine synthesis in addition to its reported interaction with autophagy-inducing proteins, RACK1 and AMPK11. These findings suggest that L-Orn could serve as a novel nutraceutical to ameliorate LACC1-associated immunological and autoimmune dysfunctions.
Methods
Expression and purification of LACC1.
mLACC1 and hLACC1 cDNA sequences were synthesized by GenScript, codon-optimized for prokaryotic expression, and cloned into a pMAL-C5x (New England BioLabs, N8108) vector18. Plasmids for human LACC1 K38E, human LACC1 I254V and human LACC1 C284R were generated using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs, E0554S). E. coli BL21(DE3) cells were transformed with the expression plasmids and grown aerobically at 37°C and 250 revolutions per minute (rpm) in ampicillin-supplemented (100 μg/mL) Luria Bertani (LB) medium. Expression was induced at an OD600 of 0.5-0.6 with 0.3 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 30°C. Cells were harvested by centrifugation (6000 × g, 4 °C, 20 min) and cell pellets were washed twice with PBS. Cells were then resuspended in 5 mL of column buffer on ice containing 20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA, pH 7.5, and cOmplete Mini EDTA-free protease inhibitor cocktail (1 tablet per 50 mL buffer, Roche, 11873580001). The cells were lysed by sonication on ice and cleared by centrifugation at 35,000 × g for 30 minutes. The protein-containing supernatant was loaded onto a pre-equilibrated amylose resin column (New England BioLabs, E8021S). The column was washed with 20 column volumes of column buffer. The protein was eluted with 10 column volumes of column buffer supplemented with 10 mM maltose. The eluted fractions were pooled and concentrated using centrifugal filters (Millipore, UFC503096) for use in further assays.
ICP-MS.
mLACC1 was expressed and purified as mentioned above. 1 mg purified protein was digested with 10 mL 5% HNO3 in a thermo digestion system, followed by the measurement of Mg, Ca, Fe, Mn, Co, Ni, Cu and Zn contents in triplicate by inductively coupled plasma mass spectrometry (ICP-MS, Perkin Elmer ICP-MS Elan DRC-e). In the averaged Zn65 quantification assay, 1 mL 10 μM purified mLACC1 was mixed with 10 mL 5% HNO3 and analyzed with Zn65 standards by ICP-MS.
In vitro enzyme screening.
To prepare the BMDM cell pellet extract, Lacc1−/− BMDMs were cultured as mentioned below and stimulated with 20 ng/mL LPS (Sigma, L4391) and 50 ng/mL IFNγ (BioLegend, 575302) for 16 hours. Cells were washed with ice-cold PBS twice and extracted with ice-cold MeOH. The extraction mixture was centrifuged and the supernatant was dried using N2 gas flow. 1 mg Lacc1−/− BMDM cell pellet extract was incubated with 10 μM mLACC1 in 100 μL PBS buffer supplemented with 1 μM ZnSO4 in triplicate for 1 h at 37°C and quenched by 200 μL ice-cold acetonitrile (AcCN). The reaction mixture was centrifuged at 20,000 × g for 10 min and the supernatant was analyzed using an Agilent iFunnel 6550 quadrupole time of flight (Q-TOF) MS instrument fitted with an electrospray ionization (ESI) source coupled to an Agilent 1290 Infinity HPLC system with an Xbridge BEH Amide XP HILIC 2.5 μm, 2.1 mm x 100 mm column (Waters, 186006091). The column was maintained at 25°C during analysis. The mobile phase A was 20 mM ammonium acetate/0.1% formic acid pH 3.5. The mobile phase B was 100% acetonitrile. The flow rate was 0.22 mL/min. The gradient elution was as follows: 0 min: 85% B; 0.5 min: 85% B; 9 min: 35% B; 11 min: 2% B; 12 min: 85% B; 25 min: 85% B. The data were acquired in positive and negative ion mode (Extended Data Fig. 1d) with a full scan range of 50-1700 m/z, respectively. The metabolomics data were processed with Mass Profiler Professional Software 14.0 (Agilent) and presented as a volcano plot showing fold change (FC) versus false discovery rate (FDR) values. Fig. 1a was created with BioRender.
Measurement of HNCO.
For the direct measurement of HNCO through NMR, 6-13C1-labelled L-Cit (1 mg) was incubated with 10 μM mLACC1 in D2O (200 μL) for 1 hour and the reaction was quenched in liquid N2. The reaction mixture was then subjected to 13C NMR to detect cyanate and its conjugate acid HNCO with reference to its central 13C chemical shift value of 128.64 ppm. The negative control was prepared as described above without mLACC1, and the chemical standard used was 1 mM NaOCN in D2O, which showed identical 13C chemical shift values as the enzyme product. Some minor spontaneous carboxylate carbamoylation of substrate L-Cit and product L-Orn was observed over extended incubation times (not shown). The measurement was performed using an Agilent 800 MHz NMR instrument equipped with a 13C-sensitivity enhanced salt-tolerant cold probe. For the indirect measurement of HNCO, an HNCO carbamoylation assay was used23. The enzymatic assay was performed as mentioned above and quenched with 200 μL MeOH. The reaction mixture was incubated with 20 mM acetic acid and 50 mM 2-aminobenzoic acid at 42°C for 30 min. Then, the mixture was centrifuged and the supernatant was analyzed by UPLC-QTOF-MS (Phenomenex Kinetex C18 (100 Å) 5 μm (250 × 4.6 mm2) column; flow rate, 0.7 mL/min; mobile phase composition, 10-100% acetonitrile in water containing 0.1% formic acid for 30 min).
Measurement of L-Orn.
The enzymatic assay was performed as mentioned above. In some conditions as noted, KH2PO4 or Na2HPO4 was used for pH adjustment while 10 μM EDTA or 10 μM TPEN was added for metal chelation. The reaction mixture was quenched with 200 μL MeOH. Then, the mixture was centrifuged and coupled with 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide, FDAA (Thermo, 48895) per the manufacturer’s protocol. The samples were analyzed by UPLC-QTOF-MS (Phenomenex Kinetex C18 (100 Å) 5 μm (250 × 4.6 mm2) column; flow rate, 0.7 mL/min; mobile phase composition, 10-100% acetonitrile in water containing 0.1% formic acid for 30 min).
Measurement of enzymatic parameters.
The enzymatic assay was performed in triplicate as mentioned above at different concentrations of L-Cit (5 μM, 10 μM, 25 μM, 50 μM, 100 μM, 200 μM and 500 μM) over time (60 s, 120 s, 300 s, and 600 s). The velocity was calculated, and the Michaelis-Menten curves were plotted for the measurement of Km and kcat using Prism 9 (GraphPad).
Mouse studies.
WT C57BL/6, Lacc1−/−, Lacc1C284R/C284R and Nos2−/− mice were used in this study. Nos2−/− mice (002609-B6.129P2-Nos2tm1Lau/J, Jackson Laboratory) were a gift from Dr. Victor Laubach44. Lacc1−/− mice were generated with CRISPR-Cas9 technology into C57/B6N embryos as previously described45. To generate Lacc1−/− mice, two sgRNAs (AAACTGCCATGAGACCTTACTGG, GTTAAGGCCATCGCGGACATGGG) were used collectively to target exon 3 and exon 7, respectively. To generate Lacc1C284R/C284R mice, an sgRNA (GAAGACGATGGGTATACAGTCGG) and an oligo donor (CCTACCGGAGTGAGCAACCCCACATGCCTTTTTCACAGGATCTGCGAAGACGATGGGTATACgGTCGGCGCCAAGAGCAGTGATTGTGACTCCTCTCTGAT TTGTGACGATCCCATCGTAAGA) were used collectively for homologous recombination. Experimental littermates were generated from heterozygote × heterozygote breeding. Sample size (n = 10) was chosen in line with pervious experimental experience and consistent with the broader literature46. 8-10-week-old male and female mice were used in equal quantities unless otherwise specified. All experiments were performed using co-housed mouse littermate controls. Isosexual male or female littermates were co-housed at 21-24°C and 40-60% humidity. A 12/12 h light/dark cycle was used. All mouse studies were performed in compliance with Yale Institutional Animal Care and Use Committee protocols. No formal blinding or randomization was conducted; however, control and treated groups were chosen arbitrarily for each experiment. Mouse weights and CFUs were measured in a blinded manner.
Cell culture studies.
BMDMs were generated from progenitor cells isolated from femurs and tibias of mice and maintained in DMEM medium (Gibco, 11965118) with 10% FBS (Sigma, F8192-500Ml), 1% penicillin/streptomycin (Gibco, 15070063) and 50 ng/mL M-CSF (R&D, 416-ML-010) for 6 days. Cells were reseeded and stimulated with 20 ng/mL LPS (Sigma, L4391) and 50 ng/mL IFNγ (BioLegend, 575302) in triplicate for 16 hours. In some experiments as indicated, 1 mM DFMO (Cayman, 16889) was supplemented during differentiation to inhibit ODC1. After a 16-hour stimulation period, culture supernatants were collected for TNFα, IL6 and IL12b ELISA assays (R&D, DY410-05, DY406-05, DY499-05) according to the manufacturer’s protocols.
S. Typhimurium infection in vivo.
As previously described47, before infection, 8-10-week-old mice (n = 10) were restricted from food and water for 4 h followed by gavage of 20 mg streptomycin. After 20 h, mice were fasted again for 4 h and infected with streptomycin resistant S. Typhimurium (SL1344 strain, provided by J. Galan). S. Typhimurium was maintained as a glycerol stock at −80°C. Before infection, bacteria were propagated overnight (37°C, 250 rpm) in LB supplemented with 100 μg/ml streptomycin. The bacterial culture was subcultured for 4 h the next day in antibiotic-free LB supplemented with 0.3 M NaCl to return it to log phase growth and increase virulence48. Using spectrophotometry, bacterial CFUs were calculated with an infection dose of 1 × 103 CFUs per mouse. To calculate faecal CFUs, faecal pellets were resuspended in PBS at 50 mg/mL and vortexed for 20 min. Bacteria containing supernatants were clarified by centrifugation at 50 × g for 10 min. Serial dilutions were conducted, and bacteria were plated in triplicate on LB streptomycin (100 μg/mL) plates. For caecal, liver and spleen CFU enumeration, organs were isolated, weighed, and added to 2 ml of PBS. Tissue was dissociated with GentleMacs C Tubes (Miltenyi Biotech) per the manufacturer’s instructions. CFUs were calculated using similar methodology as above.
S. Typhimurium infection in BMDMs.
After a 16-hour stimulation period as mentioned above, the BMDM culture medium was replaced with antibiotic-free medium. BMDMs (1 × 106) were infected in triplicate with 1 × 107 CFUs of S. Typhimurium (SL1344 strain, provided by J. Galan, MOI = 10) and centrifugated at 800 × g at 37 °C for 10 min and returned to the incubator for an additional 20 min. Medium was then removed and replaced with medium containing 100 μg/mL gentamycin to kill extracellular bacteria for 1 h (i.e., the gentamycin protection assay47). Medium was replaced with fresh medium including 25 μg/mL gentamycin. An hour later, supernatants were sampled for LDH activity with the CyQUANT™ LDH Cytotoxicity Assay (Thermo, C20300) according to the manufacturer’s protocol. After an additional 3.5-hour culture period in the incubator, cells were washed twice with PBS and lysed in a 1% Triton and 0.1% SDS buffer for 5 min under gentle agitation. Cell lysates were plated on streptomycin (100 μg/mL) containing LB plates and incubated overnight at 37°C, and then CFUs were enumerated.
13C-isotope labelling studies.
BMDMs were generated as mentioned above, reseeded at a density of 1×106 cells/mL in phenol red-free DMEM medium (Gibco, 21063029) with 10% FBS (Sigma, F8192-500Ml), 1% penicillin/streptomycin (Gibco, 15070063), and 1 mM 1,2,3,4,5,6-13C6 labeled L-Arg (Cambridge Isotope Laboratories, CLM-2265-H-0.05), 2 mM 1,2,3,4,5-13C5 labeled L-Cit (Cambridge Isotope Laboratories, CLM-8653-PK), or 1 μM 1,2,3,4,5,6-13C6, 15N4 labeled L-ArgSuc (Cambridge Isotope Laboratories, CNLM-9007-CA-0.1MG) and stimulated in triplicate with 20 ng/mL LPS (Sigma, L4391) and 50 ng/mL IFNγ (BioLegend, 575302) for 16 hours. The cells were washed twice with 10 mL ice-cold PBS, scraped in 1 mL ice-cold PBS, transferred to 1.5 mL tubes and pelleted (1 min, 6,000 × g, 4°C). The cells were washed once with 500 μL ice-cold PBS. Cellular metabolites were extracted with 50 μL of ice-cold extraction solvent (40:40:20 vol/vol/vol acetonitrile:methanol:water, 0.5% formic acid). After a 5-min incubation on ice, the acid was neutralized using NH4HCO3. After centrifugation (15 min, 20,000 × g, 4°C), the clarified supernatant was transferred to an LC-MS vial and analyzed by an Agilent iFunnel 6550 QTOF-MS instrument fitted with an ESI source or an Agilent 6490 ESI-QQQ-MS/MS instrument coupled to an Agilent 1290 Infinity HPLC system with an Xbridge BEH Amide XP HILIC 2.5 μm, 2.1 mm x 100 mm column (Waters, 186006091). The column was maintained at 25°C during the analysis. The mobile phase A was 20 mM ammonium acetate/0.1% formic acid pH 3.5. The mobile phase B was 100% acetonitrile. The flow rate was 0.4 mL/min. The gradient elution was as follows: 0 min: 95% B; 0.5 min: 95% B; 3 min: 70% B; 6 min: 40% B; 6.5 min: 0% B; 9.5 min: 0% B; 10 min: 95% B; 15 min: 95% B. MassHunter Qualitative Analysis B.07.00 (Agilent) was used to perform peak picking, peak alignment, and peak intensity integration.
Extended Data
Extended Data Fig. 1.

In vitro biochemical analysis of isolated LACC1 variants. a, ICP-MS assays showed the enrichment of Zn64 and Zn66 isotopes in recombinant mLACC1 relative to vector control. b, A quantitative ICP-MS assay for Zn showed the average Zn:mLACC1 ratio as 0.94, suggesting a 1:1 ratio in the isolated enzyme. c-d, Volcano plots from UPLC-QTOF-MS experiments showing the fold change (FC) (substrate with mLACC1 versus substrate with heat inactivated mLACC1) and false discovery rate (FDR) values collected in positive ion mode (c) and negative ion mode for the specific detection of ribose-1-phosphate (d). e, Rates of L-Orn production in enzymatic assays at different pH conditions. f, Rates of L-Orn production in enzymatic assays with the supplementation of EDTA or TPEN in the reaction buffer. g, The standard curve used for quantifying L-Orn in enzymatic assays. h, The standard curve used for quantifying 2-aminobenzoate-HNCO carbamoylation products in enzymatic assays. i, The LC-MS trace of 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide (FDAA, Marfey’s reagent)-coupled L-Orn in enzymatic assays, supporting the stereoconfiguration. j, Expanded view 13C-NMR spectroscopic data of LACC1 reaction using 6-13C1-L-Cit as substrate, showing L-Orn and HNCO as major products. The mean and SEM (error bars) are derived from three biological replicates (n = 3). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ***P<0.001; nd, not detectable.
Extended Data Fig. 2.

Kinetic analysis of isolated enzymes. Velocity plots of isolated enzyme reactions at different concentrations of L-Cit (5 μM, 10 μM, 25 μM, 50 μM, 100 μM, 200 μM and 500 μM) in enzyme kinetic analysis of WT mLACC1 (a) and hLACC1 (b) and the polymorphic human enzyme variants: K38E (c), I254V (d) and C284R (e). f, Michaelis-Menten curves of mLACC1, hLACC1, hLACC1 K38E, hLACC1 I254V, and hLACC1 C284R. The mean and SEM (error bars) are derived from three biological replicates (n = 3). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ns, not significant.
Extended Data Fig. 3.

Lacc1C284R/C284R shows an intermediate defect in antibacterial protection against S. Typhimurium infection. a, Body weight of each mouse was monitored daily after S. Typhimurium infection (n = 10). b, CFUs in fecal pellets were measured at day 4 after infection. c, d, e, CFUs in caecum, spleen and liver, respectively, were measured at day 6 after infection. f-h, ELISA measurements of TNFα, IL6 and IL12b secreted by BMDMs in the presence or absence (NT, non-treatment) of immunostimulant (LPS + IFNγ). i-k, ELISA measurements of TNFα, IL6 and IL12b secreted by BMDMs in the presence or absence (NT) of immunostimulant (LPS + IFNγ), exogenously supplied LACC1 product L-Orn (100 μM, 1 mM, 5 mM, 10 mM), and/or the conjugate base of LACC1 product HNCO, NaOCN (10 μM, 100 μM, 500 μM, 1 mM). The mean and SEM (error bars) are derived from three (f-k) or ten (a-e) biological replicates (n = 3 or n = 10). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ns, not significant.
Extended Data Fig. 4.
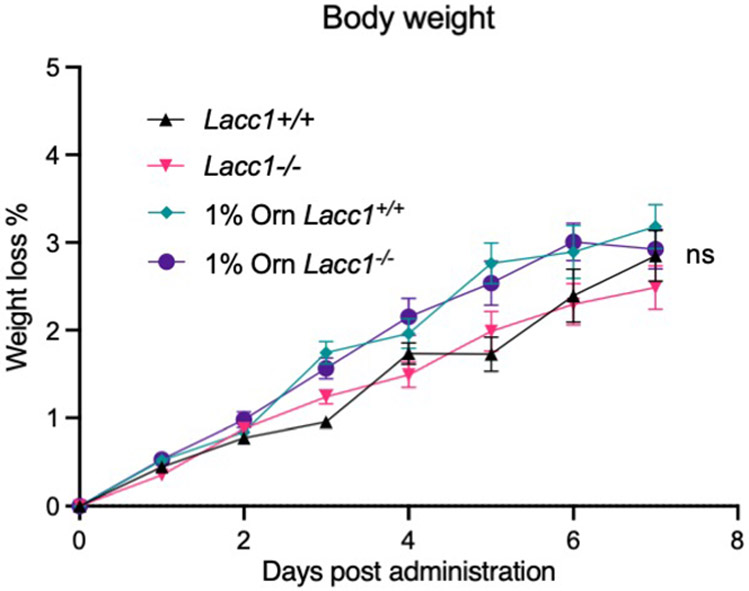
Body weight of each mouse in the absence of S. Typhimurium infection was monitored daily with or without 1% L-Orn administration in drinking water, showing no significant change in body weight. The mean and SEM (error bars) are derived from ten biological replicates (n = 10). Statistical significance (two-tailed t-test) compared to control (Ctrl): ns, not significant.
Extended Data Fig. 5.

LACC1 bridges L-Arg metabolism with polyamine synthesis in inflammatory macrophages. a, b, MS intensity of SAM and dcSAM, respectively, in inflammatory BMDMs from WT and Lacc1−/− mice. Lacc1−/− BMDMs show an elevated but non statistically significant level of dcSAM. c-e, UPLC-QTOF-MS intensity of 13C-labeled metabolites in L-Arg metabolism and polyamine synthesis with 13C-Arg feeding (c), 13C-Cit feeding (d) and 13C-ArgSuc feeding (e), respectively. These data complement LC-QQQ-MS experimental measurements shown in Fig. 3a. The mean and SEM (error bars) are derived from three biological replicates (n = 3). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ***P<0.001; ns, not significant.
Extended Data Fig. 6.

Irreversible inhibition of ODC1 phenocopied exacerbated S. Typhimurium induced cell death from Lacc1−/− inflammatory BMDMs. a, b, c, ELISA measurements of TNFα, IL6, and IL12b, respectively, secreted by BMDMs stimulated by LPS and IFNγ with or without 1 mM DFMO and/or 500 μM putrescine (Put) treatment. d, CFUs of intracellular S. Typhimurium in inflammatory BMDMs. e, LDH assay in S. Typhimurium infected inflammatory BMDMs. The mean and SEM (error bars) are derived from three biological replicates (n = 3). Statistical significance (two-tailed t-test) compared to control (Ctrl): *P<0.05; **P<0.01; ns, not significant.
Extended Data Fig. 7.

Summary of the enzymatic functions of LACC1 in inflammatory macrophages. a, LACC1’s newly described role in bridging L-Arg and polyamine metabolism in inflammatory macrophages. The carbons are highlighted with consistency to our 1,2,3,4,5-13C5-L-Cit (red) and 6-13C1-L-Cit (blue) feeding studies in inflammatory BMDMs. Carbons in spermidine and spermine highlighted in green derive from dcSAM. b, The involvement of LACC1 (a.k.a., FAMIN) in the purine nucleotide cycle as a purine nucleoside enzyme18. ADSL, adenylosuccinate lyase; ADSS, adenylosuccinate synthase; AMP, adenosine monophosphate; AMPD, AMP deaminase; GDP, guanosine diphosphate; GMP, guanosine monophosphate; GTP, guanosine triphosphate; IMP, inosine monophosphate; XMP, xanthosine monophosphate.
Acknowledgements
We thank J. Alderman, B. Cadugan, C. Lieber, C. Hughes, L. Evangelisti, E. Hughes-Picard, F. Zhang, P. Ranney, W. Philbrick and P. Maher-Rivera for help in mouse gene-editing projects and in overall administration, as well as J. Galan for the gift of S. Typhimurium SL1344 strain. We thank T. Wu at Yale West Campus Analytical Core for assistance with mass spectrometry analysis and J. Karosas at the Yale Analytical and Stable Isotope Center for assistance with ICP-MS analysis. This work was supported by the Howard Hughes Medical Institute (to R.A.F.), the Burroughs Wellcome Fund (no. 1016720 to J.M.C.), and Yale University. Z.W. was supported by the China Scholarship Council.
Footnotes
Competing interests
R.A.F. is a recipient of a grant from AbbVie, Inc, is a founder of Rheos Biomedicines and a consultant to GSK and Zai labs. The remaining authors declare no competing interests.
Data availability
Supplementary information and source data are provided within this letter. Additional data that support the findings of this study are available from the corresponding author upon reasonable request.
Code was not used in this study.
References
- 1.Heng TS, Painter MW & Consortium, I. G. P. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094, doi: 10.1038/ni1008-1091 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Skon-Hegg C et al. LACC1 Regulates TNF and IL-17 in Mouse Models of Arthritis and Inflammation. J Immunol 202, 183–193, doi: 10.4049/jimmunol.1800636 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang JW et al. Myeloid Cell Expression of LACC1 Is Required for Bacterial Clearance and Control of Intestinal Inflammation. Gastroenterology 159, 1051–1067, doi: 10.1053/j.gastro.2020.07.024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mogensen TH Pathogen recognition and inflammatory signaling in innate immune defenses. Clinical microbiology reviews 22, 240 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schieber M & Chandel NS ROS function in redox signaling and oxidative stress. Current biology : CB 24, R453–R462, doi: 10.1016/j.cub.2014.03.034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mittler R ROS are good. Trends in plant science 22, 11–19 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Martínez MC & Andriantsitohaina R Reactive nitrogen species: molecular mechanisms and potential significance in health and disease. Antioxid Redox Signal 11, 669–702, doi: 10.1089/ars.2007.1993 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Marletta MA, Yoon PS, Iyengar R, Leaf CD & Wishnok JS Macrophage oxidation of L-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry 27, 8706–8711, doi: 10.1021/bi00424a003 (1988). [DOI] [PubMed] [Google Scholar]
- 9.Nathan C & Shiloh MU Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proceedings of the National Academy of Sciences 97, 8841, doi: 10.1073/pnas.97.16.8841 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qualls JE et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell host & microbe 12, 313–323 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omarjee O et al. LACC1 deficiency links juvenile arthritis with autophagy and metabolism in macrophages. J Exp Med 218, doi: 10.1084/jem.20201006 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang C et al. Genetic Risk for Inflammatory Bowel Disease Is a Determinant of Crohn's Disease Development in Chronic Granulomatous Disease. Inflamm Bowel Dis 22, 2794–2801, doi: 10.1097/mib.0000000000000966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Assadi G et al. LACC1 polymorphisms in inflammatory bowel disease and juvenile idiopathic arthritis. Genes and immunity 17, 261–264, doi: 10.1038/gene.2016.17 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Assadi G et al. Functional Analyses of the Crohn's Disease Risk Gene LACC1. PloS one 11, e0168276, doi: 10.1371/journal.pone.0168276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kallinich T et al. Juvenile arthritis caused by a novel FAMIN (LACC1) mutation in two children with systemic and extended oligoarticular course. Pediatric rheumatology online journal 14, 63, doi: 10.1186/s12969-016-0124-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wakil SM et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol 67, 288–295, doi: 10.1002/art.38877 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Grant AV et al. Crohn's disease susceptibility genes are associated with leprosy in the Vietnamese population. J Infect Dis 206, 1763–1767, doi: 10.1093/infdis/jis588 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Cader MZ et al. FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell 180, 815, doi: 10.1016/j.cell.2020.02.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szymanski AM & Ombrello MJ Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: NOD2, PLCG2 and LACC1. Int Immunol 30, 205–213, doi: 10.1093/intimm/dxy021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearce EL & Pearce EJ Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delanghe S, Delanghe JR, Speeckaert R, Van Biesen W & Speeckaert MM Mechanisms and consequences of carbamoylation. Nature Reviews Nephrology 13, 580–593, doi: 10.1038/nrneph.2017.103 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Lundquist P, Backman-Gullers B, Kågedal B, Nilsson L & Rosling H Fluorometric determination of cyanate in plasma by conversion to 2,4(1H,3H)-quinazolinedione and separation by high-performance liquid chromatography. Anal Biochem 211, 23–27, doi: 10.1006/abio.1993.1226 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Lahiri A, Hedl M, Yan J & Abraham C Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nature communications 8, 15614, doi: 10.1038/ncomms15614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minois N, Carmona-Gutierrez D & Madeo F Polyamines in aging and disease. Aging (Albany NY) 3, 716–732, doi: 10.18632/aging.100361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardbower DM et al. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to Helicobacter pylori infection. Amino Acids 48, 2375–2388, doi: 10.1007/s00726-016-2231-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng Y et al. Helicobacter pylori-induced macrophage apoptosis requires activation of ornithine decarboxylase by c-Myc. J Biol Chem 280, 22492–22496, doi: 10.1074/jbc.C500122200 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Shiloh MU et al. Phenotype of Mice and Macrophages Deficient in Both Phagocyte Oxidase and Inducible Nitric Oxide Synthase. Immunity 10, 29–38, doi: 10.1016/S1074-7613(00)80004-7 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Roberts JM et al. Isocyanic acid in the atmosphere and its possible link to smoke-related health effects. Proceedings of the National Academy of Sciences 108, 8966, doi: 10.1073/pnas.1103352108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordes T & Metallo CMM Itaconate Alters Succinate and Coenzyme A Metabolism via Inhibition of Mitochondrial Complex II and Methylmalonyl-CoA Mutase. Metabolites 11, 117 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy MP & O'Neill LAJ Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 174, 780–784, doi: 10.1016/j.cell.2018.07.030 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T & Castegna A The metabolic signature of macrophage responses. Frontiers in immunology 10, 1462 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M et al. Itaconate is an effector of a Rab GTPase cell-autonomous host defense pathway against Salmonella. Science 369, 450–455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saveljeva S et al. A purine metabolic checkpoint that prevents autoimmunity and autoinflammation. Cell metabolism 34, 106–124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hardbower DM et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc Natl Acad Sci U S A 114, E751–e760, doi: 10.1073/pnas.1614958114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura A et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nature Communications 12, 2105, doi: 10.1038/s41467-021-22212-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaboriau F, Vaultier M, Moulinoux J-P & Delcros J-G Antioxidative properties of natural polyamines and dimethylsilane analogues. Redox Report 10, 9–18, doi: 10.1179/135100005X21561 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Ghosh I, Sankhe R, Mudgal J, Arora D & Nampoothiri M Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 83, 102083, doi: 10.1016/j.npep.2020.102083 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Jeong J-W et al. Spermidine Protects against Oxidative Stress in Inflammation Models Using Macrophages and Zebrafish. Biomolecules & therapeutics 26, 146–156, doi: 10.4062/biomolther.2016.272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisenberg T et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11, 1305–1314, doi: 10.1038/ncb1975 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Yang Q et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ 23, 1850–1861, doi: 10.1038/cdd.2016.71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puleston DJ et al. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab 30, 352–363.e358, doi: 10.1016/j.cmet.2019.05.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delporte C et al. Myeloperoxidase-catalyzed oxidation of cyanide to cyanate: A potential carbamylation route involved in the formation of atherosclerotic plaques? Journal of Biological Chemistry 293, 6374–6386, doi: 10.1074/jbc.M117.801076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laubach VE, Shesely EG, Smithies O & Sherman PA Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A 92, 10688–10692, doi: 10.1073/pnas.92.23.10688 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nowarski R et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 163, 1444–1456, doi: 10.1016/j.cell.2015.10.072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson R et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 564, 434–438, doi: 10.1038/s41586-018-0794-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen LM, Kaniga K & Galán JE Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol 21, 1101–1115, doi: 10.1046/j.1365-2958.1996.471410.x (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supplementary information and source data are provided within this letter. Additional data that support the findings of this study are available from the corresponding author upon reasonable request.
Code was not used in this study.