

Abstract
Engineered T cell therapies such as CAR-T cells and TCR-T cells have generated impressive patient responses in previously incurable diseases. In the past few years there have been a number of technical innovations that enable robust clinical manufacturing in functionally closed and often automated systems. Here we describe the latest technology used to manufacture CAR- and TCR-engineered T cells in the clinic, including cell purification, transduction/transfection, expansion and harvest. To help compare the different systems available, we present three case studies of engineered T cells manufactured for phase I clinical trials at the NIH Clinical Center (CD30 CAR-T cells for lymphoma, CD19/CD22 bispecific CAR-T cells for B cell malignancies, and E7 TCR T cells for human papilloma virus-associated cancers). Continued improvement in cell manufacturing technology will help enable world-wide implementation of engineered T cell therapies.
Graphical Abstract
INTRODUCTION
Over the last several years, engineered T cells such as chimeric antigen receptor (CAR)-T cells and TCR-engineered T cells have proven to be a powerful tool for treating B cell leukemia/lymphoma and melanoma/sarcoma, respectively [1–5]. Not only is this approach potentially curative for relapsed and/or refractory patients, engineered T cells are also more specific and can generate fewer off-target side effects than traditional approaches such as chemotherapy and radiation. At the same time, emerging evidence suggests combinatorial therapies such as anti-PDL1 can sensitize certain tumors for destruction by engineered T cells [6, 7], while advances in gene editing technology such as CRIPSR/Cas9 offer enticing possibilities for creating designer T cells in both allogeneic and autologous settings [8, 9]. It is no longer a question of whether engineered T cells have the potential to cure cancer, but how best to utilize these cells to offer each patient the greatest chance of a cure.
Indeed, the number of patients being treated with CAR- and TCR-engineered T cells is growing tremendously [10, 11]. From the 600+ registered clinical trials since 2003, it is estimated that more than 25,000 patients have been treated with CAR-T and TCR engineered T cells, the majority within the last three years [10]. Although phase I clinical trials may treat several dozen patients, as clinical trials advance through the pipeline, the need to scale manufacturing capability to meet demand is clear. Special consideration is required for both scaling up and scaling out the process, with autologous therapies in particular requiring substantial scale-out capacity.
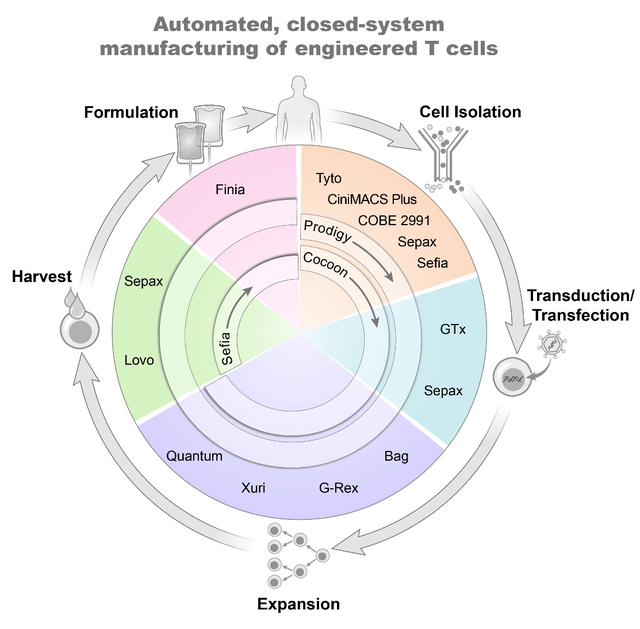
Enabling world-wide availability of engineered T cell therapies is a significant challenge. Central to that goal are advances in technology that render the manufacturing process as safe and efficient as possible. Automating key parts of the T cell manufacturing process (i.e., purification, expansion, and harvest) has garnered interest among many companies and may hold the key to improving safety and lowering overall costs. The potential advantages are many (Table 1), and yet T cells – like all cells – harbor the ability to sense and respond to their microenvironment, making the implementation of each manufacturing step critical to the performance of the final product. Here we describe some of the recent advances in automation technology for T cell manufacturing, with the caveat that it is not intended as a comprehensive list but rather to highlight the creative solutions the field has implemented.
Table 1:
Potential benefits of using automated closed-system technology for T cell manufacturing
| • Ability to maintain sterility via functionally closed disposables |
| • Reduce operator errors by relying on pre-programmed steps |
| • Increased consistency by reducing operator-to-operator variability |
| • Reduce hands-on time for technicians, which may reduce product cost |
| • Less skilled workforce required; potential cost savings in training staff |
CELL PURIFICATION
One of the first decisions clinical investigators face is whether they want to perform a T cell isolation or start from unselected peripheral blood mononuclear cells (PBMCs). PBMCs are routinely isolated in pre-clinical laboratories using open methods such as conical tubes. However, open methods are discouraged for clinical manufacturing due to the heightened potential for microbial contamination, due in part to the reduced usage of antibiotics in the culture medium. In the clinic, PBMCs are generally isolated using closed systems that are either semi-automated or fully-automated. Historically, instruments such as the COBE 2991 (Terumo BCT) have offered the ability to automate some parts of the density gradient separation [12]. More recently, Cytiva introduced the Sepax C-Pro and Sefia S-2000 technologies that can both perform fully automated density gradient separation at different scales, allowing the user to simply connect their product, buffer, and separation matrix (i.e. Ficoll) in a closed, sterile kit. The entire process takes less than 2 hours to process over a liter of product on the Sepax, with the Sefia having the ability to perform a platelet wash in tandem with density gradient separation.
A stalwart in cell therapy manufacturing, the CliniMACS Plus is often used to purify T cell populations. Anti-CD4 and anti-CD8 magnetic selection beads are typically combined to select a single T cell product on a magnetic separation column, or selections can be performed separately if specific CD4 or CD8 subsets or ratios are desired. Typically, CD3+ selections are not performed in order to leave this site available for anti-CD3 stimulation. Although more expensive than density gradient separation, the T cell purity is much improved using CliniMACS selection; in our experience, we can routinely achieve >90% purity from a fresh MNC apheresis using a CD4/CD8 combined selection [13]. While the CliniMACS Plus automates the magnetic selection process, the upstream staining and washing must be performed by hand. Miltenyi subsequently released the CliniMACS Prodigy with the same core magnetic separation technology as the CliniMACS Plus, but the addition of a culture chamber and additional input lines and valves allow the user to simply attach cells, beads, and buffer and have the entire selection process performed automatically. Although the target cell capacity is smaller (3×109 targets on the Prodigy vs. 10×109 on the CliniMACS Plus), the Prodigy saves significant hands-on time. Importantly, the method of T cell selection has a substantial impact beyond cost and labor, and that is in the actual performance of the manufactured CAR-T cells in vivo; in one recent study from our institution, switching to a CD4+CD8+ CliniMACS Plus selection from a CD3/CD28-enrichment resulted in lower doses being required for clinical efficacy [13]. This is a great example of how every step of cell therapy manufacturing, even the very first step, can have profound implications for the performance of the product in vivo.
In some cases, more complex selection schemes may be desired to isolate subsets of T cells such as stem cell memory (Tscm)-like cells [14] or regulatory T cells [15] based on multiparameter fluorescent detection. Standard droplet sorters are not GMP-compliant due to the open nature of the selection process. In contrast, the MACSQuant Tyto from Miltenyi is an elegant, GMP-compliant instrument that utilizes a single-use, closed-system cartridge with a high-speed valve that allows for the selection of 10 mL of product in 3 hours. (An updated cartridge system was released in 2021 that reduces that time to just over 1 hour.) The Tyto technology allows users to select cells based on fluorescence in up to 8 channels using standard flow cytometry gating. In our experience, purity and recovery are comparable to a standard droplet sorter, and viability of sorted T cells is excellent at ≥ 95% (data not shown).
A critical concern for developing a selection process is the availability of GMP-compliant ancillary materials (i.e., beads, antibodies), particularly for later-phase studies. Another point of consideration is the desired T cell stimulation method. While soluble anti-CD3 stimulation provides T cells with signal 1, antigen-presenting cells in the PBMC mixture are necessary to provide signal 2 to prevent T cell anergy. Thus, a PBMC starting population is important if anti-CD3 stimulation is desired, while isolated T cells require anti-CD3/anti-CD28 co-stimulatory reagents such as Dynabeads or TransAct.
CELL EXPANSION
Once a T cell-selected or PBMC-enriched population is prepared, cells are generally seeded into a closed culture system to initiate cell stimulation and expansion. Excitingly, there are a growing number of instruments and technologies available to investigators to expand their engineered T cells. The available options range from straightforward and mostly manual operations (i.e., closed culture bags, G-Rex), to entirely end-to-end automation (Prodigy, Cocoon), or expanding T cells once they reach the exponential growth phase (Xuri W25, Quantum).
Cell culture bags are one of the simplest and lowest-cost methods to grow T cells in a closed system. In contrast to open systems such as plates or flasks, which heighten the risk of contamination, bags are fully closed and utilize syringes for the manipulation of cells or media. Given that T cells prefer to be activated “low and slow,” i.e., without any shaking or manipulations that may disrupt paracrine signaling, bag culture is perfectly suited to initiating T cell cultures. However, once T cells reach exponential growth phase (generally days 4–6), the size of the culture may require time- and labor-intensive manipulations, increasing complexity and decreasing reproducibility. Because of their simplicity and relatively low costs compared to automated technologies, culture bags remain a popular choice in early-phase studies.
The Gas-permeable rapid expansion (G-Rex) bioreactor from Wilson-Wolf is a simple solution to the problem of large culture size. At the core of the technology is a gas-permeable liquid silicone rubber membrane located at the bottom of a single-use plastic culture vessel, which allows gas exchange between the outside environment and the T cells resting on the membrane. G-Rex bioreactors come in a variety of sizes, the largest being a 5L culture which can be seeded with as few as 250 million T cells. Importantly, cells should be in the exponential growth phase, but once seeded in the G-Rex they can grow in a single vessel for up to several weeks. Because T cells should ideally stay at the bottom of the vessel adjacent to the permeable membrane as much as possible, frequent sampling must be avoided. Immediately prior to harvest, the culture volume can be reduced by up to 80–90% by removing cell culture supernatant using the GatheRex system designed to work with any size G-Rex, simplifying the downstream harvesting process.
The Xuri W25 cell expansion system from Cytiva has a similar purpose to the G-Rex, in that it is designed for rapid expansion of cells following activation and/or transduction in an outside system, such as bags. Where they differ is the degree of technology and automation. Whereas the G-Rex a simple chamber device requiring manual operations, the Xuri offers a high degree of automation combined with impressive flexibility that allows investigators to tailor expansion protocols for their particular application. The heart of the Xuri technology (and its predecessor, the WAVE bioreactor) is a rocking platform that maintains temperature and gas control within an engineered culture bag, utilizing a customizable pump system that can accommodate either batch or perfusion feeding schemes. Importantly, Xuri culture bags are available in many sizes, ranging from a 1 liter culture (suitable for autologous CAR-T cells) up to 10 or 20 liters (more appropriate for allogeneic settings and/or solid tumor applications). Users can optimize the feed rates, rocking angles, and cell seeding parameters to achieve a high degree of expansion, up to 20 billion cells from a 2L culture bag [16]. As a plus, the system can monitor pH and dissolved oxygen for improved process control. Given the high degree of automation and the ease of scalability, it is no surprise that rocking platforms such as the Xuri (and its predecessor, the WAVE bioreactor) were utilized for several phase I/II CAR-T cell trials [17–20]. However, with automation comes a higher cost (including the instrument and consumables such as tubing sets), which can be a barrier to adoption.
The Quantum hollow fiber bioreactor (Terumo BCT) is another perfusion-based system with two compartments separated by a semi-permeable membrane. The T cells are typically cultured in the intra-capillary space of the hollow fibers, and the media is perfused in the extra-capillary side. The diameter of each fiber is approximately 200 μm, and with a membrane pore size of 17 kDa this keeps T cells and cytokines within the intra-capillary space while allowing gases and media components such as glucose and lactate to pass freely. This achieves a high cell density while still ensuring that adequate nutrient exchange through the recirculating media. Impressively, 10–20 billion T cells can be generated in a 9-day culture process on the Quantum [21].
New to the scene of cell therapy manufacturing is the Lonza Cocoon platform. Similar to the Xuri and the Quantum, the Cocoon provides environmental control in a compact bioreactor unit. A key difference from the Xuri and G-Rex is that the Cocoon is designed for culture of T cells from Day 0 onward; indeed, new models of the Cocoon slated for release in 2022 have been updated with magnetic selection capabilities, which could prove useful for automated T cell purification and/or magnetic-debeading to increase the utility. In contrast to the Xuri with its scalable bag sizes, the Cocoon is currently available with a fixed Proliferation Chamber volume of 180 mL and a media re-circulation volume of 460 mL. The Cocoon was designed with a vision of being able to hook up dozens of Cocoon units (being branded as a Cocoon “Orchard”) to utilize vertical space and thus reduce footprint of commercial manufacturing facilities. Certainly, the high degree of automation combined with customizable unit operations and parallelization is a remarkable advancement, although much remains to be seen as more users adopt this new technology.
The CliniMACS Prodigy from Miltenyi has been widely adopted by the cell therapy field to automate full CAR-T cell processes [22–25] from selection, transduction, expansion, to harvest – and has recently launched add-ons capable of electroporation and final formulation on the same machine. The Prodigy features a Centricult unit that functions as both culture chamber and centrifuge, which is connected to highly engineered pump and valve system that allows the user to connect various culture components at key junctures (such as selection beads, stimulation reagent, lentiviral vector, medias, and buffers) that are processed automatically with very little user interaction. Notably, this all-in-one approach provides several key advantages, including less hands-on technician time and increased reproducibility. While the Prodigy is less scalable than the Xuri or G-Rex vessels (the Prodigy is currently only available with a 250 mL culture chamber), its user-friendly touch screen programming enables easy development and robust manufacturing. However, in contrast to the other expansion systems, Prodigy users are locked into utilizing Miltenyi media and stimulation reagents, potentially limiting its use for some investigators, and potentially increasing costs as well.
TRANSDUCTION AND TRANSFECTION
Both the Prodigy and Cocoon cell expansion systems are capable of transducing cells via automated addition and washout of viral vector. Indeed, the Prodigy is even capable of performing a spinoculation via its hybrid centrifuge/culture chamber. However, when growing T cells in cell culture bags, the transduction process is typically either static or spinoculated in culture bags, which can sometimes result in bag tears that compromise product integrity. The Sepax C-Pro (Cytiva) has now been adapted to perform automated spinoculations that can match or even surpass centrifuge-based spinoculation. The speed and time can be optimized, and we have found that it takes less hands-on time with greater transduction efficiency than traditional centrifugation [26].
On the transfection front, Miltenyi has debuted the Prodigy Electroporator as an add-on feature to the traditional Prodigy that can move cells from the culture chamber, through the electroporator, and back into the culture chamber via sterile tubing system for minimal hands-on time and maximum automation. A similar set-up has been designed for the Cocoon platform with the 4D-Nucleofector (Lonza) which also feature functionally closed tubing connections. Otherwise, the GTx Electroporator from MaxCyte can transfect up to 20 billion cells in cGMP-compliant cartridges and boasts an established regulatory pathway supported by an FDA Master File [27]. Several early phase clinical trials have already utilized this technology with success [28–31].
HARVEST and FINAL FORMULATION
When faced with liters of cell-rich culture media at the end of a manufacturing process, it quickly becomes clear that automatic washing and/or harvesting technologies can provide huge time savings in addition to improved recovery. The Lovo Cell Processing System (Fresenius Kabi) features a spinning membrane technology that allows for automation of cell washing processes that can be useful at both the beginning and end of a manufacturing process. The Lovo can perform automatic cell washing of up to 22 L of culture in a closed, sterile kit, ultimately concentrating the final product down to as little as 150 mL of product. The Sepax C-Pro (Cytiva) and the Sefia S-2000 (Cytiva) also feature a spinning membrane in a sterile, closed-system disposable kit, but the addition of a temperature-controlled thermal mixer on the Sefia can be used for the addition of DMSO during final formulation (or staining reagents such as antibodies or beads for automating other parts of the cell manufacturing process). Finally, the Finia Fill and Finish system (Terumo BCT) can perform the final formulation for cryopreservation of up to 3 product bags, chilling the cells and performing the DMSO mixing step in a temperature-controlled manner, potentially improving cell viability compared to manual mixing. Automated controlled-rate freezers can freeze cryobags or cryovials to fit a variety of configurations, such as the commonly-used instruments from Planer, although newer systems have been developed that do not require the use of a liquid nitrogen, including the Viafreeze from Cytiva.
SELECTING AN EXPANSION PLATFORM: OUR APPROACH
Since 2012, our institution has helped to develop cell manufacturing processes for numerous engineered T cell therapies for phase I/II clinical trials at the NIH Clinical Center. In this section, we discuss our approach to developing a manufacturing schema for engineered T cells and present key manufacturing data from patient products. To this end, we present three case studies of engineered T cell therapies manufactured using various methods for different clinical applications (Figure 1A).
Figure 1: Key manufacturing data from patient products manufactured using various expansion systems.
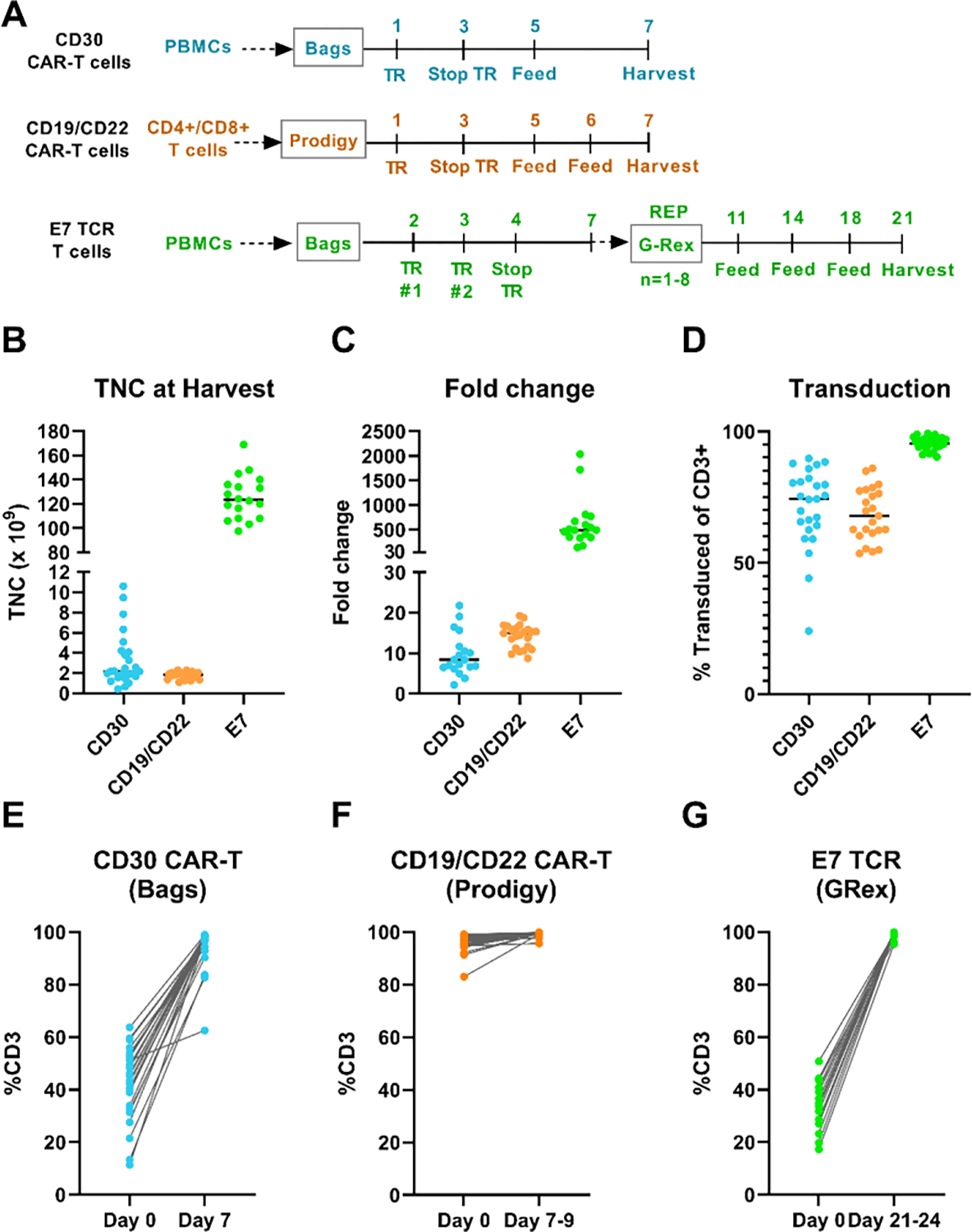
(A) Culture schematic with major processing steps. (B) TNC at harvest: CD30 CAR-T = 7 days culture, CD19/22 CAR-T = 7–9 days culture, E7 TCR = 21–24 days culture (only maximum dose level is shown). (C) CD30 CAR-T fold change = CD3+ TNC of Final (Day 7) ÷ Day 1; CD19/CD22 bispecific CAR-T fold change = CD3+ TNC of Final (Day 7–9) ÷ Day 0; E7 TCR fold change = CD3+ TNC of Final (Day 21–24) ÷ Day 2 (only dose level 3 is shown). (D) Transduction efficiencies were measured via CAR-specific flow cytometry on the day of harvest. (E) – (G) Percent CD3 in the starting product compared to final product. PBMCs = peripheral blood mononuclear cells, TNC = total nucleated cells, TR=transduction, REP = rapid expansion phase.
Case Study 1: CD30 CAR-T cells expanded in cell culture bags
In 2016, we developed a manufacturing protocol for a fully-human CD30 CAR-T cell product for the treatment of lymphoma [32], with targeted dose levels between 0.3–9×106 transduced T cells per kg. Following PBMC enrichment from an MNC apheresis using a COBE 2991 blood cell separator, approximately 800–1200×106 CD3+ cells were seeded into culture bags (PermaLife, Origen Biomedical) at a density of 1.5×106 CD3+ T cells/mL (median %CD3 = 45.3%, Table 2). On Day 1, 360–395×106 total nucleated cells were transduced for 48 hours with lentiviral vector plus protamine sulfate. The rationale behind using 360–395×106 cells on Day 1 was to use exactly one vial of lentiviral vector (which in some runs was scaled 3-fold); therefore, initiating 800–1200×106 CD3+ cells on Day 0 ensured adequate cell numbers for Day 1 transduction. Cells were expanded in media containing 50 ng/mL soluble anti-CD3 and 300 IU/mL IL-2, and cell density was adjusted on Day 3 and Day 5 to between 0.5 – 1×106 viable cells/mL, with requisite volume of media being added to the culture to achieve the target cell density. The culture was transferred to larger bag sizes as necessary, from 30 mL up to 5 liters to accommodate various cell culture volumes and growth rates. All cultures were harvested on Day 7 using a Lovo device for automatic cell washing, and cells were cryopreserved for infusion at a later time.
Table 2:
Key manufacturing data for engineered T cell products manufactured on phase I clinical trials at the NIH Clinical Center.
| CD30 CAR-T Cells | CD19/22 Bispecific CAR-T cells | E7 TCR-T Cells | |
|---|---|---|---|
| Trial Number on clinicaltrials.gov | NCT03049449 | NCT03448393 | NCT02858310 |
| Disease | Lymphoma | Recurrent or refractory B cell malignancies | Human papillomavirus-associated cancers |
| T cell expansion method | Culture Bags | CliniMACS Prodigy | G-Rex-500 (up to 8 in parallel) |
| Dose levels manufactured | 0.3, 1, 3, 9 × 106 CAR+ T cells/kg | 0.3, 1, 3 × 106 CAR+ T cells/kg | 1, 10, 100 × 109 TCR+ T cells |
| Number of patient products manufactured | 25 | 24 | 25 |
| Number of patients manufactured at maximum dose level | 7 | 16 | 18 |
| Starting cell population | PBMCs enriched via Ficoll density gradient on COBE 2991 | CD4+/CD8+ selected via CliniMACS Plus or CliniMACS Prodigy | PBMCs enriched via Ficoll density gradient on COBE 2991 |
| Media and cytokines | AIM-V, 3% AB serum, 300 IU/mL IL2 | TexMACS, 3% AB serum, 200 IU/mL IL-2 | Initial culture (D0–7)= AIM-V, 5% AB serum, 2mM GlutaMax, 300 IU/mL IL-2; REP phase (D7–21) increase IL-2 to 3000 IU/mL IL-2 |
| Stimulation | Anti-CD3 (50 ng/mL) | TransAct | Initial culture = anti-CD3 (50 ng/mL); REP phase = anti-CD3 restimulation (30 ng/mL) + irradiated PBMC feeders (100:1) |
| Culture Length, Median (Range) | 7 days | 7 days (7–9) | 22 days (21 – 24) |
| % CD3+ at Day 0, Median (Range) | 45.4% (11.4 – 63.7) | 96.5% (83.0 – 99.2) | 33.6% (17.3 – 55.2) |
| % CD3+ at harvest, Median (Range) | 97.3% (62.5 – 98.9) | 99.5% (95.7 – 99.9) | 99.6% (95.4 – 99.9) |
| Viable TNC at harvest* Median (Range) | 2.19 × 109 (0.43 – 10.60) | 1.89 × 109 (1.11 – 2.29) | 123.3 × 109 (97.5 – 169.0) |
| CAR+/TCR+ TNC at harvest* Median (Range) | 1.45 × 109 (0.19 – 7.89) | 1.26 × 109 (0.33 – 1.79) | 118.2 × 109 (96.4 – 162.4) |
| Fold-change*^, Median (Range) | 8.4-fold (2.2 – 21.8) | 15.0-fold (8.8 – 19.3) | 491.1-fold (135.4 – 2033.1) |
For E7, TNCs and fold-change are only tabulated for maximum dose level.
CD30 CAR-T fold change = CD3+ TNC of Final (Day 7) ÷ Day 1; CD19/CD22 bispecific CAR-T fold change = CD3+ TNC of Final (Day 7–9) ÷ Day 0; E7 TCR fold change = CD3+ TNC of Final (Day 21–24) ÷ Day 2.
We successfully manufactured 25 products for patients enrolled at 4 different dose levels (Table 2), only one of which was manufactured a second time from a new apheresis product due to a tubing set leak on the COBE 2991. The median total number of cells at harvest was 2.2×109 (range = 0.4–10.6×109, Figure 1B). The highest dose level enrolled was 9×106 CAR+ cells/kg (7 patients enrolled), and with a median patient weight of 83 kg this corresponds to approximately 750×106 transduced CAR+ cells required for treatment alone. The median number of transduced CAR-T cells was nearly two-fold above the number required for the maximum dose, at median 1454×106 CAR+ cells at harvest on Day 7 (range = 185–7894×106).
The use of PBMCs plus soluble anti-CD3 activation via OKT3 makes this process one of the simplest to implement; bag cultures are straightforward, and activation via soluble antibody means that there are no labor-intensive de-beading steps. Despite the variability of T cells in the initial PBMC population (11.4 – 63.7% CD3+ on Day 0), 24/25 products were ≥80% CD3+ on Day 7 harvest, and 18/25 were ≥95% CD3+ (Figure 1E).
Case Study 2: CD19/CD22 bispecific CAR-T Cells manufactured on the CliniMACS Prodigy
In 2016, we began developing a Prodigy-based manufacturing process for a CD19/CD22 bispecific CAR designed to address the emerging issue of antigen-negative relapse of CD19 CAR therapies in B cell malignancies. Early hurdles to Prodigy adoption here and elsewhere included defining the TransAct anti-CD3/28 washout step [25], as well as ensuring sterile and complete addition of important culture components such as selection beads, vector, and TransAct. A 7 to 12-day process was initially established using the Prodigy T Cell Transduction (TCT) program, which was subsequently shortened to 7 to 9 days in later clinical trials. This clinical trial, which is still open and accruing patients, is focused on treating pediatric and young adults; the dose levels targeted were 0.3, 1, and 3×106 CAR-T cells per kg (dose levels 1–3), with the majority of patients enrolled on dose level 3. With an average weight of 60 kg for the 24 patients enrolled, this corresponds to approximately 180×106 transduced cells needed for the clinical dose alone at dose level 3.
The Prodigy has proven to be very effective at manufacturing CAR-T cells at this scale. Of the 24 patients enrolled and apheresed to date, all products were successfully manufactured, with 23/24 of the manufacturing runs harvested on Day 7 and 1/24 on Day 9. Approximately 100–130×106 CD4/CD8 selected T cells were seeded on Day 0, followed by a 48-hour lentiviral transduction process on Day 1. The median TNC at harvest was 1.883×109 cells (Figure 1B), and with a median transduction efficiency of 68% at MOI = 40, this corresponded to 1.261×109 transduced T cells at harvest (Table 2). Of the 24 runs, 12 utilized selection on the Prodigy instrument, while 12 utilized the CliniMACS Plus. As expected, there was no difference in T cell purity between the CliniMACS Plus and Prodigy, with both yielding an average of 95% CD3+ (data not shown).
As expected, the final %CD3+ and TNC were more consistent on the automated Prodigy system compared to the CD30 CAR-T process described above (Table 2 and Figure 1B, 2E, 2F). Certainly, the CD30 CAR-T process has increased heterogeneity in the starting product (using PBMCs instead of selected T cells), as well as potential variability from manual technician processing in bag cultures. However, the Prodigy process had a ceiling of approximately 2.2×109 cells at harvest on Day 7, whereas the bag culture could regularly yield over 3×109 cells over the same culture duration (9/25 cultures, Figure 1B).
Case Study 3: E7 TCR T Cells expanded in G-Rex bioreactors
The targeted dose for E7 TCR-engineered T cells for the treatment of HPV-related epithelial cancers is orders of magnitude higher than CAR-T cells (1011 vs 108 cells, respectively). In this case, the use of scalable bioreactor systems is essential, and G-Rex bioreactors offered a straightforward solution given the scalability and ease of use. The E7 protocol, developed in 2015 and currently still recruiting patients, involves an initial 7 to 10-day culture with two retroviral transductions followed by a 14-day rapid expansion protocol (REP) in G-Rex bioreactors, for a total culture duration of 21–24 days [12, 33].
To start, cultures were initiated from fresh or cryopreserved PBMCs enriched from an MNC apheresis via a COBE 2991 instrument (median %CD3 = 33.6%, Figure 1G). PBMCs were cultured for 48 hours in media supplemented with anti-CD3 (50 ng/mL) and IL-2 (300 IU/mL). On Days 2 and 3, a minimum of 30–60×106 viable cells (depending on dose level) were transduced in 30 mL bags coated with retronectin followed by a 2-hour spinoculation. On Day 4, transduction was stopped, and cells were cultured in fresh media until Day 7, which could be extended to Day 10 for slower-growing cells. For the first two dose levels, at least 10×106 TCR+ T cells were required to seed into 1x G-Rex-500 bioreactor (5 L capacity), while dose level 3 required at least 60–80×106 TCR+ T cells for seeding 6–8x G-Rex-500 bioreactors.
For the REP culture phase, feeder cells were added (1×109 irradiated PBMCs per 10×106 TCR T cells, 100:1 ratio) along with anti-CD3 re-stimulation (30 μg/mL) and increased concentration of IL-2 (3000 IU/mL). To avoid over-diluting cells with media upfront, each G-Rex-500 was initiated in a volume of 200 mL, and media was added on Days 4, 7, and 11 (800 mL, 1200 mL, and 1700 mL, respectively). The target harvest day for the REP culture was Day 14, corresponding to Day 21 total culture. The harvest process was a significant undertaking involving harvest up to 40 L of culture in 3 discrete steps: (1) each individual G-Rex was volume-reduced and harvested into transfer packs via the GatheRex, (2) cells from all G-Rex bioreactors were combined, if applicable, followed by (3) automated washing on a LOVO device.
Of the 25 patients enrolled and apheresed on the E7 study, 18 were enrolled on the highest dose level corresponding to 100×109 transduced T cells. For these 18 patients, the median TNC at harvest was 123.3×109 total T cells (Table 2, Figure 1B). With transduction efficiencies ≥91% for all patients (median TE = 95%), this corresponded to a median of 118.2×109 TCR T cells. Of those 18 patients, only 2 did not reach the target of 100×109 TCR T cells, falling just short at 96.4×109 and 97.2×109 TCR T cells. Despite not meeting target dose, the products were still infused (not re-manufactured), although they were not considered as part of the calculation of maximum tolerated dose. The corresponding median fold-change at this highest dose level was 491-fold (Day 21–24 / Day 2, range = 135 – 2033-fold, Figure 1C).
Importantly, E7 TCR T cells could mediate epithelial tumor regression [34], although several challenges exist with manufacturing. Spinoculation in bags has been hampered by bag leakage at high centrifugation speeds, since bags are not necessarily designed for this task. Recently, we have found that this issue is mitigated by spinoculating in the Cytiva Sepax C-Pro device [26], which in addition to offering a closed, sterile solution is also fully automated and thus requires less hands-on time. Another challenge is the use of feeder cells, which were mixed from 3 distinct allogeneic donors in order to mitigate some of the deleterious effects of donor-to-donor variation. Later iterations of this culture for yet another distinct TCR (KK-LC-1), are focused on the elimination of feeder cells and reducing REP phase from 14 days down to 11 days to simplify the process and treat patients faster.
Selecting a manufacturing process: where the rubber meets the road
At the NIH Clinical Center, we are fortunate to have the institutional ability to pursue multiple manufacturing strategies, including bags, Prodigy, G-Rex, and others in the development pipeline. There are several concerns that drive initial conversations between our cell manufacturing team and investigators looking to bring cell therapies to patients. These discussions generally focus on scalability, comparability, and cost (Table 3).
Table 3:
Questions to consider when choosing a manufacturing platform
| • What is the target dose? How scalable does the manufacturing process need to be? |
| • How close does the investigator want to replicate pre-clinical methods? Are they intending to compare to other clinical trials using a certain system? |
| • Does the manufacturing scheme require non-standard manipulations that would be difficult to perform on an automated system? Are there significant technical limitations? |
| • How quickly can staff be trained on the new process? |
| • How does cost factor into the decision-making? |
In addition to the questions above, investigators may have different goals in initiating a clinical trial. Is the goal to treat several patients at isolated academic centers to generate deep and meaningful insights, perhaps trading a higher cost for greater robustness? Or is the focus on a low-cost translatable method that may be easier to implement but perhaps not as robust? Ultimately the field needs to bridge the gap, and automation is helping to improve robustness such that products are more consistent and less prone to manufacturing failures. Innovation in cell manufacturing technology will continue to promote consistency, and competition should drive costs downward as more companies enter the marketplace.
But before that can occur, more correlative studies are needed to identify what features lead to the best patient response, which may be different for each clinical setting. For example, lower T cell exhaustion in the infusion product correlates to better outcomes [35], driving investigators to shorten their manufacturing protocols to prevent exhaustion and treat patients faster [36–38]. As another example, several studies have identified that in vivo T cell persistence is a feature of efficacy in CD19 CAR therapies, leading investigators to hone in on Tscm-like phenotype in the infusion product [39, 40]. Importantly, correlative studies are predicated on the existence of a cohort of patient responders in order to identify those correlates of efficacy. As more correlates are identified, the field can begin to innovate manufacturing methods to drive those features in the final infusion product.
CONCLUSION
The last decade has seen an explosion of innovation in cell therapy manufacturing, with automated technologies easing key process bottlenecks while improving reliability. The most useful technologies will be flexible and customizable to allow the optimization of key process variables, while still remaining user-friendly for wide-scale adoption. While there are now several options on the market for true end-to-end automation of engineered T cells, other technologies that automate key process steps (i.e., selection, transduction, expansion, harvest) allow investigators to connect automated and/or manual processes in a modular fashion to tailor their overall process to best fit their needs.
ACKNOWLEDGEMENTS
We thank the staff of the Center for Cellular Engineering, Department of Transfusion Medicine (NIH Clinical Center) for manufacture and characterization of T cells therapies. We would like to thank Dr. James Kochenderfer (Surgery Branch, NIH Clinical Center), Dr. Nirali Shah (Pediatric Oncology Branch, NIH Clinical Center), and Dr. Christian Hinrichs (Rutgers Cancer Institute) for support in the development of the clinical protocols for CD30 CAR, CD19/CD22 Bispecific CAR, and E7 TCR T cells, respectively.
FUNDING
This research was supported by the Intramural Research Program of the NIH, Clinical Center and National Cancer Institute.
Footnotes
DECLARATION OF INTEREST
The authors declare that they have no conflict of interest.
The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
CITATIONS
- 1.Morgan RA, et al. , Cancer regression in patients after transfer of genetically engineered lymphocytes. Science, 2006. 314(5796): p. 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robbins PF, et al. , Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. Journal of Clinical Oncology, 2011. 29(7): p. 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neelapu SS, et al. , Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine, 2017. 377(26): p. 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DL, et al. , Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. New England Journal of Medicine, 2011. 365(8): p. 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, et al. , Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. New England Journal of Medicine, 2014. 371(16): p. 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherkassky L, et al. , Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest, 2016. 126(8): p. 3130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grosser R, et al. , Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell, 2019. 36(5): p. 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roselli E, Faramand R, and Davila ML, Insight into next-generation CAR therapeutics: designing CAR T cells to improve clinical outcomes. The Journal of Clinical Investigation, 2021. 131(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Mei H, and Hu Y, Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Briefings in Functional Genomics, 2020. 19(3): p. 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinte L, et al. , Global Perspective on the Development of Genetically Modified Immune Cells for Cancer Therapy. Front Immunol, 2020. 11: p. 608485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J and Wang L, The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol Cancer Res Treat, 2019. 18: p. 1533033819831068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin J, et al. , Enhanced clinical-scale manufacturing of TCR transduced T-cells using closed culture system modules. J Transl Med, 2018. 16(1): p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah NN, et al. , CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol, 2020. 38(17): p. 1938–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabatino M, et al. , Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood, 2016. 128(4): p. 519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones M, et al. , A Comparison of Automated Perfusion- and Manual Diffusion-Based Human Regulatory T Cell Expansion and Functionality Using a Soluble Activator Complex. Cell Transplant, 2020. 29: p. 963689720923578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith TA, CAR-T Cell Expansion in a Xuri Cell Expansion System W25, in Chimeric Antigen Receptor T Cells: Development and Production, Swiech K, Malmegrim KCR, and Picanço-Castro V, Editors. 2020, Springer US: New York, NY. p. 151–163. [Google Scholar]
- 17.Porter DL, et al. , Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med, 2011. 365(8): p. 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grupp SA, et al. , Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med, 2013. 368(16): p. 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brentjens RJ, et al. , Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood, 2011. 118(18): p. 4817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brentjens RJ, et al. , CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med, 2013. 5(177): p. 177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coeshott C, et al. , Large-scale expansion and characterization of CD3(+) T-cells in the Quantum(®) Cell Expansion System. J Transl Med, 2019. 17(1): p. 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lock D, et al. , Automated Manufacturing of Potent CD20-Directed Chimeric Antigen Receptor T Cells for Clinical Use. Hum Gene Ther, 2017. 28(10): p. 914–925. [DOI] [PubMed] [Google Scholar]
- 23.Vedvyas Y, et al. , Manufacturing and preclinical validation of CAR T cells targeting ICAM-1 for advanced thyroid cancer therapy. Sci Rep, 2019. 9(1): p. 10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah NN, et al. , Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med, 2020. 26(10): p. 1569–1575. [DOI] [PubMed] [Google Scholar]
- 25.Spiegel JY, et al. , CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med, 2021. 27(8): p. 1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Remley VA, et al. , High efficiency closed-system gene transfer using automated spinoculation. J Transl Med, 2021. 19(1): p. 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, et al. , Large volume flow electroporation of mRNA: clinical scale process. Methods Mol Biol, 2013. 969: p. 127–38. [DOI] [PubMed] [Google Scholar]
- 28.Beatty GL, et al. , Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res, 2014. 2(2): p. 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beatty GL, et al. , Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology, 2018. 155(1): p. 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tchou J, et al. , Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res, 2017. 5(12): p. 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah PD, et al. , Phase I trial of autologous cMET-directed CAR-t cells administered intravenously in patients with melanoma & breast carcinoma. Journal of Clinical Oncology, 2020. 38(15_suppl): p. 10035–10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi S, et al. , Design and Assessment of Novel Anti-CD30 Chimeric Antigen Receptors with Human Antigen-Recognition Domains. Hum Gene Ther, 2021. 32(13–14): p. 730–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin BY, et al. , Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight, 2018. 3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagarsheth NB, et al. , TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med, 2021. 27(3): p. 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraietta JA, et al. , Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med, 2018. 24(5): p. 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abou-El-Enein M, et al. , Scalable Manufacturing of CAR T cells for Cancer Immunotherapy. Blood Cancer Discov, 2021. 2(5): p. 408–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, et al. , A Feasibility and Safety Study of a New CD19-Directed Fast CAR-T Therapy for Refractory and Relapsed B Cell Acute Lymphoblastic Leukemia. Blood, 2019. 134(Supplement_1): p. 825–825. [Google Scholar]
- 38.Zhang C, et al. , CD19-Directed Fast CART Therapy for Relapsed/Refractory Acute Lymphoblastic Leukemia: From Bench to Bedside. Blood, 2019. 134(Supplement_1): p. 1340–1340. [Google Scholar]
- 39.Bai Z, et al. , Single-cell multiomics dissection of basal and antigen-specific activation states of CD19-targeted CAR T cells. Journal for ImmunoTherapy of Cancer, 2021. 9(5): p. e002328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaeschke F, et al. , Induction of a central memory and stem cell memory phenotype in functionally active CD4(+) and CD8(+) CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19(+) acute lymphoblastic leukemia. Cancer Immunol Immunother, 2018. 67(7): p. 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]