

Abstract
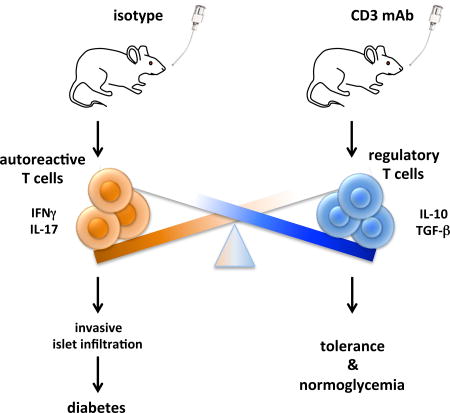
CD3-specific monoclonal antibody (mAb) treats autoimmune disease in animal models and has shown promise in clinical trials of type 1 diabetes. Whereas intravenous administration of CD3-specific mAb acts primarily by transient depletion of activated effector T cells, oral CD3-specific mAb acts primarily by the induction Tregs. We investigated whether oral CD3-specific mAb inhibits disease in non obese diabetic (NOD) mice that spontaneously develop autoimmune diabetes, closely resembling human type 1 diabetes. We found that oral CD3-specific mAb treatment delayed onset and reduced incidence of diabetes in NOD mice, inducing changes in both effector and regulatory T cell compartments. The therapeutic effect was associated with decreased T cell proliferation, decreased IFNγ and IL-17 production, and increased TGF-β and IL-10 production in vitro. In vivo transfer experiments demonstrated that oral CD3-specific mAb decreased diabetogenicity of effector T cells and increased the function of regulatory T cells. Oral OKT3, a monoclonal antibody specific for human CD3 had equivalent effects in transgenic NOD mice expressing the human CD3 epsilon chain which serves as a preclinical model for testing human CD3-specific mAb. These results suggest that oral CD3-specific mAb has the potential for treating autoimmune diabetes in humans.
Keywords: Autoimmune diabetes, immunotherapy, mucosal tolerance, CD3 monoclonal antibody, NOD, Treg
Graphical abstract
1. INTRODUCTION
The number of type 1 diabetes mellitus (T1D) patients in the US has been estimated 1–1.5 million. T1D is a chronic progressive autoimmune disease that results in the destruction of insulin producing pancreatic β-cells, leading to 3–4 times increased mortality as compared to non-diabetic individuals, most likely from renal failure and coronary heart disease. Non Obese Diabetic (NOD) mice that spontaneously develop autoimmune diabetes have been extensively used to study mechanisms underlying the development of autoimmune diabetes and therapeutic approaches to prevent or cure the disease [1]. Due to the complexity of T1D and its pathogenesis, encompassing genetic, biological and environmental factors, current treatments are limited to restoring insulin availability, normally by lifelong administration of exogenous insulin and if necessary by transplantation of pancreatic islets.
Intravenous administration of CD3-specific monoclonal antibodies (mAb) has been shown to treat ongoing disease in animal models of autoimmunity such as diabetes in NOD mice [2,3] and experimental allergic encephalomyelitis (EAE), a model for multiple sclerosis [4]. Intravenous CD3-specific mAb has shown promising results in clinical trials in patients with recent onset type 1 diabetes [4,5], though phase 3 trials did not meet their primary endpoints, most likely due to changes in dosing and altered endpoints [6,7]. Although minimal, side effects did occur. Oral administration of CD3-specific mAb is an alternative therapeutic approach targeting CD3 on the surface of T cells that has the advantage of its tolerability with virtually no side effects [8]. Whereas intravenous CD3-specific mAb induced tolerance depends on depletion of pathogenic T cells while preserving regulatory T cells [9,10] oral CD3-specific mAb relies on induction of regulatory T cells [11,12]. Oral CD3-specific mAb has demonstrated therapeutic efficacy in a number of animal models [13–15] including experimentally induced diabetes by streptozocin [16].
Since NOD mice develop spontaneous autoimmune diabetes that better resembles human type 1 diabetes and due to the availability of transgenic NOD mice expressing the human CD3 epsilon chain, allowing preclinical testing of human CD3-specific mAb, we investigated the ability of oral CD3-specific mAb to protect NOD mice from diabetes and the effect of the treatment on the immune response.
2. MATERIAL AND METHODS
2.1 Mice
NOD (purchased from Jackson Laboratory), NOD.SCID (from Charles River) and NOD-huCD3ε mice (developed by the lab of Lucienne Chatenoud) were housed in a specific pathogen-free animal facility at the Harvard Institutes of Medicine according to the animal protocol guidelines of the Committee on Animals of Harvard Medical School, which also approved the experiments. Some experiments were performed at the “Institut Necker-Enfants Malades, INSERM U1151- CNRS UMR 8253” with mice that were bred and housed there according to European Directives (86/609/EEC) and institutional guidelines. The animal facility has an agreement delivered by the Prefecture de Police of Paris, France.
2.2 Antibodies and reagents
Monoclonal anti-mouse CD3 monoclonal antibodies (CD3-specific mAb; 145-2C11 or F(ab’)2 fragments of 145-2C11), anti-human CD3 mAb (OKT3) and the respective isotype controls (hamster IgG, hamster IgG F(ab’)2 fragments and mouse IgG2a clone C1.18.4) were purchased from BioXCell. The CD8 T cell epitopes GAD65524–543, proinsulin (PI) B15–23 and the BDC2.5 mimotope1040-31 (Mimo) were purchased from SynBioSci.
2.3 Oral treatment
We fed 6-weeks old female NOD mice with 10 µg CD3-specific mAb or isotype control, respectively, diluted in 2ml PBS by gavage with stainless steel feeding needles for 5 days. For the experiment represented in figure 1B F(ab’)2 fragments of 145-2C11 were used while all other experiments were performed with the entire anti-CD3 mAb 145-2C11. Depending on the experiment, we either analyzed mice on day 6, continued feeding once a week until 16 weeks of age for follow-up of diabetes development or immunized the mice 2 days after the initial CD3-specific mAb feeding with 100µg Mimo peptide in CFA in the ventral flanks. Mice were used for in vitro assays on day 10 after immunization.
Figure 1. Diabetes incidence after oral CD3-specific mAb.

A–B. Spontaneous diabetes development in NOD mice treated with oral CD3-specific mAb (A; 145-2C11; grey triangles; n=8) as compared to hamster IgG (A; black circles; n=8) or F(ab’)2 fragments of 145-2C11 (B; grey triangles; n=8) as compared to F(ab’)2 fragments of hamster IgG (B; black circles) for 5 days and then once a week (50 µg per dose) until week 20. C. Islet infiltration of pancreata from 20 weeks old NOD mice treated with CD3-specific mAb (145-2C11; n=8) as compared to isotype control (n=7). D. Diabetes incidence after treating NOD mice with nasal CD3-specific mAb (145-2C11; grey triangles; n=8) as compared to hamster IgG (black circles; n=8) using the same protocol as in A and B. Statistically significant results are indicated in the figure: *p<0.05; **p<0.01.
2.4 Nasal treatment
For nasal treatment, we administered 10µg CD3-specific mAb or isotype control, respectively, diluted in 5µl PBS into the nostrils of 6-week old female NOD mice, using the same treatment protocol as for CD3 therapy.
2.5 Diabetes monitoring
Mice were monitored starting at 12 weeks of age or 6 weeks after adoptive transfer, respectively, once per week by measuring the glycosuria using Precision Laboratories Glucose Test Strips (Fisher Scientific). Diabetes was confirmed by testing fasting glycemia (>250 mg/dl; Bayer Contour Blood Glucose Monitoring System; Fisher Scientific).
2.6 IFNγ ELISPOT
As previously described [17], splenocytes were cultured at 2.5×105/well and pLN cells (3.0×104/well) were co-cultured with 2.0×105 irradiated (35 Gy) splenocytes/well, in presence of IL-2 (1 U/well) in ELISPOT plates (Millipore) that were coated with IFNγ capture antibody (U-CyTech Biosciences). Cells were stimulated for 20 hrs with the CD8 T cell epitopes IGRP206-14 or PPI-B15–23 (7 µM) as compared to a negative control (a mixture of viral peptides from EBV, CMV and HIV; 7 µM) and a positive control (CD3-specific mAb; 145 2C11; 1 µg/ml) and processed according to the manufacturer’s protocol (U-CyTech Biosciences). The air-dried plates were analysed using an AID reader (Autoimmun Diagnostika). Results are shown as spot-producing units (SPU) per 106 responder cells following background subtraction.
2.7 Proliferation
Single cell suspensions were prepared from spleen and pancreatic lymph nodes (pLN). To measure proliferation, cells were plated in 96-well round bottom plates (5*105 cells/well) in X-Vivo15 medium and stimulated with 1 µg/ml, 0.2 µg/ml or 0.04 µg/ml soluble CD3-specific mAb or indicated concentrations of Mimo, GAD and Insulin peptides in culture at 37°C, 5% CO2. After 72 hrs supernatants were removed and 1 µCi/well [3H]-thymidine in X-vivo15 was added. Proliferation was assessed using a 1450 Microbeta liquid scintillation counter (Perkin Elmer).
2.8 Antibodies and FACS analysis
Cells were stained in Mg2+ and Ca2+ free HBSS with 2% FCS, 0.4% EDTA (0.5 M) and 2.5% HEPES (1M) and either directly acquired or fixed in PBS containing 2.5% formaldehyde (Sigma–Aldrich, Steinheim, Germany). FoxP3 (FJK-16s) was detected by intracellular staining according to the manufacturer's instructions (eBioscience, San Diego, CA). Cells were acquired on a FACS LSRII or Fortessa (BD) and analysed using FlowJo software. Fixable viability dyes eFluor780 or eFluor506, Sytox Red, AnnexinV and Fluorochrome conjugated antibodies to mouse CD3 (145-2C11), CD4 (RM4-5), CD8a (53–6.7), GARP (YGIC86), CD62L (MEL-14), CD44 (IM7), CD25 (PC61.5), PD1 (J43), FoxP3 (FJK-16s) were purchased from eBioscience. Anti-mouse LAP (TW7-16B4) was from BioLegend.
2.9 Cytokine ELISA
Supernatants were harvested after 48 hrs of culture and the concentrations of indicated cytokines were measured by quantitative capture ELISA according to the guidelines of the manufacturer (R&D Systems).
2.10 Histology
Pancreata were fixed in AFA, embedded in paraffin, cut and stained with hematoxilin/eosin at the HMS Rodent Histopathology Core.
2.11 Adoptive transfer
We treated female, 12 weeks old, non-diabetic NOD-huCD3ε mice [18] for five consecutive days with 50µg 145-2C11, OKT3 or isotype control (5 per group). On day 6 we isolated spleen cells, and FACsorted TCR+CD62L− and TCR+CD62L+ T cells. As described in the results section we transferred 5*106 spleen cells, 5*105 TCR+CD62L− T cells (effector T cells) or 5*105 TCR+CD62L− T cells together with 1*106 TCR+CD62L+ T cells to 6 weeks old female NOD.SCID recipients that were followed-up for diabetes development for 15 weeks.
2.12 Statistical Analysis
GraphPad Prism 6.0 was used for statistical analysis. The occurrence of diabetes was plotted using the Kaplan-Meier method. Statistical comparison between the curves was performed using the logrank (Mantel-Cox) test. When appropriate, the Student’s t test or one-way analysis of variance, followed by Tukey multiple comparisons was used. A p-value < 0.05 was considered statistically significant.
3. RESULTS
3.1 Mucosal CD3-specific mAb protects NOD mice from diabetes
We orally administered 10 µg CD3 specific monoclonal antibody (CD3-specific mAb; 145-2C11) to 6 weeks old female NOD mice once daily for the first 5 days and then once per week until 20 weeks of age. Diabetes incidence was significantly reduced (50% versus 87.5%; p<0.05) as compared to isotype treated mice at 25 weeks of age (Fig. 1A). Similarly, feeding F(ab’)2 fragments of CD3-specific mAb protected NOD mice from diabetes development (Fig. 1B; 50% versus 100% diabetics; p<0.05). We stained pancreata from 20 weeks old NOD mice with hematoxylin/eosin (H&E) to assess the impact of oral CD3-specific mAb (124-2C11) on infiltration of pancreatic islets. In accordance with the reduced incidence of diabetes in CD3-specific mAb (145-2C11) treated mice, pancreata from 20 weeks old CD3-specific mAb treated mice showed significantly less islet infiltration as compared to isotype treated control (48.9% ± 14.0% versus 11.6% ± 8.3%; p<0.05) while having a tendency towards less invasive infiltrates (Fig. 1C; 22.1% ± 9.1% versus 49.9% ± 15.3%). As nasal administration of CD3-specific mAb has also been shown to induce tolerance in animal models of autoimmunity [15,19], we tested if nasal CD3-specific mAb (145-2C11) prevents diabetes onset in NOD mice. Indeed, we observed a significant decrease of diabetes incidence (Fig. 1D; 37.5% versus 87.5%; p<0.05). Given that oral CD3-specific mAb has already been given to humans [20–22] we focused our investigation on the oral route of administration.
3.2 Oral CD3-specific mAb reduces T cell proliferation and increases TGF-β and IL-10 production following non-antigen specific restimulation
To investigate mechanisms underlying the protection from diabetes development conferred by oral CD3-specific mAb we performed in vitro and in vivo experiments at different time-points after CD3-specific mAb feeding. First, we analyzed proliferation and cytokine secretion of spleen cells of 7 weeks old NOD mice 24 hours after the fifth dose of CD3-specific mAb in response to in vitro stimulation with soluble CD3-specific mAb. Feeding of NOD mice with CD3-specific mAb decreased proliferation of splenocytes after in vitro restimulation (29,873 ± 1,717 versus 55,475 ± 2,346; n=3; Fig. 2A)
Figure 2. In vitro analysis of spleen cells after five days of oral CD3-specific mAb.

A. Proliferation of splenocytes from CD3 mAb fed mice (black bars) as compared to isotype fed mice (white bars) after 72 hrs stimulation with of CD3-specific mAb in vitro (1 µg/ml). B–C. Concentration of TGF-β (B) and IL-10 (C) in the supernatant of spleen cells from CD3 mAb fed as compared to isotype fed mice after 48 hrs stimulation with CD3-specific mAb. D–G. Analysis of cell death (D, E) and apoptosis (D, G) in splenic T cells from CD3 mAb fed mice as compared to isotype fed mice after 48 hrs stimulation with of CD3-specific mAb in vitro (gated on lymphocytes). Dead cells were detected using a fixable viability dye (FVD; D, E) and apoptotic cells were defined at FVD-AnnexinV+ (F). PD-1 staining on CD4+ T cells after 48 hrs of stimulation with CD3-specific mAb. Results are shown as mean ± SEM (n=3–4). In all panels black bars represent cells from anti-CD3 mAb fed mice whereas white bars show data from isotype control fed mice. Statistically significant results are indicated: *p<0.05, ** p<0.01, ***p<0.001.
Supernatants of restimulated splenocytes isolated from CD3-specific mAb fed mice contained elevated concentrations of TGF-β (Fig. 2B; 798.5 ± 28.5 pg/ml versus 592.0 ± 25.0 pg/ml) and IL-10 (Fig. 2C; 4,149 ± 197 pg/ml versus 3,119 ± 54 pg/ml) as compared to isotype fed mice, suggesting the induction of a pro-tolerogenic environment by feeding of CD3-specific mAb.
Staining of splenic T cells with a viability dye, AnnexinV (Fig. 2D–F) and PD-1 (Fig. 2G–H) showed that hypoproliferation of spleen cells from CD3 mAb fed mice was not due to increased cell death (Fig. 2D–F) or anergy (Fig. 2G–H) after in vitro restimulation with CD3-specific mAb. T cells from CD3 mAb fed mice even showed reduced cell death after 2 days of in vitro restimulation with CD3-specific mAb (39.9 ± 6.0% versus 54.6 ± 2.2%; n=4) as compared to controls.
3.3 Oral Anti-CD3 in animals immunized with a beta-cell antigen have reduced antigen-specific T cell proliferation and IL-17 and IFNγ production
As we detected hyporesponsiveness of spleen cells from CD3-specific mAb fed mice following non-antigen specific restimulation we assessed the ability of CD3-specific mAb treatment to affect a beta-cell antigen specific immune response. We circumvented the low islet antigen specific T cell response in 8 weeks old NOD mice by immunizing the animals with the beta-cell antigen BDC2.5 mimotope1040-31 (Mimo). We fed 6 weeks old NOD mice for five days with CD3-specific antibody or isotype control, immunized the mice two days later with Mimo in CFA and restimulated spleen and pancreatic lymph node (pLN) cells ten days after immunization. Both spleen (Fig. 3A; 3,395.0 ± 554.2 cpm versus 5,448.0 ± 70.24; p<0.0001) and pLN (Fig. 3B; 264 ± 27 cpm versus 2,526 ± 297 cpm; p<0.001) cells from treated mice showed markedly lower proliferation to Mimo restimulation than the control group. CD3-specific mAb feeding reduced IL-17 secretion by pLN cells (199 ± 36 pg/ml versus 1,583 ± 140 pg/ml) and IFNγ (Fig. 3E, F) production by both splenocytes (Fig. 3E; 7.8 ± 0.1 ng/ml versus 33.2 ± 0.8 ng/ml; p<0.05) and pLN cells (Fig. 3F; 24.1 ± 1.1 ng/ml versus 50.6 ± 0.1 ng/ml; p<0.05) after restimulation with Mimo. These results demonstrate that anti-CD3 mAb feeding skews antigen-specific T cells towards a less inflammatory phenotype.
Figure 3. In vitro analysis of spleen and pLN cells after five days of oral CD3-specific mAb and immunization with the beta-cell antigen Mimo.

A–F. Analysis of proliferation (A, B) and secretion of IL-17 (C, D) and IFNγ (E, F) of spleen cells (first column) and pancreatic lymph node (pLN) cells (second column) from anti-CD3 mAb fed (black bars) or isotype fed mice (white bars), 10 days after immunization with Mimo, in response to restimulation with Mimo (100 µg/ml). Results are shown as mean ± SEM (n=3). Statistically significant results are indicated: *p<0.05; ***p<0.001; ****p<0.0001.
3.4 Oral CD3-specific mAb renders T cells less diabetogenic
Having addressed early changes in the immune response after CD3-specific mAb feeding we investigated if oral CD3-specific mAb changes the profile of autoreactive T cells at 16 weeks, an age at which many T cells infiltrate pancreatic islets. IFNγ ELISPOT demonstrated that feeding of CD3-specific antibody significantly decreased the reactivity of spleen cells (Fig. 4A; 3.5 ± 2.3 spot producing units (SPU) versus 24.3 ± 8.4 SPU; p<0.05) against the CD8 epitope proinsulin (PI) B:15–23, in contrast to cells from pancreatic lymph nodes (Fig. 4B; 5.0 ± 3.1 SPU versus 7.0 ± 4.1 SPU). Reactivity of splenocytes (Fig. 4C; 50.1 ± 22.7 SPU versus 115.1 ± 54.1 SPU) and pLN cells (Fig. 4D; 44.8 ± 16.9 SPU versus 100.1 ± 32.6 SPU) to stimulation with the CD8 epitope IGRP 206–214 was unaltered. Splenocytes from CD3-specific mAb fed mice exhibited decreased proliferation after restimulation with soluble CD3-specific mAb (Fig. 5A) as compared to the control group. We also found significantly less IFNγ (Fig. 5B; 9.1 ± 1.2 ng/ml versus 26.3 ± 3.8 ng/ml; p<0.05) and IL-17 (Fig. 5C; 394 ± 22 pg/ml versus 687 ± 135 pg/ml); p<0.05) in response to soluble CD3-specific mAb (as shown for 1 µg/ml).
Fig 4. IFNγ ELISPOT analyzing CD8+ T cell responses following oral CD3-specific mAb therapy.

A – D. IFNγ ELISPOT response of spleen (A, C) and pancreatic lymph node (pLN; B, D) cells from anti-CD3 mAb or isotype control fed mice in response to the CD8 epitopes PI B15–23 (A, B) and IGRP206–214 (C, D) in spot producing units (SPU) per 1*106 cells. Graphs show average number ± SEM of SPU per million responder cells following background subtraction (n=7–8). Results from two independent experiments were pooled. Statistically significant results are indicated: *p<0.05.
Fig 5. Analysis of effector T cells and regulatory T cells after oral CD3-specific mAb.

A–C. Proliferation (A), IFNγ (B) and IL-17 (C) production of spleen cells from 20 weeks old mice that have been fed with anti-CD3 mAb (black circles/ bars) or isotype control (white circles/ bars) in response to indicated concentrations of CD3-specific mAb in vitro. D–E. Percentage of CD4+FoxP3+ (D) and CD4+LAP+ (E) T cells in spleens and pancreatic lymph nodes (pLN) of anti-CD3 mAb (black bars) or isotype control (white bars) fed mice. F–H. Diabetes incidence of NOD.SCID mice (n=4–5 per group) having received 5*106 spleen cells (F) or 0.5*106 CD62L− effector T cells alone (G) or together with CD62L+ regulatory T cells (H) from 16 weeks old NOD-huCD3e mice that were fed for five days with the anti-mouse CD3 mAb 145-2C11 (2C11; white squares), anti-human CD3 mAb OKT3 (grey triangles; n=5) or isotype control (black or circles; n=4–5; F–H or grey circles n=5; H). Statistically significant results are indicated in the figures: *p<0.05.
Flow cytometric analysis of regulatory T cell markers revealed an increase of CD4+FoxP3+ in spleens (Fig. 5D; 14.5% ± 0.8% versus 11.3 ± 0.5%) but not in pancreatic lymph nodes (Fig. 5D; 10.6% ± 0.5% versus 11.5 ± 0.6%) of anti-CD3 mAb fed mice at 20 weeks of age. There no significant change of CD4+LAP+ T cells in either, spleen (Fig. 5E; 0.9 ± 0.1% versus 1.0 ± 0.3%) or pancreatic lymph nodes (Fig. 5E; 1.7 ± 0.4% versus 0.9 ± 0.4%) T cells.
Treatment of 12 weeks old female NOD mice expressing the human CD3 epsilon chain (NOD-huCD3ε) with five doses of either anti-mouse CD3 mAb or anti-human CD3 mAb significantly protected NOD.SCID recipients from diabetes transfer as compared to isotype control fed mice (Fig. 5F). This decreased transfer of diabetes by spleen cells from CD3-specific mAb treated mice could be due to either, decreased diabetogenicity of effector T cells and/or increased regulation by regulatory T cells.
To distinguish between these possibilities, we analyzed diabetes transfer by diabetogenic T cells that are enriched in the CD62L− T cell population [23] (Fig. 5G). Whereas CD62L− T cells from isotype control treated NOD-huCD3ε mice transferred diabetes to all recipients within 10 weeks after transfer, CD62L− T cells from anti-mouse CD3 mAb or anti-human CD3 mAb treated mice only transferred diabetes to 60% and 25%, respectively, of recipients within 15 weeks (Fig. 5G), suggesting that oral CD3-specific mAb decreases the diabetogenicity of T cells.
To test if oral administration of CD3 mAb increases the suppressive capacity of regulatory T cells we co-transferred CD62L− effector T cells from isotype fed NOD-huCD3ε mice together with CD62L+ T cells, that have been shown to preferentially inhibit diabetes development [23], from isotype or CD3-specific mAb fed mice (Fig. 5H). CD62L+ T cells from isotype fed mice significantly protected from diabetes transfer by CD62L− T cells (50% versus 100% diabetic). CD3-specific mAb feeding further increased the protective capacity of CD62L+ T cells, with none of the recipients developing diabetes within 15 weeks after transfer (Fig. 5H). We conclude that repeated feeding of CD3-specific mAb to young prediabetic NOD mice decreases the diabetogenicity of CD4+CD62L− effector T cells while increasing the regulatory function of CD62L+ T cells.
4. DISCUSSION
We found that oral and nasal administration by CD3-specific mAb decreases the incidence of diabetes in NOD mice. The therapeutic effect of oral CD3-specific mAb was independent on the Fc portion as F(ab’)2 fragments were equally effective. This has previously been observed in the EAE model [11]. Although nasal CD3-specific mAb also decreased the incidence of diabetes in NOD mice, we focused on oral CD3-specific mAb because it has shown positive biological and therapeutic effects in humans [20–22]. Histology on pancreata from 20-week old NOD mice were consistent with the protective effect of CD3-specific mAb on diabetes development as CD3-specific mAb treated mice had significantly lower islet infiltration as compared to isotype controls.
We addressed the immune mechanisms associated with the CD3-specific mAb effects using several approaches. We assessed proliferation and cytokine secretion of splenic and pLN derived T cells in response to antigen specific and non-specific stimuli. Oral CD3-specific mAb reduced proliferation as well as IL-17 and IFNγ production by T cells at early time-point after treatment, after immunization with the mimotope of diabetogenic BDC2.5 T cells and at a later time point after treatment (20 weeks of age) suggesting that oral CD3-specific mAb treatment induces long lasting hyporesponsiveness of T cells. It has been previously shown that oral administration of CD3-specific mAb doesn’t induce antigenic modulation, i.e. internalization of the CD3/TCR complex or depletion of activated T cells [11], contrarily to intravenously administered CD3-specific mAb [24]. Hyporesponsiveness was not a consequence of increased cell death or anergy after restimulation in vitro either. T cells from CD3 mAb fed mice even seemed to be more resistant to cell death, as evaluated directly after the end of anti-CD3 mAb feeding. One explanation for this long lasting hyporesponsiveness could be the induction of regulatory T cell producing the anti-inflammatory cytokines TGF-β and IL-10 that were elevated in supernatants of restimulated T cells from CD3-specific mAb treated mice.
To assess the impact of oral CD3-specific mAb on antigen-specific T cell responses we immunized CD3-specific mAb treated mice with a β-cell antigen, a mimotope of the diabetogenic T cell clone BDC2.5 (Mimo), and assessed proliferation and cytokines secretion in response to restimulation with the same antigen. We chose this approach because the frequency of antigen-specific T cells before 12–16 weeks of age is extremely low and immunization allowed us to boost responsiveness towards a chosen antigen (Mimo in this case) and to mimic the development of autoreactive T cells specific for islet antigens. Again, we detected hyporesponsiveness in CD3-specific mAb treated mice.
As Mimo is a CD4 epitope and the response against Mimo was induced by previous immunization, we addressed changes in the response of autoreactive CD8+ T cells by IFNγ ELISPOT against the two dominant CD8 epitopes PI B15–23 [25] and IGRP206–214 [26] at 20 weeks of age. Oral CD3-specific mAb decreased the number of splenic (but not pLN) CD8+ T cells responding to PI B15–23 and there was a trend towards a decreased response to IGRP206–214 in pLN. These results suggest that oral CD3-specific mAb therapy reduces the amount and/or the pathogenicity of autoreactive T cells over the long term.
Adoptive transfer of CD62L− T cells, that have been shown to be enriched in diabetogenic T cells [27], from CD3-specific mAb fed mice showed that oral CD3-specific mAb decreases the diabetogenicity of T cells which is in line with decreased proliferation and secretion of IFNγ and IL-17 in vitro.
Using NOD-huCD3ε mice that express both, the mouse and human CD3ε chain, we found that oral administration of the anti-human CD3 mAb OKT3 induced similar functional changes as compared to the anti-mouse CD3 mAb 145-2C11. We further analyzed qualitative and quantitative changes in regulatory T cells. Adoptive cotransfer showed improved function of regulatory CD62L+ T cells. Our previous studies showed that CD4+ T cells expressing latent membrane bound TGF-β (CD4+LAP+) play an important role in the ameliorating effect of oral CD3-specific mAb in EAE [11]. FACS analysis of regulatory T cell subsets in 20 weeks old NOD mice after oral treatment with CD3-specific mAb showed an increase of CD4+FoxP3+ in spleens but not pancreatic lymph nodes and we didn’t detect an increase of CD4+LAP+ T cells in either organ. The absence of an increase of CD4+LAP+ T cells may be related to alterations of regulatory T cell function in NOD mice [28,29]. Consistent with this, is has been reported that expression of the adapter protein GARP that tethers LAP membrane [31] is reduced in NOD mice [28] and the percentage of CD4+LAP+ T cells in NOD mice that we report in this article are much lower than the ones reported for other mouse strains [11,14,16]. Furthermore, IL-2 is known to increase GARP that is required for membrane bound LAP [30]. NOD mice display a qualitative diminution of IL-2 production that contributes to autoimmunity [29,31,32] and a genetic predisposing factor to development of autoimmune diabetes in humans and NOD mice is linked to IL-2/IL-2R gene polymorphisms [32].
CD3-specific mAb therapy is a promising approach for therapy of autoimmune diseases and other immune mediated disorders as it has been shown to be effective in a wide variety of autoimmune and inflammatory conditions [8]. Oral CD3-specific mAb is advantageous over intravenous administration as it stimulates natural mucosal physiologic mechanisms, is well tolerated [11,20–22] and can be given over long periods of time. Two single blind randomized placebo controlled phase 2a studies of oral OKT3 antibody in patients with treatment resistant chronic hepatitis C infection (HCV) [22] or nonalcoholic steatohepatitis (NASH) [21] reported positive effects on disease and immunological markers including an increase of regulatory T cells. Our findings suggest that oral anti-CD3 mAb does not only induce tolerance by its impact on regulatory T cells but also by modulating autoreactive T cells in a direct or indirect manner. Oral anti-CD3 may find efficacy in the treatment or prevention of type 1 diabetes.
Highlights.
Mucosal monoclonal CD3 antibody (CD3 mAb) protects NOD mice from diabetes
Oral administration of CD3 mAb decreases diabetogenicity of effector T cells
Oral CD3 mAb increases the number and function of regulatory T cells
Acknowledgments
We thank Deneen Kozoriz for her excellent technical support in cell sorting.
Funding: This work was supported by the National Institutes of Health [R01 AI43458].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 2.Chatenoud L, Thervet E, Primo J, Bach J-F. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci. U.S.a. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chatenoud L, Primo J, Bach J-F. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J. Immunol. 1997;158:2947–2954. [PubMed] [Google Scholar]
- 4.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 5.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 6.Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti-CD3 clinical trials in type 1 diabetes mellitus. Clinical Immunology. 2013;149:268–278. doi: 10.1016/j.clim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Chatenoud L, Waldmann H. CD3 Monoclonal Antibodies: A First Step Towards Operational Immune Tolerance in the Clinic. Rev Diabet Stud. 2012;9:372–381. doi: 10.1900/RDS.2012.9.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.da Cunha AP, Weiner HL. Induction of immunological tolerance by oral anti-CD3. Clinical and Developmental Immunology. 2012;2012:425021. doi: 10.1155/2012/425021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. The Journal of Immunology. 2011;187:2015–2022. doi: 10.4049/jimmunol.1100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatenoud L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat Rev Endocrinol. 2010;6:149–157. doi: 10.1038/nrendo.2009.275. [DOI] [PubMed] [Google Scholar]
- 11.Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+CD25−LAP+ T cells. Nature Medicine. 2006;12:627–635. doi: 10.1038/nm1408. [DOI] [PubMed] [Google Scholar]
- 12.Kuhn C, Weiner HL. Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy. 2016;8:889–906. doi: 10.2217/imt-2016-0049. [DOI] [PubMed] [Google Scholar]
- 13.Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso A, et al. New immunosuppressive approaches: Oral administration of CD3-specific antibody to treat autoimmunity. Journal of the Neurological Sciences. 2008;274:9–12. doi: 10.1016/j.jns.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, Center E, Tsokos G, Weiner H. Suppression of murine SLE by oral anti-CD3: inducible CD4+CD25−LAP+ regulatory T cells control the expansion of IL-17+ follicular helper T cells. Lupus. 2009;18:586–596. doi: 10.1177/0961203308100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu HY, Maron R, Tukpah AM, Weiner HL. Mucosal Anti-CD3 Monoclonal Antibody Attenuates Collagen-Induced Arthritis That Is Associated with Induction of LAP+ Regulatory T Cells and Is Enhanced by Administration of an Emulsome-Based Th2-Skewing Adjuvant. The Journal of Immunology. 2010;185:3401–3407. doi: 10.4049/jimmunol.1000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa H, Ochi H, Chen ML, Frenkel D, Maron R, Weiner HL. Inhibition of Autoimmune Diabetes by Oral Administration of Anti-CD3 Monoclonal Antibody. Diabetes. 2007;56:2103–2109. doi: 10.2337/db06-1632. [DOI] [PubMed] [Google Scholar]
- 17.Enée E, Martinuzzi E, Blancou P, Bach J-M, Mallone R, van Endert P. Equivalent specificity of peripheral blood and islet-infiltrating CD8+ T lymphocytes in spontaneously diabetic HLA-A2 transgenic NOD mice. J. Immunol. 2008;180:5430–5438. doi: 10.4049/jimmunol.180.8.5430. [DOI] [PubMed] [Google Scholar]
- 18.Kuhn C, You S, Valette F, Hale G, van Endert P, Bach J-F, et al. Human CD3 Transgenic Mice: Preclinical Testing of Antibodies Promoting Immune Tolerance. Science Translational Medicine. 2011;3:68ra10–68ra10. doi: 10.1126/scitranslmed.3001830. [DOI] [PubMed] [Google Scholar]
- 19.Wu HY, Quintana FJ, Weiner HL. Nasal Anti-CD3 Antibody Ameliorates Lupus by Inducing an IL-10-Secreting CD4+CD25−LAP+ Regulatory T Cell and Is Associated with Down-Regulation of IL-17+CD4+ICOS+CXCR5+ Follicular Helper T Cells. The Journal of Immunology. 2008;181:6038–6050. doi: 10.4049/jimmunol.181.9.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilan Y, Zigmond E, Lalazar G, Dembinsky A, Ben Ya’acov A, Hemed N, et al. Oral Administration of OKT3 Monoclonal Antibody to Human Subjects Induces a Dose-Dependent Immunologic Effect in T Cells and Dendritic Cells. J Clin Immunol. 2009;30:167–177. doi: 10.1007/s10875-009-9323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalazar G, Mizrahi M, Turgeman I, Adar T, Ben Ya’acov A, Shabat Y, et al. Oral Administration of OKT3 MAb to Patients with NASH, Promotes Regulatory T-cell Induction, and Alleviates Insulin Resistance: Results of a Phase IIa Blinded Placebo-Controlled Trial. J Clin Immunol. 2015;35:399–407. doi: 10.1007/s10875-015-0160-6. [DOI] [PubMed] [Google Scholar]
- 22.Halota W, Ferenci P, Kozielewicz D, Dybowska D, Lisovoder N, Samira S, et al. Oral anti-CD3 immunotherapy for HCV-nonresponders is safe, promotes regulatory T cells and decreases viral load and liver enzyme levels: results of a phase-2a placebo-controlled trial. J. Viral Hepat. 2015;22:651–657. doi: 10.1111/jvh.12369. [DOI] [PubMed] [Google Scholar]
- 23.Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J. Immunol. 2000;164:240–247. doi: 10.4049/jimmunol.164.1.240. [DOI] [PubMed] [Google Scholar]
- 24.You S, Candon S, Kuhn C, Bach J-F, Chatenoud L. word. Adv. Immunol. 2008;100:13–37. doi: 10.1016/S0065-2776(08)00802-X. [DOI] [PubMed] [Google Scholar]
- 25.Wong FS, Karttunen J, Dumont C, Wen L, Visintin I, Pilip IM, et al. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nature Medicine. 1999;5:1026–1031. doi: 10.1038/12465. [DOI] [PubMed] [Google Scholar]
- 26.Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc. Natl. Acad. Sci. U.S.a. 2003;100:8384–8388. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lepault F, Gagnerault MC, Faveeuw C, Bazin H, Boitard C. Lack of L-selectin expression by cells transferring diabetes in NOD mice: insights into the mechanisms involved in diabetes prevention by Mel-14 antibody treatment. Eur. J. Immunol. 1995;25:1502–1507. doi: 10.1002/eji.1830250605. [DOI] [PubMed] [Google Scholar]
- 28.D'Alise AM, Ergun A, Hill JA, Mathis D, Benoist C. A cluster of coregulated genes determines TGF-beta-induced regulatory T-cell (Treg) dysfunction in NOD mice. Proc. Natl. Acad. Sci. U.S.a. 2011;108:8737–8742. doi: 10.1073/pnas.1105364108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards JP, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM. Regulation of the Expression of GARP/Latent TGF- 1 Complexes on Mouse T Cells and Their Role in Regulatory T Cell and Th17 Differentiation. J. Immunol. 2013;190:5506–5515. doi: 10.4049/jimmunol.1300199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VES, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat. Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dendrou CA, Wicker LS. The IL-2/CD25 pathway determines susceptibility to T1D in humans and NOD mice. J Clin Immunol. 2008;28:685–696. doi: 10.1007/s10875-008-9237-9. [DOI] [PubMed] [Google Scholar]