

Abstract
Dense packing is a universal tendency of organic molecules in the solid state. Typical porous crystals utilize reticular strong intermolecular bonding networks to overcome this principle. Here, we report a solvophobicity-based methodology for assembling discrete molecules into a porous form and succeed in synthesizing isostructural porous polymorphs of an amphiphilic aromatic molecule Py6Mes. A computational analysis of the crystal structure reveals the major contribution of dispersion interaction as the driving force for assembling Py6Mes into a columnar stacking while the columns are sterically salient and form nanopores between them. The porous packing is facilitated particularly in solvents with weak dispersion interaction due to the solvophobic effect. Conversely, solvents with strong dispersion interaction intercalate between Py6Mes due to the solvophilic effect and provide non-porous inclusion crystals. The solvophobicity-directed polymorphism is further corroborated by the polymorphs of Py6Mes-analogues, m-Py6Mes and Ph6Mes.
Subject terms: Crystal engineering, Porous materials, Molecular self-assembly
Porous organic crystals can be challenging to prepare, because in the absence of guests, they often collapse into denser, non-porous polymorphs. Here the formation of porous and non-porous polymorphs in different solvents is shown to depend on a solvophobic effect driven by dispersion interactions.
Introduction
Organic molecules tend to form a dense crystal with minimal void volume so that the molecules therein can maximize the intermolecular interactions between the adjacent molecules1–3. The synthesis of a porous crystal thus requires a tailored molecular design to overcome this universal tendency. To this end, established porous crystals, such as metal–organic frameworks, typically employ organic linkers featuring multiple adhesive functional groups that can bind with each other to form a reticular framework4–8.
A fundamental question here is whether it is really unfeasible to assemble nonfunctional discrete molecules in a sparse manner. Although this question appears contradictory to the above-described tendency toward dense packing, there actually exist a handful of successful examples9–20. Organic zeolites are a well-known class of such compounds that can uptake/release guest solvent molecules efficiently, and selectively depending on the geometry and chemical affinity, yet organic zeolites are not truly porous materials because their pores readily collapse upon removing the guests21–24. More recently, several organic crystals that can retain vacant pores have been developed9–20. These compounds spontaneously assemble into a porous packing despite the fact that the packing is sustained only by weak interactions, including C–H···X bonds, π–π stacking, halogen bonds, and van der Waals (vdW) forces, whose bonding strength are much less than the conventional hydrogen bonding (15–60 kJ mol−1)25. These porous molecular crystals are intriguing not only fundamentally but also practically because of their distinct solution processability, structural flexibility, and self-healing ability, which are largely prohibited in the conventional porous crystals17–20. However, it still remains unexplored how one can drive the discrete molecules to assemble sparsely1,26. Moreover, with the existing compounds, crystallization solvent and procedure have to be carefully designed. Otherwise, the porous molecular crystals readily collapse into a densely packed polymorph, which further prohibits their development. In fact, most of the reported stable porous molecular crystals were found by chance except those composed of intrinsically porous molecular cages27–29.
Previously, we reported a porous molecular crystal Pyopen·MeCN composed of a D3h-symmetric amphiphilic aromatic compound Py6Mes (Fig. 1a)18,20. Py6Mes assembled together via multiple C–H···N bonds to form a molecular framework with one-dimensional micropores (Fig. 2a), which can maintain its porous architecture up to 202 °C. Although further heating ended up with the collapse of the pores, the resultant nonporous polymorph Pyclose spontaneously self-healed back into Pyopen·MeCN upon exposure to vapor of MeCN at ambient temperature. We anticipated that Pyopen·MeCN, featuring excellent thermal stability and compositional simplicity, could serve as a highly promising platform for investigating how discrete molecules assemble into a porous form. Along this line, we also reported, in the previous report, a plausible molecular assembly mechanism for Pyopen based on its four types of polymorphs18. However, the available crystallographic data were limited at that period and, thus, we could not establish a reliable and general design strategy toward porous molecular crystals.
Fig. 1. Schematic representations of the δD-dependent polymorphism of Py6Mes and m-Py6Mes.

a Molecular structures of Py6Mes and m-Py6Mes with their polar peripheries and nonpolar cores highlighted in violet and gray. b Crystal packing diagrams of polymorphs of Py6Mes and m-Py6Mes. Hansen dispersion cohesion parameters (δD) of the crystallization solvents are given in the parentheses.
Fig. 2. Computational analysis of the attractive and repulsive energies in Pyopen·MeCN.

a A crystal packing diagram of Pyopen·MeCN viewed along the crystallographic b-axis. b A partial crystal packing of Pyopen·MeCN highlighting the polar shell and nonpolar core. c A columnar stacking of Py6Mes in Pyopen·MeCN. d Electrostatic energy (EES), charge transfer energy with higher-order mixed terms energies (ECT + mix), and dispersion energy (EvdW) exserted along the crystallographic a- (red), b- (green), and c-axes (blue) in Pyopen·MeCN.
Here, we report a molecular strategy for synthesizing isomorphic porous molecular crystals from various organic solvents. Through a detailed computational investigation, we reveal the major contribution of dispersion force in the assembling process of the constituent Py6Mes molecules into a porous manner. Following this understanding, we crystalize Py6Mes and succeed in synthesizing porous polymorphs in solvents with less dispersion interaction due to the solvophobic effect (Fig. 1b). In contrast, solvents with larger dispersion interaction provide nonporous inclusion crystals due to the solvophilic effect. Newly synthesized Py6Mes analogs, m-Py6Mes (Fig. 1a) and Ph6Mes, also show consistent solvophobicity-directed polymorphism.
Results and discussion
Energy decomposition analysis of Pyopen·MeCN
To gain insight into how a discrete molecule assembles into a porous form, we focus on Pyopen·MeCN, whose crystal structure was previously identified18. In Pyopen·MeCN, Py6Mes stacks with each other to form a one-dimensional column along the twofold screw axis (crystallographic b-axis, Fig. 2c). The polar pyridine rings and the nonpolar benzene and mesitylene rings are spatially segregated in the column to form a polar shell and a nonpolar interior (Fig. 2b). We conduct a computational calculation of the intermolecular interactions between the adjacent Py6Mes molecules in Pyopen·MeCN. Pair interaction energy decomposition analysis (PIEDA)30 is performed for this purpose by using the fragment molecular orbital (FMO)31 method at the RI-MP2 level of theory with 6–31 + G(d) basis set (Supplementary Fig. 23 and Supplementary Table 15, see “Methods” for the details of the computational methods). A negative value represents an attractive interaction. EvdW, EES, and ECT + mix respectively represent the vdW dispersion energy, electrostatic energy, and charge transfer energy with higher-order mixed term energies. Despite the sparse and porous structure, the overall interaction between Py6Mes is relatively large (−94.3 kcal mol−1, Supplementary Table 15), explaining the excellent thermal stability of Pyopen·MeCN. The prominent energetic gain along the crystallographic b-axis (Fig. 2d) indicates the preferential formation of the columnar stacking of Py6Mes (Fig. 2c) at the expense of the energetic gain obtained from the intercolumnar packing along the crystallographic a- and c-axes. As expected from the richness of C–H···N bond, dispersion interaction is the major attractive contribution in the crystal, especially along the crystallographic a- and b-axes (Fig. 2d). Electrostatic interaction as well as dispersion interaction is prominent along the crystallographic c axis (Fig. 2d). Altogether, dispersion interaction occupies 60.9% of the total attractive energy of –134.3 kcal mol−1 in Pyopen·MeCN (Supplementary Table 15).
Subsequently, we conducted computational investigation into the effect of polarity of the crystallization solvents, which has been considered as an essential parameter for predicting the polymorphism. We calculate the total system energy of Pyopen on the assumption that the constituent Py6Mes molecules are surrounded by MeOH, CHCl3, acetone, toluene, and dichloroethane, respectively, which are available in GAMESS program32. As summarized in Supplementary Table 16, the porous architecture is stabilized more as the polarity of the surrounding solvent increases, yet the change in stabilization energy from the surrounding environment estimated by the polarized continuum model method is relatively small in comparison with the energetic gain from dispersion force. Overall, the porous assembly of Py6Mes is sustained dominantly by the dispersion forces together with the stabilization by the polarity of the surrounding media.
Crystallographic analysis of polymorphs of Py6Mes
Based on the understanding obtained from the calculation, we crystalize Py6Mes from a series of common organic solvents, and analyze their crystal structures with the aim to control the intra- and intercolumnar stacking of Py6Mes. As a typical recrystallization procedure, saturated solution of Py6Mes is poured into a small glass vial, which is loosely sealed with a cap and stood at 25 °C for several days to allow the mother solvent to sluggishly evaporate. In the previous report, we utilized MeCN, 2-propanol (iPA), tetrahydrofuran (THF), and CHCl3 as the crystallization solvents of Py6Mes. Here, we additionally utilize MeOH, EtOH, butyronitrile (BN), EtOAc, acetone, 1-chloropropane (PrCl), 1-butanol (BuOH), toluene, CH2Cl2, dimethylsulfoxide (DMSO), and γ-butyrolactone (GBL) as the crystallization solvents.
Some of the non-protic solvents (EtOAc, CH2Cl2, and toluene) successfully give crystalline precipitates of Py6Mes that are applicable for the single-crystal X-ray diffraction structure analysis. Crystallographic information and the symbols of the resultant crystals are summarized in Table 1 and Supplementary Tables 1–3. Highly polar protic solvents (MeOH and EtOH) are inappropriate for the crystallization due to their poor solubility. Other solvents yield fine crystalline powders, which are analyzed by PXRD.
Table 1.
Crystallographic information of the polymorphs of Py6Mes.
| Crystallization solvent | Symbol | Space group | ε | δD (MPa0.5) | Cell volume (Å3) | Volume per Py6Mes (Å3) | C–H···N per Py6Mes | C–H···π per Py6Mes | Sum of the contacts |
|---|---|---|---|---|---|---|---|---|---|
| MeCN | Pyopen•MeCN | P21/c | 37.5 | 15.3 | 5273 | 1318 | 5 | 2 | 7 |
| EtOAc | Pyopen•EtOAc | P21/c | 6.02 | 15.8 | 5404 | 1351 | 6 | 1 | 7 |
| iPA | Pyopen•iPA | P21/c | 18.3 | 15.8 | 5278 | 1320 | 5 | 2 | 7 |
| THF | PyVDW•THF | P21/n | 7.52 | 16.8 | 5252 | 1313 | 1 | 1 | 2 |
| CHCl3 | PyVDW•CHCl3 | P21/n | 4.81 | 17.8 | 4983 | 1246 | 2 | 2 | 4 |
| Toluene | PyVDW•C7H8 | P-1 | 2.38 | 18.0 | 4872 | 1218 | 4 | 2 | 6 |
| CH2Cl2 | PyVDW•CH2Cl2 | C2/c | 9.08 | 18.2 | 4813 | 1203 | 4 | 0 | 4 |
The space groups, cell volumes, and the number of egoistic C–H···N and C–H···π contacts per one Py6Mes molecule found in Pyopen·MeCN18, Pyopen·EtOAc, Pyopen·iPA18, PyVDW·THF18, PyVDW·CH2Cl2, PyVDW·CHCl318, and PyVDW·C7H8 together with the sum of the contacts. The relative permittivity (ε)37 and the Hansen dispersion cohesion parameters (δD)34 of the crystallization solvents are also listed.
The single crystal obtained from EtOAc (Pyopen·EtOAc) features a porous molecular packing that is virtually identical with Pyopen·MeCN and Pyopen·iPA (Fig. 2a and Supplementary Fig. 9). Pore size distribution of Pyopen·EtOAc calculated from its N2 adsorption isotherm profile (Supplementary Fig. 22) is nearly identical with that of Pyopen·MeCN18, while its BET surface area (597 m2 g−1) is larger than Pyopen·MeCN plausibly due to the higher structural integrity of Pyopen·EtOAc crystals. Trials to assign the guest solvent molecules trapped inside the pore are unsuccessful for all the porous crystals obtained from MeCN, iPA, and EtOAc (Pyopen·MeCN, Pyopen·iPA, and Pyopen·EtOAc) due to the severe disorder. Some residual electron density is detected in the pores according to the SQUEEZE program33 (66, 52, and 53 electrons for MeCN, iPA, and EtOAc, respectively), which indicates the inclusion of certain amount of the crystallization solvent molecules in the pores.
Inclusion molecular crystals PyVDW·CH2Cl2 and PyVDW·C7H8 are obtained, respectively, from CH2Cl2 and toluene. PyVDW·CH2Cl2 (Fig. 3a and Supplementary Fig. 10) belongs to a space group of C2/c, in which eight non-disordered CH2Cl2 molecules pack together with four molecules of Py6Mes in a unit cell. The H atoms in CH2Cl2 make a short contact with a N atom in Py6Mes (2.594 Å; Fig. 3b). Py6Mes molecules form C–H···N contacts (2.583 and 2.500 Å) with each other along with several C–H···H contacts. PyVDW·C7H8 (Fig. 3c and Supplementary Fig. 11) belongs to a space group of P-1, in which four toluene molecules pack together with four Py6Mes molecules in a unit cell. The guest toluene molecules form C–H···π contacts and a π–π stacking with Py6Mes (Fig. 3d, e). Py6Mes molecules form eight C–H···N contacts with each other along with C–H···π contacts and π–π stackings.
Fig. 3. Crystal packing diagrams of the polymorphs of Py6Mes and m-Py6Mes.

a, c, f, h, j Crystal packing diagrams of PyVDW·CH2Cl2 (a), PyVDW·C7H8 (c), m-PyVDW·iPA (f), m-PyVDW·CHCl3 (h), and m-PyVDW·MeCN (j). b, d, e, g, i Partial crystal structures of PyVDW·CH2Cl2 (b), PyVDW·C7H8 (d,e), m-PyVDW·iPA (g), and m-PyVDW·CHCl3 (i). The solvent–solute interactions are visualized with red dashed lines. The guest toluene molecules are colored in orange for clarity.
Precipitates obtained in other solvents are analyzed by PXRD due to the difficulty in synthesizing diffraction-quality single crystals (Supplementary Fig. 8). BN solution of Py6Mes affords a porous crystal that is isomorphic to Pyopen, while the crystals obtained from other solvents (acetone, PrCl, BuOH, DMSO, and GBL) are not isomorphic to Pyopen. The single-crystal structures, PXRD profiles, and the physical properties of the crystallization solvents (relative permittivity and Hansen parameters34) are summarized in Supplementary Table 14 along with those of mesitylene, benzene, and pyridine as the components of Py6Mes.
It is worth noting that isomorphic porous crystals were obtained from a variety of solvents (MeCN, iPA, BN, and EtOAc) that are seemingly irrelevant with each other, in terms of the polarity or hydrogen bonding capability. This is in clear contrast with the previously reported porous molecular crystals that were basically sensitive to the crystallization solvents or crystallization procedures1.
Hansen solubility parameter and polymorphism
Relative permittivity (ε) has often been regarded as the primal parameter for the prediction of the solute–solvent interactions. However, the tendency in polymorphism of Py6Mes toward ε is indistinct (Supplementary Table 14). We presume that, based on the results from the computational analysis, the capability of forming the dispersion interaction may govern the polymorphism. To prove this theory, we focus on Hansen parameters, which are the empirical values of the strength of dispersion (δD), polar (δP), and hydrogen bonding cohesion parameters (δH). Besides the conventional utility for the prediction of the solubility of organic polymers, Hansen parameters have recently been applied for the prediction of polymorphism of some pharmaceutical molecules35,36.
We apply this method to the polymorphism of Py6Mes. The crystallization solvents and Py6Mes components are plotted in the Hansen space according to their three coordinates of δD, δP, and δH (Fig. 4 and Supplementary Table 14). The Py6Mes components (green spheres in Fig. 4a) feature large δD and relatively small δP and δH. MeCN, BN, EtOAc, and iPA (red spheres in Fig. 4a) feature small δD and moderate or large δP and δH. Highly polar solvents (MeOH and EtOH, black spheres in Fig. 4a) locate at the opposite corner from the Py6Mes components. The other solvents (blue spheres in Fig. 4a) locate between the red and green spheres.
Fig. 4. Hansen space for polymorphs of Py6Mes.
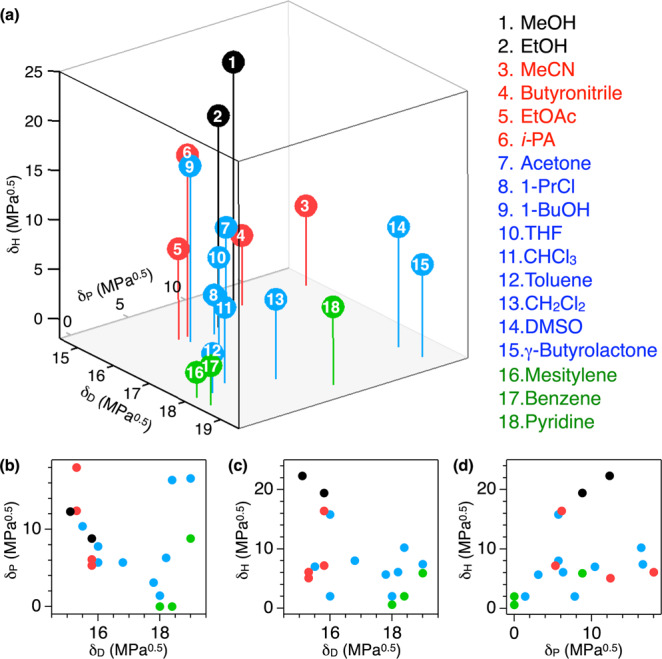
a Hansen space showing the polymorphism of Py6Mes. The solvents that poorly dissolve Py6Mes, and that afford porous crystals are respectively colored in black and red. The components of Py6Mes are colored in green. The other solvents are colored in blue. b–d The projections of the Hansen space onto the δDδP- (b), δDδH- (c), and δPδH-planes (d).
The geometrical distance in the Hansen space represents the solubility or affinity of given two substances. In line with this conventional understanding, the plot in Fig. 4a shows an explicit dependence on the distance from the Py6Mes components. Solvents that are close to the Py6Mes components yield nonporous polymorphs (blue spheres in Fig. 4a), while solvents that locate far from the Py6Mes components are poor in solubility (black spheres in Fig. 4a). The remaining slightly affinitive solvents yield the porous crystals (red spheres in Fig. 4a).
This trend can be decomposed into the basic elements by focusing on the projections of the Hansen space onto the δDδP-, δDδH-, and δPδH-planes (Fig. 4b–d). As shown in Fig. 4d, the polymorphic tendency barely correlates with δP and δH of the crystallization solvents. On the other hand, δD describes the polymorphic tendency reasonably (Fig. 4b, c and Supplementary Table 14), namely, Py6Mes crystalizes into the porous form when the crystallization solvent can partially dissolve Py6Mes, but is not affinitive with Py6Mes especially in terms of the strength of the dispersion force.
We also analyze the intermolecular short contacts and crystal packing efficiency of the single crystals, with the aim to reveal the detailed solute–solvent interactions. The egoistic C–H···N and C–H···π contacts per one Py6Mes molecule are summarized in Table 1. In the solvents with small δD, Py6Mes facilitates multiple C–H···N and C–H···π contacts with each other, while solvents with large δD suppress the egoistic contacts by intercalating between Py6Mes molecules as shown in PyVDW·CH2Cl2 and PyVDW·C7H8 (Fig. 3a, c). At the same time, the inclusion of the solvent molecules optimizes the molecular packing of Py6Mes. The averaged cell volume per one Py6Mes molecule of the three porous crystals (Pyopen·MeCN, Pyopen·EtOAc, and Pyopen·iPA) and the four inclusion crystals (PyVDW·THF, PyVDW·CH2Cl2, PyVDW·CHCl3, and PyVDW·C7H8) are 1330 and 1245 Å3, respectively (Table 1). Namely, solvents with large δD are affinitive with Py6Mes and allow the Py6Mes to assemble into a dense packing by intercalating between Py6Mes (solvophilic crystallization), while solvents with small δD are segregated from Py6Mes due to the solvophobicity and facilitate the columnar assembly of Py6Mes although the intercolumnar packing is not dense (solvophobic crystallization).
Polymorphism of m-Py6Mes and Ph6Mes
The δD-dependent solvophilic/solvophobic crystallization is further corroborated by the polymorphs of m-Py6Mes and Ph6Mes (Figs. 1a, b and 5). Meta-substituted hexapyridyl mesitylene derivative m-Py6Mes was newly synthesized by sequential Suzuki–Miyaura couplings of pyridineboronic acid, dibromo aniline, and triiodomesitylene (for details, see Supplementary Methods). Tri(terphenyl) mesitylene, Ph6Mes, was newly synthesized by Suzuki–Miyaura coupling reaction of terphenylboronic acid and triiodomesitylene (for details, see Supplementary methods). The molecular structure of the resultant m-Py6Mes and Ph6Mes are unambiguously assigned by means of 1H- and 13C-NMR spectroscopies, elemental analysis, and high-resolution mass spectrometry (Supplementary Figs. 1–7).
Fig. 5. Crystal packing diagrams of the polymorphs of Ph6Mes.

a Molecular structure of Ph6Mes. b–g Crystal packing diagrams of PhVDW·MeCN (b), PhVDW·EtOAc (c), PhVDW·THF (d), PhVDW·CHCl3 (e), PhVDW·C7H8 (f), and PhVDW·CH2Cl2 (g). The guest toluene molecules are colored in orange for clarity.
We crystalize m-Py6Mes in the same way as Py6Mes in MeCN, EtOAc, iPA, and CHCl3 to obtain diffraction-quality single crystals. Their crystal packing diagrams and crystal structure information are shown in Fig. 3f–j and Table 2, respectively. Nonporous inclusion crystals are obtained when solvents with large δD (iPA and CHCl3) are utilized (Fig. 3f, h, and Supplementary Figs. 14 and 15). The inclusion crystal obtained from iPA (m-PyVDW·iPA) belongs to a space group of P-1. The constituent m-Py6Mes molecules form multiple C–H N and C–H···π contacts with each other along with a solvophilic C–H···N contact with a guest iPA molecule (Fig. 3g). The inclusion crystal with CHCl3 (m-PyVDW·CHCl3) belongs to a space group of P-1. The constituent m-Py6Mes molecules form three C–H···N bonds with each other along with solvophilic C–H···N (Fig. 3i) and C–H···π contacts with guest CHCl3 molecules.
Table 2.
Crystallographic information of the polymorphs of m-Py6Mes.
| Crystallization solvent | Symbol | ε | δD (MPa0.5) | Space group | Cell volume (Å3) | Volume per Py6Mes (Å3) |
|---|---|---|---|---|---|---|
| MeCN | m-PyVDW•MeCN | 37.5 | 15.3 | P-1 | 4274 | 1068 |
| EtOAc | m-PyVDW•EtOAc | 6.02 | 15.8 | P-1 | 4205 | 1051 |
| iPA | m-PyVDW•iPA | 18.3 | 15.8 | P-1 | 2552 | 1276 |
| CHCl3 | m-PyVDW•CHCl3 | 4.81 | 17.8 | P21/n | 5079 | 1270 |
The crystals obtained from MeCN (m-PyVDW·MeCN) and EtOAc (m-PyVDW·EtOAc) are isomorphic with each other, featuring no apparent pores or guest solvent molecules (Fig. 3j, and Supplementary Figs. 12 and 13). The crystal packing mode is basically analogous to the nonporous polymorph Pyclose, which is obtained by thermal annealing of Pyopen (see our previous report18 for the detailed synthetic and structural information). In m-PyVDW·MeCN, the constituent m-Py6Mes molecules are solvophobically packed together to form five C–H···π contacts with each other in a unit cell. Moreover, 11 out of 12 pyridine rings in a unit cell form C–H···N bonds with the adjacent m-Py6Mes molecules.
Ph6Mes is analogously crystalized in MeCN, EtOAc, THF, CHCl3, toluene and CH2Cl2, successfully yielding diffraction-quality single crystals, whose crystal packing diagrams and crystal structure information are shown in Fig. 5b–g, Table 3, Supplementary Figs. 16–21, and Supplementary Tables 8–13, respectively. In analogy with m-Py6Mes, nonporous inclusion crystals are obtained when solvents with large δD (EtOAc, THF, CHCl3, toluene, and CH2Cl2) are utilized (Fig. 5c–g and Supplementary Figs. 17–21), while crystals from MeCN include no guest solvent molecules (Fig. 5b and Supplementary Fig. 16).
Table 3.
Crystallographic information of the polymorphs of Ph6Mes.
| Crystallization solvent | Symbol | ε | δD (MPa0.5) | Space group | Cell volume (Å3) | Volume per Py6Mes (Å3) |
|---|---|---|---|---|---|---|
| MeCN | PhVDW•MeCN | 37.5 | 15.3 | P-1 | 4348 | 1087 |
| EtOAc | PhVDW•EtOAc | 6.02 | 15.8 | P-1 | 2475 | 1238 |
| THF | PhVDW•THF | 7.52 | 16.8 | C2/c | 5207 | 1302 |
| CHCl3 | PhVDW•CHCl3 | 4.81 | 17.8 | C2/c | 5209 | 1302 |
| Toluene | PhVDW•C7H8 | 2.38 | 18.0 | P-1 | 2471 | 1236 |
| CH2Cl2 | PhVDW•CH2Cl2 | 9.08 | 18.2 | P-1 | 2464 | 1232 |
Polymorphs of m-Py6Mes not only corroborate the δD-dependency of Py6Mes polymorphs, but also tell us about the delicate energetic balance between Pyclose and Pyopen·MeCN. Geometrically, Py6Mes can assemble into a dense packing as proved by Pyclose, m-PyVDW·MeCN, or m-PyVDW·EtOAc. However, unlike m-Py6Mes, the position of the N atoms is static upon the rotation of the pyridyl rings around the single bond, which is unfavorable for the formation of multiple C–H···N bonds with each other. Therefore, Py6Mes may prefer to form a porous framework, in which Py6Mes can form multiple C–H···N and C–H···π contacts with each other at the expense of the packing efficiency.
Conclusion
In conclusion, we succeed in establishing a solvophobicity-based design strategy for the synthesis of porous molecular crystals and succeed in synthesizing porous molecular crystals by using various organic solvents. Energy decomposition analysis reveals the dominance of the dispersion energy as the attractive interaction in Pyopen·MeCN especially in the columnar stacking, which is further stabilized by the polarity of the solvent. Consistently, solvents with small δD facilitate the egoistic assembly of Py6Mes into a porous architecture via solvophobic interaction, while solvents with large δD intercalate between Py6Mes via solvophilic interaction and provide nonporous inclusion polymorphs. The dominance of the dispersion energy as the attractive interaction in Pyopen·MeCN is further supported by the polymorphism of m-Py6Mes and Ph6Mes. The combination of dispersion interaction as attractive force and solvophobicity as repulsive force, as presented in this paper, can be a conceptionally novel strategy to go beyond the conventional porous crystal engineering that largely relies on the strong affinitive bonding networks.
Methods
Materials
Commercial reagents were purchased from Sigma-Aldrich, TCI, and Wako Pure Chemical Industries, Ltd. All the chemicals are used as received unless otherwise mentioned.
Reaction, purification, and characterization techniques
All reactions were carried out under nitrogen atmosphere unless otherwise noted. Gel permeation column chromatography was performed on a Japan Analytical Industry model LC-9110 NEXT Recycling Preparative HPLC equipped with JAIGEL 2HH, by using CHCl3 as eluent. 1H and 13C NMR spectra were recorded on a JEOL model JNM-ECS-400 NMR spectrometer (1H NMR, 400 MHz, 13C NMR, 100 MHz) JMTC-400/54/SS and a Bruker model AVANCE-600 NMR spectrometer (13C NMR, 150 MHz), using the residual solvent peak as an internal standard. High-resolution MS data were obtained using a Bruker model solariX XR Mass spectrometry in the positive mode with MeCN as solvent. Elemental analysis was conducted with an Elementar model organic elemental analyzer UNICUBE. The sorption isotherm measurement for N2 (99.99995%) was performed using a Bel Japan, Inc. model BELSORP-max automatic volumetric adsorption apparatus. A known amount of Pyopen·EtOAc, placed in a glass tube, was dried under a reduced pressure at 110 °C for 3 h to remove the included guest molecules.
Typical procedure for the synthesis of single crystals of Py6Mes, m-Py6Mes, and Ph6Mes
A glass vial containing saturated solution of Py6Mes, m-Py6Mes, or Ph6Mes was placed at 25 °C with a cap loosely fastened to allow the solvent to evaporate sluggishly until some precipitates emerged. The precipitates were poured onto paraffin oil and were picked up by a loop.
Computational analysis
The FMO method31 using the second-order Møller–Plesset perturbation theory (MP2) with the resolution-of-the-identity (RI) approximation was used to elucidate the insight into the intermolecular energy between contact pairs of Py6Mes. Firstly, each molecule of Py6Mes was divided into four molecular fragments: F1, F2, F3 (1,3-di(pyridin-4-yl)benzene), and F4 (mesitylene) as shown in Supplementary Fig. 23c. The geometry optimization was then performed with the standard 6–31 + G(d) basis set implemented in GAMESS program package32. The molecular coordinates remained the same as the initial structure during the FMO calculation. Among the eight fragments of the contact pairs of Py6Mes (Supplementary Fig. 23a, b), any two fragments (I and J) were subjected to the calculation of the interaction energy decomposition analysis (PIEDA, Supplementary Table 15)30. The total of the contributed energy terms (Etotal)) is given in Eq. (1).
| 1 |
where EES is the classical electrostatic energy between Py6Mes, ECT + mix is the charge transfer energy with higher-order mixed terms energies, EvdW is the vdW dispersion energy, and EEX is the exchange repulsion between the adjacent fragments.
The total of the attractive energies (Eatt) is given in Eq. 2.
| 2 |
The total system energies of Pyopen•MeCN in a series of organic solvents with different relative permittivity ε are calculated by the conductor-like polarizable continuum model method.
Supplementary information
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Young Scientist (JP19K15334) from Japan Society for the Promotion of Science (JSPS), Kato Foundation for Promotion of Science, and The Kao Foundation for Arts and Sciences, and the Nanotechnology Platform project from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Author contributions
H.Y. designed the experiments. H.Y., M.T., and K.I. conducted the organic synthesis, crystallization, and characterizations. K.H. and Y.S. conducted the computational calculations. H.Y., M.T., and H.S. conducted single-crystal X-ray structural analysis. H.Y. and Y.Y. analyzed the data and prepared the manuscript with the feedback from the other authors.
Data availability
The data that support the findings in this study are available within the article and its Supplementary Information and/or from the corresponding authors on reasonable request. The X-ray crystallographic data reported in this article is deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers of 2072485–2072491 and 2095190–2095195. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-021-00561-8.
References
- 1.McKeown NB. Nanoporous molecular crystals. J. Mater. Chem. 2010;20:10588–10597. doi: 10.1039/c0jm01867h. [DOI] [Google Scholar]
- 2.Holst JR, Trewin A, Cooper AI. Porous organic molecules. Nat. Chem. 2010;2:915–920. doi: 10.1038/nchem.873. [DOI] [PubMed] [Google Scholar]
- 3.Cooper AI. Molecular organic crystals: from barely porous to really porous. Angew. Chem. Int. Ed. 2012;51:7892–7894. doi: 10.1002/anie.201203117. [DOI] [PubMed] [Google Scholar]
- 4.Kitagawa S, Kitaura R, Noro S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004;43:2334–2375. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]
- 5.Waller PJ, Gandara F, Yaghi OM. Chemistry of covalent organic frameworks. Acc. Chem. Res. 2015;48:3053–3063. doi: 10.1021/acs.accounts.5b00369. [DOI] [PubMed] [Google Scholar]
- 6.Lin RB, et al. Multifunctional porous hydrogen-bonded organic framework materials. Chem. Soc. Rev. 2019;48:1362–1389. doi: 10.1039/C8CS00155C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stock N, Biswas S. Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites. Chem. Rev. 2012;112:933–969. doi: 10.1021/cr200304e. [DOI] [PubMed] [Google Scholar]
- 8.Rowsell JLC, Yaghi OM. Metal-organic frameworks: a new class of porous materials. Microporous Mesoporous Mater. 2004;73:3–14. doi: 10.1016/j.micromeso.2004.03.034. [DOI] [Google Scholar]
- 9.Allcock HR. Cyclophosphazene clathrates - exploring adjustable tunnel. Acc. Chem. Res. 1978;11:81–87. doi: 10.1021/ar50123a001. [DOI] [Google Scholar]
- 10.Sozzani P, Bracco S, Comotti A, Ferretti L, Simonutti R. Methane and carbon dioxide storage in a porous van der waals crystal. Angew. Chem. Int. Ed. 2005;44:1816–1820. doi: 10.1002/anie.200461704. [DOI] [PubMed] [Google Scholar]
- 11.Msayib KJ, et al. Nitrogen and hydrogen adsorption by an organic microporous crystal. Angew. Chem. Int. Ed. 2009;48:3273–3277. doi: 10.1002/anie.200900234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bezzu CG, Helliwell M, Warren JE, Allan DR, McKeown NB. Heme-like coordination chemistry within nanoporous molecular crystals. Science. 2010;327:1627–1630. doi: 10.1126/science.1184228. [DOI] [PubMed] [Google Scholar]
- 13.McHale CM, Stegemoller CR, Hashim MI, Wang X, Miljanić OŠ. Porosity and guest inclusion in cyclobenzoin esters. Cryst. Growth Des. 2019;19:562–567. doi: 10.1021/acs.cgd.8b01774. [DOI] [Google Scholar]
- 14.Reichenbächer K, et al. Improved thermal stability of an organic zeolite by fluorination. J. Incl. Phenom. Macrocycl. Chem. 2008;61:127–130. doi: 10.1007/s10847-007-9404-2. [DOI] [Google Scholar]
- 15.Soldatov DV, Enright GD, Ripmeester JA. Polymorphism and pseudopolymorphism of the [Ni(4-methylpyridine)4(NCS)2] Werner complex, the compound that led to the concept of "organic zeolites". Cryst. Growth Des. 2004;4:1185–1194. doi: 10.1021/cg030062b. [DOI] [Google Scholar]
- 16.Nakajima S, et al. A fluorescent microporous crystalline dendrimer discriminates vapour molecules. Chem. Commun. 2018;54:2534–2537. doi: 10.1039/C7CC09342J. [DOI] [PubMed] [Google Scholar]
- 17.Baroncini M, et al. Photoinduced reversible switching of porosity in molecular crystals based on star-shaped azobenzene tetramers. Nat. Chem. 2015;7:634–640. doi: 10.1038/nchem.2304. [DOI] [PubMed] [Google Scholar]
- 18.Yamagishi H, et al. Self-assembly of lattices with high structural complexity from a geometrically simple molecule. Science. 2018;361:1242–1246. doi: 10.1126/science.aat6394. [DOI] [PubMed] [Google Scholar]
- 19.Yamagishi H, et al. Sigmoidally hydrochromic molecular porous crystal with rotatable dendrons. Commun. Chem. 2020;3:118. doi: 10.1038/s42004-020-00364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama A, Kimata H, Marumoto K, Yamamoto Y, Yamagishi H. Facile light-initiated radical generation from 4-substituted pyridine under ambient conditions. Chem. Commun. 2020;56:6937–6940. doi: 10.1039/D0CC02538K. [DOI] [PubMed] [Google Scholar]
- 21.Endo K, et al. Guest-binding properties of organic-crystals having an extensive hydrogen-bonded network - an orthogonal anthracene-bis(resorcinol) derivative as a functional organic analog of zeolites. J. Am. Chem. Soc. 1995;117:8341–8352. doi: 10.1021/ja00137a007. [DOI] [Google Scholar]
- 22.Miyata M, et al. Guest-responsive structural-changes in cholic-acid intercalation crystals. Nature. 1990;343:446–447. doi: 10.1038/343446a0. [DOI] [Google Scholar]
- 23.Tanaka T, Tasaki T, Aoyama Y. Acridinylresorcinol as a self-complementary building block of robust hydrogen-bonded 2D nets with coordinative saturation. Preservation of crystal structures upon guest alteration, guest removal, and host modification. J. Am. Chem. Soc. 2002;124:12453–12462. doi: 10.1021/ja026704l. [DOI] [PubMed] [Google Scholar]
- 24.Miyata M, Sada K, Hori S, Miki K. Intercalation phenomena and polymorphism of cholic acid crystals. Mol. Cryst. Liq. Cryst. 2006;219:71–74. doi: 10.1080/10587259208032118. [DOI] [Google Scholar]
- 25.Steiner T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002;41:49–76. doi: 10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 26.Bernstein J. Polymorphism − a perspective. Cryst. Growth Des. 2011;11:632–650. doi: 10.1021/cg1013335. [DOI] [Google Scholar]
- 27.Tozawa T, et al. Porous organic cages. Nat. Mater. 2009;8:973–978. doi: 10.1038/nmat2545. [DOI] [PubMed] [Google Scholar]
- 28.Jones JT, et al. Modular and predictable assembly of porous organic molecular crystals. Nature. 2011;474:367–371. doi: 10.1038/nature10125. [DOI] [PubMed] [Google Scholar]
- 29.Little MA, et al. Trapping virtual pores by crystal retro-engineering. Nat. Chem. 2014;7:153–159. doi: 10.1038/nchem.2156. [DOI] [PubMed] [Google Scholar]
- 30.Fedorov DG, Kitaura K. Pair interaction energy decomposition analysis. J. Comput. Chem. 2007;28:222–237. doi: 10.1002/jcc.20496. [DOI] [PubMed] [Google Scholar]
- 31.Kitaura K, Ikeo E, Asada T, Nakano T, Uebayasi M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999;313:701–706. doi: 10.1016/S0009-2614(99)00874-X. [DOI] [Google Scholar]
- 32.Schmidt MW, et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993;14:1347–1363. doi: 10.1002/jcc.540141112. [DOI] [Google Scholar]
- 33.Spek AL. Single-crystal structure validation with the program platon. J. Appl. Crystallogr. 2003;36:7–13. doi: 10.1107/S0021889802022112. [DOI] [Google Scholar]
- 34.Hansen C. M. in Hansen Solubility Parameters: A User’s Handbook, Second Edition (CRC, 2007).
- 35.Mohammad MA, Alhalaweh A, Velaga SP. Hansen solubility parameter as a tool to predict cocrystal formation. Int. J. Pharm. 2011;407:63–71. doi: 10.1016/j.ijpharm.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 36.Salem A, Nagy S, Pal S, Szechenyi A. Reliability of the Hansen solubility parameters as co-crystal formation prediction tool. Int. J. Pharm. 2019;558:319–327. doi: 10.1016/j.ijpharm.2019.01.007. [DOI] [PubMed] [Google Scholar]
- 37.Maryott, A. A. & Smith E. R. in Table of Dielectric Constants of Pure Liquids (U. S. Government Printing Office, 1951).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings in this study are available within the article and its Supplementary Information and/or from the corresponding authors on reasonable request. The X-ray crystallographic data reported in this article is deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers of 2072485–2072491 and 2095190–2095195. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.