

Abstract
Background
KRAS is a frequently mutated oncogene in human cancer. Clinical studies on the covalent inhibitors of the KRASG12C mutant have reported promising results. However, primary and acquired resistance may limit their clinical use.
Methods
Sotorasib-resistant cell lines were established. We explored the signalling pathways activated in these resistant cell lines and their roles in sotorasib resistance.
Results
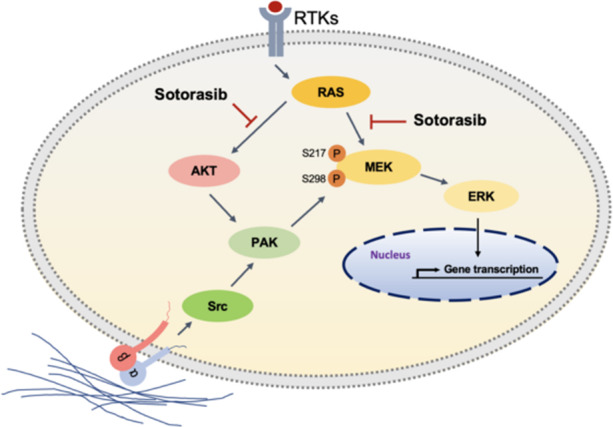
The resistant cells exhibited increased cell–matrix adhesion with increased levels of stress fibres and focal adherens. p21-activated kinases (PAKs) were activated in resistant cells, which phosphorylate MEK at serine 298 of MEK and serine 338 of c-Raf to activate the mitogen-activated protein kinase pathway. The PAK inhibitors FRAX597 and FRAX486 in synergy with sotorasib reduced the viability of KRASG12C mutant cancer cells. Furthermore, the PI3K/AKT pathway was constitutively active in sotorasib-resistant cells. The overexpression of constitutively activated PI3K or the knockdown of PTEN resulted in resistance to sotorasib. PI3K inhibitor alpelisib was synergistic with sotorasib in compromising the viability of KRASG12C mutant cancer cells. Moreover, PI3K and PAK pathways formed a mutual positive regulatory loop that mediated sotorasib resistance.
Conclusions
Our results indicate that the cell–matrix interaction-dependent activation of PAK mediates resistance to sotorasib through the activation of MAPK and PI3K pathways.
Subject terms: Targeted therapies, Oncogenes
Introduction
KRAS is a frequently mutated oncogene in human cancer, with a high mutation frequency in pancreatic ductal adenocarcinoma, colorectal adenocarcinoma, and pulmonary adenocarcinoma [1]. Most KRAS mutations occur in codons 12 and 13, whereas mutations in codons 61 and 146 are less frequent [2]. The RAS protein family members alternate between two different conformational states defined by the binding to nucleotides. In the quiescent state, RAS proteins bind to the nucleotide GDP. Upon upstream signal–induced activation, RAS proteins undergo a conformational change triggered by the exchange of GDP to GTP, which, in turn, activates the downstream pathways, including the MAPK and AKT pathways. RAS proteins have intrinsic GTPase activity, which hydrolyses GTP to GDP to return the RAS protein into a quiescent state [3]. Mutated KRAS proteins have weak associations with extrinsic GTPase-activating proteins. As a consequence, mutated KRAS proteins are trapped in the active GTP-bound state and transduce pro-growth signals continuously.
Because of the high frequency of KRAS mutations in human cancer, enormous efforts have been devoted to exploit the possibility of the direct inhibition of mutated KRAS. However, until recently, no therapeutic agent directly targeting RAS has been proven clinically useful, and mutated KRAS was considered an undruggable target. Thus, many therapeutic strategies have been developed to target downstream effector pathways, such as the MAPK pathway, but until now, these efforts have largely failed due to limited clinical benefits or toxicity [4].
Recently, several covalent inhibitors (ARS-1620, sotorasib [AMG510], adagrasib [MRTX849], JNJ-74699157, and LY3499446) targeting the KRASG12C mutant have been developed [5–7]. These inhibitors depend on mutant cysteine for irreversible binding to the switch II pocket on GDP-KRASG12C, trapping it in the inactive GDP-bound state and inhibiting KRAS-dependent signalling [8]. Several clinical trials are being conducted to evaluate the anticancer effects of the covalent inhibitors of KRASG12C. Early trials using sotorasib and MRTX849 have shown promising results. In the phase 1 study of sotorasib, 20% of the 129 patients with the KRASG12C mutation partially responded [9]. In the lung cancer cohort, 32% (19/59) and 88% (52/59) of the patients exhibited an objective response and disease control, respectively. Patients with colorectal cancer had a lower response to sotorasib; approximately 7.1% (3/42) and 73.8% (31/42) of the patients showed an objective response and disease control, respectively. In the recently published phase 2 trial, an objective response was observed in 46 of 124 lung cancer patients, which included 4 patients with a complete response and 42 patients with a partial response [10]. Therefore, these novel covalent inhibitors may provide unprecedented opportunities to target this key mutation in cancer cells. In 2021, the Food and Drug Administration granted accelerated approval to sotorasib for adult patients with KRASG12C‑mutated locally advanced or metastatic non-small cell lung cancer.
Although these studies have shown promising results, the lack of a response to these therapies and recurrence in some patients indicate that, similar to other molecular targeted therapies, the problem of intrinsic or acquired resistance is inevitably encountered for KRASG12C inhibitors, most likely due to the activation of an alternative pathway or selection of minor drug-resistant mutants. Understanding the molecular pathway of drug resistance may be helpful for developing a novel combination therapy and for maximising the therapeutic potential of drugs. The analysis of post treatment specimens of patients receiving adagrasib monotherapy showed acquired KRAS alterations and other acquired genetic bypass mechanisms in 17 of 38 (45%) patients [11]. By comparing the matched pre- and post-treatment specimens, Zhao et al. discovered genetic alterations in 27 of 43 (62%) patients after sotorasib treatment, including alterations in KRAS, NRAS, BRAF, EGFR, FGFR2, and MYC [12]. Studies have shown that the activation of receptor tyrosine kinase, epithelial–mesenchymal transition, and production of new KRASG12C proteins are possible nongenetic mechanisms for resistance to RASG12C inhibitors [13–16]. In this study, we established cancer cell lines resistant to the KRASG12C inhibitor sotorasib and found activated p21-activated kinase (PAK) and PI3K pathways in resistant cells. Our result suggests that combination treatment with the PAK or PI3K inhibitor may aid in overcoming resistance to sotorasib.
Materials and methods
Cell culture and reagents
Four cell lines harbouring the KRASG12C mutation were used in this study. Pancreatic and colorectal cancer cells MIA PaCa-2 and SW1463, respectively, were obtained from the Bioresource Collection and Research Center (Hsinchu, Taiwan). The lung cancer cell lines LU99 and LU65 were obtained from the RIKEN cell bank (Ibaraki, Japan). All cell lines used in our study were verified through short-tandem repeat profiling, tested negative for mycoplasma contamination, and were passaged in vitro for <3 months after resuscitation. MIA PaCa-2 cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco, Waltham, MA, USA). SW1463 cells was maintained in Leibovitz’s 15 medium (Gibco). The lung cancer cell lines LU99 and LU65 were cultured in RPMI 1640 medium (Gibco). Each medium was supplemented with 10% foetal bovine serum (Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, nonessential amino acids, and 1 mM sodium pyruvate. All cells were incubated at 37 °C in a water-saturated atmosphere of 5% CO2/95% air. Sotorasib-resistant cells were maintained in complete growth medium plus 5 µM sotorasib.
Sotorasib was a gift from Amgen Inc. (Thousand Oaks, CA, USA). FRAX597, FRAX486, alpelisib, ML141, and dasatinib were purchased from MedChemExpress (Monmouth Junction, NJ, USA). NSC23766 was purchased from Sigma-Aldrich (St. Louis, MO, USA). Wortmannin was purchased from Selleckchem (Houston, TX, USA). pHAGE-PIK3CA-E545K and pHAGE-PIK3CA-H1047R were gifted by Gordon Mills & Kenneth Scott (Addgene plasmid #116485 and #116500).
Cell viability assay
To measure cell viability, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) colorimetric assay was performed using MTT (Sigma-Aldrich). Cells (2000 per well) were seeded into 96-well plates and incubated at 37 °C in a humidified atmosphere of 5% CO2. At an appropriate interval, MTT (2 mg/mL in PBS) was added, and the plates were incubated for 4 h. The resultant coloured reaction product, MTT formazan, was extracted with dimethyl sulfoxide; absorbance was measured at 570 nm (n = 6 for each treatment condition).
Western blotting
Protein samples (20–50 μg each) were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then electrotransferred onto nitrocellulose membranes (Amersham, Buckinghamshire, UK). The membranes were incubated with primary and secondary antibodies at optimum dilutions, and immunoreactive signals were detected using an Immobilon Crescendo Western HRP substrate (Millipore, Burlington, MA, USA). The antibodies used and the dilution are listed in Supplementary Table 1.
Immunofluorescence
To observe cytoskeletal actin microfilaments and focal adherens, cells were fixed with 4% paraformaldehyde and permeabilized in cold (4 °C) PBS buffer containing 0.5% Triton X-100 acetone for 10 min. A 5% bovine serum albumin solution was applied to the samples to block nonspecific binding sites for 60 min at room temperature. Then, cells were incubated with anti-vinculin antibody (1:500) (GeneTex, Hsinchu, Taiwan) overnight at 4 °C and further treated with fluorescein isothiocyanate-conjugated (green fluorescence) goat antimouse secondary antibody for 1 h at room temperature (1:500; Millipore). Actin microfilaments were stained with rhodamine-conjugated phalloidin (5 μmol/L, 1:50; Molecular Probes, Eugene, OR, USA) for 60 min at room temperature. Finally, cells were washed with PBS and incubated in PBS buffer containing 4’,6-diamidine-2’-phenylindole dihydrochloride (1:5000; Sigma-Aldrich) for 30 s for nuclear staining. The samples were imaged using a fluorescent microscope.
RNA sequencing and analysis
Total RNA was extracted from cell lines and sequenced according to the manufacturer’s instructions. For mRNA sequencing, RNA quality and quantity were assessed using a Bioanalyzer 2100 system (Agilent, Santa Clara, CA, USA) and a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and RNA was then ligated to an adaptor for further amplification using the TruSeq stranded mRNA kit (Illumina, San Diego, CA, USA). All the library preparation was performed in the Translational Core Facility of Taipei Medical University. The resulting libraries were sequenced using HiSeq 2000 (Illumina, San Diego, CA, USA). After sequencing, quality control of raw sequencing data was performed with FastQC. Reads were mapped to the GRCh37 reference by using STAR, and gene expression was then calculated using RSEM. Differential genes were identified using R package, DESeq2 [17]. Gene Set Enrichment Analysis (GSEA; version 4.0.3) was used to identify key enrichment pathways. To analyse our mRNA sequencing data using GSEA, we ran our expression data set using KEGG pathways (c2.cp.kegg.v7.5.symbols.gmt) gene set databases using the following settings: 1000 permutations; collapse data set to gene symbols, false; permutation type, gene_set; enrichment statistic, weighted; and metric for ranking genes, Signal2Noise. The GSEA results were evaluated according to the following criteria: |NES| > 1 and nominal P value < 0.05. The mRNA sequencing data were deposited in the National Center for Biotechnology Information Genome Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE178479.
Drug synergy
The effects of the combination of drugs was evaluated using CompuSyn (version 2.0) software (ComboSyn Inc, Paramus, NJ, USA). This software used the Chou–Talalay combination index method, which is based on the median–effect equation derived from the mass-action law. Combination index (CI) values of <1, 1, and >1 indicate synergism, an additive effect, and antagonism, respectively [18].
Cell adhesion assay
MIA PaCa-2 cells were serum starved for 2 h. Then, cells were trypsinized and seeded into plates to allow for attachment for 30 min. Subsequently, the medium was removed, and the plate was washed with PBS. The number of remaining attached cells was counted under a phase microscope.
Lentiviral transduction
To produce lentiviral particles. pHAGE-PIK3CA-E545K and pHAGE-PIK3CA-H1047R were cotransfected with third Generation Packaging System Mix (Applied Biological Materials, Richmond, BC, Canada) into 293 T cells using the TurboFect reagent (Fermentas, Glen Burnie, MD, USA). After incubation in medium containing lentiviral particles for 2 days, the target cells were treated with puromycin (2 μg/mL, Clontech, Mountain View, CA) for 2 weeks to select cells with the stable integration of lentiviral vectors.
RNA interference
For knocking down gene expression, shRNA clones against PTEN (Clone ID: TRCN0000002746 and TRCN0000002747), PAK1 (TRCN0000002224), PAK2 ((TRCN0000002114), and PAK3 (TRCN0000003242) were obtained from RNA Technology Platform and Gene Manipulation Core (Academia Sinica, Taipei, Taiwan). An shRNA vector against lacZ (pLKO.1-shLacZ) was used as a negative control. To produce lentiviral particles, 293 T cells were transfected with 4 μg pLKO.1 lentiviral vectors in addition to 0.4 μg of envelope plasmid pMD.G and 3.6 μg of packaging plasmid pCMVΔR8.91. Viral particles were collected 40 and 64 h after transfection. To prepare cells with knocked down PTEN expression, MIA PaCa-2 cells were infected with lentiviruses for 24 h. Fresh medium containing 2 μg/mL puromycin was replaced every 3 days for 2 weeks to select drug-resistant cells.
Xenograft experiments
Animal experiments were performed following the guidelines of the Council of Agriculture, Taiwan, and were approved by the Institutional Animal Care and Use Committee of Medical School, National Taiwan University (approval number: 20201088). A total of 5 × 106 MIA PaCa-2 cells were subcutaneously implanted into both flanks of 6-week-old female NOD/SCID mice. After 3 weeks, the mice were randomised into six groups (n = 8 per group). The sample size was determined on the basis of relevant studies on drug treatment. The mice were daily treated with vehicle, sotorasib (10 mg/kg), alpelisib (30 mg/kg), FRAX597 (70 mg/kg), or the drug combination through oral gavage. Their body weights and tumour sizes were monitored every 2 days. The tumours were measured using a caliper, and tumour volume was calculated using the following formula: 1/2 × [(length in mm) × (width in mm)2]. After 14 days of treatment, the mice were euthanized and tumours were collected and weighted. The investigators were not blinded to the treatment group during the experiment.
Statistical analysis
Student’s t test was used for comparisons of the results of different groups. Two-tailed P < 0.05 was considered statistically significant.
Results
Establishment of sotorasib-resistant cells
We first tested the effects of sotorasib on the viability of a panel of pancreatic, colorectal, and lung cancer cell lines. As expected, cancer cell lines with KRASG12C mutations (MIA PaCa-2, SW1463, LU65, and LU99) were susceptible to sotorasib treatment in a dose-dependent manner, whereas the viability of cancer cell lines without KRASG12C mutations (BxPc-3, Capan-1, A549, and SW480) were not affected by sotorasib treatment (Fig. 1a, b and Supplementary Fig. 1). Among the cell lines with the KRASG12C mutation, SW1463 and MIA PaCa-2 cells were highly sensitive to sotorasib treatment, whereas LU99 cells were the most resistant. IC50 values for SW1463, MIA PaCa-2, LU65, and LU99 cells were 45.8 nM, 34.1 nM, 2.56 µM, and 22.55 µM, respectively. Sotorasib treatment reduced the phosphorylation of ERK and AKT in MIA PaCa-2 and LU65 cells (Fig. 1c, d). The phosphorylation levels of the upstream regulator MEK and the downstream target RSK of ERK were also found to be reduced in MIA PaCa-2 and LU65 cells treated with sotorasib (Supplementary Fig. 2). However, in LU99 cells, only ERK phosphorylation reduced, presumably due to the activating PIK3CA mutation. Sotorasib treatment does not affect the phosphorylation of ERK and AKT in cancer cell lines without the KRASG12C mutation.
Fig. 1. Establishment of sotorasib-resistant cancer cell lines.

a, b Effects of sotorasib treatment on (a) pancreatic and (b) lung cancer cell lines. Cell lines with KRASG12C mutations (MIA PaCa-2, LU65, and LU99) were susceptible to sotorasib treatment in a dose-dependent manner, whereas the viability of cancer cell lines without KRASG12C mutations (BxPC-3, Capan-1, and A549) was not affected by sotorasib treatment. (n = 6) c, d Sotorasib treatment downregulated ERK phosphorylation in MIA PaCa-2, LU65, and LU99 cells. Furthermore, AKT phosphorylation was downregulated in MIA PaCa-2 and LU65 cells. Sotorasib treatment did not affect the phosphorylation status of ERK and AKT in cancer cell lines without the KRASG12C mutation. e, f Establishment of sotorasib-resistant MIA PaCa-2 and LU65 cells through the incubation of tumour cells with sotorasib-containing media for 2 months. The MTT assay confirmed that cells were resistant to sotorasib. (n = 6). g Irrespective of the sotorasib treatment, the activation of ERK and AKT pathways was sustained in all three sotorasib-resistant cell lines.
To identify the mechanism underlying resistance to sotorasib treatment, cells were exposed to gradually increasing doses of sotorasib (range, 0.1–5 µM) for 2 months. SW1463 cells could not tolerate doses exceeding 0.2 µM; thus, resistant SW1463 cells could not be established. After resistant cells were established, they were incubated in sotorasib-free media for 2 weeks. The MTT assay proved that cells were persistently resistant to sotorasib (Fig. 1e, f). Sustained activation of ERK and AKT pathways was detected in all three cell lines. Sotorasib treatment reduced the phosphorylation levels of ERK and AKT in sotorasib-resistant LU65 cells, but not in sotorasib-resistant MIA PaCa-2 and LU99 cells (Fig. 1g).
Increased cell–matrix adhesion in sotorasib-resistant cells
Exome sequencing could not differentiate between treatment-naive and sotorasib-resistant cells in terms of single-nucleotide polymorphism and copy number variation in the genes associated with cell proliferation, signal transduction, and drug metabolism; this finding indicated that resistance was through nongenetic mechanisms (Supplementary Table 2). RNA sequencing was used to identify expression level changes in MIA PaCa-2 cells, which did not identify the upregulation of RAS, RAF, and receptor tyrosine kinase genes, indicating that the amplification and overexpression of these genes cannot account for the resistant phenotype. Furthermore, we did not detect the upregulation of mesenchymal markers and the downregulation of epithelial cell markers, indicating that, in contrast to a previous report [13], epithelial–mesenchymal transition was not observed in our cell model.
We observed that sotorasib-resistant cells have large flat morphology with multiple protrusions (Fig. 2a). Staining with rhodamine-phalloidin and vinculin showed increased stress fibres and focal adherens in sotorasib-resistant cells (Fig. 2b).
Fig. 2. Sotorasib-resistant cancer cells exhibit enhanced cell–matrix adhesion.

a Sotorasib-resistant cells have large flat morphology with multiple protrusions. b Rhodamine-phalloidin and vinculin staining showed increased stress fibres and focal adherens in sotorasib-resistant cells. c GSEA showed enrichment of the extracellular matrix–receptor interaction signalling pathway in sotorasib-resistant MIA PaCa-2 cells. d Adhesion assay showed that sotorasib-resistant MIA PaCa-2 cells are more adherent than untreated cells. (n = 5).
RNA sequencing was used to identify the changes in gene expression in sotorasib-resistant MIA PaCa-2 cells. GSEA was used to identify the enriched pathways (Supplementary Table 3). The extracellular matrix–receptor interaction signalling pathway was one of the most enriched pathways in sotorasib-resistant cells (Fig. 2c). An adhesion assay showed that sotorasib-resistant MIA PaCa-2 cells were more likely to be adherent than the parental cells (Fig. 2d). These results suggest that enhanced cell–matrix adhesion activates signalling pathways to circumvent the effects of sotorasib.
Consistent with the role of KRAS in mitochondrial metabolism [19], genes involved in the citric acid cycle and oxidative phosphorylation were downregulated in sotorasib-resistant cells (Supplementary Fig. 3a, b). The reduction of mitochondrial respiration was confirmed through the measurement of the oxygen consumption rate in a Seahorse bioenergic assay (Supplementary Fig. 3c). Anaerobic respiration, as determined by the extracellular acidification rate (ECAR), was also reduced in sotorasib-resistant cells (Supplementary Fig. 3d), indicating that sotorasib-resistant cells may rely on alternative energy sources to sustain cellular growth.
We also observed the downregulation of cell cycle and DNA repair genes in sotorasib-resistant cells (Supplementary Fig. 4a–d). However, sotorasib-resistant cells were not more vulnerable to the genotoxic agent cisplatin (Supplementary Fig. 4e, f). Sotorasib-resistant cells exhibited a low proliferative rate (Supplementary Fig. 4g, h) and reduced S phase progression, as evident from the results of the 5-ethynyl-2’-deoxyuridine incorporation assay (Supplementary Fig. 4i); this might have led to the downregulation of genes associated with DNA repair.
Activation of PAK in sotorasib-resistant cells
To identify the mechanism of the sustained activation of the MAPK pathway in sotorasib-resistant cells, we examined MEK phosphorylation upstream to ERK. The phosphorylation level of RAF-controlled serine 217 on MEK did not increase in sotorasib-resistant cells. By contrast, the phosphorylation level of serine 298 on MEK was highly increased in sotorasib-resistant cells (Fig. 3a, b). Because PAKs are the only known kinases that phosphorylate MEK on serine 298 [20], we explored PAK phosphorylation and noted the activation of PAK signalling (Fig. 3c, d). Enhanced phosphorylation of the serine 338 residue of CRAF, another target of PAK [21], was also observed in sotorasib-resistant cells (Fig. 3e, f).
Fig. 3. Activation of the Src/PAK/MEK axis in sotorasib-resistant cells.

a, b Phosphorylation of serine 298, but not serine 217, on MEK upregulated in sotorasib-resistant cells. c, d Phosphorylation of PAK proteins upregulated in sotorasib-resistant cells. e, f Enhanced phosphorylation of serine 338 of CRAF was observed in sotorasib-resistant cells. g, h Western blotting showed increased Src phosphorylation in sotorasib-resistant cells. i, j Treatment with dasatinib reduced the phosphorylation of PAKs and MEK1 on S298.
PAK1 can be activated by binding to the small G-proteins Cdc42 and Rac1 [22]. However, pulldown assays showed that Cdc42 and Rac1 were not activated in sotorasib-resistant cells (Supplementary Fig. 5a, b). Treatment with the Rac1 inhibitor NSC23766 paradoxically activated the phosphorylation of MEKS298 and ERK in sotorasib-resistant MIA PaCa-2 cells (Supplementary Fig. 5c), whereas treatment with the Cdc42 inhibitor ML141 slightly reduced the phosphorylation of MEKS298 and ERK in sotorasib-resistant MIA PaCa-2 cells (Supplementary Fig. 5d). This finding indicated that the activation of Cdc42 and Rac1 is not the primary mechanism underlying the activation of the PAK pathway in sotorasib-resistant cells.
A key signalling pathway that is activated by the integrin-dependent cell–matrix interaction is the focal adhesion kinase (FAK)/Src pathway, which is activated in many types of invasive cancer [23]. FAK/Src-dependent, PAK1-mediated phosphorylation of MEK1 on S298 has been reported to contribute to the cell–matrix adhesion dependence of growth factor signalling to MAPK [24, 25]. The results of western blotting revealed the enhanced phosphorylation of Src in sotorasib-resistant LU65 cells (Fig. 3h). Regarding MIA PaCa-2 cells, although the total level of Src was reduced in sotorasib-resistant cells, the total phosphorylation level of Src was similar to that of untreated cells, which indicated the enhanced phosphorylation of Src proteins (Fig. 3g). The reduction in the total protein level of Src might have resulted from the ubiquitin-mediated degradation of Src after activation [26]. Treatment with dasatinib, an inhibitor of the Src family tyrosine kinase family, reduced the phosphorylation of PAKs and MEK1 on S298 (Fig. 3i, j).
These results indicate that a cell–matrix interaction/Src/PAK/MEK axis may play a crucial role in cell resistance to sotorasib treatment.
Synergistic cytotoxicity of sotorasib and PAK inhibitors
FRAX597, a small-molecule inhibitor of group 1 PAKs, was used to study the combination effect of sotorasib treatment and PAK inhibition. The MTT assay showed that combination treatment showed a greater cytotoxic effect than sotorasib treatment alone on cancer cells. The CI was 0.34 for MIA PaCa-2 cells and 0.68 for LU65 cells (Fig. 4a, c), indicating that the two drugs synergistically attenuated cell viability. We also treated sotorasib-resistant MIA PaCa-2 and LU65 cells with FRAX597. As expected, these cells were resistant to sotorasib monotherapy. Cells were sensitive to FRAX597 monotherapy at a higher dose. The combination of FRAX597 with sotorasib further reduced the viability of sotorasib-resistant cells (Fig. 4b, d). Annexin V/propidium iodide flow cytometry assay showed that the combination of FRAX597 and sotorasib increased cell death in treatment-naive MIA PaCa-2 and LU65 cells, mainly through apoptosis mechanisms (Supplementary Fig. 6). Cell cycle analysis revealed a decreased S-phase fraction after the combined treatment with FRAX597 and sotorasib (Supplementary Fig. 7). Furthermore, FRAX486, another PAK inhibitor structurally distinct form FRAX597, showed synergy with sotorasib in treatment-naive cells and sotorasib-resistant cells (Fig. 4e–h; CI: 0.42 for MIA PaCa-2 cells and 0.42 for LU65 cells). The expression of each PAK gene was silenced using shRNA; the knockdown of PAK3 enhanced the cytotoxic effects of sotorasib on MIA PaCa-2 cells (Supplementary Fig. 8).
Fig. 4. PAK inhibitors and sotorasib synergistically kill cancer cells.

a, c Results of the MTT assay revealed that the combination of the PAK inhibitor FRAX597 and sotorasib exerted stronger cytotoxic effects than did sotorasib alone in cancer cells with KRASG12C mutations. b, d Combination treatment of FRAX597 and sotorasib reduced the viability of sotorasib-resistant cells. e–h FRAX486 also showed synergy with sotorasib treatment in treatment-naive cells and sotorasib-resistant cells. #CI < 1 at the indicated dose (n = 6 for all experiments).
We evaluated the effects of the combination of sotorasib and the MEK inhibitor trametinib or the Src inhibitor dasatinib. As shown in Supplementary Figs. 9 and 10, both trametinib and dasatinib exhibited synergy with sotorasib in treatment-naive and sotorasib-resistant cells. Therefore, the inhibition of the Src/PAK/MEK axis may enhance the cytotoxic effects of sotorasib and may overcome resistance to sotorasib treatment.
Synergistic cytotoxicity of sotorasib and PI3K inhibitor alpelisib
As mentioned previously, despite harbouring the KRASG12C mutation, LU99 cells are resistant to sotorasib treatment. LU99 cells have a T1025A mutation in the PIK3CA gene. This mutation activates AKT kinase and the propagation of a network of intracellular signals promoting tumour cell growth, proliferation, survival, and resistance to treatment [27]. Furthermore, we observed the sustained activation of AKT signalling in sotorasib-resistant cells (Fig. 1g). Therefore, we hypothesised that the activation of PI3K/AKT signalling may be a mechanism of resistance to sotorasib treatment. Alpelisib is a recently approved PI3K inhibitor for the treatment of PI3K-mutated, oestrogen receptor-positive, and Her-2 negative breast cancer [28]. Therefore, we tested the effects of the combination treatment of alpelisib and sotorasib on cancer cells. The treatment of MIA PaCa-2 cells with alpelisib reduced AKT signalling but exerted minimal effects on cell viability (Fig. 5a and Supplementary Fig. 11a). However, the addition of alpelisib augmented the effects of sotorasib at all tested doses. Alpelisib and sotorasib exerted synergistic effects on MIA PaCa-2 cells (CI = 0.12; Fig. 5a). Despite the absence of mutations in the genes encoding the proteins involved in the PI3K pathway, alpelisib treatment reduced AKT signalling and resulted in the reduced viability of LU65 cells (Supplementary Fig. 11b). Furthermore, sotorasib and alpelisib treatment synergistically acted in reducing the viability of LU65 cells (CI = 0.29; Fig. 5b). Moreover, copanlisib—another PI3K inhibitor that is structurally distinct form alpelisib—exhibited synergy with sotorasib in reducing the viability of MIA PaCa-2 and LU65 cells (CI = 0.06 and 0.12 for MIA PaCa2 and LU65, respectively; Fig. 5c, d).
Fig. 5. Activation of the PI3K pathway confers resistance to sotorasib.

a, b MTT assay showed that a combination treatment of alpelisib and sotorasib had a greater cytotoxic effect than sotorasib treatment alone on MIA PaCa-2 cells and LU65 cells. c, d Similar synergistic cytotoxic effects were observed in the combination treatment of copanlisib and sotorasib in MIA PaCa-2 cells. e Results of western blotting confirmed the efficacy of shRNA-mediated PTEN knockdown in MIA PaCa-2 cells. f Results of western blotting demonstrated the enhancement of AKT phosphorylation by the constitutively active mutants PIK3CA-E545K and PIK3CA-H1047R and the knockdown of PTEN in MIA PaCa-2 cells. g Overexpression of constitutively active mutants PIK3CA-E545K and PIK3CA-H1047R in MIA PaCa-2 cells resulted in resistance to sotorasib treatment. h PTEN knockdown significantly reduced the sensitivity of MIA PaCa-2 cells to sotorasib treatment. i, j Analysis of genetic changes occurring in pulmonary adenocarcinoma and colorectal cancer using The Cancer Genome Atlas PanCancer Atlas data revealed that KRAS mutations are nearly mutually exclusive with PIK3CA-activating or PTEN loss-of-function mutations in pulmonary adenocarcinoma; of a total of 217 colorectal cancers with KRAS mutation, 92 (42.4%) had PIK3CA-activating or PTEN loss-of-function mutations. #CI < 1 at the indicated dose (n = 6 for a–d, g, h).
To strengthen the association between PI3K pathway activation and sotorasib resistance, we overexpressed the constitutively active mutants PIK3CA-E545K and PIK3CA-H1047R through lentiviral transduction and knocked down PTEN expression using shRNA in MIA PaCa-2 cells (Fig. 5e). The results of western blotting confirmed the overexpression of the constitutively active PIK3CA mutants and the knockdown of PTEN enhanced AKT signalling (Fig. 5f). Both the overexpression of the constitutively active PIK3CA mutant and the knockdown of PTEN conferred resistance to sotorasib treatment (Fig. 5g, h).
To study the clinical implications of the alteration of the PI3K pathway genes on KRASG12C inhibitor treatment, we analysed the genetic changes occurring in pulmonary adenocarcinoma and colorectal cancer using The Cancer Genome Atlas PanCancer Atlas data. KRAS mutations were found to be nearly mutually exclusive with PIK3CA-activating or PTEN loss-of-function mutations in pulmonary adenocarcinoma (Fig. 5i). By contrast, 92 of 217 (42.4%) colorectal cancers harbouring the KRAS mutation had PIK3CA-activating or PTEN loss-of-function mutations (Fig. 5j). The concurrent mutations in KRAS and PI3K pathway genes may represent de novo sotorasib resistance mechanisms and may be a reason for the relative lack of responses to sotorasib treatment in colorectal cancer. To prove this hypothesis, we overexpressed the constitutively active PIK3CA mutants and knocked down PTEN expression in the colorectal cancer cell line SW1463. Both the overexpression of the constitutively active PIK3CA mutant and the knockdown of PTEN decreased cellular sensitivity to sotorasib treatment (Supplementary Fig. 11c).
Mutual positive regulatory loop between the PI3K and PAK pathways
PAK1 serves as a scaffold for facilitating the activation of AKT by PDK1 and the recruitment of AKT at the membrane [29]. AKT phosphorylates serine 21 on PAK1 to modulate Nck binding and cell migration [30]. Therefore, we hypothesised that the PAK and PI3K pathways would form a mutually regulated pathway to mediate resistance to sotorasib. Treatment with FRAX597 downregulated AKT phosphorylation in MIA PaCa-2 cells (Fig. 6a). Furthermore, treatment with alpelisib downregulated the phosphorylation of PAK proteins and MEKS298 (Fig. 6b). PTEN knockdown enhanced the phosphorylation of PAK proteins (Fig. 6c) The combination of sotorasib, FRAX597, and alpelisib exerted higher cytotoxic effects than those noted after treatment with a single drug or a combination of two drugs (Fig. 6d, e). These results indicate that the cell–matrix interaction–dependent activation of PAK mediates resistance to sotorasib through the activation of the MAPK (by MEKS298 phosphorylation) and PI3K/AKT pathways.
Fig. 6. Mutual positive regulatory loop between the PI3K and PAK pathways regulates cellular sensitivity to sotorasib and the antitumour effects of the combination of sotorasib with FRAX597 or alpelisib in vivo.

a Western blotting showed that FRAX597 treatment downregulated AKT phosphorylation in MIA PaCa-2 cells. b Alpelisib treatment downregulated the phosphorylation of PAK proteins and MEKS298. c PTEN knockdown enhanced the phosphorylation of PAK proteins. d, e Combination treatment with sotorasib, FARX597 (d), and alpelisib (e) resulted a greater cytotoxic effect than single or double treatment. (n = 6). f, g MIA PaCa-2 cells were injected subcutaneously into the flanks of NOD-SCID mice. After tumour establishment (mean tumour size: 150 mm3), the mice were randomised into six groups (n = 8 per group) and daily treated with the vehicle, sotorasib (10 mg/kg), FRAX597 (70 mg/kg), alpelisib (30 mg/kg), or the drug combination through oral gavage. The mice were euthanized on day 14. f Tumour volume measured over time. The combination of sotorasib with FRAX597 or alpelisib led to tumour regression. g Tumour weights at the endpoint of experiment. #CI < 1 at the indicated dose. *: P < 0.05 and **: P < 0.01 for comparison between different groups.
Antitumour effects of the combination of sotorasib with FRAX597 or alpelisib in vivo
To expand our in vitro data, we evaluated the efficacy of the combination therapy in a subcutaneous xenograft model of MIA PaCa-2. Once tumours were well established (mean volume = 150 mm3), the mice were treated with the vehicle, sotorasib, FRAX597, alpelisib, or drug combinations. At a dose of 10 mg/kg, sotorasib markedly reduced the tumour growth rate but did not induce tumour regression (Fig. 6f, g and Supplementary Fig. 12a). FRAX597 or alpelisib monotherapy reduced the tumour growth rate to an extent less than that noted for sotorasib. By contrast, the combination of sotorasib with FRAX597 or alpelisib led to noticeable tumour regression. The body weights of the mice were maintained in all groups, indicating negligible toxicity to the mice (Supplementary Fig. 12b).
Discussion
Covalent KRASG12C inhibitors have shown promising results in early clinical trials. However, on the basis of experience with previous molecular targeted therapy, despite remarkable initial responses, sustained clinical benefits often cannot be achieved due to the emergence of acquired resistance. Understanding the factors underlying drug resistance is important for developing combination therapy to overcome drug resistance.
To this end, we established cancer cell lines with acquired resistance to sotorasib by culturing cancer cells with increasing doses of sotorasib. Two major classes of resistance to molecular targeted therapy were reported previously: genetic and nongenetic. Therefore, after establishing resistant cells, we first studied whether the selection of rare genetic variants is responsible for the acquired drug resistance. Exome sequencing revealed no alterations in the genes associated with cell proliferation, signal transduction, and drug metabolism in sotorasib-resistant cells. Thus, we focused on nongenetic mechanisms underlying drug resistance.
In this report, we identified two nongenetic mechanisms of resistance to sotorasib treatment: the activation of PAK and PI3K pathways. PAKs are serine/threonine kinases that are frequently activated in cancer cells and are involved in a plethora of cancer pathogenic processes [31]. Rac1 or Cdc42 activates PAKs through the N-terminal GTPase-binding domain. In response to external signals originating from cell surface receptors and cell adhesion molecules, Rac1 and Cdc42 form macromolecular complexes with guanine nucleotide exchange factors, which convert Rac1 and Cdc42 from inactive GDP-bound forms to the active GTP-bound forms [32]. However, our results suggest that the activation of Rac1 or Cdc42 is not the main mechanism for PAK activation in sotorasib-resistant cells. Therefore, we searched for alternative pathways and found that the cell–matrix interaction–dependent activation of Src is the mechanism of PAK activation in sotorasib-resistant cells. Src cooperates with FAK to mediate the phosphorylation of ArfGAP paxillin kinase linker (PKL). The Nck-PAK-PIX-PKL protein complex is recruited to focal adhesions by paxillin on integrin engagement to transduce PAK signalling and to regulate cell spreading and protrusiveness [24]. Consistent with our result, a recent study showed that FAK inhibitors synergise with KRASG12C inhibitors in the treatment of KRASG12C-mutant cancer [33].
A major downstream target of PAKs is the MAPK cascade. PAKs activate the MAPK pathway through multiple mechanisms to maintain high ERK activity to circumvent KRAS inhibition by sotorasib. PAKs regulate both CRAF and MEK1 activities through the phosphorylation of these proteins at Ser338 and Ser298, respectively [20, 21]. Activated CRAF in turn phosphorylates MEK at Ser217. Our results revealed upregulation of the phosphorylation of PAK, MEKS298, and CRAFS338 in sotorasib-resistant cells, indicating PAK pathway activation. The lack of MEK217 phosphorylation upregulation is likely due to reduced input from KRAS signalling. Importantly, PAK kinases are amenable to small-molecule inhibitors, which have been demonstrated to have antineoplastic effects in preclinical and early clinical investigations [34, 35]. PAK1 activation is a mechanism causing resistance to BRAF inhibitors in BRAF-mutant melanomas [36]. PAK activity inhibition overcomes acquired drug resistance to the BRAF inhibitor PLX4720 [36]. PAKs mediated ERK signal reactivation in BRAF inhibitor–resistant cells through the phosphorylation of CRAF and MEK. The preset study discovered similar mechanisms to mediate sotorasib resistance. Therefore, by mitigating different upstream signals, PAK and KRAS or BRAF inhibitors may cooperate to suppress ERK signalling to induce the death of tumour cells.
We found that AKT signalling was activated soon after treatment with sotorasib, and persistently high AKT phosphorylation was observed in sotorasib-resistant cells. PI3K/AKT signalling plays crucial roles in various cellular events, such as apoptosis, cell cycle progression, and protein synthesis [37]. AKT signalling prevents apoptosis through its ability to phosphorylate and inhibit proapoptotic mediators, such as Bad and caspase-9 [38]. The activation of AKT/PI3K signalling was reported to be a mechanism underlying resistance to chemotherapy and targeted therapy [39, 40]. Our result showed that the FDA-approved PI3K inhibitor alpelisib synergises with sotorasib in treating cancer with the KRASG12C mutation, although these tumours lack the PIK3CA mutation. Recently, Misale et al. found that, among 112 drugs, alpelisib synergises the most with the KRASG12C inhibitor ARS-1620 in non-small cell lung cancer [41]. The combination of ARS-1620 and the PI3K inhibitor GDC0941 may overcome the resistance of cells to ARS-1620 in non–small-cell lung cancer [41]. In addition to small-molecule inhibitors, the tumour-derived oncogenic mutants of PIK3CA and the knockdown of PTEN induced resistance to sotorasib. A considerable proportion of colorectal cancer cells harboured concurrent KRAS and PIK3CA mutations or PTEN biallelic deletion, but these genetic events are mutually exclusive in pulmonary adenocarcinoma. This may be the underlying cause of the higher responsive rate to sotorasib treatment in lung cancer than in colorectal cancer [9]. Analysing the genetic changes in patients treated with sotorasib is needed to prove this hypothesis. If proven, genetic testing for the PIK3CA and PTEN status may be helpful in identifying patients most likely to respond to sotorasib treatment. Combination therapy of sotorasib and the PI3K inhibitor may be used for patients with concurrent mutations in KRASG12C and the PI3K pathway.
In conclusion, sotorasib-resistant cancer cells exhibit hyperactive PAK and PI3K signalling, which results in a compromised treatment effect. The drug combination comprising KRASG12C, PAK, and PI3K inhibitors exerted promising synergistic effects against cancer cells with the KRASG12C mutation. Further clinical studies are warranted to explore whether this combination strategy improves treatment outcomes in patients with cancer.
Supplementary information
Acknowledgements
We gratefully acknowledge technical support from the Second and Eighth Core Labs of National Taiwan University Hospital, the Translational Core Facility of Taipei Medical University, and RNA Technology Platform and Gene Manipulation Core, Academia Sinica.
Author contributions
C-YY and Y-MJ conceptualised the study and designed the experiments and provided financial support. C-HC, L-WC, and T-YL performed the experiments. Y-RL and T-HH collected the bioinformatics data. C-YY, L-WC, and Y-MJ interpreted the data and drafted the manuscript. All authors discussed the results; reviewed and revised the manuscript; and approved the final version of the manuscript.
Funding
This work was supported by the Ministry of Science and Technology, Taiwan [grant numbers 109-2314-B-002-084 and 110-2314-B-002-173].
Data availability
The mRNA sequencing data were deposited to the National Center for Biotechnology Information Genome Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE178479. Other data sets generated and/or analysed during this study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chien-Hui Chan, Li-Wen Chiou.
Contributor Information
Ching-Yao Yang, Email: cyang@ntuh.gov.tw.
Yung-Ming Jeng, Email: mrna0912@gmail.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-022-02032-w.
References
- 1.Rodenhuis S. Ras and human tumors. Semin Cancer Biol. 1992;3:241–7. [PubMed] [Google Scholar]
- 2.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692–6. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milburn MV, Tong L, deVos AM, Brünger A, Yamaizumi Z, Nishimura S, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–45. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 4.Nollmann FI, Ruess DA. Targeting mutant KRAS in pancreatic cancer: Futile or promising? Biomedicines. 2020;8:E281. doi: 10.3390/biomedicines8080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–89. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRASG12C inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 7.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-RasG12C inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS G12C inhibition with Sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207–17. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371–81. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med. 2021;384:2382–93. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599:679–83. doi: 10.1038/s41586-021-04065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C Inhibition. Clin Cancer Res. 2020;26:1633–43. doi: 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R, et al. Epithelial to mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C mutant non-small cell lung cancer. Clin Cancer Res. 2020;26:5962–73. doi: 10.1158/1078-0432.CCR-20-2077. [DOI] [PubMed] [Google Scholar]
- 15.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421–5. doi: 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 2020;10:1129–39. doi: 10.1158/2159-8290.CD-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzym Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 19.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beeser A, Jaffer ZM, Hofmann C, Chernoff J. Role of group A p21-activated kinase in activation of extracellular-regulated kinase by growth factors. J Biol Chem. 2005;280:36609–15. doi: 10.1074/jbc.M502306200. [DOI] [PubMed] [Google Scholar]
- 21.Chaudhary A, King WG, Mattaliano MD, Frost JA, Diaz B, Morrison DK, et al. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr Biol. 2000;10:551–4. doi: 10.1016/S0960-9822(00)00475-9. [DOI] [PubMed] [Google Scholar]
- 22.Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–6. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 23.Niit M, Hoskin V, Carefoot E, Geletu M, Arulanandam R, Elliott B, et al. Cell-cell and cell-matrix adhesion in survival and metastasis: Stat3 versus Akt. Biomol Concepts. 2015;6:383–99. doi: 10.1515/bmc-2015-0022. [DOI] [PubMed] [Google Scholar]
- 24.Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell. 2005;16:4316–28. doi: 10.1091/mbc.e05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol. 2003;162:281–91. doi: 10.1083/jcb.200212141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci USA. 1999;96:13738–43. doi: 10.1073/pnas.96.24.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl J Med. 2019;380:1929–40. doi: 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 29.Higuchi M, Onishi K, Kikuchi C, Gotoh Y. Scaffolding function of PAK in the PDK1-Akt pathway. Nat Cell Biol. 2008;10:1356–64. doi: 10.1038/ncb1795. [DOI] [PubMed] [Google Scholar]
- 30.Zhou GL, Zhuo Y, King CC, Fryer BH, Bokoch GM, Field J. Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol Cell Biol. 2003;23:8058–69. doi: 10.1128/MCB.23.22.8058-8069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rane CK, Minden A. P21 activated kinase signaling in cancer. Semin Cancer Biol. 2019;54:40–9. doi: 10.1016/j.semcancer.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Sinha S, Yang W. Cellular signaling for activation of Rho GTPase Cdc42. Cell Signal. 2008;20:1927–34. doi: 10.1016/j.cellsig.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Zhang B, Zhang Y, Zhang J, Liu P, Jiao B, Wang Z, et al. Focal adhesion kinase (FAK) inhibition synergizes with KRAS G12C inhibitors in treating cancer through the regulation of the FAK-YAP signaling. Adv Sci. 2021;8:e2100250. [DOI] [PMC free article] [PubMed]
- 34.Chung EY, Mai Y, Shah UA, Wei Y, Ishida E, Kataoka K, et al. PAK kinase inhibition has therapeutic activity in novel preclinical models of adult T-cell leukemia/lymphoma. Clin Cancer Res. 2019;25:3589–601. doi: 10.1158/1078-0432.CCR-18-3033. [DOI] [PubMed] [Google Scholar]
- 35.Semenova G, Chernoff J. Targeting PAK1. Biochem Soc Trans. 2017;45:79–88. doi: 10.1042/BST20160134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu H, Liu S, Zhang G, Wu B, Zhu Y, Frederick DT, et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature. 2017;550:133–6. doi: 10.1038/nature24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–42. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–82. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Jabbarzadeh Kaboli P, Salimian F, Aghapour S, Xiang S, Zhao Q, Li M, et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer - a comprehensive review from chemotherapy to immunotherapy. Pharm Res. 2020;156:104806. doi: 10.1016/j.phrs.2020.104806. [DOI] [PubMed] [Google Scholar]
- 40.Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med. 2016;67:11–28. doi: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 41.Misale S, Fatherree JP, Cortez E, Egan RK, McClanaghan J, Stein GT, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. 2019;25:796–807. doi: 10.1158/1078-0432.CCR-18-0368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mRNA sequencing data were deposited to the National Center for Biotechnology Information Genome Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE178479. Other data sets generated and/or analysed during this study are available from the corresponding author on reasonable request.