

Abstract
Type 2 immune responses are typically associated with protection against helminth infections and also with harmful inflammation in response to allergens. Recent advances have revealed that type 2 immunity also contributes to sterile inflammation, cancer, and microbial infections. However, the early events that initiate type 2 immune responses remain poorly defined. New insights reveal major contributions from danger-associated molecular patterns (DAMPs) in the initiation of type 2 immune responses. In this review, we examine the molecules released by the host and pathogens and the role they play in mediating the initiation of mammalian innate type 2 immune responses under a variety of conditions.
Hallmarks of Type 2 Immune Responses
The mammalian type 2 immune response is dependent on extrinsic and intrinsic signals sensed by the host. However, the stimuli and associated mechanisms influencing the course of the response remain unclear, particularly regarding the initiation of the innate type 2 immune response. This response typically includes upregulation of interleukin-4 (IL-4), IL-5, IL-9, and IL-13, collectively referred to as type 2 cytokines [1,2]. Cells mediating mammalian type 2 innate immunity include macrophages, group 2 innate lymphoid cells (ILC2s), neutrophils, mast cells, eosinophils, and basophils. The innate immune response provides an immune environment required for subsequent activation of adaptive immunity. In the type 2 immune response, adaptive immunity is mediated by CD4+ T helper (Th)2 cells producing large quantities of type 2 cytokines and B cells which secrete IgE and IgG antibodies, and also various cytokines [1,2]. Both innate and adaptive immune cells express shared and lineage-specific activation signatures, largely dependent on IL-4 receptor signaling [3]. The specific tissue in which the response occurs, including associated non-immune cells, such as epithelial cells, and the associated tissue matrix can also contribute substantially to this response [3]. Parasitic worms, or helminths, comprise the major group of pathogens consistently triggering polarized type 2 immune responses, but at least elements of this type 2 immune response are elicited by other pathogens, allergens, and also by inert sterile insults, including microparticles and trauma [3]. Of note, type 2 immune responses can also be induced during microbial infections where type 1 immune responses (see Glossary) usually predominate and can influence the host’s immune response as being either protective or associated with harmful inflammation [3]. Various pathogens can also induce type 17 immune responses, which can be associated with either type 1 or 2 immune responses [3].
The innate immune response can recognize foreign invaders such as viruses, bacteria, fungi, and parasites, in part by binding pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs). PRRs also recognize specific molecules released from stressed, damaged, or dying cells, referred to as danger-associated molecular patterns (DAMPs) [4]. These DAMPs may also have other important activities in cellular function, but when released in the context of an infection or even sterile insult, may trigger innate immunity (Box 1). Three epithelial cell-derived cytokines, thymic stromal lymphopoietin (TSLP), IL-25, and IL-33, were recently identified as important in ‘alarming’ type 2 immune responses. These cytokine alarmins are released by epithelial cells and a variety of other non-immune and immune cell types [5]. In some cases, they can play distinct and essential roles in driving type 2 immunity, while in other cases they appear to have more redundant roles [6]. There are excellent reviews discussing how these molecules might stimulate type 2 immune responses [5,7–9]. This review focuses on recently identified factors and associated signaling pathways upstream of these three cytokine alarmins, triggering innate type 2 immunity during helminth infection, sterile immunity, allergens, and cancer. This analysis reveals the ubiquitous nature of type 2 immunity and the diverse stimuli inducing its development in different tissue microenvironments.
Box 1. DAMPs/PAMPs of the Immune System.
At least five different families of PRRs have been identified, Toll-like receptors (TLRs), composed of 13 different TLRs in both humans and mice, C-type lectin receptors such as Dectin 1-2, NOD-like receptors, RIG-I-like receptors, and AIM2-like receptors [4,112]. PAMPs binding TLRs can initiate type 1 and type 17 innate immune responses through the secretion of proinflammatory cytokines and chemokines, which provides adjuvant signals for Th1 and Th17 cell development. By contrast, specific PAMPs have not been identified as playing an essential role in initiating the type 2 immune response. Instead, the innate type 2 response appears more dependent on DAMPs, also known as alarmins, which are endogenous molecules often released/secreted by stressed, damaged, or dying cells. These DAMPs can include ATP, adenosine, and UA crystals. Many of these DAMPs, for example, ATP, may also play crucial roles in mediating various cellular functions during homeostasis. However, their release following cellular damage and their subsequent recognition by cell surface receptors are an essential mechanism used by the host to sense danger and activate the immune response. Along with TLRs, DAMP signaling can be mediated through the receptor for advanced glycation end-products, triggering receptors expressed on myeloid cells, and several ionotropic or G-protein-coupled receptors [113]. DAMPs can initiate innate immune responses through stimulation of non-immune cells and immune cells including macrophages, DCs, neutrophils, and mast cells, leading to the production of proinflammatory cytokines, recruitment of inflammatory cells, and activation of adaptive immunity [113,114].
Helminth Infections
Large multicellular helminths can trigger potent and polarized type 2 immune responses. Considerable advances in our understanding of the initiation phase of the immune response to helminth infections have been made over the past few years, using experimental models of helminth infection, including Heligmosomoides polygyrus bakeri (Hpb), Schistosoma mansoni (Sm), Nippostrongylus brasiliensis (Nb), Trichinella spiralis (Ts), and Trichuris muris (Tm) [10]. DAMPs are often released by damaged host cells, as a consequence of these large multicellular parasites trafficking through tissues and causing cellular damage by their sheer physical size and release of proteases. Several DAMPs shown to trigger innate immunity in the context of helminth infection include ATP, chitinase-like proteins, and trefoil factors. In many cases each of these different stimuli are essential for the development of this type 2 immune response, indicating the high threshold required for its initiation.
ATP
ATP functions as a biological energy currency, but when released by stressed or dying cells ATP can also promote initiation of type 2 immune responses. Recent studies have shown that after Hpb infection in mice, extracellular ATP (eATP) is released by apoptotic intestinal epithelial cells (IECs) [11]. eATP can bind the type 2 purinergic receptor, P2X7 on mast cells, leading to their activation, IL-33 production, and the development of type 2 responses [11]. ATP can also be catabolized to adenosine by the cell surface ectonucleotidases CD39 and CD73 [12,13], which are elevated in intestinal epithelial lymphocytes following Hpb infection in mice [14]. Furthermore, A2B adenosine receptor (A2BAR) deficient mice (A2BAR−/−) have impaired type 2 immunity and delayed worm expulsion relative to wild type (WT) mice [14] (Figure 1). Whether adenosine is derived from eATP or other sources remains unclear. Taken together, these studies indicate that ATP and its metabolites can function as essential DAMPs in driving helminth-induced immune responses. However, the mechanism of ATP release, the potential role of other cell sources of ATP, and whether extracellular and/or intracellular ATP/adenosine interactions initiate type 2 immune responses remains unclear.
Figure 1. Helminths and Allergens Initiating Type 2 Immune Responses.
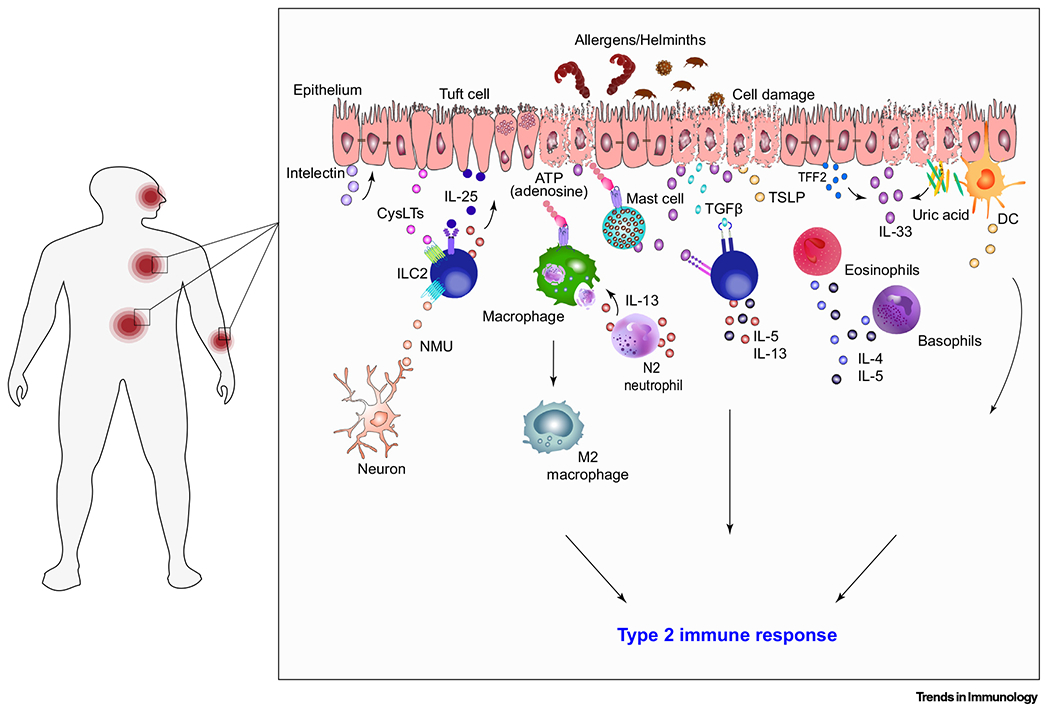
The schematic illustrates multiple often nonoverlapping pathways that together trigger type 2 immune responses in mice. DAMPs, such as extracellular ATP released from damaged cells, and the release of preformed IL-33, activate ILC2s and myeloid cells resulting in type 2 cytokine production, alternatively activated (M2) macrophages, and development of type 2 responses. Efferocytosis of neutrophils can also contribute to M2 macrophage activation [39]. Furthermore, damaged and dying cells secrete uric acid which binds and activates DCs, to initiate the type 2 immune response [125]. TGF-β released from protease damaged cells stimulates the production of IL-5 and IL-13 from ILC2s [126]. At early stages of infection, tuft cell chemosensing of larvae triggers the production of cysLTs, which activate ILC2s in the presence of constitutive IL-25 production by tuft cells [120]. Activated ILC2s produce IL-13, in turn leading to tuft cell hyperplasia and associated increases in IL-25 [116,118,119]. TFF2 secreted by epithelial cells stimulates the release of IL-33 and stimulates CD4+ T cells to produce IL-4 and IL-13 [16]. Stimulation of epithelial cells by IL-13 also induces the production of intelectin which in turn increases the production of alarmins IL-25, IL-33, and TSLP. Although not shown, recent studies also suggest that IL-17 and type I interferons also contribute to the initiation of type 2 immunity [24,25,28]. Abbreviations: cysLT, cysteinyl leukotriene; DAMP, danger-associated molecular pattern; DC, dendritic cell; IL, interleukin; ILC2, group 2 innate lymphoid cell; N2, alternative activated neutrophil; NMU, neuromedin U; TFF2, trefoil factor 2; TSLP, thymic stromal lymphopoietin; TGF-β, transforming growth factor-β.
Trefoil factors
Trefoil factor family (TFF) members 1–3 are epithelial protease-resistant proteins expressed in humans and mice that have protective functions and are involved in wound healing but also can contribute to the initiation of the type 2 response [15,16]. TFF2, a molecule secreted by epithelial cells, stimulates the release of IL-33 from lung and gut epithelial cells to promote type 2 immunity during Nb infection in mice [16]. TFF3 secreted by goblet cells enhances epidermal growth factor receptor (EGFR) signaling through the disruption of leucine-rich repeats and Ig-like domain-containing nogo receptor-interacting protein 2–EGFR complexes. This disruption leads to EGFR signaling and enhancement of type 2 immune responses to Hpb and Nb infections in mice [17,18].
Chitinase and Chitin
Chitins are polysaccharides that help form the protective barriers of many fungi and the exoskeletons of insects and parasites. Chitinases are chitin-degrading enzymes, expressed by epithelial cells and macrophages. They are likely important in digestion, and recent studies suggest they may prime type 2 responses to helminths [19,20]. Epithelial-cell-derived acidic mammalian chitinase (AMCase) can initiate type 2 immune responses in the gastrointestinal tract of mice during Nb and Hpb infections [20]. Epithelial-cell-derived cytokines, TSLP, IL-25, and IL-33, released in response to chitin stimulation, can induce ILC2s to produce IL-5/IL-13, potentially driving eosinophilia and accumulation of alternatively activated (M2) macrophages in murine lungs [21]. By contrast, chitinase-like proteins (CLPs) share homology with chitinase but are enzymatically inactive. During infection of mice with Nb, the CLP Ym-1 is released as larvae invade the lungs, inducing the expansion of γδ T cells and their production of IL-17 [22]. IL-17 then recruits neutrophils to the lungs, contributing to acute lung injury in the infected mouse [23]. IL-17 can also suppress early interferon (IFN)γ production in mice, and potentially have other effects which promote type 2 responses [24–26]. Thus, although DAMPs play a major role in driving type 2 immunity, chitin, and potentially other helminth-derived molecules may function as PAMPs, contributing to the overall response.
Early Cytokines
Type 1 IFNs (IFN-Is) are cytokines secreted by infected cells and through their binding to their specific receptor, induce the transcription of IFN-stimulated genes (ISGs), leading to a protective response during viral and certain bacterial infections [27]. Recent work has shown that IFN-Is may also play an important role during initiation of type 2 immunity. Specifically, using Sm egg antigen injection as a model to induce type 2 immune responses in mice, IFN-I signaling was required to drive dendritic cell (DC) activation, migration, and localization with CD4+ T cells, in the draining lymph nodes in WT mice compared with Ifnar1−/− mice [28]. In another study, IFN signaling promoted Th2 cell activation after Nb infection: IFN-I signaling in DCs from Nb-infected mice promoted the optimal activation of IL-4-producing CD4+ T cells. However, blockade of IFN-I signaling, by administering anti-interferon (α and β) receptor (IFNAR) antibody at the time of Nb inoculation, did not affect DC migration, nor DC antigen transport into the skin draining lymph node [29]. Thus, though not yet well-defined, type I IFNs and IL-17 need to be included as potential early players in the initiation of type 2 responses.
Parasite Products
Excretory/secretory (ES) products produced by helminths are diverse and can modulate various components of the host immune response. Recent studies suggest that ES products induce production of the neuropeptide neuromedin U (NMU) by neuronal cells; NMU then binds the receptor NMUR1 on ILC-2s in mice, driving IL-5 and IL-13 production [30,31]. In the intestine, ES products likely activate the tuft cell to promote type 2 responses (Box 2). Furthermore, the helminth secretory product, IL-4-inducing principle of Sm eggs (IPSE)/α-1, can induce the production of IL-4 from basophils via IgE receptor crosslinking in mice [32]. In contrast, Omega-1, a ribonuclease also produced by Sm, suppresses Th1 responses and conditions mouse DCs to polarize CD4+ T cells into Th2 cells and regulatory T cells [33–35]. ES molecules also add a significant regulatory component to the helminth-induced type 2 immune response, which may modulate initiation of the response [36].
Box 2. Tuft Cells: Mucosal Tissue Guards.
Tuft cells are taste-chemosensory epithelial cells present in small numbers in the small intestine, and in the lungs where they are also referred to as brush cells [115]. Recent studies have demonstrated a role for taste receptors expressed by these cells in orchestrating type 2 immune responses [116–118]. Tuft cells are the primary source of IL-25 in the gut, produced constitutively at readily detectable amounts [118,119]. During the early stages of Hpb infection in mice, tuft cell chemosensing of Hpb larvae leads to their secretion of cysteinyl leukotrienes (cysLTs), which rapidly activate ILC2s in the presence of homeostatic IL-25 [120]. ILC2 activation and secretion of IL-13 induce goblet and tuft cell hyperplasia in mouse intestinal tract, leading to increases in IL-25, through a tuft cell IL-25-ILC2 circuit, thereby promoting a type 2 immune response and worm expulsion [116,118,119] (see Figure 1 in main text). Although ATP can activate airway tuft cells and induce production of cysLTs [121], it has not been shown to induce cysLT release in intestinal tuft cells in mice [120]; this suggests that another ligand might initiate cysLTs release, which remains to be tested. Tuft cells also express the taste-chemosensory succinate receptor (SUCNR1) in the small intestine where the microbial metabolite succinate secreted by Tritrichomonas protists binds SUCNR1 on tuft cells driving the tuft-ILC2 circuit and thereby promoting tuft cell hyperplasia and type 2 immune responses [122,123]. Although the metabolite succinate is secreted during helminth infection such as with Nb, the sensing of Nb is SUCNR1 independent, suggesting a redundant or a different signaling pathway being required for their activation during Nb infection [122]. Furthermore, both helminth-secreted excretory/secretory products and extract of Ts can stimulate another group of bitter-taste receptors (Tas2rs) on tuft cells enhancing the tuft-ILC2 circuit [124]. As the infection progresses, tuft cell expansion and secretion of IL-25 during the later effector phase of Hpb infection appear to be essential factors involved in worm clearance [117].
Early Innate Cell Interactions
Various innate immune cells contribute to the initiation of type 2 immune responses to helminth infection. Neutrophils are the most abundant leukocytes in circulation and during Nb infection in mice, neutrophils swarm L3 larvae immediately after they penetrate the skin, deploying neutrophil extracellular traps (NETs) capable of damaging the larvae [37]. During Nb infection, neutrophils can also promote M2 macrophage differentiation, with recent studies suggesting the involvement of macrophage efferocytosis of apoptotic neutrophils [38] and also neutrophil production of cytokines, including IL-13 [39]. In further studies, surfactant proteins also promoted M2 macrophages during helminth infection, suggesting an important role for the cellular matrix in the tissue microenvironment in shaping macrophage differentiation and the associated type 2 response [40,41]. Basophils and eosinophils are recruited to the lung tissue after primary infection with Nb and can produce IL-4 and IL-5 [42,43]. Basophils can also play a role in the initiation of type 2 immune responses during secondary infection to Nb in mice, where they can promote the alternative activation of macrophages through the production of IL-4 [44]. In addition, a mouse dermal DC subset expressing CD301b primes CD4+ T cells in the draining lymph nodes to produce IL-4, favoring the initiation of type 2 immune responses in Nb infections in mice [45]. Also, in humans and mice, ILC2s can produce IL-5 and IL-13, and are thus potentially important as early initiators of type 2 immune responses [46–48]. Overall, ILC2s may play an important role in barrier immunity, being activated by skin, lung, and gut epithelial cells after helminth infection in humans and mice [49]. In the context of helminth infection in mice, stromal cell IL-33 release can trigger ILC2 production of IL-5 and IL-13 in white adipose tissue, thereby likely promoting metabolic homeostasis. Specifically, IL-33 was shown to be essential in this process, as cytokine production was blocked when ILC2s lacked the ST2/IL-33 receptor in mice (Il1rl1−/−) [50]. However, the actual role of ILC2s in driving type 2 immunity in humans and in intact mice remains unclear, raising the possibility that they might have other effects, which remain to be assessed [51]. Specific interactions between these different innate immune cell populations and also their communication with non-immune cells and effects of signals produced by the tissue-specific cell matrix remains understudied and are likely to become a major focus of future research.
Sterile Inflammation
Sterile inflammation (SI) occurs in response to mechanical trauma, irritants, cell death, tissue injury, or chemically induced injury in the absence of pathogenic microbes. Numerous particulates have been identified that promote SI including: uric acid (UA), silica crystals, aluminum salts, asbestos, pollutants, prosthetic implant wear debris, and cholesterol crystals [52–56]. SI plays an important role in tissue repair, but dysregulation of this immune response can also lead to chronic inflammatory diseases, such as gout [57]. Although the triggers that initiate SI are poorly understood, a number of recent studies indicate a significant role for type 2 immune responses (Figure 2).
Figure 2. Signaling Molecules Initiating Type 2 Immune Response in Sterile Inflammation.
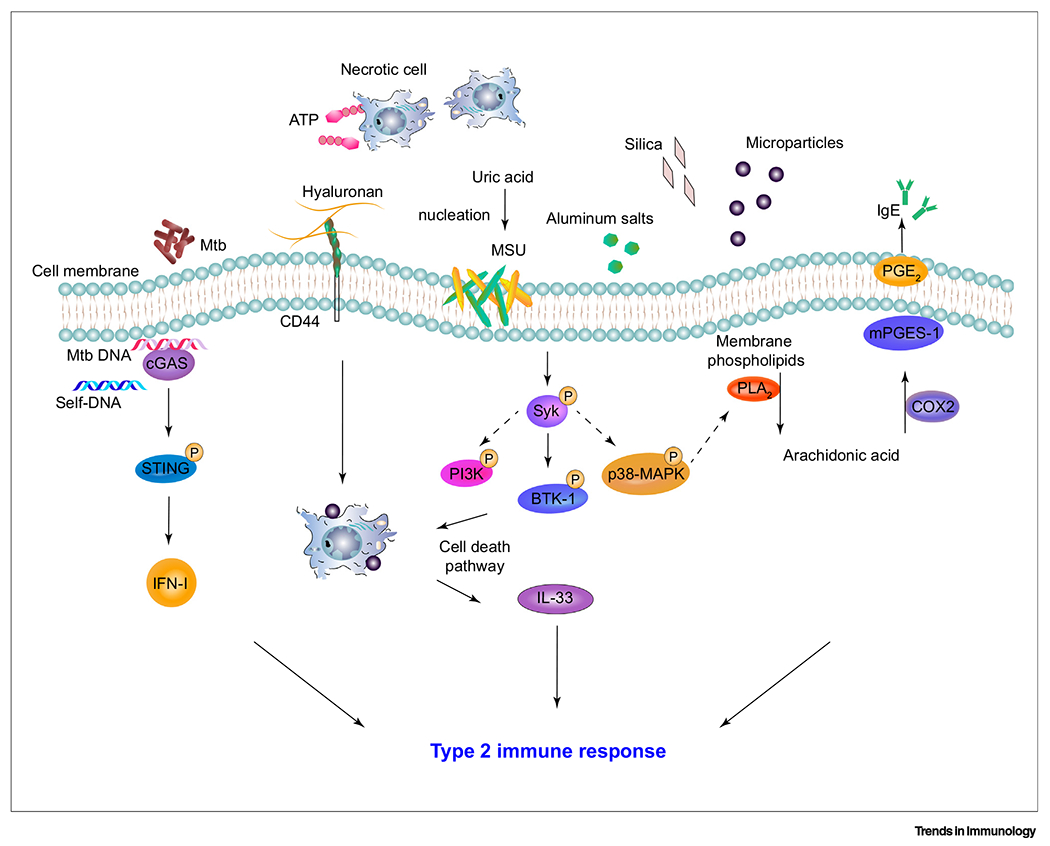
Microparticles, trauma, and tissue injury can trigger sterile inflammatory responses including innate type 2 immunity. In mouse models of liver injury, ATP recruitment of GATA-6+ macrophages and the activation via hyaluronan–CD44 can lead toM2 macrophage phenotype polarization [69]. Microparticles induce Syk and downstream BTK-1 signaling leading to the release of IL-33 in mice [62]. Silica has also been shown to prime type 2 immune responses by triggering the release of DNA by damaged cells, thereby activating the cGAS/STING pathway and increased IFN-I in mice [68]. Type 2 immunity is also mediated by activation of p38 MAPK and PLA2, resulting in the release of arachidonic acid from membrane lipids. Arachidonic acid leads to the production of COX2 and mPGES-1, converting arachidonic acid to PGE2, which can enhance IgE production in mice [54]. Abbreviations: BTK-1, Bruton’s tyrosine kinase-1; cGAS, cyclic GMP-AMP synthase; COX2, cyclooxygenase-2; IFN-I, interferon-I; MAPK, mitogen-activated protein kinase; mPGES-1, membrane-associated PGE synthase-1; MSU, monosodium urate crystal; Mtb, Mycobacterium tuberculosis; PGE2, prostaglandin E2; PI3K, phosphoinositide 3-kinase; PLA2, phospholipase A2; STING, stimulator of interferon genes; Syk, spleen tyrosine kinase.
Particle-Induced Inflammation
A diverse group of particulates triggers SI and skews the response towards type 2 immunity. For example, UA crystals, the end product of purine metabolism, which contributes to gout and acute gout arthritis [58,59], is released from dying or stressed cells. UA crystals, produced by airway epithelial cells after internasal house dust mite (HDM) allergen inoculation with ovalbumin (OVA), can directly induce type 2 immunity in mice, as measured by increased production of type 2 cytokines and serum IgE relative to controls. In this model, inflammatory DCs were activated through the spleen tyrosine kinase (Syk) and PI3-kinase δ signaling pathway independently of the NLRP3 inflammasomes [60]. Silica crystals and alum administered to mice could also trigger type 2 immune responses through Syk-dependent and inflammasome-independent pathways leading to the production of prostaglandin (PG)E2 by macrophages and elevated serum IgE [54,60]. Of note, in these studies, microparticles did initially trigger activation of the NLPR3 inflammasome and production of biologically active IL-1β; however, this parallel response was not required for the subsequent development of the type 2 response [54,55,60–62]. Also, particulates, including pollutants such as diesel exhaust particles (DEPs), could trigger potent type 2 responses in mice, enhancing emphysema and allergen-induced inflammation relative to controls [63–65]. Recently, micron-sized particles released as wear debris by prosthetic joint replacements have been linked to increases in SI and type 2 immunity, resulting in osteolysis and implant failure in humans [62,66]. In peritoneal and knee joint mouse models, sterile metallic microparticles, similar in size and composition to wear debris, stimulate the recruitment of innate immune cells, including neutrophils, eosinophils, and M2 macrophages, resulting in elevated type 2 cytokines relative to controls. These macrophages produce IL-33 through a Syk- and Bruton’s tyrosine kinase (BTK)-dependent pathway, which promotes inflammasome-independent initiation of type 2 immunity and requires activation of cell death pathways [62,67].
Sterile particulates, including pollutants, can also enhance inflammation during infectious disease. Recently, silica exposure in the mouse lung induced type 2 immune responses, exacerbating infection by Mycobacterium tuberculosis [68]. Associated tissue damage triggered the release of extracellular DNA, which stimulated the cyclic GMP–AMP synthase (cGAS)/STING (stimulator of interferon genes) pathway, as demonstrated by the increased expression of the STING-coding gene Tmem173 and the cGAS-coding gene Mb21d1. Associated increases in IFN-I promoted harmful type 2 inflammation, which was neutralized in Ifnar−/− mice [68]. Taken together, these studies indicated that sterile inert micrometer sized particles of varying composition can trigger type 2 innate inflammatory responses in different tissues. Increased particle exposure in the external environment, ranging from diesel exhaust particles to silica exposure, and also the release of microparticles from various internal prosthetic implants, can potentially impact immune function, and thus, this understudied area merits further attention.
Trauma-Induced Inflammation
Mechanical/hypoxic traumatic injuries can also trigger SI and type 2 immunity. During sterile liver injury in experimental mouse models, necrotic cell release of ATP induced the recruitment of GATA-6+ macrophages from the peritoneal cavity. At the site of injury, these macrophages differentiated into M2 macrophages, where they contributed to tissue repair by ingesting necrotic nuclei of cells at the site of injury [69]. In a different study, pericardial fluid samples, taken from patients undergoing valve replacement surgery, were elevated in M2 macrophages, suggesting that they might contribute to cardiac repair after injury [70]. Also, clinical studies of patients with severe blunt force trauma injuries, excluding brain and head traumas, revealed elevated concentrations of IL-33 and type 2 cytokines in human blood relative to controls [71]. As such, type 2 immunity might be a major component of SI resulting from tissue injury caused by a variety of stimuli ranging from solid particulates to mechanical injury.
Allergens
The type 2 immune response plays a major role in allergic inflammation including immediate hypersensitivity reactions. Allergic reactions can be associated with infectious disease or can occur following exposure to allergens under sterile conditions. Allergens include pollen, fungi, animal dander, pollutants, and HDM, many of which can have enzymatic activities contributing to tissue damage. Unlike inert particulates, allergens may also have epitopes that can activate adaptive immunity, including both T and B cells, exacerbating the inflammatory response to challenges after sensitization [72].
Purinergic Signaling
Recent studies have begun to unravel how allergens stimulate innate type 2 immune responses. Infection with the fungal allergen, Alternaria alternata, can drive allergic lung inflammation: airway epithelial cell exposure to this allergen in mice increased calcium signaling and their release of eATP [73]. Subsequent signaling through the cell surface purinergic receptor, P2Y2, triggered the release of full-length IL-33 from airway epithelial cells, inducing the production of IL-5 and IL-13 and a downstream type 2 response in murine lungs [73,74]. Staphylococcus aureus serine-protease-like proteins also increase the release of full-length IL-33, through pathways that are independent of either apoptosis or necrosis [75]. Similarly, in OVA peptide and HDM asthma mouse models, eATP signaling through the P2X7 receptor on macrophages stimulates an M2 macrophage phenotype [76]. Adenosine can also contribute to chronic allergic inflammation in mice, as the response to OVA plus alum is attenuated by blocking A2BAR signaling relative to controls [77]. These studies have demonstrated the importance of eATP release and downstream adenosine in driving allergic responses in several different mouse models. Of note, cellular stress triggering calcium flux and eATP release from intact cells might in some cases be sufficient to drive the response in the absence of significant cell death.
Other Signaling Pathways Leading to Allergy
Several other mechanisms besides eATP have been implicated in allergic inflammation. As already discussed, UA crystals can also promote type 2 responses. When released in the airways they can trigger allergic inflammation in HDM-allergen-challenged mice, and increases in UA are correlated with increased allergic inflammation in asthmatic subjects following allergen provocation [60]. Also, pollen proteases damage the epithelial tight junction occludin, claudin-1, and ZO-1 proteins, likely triggering the release of DAMPs and allowing translocation of allergens in the airway epithelium, inducing allergic reactions [78]. In an HDM mouse model, the allergen protease Derp1 of HDM can enhance initiation of the type 2 immune response through cleavage of full-length IL-33 to more active forms, thereby directly promoting type 2 immunity, including activation of ILC2s to produce IL-5 and IL-13 [79]. Of note, full-length IL-33 may be also cleaved extracellularly by enzymes released by both innate immune cells and epithelial cells, resulting in its inactivation, or the formation of more active fragments, which can enhance the initiation of type 2 immunity [80–82]. Additionally, proteases such as papain activate type 2 immune responses by damaging IECs, triggering the release of IL-33 [83]. In another mouse model, alkaline protease (Alp)1 from the fungus Aspergillus fumigatus induces damage to epithelial cell junctions of bronchiolar club cells in the lungs. Specifically, this mechanical damage is recognized by transient receptor potential channel 4 on club cells, initiating calcium flux and inducing calcineurin; this promotes IL-5 production by GATA3+ Th cells and eosinophilia, thereby triggering allergic inflammation [84].
Mechanisms other than cell damage may also contribute to the type 2 immune response. In a HDM mouse model of allergy, ILC2 production of IL-13 is instead driven by NMU, which amplifies IL-25-mediated inflammation by binding its receptor NMUR1 on ILC2s [85] (Figure 1). How neurons and sensory cells sense the allergen and induce the expression of NMU remains unknown. Also, the carbohydrate-binding lectin, intelectin, expressed in humans and mice, is upregulated in airway and skin epithelial cells in OVA or HDM mouse models, and is required for the upregulation of IL-25, IL-33, and TSLP [86–88]. Taken together, several different pathways have now been implicated in triggering allergic reactions. Accordingly, various early signals ranging from eATP to NMU may play essential roles in initiating the type 2 immune responses. Taken together, studying how these different pathways can interact to drive allergic inflammation is an important next step to understand how these allergic responses are initiated. Although they remain understudied, death signaling pathways might also play a significant role in allergic inflammation and thus, this connection might potentially be an important area of future investigation.
Cancer
A hallmark of cancer development is tumor-promoting inflammation, which may occur at various stages of tumorigenesis [89]. Type 1 immunity plays a predominant role during the tumor initiation phase with natural killer (NK) and CD8+ T cells recognizing cancer cells and eliminating them [89]. As the tumor progresses, a type 2 immune response associated with immunosuppression and immune tolerance can develop, resulting in enhanced tumor growth and/or progression, thereby promoting tumor malignancy (Figure 3) [90]. Myeloid cell plasticity is characteristic of this transition [91], as the tumor microenvironment (TME) progressively develops into a complex network of host cells including fibroblasts, endothelial cells, immune cells, and the extracellular matrix, interacting together to support tumor growth [91]. Moreover, tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), monocytic myeloid-derived suppressor cells (M-MDSCs), and DCs are all key innate immune cell players [89].
Figure 3. The Tumor Microenvironment Can Suppress Immune Responses by Initiating Type 2 Immunity.
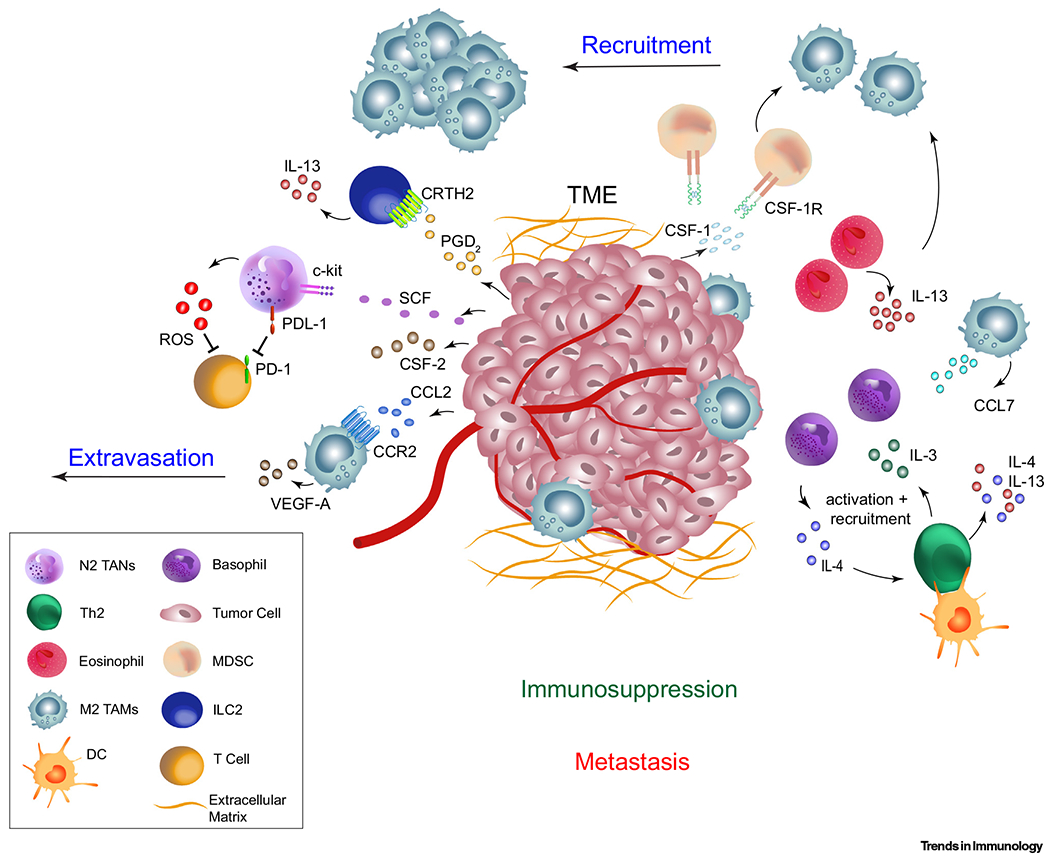
Tumor cell secretion of PGD2 stimulates CRTH2 signaling on ILC2s and their consequent IL-13 production and recruitment of MDSCs in mice [106,107]. Secreted CSF-1 can promote the differentiation of alternative activated (M2) macrophages, while CCL2 secreted from tumor and stromal cells and eosinophil secretion of IL-13 can promote M2 macrophage recruitment [95]. VEGF-A secreted by these M2 macrophages can lead to extravasation of tumor cells and metastasis in certain models [95–97]. Mouse M2 macrophage release of CCL7 can lead to the recruitment and activation of IL-4-producing basophils, in turn activating Th2 CD4+ T cells to produce IL-4/IL-13 [110]. Furthermore, tumor-derived CSF-2 can activate N2 TANs and PD-L1 expression can suppress T cell function in certain APL and bladder cancer human patients [104]. Tumor derived SCF can also drive the production of ROS from TANs, leading to suppression of T cell function in mice [105]. Abbreviations: CCL, C-C motif chemokine ligand; CCR2, C-C chemokine receptor type 2; CRTH2, chemoattractant receptor-homologous molecule 2; CSF, colony-stimulating factor; CSF-1R, CSF-1 receptor; DC, dendritic cell; ILC2, group 2 innate lymphoid cell; M2 TAM, M2 tumor-associated macrophage; MDSC, myeloid-derived suppressor cell; N2 TAN, N2 tumor-associated neutrophil; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PGD2, prostaglandin D2; ROS, reactive oxygen species; SCF, stem cell factor; Th2, T helper type 2; TME, tumor-microenvironment; VEGF-A, vascular endothelial growth factor A.
Tumor-Secreted Factors – Tumorigenic M2 Macrophages
Type 2 immunity is in part initiated within the TME by tumor cell production of colony-stimulating factor (CSF)-1, which promotes M2 macrophage differentiation [92,93]. In a mouse mammary cancer model, CSF-1, expressed by the mammary epithelium, binds CSF1 receptor on macrophages, promoting tumor metastasis in part by regulating macrophage function and infiltration into the TME [94]. In a PyMT mammary cancer mouse model, type 2 immunity is also driven by tumor and stromal cell secretion of CCL2, which induces CCR2-dependent recruitment of M2 macrophages [95]. Activated M2 macrophages, within the TME, secrete vascular endothelial growth factor A (VEGFA), which enhances tumor cell extravasation promoting metastasis [95] and tumor angiogenesis [96,97]. By contrast, tumor necrosis factor (TNF) has antagonized M2 macrophage polarization, as Tnfrsf1a−/− mice implanted with thymoma (EG7) lymphomas, show enhanced M2 macrophage polarization, which is further promoted by eosinophils producing IL-13, relative to controls [98]. Thus, a balance between type 1 inflammation associated with TNF, and type 2 inflammation associated with IL-13 and CSF-1 could influence the development of M2 macrophages and associated tumor progression [98]. Lastly, although controversial, M-MDSCs can give rise to TAMs and have been associated with shaping the metabolic state of the TME. This metabolic axis has been deemed to include lipid, glucose, and amino acid metabolism, which can in turn inhibit antitumor responses [99].
Tumor-Secreted Factors – Tumorigenic N2 Neutrophils
Similar to M2 macrophages, neutrophils can also exhibit a protumor, alternatively activated phenotype [100]. Such neutrophils can express tumor-associated factors that may promote metastasis, carcinogenesis, immunosuppression, and angiogenesis [101–103]. Factors secreted by tumor cells such as tumor-derived CSF-2 within the TME of human gastric cancer patients can activate these neutrophil types [104]. In vitro studies have shown that surface expression of programmed death ligand 1 on neutrophils can suppress T cell function in vitro [104] (Figure 3). Within the local TME, another study in 4T1 tumor-bearing mice has shown that c-kit+ tumor-elicited neutrophils can prime a low glucose metabolic state that sustains an immunosuppressive type 2 microenvironment while inhibiting a type 1 immune response [105]. This N2 neutrophil phenotype is similar to the N2 neutrophil phenotype that was recently described in the context of helminth infections [39]. It will be interesting in further studies to compare these alternatively activated neutrophils that seem to arise in response to different stimuli and associated tissue microenvironments.
ILC2s: Pro- or Antitumorigenic?
In patients with acute promyelocytic leukemia (APL), type 2 immunity promotes tumor growth and progression, potentially through the involvement of tumorigenic ILC2s [106]. Recent studies using human APL and bladder cancer samples, as well as APL mouse models, suggest that the type 2 immune response may be initiated by the binding of tumor derived factors, such as PGD2 and B7H6, which bind to their receptors CRTH2 and NKp30m, respectively, on ILC2s, driving an IL-13 response and the recruitment and activation of M-MDSCs into the tumor [106,107]. Furthermore, in a mouse B16-tumor-bearing model, ILC2s are rendered protumorigenic by suppressing NK cell IFN-γ production [108]. Conversely, ILC2s have also been reported to be antitumorigenic in specific TMEs. For example, in a B16 melanoma mouse tumor model of decreased lactate dehydrogenase A expression (using small hairpin RNAs complementary to Ldha in melanoma cells), stimulation of ILC2s with IL-33 leads to recruitment of eosinophils to the tumor site, thus minimizing tumor growth relative to controls [109]. Thus, recent studies have suggested that the particular TME may influence whether ILC2s are pro- or antitumorigenic.
Other Immune Cells
In a mouse model of pancreatic ductal adenocarcinoma TME, M2 macrophages release CCL7, which promotes the recruitment of basophils secreting IL-4, thus stimulating M2 macrophage recruitment and type 2 inflammatory responses in the tumor-draining lymph nodes [110]. Moreover, in a transgenic mouse model of mammary adenocarcinoma, CD4+ T cell-derived IL-4 stimulates the polarization of TAMs and enhanced EGF/EGFR signaling, resulting in tumor invasion and metastasis, relative to controls [111]. Taken together, these studies suggest that tumor-secreted factors can act as prerequisites for the initiation of type 2 immune responses [91], although their individual roles in initiating type 2 immunity in the TME remain elusive.
Concluding Remarks
Our understanding of the complex network of initiating factors involved in the burgeoning field of type 2 immunity has advanced considerably but remains at an early stage of investigation (see Outstanding Questions). The diversity of stimuli that trigger type 2 immune responses is considerable. However, it is also clear that many common pathways are shared in the initiation of the ensuing response. Overall, DAMPs released because of cell stress or death appear to be generally significant in driving the initiation of type 2 immune responses. In worm infections, even though helminth-secreted ES factors certainly contribute to activation of the immune response, cell damage resulting from these multicellular parasites trafficking through tissues is likely a major factor in the initiation of the response. However, the plasticity of type 2 immune response, including its effects in the context of microbial infections, underscores its previously underappreciated ubiquitous nature. In certain cases, the apparent variation in response to these stimuli might be partly due to what has been initially investigated in different model systems. In this regard, it will be important to conduct more direct comparisons between type 2 responses elicited by allergens, helminths, cancer, particulates, trauma, or other stimuli. Also, the tissue microenvironment might play a significant role in these observed differences. The initiation of the type 2 immune response at barrier surfaces might preferentially depend on specialized epithelial cells and associated innate myeloid and lymphoid cells triggering the response, while non-epithelial responses might utilize different pathways. As an example, a recent study has shown that the initiation of joint and peritoneal type 2 responses to microparticles are dependent on BTK signaling in macrophages, while enteric responses to helminths are BTK independent, and such findings might exemplify such differences [62]. Finally, it also clear that the ubiquitous type 2 response can have beneficial effects associated with tissue repair, versus harmful effects associated with fibrosis and increased susceptibility to infection. What tips the balance in these outcomes may be context dependent, including the tissue microenvironment, the genetic background, as well as previous exposure to pathogenic and commensal microbes and parasites, resulting in a trained innate immune response with a bias toward a specific type of response, although this remains conjectural. Further elucidation of the triggers that initiate type 2 immunity could reveal novel checkpoints that might be targeted for future therapies.
Outstanding Questions.
How do specific recently identified factors and cell populations triggering type 2 immunity interact to orchestrate the overall response? Individual cell populations and molecules identified as essential may not be sufficient. Research is needed to reveal how these different factors together initiate the in vivo response.
What are the common and distinct features characterizing type 2 responses that develop in different TMEs and in response to different stimuli? Few studies have compared how type 2 responses develop and differ in response to distinct stimuli and microenvironments. Insights might reveal new candidate checkpoints that might be targeted to influence type 2 immunity initiation and progression.
How do cell death signaling pathways activated in response to tissue damage influence the initiation of type 2 responses? Given the importance of DAMPs in initiating type 2 immunity, understanding the nature of the cell death pathways involved might provide new insights into how to promote or control this response.
What is the role of helminth ES products in influencing the initiation of type 2 responses and associated immune regulatory circuits? ES products might provide an important source for the development of novel potential therapeutics to regulate specific components of harmful inflammation, or for the identification of new adjuvants to promote type 2 immunity.
Given the importance of ATP and its metabolites in initiating type 2 responses, what are the cellular sources of ATP and the mechanisms that regulate ATP and adenosine signaling? It will be important to tease apart the specific purinergic signaling pathways that contribute to the initiation of type 2 immunity, as well as how to potentially modulate its heterogeneity. These insights might provide a platform for the development of novel candidate treatments to treat a variety of type 2 immunity-related pathological responses.
Highlights.
Danger associated molecular patterns (DAMPs) released in response to tissue damage are likely to play a major role in initiating type 2 responses in humans and mice.
The initiating factors of type 2 immunity can vary with the specific tissue microenvironment. Epithelial cells are of importance in barrier responses, while other cell types such as macrophages or stromal cells may be preferentially important at other tissue sites.
As the initiation of a type 2 immune response progresses, a variety of innate immune cells including macrophages and granulocytes, as well as innate lymphoid cells, are instrumental and often interact to shape the development of type 2 immunity.
In many cases, these initial interactions result in the production of cytokine alarmins, interleukin (IL)-33, IL-25, and thymic stromal lymphopoietin (TSLP), which then promote and amplify the type 2 immune response.
Acknowledgments
National Institutes of Health (NIH) grants R01GM066189 (G.H.) and R01DK113790 (W.C.G. and G.H.), R01DK113790 (W.C.G), R01AI131634 (W.C.G), and T32AI125185 to D.W.E.
Glossary
- Alternative activated (M2) macrophages:
historically, this term was used to describe macrophages activated by IL-4 and/or IL-10 in vitro, with further attempts to categorize according to specific stimulating cytokines. However, macrophage phenotypes are more heterogeneous in vivo, as additional stimuli besides cytokines contribute to their activation. As such, the term M2 macrophage, although still debated, is now often used to describe a generalized macrophage activation phenotype expressing a broad set of shared markers in the context of type 2 immune responses
- cGAS/Sting pathway:
self and non-self DNA-sensing nucleotidyl transferase enzyme cyclic GMP–AMP synthase (cGAS), stimulator of interferon genes (STING). Pathway involved in initiating IFN induced responses to the accumulation of cytosolic DNA
- Cysteinyl leukotrienes (cysLTs):
lipid mediators synthesized from arachidonic acid and released upon demand by tuft cells, mast cells, eosinophils, basophils, and macrophages. cysLTs initiate immune responses and contribute to maintain tissue homeostasis
- Cytokine alarmins:
subset of cytokines, including epithelial and myeloid cell-derived IL-25, TSLP, and IL-33. They are often released or secreted by stressed or damaged epithelial cells and can act to initiate and sustain innate and adaptive type 2 immune responses
- Danger-associated molecular patterns (DAMPs):
also known as alarmins; endogenous molecules released from stressed/damaged cells that can promote immune cell activation
- Efferocytosis:
the process where phagocytic cells engulf dead or dying cells
- Neutrophil extracellular traps (NETs):
chromatin mesh that includes proteinases and peptidases capable of trapping and killing extracellular pathogens
- Pathogen-associated molecular patterns (PAMPs):
conserved microbial structures that can activate innate responses
- Pattern recognition receptors (PRRs):
receptors that recognize PAMPs and DAMPs, and primarily mediate the activation of innate immunity
- Surfactant proteins:
innate immune molecules that are components of a surfactant and can contribute to pathogen resistance and modulating immune function
- Trained innate immune response:
the conserved reprogramming of innate immune cells following stimulation, which results in an altered response after subsequent challenge
- Tuft cells:
chemosensory epithelial cells lining the mucosal surfaces including the small intestine and lungs (brush cells)
- Type 1 immune response:
triggered by certain microbial pathogens, resulting in the production of IFN-γ and other cytokines
- Type 17 immune responses:
often triggered by extracellular pathogens and characterized by IL-17 production
Footnotes
Disclaimer Statement
G.H. owns stock in Purine Pharmaceuticals, Inc. and has patents related to purinergic signaling in sepsis.
References
- 1.Allen JE and Maizels RM (2011) Diversity and dialogue in immunity to helminths. Nat. Rev. Immunol 11,375–388 [DOI] [PubMed] [Google Scholar]
- 2.Gause WC et al. (2013) Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol 13, 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gause WC et al. (2020) Heterogeneity in the initiation, development and function of type 2 immunity. Nat. Rev. Immunol 20, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeuchi O and Akira S (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 5.Roan F et al. (2019) Epithelial cell-derived cytokines: more than just signaling the alarm. J. Clin. Invest 129, 1441–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vannella KM et al. (2016) Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci. Transl. Med 8, 337ra65. [DOI] [PubMed] [Google Scholar]
- 7.Hammad H and Lambrecht BN (2015) Barrier epithelial cells and the control of type 2 immunity. Immunity 43, 29–40 [DOI] [PubMed] [Google Scholar]
- 8.Liew FY et al. (2016) Interleukin-33 in health and disease. Nat. Rev. Immunol 16, 676–689 [DOI] [PubMed] [Google Scholar]
- 9.Kabata H et al. (2018) The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol. Rev 286, 37–52 [DOI] [PubMed] [Google Scholar]
- 10.Anthony RM et al. (2007) Protective immune mechanisms in helminth infection. Nat. Rev. Immunol 7, 975–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimokawa C et al. (2017) Mast cells are crucial for induction of group 2 innate lymphoid cells and clearance of helminth infections. Immunity 46, 863–874 e4 [DOI] [PubMed] [Google Scholar]
- 12.Antonioli L et al. (2019) The purinergic system as a pharmacological target for the treatment of immune-mediated inflammatory diseases. Pharmacol. Rev 71, 345–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonioli L et al. (2013) CD39 and CD73 in immunity and inflammation. Trends Mol. Med 19, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel N et al. (2014) A2B adenosine receptor induces protective antihelminth type 2 immune responses. Cell Host Microbe 15, 339–350 [DOI] [PubMed] [Google Scholar]
- 15.Taupin D and Podolsky DK (2003) Trefoil factors: initiators of mucosal healing. Nat. Rev. Mol. Cell Biol 4, 721–732 [DOI] [PubMed] [Google Scholar]
- 16.Wills-Karp M et al. (2012) Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J. Exp. Med 209, 607–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minutti CM et al. (2017) Epidermal growth factor receptor expression licenses type-2 helper T Cells to function in a T cell receptor-independent fashion. Immunity 47, 710–722 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belle NM et al. (2019) TFF3 interacts with LINGO2 to regulate EGFR activation for protection against colitis and gastrointestinal helminths. Nat. Commun 10, 4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohno M et al. (2016) Acidic mammalian chitinase is a proteases-resistant glycosidase in mouse digestive system. Sci. Rep 6, 37756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vannella KM et al. (2016) Acidic chitinase primes the protective immune response to gastrointestinal nematodes. Nat. Immunol 17, 538–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Dyken SJ et al. (2014) Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gamma delta T cells. Immunity 40, 414–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutherland TE et al. (2014) Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat. Immunol 15, 1116–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen F et al. (2012) An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med 18, 260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ajendra J et al. (2020) IL-17A both initiates, via IFNgamma suppression, and limits the pulmonary type-2 immune response to nematode infection. Mucosal Immunol. 13, 958–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen JE et al. (2015) IL-17 and neutrophils: unexpected players in the type 2 immune response. Curr. Opin. Immunol 34, 99–106 [DOI] [PubMed] [Google Scholar]
- 26.Sutherland TE et al. (2018) Ym1 induces RELMalpha and rescues IL-4Ralpha deficiency in lung repair during nematode infection. PLoS Pathog 14, e1007423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivashkiv LB and Donlin LT (2014) Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webb LM et al. (2017) Type I interferon is required for T helper (Th) 2 induction by dendritic cells. EMBO J. 36, 2404–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor LM et al. (2017) Th2 responses are primed by skin dendritic cells with distinct transcriptional profiles. J. Exp. Med 214, 125–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klose CSN et al. (2017) The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cardoso V et al. (2017) Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schramm G et al. (2007) Cutting edge: IPSE/alpha-1, a glycoprotein from Schistosoma mansoni eggs, induces IgE-dependent, antigen-independent IL-4 production by murine basophils in vivo. J. Immunol 178, 6023–6027 [DOI] [PubMed] [Google Scholar]
- 33.Steinfelder S et al. (2009) The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1). J. Exp. Med 206, 1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Everts B et al. (2009) Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J. Exp. Med 206, 1673–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaccone P et al. (2011) The S. mansoni glycoprotein omega-1 induces Foxp3 expression in NOD mouse CD4(+) T cells. Eur. J. Immunol 41, 2709–2718 [DOI] [PubMed] [Google Scholar]
- 36.Maizels RM et al. (2018) Modulation of host immunity by helminths: the expanding repertoire of parasite effector molecules. Immunity 49, 801–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouchery T et al. (2020) Hookworms evade host immunity by secreting a deoxyribonuclease to degrade neutrophil extracellular traps. Cell Host Microbe 27, 277–289 e6 [DOI] [PubMed] [Google Scholar]
- 38.Bosurgi L et al. (2017) Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356, 1072–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen F et al. (2014) Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat. Immunol 15, 938–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thawer S et al. (2016) Surfactant protein-D is essential for immunity to helminth infection. PLoS Pathog. 12, e1005461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minutti CM et al. (2017) Local amplifiers of IL-4Ralpha-mediated macrophage activation promote repair in lung and liver. Science 356, 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min B et al. (2004) Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med 200, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voehringer D (2004) K. et al. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity 20, 267–277 [DOI] [PubMed] [Google Scholar]
- 44.Obata-Ninomiya K et al. (2013) The skin is an important bulwark of acquired immunity against intestinal helminths. J. Exp. Med 210, 2583–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumamoto Y et al. (2013) CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 39, 733–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price AE et al. (2010) Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. U. S. A 107, 11489–11494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moro K et al. (2010) Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 [DOI] [PubMed] [Google Scholar]
- 48.Neill DR et al. (2010) Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbert DR et al. (2019) Group 2 innate lymphoid cells (ILC2): type 2 immunity and helminth immunity. Int. J. Mol. Sci 20, 2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahlakoiv T et al. (2019) Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol 4, eaax0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kotas ME and Locksley RM (2018) Why innate lymphoid Cells? Immunity 48, 1081–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mossman BT and Churg A (1998) Mechanisms in the pathogenesis of asbestosis and silicosis. Am. J. Respir. Crit. Care Med 157, 1666–1680 [DOI] [PubMed] [Google Scholar]
- 53.Kono H et al. (2010) Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Invest 120, 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuroda E et al. (2011) Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity 34, 514–526 [DOI] [PubMed] [Google Scholar]
- 55.McKee AS et al. (2009) Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J. Immunol 183, 4403–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duewell P et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen GY and Nunez G (2010) Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol 10, 826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desai J et al. (2017) Molecular pathophysiology of gout. Trends Mol. Med 23, 756–768 [DOI] [PubMed] [Google Scholar]
- 59.So A and Thorens B (2010) Uric acid transport and disease. J. Clin. Invest 120, 1791–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kool M et al. (2011) An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity 34, 527–540 [DOI] [PubMed] [Google Scholar]
- 61.Martinon F et al. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 [DOI] [PubMed] [Google Scholar]
- 62.Mishra PK et al. (2019) Sterile particle-induced inflammation is mediated by macrophages releasing IL-33 through a Bruton’s tyrosine kinase-dependent pathway. Nat. Mater 18, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moreira AR et al. (2020) Chronic exposure to diesel particles worsened emphysema and increased M2-like phenotype macrophages in a PPE-induced model. PLoS One 15, e0228393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Grove KC et al. (2017) Dysregulation of type 2 innate lymphoid cells and TH2 cells impairs pollutant-induced allergic airway responses. J. Allergy Clin. Immunol 139, 246–257 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manners S et al. (2014) A mouse model links asthma susceptibility to prenatal exposure to diesel exhaust. J. Allergy Clin. Immunol 134, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koulouvaris P et al. (2008) Expression profiling reveals alternative macrophage activation and impaired osteogenesis in periprosthetic osteolysis. J. Orthop. Res 26, 106–116 [DOI] [PubMed] [Google Scholar]
- 67.Mishra PK et al. (2011) Micrometer-sized titanium particles can induce potent Th2-type responses through TLR4-independent pathways. J. Immunol 187, 6491–6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benmerzoug S et al. (2019) Sterile lung inflammation induced by silica exacerbates Mycobacterium tuberculosis infection via STING-dependent type 2 immunity. Cell Rep. 27, 2649–2664 e5 [DOI] [PubMed] [Google Scholar]
- 69.Wang J and Kubes P (2016) A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 165, 668–678 [DOI] [PubMed] [Google Scholar]
- 70.Deniset JF et al. (2019) Gata6(+) Pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 51, 131–140 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu J et al. (2017) IL33-mediated ILC2 activation and neutrophil IL5 production in the lung response after severe trauma: a reverse translation study from a human cohort to a mouse trauma model. PLoS Med. 14, e1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.von Moltke J and Pepper M (2018) Sentinels of the type 2 immune response. Trends Immunol. 39, 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kouzaki H et al. (2011) The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol 186, 4375–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Snelgrove RJ et al. (2014) Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J. Allergy Clin. Immunol 134, 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Teufelberger AR et al. (2018) The IL-33/ST2 axis is crucial in type 2 airway responses induced by Staphylococcus aureus-derived serine protease-like protein D. J. Allergy Clin. Immunol 141, 549–559 e7 [DOI] [PubMed] [Google Scholar]
- 76.Li R et al. (2020) ATP/P2X7r axis mediates the pathological process of allergic asthma by inducing M2 polarization of alveolar macrophages. Exp. Cell Res 386, 111708. [DOI] [PubMed] [Google Scholar]
- 77.Zaynagetdinov R et al. (2010) Attenuation of chronic pulmonary inflammation in A2B adenosine receptor knockout mice. Am. J. Respir. Cell Mol. Biol 42, 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Runswick S et al. (2007) Pollen proteolytic enzymes degrade tight junctions. Respirology 12, 834–842 [DOI] [PubMed] [Google Scholar]
- 79.Cayrol C et al. (2018) Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol 19, 375–385 [DOI] [PubMed] [Google Scholar]
- 80.Clancy DM et al. (2018) Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 22, 2937–2950 [DOI] [PubMed] [Google Scholar]
- 81.Lefrancais E et al. (2014) Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. U. S. A 111, 15502–15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scott IC et al. (2018) Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci. Rep 8, 3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hara K et al. (2014) Airway uric acid is a sensor of inhaled protease allergens and initiates type 2 immune responses in respiratory. J. Immunol 192, 4032–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wiesner DL et al. (2020) Club cell TRPV4 serves as a damage sensor driving lung allergic inflammation. Cell Host Microbe 27, 614–628.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wallrapp A et al. (2017) The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gu N et al. (2010) Intelectin is required for IL-13-induced monocyte chemotactic protein-1 and −3 expression in lung epithelial cells and promotes allergic airway inflammation. Am. J. Physiol. Lung Cell Mol. Physiol 298, L290–L296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhen G et al. (2007) IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am. J. Respir. Cell Mol. Biol 36, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yi L et al. (2017) Intelectin contributes to allergen-induced IL-25, IL-33, and TSLP expression and type 2 response in asthma and atopic dermatitis. Mucosal Immunol. 10, 1491–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Galon J and Bruni D (2020) Tumor immunology and tumor evolution: intertwined histories. Immunity 52, 55–81 [DOI] [PubMed] [Google Scholar]
- 90.Mantovani A et al. (2017) Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 14, 399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gonzalez H et al. (2018) Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pyonteck SM et al. (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 19, 1264–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lin EY et al. (2002) The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J. Mammary Gland Biol. Neoplasia 7, 147–162 [DOI] [PubMed] [Google Scholar]
- 94.Lin EY et al. (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med 193, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qian BZ et al. (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Halin S et al. (2009) Extratumoral macrophages promote tumor and vascular growth in an orthotopic rat prostate tumor model. Neoplasia 11, 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin EY et al. (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66, 11238–11246 [DOI] [PubMed] [Google Scholar]
- 98.Kratochvill F et al. (2015) TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep. 12, 1902–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yan D et al. (2019) Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front. Immunol 10, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fridlender ZG et al. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nozawa H et al. (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. U. S. A 103, 12493–12498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Verbeke H et al. (2011) Isotypic neutralizing antibodies against mouse GCP-2/CXCL6 inhibit melanoma growth and metastasis. Cancer Lett. 302, 54–62 [DOI] [PubMed] [Google Scholar]
- 103.Coffelt SB et al. (2015) IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang TT et al. (2017) Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut 66, 1900–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rice CM et al. (2018) Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun 9, 5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trabanelli S et al. (2017) Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun 8, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chevalier MF et al. (2017) ILC2-modulated T cell-to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig 127, 2916–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Long A et al. (2018) Type 2 innate lymphoid cells impede IL-33-mediated tumor suppression. J. Immunol 201, 3456–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wagner M et al. (2020) Tumor-derived lactic acid contributes to the paucity of intratumoral ILC2s. Cell Rep. 30, 2743–2757 e5 [DOI] [PubMed] [Google Scholar]
- 110.De Monte L et al. (2016) Basophil recruitment into tumor-draining lymph nodes correlates with Th2 inflammation and reduced survival in pancreatic cancer patients. Cancer Res. 76, 1792–1803 [DOI] [PubMed] [Google Scholar]
- 111.DeNardo DG et al. (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kawasaki T and Kawai T (2014) Toll-like receptor signaling pathways. Front. Immunol 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang Z et al. (2017) Alarmins and immunity. Immunol. Rev 280, 41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gong T et al. (2020) DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol 20, 95–112 [DOI] [PubMed] [Google Scholar]
- 115.Gerbe F et al. (2011) Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol 192, 767–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.von Moltke J et al. (2016) Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smith KA et al. (2018) Concerted IL-25R and IL-4Ralpha signaling drive innate type 2 effector immunity for optimal helminth expulsion. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Howitt MR et al. (2016) Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gerbe F et al. (2016) Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McGinty JW et al. (2020) Tuft-cell-derived leukotrienes drive rapid anti-helminth immunity in the small intestine but are dispensable for anti-protist immunity. Immunity 52, 528–541 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ualiyeva S et al. (2020) Airway brush cells generate cysteinyl leukotrienes through the ATP sensor P2Y2. Sci. Immunol 5, eaax7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nadjsombati MS et al. (2018) Detection of succinate by intestinal tuft cells triggers a type 2 innate immune circuit. Immunity 49, 33–41 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schneider C et al. (2018) A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 174, 271–284 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo XC et al. (2019) Infection by the parasitic helminth Trichinella spiralis activates a Tas2r-mediated signaling pathway in intestinal tuft cells. Proc. Natt. Acad. Sci. U. S. A 116, 5564–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kool M et al. (2011) An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity 34, 527–540 [DOI] [PubMed] [Google Scholar]
- 126.Denney L et al. (2015) Pulmonary epithelial cell-derived cytokine TGF-beta1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity 43, 945–958 [DOI] [PMC free article] [PubMed] [Google Scholar]