

Abstract
Polymer beads have been used as the core of magnetic particles for around twenty years. Here we report studies to attach polymetallic complexes to polymer beads for the first time, producing beads of around 115 microns diameter that are attached to 1014 hybrid inorganic-organic [2]rotaxanes. The bead is then formally a [1014] rotaxane. The number of complexes attached is counted by EPR spectroscopy after including TEMPO radicals within the thread of the hybrid [2]rotaxanes.
Subject terms: Interlocked molecules, Coordination chemistry
Polymer beads are used in the core of magnetic particles, and beads functionalised with paramagnetic molecules are promising as agents for dynamic nuclear polarization. Here, the authors use conventional click chemistry to decorate a polymer bead with 1014 [2]rotaxanes containing paramagnetic {Cr7Ni} rings.
Introduction
Since 1999 polymer beads have been used as the core of magnetic particles, with Caruso et al. showing that magnetite could be grown on the exterior of polystyrene1. This hugely influential work involved use of a polyelectrolyte attached to the polystyrene core before attachment of Fe3O4 nanoparticles. Similar approaches have been used to make core-shell structures with other inorganic materials. ZnS has been grown around polystyrene as a route to photonic crystals2 and metal-organic framework shells have been grown as a route to particles that can be used to adsorb pollutants3,4.
Beads functionalised with paramagnetic molecules have been used in catalysis5 and studied as agents for dynamic nuclear polarisation (DNP)6. Further functionality has been introduced by blending these properties on or in polymer hosts, and such magnetic polymer beads have been exploited in many areas, including sensing, imaging and separations7. Radical-bead functionalisation has been used to probe the macromolecular structures of the beads and their swelling on solvation8.
We wondered if it was possible to attach polymetallic compounds to a polystyrene bead. This could then be developed in two distinct directions: the attached compound could be decomposed to give a core-shell particle with great control of the metal shell, and perhaps allow formation of different metal-shells than are accessible from synthesis from simple precursors. Secondly, decorating a bead with a very large number of highly paramagnetic molecules could lead to a use in DNP or as a contrast agent or even as a ferrofluid. However, before speculating on possible applications we needed to establish that the chemistry itself is possible. This is purpose of this report.
Well established chemistry was chosen. To form the link from the bead to the paramagnetic molecule we use conventional copper-catalysed azide-alkyne cycloaddition (CuAAC) click chemistry9. To enable this to work we functionalised commercially available polystyrene microspheres with pendant azides (Fig. 1). As the polymetallic unit we use {Cr7Ni} rings that we have studied over a period of years10. Here, we use the rings as part of a hybrid inorganic–organic [2]rotaxane11,12 and then perform the click chemistry at one end of a carefully designed thread. We find that the chemistry is sufficiently flexible to allow several different [2]rotaxanes to be clicked onto the bead, including [2]rotaxanes that contain organic radicals in addition to the {Cr7Ni} rings. This has allowed us to perform an experiment where we count the spins from the organic radicals and that allows us to calculate that 1014 [2]rotaxanes are attached to the bead. As the bead is a cross-linked polymer, it means that each bead formally corresponds to a [1014]rotaxane.
Fig. 1. Synthesis of a decorated bead.

a General overview. b Reaction conditions: (i) Pivalic acid, 2NiCO3·3Ni(OH)2·4H2O, CrF3·4H2O, 140 °C, 24 h; (ii) Pentynoic acid, DCC, DMAP, THF, 50 °C, 16 h; (iii) 4-Carboxy-TEMPO, DCC, DMAP, CH2Cl2, RT, 48 h. (iv) Azide-terminated polystyrene beads, [Cu(MeCN)4]PF6, CH2Cl2:DMF (3:2), RT, 72 h. † - 115 ± 35 μm. ‡ - 10 ± 1 μm. TEMPO 2,2,6,6-Tetramethylpiperidine 1-oxyl, DCC N,N′-Dicyclohexylcarbodiimide, DMAP 4-Dimethylaminopyridine, THF tetrahydrofuran.
We note that the first reported [2]rotaxane was prepared by repetitive statistical threading-and-capping of a polymer-supported macrocycle13.
Results
Synthesis
The approach is summarised in Fig. 1 and is based on the {Cr7Ni} rings forming around a secondary ammonium cation template, while the exterior of the ring is covered by sixteen pivalate groups, giving [(R2NH2)Cr7NiF8(tBuCO2)16]. Full details are given in the Methods Section and the Supplementary methods sections S1, S3 and S4.
The thread (HOC6H4CH2CH2)2NH 1 is formed from a primary amine and an alkyl bromide, each bearing a terminal phenolic group, and is the neutral precursor to the ammonium template. Choice of the distance between the ammonium and phenol unit is important. If the distance is too short, the phenol will be too sterically hindered for further functionalisation. If the distance is too long, the phenol is esterified by reaction with the pivalic acid used during the formation of the ring12. Here, we aimed for an intermediate distance, but a small amount of esterified product is always obtained when 1 is used to form [2]rotaxanes. This ability to control the reactivity of the phenol is important as it allows difunctionalisation of the thread.
Rotaxane 2 was prepared by reacting 1 with hydrated chromium(III) fluoride and basic nickel carbonate in pivalic acid (See Methods section for preparative details) and was purified by column chromatography. X-ray quality single crystals of 2 were grown from acetone by slow evaporation. The crystal structure of 2 shows the inorganic–organic [2]rotaxane with the axle terminated by phenols.
To introduce a terminal alkyne for CuAAC click chemistry, rotaxane 2 was reacted with 4-pentynoic acid under Steglich esterification conditions to yield 3. A second esterification reaction with 4-carboxy-TEMPO gave 4. Formation of rotaxane 4 was confirmed by electrospray mass spectrometry (ESI-MS), elemental analysis and single-crystal X-ray crystallography.
Rotaxane 4 was reacted with azide-functionalised polymer beads 5 or 6 (Fig. 1, see Methods section for preparative details) in a heterogeneous click reaction. After reaction with the rotaxanes, the initially colourless beads turned deep green giving the decorated beads 7 and 8. To remove any unreacted [2]rotaxane and the catalyst, the beads were then washed repeatedly with CH3CN, CH3OH and CH2Cl2 following recommended methods14.
Product formation was monitored by FT-IR spectroscopy. The IR absorption peak for an azide group around 2090 cm−1 was used as a reference. After reaction with [2]rotaxane 4, a significant decrease in the intensity of the azide band was observed (see SI, section 4). We attribute the small, residual azide band in the IR spectrum to azide functionalities present in inner, inaccessible regions of the bead15.
To estimate the number of solvent-accessible sites we made the following assumptions. Firstly, that the specification given for the beads is accurate (https://www.sigmaaldrich.com/GB/en/product/mm/855011, http://www.rapp-polymere.com/index.php?id=893¤cy=968, http://www.rapp-polymere.com/index.php?id=1219¤cy=498), and that the polymer bead is spherical. Secondly, that the density of accessible sites is uniform across the polymer bead, and that the number of accessible surface sites is not limited to the surface of the bead as there is a degree of swelling and ‘depth’ in which molecules can react at sites further inside the sphere. We then have to estimate a lower bound depth and we chose 0.5 µm. Thirdly, that the surface volume can be determined from the volume of two spheres of different diameters and that the density of polymer sites is uniform at the surface. Given these assumptions and assuming that all azide sites within 0.5 μm of the surface react, the number of rotaxanes on a single bead of diameter 115 µm, 5, was estimated to be at least 2 × 1013 and for the smaller beads, 6, we estimate that 8 × 1010rotaxanes could be attached (see SI, section 2, Fig. S11). The upper bound of these calculations, if all sites within the polymer beads react was 7 × 1014 rotaxanes for 5 and 3 × 1011 rotaxanes for 6. We also assume that the azide formation and click reactions proceed quantitatively16,17.
X-ray crystal structures
Both 2 and 4 crystallise in monoclinic space groups: P21/c and C2/c respectively (Table S1). The structures are shown in Fig. 2. In both structures the eight metals are arranged in an octagon, and the single divalent metal site is not identified by the crystallography but it is disordered about the octagon. Each metal…metal edge is bridged by two carboxylates outside the ring, and by a single fluoride, which lies within a cavity. The eight fluorides therefore provide hydrogen bond acceptor sites, and these form H-bonds to the protonated nitrogen of the threads. The metal sites are all six-coordinate, bound to four O- and two F-donors. The metal–ligand distances are very similar to those found in previous {Cr7Ni} rings10. For attachment to a bead the key metric is the distance from the terminal C-atom of the alkyne to the N-atom at the ring centre and it is around 1.36 nm, which allows the CuAAC click reaction to proceed.
Fig. 2. X-Ray crystal structures of compounds 2, 4 and 10.

Ball and stick representations: a 2; b 4; c 10. Space-filling representations of 4: d into the edge of the {Cr7Ni} ring; e perpendicular to the Cr7Ni} ring. Colours in a, b and c: Cr-green, Ni-violet, F-yellow, O-red, N-blue and C-grey. Colours in d and f: thread shown in silver with pivalate groups on rings excluded for clarity. H-atoms excluded for clarity throughout.
Electron paramagnetic resonance (EPR) spectroscopy and spin counting
Continuous wave (CW) Q-Band (ca. 34 GHz) EPR spectra of 4 and 7 were recorded at 5 K. There is a resonance at g = 1.78, which is only observed at low temperature, which arises from the S = ½ ground state of the {Cr7Ni} ring (Fig. 3 and S19)18. This signal is not perturbed between the rotaxane 4 and bead 7. All spectra were simulated using parameters given in Table 1.
Fig. 3. CW Q-Band (ca. 34 GHz) EPR spectra.
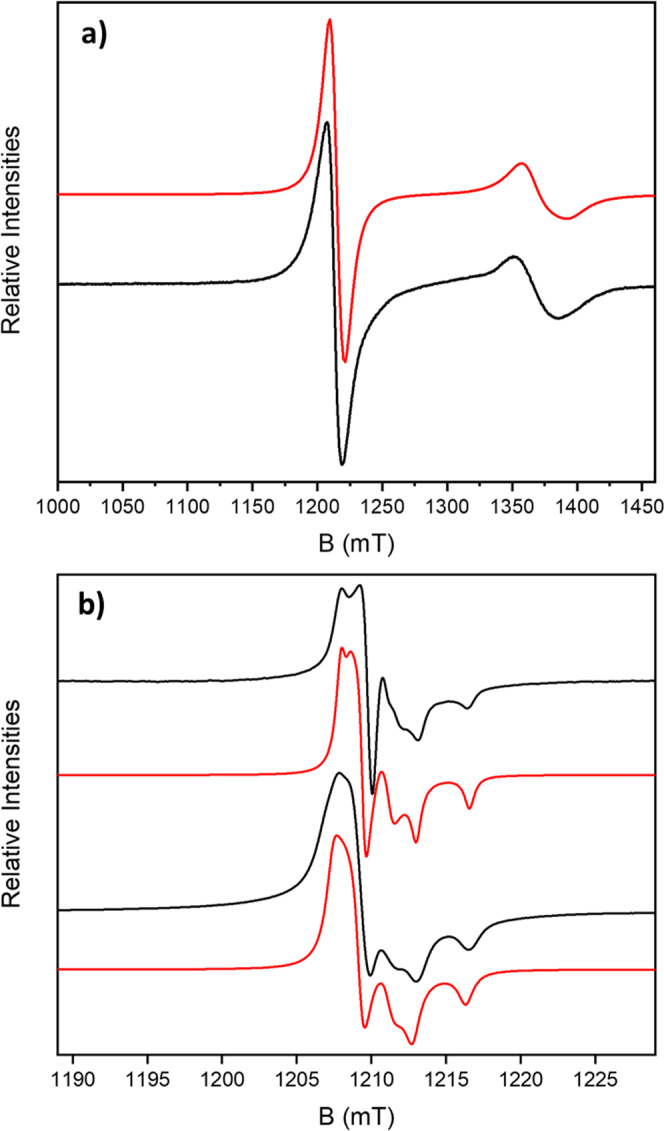
a Powder sample of 4 at 5 K (black) and simulation (red), with the nitroxide and {Cr7Ni} resonances. b Multiple bead samples of 7 at 280 K (black top) and 5 K (black bottom), with their respective simulations (red), focused at the nitroxide signal. The experimental frequencies are a 34.032921 GHz and b 34.033889 GHz (280 K) and 34.040610 GHz (5 K).
Table 1.
EPR simulation parameters for compounds 4, 7 and 8.
| T/ K | NO˙ g values | NAz/MHz | Line width NO˙/mT | {Cr7Ni} g values |
Line width {Cr7Ni}/mT | |
|---|---|---|---|---|---|---|
| 4 | 5 | 2.0106, 2.0060, 2.0030 | 100 | 10 | 1.788, 1.788, 1.748 | 20 |
| 7 | 5 | 2.0106, 2.0060, 2.0030 | 100 | 1.2 | 1.775, 1.775, 1.737 | 20 |
| 7 | 280 | 2.0097, 2.0057, 2.0026 | 100 | 0.8 | N/A | N/A |
| 8 | 5 | 2.0106, 2.0060, 2.0030 | 100 | 5 | 1.775, 1.775, 1.737 | 15 |
Rotaxane 4 and beads 7 and 8 contain TEMPO radicals terminating the thread, and the EPR spectra show resonances for the nitroxides that are observed at all temperatures (Fig. 3). In powdered [2]rotaxane 4, the nitroxide spectrum is broad due to intermolecular interactions, whilst for bead 7 the signal is sharper, with partial resolution of the 14N hyperfine coupling, consistent with greater dilution of the rings on the bead surface. There are several previous examples of rotaxanes that involve TEMPO radicals19–21.
The nitroxide signal at RT for 7 makes it possible to quantify the number of nitroxides, and hence the number of threaded rings, on an individual bead, although the values approach the limit of EPR sensitivity22. Performing the calculation at lower temperatures is far more demanding, especially due to fluctuations in temperature that could influence the intensity of the signal. It is far easier to perform this at the nitroxide resonance than at the {Cr7Ni} resonance as the nitroxide resonance is visible at room temperature (Fig. 3b) and is far sharper than the broad resonance due to the heterometallic ring.
EPR signal intensity calibration curves were constructed from measurements on the [2]rotaxane 4 in toluene solution at room temperature (Fig. S19a, see SI, section 5), using twelve concentrations between 0.005 and 0.20 mM (Figs. S21 and S22). However, in repeated calibrations we found a discontinuity in the signal intensity as a function of concentration at around 0.07 mM, hence we have restricted spin counting on the beads to calibrant concentrations between 0.005 and 0.06 mM.
Spin counting measurements were then performed on beads of 7 (Fig. 4b), using increasing numbers of individual beads from one to fifteen (see SI, section 5 for full details). The signal intensities for the different numbers of beads are mapped onto the calibration curve in Fig. 4c.
Fig. 4. Spin counting on decorated beads.

All spectra recorded at Q-band at room temperature. a First integral of Q-band EPR calibration solutions of 4 in toluene. b First integral of Q-band EPR spectra varying number of beads of 7. c Spin counting on different numbers of beads 7, calibrated against a solution of [2]rotaxane 4. Black figures indicate the number of beads. Measurements for 2, 5, 10, 11 and 13 beads were omitted due to difficulties in obtaining accurate baseline corrections.
The number of nitroxide spins per bead was found to be of the order 1014 (Table 2). Although the signal intensity increases with number of beads, it does not do so linearly. The detection limit of EPR at these frequencies, temperatures and microwave powers is ca. 1012 spins/mT line-width22. Hence, at the lower end of our concentration scale the accuracy of the spin counting experiment is very limited. Moreover, (i) with a single (or a few) beads, positioning of the sample in the EPR tube becomes more crucial, and (ii) there is a distribution of bead sizes with bead diameters between 80 and 150 μm. Hence, we consider the average number of nitroxide spins per bead from the larger numbers of beads to be the most meaningful. The conclusion is that there are ~1014 spins per bead, which is within an order of magnitude of the lowest estimate of the number of rotaxanes that could be bound (ca. 2 × 1013), calculated from the number of solvent-accessible surface sites (See SI, section 2).
Table 2.
Spin counting of the TEMPO radical signal in 7 measured at 34 GHz and room temperature.
| No. of beads | Total no. of spins/1014 | No. of spins per bead/1014 |
|---|---|---|
| 6 | 2.2 | 0.4 |
| 7 | 3.5 | 0.5 |
| 8 | 4.7 | 0.6 |
| 9 | 6.8 | 0.8 |
| 12 | 10.4 | 0.9 |
| 14 | 12.9 | 0.9 |
| 15 | 16.3 | 1.1 |
In addition, 10 µm diameter beads 6 were decorated with 4 giving 8, and these beads were also studied (Fig. S20). The mass of the beads measured (1.35 mg) equates to ~7.1 × 105 beads. The signal intensity observed was compared with our calibration and this leads to 7.2 × 1010 spins/bead, which is within an order of magnitude from the estimate of the number of rotaxanes that could be bound (ca. 8 × 1010), calculated from the number of solvent-accessible surface sites (see SI, section 2). The nitroxide resonance in 4 and 8 is slightly broader than in 7.
Magnetic measurements
Measuring the magnetic properties of samples containing polystyrene beads presents significant challenges, particularly as there is considerable static between the beads within samples. Nonetheless we have attempted to measure the magnetic properties of three samples of beads: an azide-decorated bead 5, 7 and a bead decorated with crystallographically characterised (see Table S1) [2]rotaxane 10 [{(HC≡CCH2CH2C(O)O-Ph-CH2)2NH2}][Cr7NiF8(O2CtBu)16], forming 11 that does not contain the nitroxide radical on the thread (See Methods section for preparative details). The results are shown in Fig. 5 plotted per bead. The values per bead are based on counting ~830 beads per mg in a sample of 11.
Fig. 5. Magnetic properties of decorated beads.

a Variable temperature moment (m) at 1000 Oe for beads scaled to one bead: 5 (grey), 7 (black), 11 (green), 12 calculated with 1 × 1014 rotaxanes (purple squares), 12 calculated with 3 × 1014 rotaxanes (blue triangles). b Variable field moment (m) at 2 K for beads scaled to one bead: 5 (grey), 7 (black), 11 (green), 12 calculated with 1 × 1014 rotaxanes (purple squares), 12 calculated with 3 × 1014 rotaxanes (blue triangles).
At room temperature the susceptibility of 7 and 11 are similar, especially given the limitations of the experiment, and higher than that of the beads alone. As the temperature is lowered the behaviour diverges (Fig. 5a). Both show a maximum at around 25 K in the moment, m, (equivalent to susceptibility, χ), which is found in pure samples of the {Cr7Ni} rings10. This indicates that the predominant paramagnetic species present are intact {Cr7Ni} rings. This led us to compare 7 and 11 with the magnetic behaviour of [Pr2NH2][Cr7NiF8(O2CtBu)16] 12. In Fig. 5a, b we include the molar value for moment for 12, scaled by both 1014/NA and 3 × 1014/NA giving the value for 1014 and 3 × 1014 molecules.
At the highest temperatures the magnetic susceptibilities of both 7 and 11 match the scaled value for 1014 molecules of 12 well. At low T both the temperature and field dependence of the moment for 7 match that for 1014 molecules of 12; this is unexpected as 7 contains an additional s = ½ centre per rotaxane due to the radical. When comparing with 11, the moment in both variable T and H fits with 3 × 1014 molecules of 12. The susceptibility of 11 diverges after the maximum at 25 K.
We stress there must be considerable errors in these preliminary experiments and it is important we do not over interpret. However, both the EPR and magnetic measurements indicate around 1014 rotaxanes per bead; this is close to the lowest estimate of 2 × 1013 we calculate based on solvent-accessible sites (see section S2 and Fig. S11). Given the limitations of the experiments the agreement is excellent. The difference seen between 7 and 11 may indicate some weak interactions are present in the beads but the evidence is inconclusive.
Conclusions
Highly paramagnetic microparticles have been prepared using a very different route to traditional paramagnetic nanoparticles. We have shown we can attach around 1014 rotaxanes on beads of average diameter 115 μm, measuring the number of rotaxanes attached by both EPR spectroscopy and magnetometry; the number matches well with our estimate that at least 2 × 1013 sites are available on a 115 μm bead. This also demonstrates that CuAAC click chemistry could be generally useful for making highly unusual nanostructures containing hybrid inorganic–organic molecules and supramolecules. In the future we will examine whether such nanoparticles could be used in applications such as DNP6.
Methods
Materials
All chemicals were of reagent grade quality, and they were purchased from commercial sources and used as received. The azide functionalized polystyrene beads were prepared by literature procedures16,17 (see SI section 1.2). Full characterisation details of all compounds are given in the section S1 and S3 of the Supplemental Information.
Physical techniques
1H NMR spectra were recorded on a Bruker AVIII HD 400 MHz at 298 K. Chemical shifts are reported in parts per million (ppm) from high to low frequency using the residual solvent peak as the internal reference (DMSO-d6 = 2.50 ppm). All 1H resonances are reported to the nearest 0.01 ppm. Infrared spectra were recorded neat on a Bruker FT-IR Alpha II Platinum ATR spectrometer. Fully characterised compounds were chromatographically homogeneous. Flash column chromatography was carried out using Silica 60 Å (particle size 40–63 μm, Sigma Aldrich, UK) as the stationary phase. Analytical TLC was performed on precoated silica gel plates (0.25 mm thick, 60 F254, Merck, Germany) and visualised using both short and longwave ultraviolet light in combination with standard laboratory stains (acidic potassium permanganate, acidic ammonium molybdate and ninhydrin). Low-resolution ESI mass spectrometry and high-resolution mass spectrometry was carried out by staff at the Mass Spectrometry Service, Department of Chemistry, The University of Manchester. Elemental analysis was carried out by the Analytical Service of the University of Manchester; metals analysis by Thermo iCap 6300 inductively coupled plasma optical emission spectroscopy (ICP-OES).
Synthesis of thread 1 (see Fig. 6a)
Fig. 6. Synthetic reactions.

a Synthesis of thread 1. b Synthesis of 2. c Synthesis of 4. d Synthesis of 7. e Synthesis of 8. f Synthesis of 10. g Synthesis of 11.
To a solution of 2-(4-hydroxyphenyl)−1-bromoethane (2.0 g, 10 mmol) in anhydrous DMF (45 mL) was added 2-(4-hydroxyphenyl)ethylamine (6.0 g, 44 mmol, 4.4 eq.) and N-ethyldiisopropylamine (5.2 mL, 30 mmol, 3 eq.). The reaction mixture was heated at 75 °C for 16 h. When the reaction was complete, the mixture was cooled to room temperature, diluted with EtOAc and washed with brine. The organic phase was dried (MgSO4), filtered, and the solvent removed under reduced pressure. The solid residue was dissolved in hot CH3OH (100 mL) and precipitated by addition of water (20 mL). The product was filtered and washed repeatedly with CHCl3 to yield the title compound as brown solid (1.8 g, 7.0 mmol, 70%).
Synthesis of rotaxane 2 (see Fig. 6b)
To pivalic acid (45.0 g, 440 mmol) at 100 °C in a Teflon™ flask was added 1 (2.5 g, 9.7 mmol). The reaction mixture was stirred at 100 °C for 10 min then [CrF3·4H2O] (6.0 g, 33 mmol) and [2NiCO3·3Ni(OH)2·4H2O] (1.0 g, 1.7 mmol) were added. The reaction mixture was stirred at 140 °C for 24 h. The reaction mixture was cooled to room temperature and the crude product was extracted with CHCl3. The solvent was removed under reduced pressure and the crude residue was purified by column chromatography (SiO2, CHCl3:EtOAc, 90:10) to yield rotaxane 2 (3.5 g, 1.4 mmol, 15%).
Synthesis of rotaxane 4 (see Fig. 6c)
To prepare molecule 4, a two-step esterification was performed. Firstly, to a solution of rotaxane 2 (3.0 g, 1.2 mmol), in THF (100 mL) was added pentynoic acid (0.12 g, 1.2 mmol), DCC (0.90 g, 4.3 mmol) and DMAP (0.75 g, 6.1 mmol). The reaction mixture was stirred at 50 °C for 16 h. The solvent was removed under reduced pressure and the crude residue was purified by column chromatography (SiO2, CHCl3:EtOAc, 95:5) to yield mono-esterified rotaxane 3 (0.75 g, 0.30 mmol, 25%).
To a solution of rotaxane 3 (0.75 g, 0.30 mmol) in CH2Cl2 (50 mL) was added 4-carboxy-TEMPO (0.18 g, 0.90 mmol), DCC (0.55 g, 2.6 mmol) and DMAP (0.55 g, 4.5 mmol). The reaction mixture was stirred at room temperature for 16 h. The solvent was removed under reduced pressure and the crude residue was purified by column chromatography (SiO2, CHCl3:CH3OH, 95:5) to yield rotaxane 4 (0.65 g, 0.24 mmol, 80%).
Synthesis of bead-rotaxane 7 (see Fig. 6d)
Azide polystyrene beads 5 (3.0 mg, 1.3 mmol g−1, 3.9 μmol) were added to [(C31H40N2O5)Cr7NiF8(C5H9O2)16] 4 (30 mg, 11 μmol, 3 eq.) and Cu(MeCN)4PF6 (4.0 mg, 11 μmol, 3 eq.) under argon. A CH2Cl2:DMF mixture (3:2, 5 mL, degassed with argon) was added and the reaction was stirred for 72 h at room temperature. The reaction mixture was then filtered and washed with CH3CN (3 × 10 mL), CH3OH (3 × 10 mL), and CH2Cl2 (3 × 10 mL) to afford [2]rotaxane–polystyrene beads 7 (13.0 mg, 3.7 μmol).
Synthesis of bead-rotaxane 8 (see Fig. 6e)
Azide polystyrene beads 6 (2.5 mg, 1.0 mmol g−1, 2.5 μmol) were added to [(C31H40N2O5)Cr7NiF8(C5H9O2)16] 4 (30 mg, 11 μmol, 4 eq.) and Cu(MeCN)4PF6 (5.0 mg, 13 μmol, 5 eq.) under argon. A CH2Cl2:DMF mixture (3:2, 5 mL, degassed with argon) was added and the reaction was stirred for 72 h at room temperature. The reaction mixture was then filtered and washed with CH3CN (3 × 10 mL), CH3OH (3 × 10 mL), and CH2Cl2 (3 × 10 mL) to afford [2]rotaxane–polystyrene beads 8 (8.3 mg, 2.4 μmol).
Synthesis of rotaxane 10 (see Fig. 6f)
To 3,3-dimethylacrylic acid (15.0 g, 150 mmol) in a Teflon™ flask was added 1 (1.5 g, 5.8 mmol) and 5.0 mL of 1,2-dichlorobenzene and heated to 140 °C for 10 min. To this solution [CrF3·4H2O] (3.0 g, 16 mmol) and [2NiCO3·3Ni(OH)2·4H2O] (0.75 g, 1.2 mmol) were added and stirred at 140 °C for 24 h. The reaction mixture was cooled to room temperature, transferred in to a 500 mL flask and 300 mL of hexane was added. The crude product was extracted with diethyl ether and purified by column chromatography (Silica, toluene/EtOAc) to yield rotaxane 9 (2.5 g). Single-crystal X-ray quality crystals were grown from acetone by slow evaporation method.
To prepare rotaxane 10, to a solution of rotaxane 9 (2.0 g, 0.8 mmol), in THF (100 mL) was added pentynoic acid (0.2 g, 2.0 mmol), DCC (0.3 g, 1.4 mmol) and DMAP (0.25 g, 2.0 mmol). The reaction mixture was stirred at 50 °C for 16 h. The solvent was removed under reduced pressure and the crude residue was purified by column chromatography (SiO2, CHCl3:EtOAc, 95:5) to yield 10. Single-crystal X-ray quality crystals were grown from acetone by slow evaporation method.
Synthesis of bead-rotaxane 11 (see Fig. 6g)
Azide polystyrene beads 5 (7.0 mg, 1.3 mmol g−1, 9.0 μmol) were added to [(C26H28NO4)Cr7NiF8(C5H7O2)16] 10 (72 mg, 27 μmol, 3 eq.) and Cu(MeCN)4PF6 (10 mg, 27 μmol, 3 eq.) under argon. A CH2Cl2:DMF mixture (3:2, 5 mL, degassed with argon) was added and the reaction was stirred for 72 h at room temperature. The reaction mixture was then filtered and washed with CH3CN (3 × 10 mL), CH3OH (3 × 10 mL), and CH2Cl2 (3 × 10 mL) to afford [2]rotaxane–polystyrene beads 11 (28 mg, 8.8 μmol).
Single-crystal X-ray diffraction
X-Ray data for compounds 2, 4 and 9 were collected at a temperature of 100 K using a dual wavelengh Rigaku FR-X with Cu-Kα radiation equipped with a HypixHE6000 detector and an Oxford Cryosystems nitrogen flow gas system. X-ray data for compound 10 was collected at a temperature of 100 K using synchrotron radiation at beamline I19 in Diamond Light Source equipped with a Pilatus 2 M detector and an Oxford Cryosystems nitrogen flow gas system. Data were measured using GDA and CrysAlisPro suite of programmes.
X-Ray data were processed and reduced using CrysAlisPro suite of programmes. Absorption correction was performed using empirical methods (SCALE3 ABSPACK) based upon symmetry-equivalent reflections combined with measurements at different azimuthal angles23. The crystal structure was solved and refined against all F2 values using the SHELXL24 and Olex 2 suite of programmes25.
All atoms in crystal structures were refined anisotropically with the exception of the hydrogen atoms, that were placed in the calculated idealised positions for all crystal structures. The pivalate ligands, and threads in crystal structures were disordered and modelled over two positions, using structural same distance (SADI) and distance fix (DFIX) Shelxl restraints commands. The atomic displacement parameters (adp) of the ligands have been restrained using similar Ueq and rigid bond (SIMU) and Similar Ueq (SIMU) restraints.
Spectroscopic measurements
Q-band (ca. 34 GHz) EPR spectra were recorded with a Bruker EMX580 spectrometer at the EPRSC UK National EPR Research Facility at The University of Manchester. The data were collected on polycrystalline powders and on solid polymeric beads at 280 K, and 5 K using liquid helium cooling. Spectral simulations were performed using the EasySpin 4.5.5 simulation software26.
Magnetic measurements
Magnetic properties were measured on a Quantum Design MPMS3. Susceptibility was measured from 300 to 2 K in a 1000 Oe magnetic field. Magnetisation was measured at 2 K to a maximum field of 7 T. Samples were mounted in gelatine capsules. The mass of samples measured were: 5, 21.83 mg; 7, 7.04 mg; 11, 14.28 mg; 12. 15.01 mg. No diamagnetic correction has been applied to data. For 7, beads were counted and weighed and this leads to an estimate of 831 beads per mg, which we have used in calculations.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by the EPSRC (EP/R043701/1/) including an Established Career Fellowship (EP/R011079/1) to R.E.P.W. R.E.P.W. also thanks the European Research Council for an Advanced Grant (ERC-2017-ADG-786734). D.A.L. thanks the Engineering and Physical Sciences Research Council (EP/P027067/1) and the European Research Council (Advanced Grant No. 786630) for funding. We also thank the EPSRC for an X-ray diffractometer (EP/K039547/1) and magnetometer (EP/S033181/1), and for access to the EPR National Research Facility (NS/A000055/1). We thank the University of Manchester for a President’s Scholarship (to D.T.).
Author contributions
D.A. and D.T. performed the synthetic chemistry, with advice from G.A.T. S.J.L. measured the EPR spectra, advised by D.C. and E.J.L.McI. S.J.L. also solved and refined the X-ray structures, with more challenging refinements completed by I.J.V.-Y. and G.F.S.W. A.B. performed the magnetic measurements, with data work up by S.J.L. D.A.L. and R.E.P.W. devised the project. The manuscript was written by D.A., D.T., S.J.L. and R.E.P.W. with input from all authors.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
Supplementary data are available in the online version of the paper. This includes Supplementary data 1, which includes the crystallographic data for this paper. These data have also been deposited as CCDC 2058841, 2058842, 2058844 and 2058846 numbers. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk). Reprints and permissions information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to R.E.P.W.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-022-00689-1.
References
- 1.Caruso F, Susha AS, Giersig M, Möhwald H. Magnetic core-shell particles: preparation of magnetite multilayers on polymer latex microspheres. Adv. Mater. 1999;11:950–953. doi: 10.1002/(SICI)1521-4095(199908)11:11<950::AID-ADMA950>3.0.CO;2-T. [DOI] [Google Scholar]
- 2.Breen ML, Dinsmore AD, Pink RH, Qadri SB, Ratna BR. Sonochemically produced ZnS-coated polystyrene core-shell particles for use in photonic crystals. Langmuir. 2001;17:903–907. doi: 10.1021/la0011578. [DOI] [Google Scholar]
- 3.Del Rio M, et al. Metal oxide assisted preparation of core-shell beads with dense metal-organic framework coatings for the enhanced extraction of organic pollutants. Chem. Eur. J. 2016;22:11770–11777. doi: 10.1002/chem.201601329. [DOI] [PubMed] [Google Scholar]
- 4.Kuo Y-C, Pal S, Li F-Y, Lin C-H. Polystyrene-supported core-shell beads with aluminium MOF coating for extraction of organic pollutants. Chem. Asian J. 2019;14:3675–3681. doi: 10.1002/asia.201900737. [DOI] [PubMed] [Google Scholar]
- 5.Bukowska A, Bester K, Pytel M, Bukowski W. Polymer beads decorated with dendritic systems as supports for A3 coupling catalysts. Catal. Lett. 2021;151:422–4434. doi: 10.1007/s10562-020-03301-0. [DOI] [Google Scholar]
- 6.Zhang Y, Baker PJ, Casabianca LB. BDPA-doped polystyrene beads as polarization agents for DNP-NMR. J. Phys. Chem. B. 2016;120:18–24. doi: 10.1021/acs.jpcb.5b08741. [DOI] [PubMed] [Google Scholar]
- 7.Philipova O, Barabanova A, Molchanov V, Khokhlov A. Magnetic polymer beads: recent trends and developments in synthetic design and applications. Eur. Polym. J. 2011;47:542–559. doi: 10.1016/j.eurpolymj.2010.11.006. [DOI] [Google Scholar]
- 8.Marchetto R, Cilli EM, Jubuilut GN, Schreier S, Nakaie CR. Determination of site−site distance and site concentration within polymer beads: a combined swelling-electron paramagnetic resonance study. J. Org. Chem. 2005;70:4561–4568. doi: 10.1021/jo0483318. [DOI] [PubMed] [Google Scholar]
- 9.Liang L, Astruc D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011;255:2933–2945. doi: 10.1016/j.ccr.2011.06.028. [DOI] [Google Scholar]
- 10.McInnes EJL, Whitehead GFS, Timco GA, Winpenny REP. Heterometallic rings as a playground for physics and supramolecular building blocks. Angew. Chem. Int. Ed. 2015;54:14244–14269. doi: 10.1002/anie.201502730. [DOI] [PubMed] [Google Scholar]
- 11.Lee C-F, et al. Hybrid organic-inorganic rotaxanes and molecular shuttles. Nature. 2009;458:314–318. doi: 10.1038/nature07847. [DOI] [PubMed] [Google Scholar]
- 12.Ballesteros B, et al. Synthesis, structure and dynamic properties of hybrid organic-inorganic rotaxanes. J. Am. Chem. Soc. 2010;132:15435–15444. doi: 10.1021/ja1074773. [DOI] [PubMed] [Google Scholar]
- 13.Harrison IT, Harrison S. Synthesis of a stable complex of a macrocycle and a threaded chain. J. Am. Chem. Soc. 1967;89:5723–5724. doi: 10.1021/ja00998a052. [DOI] [Google Scholar]
- 14.Townsend JB, Shaheen F, Liu R, Lam KS. Jeffamine derivatized tentagel beads and poly(dimethylsiloxane) microbead cassettes for ultrahigh-throughput in situ releasable solution-phase cell-based screening of one-bead-one-compound combinatorial small molecule libraries. J. Comb. Chem. 2010;12:700–712. doi: 10.1021/cc100083f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherrington DC. Preparation, structure and morphology of polymer supports. Chem. Commun. 1998;21:2275–2286. doi: 10.1039/a803757d. [DOI] [Google Scholar]
- 16.Bettinetti L, Lober S, Hubner H, Gmeiner P. Parallel synthesis and biological screening of dopamine receptor ligands taking advantage of a click chemistry based BAL linker. J. Comb. Chem. 2005;7:309–316. doi: 10.1021/cc049860s. [DOI] [PubMed] [Google Scholar]
- 17.Arseniyadis S, Wagner A, Mioskowski C. A straightforward preparation of amino-polystyrene resin from Merrifeld resin. Tetrahedron Lett. 2002;43:9717–9719. doi: 10.1016/S0040-4039(02)02192-5. [DOI] [Google Scholar]
- 18.Piligkos S, et al. EPR spectroscopy of a family of CrIII7MII (M = Cd, Zn, Mn, Ni) “wheels”: studies of isostructural compounds with different spin ground states. Chem. Eur. J. 2009;15:3152–3167. doi: 10.1002/chem.200801895. [DOI] [PubMed] [Google Scholar]
- 19.Peng Z, et al. Rotaxane-branched radical dendrimers with TEMPO termini. Chem. Commun. 2022;58:2006–2009. doi: 10.1039/D1CC06158E. [DOI] [PubMed] [Google Scholar]
- 20.Boulon M–E, et al. Measuring spin…spin interactions between heterospins in a hybrid [2]rotaxane. Angew. Chem. Int. Ed. 2017;56:3876–3879. doi: 10.1002/anie.201612249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casati C, Franchi P, Pievo R, Mezzina E, Lucarini M. Unraveling unidirectional threading of α-cyclodextrin in a [2]rotaxane through spin labelling approach. J. Am. Chem. Soc. 2012;134:19108–19117. doi: 10.1021/ja3073484. [DOI] [PubMed] [Google Scholar]
- 22.Möser J, et al. Using rapid-scan EPR to improve the detection limit of quantitative EPR by more than one order of magnitude. J. Magn. Reson. 2017;281:17–25. doi: 10.1016/j.jmr.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rigaku Corporation. Rigaku Oxford Diffraction, CrysAlisPro Software system version 1.171.37.33 (Rigaku Corporation, Oxford, UK, 2018).
- 24.Sheldrick GM. A short history of SHELX. Acta Crystallogr. 2015;A71:3–8. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 25.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009;42:339–341. doi: 10.1107/S0021889808042726. [DOI] [Google Scholar]
- 26.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Supplementary data are available in the online version of the paper. This includes Supplementary data 1, which includes the crystallographic data for this paper. These data have also been deposited as CCDC 2058841, 2058842, 2058844 and 2058846 numbers. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk). Reprints and permissions information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to R.E.P.W.