

Abstract
Adiabatic state preparation (ASP) can generate the correlated wave function by simulating the time evolution of wave function under the time-dependent Hamiltonian that interpolates the Fock operator and the full electronic Hamiltonian. However, ASP is inherently unsuitable for studying strongly correlated systems, and furthermore practical computational conditions for ASP are unknown. In quest for the suitable computational conditions for practical applications of ASP, we performed numerical simulations of ASP in the potential energy curves of N2, BeH2, and in the C2v quasi-reaction pathway of the Be atom insertion to the H2 molecule, examining the effect of nonlinear scheduling functions and the ASP with broken-symmetry wave functions with the S2 operator as the penalty term, contributing to practical applications of quantum computing to quantum chemistry. Eventually, computational guidelines to generate the correlated wave functions having the square overlap with the complete-active space self-consistent field wave function close to unity are discussed.
Subject terms: Quantum chemistry, Method development
Adiabatic state preparation (ASP) can generate correlated wave functions for quantum chemical calculations, but is inherently unsuitable for studying strongly correlated systems. Here, the authors perform numerical simulations of ASP for the ground state wave functions of molecules with strongly correlated electrons and propose practical conditions for preparation of close-to-exact correlated wave functions.
Introduction
Quantum computing and quantum information processing (QC/QIP) is one of the most innovative research fields in the current central science1 and it has a potential to bring a paradigm shift in chemistry research. Among the diverse topics in the field of QC/QIP, sophisticated quantum chemical calculations have attracted attention as the near-future applications of quantum computers. Quantum chemical calculations are based on the Schrödinger equation that is a fundamental equation in quantum mechanics, and methods for accurate quantum chemical calculations potentially pave the way toward predictive quantum chemistry. Variationally best possible wave functions within the Hilbert space spanned by the basis set being used can be obtained by employing the full-configuration interaction (full-CI) calculations. However, the computational cost of full-CI methods scales exponentially against the number of basis functions relevant to the system size under study, and it easily reaches astronomical figures even for small molecules2,3.
To date, two major approaches for the quantum chemical calculations on quantum computers have been widely investigated, namely quantum phase estimation (QPE)-based full-CI calculations and a variational quantum eigensolver (VQE). The QPE-based approach was proposed in 20054. It is based on the quantum simulations of the time evolution of an approximated wave function and projective measurement to the eigenstate of a given Hamiltonian. The quantum circuit for the QPE-based full-CI is too deep to be executed on noisy intermediate-scale quantum (NISQ) devices currently available. The QPE-based approach, however, is expected to be a powerful tool when fault-tolerant quantum computing (FTQC) becomes available, because the computational cost of the QPE-based full-CI scales polynomially against the system size, and therefore exponential improvement of the computational cost scaling from the classical counterpart is guaranteed theoretically. In the QPE-based methods the time evolution of wave functions should be simulated conditionally on the ancillary qubit, but this requirement is recently removed by the appearance of the approach based on the Bayesian phase difference estimation (BPDE) algorithm5. VQE is a quantum–classical hybrid algorithm, and it utilizes parametrized quantum circuits to prepare correlated wave functions and computes energy expectation values by statistical sampling of the measurement outcome6. Classical computers are used to perform the variational optimizations of parameters relevant to the quantum circuits. VQE has been extensively studied from both the experimental and theoretical sides, because the parametrized quantum circuit used for wave function preparation is usually very shallow enough to be executed on NISQ devices. It should be noted that, however, the computational cost scaling of the VQE-based approaches has not been sorely elucidated yet, and it is unclear whether quantum chemical calculations can be accelerated by using VQE. For example, VQE calculations with the unitary coupled cluster with singles and doubles (UCCSD) ansatz scale polynomial. The approximate UCCSD calculations, however, can also be done with the polynomial cost on a classical computer, by solving the amplitude equation of the similarity transformed Hamiltonian. Thus, the computational cost scaling becomes polynomial vs. polynomial for the UCCSD calculations on quantum and classical computers. Also, because the full-CI wave function contains an exponentially large number of variables, solving the full-CI using VQE with naïve implementation scales exponential. Recent reviews on quantum chemical calculations on quantum computers including QPE and VQE can be found elsewhere7–13.
In the QPE-based full-CI calculations, the preparation of a “good” approximated wave function having sufficiently large overlap with the exact wave function of the target electronic state is crucial. This is because the probability of which eigenenergy of the electronic state can be obtained in the QPE is proportional to the square overlap between the approximated and the exact wave functions. If the approximated wave function has exponentially small overlap with the eigenfunction of the target electronic state, an exponentially large number of QPE experiments are required to acquire the correct results, which spoils the advantages of a quantum speedup. In the equilibrium geometry of typical closed-shell singlet molecules and open-shell high-spin molecules carrying no unpaired electrons of spin-β, the Hartree–Fock (HF) wave function |ΨHF〉 dominantly contributes to the full-CI wave function of the electronic ground state, and the use of |ΨHF〉 is generally a good choice. In the low-spin states of open-shell molecules, by contrast, the wave function is approximated by the linear combination of several Slater determinants so as to be an eigenfunction of the S2 operator, and the overlap between |ΨHF〉 and |Ψfull-CI〉 becomes small. We demonstrated that the symmetry-adapted configuration state function (CSF) can have a large overlap with the full-CI wave function of open-shell low-spin systems, and proposed quantum circuits to prepare the |ΨCSF〉 on a quantum computer14,15. We also reported an approach to generate multi-configurational wave functions on quantum computers without performing any post-HF calculations16, which is suitable for the study of the systems with intermediate open-shell characters like molecules under covalent bond dissociations. These approaches can effectively consider static (nondynamical) electron correlation effects.
Although these approaches are useful to treat electronic states of open-shell molecules, the overlap with the full-CI wave function becomes small when dynamical electron correlation effects are also significant. Molecules having electronic structures too complicated to deal with facile approaches like density functional theory (DFT) are naturally one of the main targets of sophisticated quantum chemical calculations, and the development of theoretical methods to generate correlated wave functions considering both static and dynamical electron correlation effects on quantum computers is an important task. Note that one of the anticipated usage of VQE is the preparation of approximate wave functions used as the input for QPE17, but recent numerical simulations of VQE of the C2v quasi-reaction pathway of Be + H2 → BeH2 reaction revealed that the variational parameter optimization converges very slowly for strongly correlated systems18.
Noticeably, recent years have witnessed that these promising approaches are relevant to innovative development in simulating quantum systems, such as Hamiltonian simulation. Adiabatic state preparation (ASP)4 is an approach to generate correlated wave functions based on an adiabatic theorem19. ASP belongs to an adiabatic quantum algorithm20,21, in which the wave function of the ground state of a problem Hamiltonian HP is generated adiabatically, by starting from the ground state wave function of an initial Hamiltonian HI and slowly varying the Hamiltonian by using a scheduling function s(t) in Eq. 1 from 0 to 1.
| 1 |
The scheduling function is often set as s(t) = t/T, where T is a total evolution time length. By using |ΨHF〉 and a Fock operator as the initial wave function and Hamiltonian, respectively, and the full Hamiltonian as HP, we can obtain |Ψfull-CI〉 if the evolution time T is long enough4. Note that an approach using the maximum commuting Hamiltonian as the initial Hamiltonian HI was proposed recently22. ASP was adopted for the QPE-based full-CI of H2 molecule using an NMR quantum computer23. ASP numerical simulations of the 1 1A1 state of methylene (CH2) molecule was reported in 201424, and the optimization of the scheduling procedure by using VQE was discussed in 202125. Application of the complete active space configuration interaction (CASCI) wave function as the initial wave function and the nonlinear scheduling functions s(t) = (t/T)c (0 < c < 1) was also reported in 202126.
Although ASP is a long-term (or FCTC) algorithm because the quantum circuit for ASP is usually too deep to execute on NISQ devices, it is promising because it does not require detailed a priori knowledge of the electronic structure of the system being studied. However, ASP is potentially unsuitable for strongly correlated systems, because the evolution time length T should be set longer when it is applied to the systems with smaller energy gaps between the ground and excited states. We also emphasize that the number of ASP studies so far documented are very few, and there has been little knowledge on the optimal computational conditions of ASP, such as the setting of the evolution time length and the selection of scheduling functions. Then, it is essential to explore suitable computational conditions which help make ASP as a practical tool for the preparation of correlated wave functions, which is the main subject of this work. Note that variational quantum imaginary time evolution (QITE) is also available for the preparation of correlated wave functions27–30. However, the total length of imaginary time propagation is determined by the spectrum of the Hamiltonian and the initial overlap with the exact wave function, and the application of QITE to strongly correlated systems is also an important issue.
In this work, we have carried out numerical simulations of ASP for the generation of correlated wave functions in the triple bond dissociation of N2 molecule, the symmetric bond dissociation of BeH2 molecule, and the C2v quasi-reaction pathway of beryllium atom insertion to H2 molecule. We have explored the effects of nonlinear scheduling functions and the ASP starting with a broken-symmetry (BS) wave function |ΨBS〉 by using the electron spin S2 operator as the penalty term in the time-dependent Hamiltonian. As the scheduling function s(t) in Eq. 1, we examined five different functions listed in Table 1 and plotted in Fig. 1; all of them were studied as the scheduling function in adiabatic algorithms by Hu and Wu31. An anticipated application of ASP is the wave function preparation for QPE, and thus in this study the quality of the wave functions obtained from ASP is evaluated by means of the square overlap with the CASCI wave function, |〈ΨASP|ΨCASCI〉|2. Note that to use ASP for the wave function preparation in QPE, the evolution time required for ASP must be significantly shorter than that needed in QPE (T ~ 2000 atomic unit to achieve 1 kcal mol−1 of energy precision16). Eventually, we attempt to propose guidelines for the evolution time length and the selection for starting wave functions toward the practical use of ASP.
Table 1.
Scheduling functions s(x) (x = m/M) tested in Eq. 1.
| Name and abbreviation | Function |
|---|---|
| Linear (Lin) | |
| Square (Squ) | |
| Sinusoidal (Sin) | |
| Sinusoidal cubic (SinCub) | |
| Cubic (Cub) |
Fig. 1. Plots of the scheduling functions tested in Eq. 1.
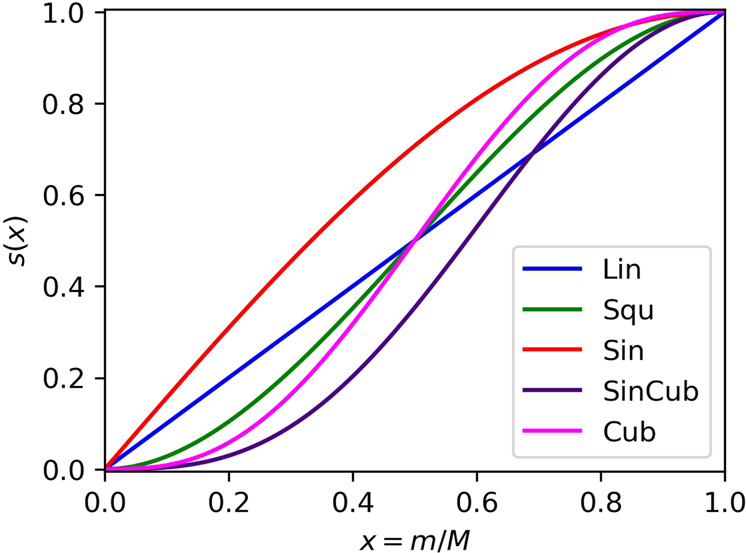
Mathematical definitions of the scheduling functions are provided in Table 1.
Results and discussion
Scheduling function and evolution time length dependences of ASP in the potential energy curve of N2
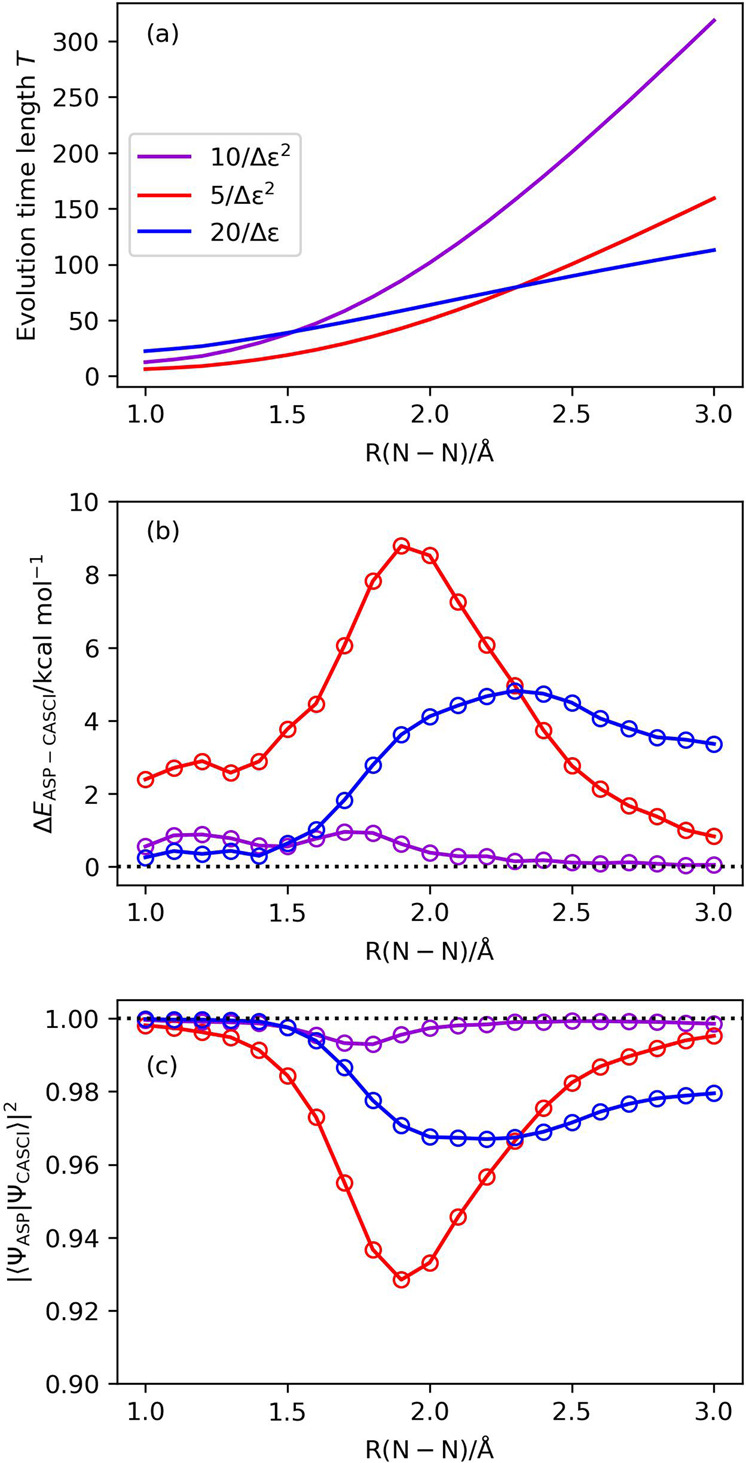
First, we explored the scheduling function and the evolution time length dependences of ASP in N2 molecule with R(N–N) = 1.0, 2.0, and 3.0 Å. These geometries are selected as the representatives of the structures close to the equilibrium geometry where |〈ΨHF|ΨCASCI〉|2 is large, in the intermediate bond dissociation region where neither the |ΨHF〉 nor the |ΨCSF〉 are good approximation of the ground state, and in the bond dissociated region where the |ΨCASCI〉 can be approximate to a |ΨCSF〉. The numerical quantum circuit simulation results with T = 10–100 are summarized in Fig. 2. The number of quantum gates required for a single Trotter step is about 6900. At the geometry R(N–N) = 1.0 Å, ASP gives the wave function close to the |ΨCASCI〉 even for the shortest evolution time length being tested (T = 10) regardless of the choice of the scheduling functions. The scheduling function and evolution time length dependences become significant for the longer N–N distances. Among the five scheduling functions being tested, the sinusoidal function exhibits the fastest convergence to the CASCI wave function against the evolution time length T. By employing the sinusoidal function as the scheduling function, we need T = 50 and 70 for R(N–N) = 2.0 and 3.0 Å, respectively, to achieve |〈ΨASP|ΨCASCI〉|2 > 0.9. Longer evolution time is required to achieve the same magnitude of the square overlap for the elongation of the N–N bond, because the HOMO–LUMO gap and the energy gap between the ground and excited states become smaller for the elongated N–N distances. Plots of the S1−S0 energy gap of the instantaneous Hamiltonian (Fig. 3) indicate that the s(x) value giving the minimum ΔE(S1−S0) value becomes larger for the shorter R(N−N) values.
Fig. 2. Results of the numerical quantum circuit simulation of N2 molecule.

a,c,e The energy deviations from the CASCI values in (a) R(N–N) = 1.0 Å, (c) R(N–N) = 2.0 Å, and (e) R(N–N) = 3.0 Å. b, d, f The square overlaps with the CASCI wave function in (b) R(N–N) = 1.0 Å, (d) R(N–N) = 2.0 Å, and (f) R(N–N) = 3.0 Å.
Fig. 3. The S1−S0 energy gap of the instantaneous Hamiltonian H(s(x)) of N2 molecule with the bond lengths R(N−N) = 1.0, 2.0, and 3.0 Å.
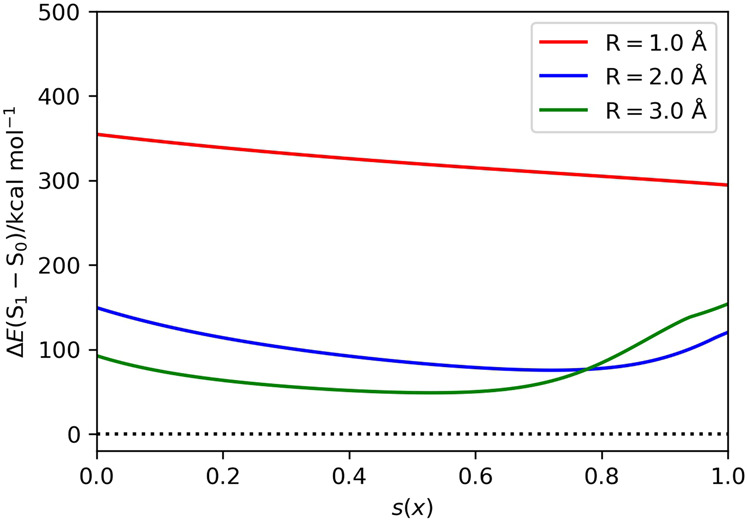
The ΔE(S1 − S0) values at s(x) = 0.0 and 1.0 corresponds to the energy gap of the Fock operator and the full electronic Hamiltonian, respectively.
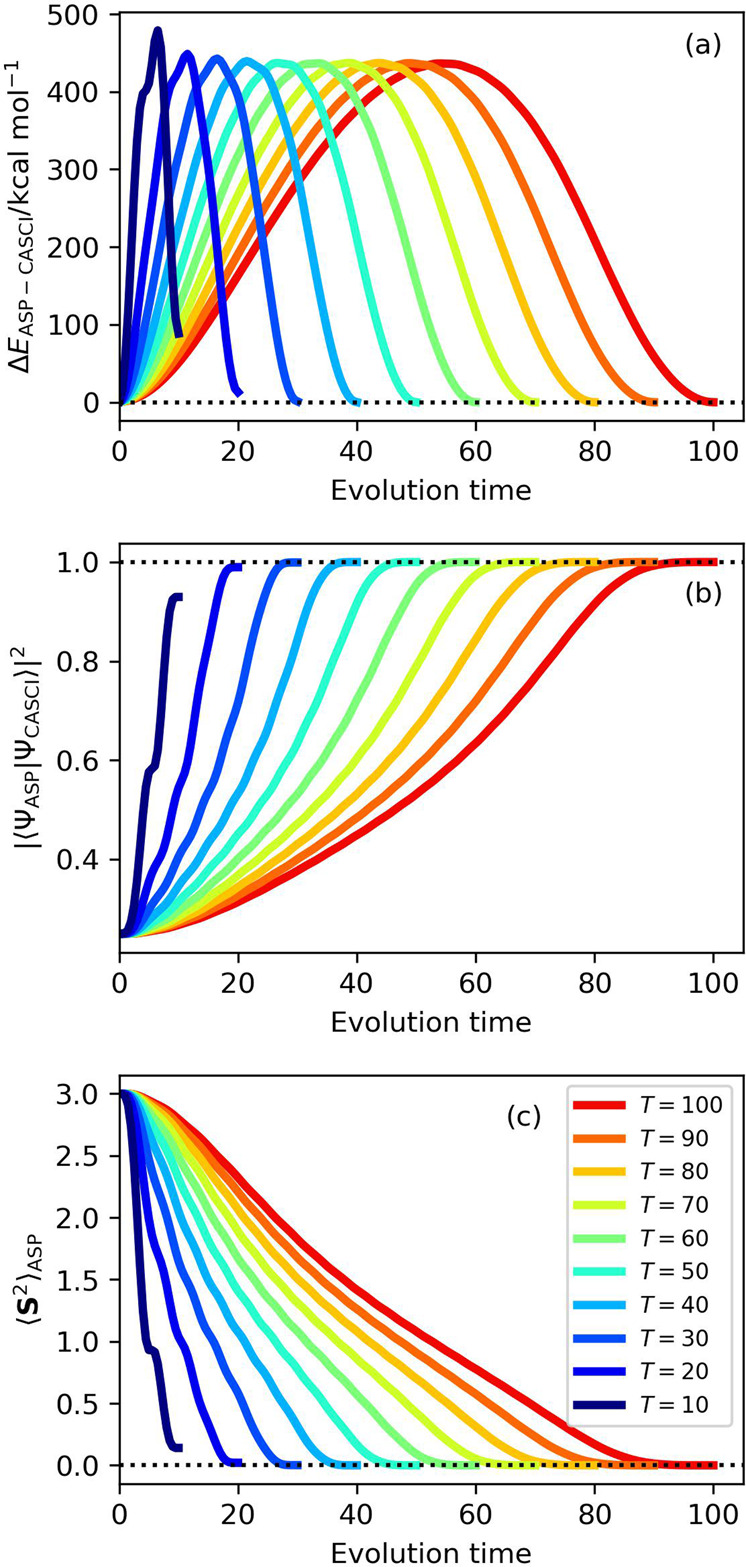
The trajectories of ASP of N2 in the geometry R(N–N) = 3.0 Å with the evolution time length T = 100 are plotted in Fig. 4. Among the five scheduling functions, the sinusoidal function gives the smallest ΔEASP−CASCI value and the largest square overlap at each time step. Note that the trajectories of the square overlap calculated by using the linear and sinusoidal functions oscillate in the beginning of ASP. This originates from the fact that the gradient ∂E(t)/∂s(t) ≠ 0 at t = 031. The same trends were also observed in the other geometries and molecules under study.
Fig. 4. Trajectories of ASP with T = 100 in N2 molecule with R(N–N) = 3.0 Å.

a The energy deviations from the CASCI value. b The square overlaps with the CASCI wave function.
The reason why the sinusoidal function gave the best results among the five scheduling functions can be explained by the structure of the HF wave function and the energy landscape of the instantaneous Hamiltonian. One has to set the evolution time length prior to computation to apply ASP. The evolution time length T is usually set to satisfy the condition in Eq. 232.
| 2 |
Here, |Ψg(s)〉 and |Ψe(s)〉 are the wave functions of the electronic ground and excited states, respectively, of the instantaneous Hamiltonian H(s), and εg(s) and εe(s) are the corresponding energy eigenvalues. From Eq. 2, we can sweep the time-dependent Hamiltonian faster if the energy gaps between the ground and the excited states are large and if the numerator of Eq. (2) is small. The |ΨHF〉 can be rewritten as |ΨHF〉 = Σj cj|Ψj〉, where |Ψj〉 is the jth eigenfunction and cj is the corresponding coefficient. Because the |ΨHF〉 is spin and spatial symmetry-adapted, only the excited states belonging to the same spin and spatial symmetry can have non-negligible contribution to the numerator of Eq. 2 in the absence of Trotter decomposition errors and noises. The excitation energies of the corresponding excited states in the weakly correlated regime are generally larger in the earlier stage of ASP. This is because the Fock operator used as the initial Hamiltonian contains the terms with occupied orbitals only, and the excited states cannot be sufficiently stabilized under the Fock operator. Thus, we can sweep the Hamiltonian faster in the earlier stage of ASP, and the sweep speed must be attenuated by the time evolution. The sinusoidal function has such a structure.
Note that Eq. 2 is insufficient, and it does not guarantee the validity of the adiabatic approximation33,34. In fact, Marzlin and Sanders claimed that the application of the adiabatic theorem may lead to an inconsistency no matter how slowly the Hamiltonian is varied33. They also pointed out that the inconsistency becomes a potential problem whenever |Ψ(T)〉 deviates greatly from the initial state |Ψ(0)〉. Strongly correlated systems can be the case of such a small overlap |〈Ψ(0)|Ψ(T)〉| if the |ΨHF〉 is employed as |Ψ(0)〉, and therefore the application of ASP to strongly correlated systems is a challenging problem. The necessary and sufficient condition for adiabatic evolution was discussed by Wang and Plenio, by decomposing the diabatic propagator into the geometric functions determined by the eigenstates and the modulation functions determined by the energy gaps and the speed of sweeping35. By utilizing these necessary and sufficient conditions for adiabatic condition, Xu and coworkers demonstrated the adiabatic evolution in the presence of vanishing energy gaps, using a nitrogen-vacancy center in diamond36. It should be also noted that approaches based on the shortcuts to adiabaticity (STA) have been eagerly studied to manipulate the quantum system on timescales shorter than decoherence time37, but it requires some non-physical Hamiltonian in order to make it work, and it is still an open and challenging problem.
Although Eq. 2 may lead to an inconsistency of the adiabatic evolution33,34, it can be used as the guideline of the evolution time length. Indeed, the variation of the wave function under the time-dependent Hamiltonian, which is responsible for the inconsistency, may be difficult to estimate in advance of the ASP simulation, but the energy gap between the ground and the first excited states can be roughly evaluated from the HOMO–LUMO gap Δε = ε(LUMO) − ε(HOMO), where ε(HOMO) and ε(LUMO) are the orbital energies of HOMO and LUMO, respectively. We note that the HOMO–LUMO gap estimation is only a practical approach. The exact energy landscape is required to fully adjust the scheduling function s(x), which corresponds to solving the problem altogether. However, the HOMO–LUMO gap can be used for crude estimation of the excitation energy. These orbital energies can be obtained from the HF calculations, and therefore using the HOMO–LUMO gap for the determination of the evolution time length does not need additional computation. Excitation energy estimations based on the single excitation CI or time-dependent DFT may be more reliable, but such computations inherently raise the computational cost. Based on this strategy, we examined the following evolution time determination methods: T = 5/Δε2 and T = 10/Δε2. In addition to them, we also tested the evolution time length T = 20/Δε. The results of the quantum circuit simulations with different evolution time length determination strategies with the sinusoidal scheduling function are summarized in Fig. 5. If we adopt the strategy T = 10/Δε2, we can obtain the correlated wave function with the square overlap |〈ΨASP|ΨCASCI〉|2 > 0.99 at all bond lengths under study, but the evolution time for the longer N–N distance is quite long. The strategy T = 5/Δε2 gives fairly good results except for intermediate bond dissociation region. Considering the evolution time lengths and the square overlap, the strategy T = 20/Δε seems to be most suitable for ASP of the triple bond dissociation in N2. The same trend was observed for the other scheduling functions, although the deviations from the CASCI result are larger for the other functions than the sinusoidal one (see Supplementary Fig. S2).
Fig. 5. Results of the numerical simulation of ASP with different evolution time length determination strategies in N2.
Sinusoidal function is used for the scheduling function. a Evolution time length being tested. b The energy deviations from the CASCI values. c The square overlaps with the CASCI wave functions.
ASP with broken-symmetry wave functions and the S2 penalty term in the potential energy curve of N2
It is naturally expected that the use of multiconfigurational wave functions gives a plausible choice for the starting wave function in strongly correlated systems. In fact, Kremenetski and coworkers reported that substantial speedup of ASP can be achieved by using the CASCI wave function as the starting wave function26. However, encoding the CASCI wave function on a quantum register becomes nontrivial when the active space of CASCI is large. Here, we examine an alternative and facile approach of ASP for strongly correlated systems by adopting the BS wave function |ΨBS〉 as the starting wave function with the S2 operator as the penalty term in the time-dependent Hamiltonian. The BS methods have been widely used in the DFT calculations of open-shell low-spin states such as spin-singlet states of biradicals38–40. The |ΨBS〉 is a single Slater determinant with spin-β unpaired electrons in the localized singly occupied molecular orbitals (SOMOs). The |ΨBS〉 is an eigenfunction of the Sz operator but is not an eigenfunction of the S2 operator, and thus the |ΨBS〉 is expressed by a linear combination of wave functions having different spin quantum numbers S, as in Eqs. 3 and 4 for four- and six-spin MS = 0 BS wave functions, respectively, for example.
| 3 |
| 4 |
The coefficients in the right-hand side of Eqs. 3 and 4 can be derived from the structure of spin eigenfunctions41. The eigenvalue of the S2 operator is S(S + 1), and the expectation values of the S2 operator of the wave functions given in Eqs. 3 and 4 are calculated to be 2.0 and 3.0, respectively.
Because the quantum state corresponding to the |ΨBS〉 can be prepared on a quantum computer with the same cost as the preparation of |ΨHF〉 regardless of the number of singly occupied orbitals, it is possible to use the |ΨBS〉 as the starting wave function in ASP. The wave function components having the spin quantum number different from the target electronic state can be eliminated by introducing the S2 operator as the penalty term in the time-dependent Hamiltonian H(t), as in Eq. 5.
| 5 |
Here, c is a coefficient that controls the strength of the penalty term. The S2 operator as the penalty term works to raise the energy of the wave function with the spin quantum number S by cS(S + 1), and therefore spin contaminants can be readily eliminated during ASP. We expect that ASP starting from |ΨBS〉 can generate multiconfigurational wave functions efficiently, just as the spin-projected extended Hartree–Fock (EHF) method42 in classical computation that applies the spin projection operator to the spin contaminated UHF wave function. In Eq. 3, larger c values can shift the energies of the spin contaminants greatly, but too large c values will result in departure from the adiabatic pathway. Note that we have already proposed a method to construct a quantum circuit for the time evolution operator exp(−is(t)cS2t) by utilizing a generalized spin coordinate mapping43, which can be directly used for the present ASP study.
The results of the ASP starting from the six-spin |ΨBS〉 (the wave function in Eq. 4) with c = 0.5, the square scheduling function, and the evolution time length T = 10–100 of N2 molecule at R(N–N) = 3.0 Å are summarized in Fig. 6, and the results obtained by using the other scheduling functions are given in Supplementary Fig. S3–S6. Interestingly, ASP starting with the |ΨBS〉 gave the square overlap |〈ΨASP|ΨCASCI〉|2 larger than 0.995 even for T = 20. The scheduling function dependence on ASP with the |ΨBS〉 with the total evolution time T = 50 were plotted in Fig. 7. The |〈ΨASP|ΨCASCI〉|2 values calculated by using the square, sinusoidal cubic, and cubic scheduling functions asymptotically approach unity for longer N–N distances. These three scheduling functions exhibit the best square overlap values in the bond dissociation region. No significant differences were observed among these scheduling functions, but the square function gave larger square overlap than the sinusoidal cubic and cubic functions for shorter N–N bond lengths. Importantly, the square, sinusoidal cubic, and cubic scheduling functions gave almost spin-pure wave functions with 〈S2〉ASP ≈ 0, although spin contaminations were not eliminated completely in the |ΨASP〉 generated by employing the sinusoidal and linear scheduling functions. In the following discussions we used the square scheduling function for ASP with the |ΨBS〉 wave functions.
Fig. 6. Results of the numerical simulation of ASP with |ΨBS〉 as the starting wave function and square function as the scheduling function in N2 molecule at R(N–N) = 3.0 Å.
a The energy deviations from the CASCI values. b The square overlaps with the CASCI wave functions. c The 〈S2〉 values.
Fig. 7. Numerical simulation results of the scheduling function dependence on ASP with the |ΨBS〉 as the starting wave function and T = 50 in N2 molecule.
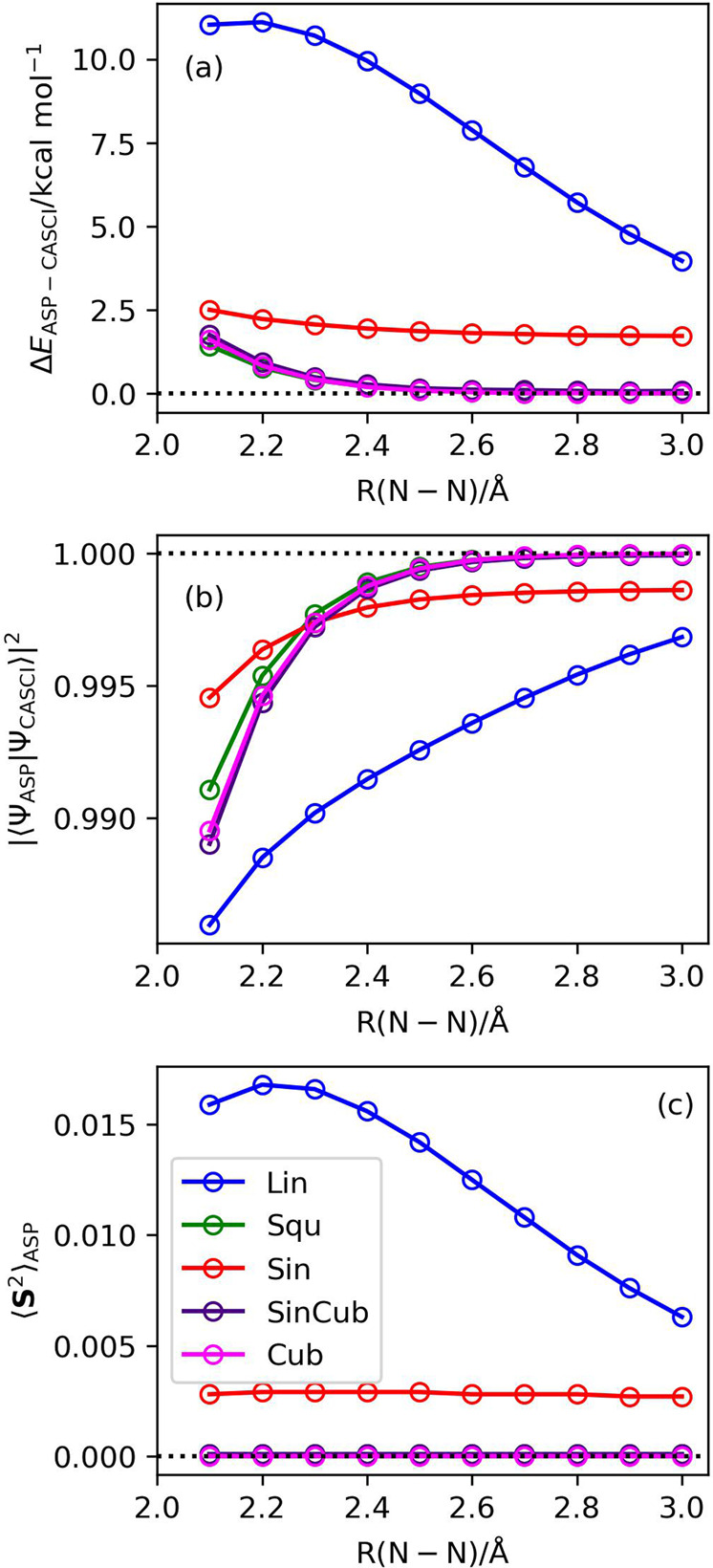
a The energy deviations from the CASCI values. b The square overlaps with the CASCI wave functions. c The 〈S2〉 values.
Again, the reason why the square, sinusoidal cubic, and cubic scheduling functions gave good results can be explained by the structure of wave functions and the energy landscape of the adiabatic evolution pathway. As given in Eqs. 3 and 4, the |ΨBS〉 is expressed by the linear combination of electronic states belonging to different spin multiplicities. These electronic states have similar energies in the beginning of ASP, but the quasi-degeneracy is gradually lifted as the ASP proceeds, owing to the S2 operator introduced as the penalty term in the Hamiltonian. Therefore, variation of the Hamiltonian must slow in the earlier stage of ASP, if the |ΨBS〉 is used as the starting Hamiltonian. All the square, sinusoidal cubic, and cubic functions have zero gradient ds(x)/dx = 0 at x = 0, which is very important to acquire good wave functions in the ASP with the |ΨBS〉.
Note that ASP with the |ΨBS〉 has not only advantages described above but also disadvantages. The BS method generally breaks the spatial symmetry as well as the spin symmetry, because the localized orbitals are prepared by taking a linear combination of HOMO – i and LUMO + i orbitals. In the ASP starting from |ΨHF〉, we can utilize the point group symmetry to reduce the number of the nonzero Hamiltonian terms, but such reduction is not applicable for the |ΨBS〉 with localized MOs. In addition, the computational cost of the time evolution operator under the S2 operator, exp(−iS2t) also pushes the computational cost of ASP. The scaling of the computational cost of simulating exp(−iS2t) is O(nMO2M), where nMO is the number of molecular orbitals in the active space and M refers to the number of ASP steps. As a result, the computational cost of each ASP step is larger by using the |ΨBS〉 than that for using the |ΨHF〉, especially for the molecular systems with high point group symmetry. However, for large molecular systems, it is possible to take advantage of the locality of MOs to reduce the computational cost, by ignoring the Hamiltonian terms having the norms smaller than the threshold44. It should be also noted that our numerical simulations suggest QITE can be also accelerated by using the |ΨBS〉 as the starting wave function in conjunction with the S2 penalty term in the Hamiltonian.
A criterion for the switching of the starting wave function
In the previous section, we demonstrated that ASP using the |ΨBS〉 converges quickly in the region of the bond dissociation. However, we can expect that the |ΨHF〉 is more suitable for the starting wave function of ASP around the equilibrium geometry. It should be also noted that the BS-UHF calculation converges to the RHF solution for shorter bond lengths. Constructing a guideline for the switching of the starting wave function is important toward practical application of ASP to other molecular systems.
Here, we have examined a criterion for selecting the starting wave function based on diradical characters y45,46. Diradical characters are used as the measure of open shell electronic configurations. At the spin-projected UHF level, they can be calculated from the occupation number of the unoccupied natural orbitals nLUNO+i, using Eq. 646.
| 6 |
In the triple bond dissociation of N2 molecule, two types of diradical characters can have significant values: y(π) = y0 = y1 and y(σ) = y2, those reflect open shell electronic configurations in the valence π and σ bonds, respectively. Thus, there are two possible choices of the |ΨBS〉 in the ASP study of the potential energy curve of N2 molecule, |ΨBS2〉 and |ΨBS3〉. The |ΨBS2〉 is the four-spin BS wave function where two π orbitals are treated by means of the BS approach. The |ΨBS3〉 is a six-spin BS wave function and both the σ and π orbitals are dealt with the BS framework.
We assume that the BS treatment is more feasible if the corresponding diradical character is larger. Following this assumption, we explored the threshold value of the diradical characters for the switching of the starting wave function. By setting the evolution time length T = 20/Δε for the ASP with the |ΨHF〉 and T = 50 for that with the |ΨBS〉, we found that setting the threshold value for the diradical characters to be 0.6 gives fairly good results, from the viewpoints of both the evolution time length and quality of the |ΨASP〉, in the potential energy curve of N2 molecule (Fig. 8). This is the result of particular ASP conditions, N2 molecule with (6e,6o) active space and STO-3G basis set with the sinusoidal and square scheduling functions for the |ΨHF〉 and the |ΨBS〉, respectively, and it is natural that different molecular systems and different computational conditions will give different optimal threshold values for the switching of the starting wave function. We also carried out the ASP simulations of N2 molecule using the 6-31G* and 6-311G* basis sets with (6e,6o), (10e,8o), (6e,8o), and (10e,10o) active spaces, obtaining qualitatively the same results (see Supplementary Note 4). Nevertheless, the exploration of other molecular systems is necessary to get further insight of ASP. In the following sections, we describe the results of ASP in the potential energy curve under the symmetric Be–H bond dissociation in the linear BeH2 molecule, and the C2v quasi-reaction pathway of the Be + H2 → BeH2 reaction.
Fig. 8. Results of the numerical simulation of ASP of the potential energy curve for N2 molecule, using the diradical characters as the indicator for selecting the initial wave function.

Background colors specify the regions of the initial wave function recommended from the criterion based on the diradical character y > 0.6. Sinusoidal and square functions were adopted as the scheduling functions with the |ΨHF〉 and |ΨBS〉, respectively, as the starting wave function. a Diradical characters calculated using Eq. 6. b Evolution time lengths. c The energy differences from the CASCI values. (d) The square overlaps with the CASCI wave functions.
Potential energy curve of the symmetric bond dissociation in the linear BeH2
BeH2 is a linear molecule with R(Be–H) = 1.326 Å in the equilibrium geometry47. Symmetric elongation of two Be–H bonds generates a Be atom in the (1s)2(2s)2 closed-shell singlet electron configuration and two H atoms. In this work, we have explored the potential energy curve in the range of R(Be–H) from 0.7 to 4.0 Å. The BS-UHF/STO-3G calculations converged to the RHF solution at the bond length R(Be–H) = 1.9 Å and shorter. The calculated diradical character y is plotted in Fig. 9a. By adopting the criterion for selecting the starting wave function discussed in the previous section, we expect that the |ΨBS〉 is suitable for the starting wave function of ASP at the bond length R(Be–H) = 2.5 Å and longer. The quantum circuit simulation results are summarized in Fig. 9. By switching the starting wave function from the |ΨHF〉 to the |ΨBS〉 at the bond length R(Be–H) = 2.5 Å, we obtained the |ΨASP〉 with the square overlap |〈ΨASP|ΨCASCI〉|2 > 0.98 for all the bond lengths being studied. These results exemplify the usefulness of the diradical character as the indicator of the starting wave function switching. However, the |〈ΨASP|ΨCASCI〉|2 values are slightly larger in the geometries with intermediate diradical characters (R(Be–H) ~ 2.4 Å). The ASP simulations with the longer evolution time revealed that we can achieve |〈ΨASP|ΨCASCI〉|2 > 0.998 for T = 200 and 100 with |ΨHF〉 and |ΨBS〉, respectively, as the starting wave function (see Supplementary Figs. S16 and S17). We also examined the numerical simulations by using the other scheduling functions for ASP with the |ΨHF〉 and the evolution time length T = 20/Δε. The results are summarized in Supplementary Fig. S18, insisting that the sinusoidal function is suitable for the scheduling function.
Fig. 9. Results of the numerical simulation of ASP of the potential energy curve of the symmetric bond dissociation in BeH2 molecule, using the diradical character as the indicator for selecting the initial wave function selection.

Background colors specify the regions of the initial wave function recommended from the criterion based on the diradical character y > 0.6. Sinusoidal and square functions were adopted as the scheduling functions with the |ΨHF〉 and |ΨBS〉, respectively, as the starting wave function. a Diradical characters calculated using Eq. 6. b Evolution time lengths. c The energy differences from the CASCI values. d The square overlaps with the CASCI wave functions.
The C2v quasi-reaction pathway of Be atom insertion to H2
Finally, we have examined ASP in the C2v quasi-reaction pathway of the Be atom insertion to H2 molecule. This system has been widely studied as the model of strongly correlated electronic structures48–52. Cartesian coordinates of H atoms are summarized in Table 2. The reaction pathway contains the S0–S1 avoided crossing at the transition structure (point E in Table 2), and the energy gap between the S0 and S1 states becomes small around the transition structure.
Table 2.
Cartesian coordinates of H atoms for the points being studied in the Be + H2 → BeH2 reaction, in units of Bohr[a].
| Point | X | Y | Z |
|---|---|---|---|
| A | 0.000 | ±2.540 | 0.000 |
| B | 0.000 | ±2.080 | 1.000 |
| C | 0.000 | ±1.620 | 2.000 |
| D | 0.000 | ±1.390 | 2.500 |
| E | 0.000 | ±1.275 | 2.750 |
| F | 0.000 | ±1.160 | 3.000 |
| G | 0.000 | ±0.930 | 3.500 |
| H | 0.000 | ±0.700 | 4.000 |
| I | 0.000 | ±0.700 | 6.000 |
[a]Be atom is located at the origin of coordinates.
Results of the numerical quantum circuit simulations are summarized in Fig. 10. By using the |ΨHF〉 as the starting wave function and setting the evolution time length as T = 20/Δε and using the sinusoidal scheduling function, we obtained the correlated wave function with the square overlap close to unity, except for point E. To disclose the convergence behavior of ASP at point E, we examined the ASP simulations with longer evolution time lengths T = 200, 300, 400, and 500. The square overlaps were calculated to be |〈ΨASP|ΨCASCI〉|2 = 0.9308, 0.9720, 0.9894, and 0.9962, respectively, and thus the convergence of the square overlap against the evolution time length T is very slow. The simulations around point E with finer geometrical changes revealed that the square overlap is smaller for the geometry closer to the transition structure (see Supplementary Note 7). We also computed the energy landscape of the instantaneous Hamiltonian at point E, finding that the S1 and S0 states become almost gapless around s(x) = 0.9 (see Supplementary Fig. S19). The 16, 14, and 12 qubit ASP simulations by removing the highest virtual orbitals one by one gave the square overlaps |〈ΨASP|ΨCASCI〉|2 = 0.8464, 0.8538, and 0.9751, respectively. These results exemplify that selecting appropriate active orbitals is essential to obtain sophisticated wave functions from ASP. In fact, 16 qubit ASP simulations using the natural orbitals constructed from CISD/6-31G* calculations gave the square overlap larger than 0.98 for all points being studied (see Supplementary Fig. S21).
Fig. 10. Results of the numerical simulation of ASP of the C2v quasi-reaction pathway of Be + H2 → BeH2, using the diradical character as the indicator for selecting the initial wave function.

Background colors specify the regions of the initial wave function recommended from the criterion based on the diradical character y > 0.6. Sinusoidal and square functions were adopted as the scheduling functions with the |ΨHF〉 and |ΨBS〉, respectively, as the starting wave function. a Diradical characters calculated using Eq. 6. b Evolution time lengths. c The energy differences from the CASCI values. d The square overlaps with the CASCI wave functions.
The BS-UHF calculations revealed that points D, E, and F have non-negligible diradical characters. The diradical characters were calculated to be y = 0.2991, 0.7851, and 0.6125 for points D, E, and F, respectively. The diradical character indicates that the |ΨBS〉 is suitable for the starting wave function of ASP in points E and F. Our numerical simulations revealed that ASP with the |ΨBS〉 gave larger square overlap values with the CASCI wave functions not only at points E and F but also at point D, with shorter evolution time length than ASP with the |ΨHF〉. More extended studies are necessary to thoroughly understand the performance of ASP, which is left as future work.
Conclusions
In this work, we have examined the numerical quantum circuit simulations of ASP in N2 and BeH2 molecules, seeking practical computational conditions for the generation of correlated wave functions having the square overlap with the CASCI wave function close to unity. The numerical quantum circuit simulations revealed that if the |ΨHF〉 is employed as the starting wave function, the sinusoidal function s(x) = sin(πx/2), where x = t/T, gives the wave function having large square overlaps for shorter evolution time lengths among the five scheduling functions being tested. By using the |ΨHF〉 as the starting wave function, the evolution time length required to achieve sufficiently large square overlap with the CASCI wave function increases with increasing the open-shell character and decreasing the HOMO−LUMO gap. By setting the evolution time length T = 20/Δε, where Δε denotes the HOMO−LUMO gap, we can obtain the correlated wave function with the square overlap larger than 0.95, except for point E as given in the Be + H2 → BeH2 reaction pathway. By contrast, by using the |ΨBS〉 as the starting wave function and introducing the S2 operator as the penalty term in the time-dependent Hamiltonian, the ASP simulations with the square function s(x) = 3x2 − 2x3, sinusoidal cubic function s(x) = sin3(πx/2), and cubic function s(x) = 6x5 − 15x4 + 10x3 gave the correlated wave function with the square overlap close to unity with substantially shorter evolution time length than ASP with the |ΨHF〉 when the diradical character y is large. The results of the present numerical quantum circuit simulations indicate that ASP is capable of generating the correlated wave functions with sufficiently large square overlap with the CASCI wave function by switching the starting wave function from the |ΨHF〉 to the |ΨBS〉 and simultaneously changing the scheduling function from the sinusoidal to the square functions, when the diradical character exceeds 0.6. ASP numerical simulations with larger basis sets revealed the importance of the appropriate active space selections based on the occupation number of natural orbitals to acquire sophisticated wave functions from ASP.
It should be emphasized that the computational conditions described in this paper do not have to be optimal for any molecular systems. The number of theoretical and experimental studies of ASP is not many and more elaborated investigations are necessary to shed light on the performance of ASP. Another important direction of the study of ASP is to connect to the QPE algorithms including the Bayesian phase difference estimation algorithm, which is a controlled-time evolution-free algorithm5,53. The relevant studies are underway and will be published in the forthcoming paper.
Methods
Adiabatic state preparation
Assume that the quantum system is in the ground state of an initial Hamiltonian HI at t = 0, and the Hamiltonian of the system changes slowly. If the variation of the Hamiltonian is sufficiently slow, the system remains in the ground state of the instantaneous Hamiltonian at t > 0, which is known as an adiabatic theorem19. ASP utilizes the adiabatic theorem to obtain the full-CI wave function, by using the Fock operator F defined in Eq. 7 as HI and the electronic Hamiltonian H in Eq. 8 as HP.
| 7 |
| 8 |
Throughout this paper, we used indices i and j for the occupied spin orbitals in the starting wave function, a and b for the unoccupied spin orbitals, and p, q, r, and s for general spin orbitals. hpq and hpqrs are one- and two-electron molecular orbital (MO) integrals defined in Eqs. 9 and 10, respectively. ap† and ap are creation and annihilation operators, respectively, acting on the pth spin orbital.
| 9 |
| 10 |
In Eq. 9, A runs over atoms, and ZA and RA are the atomic number and spatial coordinates, respectively, of atom A. ϕp is the spatial part of the spin orbital of p.
In order to implement ASP on a gate model quantum computer, the adiabatic evolution is usually discretized: the evolution time length T is divided into M steps, and the time evolution under the time-independent Hamiltonian Hm in Eqs. 11 and 12 is simulated.
| 11 |
| 12 |
There are several error sources in ASP. For example, if the evolution time length T is too short to follow the adiabatic path or if the step number M is too small and the Hamiltonian of the (m + 1)th step is too different from that of the mth step, the probability to cause nonadiabatic transitions to other electronic states becomes large and the wave function obtained from ASP is expected to have smaller overlap with the exact wave function. Error sources inherent in quantum computing such as decoherence and errors arising from Trotter decomposition, as discussed in the next section, also affect the quality of the wave function. Note that the computational cost of ASP increases linearly to the step number M, although the finer time steps generally give the wave function with the larger overlap with the exact wave function.
Quantum chemical calculations on a quantum computer
To execute ASP on a gate model quantum computer, wave functions are mapped onto qubits by using a fermion–qubit transformation technique, and the quantum circuit corresponding to the time evolution operator is constructed by using basic quantum gate sets54. Several fermion–qubit transformation techniques have been proposed55–58, and in this work we adopted a Jordan–Wigner transformation (JWT)4,55. In the JWT, the wave function of the N spin-orbital systems is mapped onto N qubits, and each qubit stores an occupation number of a particular spin orbital: the qubit is in the |1〉 state if the corresponding spin orbital is occupied by an electron, otherwise in the |0〉 state. The creation and annihilation operators appearing in the second quantized Hamiltonian (Eqs. 7 and 8) are transformed onto the direct products of Pauli operators (Pauli strings) using Eqs. 13 and 14, respectively.
| 13 |
| 14 |
Here, Xp, Yp, and Zp are Pauli operators defined in Eqs. 15–17, acting on the pth qubit.
| 15 |
| 16 |
| 17 |
By applying the JWT, the second quantized Hamiltonians in Eqs. 7 and 8 are transformed onto qubit Hamiltonians consisting of a linear combination of Pauli strings, as in Eqs. 18 and 19.
| 18 |
| 19 |
The time evolution operator U is defined as in Eq. 20. Trotter–Suzuki decomposition59,60 is usually used to decompose the time evolution operator and to construct the corresponding quantum circuits. The time evolution operators obtained by applying the first- and second-order Trotter–Suzuki decompositions are given in Eqs. 21 and 22, respectively61.
| 20 |
| 21 |
| 22 |
Note that magnitude of the Trotter decomposition error depends on the ordering of terms. It is known that the magnitude ordering in which Hamiltonian terms are applied in the descending order of the absolute value of the coefficient |ωk| often gives smaller Trotter decomposition errors than the lexicographical ordering that is an ordering scheme, which maximizes the similarity of the Pauli strings of adjacent terms62,63. Once the Trotter decomposition is applied, the quantum circuit corresponding to the operator exp(−iωkPkt/L) is constructed by following the literature54. Supplementary Fig. S1 illustrates the quantum circuit corresponding to the operator exp(−iωX0Z1Z2X3t) as an example. Definitions of the quantum gates are also given in the Supplementary Note 1.
Implementation of ASP
ASP can be implemented by the following procedures. (1) Perform the RHF or BS-UHF calculations using conventional quantum chemistry program packages and compute the molecular integrals defined in Eqs. 9 and 10. (2) Divide the electronic Hamiltonian (Eq. 8) into the Fock operator (Eq. 7) and the rest terms, and apply fermion–qubit transformation to them to obtain corresponding qubit Hamiltonians. (3) Set the computational conditions for ASP. Select the initial wave function, scheduling function for adiabatic evolution, total evolution time, time for the single Trotter step, strategy for Trotter term ordering, and so on. (4) Compute the instantaneous Hamiltonian at each discretized time step, and construct the quantum circuit corresponding to the time evolution operator. (5) Encode the starting wave function on the Norb of qubits, where Norb is the number of spin orbitals in the active space, and execute the quantum circuit constructed in the step 4.
The steps 1–4 above are performed on a classical computer, and the step 5 is the main part of the ASP computation. If we adopted |ΨHF〉 as the starting wave function and the JWT for fermion–qubit transformation, the wave function encoding described in the step 5 can be done by applying the Pauli-X (NOT) gates to the qubits storing the occupation number of occupied orbitals in the |ΨHF〉 to the quantum states initialized to the |00…0〉 state.
Note that in most of adiabatic quantum computing problems the initial Hamiltonian contains the terms those are not included in the final Hamiltonian. On the contrary, all the terms in the Fock operator in Eq. 7 are included in the electronic Hamiltonian in Eq. 8. Therefore, the instantaneous Hamiltonian H(m) defined in Eq. 12 can be rewritten as in Eq. 23, where Fock specifies the qubit Hamiltonian corresponding to the Fock operator.
| 23 |
In the quantum circuit level, the quantum circuit at each time step has exactly the same structure with different rotational angles θ of the Rz gate (see Supplementary Fig. S1).
Computational conditions
In this work, we have focused on three molecular systems: the potential energy curve of the triple bond dissociation of N2 molecule, the symmetric Be–H bond dissociations in linear BeH2 molecule, and the C2v quasi-reaction pathway of a Be atom insertion to a H2 molecule, at the CASCI level of theory. For the study of the potential energy curve of N2 molecule under the triple bond dissociation, we used the STO-3G basis set in conjunction with the six electrons in the six orbital (6e, 6o) active space consisting of valence σ/σ* and π/π* orbitals. The potential energy curve of BeH2 molecule under the symmetric Be–H bond cleavage was studied by using the STO-3G basis set and the full-valence (4e, 6o) active space. The quasi-reaction pathway of Be + H2 → BeH2 was investigated by using the basis set comprised of (10s 3p)/[3s 1p] for Be and (4s)/[2s] for H, which was used by Purvis and coworkers44 for the study of the same system. Frozen core orbital approximation was adopted for the CASCI calculations and thus the active space is (4e, 9o).
For the numerical quantum circuit simulations of ASP executable on classical computers, we developed a python program by utilizing Cirq64 and OpenFermion65 libraries. The step number M in Eqs. 11 and 12 was set to be using a ceil function, and the quantum circuit for the time evolution operator of each step was constructed by adopting the second-order Trotter–Suzuki decomposition with L = 1 in Eq. 22. We used the magnitude ordering for the Trotterized time evolution operators, and the ordering of the terms were optimized for every time step.
For the preparation of the starting wave functions of ASP, we performed the RHF and the BS-UHF calculations using GAMESS-US program package66. One- and two-electron atomic orbital integrals were also computed using GAMESS-US software, and MO integrals hpq and hpqrs in Eqs. 9 and 10 were generated by using our own AO → MO integral transformation program.
Supplementary information
Acknowledgements
This work was supported by JST PRESTO “Quantum Software” project (Grant No. JPMJPR1914), Japan, and KAKENHI Scientific Research C (Grant No. 18K03465 and 21K03407) from JSPS, Japan. This work was partially supported by AOARD Scientific Project on “Molecular Spins for Quantum Technologies” (Grant FA2386-17-1-4040, 4041), USA. K.Su. thanks Prof. Y. Mochizuki (Rikkyo University) for helpful discussions.
Author contributions
K.Su., K.Sa., and T.T. planned and conducted the project. K.Su. developed the numerical simulation programs and performed quantum chemical calculations and numerical simulations. K.T. and D.S. discussed the results. K.Su. and T.T. wrote the paper.
Peer review
Peer review information
Communications Chemistry thanks Libor Veis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The data that support the findings on this study are available from the corresponding authors on reasonable request.
Code availability
The python source code for the numerical quantum circuit simulations of ASP and integral files are available via github repository: https://github.com/Kenji-Sugisaki/ASP.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kenji Sugisaki, Email: sugisaki@osaka-cu.ac.jp.
Kazunobu Sato, Email: sato@osaka-cu.ac.jp.
Takeji Takui, Email: takui@sci.osaka-cu.ac.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-022-00701-8.
References
- 1.Nielsen, M. A. & Chuang, I. L. Quantum Computation and Quantum Information, 10th Anniversary ed. (Cambridge University Press, 2010).
- 2.Rossi E, Bendazzoli GL, Evangelisti S, Maynau D. A full-configuration benchmark for the N2 molecule. Chem. Phys. Lett. 1999;310:530–536. doi: 10.1016/S0009-2614(99)00791-5. [DOI] [Google Scholar]
- 3.Gan Z, Grant DJ, Harrison RJ, Dixon DA. The lowest energy states of the group-IIIA–group-VA heteronuclear diatomics: BN, BP, AlN, and AlP from full configuration interaction calculations. J. Chem. Phys. 2006;125:124311. doi: 10.1063/1.2335446. [DOI] [PubMed] [Google Scholar]
- 4.Aspuru-Guzik A, Dutoi AD, Love PJ, Head-Gordon M. Simulated quantum computation of molecular energies. Science. 2005;309:1704–1707. doi: 10.1126/science.1113479. [DOI] [PubMed] [Google Scholar]
- 5.Sugisaki K, et al. Quantum algorithm for full configuration interaction calculations without controlled time evolutions. J. Phys. Chem. Lett. 2021;12:11085–11089. doi: 10.1021/acs.jpclett.1c03214. [DOI] [PubMed] [Google Scholar]
- 6.Peruzzo A, et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 2014;5:4213. doi: 10.1038/ncomms5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Hu J, Zhang X-M, Song Z, Yung M-H. Variational quantum simulation for quantum chemistry. Adv. Theory Simul. 2019;2:1800182. doi: 10.1002/adts.201800182. [DOI] [Google Scholar]
- 8.Cerezo M, et al. Variational quantum algorithms. Nat. Rev. Phys. 2021;3:625–644. doi: 10.1038/s42254-021-00348-9. [DOI] [Google Scholar]
- 9.Tilly, J. et al. The variational quantum eigensolver: a review of methods and best practices. arXiv:2111:05176, https://arxiv.org/abs/2111.05176.
- 10.Bharti K, et al. Noisy intermediate-scale quantum (NISQ) algorithms. Rev. Mod. Phys. 2022;94:015004. doi: 10.1103/RevModPhys.94.015004. [DOI] [Google Scholar]
- 11.Cao Y, et al. Quantum chemistry in the age of quantum computing. Chem. Rev. 2019;119:10856–10915. doi: 10.1021/acs.chemrev.8b00803. [DOI] [PubMed] [Google Scholar]
- 12.McArdle S, Endo S, Aspuru-Guzik A, Benjamin SC, Yuan X. Quantum computational chemistry. Rev. Mod. Phys. 2020;92:015003. doi: 10.1103/RevModPhys.92.015003. [DOI] [Google Scholar]
- 13.Head-Marsden K, Flick J, Ciccarino CJ, Narang P. Quantum information and algorithms for correlated quantum matter. Chem. Rev. 2021;121:3061–3120. doi: 10.1021/acs.chemrev.0c00620. [DOI] [PubMed] [Google Scholar]
- 14.Sugisaki K, et al. Quantum chemistry on quantum computers: a polynomial-time quantum algorithm for constructing the wave functions of open-shell molecules. J. Phys. Chem. A. 2016;120:6459–6466. doi: 10.1021/acs.jpca.6b04932. [DOI] [PubMed] [Google Scholar]
- 15.Sugisaki K, et al. Open shell electronic state calculations on quantum computers: a quantum circuit for the preparation of configuration state functions based on Serber construction. Chem. Phys. Lett. 2019;737S:100002. doi: 10.1016/j.cpletx.2018.100002. [DOI] [Google Scholar]
- 16.Sugisaki K, et al. Quantum chemistry on quantum computers: a method for preparation of multiconfigurational wave functions on quantum computers without performing post-Hartree–Fock calculations. ACS Cent. Sci. 2019;5:167–175. doi: 10.1021/acscentsci.8b00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halder, D., Prasannaa, S. V., Agarawal, V. & Maitra, R. Digital quantum simulation of strong correlation effects with iterative quantum phase estimation over the variational quantum eigensolver algorithm: H4 on a circle as a case study. arXiv:2110.02864, https://arxiv.org/abs/2110.02864.
- 18.Sugisaki K, Kato T, Minato Y, Okuwaki K, Mochizuki Y. Variational quantum eigensolver simulations with the multireference unitary coupled cluster ansatz: a case study of the C2v quasi-reaction pathway of beryllium insertion into a H2 molecule. Phys. Chem. Chem. Phys. 2022;24:8439–8452. doi: 10.1039/D1CP04318H. [DOI] [PubMed] [Google Scholar]
- 19.Bohn M, Fock V. Beweis des adiabatensatzes. Z. Phys. 1928;51:165–180. doi: 10.1007/BF01343193. [DOI] [Google Scholar]
- 20.Farhi, E., Goldstone, J., Gutmann, S. & Sipser, M. Quantum computation by adiabatic evolution. arXiv:quant-ph/0001106, https://arxiv.org/abs/quant-ph/0001106.
- 21.Albash T, Lidar DA. Adiabatic quantum computation. Rev. Mod. Phys. 2018;90:015002. doi: 10.1103/RevModPhys.90.015002. [DOI] [Google Scholar]
- 22.Yu H, Wei T-C. Quantum Zeno approach for molecular energies with maximum commuting initial Hamiltonians. Phys. Rev. Res. 2021;3:013104. doi: 10.1103/PhysRevResearch.3.013104. [DOI] [Google Scholar]
- 23.Du J, et al. NMR implementation of a molecular hydrogen quantum simulation with adiabatic state preparation. Phys. Rev. Lett. 2010;104:030502. doi: 10.1103/PhysRevLett.104.030502. [DOI] [PubMed] [Google Scholar]
- 24.Veis L, Pittner J. Adiabatic state preparation study of methylene. J. Chem. Phys. 2014;140:214111. doi: 10.1063/1.4880755. [DOI] [PubMed] [Google Scholar]
- 25.Matsuura S, Buck S, Senicourt V, Zaribafiyan A. Variationally scheduled quantum simulation. Phys. Rev. A. 2021;103:052435. doi: 10.1103/PhysRevA.103.052435. [DOI] [Google Scholar]
- 26.Kremenetski V, Mejuto-Zaera C, Cotton SJ, Tubman NM. Simulation of adiabatic quantum computing for molecular ground states. J. Chem. Phys. 2021;155:234106. doi: 10.1063/5.0060124. [DOI] [PubMed] [Google Scholar]
- 27.McArdle S, et al. Variational ansatz-based quantum simulation of imaginary time evolution. npj Quantum Info. 2019;5:75. doi: 10.1038/s41534-019-0187-2. [DOI] [Google Scholar]
- 28.Motta M, et al. Determining eigenstates and thermal states on a quantum computer using quantum imaginary time evolution. Nat. Phys. 2020;16:205–210. doi: 10.1038/s41567-019-0704-4. [DOI] [Google Scholar]
- 29.Yater-Aydeniz K, Pooser RC, Siopsis G. Practical quantum computation of chemical and nuclear energy levels using quantum imaginary time evolution and Lanczos algorithms. npj Quantum Info. 2020;6:63. doi: 10.1038/s41534-020-00290-1. [DOI] [Google Scholar]
- 30.Gomes N, et al. Adaptive variational quantum imaginary time evolution approach for ground state preparation. Adv. Quantum Technol. 2021;4:2100114. doi: 10.1002/qute.202100114. [DOI] [Google Scholar]
- 31.Hu H, Wu B. Optimizing the quantum adiabatic algorithm. Phys. Rev. A. 2016;93:012345. doi: 10.1103/PhysRevA.93.012345. [DOI] [Google Scholar]
- 32.Amin MHS. Consistency of the adiabatic theorem. Phys. Rev. Lett. 2009;102:220401. doi: 10.1103/PhysRevLett.102.220401. [DOI] [PubMed] [Google Scholar]
- 33.Marzlin K-P, Sanders BC. Inconsistency in the application of the adiabatic theorem. Phys. Rev. Lett. 2004;93:160408. doi: 10.1103/PhysRevLett.93.160408. [DOI] [PubMed] [Google Scholar]
- 34.Tong DM, Singh K, Kwek LC, Oh CH. Quantitative conditions do not guarantee the validity of the adiabatic approximation. Phys. Rev. Lett. 2005;95:110407. doi: 10.1103/PhysRevLett.95.110407. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z-Y, Plenio MB. Necessary and sufficient condition for quantum adiabatic evolution by unitary control fields. Phys. Rev. A. 2016;93:052107. doi: 10.1103/PhysRevA.93.052107. [DOI] [Google Scholar]
- 36.Xu K, et al. Breaking the quantum adiabatic speed limit by jumping along geodesics. Sci. Adv. 2019;5:eaax3800. doi: 10.1126/sciadv.aax3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guéry-Odelin D, et al. Shortcuts to adiabaticity: Concepts, methods, and applications. Rev. Mod. Phys. 2019;91:045001. doi: 10.1103/RevModPhys.91.045001. [DOI] [Google Scholar]
- 38.Yamaguchi K. The electronic structures of biradicals in the unrestricted Hartree–Fock approximation. Chem. Phys. Lett. 1975;33:330–335. doi: 10.1016/0009-2614(75)80169-2. [DOI] [Google Scholar]
- 39.Shoji M, et al. A general algorithm for calculation of Heisenberg exchange integrals J in multi-spin systems. Chem. Phys. Lett. 2006;432:343–347. doi: 10.1016/j.cplett.2006.10.023. [DOI] [Google Scholar]
- 40.Bally, T. & Borden, W. T. Calculations on open-shell molecules: a beginner’s guide in Reviews in Computational Chemistry, vol. 13, (eds. Lipkowitz, K. B. & Boyd, D. B.) 1–97 (Wiley-VCH, 1999).
- 41.Pauncz, R. The Construction of Spin Eigenfunctions. An Exercise Book. (Kluwer Academic/Plenum Publishers, 2000).
- 42.Mayer, I. The spin-projected extended Hartree–Fock method in Advances in Quantum Chemistry, vol. 12, (ed. Löwdin, P. O.) 189–262 (Elsevier, 1980).
- 43.Sugisaki K, et al. Quantum chemistry on quantum computers: quantum simulations of the time evolution of wave functions under the S2 operator and determination of the spin quantum number. S. Phys. Chem. Chem. Phys. 2019;21:15356–15361. doi: 10.1039/C9CP02546D. [DOI] [PubMed] [Google Scholar]
- 44.McClean JR, Babbush R, Love PJ, Aspuru-Guzik A. Exploiting locality in quantum computation for quantum chemistry. J. Phys. Chem. Lett. 2014;5:4368–4380. doi: 10.1021/jz501649m. [DOI] [PubMed] [Google Scholar]
- 45.Hayes EF, Siu AKQ. Electronic structure of the open forms of three-membered rings. J. Am. Chem. Soc. 1971;93:2090–2091. doi: 10.1021/ja00737a064. [DOI] [Google Scholar]
- 46.Döhnert D, Koutecky J. Occupation numbers of natural orbitals as a criterion for biradical character. Different kinds of biradicals. J. Am. Chem. Soc. 1980;102:1789–1796. doi: 10.1021/ja00526a005. [DOI] [Google Scholar]
- 47.Shayesteh A, Tereszchuk K, Bernath PF, Colin R. Infrared emission spectra of BeH2 and BeD2. J. Chem. Phys. 2003;118:3622–3627. doi: 10.1063/1.1539850. [DOI] [Google Scholar]
- 48.Purvis GD, III, Shepard R, Brown FB, Bartlett RJ. C2v insertion pathway for BeH2: a test problem for the coupled-cluster single and double excitation model. Int. J. Quantum Chem. 1983;23:835–845. doi: 10.1002/qua.560230307. [DOI] [Google Scholar]
- 49.Nakano H. Quasidegenerate perturbation theory with multiconfigurational self-consistent-field reference functions. J. Chem. Phys. 1993;99:7983–7992. doi: 10.1063/1.465674. [DOI] [Google Scholar]
- 50.Mahapatra US, Datta B, Mukherjee D. Molecular applications of a size-consistent state-specific multireference perturbation theory with relaxed model-space coefficients. J. Phys. Chem. A. 1999;103:1822–1830. doi: 10.1021/jp9832995. [DOI] [PubMed] [Google Scholar]
- 51.Mahapatra US, Datta B, Mukherjee D. A size-consistent state-specific multireference coupled-cluster theory: formal developments and molecular applications. J. Chem. Phys. 1999;110:6171–6188. doi: 10.1063/1.478523. [DOI] [Google Scholar]
- 52.Evangelista FA. Alternative single-reference coupled cluster approaches for multireference problems: The simpler, the better. J. Chem. Phys. 2011;134:224102. doi: 10.1063/1.3598471. [DOI] [PubMed] [Google Scholar]
- 53.Sugisaki K, et al. Bayesian phase difference estimation: a general quantum algorithm for the direct calculation of energy gaps. Phys. Chem. Chem. Phys. 2021;23:20152–20162. doi: 10.1039/D1CP03156B. [DOI] [PubMed] [Google Scholar]
- 54.Whitfield JD, Biamonte J, Aspuru-Guzik A. Simulation of electronic structure Hamiltonians using quantum computers. Mol. Phys. 2011;109:735–750. doi: 10.1080/00268976.2011.552441. [DOI] [Google Scholar]
- 55.Jordan P, Wigner E. Über das Paulische äquivalenzverbot. Z. Phys. 1928;47:631–651. doi: 10.1007/BF01331938. [DOI] [Google Scholar]
- 56.Verstraete, F. & Cirac, J. I. Mapping local Hamiltonians of fermions to local Hamiltonians of spins. J. Stat. Mech. P09012 (2005).
- 57.Seeley JT, Richard MJ, Love PJ. The Bravyi–Kitaev transformation for quantum computation of electronic structure. J. Chem. Phys. 2012;137:224109. doi: 10.1063/1.4768229. [DOI] [PubMed] [Google Scholar]
- 58.Setia K, Whitfield JD. Bravyi–Kitaev superfast simulation of electronic structure on a quantum computer. J. Chem. Phys. 2018;148:164104. doi: 10.1063/1.5019371. [DOI] [PubMed] [Google Scholar]
- 59.Trotter HF. On the product of semi-groups of operators. Proc. Am. Math. Soc. 1959;10:545–551. doi: 10.1090/S0002-9939-1959-0108732-6. [DOI] [Google Scholar]
- 60.Suzuki M. Relationship between d-dimensional quantal spin systems and (d + 1)-dimensional Ising systems: equivalence, critical exponents and systematic approximants of the partition function and spin correlations. Prog. Theor. Phys. 1976;56:1454–1469. doi: 10.1143/PTP.56.1454. [DOI] [Google Scholar]
- 61.Hatano, N. & Suzuki, M. Finding exponential product formulas of higher orders in Quantum Annealing and Other Optimization Methods, Lecture Notes in Physics, vol. 679, (eds. Das, A. & Chakrabarti, B. K.) 37–68 (Springer, 2005).
- 62.Tranter A, Love PJ, Mintert F, Coveney PV. A comparison of the Bravyi–Kitaev and Jordan–Wigner transformations for the quantum simulation of quantum chemistry. J. Chem. Theory Comput. 2018;14:5617–5630. doi: 10.1021/acs.jctc.8b00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tranter A, Love PJ, Mintert F, Wiebe N, Coveney PV. Ordering of Trotterization: impact on errors in quantum simulation of electronic structure. Entropy. 2019;21:1218. doi: 10.3390/e21121218. [DOI] [Google Scholar]
- 64.Quantum AI team and collaborators. (March 5, 2021). quantumlib/Cirq: Cirq, Version v0.10.0. Zenodo. 10.5281/zenodo.4586899.
- 65.McClean JR, et al. OpenFermion: the electronic structure package for quantum computers. Quantum Sci. Technol. 2020;5:034014. doi: 10.1088/2058-9565/ab8ebc. [DOI] [Google Scholar]
- 66.Barca GMJ, et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020;152:154102. doi: 10.1063/5.0005188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings on this study are available from the corresponding authors on reasonable request.
The python source code for the numerical quantum circuit simulations of ASP and integral files are available via github repository: https://github.com/Kenji-Sugisaki/ASP.