

Abstract
Key to the fragment optimisation process within drug design is the need to accurately capture the changes in affinity that are associated with a given set of chemical modifications. Due to the weakly binding nature of fragments, this has proven to be a challenging task, despite recent advancements in leveraging experimental and computational methods. In this work, we evaluate the use of Absolute Binding Free Energy (ABFE) calculations in guiding fragment optimisation decisions, retrospectively calculating binding free energies for 59 ligands across 4 fragment elaboration campaigns. We first demonstrate that ABFEs can be used to accurately rank fragment-sized binders with an overall Spearman’s r of 0.89 and a Kendall τ of 0.67, although often deviating from experiment in absolute free energy values with an RMSE of 2.75 kcal/mol. We then also show that in several cases, retrospective fragment optimisation decisions can be supported by the ABFE calculations. Comparing against cheaper endpoint methods, namely Nwat-MM/GBSA, we find that ABFEs offer better ranking power and correlation metrics. Our results indicate that ABFE calculations can usefully guide fragment elaborations to maximise affinity.
Subject terms: Computational chemistry, Lead optimization
Fragment-based drug design is an effective approach to identifying potential binders for a given target protein, but accurately capturing changes in affinity associated with a given set of chemical modifications remains challenging. Here, the authors evaluate the use of absolute binding free energy calculations in guiding fragment optimisation decisions, finding that such calculations can usefully guide fragment elaborations to maximise affinity.
Introduction
Over the last few decades, the fragment-based drug design (FBDD) process has matured into a popular and effective approach to designing novel binders1. Indeed, during the 2015–2019 period, over 131 successful fragment-to-lead campaigns were published2–6. With an intuitive structure-based approach, and ability to more easily sample a large chemical space, FBDD has become a strong contender to more traditional high-throughput screening methods.
In the FBDD process, a library of fragments is first screened against a given protein target in order to identify potential binders. These fragments usually adhere to the so-called “rule of three”7, having a molecular weight ≤ 300 Da, ClogP ≤ 3, a number of hydrogen bond donors ≤3 and a number of hydrogen bond acceptors ≤3. From this initial screen, binders are then identified through structural (e.g. X-ray crystallography, nuclear magnetic resonance (NMR)), biochemical, or biophysical (e.g. NMR, surface plasmon resonance, isothermal titration calorimetry (ITC)) characterisation8–10. Confirmed binders are then optimised, heavily relying on structure activity relationships to create bespoke high affinity binders. Optimising strategies either concentrate on improving the affinity of a single binder through fragment growing, or by combining multiple fragments occupying distinct binding sites through linking and merging decisions11,12.
Accurately characterising fragment-protein interactions is therefore central to the FBDD process. Despite substantial improvements in leveraging in vitro methods for fragment screening1, this remains a challenging task. By their nature fragments tend to be low affinity binders, somewhere in the millimolar to micromolar range, with a propensity to access multiple binding sites in a protein target. Not only do highly sensitive affinity measurements need to be employed, something that cannot always be readily used for large fragment screens, but multiple orthogonal methods are often required to validate low affinity hits1,13.
As a consequence, in silico methods have become increasingly popular in helping guide and support FBDD decisions14,15. A variety of approaches have been employed for this task, which can roughly be separated into two categories; (i) methods to identify fragment interaction sites, and (ii) methods to characterise fragment binding affinities. For the former, both simple methods such as molecular docking16, and more complex molecular dynamics-based approaches such as hotspot mapping17–21 or unbiased molecular dynamics (MD) with Markov-state modelling22 have shown success in identifying potential fragment binding sites on protein targets. In terms of characterising affinity, alchemical relative binding free energy (RBFE) methods have been particularly successful in ranking fragment affinities15,23–26. Of particular note is a 2015 study by Steinbrecher et al.26, which demonstrated that the FEP + RBFE tool could be used to successfully rank fragment affinities, achieving an RMSE of 1.14 kcal/mol for 96 ligands across eight fragment optimisation campaigns. Whilst RBFEs are a powerful and relatively cheap tool for this purpose, there are some disadvantages that limit its applicability in FBDD. Firstly, RBFEs do not give a direct measure of ligand binding affinities on an absolute scale, instead the method relies on other methods to normalise output free energy values. Often this would be done via an in vitro experimental measurement of a few select compounds, however as discussed above it can often be difficult to do so accurately for fragments and therefore is not always tractable in FBDD. The second major disadvantage is that RBFE protocols are generally developed to investigate small chemical perturbations on a given common chemical scaffold. This can have limited applicability in some FBDD campaigns, which will often investigate several fragments with different chemical scaffolds, sometimes even in different binding site locations. Nevertheless, we note some recent success in using RBFE methods for fragment linking purposes, although requiring several extra intermediate steps to achieve good results27.
As an alternative to RBFE methods, we propose that absolute binding free energies (ABFE)28–32 could instead be used to directly investigate fragment affinities. Despite larger computational costs, ABFEs offer a direct solution to the above described limitations of RBFEs by directly calculating the absolute free energy of binding for each individual ligand, and not requiring a transformation to another chemical entity. Previous works have shown ABFEs to offer highly accurate estimates of binding free energies across a variety of target systems, although often at increased computational costs30,33. Indeed, the idea of using ABFEs in fragment binding is not a novel one, and we note several other investigations employing such methods for fragment-sized molecules15,29,34,35. That being said, these have been mostly limited in scope and to our knowledge there have yet to be any large-scale analyses of the applicability of ABFEs to the FBDD process.
Here we specifically look at evaluating the use of ABFEs in the fragment optimisation process. Retrospectively calculating the binding free energies for 59 ligands across four FBDD campaigns (Fig. 1), we aim to investigate whether; (a) ABFEs offer comparable results to in vitro affinity measures, and (b) ABFEs could be used to achieve similar synthetic decisions in fragment optimisation. We also look at how ABFEs compare against cheaper methods, namely Nwat-MM/GBSA36,37.
Fig. 1. Overview of the fragment elaboration datasets.

A total of 59 ligands from four different elaboration studies38–41 with affinities spanning the millimolar to nanomolar range are investigated here.
Results and discussion
ABFE results across all system
The ABFE (Fig. 2) of 59 ligands (Figs. S1, S2, S4 and S5) for fragment optimisation campaigns of the PWWP138, HSP9039, MCL-140 and Cyclophilin D41 receptors were computed (see Methods). As shown in Fig. 3a, we see very good agreement between the calculated and experimental affinities, with a Pearson r of 0.89 ± 0.03, and a Kendall τ of 0.67 ± 0.05. However, we observe a RMSE of 2.75 ± 0.20 kcal/mol. As outlined in Fig. 3b–e, each dataset deviates to varying degrees from experiment, with only PWWP1 showing an RMSE close to 1 kcal/mol. The correlation shown here is on par, if not better, than other comparable alchemical free energy studies26,42. Whilst the RMSE is larger than the 1 kcal/mol limit shown by some other ABFE studies32,42, similar system dependent shifts have been reported previously33.
Fig. 2. Absolute binding free energy thermodynamic cycle employed.

The free energy of binding, i.e. going from a ligand in solution (state a) to a protein-ligand complex (state e), is captured through a non-physical path. First, the electrostatics are annihilated to zero (state b) over 11 λ windows. This is followed by a further 21 λ windows which decouple the ligand van der Waals interaction from the solvent (state c). The decoupled ligand is then analytically restrained as defined by ref. 102 (state d). By accounting for this restraint the ligand state is then equivalent to a non-interacting ligand in a protein-ligand complex (state h). The ligand interactions with the environment are then turned back on, first re-coupling the van der Waals interactions over 21 λ windows (state g), followed by a further 11 λ windows to add back electrostatics (state f). Finally, the orientational restraints are turned off over 12 λ windows resulting in a fully interacting protein-ligand complex (state e).
Fig. 3. ABFE calculations results for different systems.

a All four datasets, b PWWP1, c HSP90, d MCL-1, e Cyclophilin D. Free energy estimates are the means of the estimates across replicas, with error bars as their standard deviation. Correlation metrics calculated from the mean estimate values, with error bars derived from bootstrap resampling. All free energy results, including RMSE values, have units of kcal/mol.
Sampling of free energies agrees well between replicas with a mean error of 0.79 ± 0.38 kcal/mol, although ~22% of the estimates show an error bar >1 kcal/mol, with the highest value being 1.93 kcal/mol. This is generally on a par with sampling errors shown by other absolute alchemical free energy studies32,33,43 and matching a similar level of uncertainty as the cycle closure errors shown in the Steinbrecher et al. 2015 fragment FEP + study26. Whilst the error in experimental measurements was not provided for all our systems, our sampling errors are for the most part approximately twice as large as the ~0.5 kcal/mol limit which we might expect from experimental measurements44.
ABFEs of fragments for the PWWP1 domain of NSD3
Specifically looking at each individual case, we start with a fragment elaboration for the PWWP1 domain of NSD3 by Böttcher et al.38. Here 11 ligands were elaborated from an initial 160 µM fragment hit (ligand 8), eventually resulting in the 170 nM ligand BI-9321 (SI Fig. S1, Table S1). As shown in Fig. 3b, the ABFE results correlate very well with experiment, with an RMSE of 1.14 ± 0.16 kcal/mol, the lowest of all four optimisation campaigns investigated. In the original elaboration study, seven specific elaboration decisions were outlined (SI Table S2). Of these, only two would have been definitively supported by our ABFE results, that is to say—there is a greater-than-error difference between the calculated ligand binding energies in each decision. Whilst the ABFE results accurately predict the direction of the affinity change for six of the seven decisions, the majority of the changes in free energy remained within the range of the errors of the estimates. We note that the one decision where the wrong sign is predicted involves experimental ΔΔG values of −0.12 and −0.23 kcal/mol, which is well within the limit of both experimental45,46 and force field47 accuracy.
ABFEs of a fragment optimisation for HSP90
Next, we look at a fragment elaboration study by Murray et al.39 elaborating 17 ligands from an initial fragment hit (ligand 3, SI Fig. S2, SI Table S4). Whilst the original study does outline two separate fragment elaboration campaigns, we specifically looked at the second elaboration set which involved a larger number of ligands. As shown in Fig. 3c, the free energy estimates correlate very well with the experimental values with a Pearson r of 0.96 ± 0.03 and a Kendall τ of 0.73 ± 0.12. However, we do see a progressive deviation from experiment as the affinity of the ligands increases leading to a RMSE of 3.82 ± 0.33 kcal/mol.
This difference between calculated and experimental values has been observed by other free energy studies of HSP90 and as detailed by Baumann et al.48, is possibly explained by several slow degrees of freedom associated with HSP90 binding. These include; the presence of varying waters in the binding site, ligand re-orientation, and side-chain motions. Interestingly, Baumann et al.48 identify that the presence of waters in the binding site worsened free energy estimates for their HSP90 test case, although it offered improved convergence in results. Whilst we have attempted to optimise the waters in the binding site using the MC/MD sampling steps in our equilibration procedure, it is possible that similar effects are impacting the accuracy of our results during the decoupling stages of our ABFE calculations. We note that in all but one ligand, ABFE calculations started with three buried waters present in the binding site (SI Fig. S3). In the case of ligand 24, for which the MC/MD procedure only added one buried water, we attempted to investigate the impact of these missing waters by calculating an ABFE with all three waters manually added to the binding site. This led to a within error change in the free energy from −12.20 ± 0.71 kcal/mol (1 water) to −11.33 ± 0.52 kcal/mol (3 waters) (SI Tables S4 and S5). From this single result, it is unclear as to how much of an influence the initial presence of waters has on the binding affinity. Future work, possibly by combining water MC steps49 as part of the free energy procedure (as done for RBFE in works such as those of Ben-Shalom et al.50 would be required to further investigate this issue).
One slow motion which could have an impact here is the re-arrangement of one of the binding site loops in the region of residues ASN106 through to SER113. As shown in Fig. 4, the helicity of this loop region changes between the initial models used for ligands 3–20, 28 and 31 (PDB IDs: 2XDL and 2XAB) to the ones used for ligands 21–27, 29–30 (PDB ID: 2XHT and 2XHX). The re-arrangement of this loop from a helix-loop-helix, akin to the 2XHT conformation, to a continuous helix is well documented and previous work has shown it to be variably induced depending on the binding ligand51–53. In their work Murray et al. only crystalised a small subset of their ligands in either one or the other crystal forms39. As a result, the initial models chosen for each ligand were purely based on chemical similarity with ligands from one of the resolved structures. To investigate the impact of this loop re-arrangement on the free energies, ligands 21–27 and 29–30 were re-calculated using the 2XDL crystal as a starting conformation. As can be seen in Fig. 4d. and SI Tables S4 and S5, estimated free energies remain within error of each other despite a small 0.33 kcal/mol improvement in the mean RMSE value. Whilst not having a major impact on the results presented here, it is still possible that there are long timescale influences of this loop motion to the binding free energy which are not captured in this work. Further work using enhanced sampling methods, coupled with experimental validation of loop preference (e.g. through ATR-FTIR spectroscopy53) may be required to investigate the true extent of the impact of this loop motion on ABFE results.
Fig. 4. Impact of conformation on ABFE calculations for HSP90.

a Comparison of the structures of PDB IDs 2XHT (blue) and 2XDL (red) demonstrating the difference in helicity between the two models and b, c a zoomed view of the affected helix. Residues 100–124 have been highlighted in a darker colour to aid in visualisation. PDB ID 2XAB, which is similar in structure to 2XDL and 2XHX which is similar in structure to 2XHT are not shown. d Absolute binding free energies of the HSP90 ligands all starting from the 2XDL-like helix loop conformation. Free energy estimates are the means of the estimates across replicas, with error bars as their standard deviation. Correlation metrics calculated from the mean estimate values, with error bars derived from bootstrap resampling. All free energy results, including RMSE values, have units of kcal/mol.
The 2XAB and 2XDL conformations are close to those of the apo structure shown in PDB ID 5J2V54 with an all-atom alignment RMSD of 0.92 Å. We therefore do not anticipate that differences between the apo and holo structures are leading to the large overpredictions of the binding affinities seen in this fragment optimisation set, particularly those seen in the stronger binders. To verify this an ABFE of the strongest binder, ligand 31, was calculated using the 5J2V crystal as the initial protein conformer. Doing so led to a change of free energies from −18.28 ± 0.80 kcal/mol (2XAB initial structure) to −16.93 ± 0.92 kcal/mol (5J2V initial structure). Whilst indicating a subtle crystal structure dependent influence on the free energies, these two results remain within error of each other and do not account for the ~5 kcal/mol overprediction in the binding affinity compared to experiment.
In this study six elaboration decisions were outlined, of these three would have been clearly supported by our ABFE results (SI Table S6). Of the three failed decisions only one (decision 2), would have been fully miscalculated. For the other two, one involved a small change in experimental affinity (decision 5) with ΔΔG values ~0.6–0.8 kcal/mol. This likely still falls within the accuracy of ITC measurement but is too small a change to be properly captured by ABFE given the size of sampling errors. The other elaboration decision clearly identified improvements in affinity but failed to identify ligand 24 as a better binder than ligands 25 and 26 (decision 4).
ABFEs of a fragment optimisation for MCL-1
The third system presented here is an elaboration of fragments for MCL-1 by Friberg et al40. In this study, two fragment series are grown in parallel, with an eventual merge to form a nanomolar compound (SI Fig S4 and Table S8). Here we have one of the lowest correlations between calculated and experimental ΔG values with a Pearson r of 0.81 ± 0.08 and a Kendall τ of 0.56 ± 0.15. We also see a shift from experiment in the absolute values, with an RMSE of 2.33 ± 0.24 kcal/mol. It should also be noted that in this series only an upper bound Ki of >1000 µM was assigned for three of the ligands (ligands 1, 6, and 12, SI Table S8). Disregarding these worsens correlation with experiment, yielding a Pearson r of 0.72 ± 0.16, Kendall τ of 0.43 ± 0.19, and a RMSE of 2.50 ± 0.24 kcal/mol. This rather poor correlation by comparison to the other datasets may in part be explained by the narrower activity range covered by the ligands in this series (Fig. 3d). Except from two (ligands 60 and 65), the ligands cover an ~2 kcal/mol activity range. This is possibly too narrow a range to distinguish between binding, especially given the size of the uncertainties in our calculated estimates. No apparent causes could be identified for the ~2 kcal/mol systematic shift from experiment seen in these results. Whilst no specific slow conformational changes or binding waters were identified, it is possible that unobserved long timescale motions, such as changes between apo and holo conformations, or force field inaccuracies in dealing with charged compounds may play a role here. Unfortunately to our knowledge, at the time of this work no adequate apo structure of MCL-1 was available for us to verify the difference in calculated absolute binding free energies between the two states.
In their paper Friberg et al.40 did not specifically outline a set of synthetic decisions for these ligands, but instead demonstrated four activity cliffs (SI Table S9) based on the growing of >1 mM class I fragments to micromolar ligands (decisions 1 through 3), and the merging of the class I and II ligands occupying distinct parts of the MCL-1 binding site (decision 4). The majority of fragment growing activity cliffs are clearly captured by the ABFE results, demonstrating greater than error ΔΔG values. However, both decisions 1 and 3 have one ligand pair (ligands 1–5, and 3–13) which are within uncertainty of each other. In addition, the predicted affinities for the optimised ligands are at times predicted to be several kcal/mol away from each other, even though they are experimentally determined to be within a ~1 kcal/mol range. The fragment merging case (decision 4) could be clearly captured by our calculated results, despite large errors in the binding free energy estimates of the merged scaffold ligands 60 and 65. Indeed, the MCL-1 dataset has on average the highest standard deviations between ABFE replicates. These large sampling errors are reflected by the charged nature of the ligands and the large conformational space accessible to the ligands within the binding site. As demonstrated for ligand 60 in Fig. 5, the ligand can effectively roll within the binding site, resulting in a pose with an RMSD > 4 Å from the others. The flexibility of binding modes for MCL-1 was also noted by Steinbrecher et al.26, and indicates a need for either longer simulation times or enhanced sampling schemes to reach sufficient convergence for systems such as these.
Fig. 5. Overlay of the starting configurations of each replica of the ABFE calculations for ligand 60 in MCL-1.
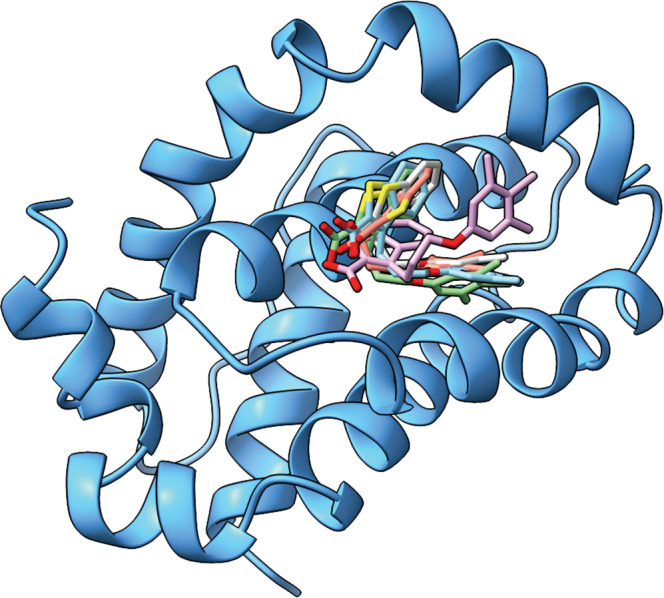
The re-arrangement (pink coloured ligand) of the merged ligand 60 in the MCL-1 binding site can clearly be seen.
ABFEs of a fragment optimisation for Cyclophillin D
Our final elaboration dataset is a set of fragment merging decisions for a fragment screen of Cyclophilin D by Gradler et al41. Here we simulate a subset of the ligands investigated in the original study, covering a total of 5 merging decisions (SI Fig. S5 and SI Table S12). We again find good correlation between the calculated and experimental free energies, with a Pearson r of 0.91 ± 0.10 and a Kendall τ 0.69 ± 0.19 albeit with a relatively large RMSE of 2.57 ± 0.33 kcal/mol. For the most part (with ligand 2 being an exception) this seems to represent a systematic shift in the free energy by ~2.5 kcal/mol (Fig. 3e). The exact cause of this shift is unclear, buried binding site waters of concern were not observed and no significant conformational differences between the apo and holo protein conformations were found. To verify the latter, the ABFE for ligand 27 was re-calculated using the apo crystal, PDB ID 3QYU55. As seen in SI Table S12, the 0.29 kcal/mol difference between the apo and holo calculated free energies sits is well within the >1 kcal/mol sampling error. Ligand conformational flexibility or force field accuracy may also play a role here, however we were unable to specifically identify the exact cause.
Of the five merging decisions captured by the simulated ligands, four would have been clearly supported by the ABFE results (SI Table S13). The large activity cliffs seen in these merging decisions, often exceeding 5 kcal/mol, are easily captured by the ABFE simulations even in cases where uncertainty exceeds 1 kcal/mol. The only miscalculated merging decision involves a relatively small ΔΔG change of 0.64 kcal/mol in experimental affinities (ligand 2 to ligand 16), which is miscalculated through ABFE by 3 kcal/mol in the opposite direction. Beyond merging decisions, whilst the estimates can differentiate between low affinity fragments (within the limit of error), the higher affinity merged ligands cannot be clearly distinguished from each other. For example, the 6 nM ligand 14, is estimated as within 0.3 kcal/mol of the 660 nM ligand 39.
Comparison to other methods
The computational cost of ABFEs is significant, being easily orders of magnitude higher than cheaper endpoint methods. Previous works26,56,57 have shown that endpoint methods such as MM/PBSA and MM/GBSA can often be competitive with alchemical methods, especially given the much-reduced computational costs. Here we compare our ABFE results with those calculated using Nwat-MM/GBSA. As shown in Figs. 3 and 6, we find that overall ABFE calculations yield improved free energy estimates compared to Nwat-MM/GBSA, with up to 0.2 improvements in correlation metrics (Pearson r and Kendall τ) for all datasets except MCL-1 where Nwat-MM/GBSA shows a higher Pearson r of 0.91 ± 0.11. As is usual for methods like Nwat-MM/GBSA56, in part as a consequence of not directly accounting for entropy58, the calculated absolute free energies tend to be overestimated and therefore a meaningful comparison of RMSEs between the two methods cannot be done. Analysis of the correlation of signed errors between both methods (Supplementary Note S4 and SI Fig. S8) shows that for PWWP1 and HSP90 the same ligands seem to lead to largest deviations from experiment. This could indicate that for these systems errors may predominantly stem from inaccuracies in the model (e.g. force field) rather than purely sampling errors. For the other systems, Cyclophilin D and MCL-1, no such trend is observed. However, we also note that an analysis of potential outliers (Supplementary Note S5 and SI Figs. S10–S12) does not show much overlap in identified outliers between the two methods, with more outliers identified for Nwat-MM/GBSA, in part due to a wider range in predicted free energy values.
Fig. 6. Binding free energies estimated by Nwat-MM/GBSA.

a PWWP1, b HSP90, c MCL-1, and d Cyclophilin D datasets. Free energy estimates are the means of the estimates across replicas, with error bars as their standard deviation. Correlation metrics calculated from the mean estimate values, with error bars derived from bootstrap resampling. All free energy results, have units of kcal/mol.
Despite a lower predictive performance in ranking binders, the Nwat-MM/GBSA method shows a reasonably good accuracy in ranking fragment optimisations, being able to clearly predict several of the elaboration decisions (SI Tables S3, S7, S10, S14). Surprisingly, a similar amount of decisions to the ABFE calculations are predicted here. Whilst the Nwat-MM/GBSA results predict one fewer decision for the HSP90 dataset, two more decisions are predicted for PWWP1. In addition, for MCL-1 whilst the fragment optimisation activity cliffs for decision 1 cannot be clearly distinguished due to large uncertainties, the optimisation of ligand 12 into ligand 13 (decision 3), which could not be supported by the ABFE results can be clearly identified using Nwat-MM/GBSA.
We can also compare some of the results with previously published RBFE data. Specifically, a subset of the MCL-1 dataset investigated here was also calculated using FEP + in Steinbrecher et al.’s 2015 fragment optimisation study26. Whilst only offering a limited comparison, we find the two methods to give comparable results (Fig. 7), with the two methods offering within error values for Pearson r and Kendall τ. Looking at the signed errors from experiment (SI Fig. S9), there appears to be correlation between the two methods indicating that similar ligands deviate the most from experiment. This concurs with the observation from the Nwat-MM/GBSA results that indicate that errors in the model (e.g. force field) may have a stronger influence in the MCL-1 set than sampling errors.
Fig. 7. Comparison of a subset of the MCL-1 dataset.

a ABFE simulations and b the 2015 FEP + study by ref. 26 Free energy estimates are the means of the estimates across replicas, with error bars as their standard deviation. Correlation metrics calculated from the mean estimate values, with error bars derived from bootstrap resampling. All free energy results, including RMSE values, have units of kcal/mol.
Comparison of retrospective elaboration decisions (SI Tables S9 and S11) shows that FEP + is also able to accurately predict the correct changes in free energies for the elaborations in this subset of MCL-1. Although an analysis of errors cannot be made as errors between repeats were not provided in the Steinbrecher et al. study26 the relatively low cycle closure errors for these ligands (for the most part lower than 1 kcal/mol) indicate that FEP + would likely outperform the ABFE results shown here (with errors exceeding 1 kcal/mol) in clearly identifying these elaborations. Given the narrow activity range and the relatively large uncertainties in the estimates, further comparison of correlation between calculated and experimental values is unlikely to yield much insight. We do note that the reported RMSE for FEP + is much lower at 0.93 kcal/mol, however the absolute numbers in this FEP + study were normalised based on the experimental affinity and therefore cannot be directly compared to the ABFE absolute results which required no a priori experimental affinity data.
Conclusions
In this study, we demonstrate that absolute binding free energy calculations can successfully estimate affinity changes in the fragment optimisation process. Whilst not always being able to clearly support elaboration decisions, the right direction in the affinity change was captured for the vast majority of cases (46 of the 52 ligand pairs involved in the 22 elaboration decisions). Despite occasional challenges in accurately matching experimentally derived binding free energies, the predictive power displayed here shows that ABFEs can be suitably used to not only act to orthogonally validate fragment hits but also to guide fragment optimisation.
We also show that ABFE calculations outcompete cheaper endpoint methods such as Nwat-MM/GBSA. Nevertheless, the Nwat-MM/GBSA method employed here showed reasonably good predictive ability, particularly in supporting retrospective elaboration decisions, and could easily be used as a low computational cost prefilter to more expensive ABFE calculations. The Nwat-MM/GBSA method employed here is also a very simplistic one, accounting directly for entropy59–61 and using methods employing independent trajectories for the complex, protein and ligand components may yield improved results. For one system in our benchmark set, MCL-1, we also found that ABFEs offer comparable results to RBFE methods Given the lower computational costs of relative binding free energies, the decision as to whether or not they should be used preferentially to absolute calculations is likely to depend on the use case. In cases where one attempts to elaborate from a common core structure, RBFEs are likely to offer better convergence at much cheaper compute requirements. However, when dealing with issues such as scaffold hopping and fragment linking (as shown in the Cyclophilin D set), although we note recent success in employing FEP + for such cases27, ABFEs become a lot simpler, particularly in the number of intermediate steps involved. The comparison between the two methods shown here is too limited to offer a complete evaluation of the differences in efficiency between RBFEs and ABFEs in fragment optimisation, particularly in fragment linking cases. Such an evaluation may form the basis of future work in this area. It is also worth mentioning that whilst ABFEs do have a much larger computational cost compared to end-point or RBFE methods, advances in GPU utilisation are making the high-throughput use of such a technique more tractable.
Our results demonstrated cases of both precision and accuracy limitations. This was either seen through poor inter-replica convergence (as shown in some of the MCL-1 ligands), or large deviations from experimental affinities (as shown in HSP90). The exact causes of some of the large deviations seen in these benchmarks, particularly for HSP90 and Cyclophilin D, could not be identified. Whilst still likely playing a role, the influence of bound waters and apo-to-holo protein conformational changes did not appear to be main contributors to these free energy deviations. One potential cause could be inadequate sampling of slow processes not easily identifiable from the short 20 ns simulations employed here. Indeed, we note that the ABFE protocol used here is reasonably simple and could easily be improved to address some of these sampling limitations62. This could not only improve the precision of our results, but also possibly the accuracy by more readily capturing rare events occurring during the ligand binding process. For example, recent work by Khalak et al.30 demonstrates that non-equilibrium absolute binding free energy can be used to bridge differences between large conformational differences between bound and unbound states. The use of higher quality force fields, especially via bespoke ligand parameterisations63,64, may also be key to alleviating some of the accuracy limitations identified here. We hope that future work will focus on improving both the precision and accuracy of ABFE results for fragment optimisation.
It is important to also note that a priori system specific knowledge remains crucial in ensuring the performance of alchemical methods. Whilst one of the main advantages of ABFE calculations is the direct estimate of binding affinity, having access to experimental affinities for a subset of the calculated binders to the same target or a structurally similar one, would significantly help in identifying and adjusting issues in a model. For example, we were able to confidently use an N-terminal truncated model of the PWWP1 system (see Supplementary Note 2) after observing that initial calculated free energies were close to those obtained from experiment. Similarly, the overestimations in the free energy estimates seen here are sufficiently large that they could mislead synthetic efforts, but also could easily be adjusted through prior affinity data on a few of the data points.
Despite the positive results displayed here, it is important to make clear that in many ways this benchmark only tackles a simplified set of tasks in the FBDD process and that several challenges remain ahead in validating and using ABFE. Of particular note, this dataset solely looked at cases where most binding poses were known and as a consequence were likely to not include ligands occupying more than one binding pose significantly contributing to the total free energy. Thus, our use of orientational restraints in this study could be justified. Whilst one could envision using ABFEs to identify optimal poses, an initial assessment (Supplementary Note S6, Table S16) indicate that this is unlikely to be a simple task. Indeed there are FBDD cases where accounting for multiple binding poses either through less restrictive restraints, such as center of mass restraints48,65–68 or through enhanced sampling means (e.g. NCMC69 or metadynamics70), or by explicitly accounting for multiple known poses71,72 will be required. In addition, as mentioned in our methods we also specifically attempted to avoid known cases where issues may arise in ABFE calculations, such as the presence of structural ions, membrane proteins, or ambiguous protonation states. Future work will look at tackling some of these more complex issues, particularly within the context of FBDD and see what type of solutions can be leveraged to ensure the accurate calculation of ABFE. There is still a lot of work to be done if these methods are to be employed routinely in design decisions in future, although progress is encouraging and not just in simple systems73.
Methods
System selection
Four previously published FBDD campaigns38–41 (Fig. 1), each with a different protein target, were selected for this benchmark. The systems were selected based on the availability of high-quality experimental measures, with the presence of both crystallographic and affinity data. We also looked to cover a wide activity range, spanning from millimolar to nanomolar affinities, and various different fragment optimisation cases, such as the presence of multiple distinct binding sites, changes in net charge, fragment growing and fragment merging (but no fragment linking cases). Note that some cases were specifically avoided to ensure this study remained tractable. For example, large multimeric protein targets were not included to reduce computational costs. Similarly, membrane proteins and systems with binding site metals or known protonation issues were not chosen to avoid introducing additional complexity in this initial dataset. Ideally, the experimental measurements across all four datasets would be the same (e.g. all ITC measurements of affinity) as this is potentially a source of systematic error. This unfortunately was not the case here (Fig. 1). Some of the pitfalls associated with experimental equilibrium binding measurements have recently been highlighted74.
System preparation and simulation details
As structural data was not available for all protein-ligand complexes, the initial configurations were generated by modifying a chemically close protein-ligand crystal structure using open source PyMOL75 (see Supplementary Note S1 for full details). If alternate states were available in the starting crystal, state A was always retained. Where appropriate, acetyl and N-methyl caps were also added to protein structures using PyMOL. Protons were assigned using protoss76,77, as made available within the ProteinPlus web server78,79. In the case of the PWWP1 domain, missing loop residues were modelled using modeller 9v2180 and the DOPE-HR scoring method. Of the 500 generated models, the top 10 models were rescored using the QMEAN81 scoring function as implemented in SWISS-MODEL82, with the final model selected as the best QMEAN-scored model. The PWWP1 N-termini was also truncated at isoleucine 393 to avoid long timescale interactions between residues in the disordered N-termini region and the ligand binding site (see Supplementary Note S2 and SI Fig. S6).
Solvation and force field assignment was achieved through a combination of AmberTools 1883 and GROMACS 201984. Ligand parameters and partial charges were assigned using the GAFF2 force field and the AM1-BCC partial charge model. The ff99SB-ILDN force field was used for protein parameters and the complexes were solvated in TIP3P85 cubic boxes with a minimum distance of 12 Å from the solute to the box edge using GROMACS’ solvate module. Sodium and chloride ions were added to neutralise the systems and achieve a concentration of 150 mM using the genion module of GROMACS. ParmEd version 3.2.0 (https://github.com/ParmEd/ParmEd) was used to convert input topologies and coordinates between AMBER and GROMACS file formats.
Unless otherwise mentioned, a hydrogen mass repartitioning scheme (HMR)86,87 was used to achieve a 4 fs integration timestep for our simulations. Due to differences in how MD engines handle hydrogens with analytical constraints87, hydrogen masses, except those of waters, were increased to 3 and 4 atomic mass units for AMBER and GROMACS respectively. Whilst HMR has been employed before in alchemical free energy calculations88–90, there is limited data on its use in ABFE in GROMACS. To this end, a small validation using the Cyclophilin D dataset is shown in Supplementary Information (see Supplementary Note S3, SI Fig. 7 and SI Table S15). Water hydrogen motions were constrained using SETTLE91 and either SHAKE92 or LINCS93,94 for other constraints in AMBER or GROMACS respectively. In all cases, simulation temperature was maintained at 298.15 K though Langevin dynamics with a collision frequency of 2 ps−1. A simulation pressure of 1 atmosphere was maintained using various barostats. For AMBER simulations, a Monte Carlo barostat95 was employed with a volume exchange attempt frequency of 100 ps. For GROMACS, the initial equilibration steps used the Berendsen barostat96 with a time constant of 1 ps, followed by the Parrinello-Rahman barostat97 with a time constant of 2.0 ps for all follow-on equilibration and production simulations. In all cases, a cut-off of 1 nm was used for short range interactions, and long range electrostatics are handled via PME98,99. Input topologies, coordinates, and simulation control files are provided as Supplementary Information (zenodo: 10.5281/zenodo.5913469 and https://github.com/bigginlab/fragment-opt-abfe-benchmark).
System equilibration
A two-step equilibration procedure is followed here. First, binding site waters were equilibrated using the AMBER MC/MD procedure49 as implemented in AMBER18’s pmemd.cuda engine100,101. The system first underwent a short initial equilibration phase consisting of 10,000 steps of minimisation, followed by a 500 ps NVT phase, and then 5 ns of NPT. During this equilibration, protein backbone atoms and non-hydrogen ligand atoms were restrained using a 5 kcal/mol/Å2 force constant. The system then underwent a 5 ns MC/MD with the same positional restraints with 25 000 MC attempts every 1000 MD steps. This was followed by a further 10 ns of MC/MD water exchange with position restraints on the ligand removed. In all MC/MD simulations, the NVT ensemble was sampled and the MC swap region box was trimmed to ensure as many exchanges with binding site waters as possible.
Once complete, the final frame from the MC/MD procedure was converted to GROMACS using ParmEd. The system was then further equilibrated using the mdrun engine in GROMACS 201984. This included an initial 10,000 step minimisation, followed by 1 ns of restrained (2.39 kcal/mol/Å2 applied to the protein backbone and ligand non-hydrogen atoms) NVT and NPT equilibration. The latter step employed the Berendsen barostat96 as detailed above. The system was then relaxed using 5 ns of unrestrained NPT simulation using the Parrinello-Rahman barostat97. Finally, a further 20 ns of NPT simulation was generated. This final 20 ns simulation was used to both derive Boresch-style102 orientational restraint parameters and for Nwat-MM/GBSA analysis.
Absolute binding free energy calculations
Here we employed an ABFE protocol similar to the one previously described by Aldeghi et al.31,32. Following the equilibration phase, a partial decoupling scheme is employed to trace the alchemical path from a fully interacting protein-ligand complex to a ligand in solution as shown in Fig. 2. This partial decoupling scheme involves annihilating ligand partial charges through 11 windows spaced at λ intervals of 0.1 from each other. A charge annihilation scheme was used here in order to avoid known issues with nonbonded exclusions when using the free energy code in GROMACS 2021 (see https://manual.gromacs.org/2021-current/). The charge decoupling is then followed by 21 Van der Waals decoupling windows spaced with the following λ schedule [0.0, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.55, 0.6, 0.65, 0.7, 0.75, 0.8, 0.85, 0.9, 0.95, 1.0]. A soft-core potential for decoupled Van der Waals interactions was used103. In addition, to restrict ligand motion in the complex phase an orientational restraint, as defined by Boresch et al.102, was employed. This restraint was applied over 12 windows in the complex decoupling phase with the following schedule [0.0 0.01, 0.025, 0.05, 0.075, 0.1, 0.15, 0.2, 0.35, 0.5, 0.75, 1.0]. In the solvent phase, the influence of this restraint was accounted for analytically.
Appropriately choosing which 6 atoms to involve in Boresch-style102 orientational restraints can be a complex issue, here we use the procedure as implemented in MDRestraintsGenerator (10.5281/zenodo.4570555). Briefly, we pick out the least mobile ligand atoms from our final 20 ns equilibration simulation (see System Equilibration above) as potential anchor points for our orientational restraint. We then analyse the trajectory to select all available alpha carbon protein anchor atoms within an 8 Å cut-off of the ligand anchor atoms, generating a list of potential orientational restraints (where the nearest bonded ligand heavy atoms and protein backbone atoms are selected as the remaining atoms involved in the orientational restraint). Bond, angle and dihedral timeseries for all identified restraints are obtained and the restraint with the lowest standard deviation across all values is picked as our orientational restraint of choice. The frame closest to the mean bond, angle and dihedral values of the restraint over the 20 ns simulation is then used as the starting point for the ABFE cycle. With each replica undergoing independent equilibrations, although starting from the same initial structure, we obtain a different restrained conformation for each replica, helping us better capture the impact of conformational flexibility on the calculated free energy.
Each ABFE window consists of a short equilibration similar in protocol to the previously detailed equilibration cycles, although using 10 ps for the restrained NVT equilibration, 100 ps for the restrained NPT Berendsen barostat equilibration, and 500 ps for the unrestrained NPT Parinnello-Rahman barostat equilibration. This is followed by 20 ns of production NPT simulation. A total of five independent replicas of the ABFE cycle are simulated. For the protein-ligand complexes, each replica is independently equilibrated and uses different orientational restraints, allowing the replicas to more representatively sample the available conformational space within the binding site. GROMACS 2021 was used for all ABFE calculations. For charged ligands, an analytical correction was used to account for finite size errors as detailed by Rocklin et al.104. To achieve this, the ABFE simulations were carried out with a net system charge in the fully coupled state by adding or removing a counterion as necessary. Files containing the sampled ΔH and ΔH/Δλ energies have been made publicly available at zenodo: https://zenodo.org/record/5906262, https://zenodo.org/record/5906110, https://zenodo.org/record/5904110, https://zenodo.org/record/5906019 and https://zenodo.org/record/5906805)
Nwat-MM/GBSA calculations
The last 20 ns of our equilibration procedure was analysed using the Nwat-MM/GBSA method36,37. Here the N = 20 nearest waters to the ligand in the binding site are included as part of the protein in the MM/GBSA calculation. As is typical for Nwat-MM/GBSA calculations, a single trajectory approach was employed, and no entropy corrections were included. The Nwat-MM/GBSA method was chosen here due to its simplicity, low computational costs, and previous work demonstrating Nwat-MM/PBSA to have good accuracy in ranking Bromodomain-binding ligands compared ABFE calculations56. GROMACS XTC files were converted to AMBER NETCDF format using MDAnalysis 1.1.1105,106, and the trajectory and topologies manipulated to remove excess waters using cpptraj v5.1.0107. The mmbondi2 radii with GB model 2 parameters108 were employed with a GB salt concentration of 150 mM. Frames were sampled every 25 ps from the 20 ns trajectories. The AmberTools21 versions of MMPBSA.py and sander were used for the MM/GBSA calculations109.
Analysis
Analysis of the alchemical simulations was achieved using the alchemlyb v0.3.0 library (https://zenodo.org/record/3361016) 110 and the MBAR estimator as implemented in pyMBAR v3.0.3111. The first 1 ns of each production window was discarded as extra equilibration time and samples were decorrelated based on the derivative of the potential with respect to λ up to a maximum frequency of 100 ps−1. Sampling error is shown as the standard deviation of the mean free energy estimates across all five repeats. Where appropriate, an analytical correction was included to account for finite size errors as defined by Rocklin et al.104 and implemented in rocklinc (https://github.com/xiki-tempula/rocklinc), using ABPS v3.0112,113 for Poisson-Botlzmann calculations113. It should be noted that no additional long range dispersion corrections (e.g. EXP-LR)114 were employed here.
All other simulation analyses were carried out using MDAnalysis 1.1.1105,106, spyrmsd for symmetry corrected RMSDs115, and the numpy116, scipy117, and scikit-learn118 libraries. Plotting was done using the matplotlib library119, and images of atomic coordinates through ChimeraX120. The correlation between the calculated and experimental affinities are analysed via Spearman r, Kendall τ, and RMSE, with error bars obtained as the standard deviation of the means generated through bootstrap resampling (100,000 iterations).
Evaluation of synthetic decisions
As part of our analysis we attempt to retrospectively evaluate the influence our predicted values would have had on the original synthetic decisions undertaken in the fragment optimisations. Overall this is a rather difficult task with many caveats, including; synthetic decisions not always being well defined, steps involving testing affinity changes between more than one ligand pair, and inequalities in the difficulty between synthetic steps. For example; for PWWP1 Böttcher et al.38 first detail investigating the impact of adding a 3,5-dimethyl-1,2-oxazole moiety on ligand 8 into order to form ligand 9. The authors then detail their next step as testing 2-methyl- and 2,6-dimethyl-phenyl substituents instead of the dimethyl-1,2-oxazole, showing moderately improve potency (ligands 10 and 11). Given the context of the tested substituents, one could look at these as either two or three optimisation decisions, based on whether one considers the synthesis of ligands 10 and 11 as a combined test of affinity change in the optimisation process. Here we have, to the best of our ability, attempted to group these syntheses into decisions based on the text outlined in the source reference (see SI Tables S2–3, S6–S7, S9–S11, and S13–14. This was easily done for HSP9039 and PWWP138 but was more complex for MCL-140 and Cyclophilin D41. For MCL-1 we opted to group the decisions into three sets of substitutions, each for a specific fragment core, and one fragment merging decision. For Cyclophilin D, five clear fragment merging cases could be identified from the simulated ligands and these were used as the synthetic decisions. One ligand from the PWWP1 set, ligand 14, was not included in the elaboration decisions. This is due to the main text rationalising the synthesis of this ligand based on the affinity of a ligand which was not included in our calculated set due to its large size (identified as ligand 7 by Böttcher et al.38). We nevertheless kept ligand 14 in our calculated set as it was structurally similar to the other PWWP1 ligands and offers a convenient extra data point towards evaluating the accuracy of ABFE calculations relative to experiment. It is also worth noting that the non-equal number of syntheses per decision, combined with our assessment that a failed decision occurs when any one ΔΔG difference in that set is insufficiently estimated, makes it such that some decisions have a much higher predictive difficulty than others.
Supplementary information
Acknowledgements
We thank Vytautas Gapsys, Rocco Meli, Zhiyi Wu, Charlie Cook, and Joseph Bluck for helpful discussions. This project made use of time on ARCHER granted via the UK High-End Computing Consortium, HECBioSim (www.hecbiosim.ac.uk) supported by EPSRC (grant no. EP/R029407/1).
Author contributions
D.S. and P.C.B. conceived the project. I.A. performed the simulations. I.A., A.M. and P.C.B. analysed the data. I.A. and P.C.B. wrote the paper.
Peer review
Peer review information
Communications Chemistry thanks Cris Oostenbrink, Mary Pitman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Input topologies, coordinates, and simulation control files are provided at https://zenodo.org/record/5913469 and https://github.com/bigginlab/fragment-opt-abfe-benchmark.
The implementation of the MDRestraintsGenerator can be found here: https://zenodo.org/record/4570556 Files containing the sampled ΔH and ΔH/Δλ energies have been made publicly available at zenodo: https://zenodo.org/record/5906262, https://zenodo.org/record/5906110, https://zenodo.org/record/5904110, https://zenodo.org/record/5906019 and https://zenodo.org/record/5906805.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-022-00721-4.
References
- 1.Lamoree B, Hubbard R. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017;61:453–464. doi: 10.1042/EBC20170028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson CN, Erlanson DA, Jahnke W, Mortenson PN, Rees DC. Fragment-to-Lead Medicinal Chemistry Publications in 2016. J. Med. Chem. 2018;61:1774–1784. doi: 10.1021/acs.jmedchem.7b01298. [DOI] [PubMed] [Google Scholar]
- 3.Mortenson PN, Erlanson DA, de Esch IJP, Jahnke W, Johnson CN. Fragment-to-Lead Medicinal Chemistry Publications in 2017. J. Med. Chem. 2019;62:3857–3872. doi: 10.1021/acs.jmedchem.8b01472. [DOI] [PubMed] [Google Scholar]
- 4.Erlanson DA, de Esch IJP, Jahnke W, Johnson CN, Mortenson PN. Fragment-to-Lead Medicinal Chemistry Publications in 2018. J. Med. Chem. 2020;63:4430–4444. doi: 10.1021/acs.jmedchem.9b01581. [DOI] [PubMed] [Google Scholar]
- 5.Jahnke W, et al. Fragment-to-Lead Medicinal Chemistry Publications in 2019. J. Med. Chem. 2020;63:15494–15507. doi: 10.1021/acs.jmedchem.0c01608. [DOI] [PubMed] [Google Scholar]
- 6.Johnson CN, Erlanson DA, Murray CW, Rees DC. Fragment-to-Lead Medicinal Chemistry Publications in 2015. J. Med. Chem. 2017;60:89–99. doi: 10.1021/acs.jmedchem.6b01123. [DOI] [PubMed] [Google Scholar]
- 7.Congreve M, Carr R, Murray C, Jhoti H. A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov. Today. 2003;8:876–877. doi: 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 8.Li Q. Application of fragment-based drug discovery to versatile targets. Front. Mol. Biosci. 2020;7:180. doi: 10.3389/fmolb.2020.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coyle J, Walser R. Applied biophysical methods in fragment-based drug discovery. SLAS Disco. 2020;25:471–490. doi: 10.1177/2472555220916168. [DOI] [PubMed] [Google Scholar]
- 10.Jacquemard C, Kellenberger E. A bright future for fragment-based drug discovery: what does it hold. Expert Opin. Drug Disco. 2019;14:413–416. doi: 10.1080/17460441.2019.1583643. [DOI] [PubMed] [Google Scholar]
- 11.de Souza Neto LR, et al. In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 2020;8:93. doi: 10.3389/fchem.2020.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bian Y, Xie X-Q. Computational fragment-based drug design: current trends, strategies, and applications. AAPS J. 2018;20:59. doi: 10.1208/s12248-018-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gossert AD, Jahnke W. NMR in drug discovery: a practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nuc. Mag. Res. Spec. 2016;97:82–125. doi: 10.1016/j.pnmrs.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Bissaro M, Sturlese M, Moro S. The rise of molecular simulations in fragment-based drug design (FBDD): an overview. Drug Discov. Today. 2020;25:1693–1701. doi: 10.1016/j.drudis.2020.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Georgiou C, et al. Pushing the limits of detection of weak binding using fragment-based drug discovery: identification of new cyclophilin binders. J. Mol. Biol. 2017;429:2556–2570. doi: 10.1016/j.jmb.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Verdonk ML, et al. Docking performance of fragments and druglike compounds. J. Med. Chem. 2011;54:5422–5431. doi: 10.1021/jm200558u. [DOI] [PubMed] [Google Scholar]
- 17.Defelipe LA, et al. Solvents to fragments to drugs: MD applications in drug design. Molecules. 2018;23:3269. doi: 10.3390/molecules23123269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guvench O. Computational functional group mapping for drug discovery. Drug Discov. Today. 2016;21:1928–1931. doi: 10.1016/j.drudis.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakan A, Nevins N, Lakdawala AS, Bahar I. Druggability assessment of allosteric proteins by dynamics simulations in the presence of probe molecules. J. Chem. Theory Comput. 2012;8:2435–2447. doi: 10.1021/ct300117j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lexa KW, Carlson HA. Full protein flexibility is essential for proper hot-spot mapping. J. Am. Chem. Soc. 2011;133:200–202. doi: 10.1021/ja1079332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guvench O, MacKerell AD., Jr. Computational fragment-based binding site identification by ligand competitive saturation. PLOS Comput. Biol. 2009;5:e1000435. doi: 10.1371/journal.pcbi.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linker SM, Magarkar A, Köfinger J, Hummer G, Seeliger D. Fragment binding pose predictions using unbiased simulations and Markov-state models. J. Chem. Theory Comput. 2019;15:4974–4981. doi: 10.1021/acs.jctc.9b00069. [DOI] [PubMed] [Google Scholar]
- 23.Matricon P, et al. Fragment-based design of selective GPCR ligands guided by free energy simulations. Chem. Comm. 2021;57:12305–12308. doi: 10.1039/D1CC03202J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Ranganathan A, Ijzerman AP, Siegal G, Carlsson J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2a adenosine receptor. J. Chem. Inf. Model. 2013;53:2701–2714. doi: 10.1021/ci4003156. [DOI] [PubMed] [Google Scholar]
- 25.Matricon P, et al. Fragment optimization for GPCRs by molecular dynamics free energy calculations: probing druggable subpockets of the A2Aadenosine receptor binding site. Sci. Reps. 2017;7:6398. doi: 10.1038/s41598-017-04905-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinbrecher TB, et al. Accurate binding free energy predictions in fragment optimization. J. Chem. Inf. Model. 2015;55:2411–2420. doi: 10.1021/acs.jcim.5b00538. [DOI] [PubMed] [Google Scholar]
- 27.Yu HS, et al. General theory of fragment linking in molecular design: why fragment linking rarely succeeds and how to improve outcomes. J. Chem. Theory Comput. 2021;17:450–462. doi: 10.1021/acs.jctc.0c01004. [DOI] [PubMed] [Google Scholar]
- 28.Deng Y, Roux B. Calculation of Standard Binding Free Energies:Aromatic Molecules in the T4 Lysozyme L99A Mutant. J. Chem. Theory Comput. 2006;2:1255–1273. doi: 10.1021/ct060037v. [DOI] [PubMed] [Google Scholar]
- 29.Mobley DL, et al. Predicting absolute ligand binding free energies to a simple model site. J. Mol. Biol. 2007;371:1118–1134. doi: 10.1016/j.jmb.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalak Y, et al. Alchemical absolute protein–ligand binding free energies for drug design. Chem. Sci. 2021;12:13958–13971. doi: 10.1039/D1SC03472C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldeghi M, Heifetz A, Bodkin MJ, Knapp S, Biggin PC. Predictions of ligand selectivity from absolute binding free energy calculations. J. Am. Chem. Soc. 2017;139:946–957. doi: 10.1021/jacs.6b11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aldeghi M, Heifetz A, Bodkin MJ, Knapp S, Biggin PC. Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 2016;7:207–218. doi: 10.1039/C5SC02678D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gapsys V, et al. Accurate absolute free energies for ligand–protein binding based on non-equilibrium approaches. Comm. Chem. 2021;4:61. doi: 10.1038/s42004-021-00498-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyce SE, et al. Predicting ligand binding affinity with alchemical free energy methods in a polar model binding site. J. Mol. Biol. 2009;394:747–763. doi: 10.1016/j.jmb.2009.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rizzi A, et al. Overview of the SAMPL6 host–guest binding affinity prediction challenge. J. Comp.-aided Mol. Des. 2018;32:937–963. doi: 10.1007/s10822-018-0170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maffucci I, Contini A. Improved Computation of Protein–Protein Relative Binding Energies with the Nwat-MMGBSA Method. J. Chem. Inf. Model. 2016;56:1692–1704. doi: 10.1021/acs.jcim.6b00196. [DOI] [PubMed] [Google Scholar]
- 37.Maffucci I, Contini A. Explicit ligand hydration shells improve the correlation between MM-PB/GBSA binding energies and experimental activities. J. Chem. Theor. Comput. 2013;9:2706–2717. doi: 10.1021/ct400045d. [DOI] [PubMed] [Google Scholar]
- 38.Böttcher J, et al. Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol. 2019;15:822–829. doi: 10.1038/s41589-019-0310-x. [DOI] [PubMed] [Google Scholar]
- 39.Murray CW, et al. Fragment-based drug discovery applied to HSP90. Discovery of two lead series with high ligand efficiency. J. Med. Chem. 2010;53:5942–5955. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- 40.Friberg A, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grädler U, et al. Discovery of novel Cyclophilin D inhibitors starting from three dimensional fragments with millimolar potencies. Bioorg. Med. Chem. Letts. 2019;29:126717. doi: 10.1016/j.bmcl.2019.126717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khalak Y, Tresadern G, de Groot BL, Gapsys V. Non-equilibrium approach for binding free energies in cyclodextrins in SAMPL7: force fields and software. J. Comput. -Aided Mol. Des. 2021;35:49–61. doi: 10.1007/s10822-020-00359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gapsys V, et al. Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chem. Sci. 2020;11:1140–1152. doi: 10.1039/C9SC03754C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown SP, Muchmore SW, Hajduk PJ. Healthy skepticism: assessing realistic model performance. Drug Discov. Today. 2009;14:420–427. doi: 10.1016/j.drudis.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Myszka DG, et al. The ABRF-MIRG’02 study: assembly state, thermodynamic, and kinetic analysis of an enzyme/inhibitor interaction. J. Biomol. Tech. 2003;14:247–269. [PMC free article] [PubMed] [Google Scholar]
- 46.Tellinghuisen J, Chodera JD. Systematic errors in isothermal titration calorimetry: concentrations and baselines. Anal. Biochem. 2011;414:297–299. doi: 10.1016/j.ab.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 47.Henriksen NM, Gilson MK. Evaluating force field performance in thermodynamic calculations of cyclodextrin host–guest binding: Water models, partial charges, and host force field parameters. J. Chem. Theory Comput. 2017;13:4253–4269. doi: 10.1021/acs.jctc.7b00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baumann, H. M., Gapsys, V., de Groot, B. L. & Mobley, D. L. Challenges encountered applying equilibrium and nonequilibrium binding free energy calculations. J. Phys. Chem. B. 125, 4241–4261 (2021). [DOI] [PMC free article] [PubMed]
- 49.Ben-Shalom IY, Lin C, Kurtzman T, Walker RC, Gilson MK. Simulating water exchange to buried binding sites. J. Chem. Theory Comput. 2019;15:2684–2691. doi: 10.1021/acs.jctc.8b01284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ben-Shalom IY, et al. Accounting for the central role of interfacial water in protein–ligand binding free energy calculations. J. Chem. Theory Comput. 2020;16:7883–7894. doi: 10.1021/acs.jctc.0c00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright L, et al. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem. Biol. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 52.Immormino RM, Kang Y, Chiosis G, Gewirth DT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class hsp90 inhibitors. J. Med. Chem. 2006;49:4953–4960. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- 53.Güldenhaupt J, et al. Ligand-induced conformational changes in HSP90 monitored time resolved and label free—towards a conformational activity screening for drug discovery. Ang. Chem. Int. Ed. 2018;57:9955–9960. doi: 10.1002/anie.201802603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amaral M, et al. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 2017;8:2276. doi: 10.1038/s41467-017-02258-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.le Maire A, et al. In-plate protein crystallization, in situ ligand soaking and X-ray diffraction. Acta Crysta. D. 2011;67:747–755. doi: 10.1107/S0907444911023249. [DOI] [PubMed] [Google Scholar]
- 56.Aldeghi M, Bodkin MJ, Knapp S, Biggin PC. A statistical analysis on the performance of MMPBSA versus absolute binding free energy calculations: bromodomains as a case study. J. Chem. Inf. Model. 2017;57:2203–2221. doi: 10.1021/acs.jcim.7b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Disco. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tuccinardi T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Disco. 2021;16:1233–1237. doi: 10.1080/17460441.2021.1942836. [DOI] [PubMed] [Google Scholar]
- 59.Genheden S, Kuhn O, Mikulskis P, Hoffmann D, Ryde U. The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012;52:2079–2088. doi: 10.1021/ci3001919. [DOI] [PubMed] [Google Scholar]
- 60.Kongsted J, Ryde U. An improved method to predict the entropy term with the MM/PBSA approach. J. Comput-Aid Mol. Des. 2008;23:63. doi: 10.1007/s10822-008-9238-z. [DOI] [PubMed] [Google Scholar]
- 61.Ekberg V, Ryde U. On the use of interaction entropy and related methods to estimate binding entropies. J. Chem. Theory Comput. 2021;17:5379–5391. doi: 10.1021/acs.jctc.1c00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Procacci P. Methodological uncertainties in drug-receptor binding free energy predictions based on classical molecular dynamics. Curr. Opin. Struct. Biol. 2021;67:127–134. doi: 10.1016/j.sbi.2020.08.001. [DOI] [PubMed] [Google Scholar]
- 63.Nelson L, et al. Implementation of the QUBE force field in SOMD for high-throughput alchemical free-energy calculations. J. Chem. Inf. Model. 2021;61:2124–2130. doi: 10.1021/acs.jcim.1c00328. [DOI] [PubMed] [Google Scholar]
- 64.Cole DJ, Cabeza de Vaca I, Jorgensen WL. Computation of protein–ligand binding free energies using quantum mechanical bespoke force fields. Med. Chem. Comm. 2019;10:1116–1120. doi: 10.1039/C9MD00017H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cabeza de Vaca I, Qian Y, Vilseck JZ, Tirado-Rives J, Jorgensen WL. Enhanced Monte Carlo methods for modeling proteins including computation of absolute free energies of binding. J. Chem. Theory Comput. 2018;14:3279–3288. doi: 10.1021/acs.jctc.8b00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duboué-Dijon E, Hénin J. Building intuition for binding free energy calculations: Bound state definition, restraints, and symmetry. J. Chem. Phys. 2021;154:204101. doi: 10.1063/5.0046853. [DOI] [PubMed] [Google Scholar]
- 67.Mobley DL, Chodera JD, Dill KA. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 2006;125:084902. doi: 10.1063/1.2221683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Y, Laury ML, Wang Z, Ponder JW. AMOEBA binding free energies for the SAMPL7 TrimerTrip host–guest challenge. J. Comput-Aid Mol. Des. 2021;35:79–93. doi: 10.1007/s10822-020-00358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gill SC, et al. Binding Modes of Ligands Using Enhanced Sampling (BLUES): Rapid Decorrelation of Ligand Binding Modes via Nonequilibrium Candidate Monte Carlo. J. Phys. Chem. B. 2018;122:5579–5598. doi: 10.1021/acs.jpcb.7b11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanida Y, Matsuura A. Alchemical free energy calculations via metadynamics: application to the theophylline-RNA aptamer complex. J. Comput. Chem. 2020;41:1804–1819. doi: 10.1002/jcc.26221. [DOI] [PubMed] [Google Scholar]
- 71.Kaus JW, et al. How to deal with multiple binding poses in alchemical relative protein–ligand binding free energy calculations. J. Chem. Theory Comput. 2015;11:2670–2679. doi: 10.1021/acs.jctc.5b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stjernschantz E, Oostenbrink C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys. J. 2010;98:2682–2691. doi: 10.1016/j.bpj.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paulsen JL, et al. Evaluation of free energy calculations for the prioritization of macrocycle synthesis. J. Chem. Inf. Model. 2020;60:3489–3498. doi: 10.1021/acs.jcim.0c00132. [DOI] [PubMed] [Google Scholar]
- 74.Jarmoskaite I, AlSadhan I, Vaidyanathan PP, Herschlag D. How to measure and evaluate binding affinities. eLife. 2020;9:e57264. doi: 10.7554/eLife.57264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.The PyMOL molecular graphics system. (2004).
- 76.Bietz S, Urbaczek S, Schulz B, Rarey M. Protoss: a holistic approach to predict tautomers and protonation states in protein-ligand complexes. J. Cheminform. 2014;6:12. doi: 10.1186/1758-2946-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lippert T, Rarey M. Fast automated placement of polar hydrogen atoms in protein-ligand complexes. J. Cheminform. 2009;1:13. doi: 10.1186/1758-2946-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fährrolfes R, et al. ProteinsPlus: a web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017;45:W337–W343. doi: 10.1093/nar/gkx333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schöning-Stierand K, et al. ProteinsPlus: interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020;48:W48–W53. doi: 10.1093/nar/gkaa235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Webb B, Sali A. Comparative protein structure modeling using modeller. Curr. Prot. Bioinf. 2014;5:5.61–65.66.32. doi: 10.1002/0471250953.bi0506s47. [DOI] [PubMed] [Google Scholar]
- 81.Benkert P, Tosatto SCE, Schomburg D. QMEAN: a comprehensive scoring function for model quality assessment. Proteins: Struct. Funct. Bioinforma. 2008;71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 82.Waterhouse A, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.AMBER 2018 (University of California, 2018).
- 84.Abraham MJ, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 85.Jorgensen WL. Transferable intermolecular potential functions for water, alcohols, and ethers, application to liquid water. J. Am. Chem. Soc. 1981;103:335–340. doi: 10.1021/ja00392a016. [DOI] [Google Scholar]
- 86.Feenstra KA, Hess B, Berendsen HJC. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comp. Chem. 1999;20:786–798. doi: 10.1002/(SICI)1096-987X(199906)20:8<786::AID-JCC5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 87.Hopkins CW, Le Grand S, Walker RC, Roitberg AE. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015;11:1864–1874. doi: 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- 88.Heinzelmann G, Gilson MK. Automation of absolute protein-ligand binding free energy calculations for docking refinement and compound evaluation. Sci. Rep. 2021;11:1116. doi: 10.1038/s41598-020-80769-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee T-S, et al. Alchemical binding free energy calculations in AMBER20: advances and best practices for drug discovery. J. Chem. Inf. Model. 2020;60:5595–5623. doi: 10.1021/acs.jcim.0c00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang H, et al. CHARMM-GUI free energy calculator for practical ligand binding free energy simulations with AMBER. J. Chem. Inf. Model. 2021;61:4145–4151. doi: 10.1021/acs.jcim.1c00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miyamoto S, Kollman PA. SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water molecules. J. Comp. Chem. 1992;18:1463–1472. [Google Scholar]
- 92.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 93.Hess BP-lincs. A parallel linear constraint solver for molecular simulation. J. Chem. Theor. Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 94.Hess B, Bekker J, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 95.Allen, M. P. & Tildesley, D. J. Computer Simulation of Liquids. (Oxford University Press, 1987).
- 96.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 97.Parrinello M, Rahman A. Polymorphic transitions in single crystals - a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 98.Essman U, et al. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 99.Darden T, Pearlman D, Pedersen LG. Ionic charging free energies: spherical versus periodic boundary conditions. J. Chem. Phys. 1998;109:10921–10935. doi: 10.1063/1.477788. [DOI] [Google Scholar]
- 100.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 101.Le Grand S, Götz AW, Walker RC. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013;184:374–380. doi: 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- 102.Boresch S, Tettinger F, Leitgeb M, Karplus M. Absolute binding free energies:A quantitative approach for their calculation. J. Phys. Chem. B. 2003;107:9535–9551. doi: 10.1021/jp0217839. [DOI] [Google Scholar]
- 103.Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem. Phys. Lett. 1994;222:529–539. doi: 10.1016/0009-2614(94)00397-1. [DOI] [Google Scholar]
- 104.Rocklin GJ, Mobley DL, Dill KA, Hünenberger PH. Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: an accurate correction scheme for electrostatic finite-size effects. J. Chem. Phys. 2013;139:184103. doi: 10.1063/1.4826261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gowers, R. J. et al. In Proceedings of the 15thPython in Science Conference. (eds. Benthall, S. & Rostrup, S.) 98–105.
- 106.Michaud-Agrawal N, Denning EJ, Woolf T, Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 108.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins: Struct. Funct. Bioinforma. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 109.AMBER 2021 (University of California, 2020).
- 110.https://github.com/alchemistry/alchemlyb.
- 111.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008;129:124105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jurrus E, et al. Improvements to the APBS biomolecular solvation software suite. Prot. Sci. 2018;27:112–128. doi: 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shirts MR, Mobley DL, Chodera JD, Pande VS. Accurate and efficient corrections for missing dispersion interactions in molecular simulations. J. Phys. Chem. B. 2007;111:13052–13063. doi: 10.1021/jp0735987. [DOI] [PubMed] [Google Scholar]
- 115.Meli R, Biggin PC. spyrmsd: symmetry-corrected RMSD calculations in Python. J. Cheminf. 2020;12:49. doi: 10.1186/s13321-020-00455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harris CR, et al. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Virtanen P, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Meth. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pedregosa F, et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 119.Hunter JD. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 120.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Input topologies, coordinates, and simulation control files are provided at https://zenodo.org/record/5913469 and https://github.com/bigginlab/fragment-opt-abfe-benchmark.
The implementation of the MDRestraintsGenerator can be found here: https://zenodo.org/record/4570556 Files containing the sampled ΔH and ΔH/Δλ energies have been made publicly available at zenodo: https://zenodo.org/record/5906262, https://zenodo.org/record/5906110, https://zenodo.org/record/5904110, https://zenodo.org/record/5906019 and https://zenodo.org/record/5906805.