

Abstract
Recent advances in neoantigen research have accelerated the development and regulatory approval of tumor immunotherapies, including cancer vaccines, adoptive cell therapy and antibody-based therapies, especially for solid tumors. Neoantigens are newly formed antigens generated by tumor cells as a result of various tumor-specific alterations, such as genomic mutation, dysregulated RNA splicing, disordered post-translational modification, and integrated viral open reading frames. Neoantigens are recognized as non-self and trigger an immune response that is not subject to central and peripheral tolerance. The quick identification and prediction of tumor-specific neoantigens have been made possible by the advanced development of next-generation sequencing and bioinformatic technologies. Compared to tumor-associated antigens, the highly immunogenic and tumor-specific neoantigens provide emerging targets for personalized cancer immunotherapies, and serve as prospective predictors for tumor survival prognosis and immune checkpoint blockade responses. The development of cancer therapies will be aided by understanding the mechanism underlying neoantigen-induced anti-tumor immune response and by streamlining the process of neoantigen-based immunotherapies. This review provides an overview on the identification and characterization of neoantigens and outlines the clinical applications of prospective immunotherapeutic strategies based on neoantigens. We also explore their current status, inherent challenges, and clinical translation potential.
Subject terms: Predictive markers, Tumour immunology, Cancer, Predictive medicine, Biomaterials
Introduction
Accumulating genetic alterations in cancers result in the production of tumor-specific antigens (TSAs) or neoantigens, which can be presented by major histocompatibility (MHC) molecules of tumor cells.1–6 These tumor-specific peptide-MHC (pMHC) complexes are recognized by T cells and trigger an anti-cancer immune response in patients. However, it has been discovered that cancer cells have evolved resistance to anti-cancer immunity.7 These immune escape mechanisms can be reversed by cancer immunotherapies, including the use of tumor vaccines to improve antigen presentation, the increase of anti-tumor T cells via adoptive transferring of tumor-infiltrating lymphocytes (TILs) and T cell receptor (TCR)-transduced T cells, restoring the effector capacity of CD8+ T cells by immune checkpoint blockades (ICBs), increasing the immune recognition of tumors with bispecific antibodies (bsAbs) and chimeric antigen receptor (CAR)-transduced T cells, and modulating the tumor immune microenvironment.8–19 A variety of clinical studies examined the efficacy of immunotherapies targeting tumor-associated antigens (TAAs), like vaccines against ERBB2, MUC1, and hTERT. TAAs exhibit abnormal expression in malignancies or are only produced during specific stages of differentiation, whereas their expression in normal tissues is extremely limited. The prevalence of TAAs among cancer patients makes them public targets for off-the-shelf immunotherapies. However, as TAAs are non-mutated self-antigens, central T cell tolerance may contribute to the largely poor T cell responses observed in clinical trials.20,21 Nonetheless, the widespread use of tumor immunotherapies has been hindered by a shortage of targetable antigens in various cancers.22
Neoantigens are self-antigens generated by tumor cells because of genomic mutations. Besides, neoantigens can also derive from unique proteins or peptides produced by dysregulated RNA splicing and disordered post-translational protein modification in non-virus-associated malignancies. For cancers with a viral infection, such as HPV-positive cervical cancer and EBV-associated nasopharyngeal carcinoma, neoantigens can also be created by virally encoded open reading frames (ORFs).22–24 Compared with other types of tumor antigens, such as cancer-testis antigens (CTAs) and TAAs, neoantigens offer a distinct advantage in their unique tumor-specific and absence in normal tissues, presenting ideal targets for effectively personalized treatment of tumors (Table 1).25,26 Notably, T cells specialized for neoantigens can bypass negative selection effects in the thymus due to the highly antigenic neoantigens acquired through somatic tumor mutations. Increasing the pool of neoantigen-specific T cells due to this ability to avoid T cell central tolerance makes it possible to enhance tumor-specific immune responses.27–29 Furthermore, the capacity of immunotherapy-enhanced neoantigen-specific T cell responses in enduring and giving post-treatment immunological memory offers hope for long-term protection against disease recurrence.30
Table 1.
The characteristics of TAAs and neoantigens
| Tumor antigens | Specifical expression | Central tolerance | Autoimmune toxicities | |
|---|---|---|---|---|
| Tumor-associated antigens (TAAs) |
Overexpressed proteins, lineage-specific differentiation markers |
Tumors | The central and peripheral tolerance | Colitis, renal impairment, severe hepatitis, rapid respiratory failure, treatment-related death |
| Cancer germline antigens (CGAs) | Tumors, testes, fetal ovaries, trophoblasts | The central and peripheral tolerance | Colitis, renal impairment, severe hepatitis, rapid respiratory failure, treatment-related death | |
| Tumor-specific-antigens (TSAs) | Oncoviral antigens | Virus-associated tumors | Non-central tolerance | Less |
| Neoantigens | Tumors | Non-central tolerance | Less |
Currently, advanced techniques, including tumor gene sequencing, neoantigen discovery, and neoantigen-based immune therapeutic product preparation, play a significant role in the development of personalized cancer vaccines (PCVs) and adoptive cell therapy (ACT).19 Next-generation sequencing (NGS) has permitted the fast and cost-effective detection of tumor-specific mutations in individual patients. In addition, the development of algorithms for predicting MHC molecules-binding epitopes has made it possible to identify possibly immunogenic neoepitopes.31 These technological developments have enabled the production of personalized immunotherapy, specifically targeting tumors in individual patients (Fig. 1). However, some limitations such as the costs and time in the process of personalized immunotherapeutic products, and the ideal platform for neoantigen identification, need further improvement. With the continuous development and wide cross-integration of biotechnology, immunology, materials science, chemistry, and artificial intelligence, additional neoantigens will be identified and employed in tumor immunotherapy.13,32
Fig. 1.
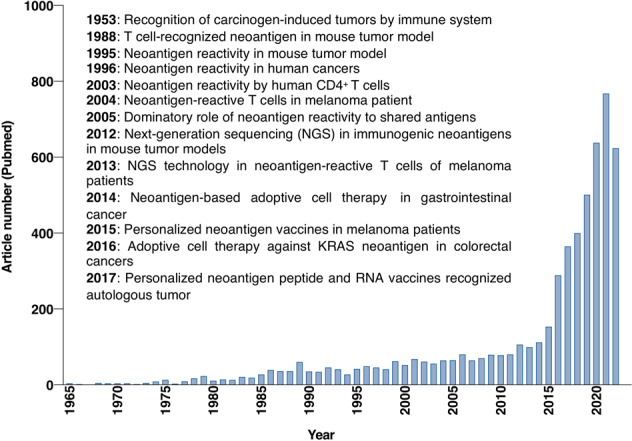
Historical overview of tumor-specific neoantigens. Based on keyword searches in the PubMed database using the terms "neoantigen" or "neoepitope", the number of articles from 1965 to 2022 is displayed in the column chart
Herein, we provide a comprehensive summary of the source and biological function of neoantigens, potential neoantigen prediction tools, and clinical applications of neoantigen-based immunotherapy strategies. Moreover, we also discuss the opportunities and limitations associated with the clinical application of immunotherapies based on neoantigens and propose some potential solutions.
The source of neoantigens
Neoantigens are identified as foreign proteins that are absent in normal tissues, but can arise from tumors through various mechanisms, such as genomic mutation, aberrant transcriptomic variants, post-translational modifications (PTMs), and viral ORFs (Fig. 2).27,33
Fig. 2.

Overview of the neoantigen production and presentation. Neoantigens can develop at the genomic level through SNVs, base INDELs and gene fusions, at the transcriptomic level through alternative splicing, polyadenylation (pA), RNA editing and allegedly non-coding regions, and at the proteomic level through dysregulated translation and PTMs. The integrated viral ORF is another source of neoantigens for cancers linked to viruses. The mutant peptides created by the proteasome-mediated breakdown of endogenous proteins are subsequently transported to the endoplasmic reticulum (ER) via transporters associated with antigen processing (TAP), where they may be loaded onto MHC-I. MHC-II dimers are assembled and bound to the invariant chain (Ii) in the ER. The Ii-MHC-II complex can be directly transported or sometimes indirectly internalized from the cell surface to the MHC-II compartment (MIIC), where the degradation of Ii by a series of endosomal proteases releases the MHC-II for binding a specific peptide derived from a mutant protein broken down in the endosomal pathway. These pMHC complexes will then traffic to the cell surface where they are recognized by T cells
Genomic variants
Somatic genomic alterations, including single-nucleotide variants (SNVs), base insertions and deletions (INDELs) and gene fusions, are the main factors that promote the production of tumor neoantigens.8,34–37
SNVs
SNVs are the most prevalent type of mutation at the genomic level in tumor cells; they can yield variant peptides distinct from wild-type proteins that are presented by MHC-I as specific antigens.19,38 Up to hundreds of non-synonymous somatic mutations per cancer patient have been recorded, resulting in an average of 150 potential neoantigenic peptides per individual. For example, a total of 231 non-synonymous SNVs, 13 gene fusions and 21 INDELs have been identified in Ph-negative myeloproliferative neoplasms (MPNs).39 Using The Cancer Genome Atlas (TCGA) database, 933,954 expressed neoantigens in 20 solid tumors have been characterized, which originates from 893,960 somatic mutations with a varied median frequency of neoantigens across cancers. Only 24 of these neoantigens, including those arising from mutations in driver genes like PIK3CA, RAS, and BRAF, are shared by at least 5% of patients with the same or distinct cancers.38,40 Notably, relapsed populations may have greater tumor mutation burden (TMB) and more novel potential neoantigens than newly diagnosed patients. In patients with multiple myeloma, only two potential neoantigens, UBR4 and PRKDC, were detected in both relapsed and newly diagnosed patients.41 Therefore, the SNV neoantigens landscape is highly variable between different cancer types and different stages of the same cancer type (Table 2).
Table 2.
Advantages, disadvantages and relevant cancers for each class of neoantigens
| Neoantigen classification | Sources | Advantages | Disadvantages | Relevant cancer |
|---|---|---|---|---|
| Genomic variants | Single-nucleotide variants (SNVs) | Simple prediction; relatively high burden |
Similar to self-antigen; rarely shared between patients |
Melanoma, glioblastoma, lung cancer (adeno and squamous), bladder cancer |
| Insertions and deletion (INDEL) frameshift |
More potential targets per mutation; more dissimilar from self-antigen; more immunogenic |
Relatively low burden | MSI-H tumors, renal cell carcinomas (clear cell, papillary, and chromophobe) | |
| Fusion genes |
More dissimilar from self-antigen; shared targets between tumors; more potential targets per mutation; more immunogenic |
Relatively low burden | Acute myelocytic leukemia, acute lymphocytic leukemia, chronic myeloid leukemia, sarcomas | |
| Chromosomal rearrangements | High immunogenicity | Less well studied | Malignant pleural mesothelioma | |
| Transcriptomic variants | RNA splicing |
A large number of predicted targets; More dissimilar from self-antigen |
Fewer tools available; not well validated in pre-clinical models; current tools do not account for nonsense mediated decay (NMD) |
Acute myelocytic leukemia, chronic myelomonocytic leukemia, chronic lymphocytic leukemia, myelodysplastic syndrome |
| Polyadenylation (pA) and RNA editing | Easy prediction | Less well studied | Chronic lymphocytic leukemia | |
| Allegedly non-coding regions |
Relatively high burden; more potential targets |
Less well studied; fewer tools available |
Acute lymphoblastic leukemias, lung cancers | |
| Proteomic variants | Post-translational modifications (PTMs) | Shared between patients | Less well studied | leukemia, renal cancer, non-small cell lung cancer |
| Proteasome processing | High specificity |
Less well studied; fewer tools available |
Acute myeloid leukemia | |
| T cell epitopes associated with impaired peptide processing (TEIPP) | TEIPP-specific T cells can escape thymic selection |
Less well studied; limited in HLA-I low or TAP-deficient tumors |
Lung cancer | |
| Viral-derived neoantigens | Viral open reading frames |
High immunogenicity; more dissimilar from self-antigen; shared between patients; without apparent toxicity to normal tissues. |
Limited in specific tumors | Hepatocellular carcinoma, Merkel cell carcinoma, nasopharyngeal carcinoma, head and neck cancer, cervical cancer, anal cancers |
SNVs can also arise in mitochondrial DNA (mtDNA), which is found in most cancer cells and correlates with alterations in tumor metabolic profiles and cancer cell metastatic capacity.42–45 Despite the fact that the compact normal human mitochondrial genome, a 16,569 base-pair circular DNA, encodes only 13 protein subunits of the electron transport chain, it may account for around 30% of total mRNA transcripts in certain organs.46 mtDNA has a 10- to 20-fold greater mutation rate than nuclear DNA.47,48 Both mouse and human immune systems were able to recognize and respond to mtDNA single nucleotide polymorphisms (SNPs)-derived peptides, suggesting that individual SNPs in mtDNA are adequate to generate immunogenic neoantigens. Thus, non-synonymous SNPs in mtDNA may yield a substantial quantity of mutant peptides, offering an additional source of neoantigens.49–51
INDELs
INDEL mutations are mainly caused by the insertion or deletion of base pairs in the genome, which frequently lead to non-synonymous novel ORFs, also known as frameshift mutations.27,52 Frameshift INDELs can generate more types of neoantigens with increased MHC-I binding affinity, suggesting a higher immunogenic mutation type compared to SNVs (Table 2).53,54 Especially in renal cell carcinoma with a medium-range mutational burden, about 16% of predicted neopeptides are derived from frameshift INDELs, whereas 21% of T cell-recognized neoepitopes are arising from frameshift INDELs, indicating that frameshift-derived neoepitopes have a greater immunogenic potential.53,55
Similar to SNV neoantigens, INDEL neoantigens are more prevalent in cancers with microsatellite instability-high (MSI-H) due to the lack of DNA mismatch repair (MMR) mechanisms.27,56–58 As the evolution of MMR-deficient cancers is mainly triggered by mutations that inactivate tumor suppressor genes (TSGs) containing coding microsatellites, frameshift peptide neoantigens are more frequently shared among MMR-deficient cancers (e.g., endometrial, colorectal, and gastric) than missense mutation-derived neoantigens.59–64 Frameshift INDEL neoantigen burden has a strong correlation with immunological response.38 MSI colorectal cancers with frameshift mutations have a larger proportion of TILs than other colorectal cancers.64–66 Similarly, shared immunogenic frameshift peptide neoantigens can be produced as a result of recurrent frameshift mutations, offering excellent candidates for immunotherapy against MSI cancers.59,61–64 The combination of four frameshift peptide neoantigens dramatically boosts neoantigens-specific adaptive immunity, decreases intestinal tumor burden, and prolongs the overall survival in the VCMsh2-driven intestinal cancer mouse model, which can be further strengthened by naproxen.67,68 According to a clinical phase I/IIa trial, the frameshift peptide neoantigenic vaccine is well tolerated systemically and triggers immune responses regularly, representing a promising new strategy for the treatment and even prevention of MMR-deficient malignancy.60 These findings showed that an off-the-shelf vaccine is feasible for treating and preventing cancers with frameshift mutations and neoantigenic peptides because of MSI.69
The frameshift INDEL neoantigen burden is also a novel biomarker for ICB response.27,38,70–72 INDEL frameshift mutations are supposed to produce more immunogenic neoantigens, hence improving response to ICBs. When frameshift mutations are present, the progression-free survival of patients receiving ICBs is significantly prolonged. Further evidence that frameshift mutations may play a predictive role in ICB response comes from the considerable discrepancies in overall response rates and disease control rates observed in non-small cell lung cancer (NSCLC) patients with frameshift mutations.73 In addition, ICBs can also strengthen the immune response to frameshift neoantigens. The frameshift mutation in CALR elicits both CD4+ and CD8+ T cell responses, which are inhibited by the expression of PD-1 or CTLA4. Importantly, blocking PD-1 and CLTA4 ex vivo and PD-1 in vivo with pembrolizumab restores frameshift neoantigen-specific T cell immunity in myeloproliferative neoplasms.39,74
Gene fusions
Gene fusion is another important type of mutation in tumors that may provide many neoantigens, which can be generated by mesenchymal deletion, chromosomal translocation or chromosomal inversion.28,75–77 Studies have shown that polypeptides derived from the different fusion regions of the proteins can be recognized by the patient’s own T cells, such as the BCR-ABL fusion protein produced by the translocation between chromosomes 9 and 22 in chronic myeloid leukemia (CML) patients and SYT-SSX1 fusion proteins produced by X and 18 chromosomal translocations in synovial sarcoma patients. Even in some tumors with low TMB and limited immune infiltration, neoantigens generated by gene fusion are still able to activate cytotoxic T cells.21,78,79 In a comprehensive study of fusion neoantigens in tumors, analyses of three datasets from the TCGA database found that fusion mutations could generate more novel ORFs, yielding 6-fold neoantigens and 11-fold specific candidate neoantigens more than SNVs and INDELs. The fusion neoantigens are more likely to induce stronger immune response than the neoantigens produced by SNVs and INDELs, and the neoantigen produced by frameshift fusion has better immunogenicity than the in-frame fusion neoantigen (Table 2). Similar to the candidate neoantigen burden of SNVs and INDELs, fusion neoantigen burden was closely related to fusion mutation burden, especially in microsatellite stable tumors with higher fusion mutation burden.19,80 An expanded study of 30 different tumor types revealed that 24% of fusion protein-expressing cancers contained neoepitopes resulting from the fusion, and these neoantigens were predicted to bind to patient-specific MHC-I.78,81 It is worth noting that the repetition rate of fusion neoantigens between different patients is extremely low. According to statistics, only 5.8% of fusion neoantigens in the TCGA database repeat between patients, and these neoantigens usually have very low immunogenic potential.80 In addition, malignancies with greater immune-depleted microenvironments or human leukocyte antigen (HLA) loss exhibited fusion neoantigens more frequently. In melanomas treated with anti-PD-1 therapy, the removal of tumor cells carrying fusion-derived neoantigens demonstrated a negative immune surveillance selective pressure on these neoantigens.78,82 According to FACETS analysis of the TCGA exome data, 18.4% of cases had a loss of heterozygosity (LOH) in the HLA, which increased the possibility that a fusion neoantigen would be present.78,83 These results demonstrate the significance of gene fusions as a source of tumor-specific neoantigens.78,81
Gene fusion-derived neoantigens can elicit specific immune responses against tumors.84,85 The fusion neoantigens, such as BCR-ABL, SYT-SSX1/SSX2, PAX3-FOXO1, TPM3/TPM4-ALK, and EWS-FLI1, showed immunogenic potential, providing the possible targets for immunotherapy to treat tumors.86,87 CBFB-MYH11 fusion neoantigen is distributed on acute myeloid leukemia (AML) cells, which activates T cells and induces specific killing against AML, a cancer with low mutation frequency.88–91 Two neoantigens, SS393 (GYDQIMPKK) and SS391 (PYGYDQIMPK), are derived from the SYT-SSX fusion neoantigen that is common in synovial sarcoma. These neoantigen peptides successfully induced synovial sarcoma-specific cytotoxic T lymphocytes (CTLs) that specifically killed HLA-A24-positive synovial sarcoma cells containing the SYT-SSX neoantigen as well as the target cells pulsed with these peptides.92–94 A study on head and neck squamous cell carcinomas (HNSCC) found that the tumor’s immune response to anti-PD-1 therapy was mediated by neoantigens generated by DEK-AFF2 fusion. A DEK-AFF2-derived peptide (DKESEEEVS) enhanced T cell activation depending on MHC class when delivered to autologous peripheral blood mononuclear cells (PBMCs).78 A comprehensive study of 33 tumor types found that various common recurrent fusion neoantigens, including TMPRSS2-ERG, MYB-NFIB, FGFR3-TACC3, EML4-ALK and CCDC6-RET.95 TMPRSS2-ERG is the most common recurrent gene fusion that occurred in 38.2% of prostate cancer patients. Several high-affinity HLA-restricted epitopes were identified from the recurrent TMPRSS2-ERG type VI fusion, which could bind to HLA-A*02:01 in vitro and were recognized by CD8+ T cells.96 The fusion of the proto-oncogene MYB with the transcription factor NFIB serves as a biomarker for adenoid cystic carcinoma, which occurs in 60% of cases. Three MYB-NFIB-derived peptides (QFIDSSWYL, SLASPLQPT and SLASPLQSWYL) and one NFIB-MYB-derived peptide (MMYSPICLTQT) can bind to HLA-A*02:01 to activate the immune system.78,97 The EML4-ALK fusion gene is predominantly found in young, rarely/never smoker NSCLC patients, and ~5% of NSCLC patients have this fusion mutation. The use of EML4-ALK-derived peptides can stimulate specific CTL responses and have the potential to treat EML4-ALK-positive NSCLC.98 Therefore, the neoantigens generated by fusion mutations greatly increase the capacity of the tumor-specific neoantigen repertoire, providing more potential targets or predictors for cancer immunotherapies.8,78,81,88,96,99–101
Structural variants
Structural variant (SVs) is one of the most frequent forms of driver mutations in tumors, which can result in alterations in genome structure and then change the expression or function of genes to promote malignant transformation. SVs generally refer to genetic variants that are larger than 50 base pairs, such as insertions, deletions, inversions, translocations, duplications/amplifications, and chromosomal additions and deletions, as well as chromosomal rearrangements.5,102–109 Among them, chromosomal rearrangement is the most complex (such as chromothripsis and chromoplexy), which is a prominent feature of tumors and plays a crucial role in the occurrence and immune recognition of various malignant tumors.110–112 Chromosomal rearrangements are not easily detected by traditional DNA sequencing techniques but can be screened by WES methods like mate pair sequencing (MPseq). Potential neoantigens generated by chromosomal rearrangements have been identified by a combination of MPseq and RNA sequencing (RNA-seq) in malignant pleural mesothelioma (MPM) patients. Rearrangement-related neoantigens may generate MPM-specific immune responses in a manner similar to frameshift INDEL neoantigens. Specifically, neoantigens produced by SVs are predicted to be presented by tumors on MHC proteins, which are closely related to clonal expansion of TILs, and effector T cells against these neoantigens are found in the circulation of cancer patients.110,113 Therefore, SV-derived neoantigens may also serve as valuable targets for anti-tumor immunotherapy.
Transcriptomic variants
Post-transcriptionally events offer the potential of a broadened neoantigen space. Alternative processing of mRNA, including alternative splicing events, polyadenylation (pA), RNA editing and allegedly non-coding regions, contributes to the diversity of tumor-specific neoantigens.19,81,114–116
Transcript alternative splicing
The abnormal alternative mRNA splicing is another potential source of tumor-specific neoantigens.22,117 RNA splicing process the premature mRNA into mature RNA with high efficiency and fidelity in normal cells. However, it may be induced by mutations in RNA cis-regulatory elements, trans-acting regulators or the core spliceosome.117–119 The highly aberrant splicing events in tumors expand the scope of tumor-specific neoantigens, especially in tumors with low rates of copy number variation and somatic mutations.23,117,120
Cis-acting mutations
Mutation at cis-acting elements generates potential neoantigens through altered splicing, including alternative 5’ and 3’ splice site determination, intron retention, exon skipping and mutually exclusive exons.23,33,121,122 Intron retention is more prevalent in nearly all cancers compared with normal control tissues, even in the absence of mutations in genes encoding splicing factors. Normally, intron retention transcripts will be degraded by nonsense-mediated RNA decay (NMD). Neoantigens can still be generated from intron retention during their pioneer round of translation before being subjected to NMD.33,117,123,124 Numerous exon-exon junctions that are unique in tumors have been identified through extensive study of the TCGA, most of which can express neoantigens.117,125,126 The production of neoantigens from skipped exons, also known as “neojunctions”, occurs more frequently and is more likely to be shared among patients than those generated from SNV mutations.29,117,127,128 A recent study has identified exitron splicing, a non-canonical splicing mechanism, as a new source of tumor neoantigens. Exitrons are exon-embedded cryptic introns distinguished from conventional introns in that they have both splicing (intron) and protein-coding (exon) potential while lacking stop codons or premature termination codons. Because tumor-specific exitron-spliced transcripts are far more likely to escape NMD than intron retentions, their overall expression is higher than retained introns. Accordingly, exitrons splicing creates more validated neoantigens with higher immunogenicity in malignancies with low TMB.128,129
Trans-acting alterations in splicing factors
Trans-acting alterations, in which a somatic mutation in a splicing factor results in an altered splicing variant, induce the production of neoantigens throughout the genome.130 In hematological malignancies, common mutations in spliceosomal components, including SRSF2, SF3B1, and U2AF1/2, raise the expression of splice variant mRNAs, resulting in the translation of TSAs and neoantigens. In addition to hematological tumors, a recent reassessment of pan-cancer data in the TCGA database has shown that the somatic alterations of splicing factors, including SF3B1, U2AF1, SRSF2 and Zinc Finger CCCH-Type, ZRSR2, U2AF2, SF1, PRPF8, and SF3A1, leading to the production of splicing variant-derived neoantigens across the genome in solid tumors.22,27,117,119,131–138 Moreover, epigenetic alterations and PTMs of splicing factors might promote global splicing dysregulation.139 Neoantigens derived from splicing variants due to mutation and dysregulated expression of splicing factors have facilitated the development of novel therapeutics for tumors. For example, mutated SF3B1 (a splicing factor in the spliceosome) in uveal melanoma generates tumor-specific neoantigens that activate specific CD8+ T cells to kill tumor cells.140
Nonsense-mediated RNA decay (NMD)
Another important determinator for tumor-specific splicing variants is NMD, a highly conserved RNA turnover mechanism that preferentially destroys RNAs carrying premature translation termination codons. In cells with normal NMD function, a pioneer round of translation is required for initiating the NMD-mediated degradation of aberrant transcripts, which can lead to the production of small amounts of neoantigens.23,123,141,142 Moreover, the NMD regulatory mechanism is frequently impaired in tumor cells, enabling aberrant transcripts to avoid degradation and potentially produce large amounts of neoantigens. For example, mutations in the highly conserved core NMD factor UPF1 are prevalent in pancreatic squamous cell carcinoma and lung adenocarcinoma, increasing the frequency of aberrant transcripts and neoantigen production.143–145 A recent study demonstrated that NMD regulates the mutational profile of malignancies by preferentially suppressing the expression of TSGs rather than oncogenes. Further evidence for the beneficial effect of NMD on tumors comes from the observation that NMD frequently degrades mRNA encoding immunogenic neoantigen peptides. Accordingly, NMD inhibitory therapy may be beneficial in the treatment of a variety of cancers, including those capable of producing large numbers of mutated neoantigens.146,147
Altogether, these studies highlight that alternative splicing of transcripts could promote the production of neoantigens. Even though the application of splicing variant neoantigens in personalized therapies has not yet been thoroughly investigated, screening new alternative splicing-based neoantigens as immunotherapeutic targets will benefit tumor patients.22,23,27
Polyadenylation (pA) and RNA editing
Similar to RNA splicing, polyadenylation (pA) and RNA editing can alter the proteomic profile of tumor cells, thereby increasing the pool of potential immunotherapeutic targets.81,148
Polyadenylation plays a critical role in the processing and maturation of most eukaryotic mRNAs, primarily by cleaving and adding a poly(A) tail at the 3’ end. Most alternative polyadenylation (APA) events occur in the 3ʹ untranslated region (UTR) of mRNA. APA can significantly affect post-transcriptional gene regulation in several aspects, including transcript stability, translation, cellular localization, and nuclear export.149–154 Nonetheless, some APA events occur in the intronic region upstream of the last exon, which is called intronic polyadenylation (IPA).155,156 IPA can result in the production of truncated or non-coding transcripts that have the potential to generate tumor-specific immunotherapeutic targets. A recent study used 3’ end sequencing technology to analyze normal and malignant B cells of 59 patients with chronic lymphocytic leukemia (CLL), the study found that IPA-induced mRNA and protein truncations are prevalent in CLL cells, mainly involving TSGs such as DICER and FOXN3, and even some oncogenes such as CARD11, MGA, and CHST11.157 It is worth noting that 72% of the 190 TSGs found in hematological tumors are only truncated in solid tumors.158 In tumors, when a specific IPA event occurs in the coding region, genes upstream of the new pA site and downstream of the closest 5ʹ splice site are translated, creating neoantigens that can be presented by MHC and recognized by the immune system.81 By comparing RNA-seq data between tumor and normal tissue samples from various cancers, more neoantigens created by IPA might be identified, providing prospective targets for cancer immunotherapy.
RNA editing is an important pre-mRNA processing method that can induce non-synonymous substitutions by altering specific nucleotides in the RNA sequence, resulting in the production of new proteins.38,159 Similar to splicing and polyadenylation, RNA editing events frequently occur in a variety of tumors.160–164 Adenosine-to-inosine (A-to-I) editing is the most prevalent type of RNA editing in mammals, and millions of such sites have been found in human genes. Protein peptides produced by A-to-I editing can be presented by MHC-I molecules, which further induce the activation of specific CD8+ T cells, suggesting that these novel peptides are immunogenic and can activate the immune system.165–169 Nevertheless, it must be emphasized that these peptides are not necessarily tumor-specific, as RNA editing can also occur in normal tissues. Therefore, more in-depth research and advanced prediction methods are needed to identify tumor-specific RNA editing protein products for immunotherapy.81,170
Allegedly non-coding regions
Given that 99% of tumor-specific mutations occur in non-coding regions of genes, and exonic regions account for only 2% of the entire human genome, the screening for neoantigens only derived from mutations in exonic regions is limited. Recent studies showed that many regions previously defined as non-coding are now found to have coding functions. Therefore, by studying these newly defined genes with coding capacity, researchers have discovered many novel antigenic peptides that can be presented by MHC-I, and some antigens have been confirmed as target for TIL immunotherapy.171–173 These MHC-I-associated peptides (MAPs) derived from genes at non-coding regions expand the range of CD8+ T cell immune surveillance from 2% (the proportion of the human genome in exons) to 75%.52,172,174,175 According to a proteogenomic profiling of non-canonical proteins, 60% of non-canonical proteins are encoded by genes that were considered to be located at non-coding regions previously. More recently, using the mass spectrometry (MS) methods, many sorts of non-coding regions have been identified to produce large amounts of aberrantly expressed tumor-specific antigens, the bulk of which originate from epigenetic modifications in atypical translation events rather than mutations.176 These aberrantly expressed tumor-specific neoantigens are more prevalent than neoantigens created by mutations in coding areas and can be shared between tumor patients.27,172,177 Numerous such cryptic peptides were found in tumor immunopeptidomes using Peptide-PRISM. The presentation of cryptic peptides is HLA-I allele dependent, with HLA-A*03 and HLA-A*11 showing the largest proportion of cryptic peptides.178 Critically, cryptic proteins create MHC-I peptides five times more efficiently per translation event than canonical proteins do, due to their more predicted disordered residues and lower stability.179 No studies have reported that MHC-II-associated neoantigens generated from non-coding regions may activate CD4+ T cells. Compared with other mutations at the genome and transcriptome level mentioned in this review, neoantigens derived from the translation of non-coding regions are rarely clearly understood. Therefore, it is urgent to develop fast and efficient computational algorithms to screen these potential neoantigens and to verify their feasibility for immunotherapy.38
Proteomic variants
Dysregulated translation that is a characteristic of carcinogenesis offers an important new source of tumor-specific neoantigens.180 In addition, the proteomic variants also come from the aberrant function of PTMs, proteasome processing, and transporter associated with antigen processing (TAP).181–184
Neoantigen presentation by MHC molecules to T cells can maintain specific PTMs.181,182 Aberrant PTMs, including glycosylation, O-linked β-N-acetylglucosamine (O-GlcNAc) and phosphorylation, can create neoantigenic peptides presented by MHC complexes in tumors.185 For example, the neoantigen arising from post-translationally modified MUC1 was presented by MHC-I and exclusively recognized by a glycoform-specific TCR.182 Moreover, an unusually large proportion of mutations may enhance the formation of novel N-glycosylation sites, resulting in generation of neoantigens.186 Five O-GlcNAc modified peptides in leukemia were found to induce multifunctional memory T cell responses in healthy donors. Neoantigens derived from O-GlcNAc modified proteins explain why leukemias are highly immunogenic despite having a low mutational load, thereby offering prospective therapeutic targets.187 Dysregulated phosphorylation can generate neoantigens by promoting the binding of epitopes to MHC molecules or by altering the antigenic features of presented epitopes.188 The cancer-associated phosphopeptides derived from insulin receptor substrate 2 (pIRS2) and breast cancer antiestrogen resistance 3 (BCAR3) were immunogenic in vivo in mice, and in vitro in normal human donors.189,190 Several T cell lines have demonstrated a specifically recognition of the post-translationally modified peptide but not the unmodified peptide, indicating that the aberrant PTMs results in a different neoantigen and cognate TCR.182,187 Notably, immunogenic peptides derived from dysregulated PTMs in cancer cells constitute an unexplored class of tumor-specific neoantigens that could serve as off-the-shelf targets for cancer immunotherapy. PTMs can also be employed to produce unique neoantigens to improve the immune recognition of cancer cells. Covalent KRAS-G12C inhibitors, like ARS1620, result in covalently modified peptides, which can be presented on MHC-I to elicit T cell response. These tumor-specific PTMs, which involve the covalent drug-mediated alkylation of mutant cysteine residues on oncoproteins, provide a novel source of neoantigens that can be readily targeted by immunotherapies.191,192
Another repertoire of neoantigenic epitopes is derived from impaired proteasome processing or TAP complexes. The proteasome processes proteins and converts them into peptides, which is particularly critical for transforming proteins into MHC-restricted epitopes. The oxidants like peroxynitrite generated by myeloid cells in tumor microenvironment (TME) inhibit the activity of proteasome, thereby decreasing the production of MHC-I peptides.193,194 Protein splicing significantly increases the proteome complexity of malignancies, which alters the hierarchy of antigenic epitopes.195,196 Studies have also revealed that the proteasome can produce novel immunoreactive spliced epitopes (splicetopes) by fusing with peptide fragments excised by reverse proteolysis during proteasome-catalyzed peptide splicing (PCPS), which differ from the original substrate protein sequence.196–198 According to preliminary statistical analysis, the proteasome is responsible for splicing around one-third of MHC-I-related immune peptides.199
There is evidence that neoantigens involving the linkage of existing individual peptides can activate CD4+ T cells in type 1 diabetes (T1D), indicating that proteomic variants processes may generate MHC-II-associated neoantigens.200 Several studies have reported that the spliced peptides produced by the proteasome are able to activate CD8+ T cells.198,201,202 Splicetope-specific CD8+ T cells from TILs isolated from human AML patients inhibited the growth of their corresponding tumor cells in severe combined immunodeficient (SCID) mice model.203 Combining in vitro confirmation of proteasome-dependent splicetope with screening of specific anti-tumor CD8+ T cells enables monitoring of HLA class I binding and immune recognition processes, which will help to obtain more novel tumor-associated splicetope.200 Epitopes such as FGF-5, SP110, and gp100-derived splicetope have been identified during in vitro PCPS approach, which could be recognized by CD8+ T cells. However, the current research strategy to discover new tumor-specific splicetope needs to be further developed and refined in the future. Protein splicing-derived neoantigens could provide more yet-to-be-developed or identified neoantigens for anti-tumor vaccines and cancer immunotherapy.38,199
Most tumor antigens require proteasome processing and TAP-mediated peptide transport. However, most tumors eventually acquire drug resistance and immune escape. It has been reported that tumors can avoid recognition by T cells by producing defective HLA-I antigen processing pathways or downregulating related gene expression. Notably, a class of neoantigens called T cell epitopes associated with impaired peptide processing (TEIPP) have been identified in some HLA-I low/TAP-deficient tumors. They are a class of unmutated antigens derived from the tumor’s own housekeeping proteins that activate TEIPP-specific CD8+ T cells and specifically kill these TAP-deficient cancer cells. It is currently believed that TEIPP peptides are immunogenic because they cannot be presented by normal cells, and TEIPP-specific T cells are not negatively selected in the thymus. A TEIPP peptide derived from Lass5 protein, also known as Trh4, was able to activate specific T lymphocytes and inhibit the growth of MHC-I low/TAP-deficient tumors in a TCR transgenic mouse model. In addition, several TEIPP non-mutated tumor epitopes have been identified in humans, including the procalcitonin (ppCT) signal peptide (ppCT16-25, ppCT9-17) regions, and the procalcitonin (pCT) precursor protein (ppCT50-59 and ppCT91-100) regions. Further studies confirmed that these TEIPP-based antigenic peptides can effectively induce anti-tumor CTL effects and inhibit tumor growth. Therefore, targeting these TEIPP neoantigens will potentially provide a promising new immunotherapeutic approach for the treatment of TAP-deficient/HLA-I-low tumors.27,183,184,204–207
Viral-derived tumor antigens (Viral ORFs)
Viral proteins may be considered as another class of neoantigens in tumors caused by viruses because they are almost completely different from normal cellular proteins, and they can elicit high-affinity TCR responses. Some solid tumors are directly caused by viral infection, including Merkel cell carcinoma (MCC) caused by Merkel cell polyoma virus (MCPyV) infection and nasopharyngeal carcinoma caused by Epstein-Barr virus (EBV) infection.208–213 In other tumors, viral genes with oncogenic properties can integrate into the cellular genome, promoting the continuous expression of viral genes and leading to tumorigenesis. For example, the expression of E6 and E7 genes from HPV promotes the development and progression of human papillomavirus (HPV)-related cervical, anal, head and neck cancers.214–217
Numerous immunotherapy studies have focused on virus-derived tumor antigens. Two of nine HPV-positive patients with metastatic malignancies achieved sustained tumor regressions in ACT research using TILs chosen for their reactivity against viral antigens.218 A further investigation revealed that the number of HPV-reactive cells in the reinfused product exceeded those that recognized other types of tumor antigens.219 In two separate clinical trials, autologous T cells transduced with anti-E7 TCR responded in 4 of 12 patients, while T cells transduced with anti-E6 TCR responded in all 12 patients.220,221 The NCT02280811 and NCT02858310 trials using these TCRs are currently ongoing and should yield more conclusive proof about the value of focusing on HPV epitopes. Treatment of the corresponding tumors with ACT therapy targeting MCPyV and EBV also achieved clinical results, although other effective therapies were also administered in these experimental regimens. Notably, none of these clinical trials occurred with any apparent toxicity to normal tissues. Collectively, these trials demonstrate the safety and efficacy of targeting oncogenic viral proteins to treat related tumors, supporting the development of further comprehensive treatment regimens. Given their critical function in oncogenesis and the fact that patients share them, these neoantigens continue to be desirable targets for cancer immunotherapy.21,221–223
The neoantigens are generated as a result of alterations at genomic, transcriptomic and proteomic levels (Fig. 2). Current studies mainly focus on SNVs and INDELs, the most prevalent types of mutations at the genome level in tumor cells. However, the clinical application of neoantigens produced from SNVs and INDELs is limited by their patient specificity and poor immunogenicity, which results in less clinical benefit for cancer patients. Accumulating evidence suggests that alternative sources of cancer neoantigens, such as gene fusions, alternative splicing variants and PTMs, may be attractive novel targets for immunotherapy. The neoantigens produced by gene fusion, particularly the frameshift fusion, have better immunogenicity than the SNV- and INDEL-neoantigens, which were included in numerous clinical trials. Furthermore, neoantigens generated from gene fusion, recurrent mutations in cancer driver genes, non-coding regions and abnormal PTMs have a higher likelihood of being shared among patients, providing readily public neoantigens for immunotherapy.27,35,38
Identification, prediction, and validation of immunogenic neoantigens
Identification of immunogenic neoantigens from the numerous sources mentioned above is a crucial step in the development of effective immunotherapies.177 Neoantigens may now be thoroughly screened across the entire cancer spectrum thanks to the convergence of whole-exome sequencing (WES), RNA-seq, and proteomic data from TCGA.120,224 However, given the wide variations in tumor types, tumor lesions, and patients, customized immune treatments necessitate the detection and prediction of neoantigens based on distinct patient and tumor characteristics. The identification of genome-expressed mutations as well as details on MHC types of patients are required for the prediction of immunogenic neoantigens, as the sequential stimulation of immune response by tumor neoantigens from mutations depends on several variables, including the translation and processing of peptides, the presentation of the mutated peptides by the MHC molecules and the affinity of the pMHC complexes with the TCRs.177,225,226 Two main strategies for identifying neoantigen epitopes are developed: the immunogenomic approach can create virtual peptidomes by in silico methods based on NGS, and the immunopeptidomic strategy use MS to analyze the MHC-loaded peptides.227 Several TCR-guided neoantigen discovery strategies have recently been developed to systematically map the immunogenic neoantigens.
Identification of somatic mutations
The immunogenomic strategies were greatly hastened by comparing the genetic changes between tumor and normal tissue using NGS. Currently, the initial stage in the process of detecting possible neoantigens from NGS data is mapping tumor-specific genetic abnormalities using WES of the tumor and normal DNA. RNA-seq data may be combined with WES to determine whether a mutant gene is expressed in the tumor. In addition, more hidden biological information, such as information about copy number changes, microbial contamination, transposable elements, cell type, and the existence of neoantigens, can be found in RNA-seq.228,229 RNA-seq can also be used to detect alternative splicing events and estimate the relative frequency of the mutant allele’s expression.230 By using methods like mate-pair sequencing that may detect chromosomal rearrangements, the predictive values of NGS-based TMB measures may be greatly improved.110 Recent studies have shown that antigenic peptides are produced by transcripts with frameshift mutations and atypical splicing patterns when NMD is assumed to be present. Exact peptide sequences from full-length transcript structures are required in order to fully identify the neoantigens that resulted from frameshift mutations and aberrant isoforms.231 Using the Oxford Nanopore Technologies nanopore-type sequencer MinION, full-length transcriptome sequencing may cover the whole transcript at the proper sequencing depth with an accuracy of roughly 90%, providing complementary information to the current RNA-seq to identify allele-specific transcription and splicing.143,232
Based on cancer genomic data, the immunogenomic technique predicted millions of possible mutation-derived neoantigens, but the vast majority of them did not manifest in proteomic profiling of HLA-bound peptides.233,234 The high-throughput identification of peptides attached to MHC is made possible by immunopeptidomics techniques, which use MS to directly examine the immunoprecipitated and extracted MHC-bound peptides.230,235–239 MS has advanced in verifying in silico predicted neoantigens.38 Comparing the tandem mass spectra of the sample with that of the synthetic peptide can verify the neoantigens that are predicted by immunogenomic approaches.240,241 Particularly for rare HLA allotypes and HLA-II ligands, mapping the tumor HLA ligandome has helped to uncover targets for the neoantigen-specific cancer immunotherapies in clinical trials.38 In addition to validate the neoantigens arising from aberrant DNA sequence or RNA expression, MS-based proteomics provide the "gold standard" for neoantigen detection at the protein level, which cannot be discovered from DNA and RNA studies. For instance, MS can be used to detect novel MHC-associated neoantigens resulting from PTMs that are dysregulated during cellular transformation.127,188,190,242–244 Moreover, MS is also integrated with NGS to further detect the tumor-specific neoantigens created by somatic mutations, non-coding RNA and proteasome splicing, which is omitted by whole-exome or transcriptome-based sequencing technology.38,172,236 To allow a deeper knowledge of neoantigens in protein levels, more user-friendly and practical tools that integrates genomic, transcriptomic and proteomic data for immunopeptidomic-based neoantigen detection should be created.
In silico neoantigen prediction
Based on the NGS data, virtual peptidomes have been created and potential neoantigens have been discovered by in silico methods.177,245 Briefly, a typical workflow for neoantigen prediction can be summarized into the following steps: (i) mutation calling, (ii) HLA typing, (iii) neoantigen filtering and prioritization based on HLA binding affinity, and (iv) experimental validation of immunogenic neoantigens using T cell-based assays (Fig. 3).177,246,247
Fig. 3.

Computational workflow for neoantigen prediction. Current available bioinformatic pipelines for neoantigen prediction from somatic mutations share four main computational modules: (i) HLA typing from tumor WGS, WES data and RNA-seq; (ii) mutant peptide calling using a set of somatic mutations and splicing variants; (iii) HLA binding prediction; and (iv) T cell recognition prediction. The in silico tools for mutation calling are listed as follows. Mutation calling: INTEGRATE-neo,561 neoFusion,80 pVACtools,562 Epidisco,563 GATK564 and Antigen.garnish,565,566 Spliceman,567 MutPred,568 REVEL,569 rMATS,570 pVACseq,240 Neopepsee,571 MuPeXI,572 RepeatMasker, CloudNeo,573 Tlminer, MuTect/MuTect2, Strelka/Strelka2,574 SMUFIN, VarScan2, SomaticSniper, CaVEMan, MuSE, cgpPindel, SvABA, RADIA, NeuSomatic, NeoantigenR, MutPred, JuncBase, Splice, SpliceGrapher, rMATS, SplAdder, ASGAL, REVEL, TSNAD,575 HERVd,569 HESAS576 and EnHERV,577 hervQuant.578 HLA typing: Polysolver,254 OptiType,253 HLAreporter,579 PHLAT,580 HLAScan,260,581 HLAProfiler.260 HLA binding affinity: NetMHCpan,265 NetMHCIIpan4.0,267 MixMHC2pred,582 MARIA,268 neomhc2,583 pVAC-Seq, TIminer, HLAthena, DeepHLApan, TEPITOPEpan, NetMHCIIpan, SYFPEITHI, RNAKPEP, MULTIPRED2, ProPred, MHCPred, MARIA, Neonmhc2, EDGE.38,238 T cell recognition: NetCTL/NetCTLpan, POPISK, PAComplex, CTLPred, EpiMatrix, TCRMatch
HLA typing
Neoantigens are often presented in a cell-specific way by MHC-I for CD8+ T cells and MHC-II for CD4+ T cells, much like other antigens. Humans have more than 24,000 distinct HLA-I (HLA-A, -B, and -C) and HLA-II (HLA-DR, HLA-DQ, and HLA-DP) alleles, and their admixture results in polymorphism diversity.248–251 The HLA alleles of the patient determine their tumor-specific neoantigen repertoire that will be presented for T cell recognition. In addition, HLA-LOH, which occurs in 40% of NSCLC, impairs the presentation of neoantigens, facilitating immune evasion. Therefore, one of the most important initial steps in neoantigen prediction is determining the patient’s HLA genotypes.83,252 Several computational methods can now be applied with NGS data to achieve this goal. Most methods rely on DNA-derived NGS data acquired from WES or WGS. For example, Optitype253 and Polysolver254 are well performing tools for identification of class I HLA alleles. A bioinformatics tool, LOHHLA, is developed for accurate measurement of allele-specific HLA copy numbers. The tools, including HISAT genotype,255 ATHLATES,256 and HLA-HD257 can be used for both class I and class II typing.83 RNA-seq data can also be used by tools, such as arcasHLA,258 seq2HLA259 and HLAProfiler,260 to type HLA alleles with advantage of the unbiased dataset that covers both fully expressed parental alleles equally.260 The newly developed RNA-seq data-based methods bring a new dimension to HLA typing and biomarker investigations, even though Optitype discovered that WES produced superior results for HLA typing than RNA-seq data.230,253
Mutation and variant calling
By comparing NGS data of tumor and normal tissues from the same patient, mutant peptides resulting from somatic mutations can be predicted.261 WES is the recommended source of NGS data for neoantigen prediction because it offers the highest mutation coverage through focusing on the protein-coding regions of the genome. The computational analysis consists of data pre-processing and quality control, variant calling for somatic mutations, and prediction of the altered proteins and functional impact utilizing public genomic, transcriptomic, and proteomic sequence databases. For various neoantigen sources, a variety of integrated techniques have been developed for neoantigen identification and prioritization.29 Based on the strategy employed to screen putative neoantigens, these technologies can be classified into two groups: stepwise-analyses-based filtering strategy and integrative-scoring-system-based filtering strategy. The efficient one-stop tools accept WES/WGS and RNA-seq data as input and perform a series of filtering steps based on selected cutoff metrics, such as the binding affinity of peptides and MHC molecules, sequence coverage, variant allele frequency and gene expression, to remove false positives and generate a list of potential neoantigens. An integrated scoring system-based filtering technique assesses the immunogenicity of neoantigens by a quantitative score based on significant neopeptide characteristics, including the rank affinity of the mutant and normal peptides, the frequency of mutant alleles, and the amount of gene expression to experimentally assess the immunogenicity of the discovered neopeptides.38,262,263 Recently, a scoring method for evaluating immunogenicity that is based on machine learning models has also been suggested, optimizing the accurate prediction of neoantigens and reducing false positives.177 For a review and extensive discussion of these methods, we refer to prior literatures.246,249
Prediction of HLA binding and neoantigen presentation
Numerous computer prediction tools have been created for the in silico discovery of neoantigens based on MHC molecule processing and presentation, including NetChop, NetCTL and NetCTLpan264,265 (Fig. 3). The prediction capacity is actively improved by incorporating HLA-ligandome data into machine learning algorithms, such as linear regression and artificial neural networks.230,249 In vitro peptide-HLA binding dataset is used to train machine learning models by NetMHCpan265 and MHCflurry266 that are the main component of current HLA ligand identification pipelines.38 It is noteworthy that NetMHCpan, in contrast to state-of-the-art methods, improves the prediction performance of tumor neoantigens by combining information from binding affinity data with MS peptidome data to give a "panspecific" machine-learning strategy for MHC-I alleles.230,264,267 Two recent studies created computational frameworks called MSIntrinsic and EDGE that are highly effective in predicting HLA antigens using HLA peptides acquired from RNA-seq and liquid chromatography tandem MS (LC-MS/MS) data. Based on 24,000 HLA-I peptides collected by LC-MS/MS, the neural-network prediction algorithm, MSIntrinsic, outperformed previous affinity-based predictors by an average of 30% in positive predictive value (PPV).251 Similar findings were made by EDGE, which found that adopting a deep-learning architecture to identify HLA ligands using proteomic and transcriptomic data can improve the accuracy of HLA antigen prediction by up to ninefold.38,238
Emerging evidence has proved the significance of MHC-II neoantigens in anti-tumor immune response.234,268–271 A wide range of computational techniques for predicting MHC-II binding epitopes have been developed using artificial neural networks, including NetMHCII, NetMHCIIpan,272,273 SYFPEITHI, RNAKPEP, MULTIPRED2, ProPred, and MHCPred. However, compared to MHC-I molecules, computational prediction of the MHC-II-peptide binding affinity are currently less precise. First, compared to MHC-I molecules, MHC-II-binding peptides are more promiscuous in terms of peptide length and binding sequence motifs. Second, the polymorphism of the α and β chains in MHC-II molecules also considerably expands the diversity of peptide binding specificity.38,230 Recently, computational methods based on transcriptome and MS data have been developed. The deep learning model trained by MARIA, which incorporates both sequencing data with naturally occurring MHC-II ligandomes, was demonstrated to outperform the most widely used predictor NetMHCIIpan3.1 in the lymphoma dataset when cross validated against known MHC-II ligands. However, more study using significant datasets is necessary to demonstrate its robustness and effectiveness.38,268
Given multiple processes control the neoantigen presentation, it can be inferred that improving binding affinity alone does not accurately reflect cellular processing and CD8+ T cell responses. Additional properties, including proteasomal cleavage, transportation of peptides into the endoplasmic reticulum, and HLA alleles, are in conjunction with binding affinities between the peptide and the MHC molecules to prioritize possible neoantigens.230
Evaluation and validation of candidate neoantigens’ immunogenicity
It is well known that an immunogenic neoantigen must satisfy two or more requirements, the main bottlenecks are appropriate MHC molecule presentation and effective TCR recognition. According to recent studies, the majority of predicted neoantigens via MHC molecule presentation do not trigger an immune response.234,274,275 Therefore, while assessing the immunogenicity of potential neoantigens, it is crucial to take the TCR recognition of pMHC complexes into account. There are many in silico techniques that can forecast neoantigen-specific T cell recognition. The most used method is NetCTL/NetCTLpan, which generates a composite score rather than predicting T cell binding directly by combining MHC binding, C-terminal cleavage affinity and TAP transport.38 Recent studies use machine learning or deep learning techniques to predict TCR-peptide/-pMHC binding. The batch of TCR repertoire annotation in several manually curated databases, including McPAS-TCR and VDJdb, allows for the training of TCR specificity predictors and match against TCRs of interest.276–278 McPAS-TCR provides a list of TCR sequences linked with various pathologies, while VDJdb offers a detailed description of TCR:pMHC interactions based on epitope-centric approach for TCR annotation rather than the underlying biological context.279,280 Besides identification of TCR-pMHC pairings, clustering methods, like pMTnet and GLIPH, can also cluster TCRs that recognize the same epitope and predict their HLA restriction.281–285 Nevertheless, the prediction for binding affinity of TCR and pMHC in silico is still challenging due to the low affinities of TCRs for their pMHC ligands.230,246,249,286,287
For a more precise assessment of the possible application of neoantigens in immunotherapy, experimental validation of their T cell reactivity is essential. Neoantigen-reactive T cells have been validated or screened using T cell-based assays, multicolor-labeled MHC tetramers, the enzyme-linked immunosorbent spot (ELISpot) and T-cell repertoire profiling.33,288 T cell immunogenicity assay is the most direct way to evaluate the immunogenicity of candidate neoantigens. The entire set of possible mutant peptides discovered by cancer exome/RNA-seq can be tested using T cells from either cancer patients or healthy donors. After peptide stimulation, the in vitro expanded neoantigen-specific T cell reactivity is measured by flow cytometric measurement of the T-cell activation markers 4-1BB and OX-40 and IFN-production on the ELISpot assay.62,289 Multicolor-labeled MHC tetramers allow for the highly sensitive and minimally material-required evaluation of T cell reactivity against a wide range of potential epitopes using DNA barcoding, lanthanide coding, or fluorochrome coding of peptides. These technologies rely on epitope predictions and are low throughput since they can only efficiently generate a subset of the human MHC class I alleles. Integrating single-cell RNA sequencing (scRNA-seq) with TCR sequencing of responsive cell groups may be used to boost the sensitivity of detection. The scRNA-seq was used to discover paired TCR sequences linked with cells expressing high levels of IFN- γ and IL-2 in TILs co-cultured with tandem minigene (TMG)-transfected or peptide-stimulated antigen-presenting cells (APCs).290,291 Based on WES-guided prediction of neoantigens and TCR sequencing of short-term peptide-stimulated T cell cultures, the Mutation-Associated Neoantigen Functional Expansion of Specific T cells (MANAFEST) assay sensitively characterizes neoantigen-specific TCR Vβ clonotypes. The MANAFEST assay is compatible with all HLA haplotypes and can track neoantigen-specific T cells in formalin-fixed paraffin-embedded (FFPE) and/or frozen tissues. In addition to assess the tumor specificity of TCR Vβ clonotypes, MANAFEST can also look into the dynamics of the neoantigen-specific T cell response over time and monitor the efficacy of immunotherapy using liquid biopsies obtained before or after treatment.292
Several unbiased TCR-guided neoantigen discovery strategies have been developed to systematically profile neoantigen-specific TCRs. A yeast-displayed pMHC library can be used to discover neoantigen-specific TCRs. However, it is a time-consuming process to make soluble TCR reagents. Without endogenous processing of neoantigens or functional activation of T cells, the identified random peptides may not represent the physiological TCR-pMHC interaction.293 To overcome these drawbacks, two innovative strategies make use of different biological processes to mark the target cells in a co-culture system. One approach utilizes the chimeric receptors known as signaling and antigen-presenting bifunctional receptors (SABRs), which can induce a TCR‐like signal following pMHC-TCR interactions. SABRs enable the successful identification of TCR-pMHC interaction, which can be used for both known public TCRs and private neoantigen-specific TCRs.294 Trogocytosis, a membrane transfer process, is exploited by a cell-based selection platform for TCR ligand discovery. The TCR-pMHC interactions result in specific labeling of cognate target cells, which are then isolated and sequenced to identify the neoantigen-specific TCRs.295 In addition, putative pMHCs are displayed on spectrally encoded beads in BATTLES, facilitating the investigation of neoantigen-specific T cell responses under physiological force.296 T-Scan, a method for TCR epitope scanning independent of predictive algorithms, relies on the physiological activity of T cell killing rather than just assessing TCR-pMHC binding affinity, enabling the interrogation of a significantly larger antigen space than previous methods.297 Thus, these emerging approaches for TCR ligand discovery will be useful for studying the immunogenicity of candidate neoantigens, providing new targets for immunotherapy.
Neoantigens-based therapeutic strategies
As previously mentioned, tumor-specific neoantigens arising from genetic alterations elicit high-avidity T cells due to the absence of thymic selection and central tolerance. Based on their advantages of tumor-specific and immunogenetic, neoantigens may serve as emerging targets for cancer immunotherapies, including tumor vaccines, ACTs and antibody-based therapies, as well as potential predictors for ICBs (Fig. 4).8,226,298,299 The neoantigens consist of either personalized neoantigens found specifically for each patient or shared neoantigens expressed in numerous patient cancers. The off-the-shelf therapies based on public neoantigens are less resource- and time-intensive than individualized neoantigen therapies. Because personalized neoantigens are patient-specific, they cannot be used to target a large number of patients. With the recent advance in high-throughput sequencing, personalized neoantigens enable the immune system to target appropriately immunogenic epitopes on malignancies without predefined public antigens.300,301
Fig. 4.

Classification of neoantigen-based therapies. Immunotherapies that target neoantigens mainly include ACTs, bispecific antibodies and cancer vaccines. Cancer vaccines stimulate a specific immune response to tumor neoantigens using nucleic acids, peptides and DCs. The ACT utilizes the neoantigen-specific TCR or CAR engineered T cells to selectively recognize and kill tumor cells. The bispecific antibodies have one arm that targets neoantigens presented by tumor cells and one arm that targets CD3 on the surface of T cells
Neoantigen-based therapeutic vaccines
Neoantigen vaccines are an effective approach for stimulating, enhancing, and diversifying anti-tumor T cell responses, with their high feasibility, general safety and easier to manufacture. Various forms of neoantigen-based vaccines, such as peptide, nucleic acid and dendritic cell (DC) vaccines are being evaluated in clinical trials on patients with different types of tumors (Fig. 5).9,15,245,302 Current peptide and nucleic acid vaccines mainly target the predicted neoantigens derived from somatic mutations, including SNVs, frameshift INDELs and gene fusions. DC vaccines can target both selected neoantigens via pulsing with synthetic peptides or nucleic acids and overall TSAs by introducing with whole cell lysates (WCL).
Fig. 5.

Schematic illustration of neoantigen-based cancer immunotherapy production. The individualized neoantigens are identified using blood cells and tumor tissues from patient. These patient-specific neoantigens are used to develop immunotherapies, such as cancer vaccines and ACTs. Cancer vaccines in the form of peptides, DNA or mRNA, and dendritic cells are generated and administered to the same patient. For ACTs, T cells are extracted from the peripheral blood or tumor tissues of a patient and then induced to proliferate by cytokines, monoclonal antibodies against CD3 and CD28, and other reagents. The development of neoantigen-specific T lymphocytes with neoantigen-specific targeting requires co-culturing T cells with primed APCs and genetic engineering of immune cells with TCRs or CARs. After sufficient T cell expansion, T cell products are injected into lymphodepleted patients with the hope of eliciting an immune response that attacks the tumors
Peptide vaccines
Peptide-based neoantigen vaccines have received most of the attention in the research area of personalized neoantigen vaccines due to their high specificity, economical manufacture and established safety record (Table 3).177,303,304 The neoantigen peptides can be produced as genetically encoded long peptides or fused polypeptides and chemically synthesized short peptides. The peptides are subjected to affinity chromatography, size-exclusion chromatography (SEC) or high-pressure liquid chromatography (HPLC) to obtain sterile, endotoxin-free products with a purity of >98%. Following verification by MS, the peptides are mixed with appropriate adjuvants for subcutaneous injection immunization.99,305 In a phase I immunotherapy clinical trial in patients with disseminated synovial sarcoma, an SYT-SSX neoantigen peptide-based vaccine prevented disease progression in one patient and successfully induced specific CTL responses in four patients, and no serious adverse reactions or delayed-type hypersensitivity (DTH) reactions occurred throughout the treatment.305 The peptides, such as KQSSKALQR, produced from the breakpoint of BCR-ABL can be processed in the cytosol and loaded onto MHC molecules, which will be transferred to the CML cell surface for potential T cell recognition.86 In an initial clinical trial, this neoantigen-based vaccine elicits a BCR-ABL peptide-specific T cell immune response, while has no significant toxic effects.99,306 Selected neoantigens containing T cell epitopes can be produced in the form of single epitopes, polypeptide chains, or peptide pools. To overcome issues like tumor heterogeneity, HLA haplotype diversity and antigen down-regulation, overlapping peptides or long multi-epitope peptides rather than short single-epitope peptides are typically used to stimulate a powerful immune response in T cells.307 In addition, immunostimulatory adjuvants and multimeric formulation techniques are being developed to boost the immunogenicity of personalized peptide vaccinations. Therefore, personalized neoantigen vaccines based on synthetic peptides have been evaluated in clinical studies on patients with various types of cancers, including lung cancer, breast cancer, bladder cancer, pancreatic cancer, pediatric brain tumor, melanoma, and colorectal cancer (Table 4).
Table 3.
Advantages and disadvantages for neoantigen-based immunotherapies
| Immunotherapy | Formulation | Advantages | Disadvantages |
|---|---|---|---|
| Adoptive cell therapy | Tumor-infiltrating lymphocytes (TILs) |
High specificity, low toxicity; direct and continuous killing of target cells |
Expensive, time- and labor-intensive process; MHC-restricted; high proportion of tumor unrelated bystander TILs |
| T cell receptors engineered T cells (TCR-T) |
Recognize intracellular neoantigens with a high mutation rate; poor affinity but high neoantigen sensitivity; natural protein with low immunogenicity |
Expensive, time- and labor-intensive process; MHC-dependent neoantigen presentation; non-specific efficacy due to mispairing with endogenous TCRs; limited patient applicability, especially in low mutation rate cancers |
|
| Chimeric antigen receptor T cells (CAR-T) |
MHC-independent neoantigen detection; high neoantigen affinity; precisely controlling of neoantigen response by modular design; recognizing proteins, carbohydrates and glycolipids |
Expensive, time- and labor-intensive process, toxic effects; unnatural protein with potential immunogenicity; limited recognition of cell-surface neoantigen due to competing soluble neoantigen; on-target CAR-T cell activation by soluble antigens |
|
| Vaccine | Peptide vaccines |
Inexpensive and easy to produce; high specificity; low toxicity |
Restricted to the HLA subtype; low/moderate immunogenicity |
| Nucleic acid vaccines (RNA vaccine and DNA vaccine) |
Inexpensive to produce; easy delivery of multiple antigens; not restricted to HLA-patient type; activation of both cellular and humoral immunity |
Poorly immunogenic in humans; RNA vaccines require specific transportation or storage conditions |
|
| Dendritic cell (DC) Vaccines |
High immunogenicity; control of antigen presentation |
Expensive and difficult to produce; risk of leukapheresis (vascular injury, electrolyte imbalance) |
|
| Antibody-based therapy |
Full-length antibodies, antibody-drug conjugates, bispecific antibodies |
Synergy helps circumvent drug resistance | Costly for widespread use targeting individual tumor neoantigens |
Table 4.
The clinical studies of neoantigen-based immunotherapies
| Neoantigen-based immunotherapies | Therapeutic strategies | Cancer types | Clinical Trail Numbers (Primary Outcome Measures) |
|---|---|---|---|
| Cancer vaccines | |||
| Dendritic cell vaccine | Dendritic cell vaccine | Breast cancer, colorectal cancer, diffuse intrinsic pontine glioma, glioblastoma, gastric cancer, hepatocellular carcinoma, non-small cell lung cancer, esophageal squamous cell carcinoma, esophagus cancer, HPV-positive cancer, melanoma, gastrointestinal cancer, ovarian cancer, pancreatic cancer | NCT04105582 (One-year safety and adverse events (AEs)), NCT04879888 (AEs), NCT01885702 (5-years safety), NCT03914768 (2-years safety and overall survival (OS) at 12 months (OS12)), NCT05317325 (AEs), NCT05023928 (AEs), NCT04147078 (5-years disease-free survival (DFS)), NCT03674073 (One-year safety and AEs), NCT03870113 (AEs and immunogenicity), NCT03300843 (Clinical response rates), NCT03871205 (AEs and immunogenicity), NCT02956551 (AEs), NCT04078269 (Safety and tolerability), NCT03205930 (AEs), NCT05270720 (AEs), NCT01278940 (Safety and toxicity) |
| Dendritic cell vaccine + ACTs | Melanoma, bladder cancer, colorectal cancer | NCT05235607 (AEs) | |
| Dendritic cell vaccine + chemical drug | Advanced biliary tract malignant tumor, acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, myelodysplastic syndrome, non-hodgkin’s lymphoma, glioblastoma multiforme | NCT02632019 (2-years OS), NCT00923910 (AEs), NCT04968366 (AEs) | |
| Dendritic cell vaccine + ICBs | Hepatocellular carcinoma, colorectal cancer liver metastases | NCT04912765 (2-years relapse free survival) | |
| Dendritic cell vaccine + chemical drug + ICBs | Lymphocytic leukemia | NCT03219450 (One-year safety) | |
| Dendritic cell vaccine + TILs | Fallopian tube cancer, ovarian cancer, primary peritoneal cancer | NCT03735589 (AEs and dose-limiting toxicities (DLTs)) | |
| DNA vaccine | DNA vaccine | Glioblastoma, melanoma, pancreatic cancer, pediatric recurrent brain tumor, lynch syndrome, non-small cell lung carcinoma | NCT04015700 (Safety and tolerability), NCT03655756 (Serious adverse events (SAEs) and DLTs), NCT03122106 (Safety), NCT03988283 (Safety and tolerability), NCT05078866 (AEs and immunogenicity), NCT04990479 (Safety and tolerability) |
| DNA vaccine + ICBs | HPV 16-positive oropharynx cancer, small cell lung cancer, prostate cancer, renal cell carcinoma, triple-negative breast cancer, solid tumors, hepatocellular carcinoma | NCT04001413 (5 years clearance rates of HPV), NCT04397003 (Safety and tolerability), NCT03532217 (Safety, tolerability, and clinical response rates), NCT03598816 (Safety), NCT03199040 (Safety), NCT04251117 (AEs and immunogenicity), NCT05354323 (AEs) | |
| mRNA vaccine | mRNA vaccine | Melanoma, colon cancer, gastrointestinal cancer, genitourinary cancer, hepatocellular cancer, esophageal cancer, non-small cell lung cancer, triple negative breast cancer | NCT02410733 (AEs), NCT03480152 (Clinical response rates and AEs), NCT05198752 (DLTs), NCT02035956 (Safety and tolerability), NCT03908671 (AEs), NCT02316457 (AEs) |
| mRNA vaccine + ICBs | Melanoma, non-small cell lung cancer, bladder cancer, colorectal cancer, triple negative breast cancer, renal cancer, head and neck cancer, pancreatic cancer, solid tumors | NCT03289962 (DLTs and AEs), NCT04267237 (62-months DFS), NCT03897881 (3-years recurrence-free survival (RFS)), NCT03948763 (DLTs and AEs), NCT03313778 (AEs) | |
| mRNA vaccine + ICBs + Chemical drug | Pancreatic cancer | NCT04161755 (Toxicity) | |
| Peptide vaccine | Peptide vaccine | Melanoma, non-small cell lung cancer, pancreatic cancer, pediatric brain tumor, colorectal cancer, breast cancer, head and neck squamous cell carcinoma, liver cancer, diffuse intrinsic pontine glioma, glioblastoma, glioblastoma multiforme, astrocytoma, acute lymphoblastic leukemia, esophageal cancer, lynch syndrome, bladder urothelial carcinoma | NCT03662815 (Clinical response rates and AEs), NCT01970358 (AEs), NCT04509167 (WBC and RBC changes), NCT04397926 (Safety), NCT05013216 (Toxicity), NCT03956056 (Safety), NCT05111353 (Safety), NCT03068832 (Safety and tolerability), NCT04087252 (Safety), NCT05238558 (Delayed type hypersensitivity (DTH), proliferative T-cell responses and AEs), NCT03552718 (AEs), NCT04749641(Safety and one-year survival rate), NCT05356312 (NA), NCT02510950 (Safety and tolerability), NCT03559413 (Clinical response), NCT02992977 (AEs), NCT05307835 (One-year relapse-free survival and AEs), NCT04943718 (AEs), NCT03807102 (AEs and 2-years DFS), NCT03715985 (AEs), NCT04998474 (Immunogenicity), NCT04810910 (AEs and 4 years-relapse free survival), NCT03673020 (AEs), NCT03645148 (Objective response rate (ORR) and AEs), NCT03558945 (2-years overall survival) |
| Peptide vaccine + ICBs | Breast cancer, colorectal cancer, pancreatic cancer, melanoma, advanced solid tumor, diffuse intrinsic pontine glioma, diffuse midline glioma, non-small cell lung cancer, head and neck squamous cell, fibrolamellar hepatocellular carcinoma, follicular lymphoma, glioblastoma, hepatocellular carcinoma, urothelial carcinoma, renal cell carcinoma, bladder urothelial cancer, gastrointestinal tract cancer, myeloproliferative neoplasms, squamous cell lung cancer, gastroesophageal adenocarcinoma, urothelial carcinoma, ovarian cancer, platinum-resistant fallopian tube carcinoma, primary peritoneal carcinoma | NCT04864379 (ORR and AEs), NCT05269381 (AEs), NCT05098210 (AEs), NCT03606967 (6- and 12-months progression-free survival (PFS)), NCT02600949 (AEs), NCT03597282 (AEs), NCT04072900 (AEs and Clinical response), NCT04117087 (AEs), NCT04943848 (Safety and Tolerability), NCT03633110 (AEs), NCT04248569 (Toxicity), NCT03361852 (Clinical response), NCT03422094 (Safety and Tolerability), NCT03568058 (AEs), NCT03359239 (AEs), NCT05153304 (AEs), NCT02950766 (DLTs), NCT03929029 (DLTs), NCT04930783 (DLTs), NCT04364230 (Safety and immunogenicity), NCT05444530 (DLTs and AEs), NCT03166254 (Safety), NCT04266730 (AEs), NCT03639714 (AEs, SAEs, DLTs and ORR), NCT03953235 (AEs, SAEs, DLTs and ORR), NCT04024878 (AEs and SAEs), NCT04799431 (Toxicity), NCT03206047 (AEs and one-year PFS), NCT02897765 (AEs and SAEs) | |
| Peptide vaccine + chemical drug | Non-small cell lung cancer, smoldering plasma cell myeloma | NCT04487093 (AEs), NCT03631043 (AEs) | |
| Peptide vaccine + ICBs + chemical drug | Non-small cell lung cancer | NCT03380871 (AEs and SAEs) | |
| Peptide vaccine + ICBs + chemical drug + Radiotherapy | Glioblastoma | NCT02287428 (AEs) | |
| Adoptive cell therapy (ACT) | |||
| TCR-T cell | TCR-T cell | Gynecologic cancer, colorectal cancer, pancreatic cancer, non-small cell lung cancer, cholangiocarcinoma, endometrial cancer, ovarian carcinoma, squamous cell lung cancer, adenocarcinoma of lung, adenosquamous cell lung cancer, hepatocellular carcinoma, advanced malignant solid tumor, melanoma, advanced solid tumor | NCT05194735 (DLTs, ORR and AEs), NCT05105815 (18-months DFS), NCT04625205 (AEs and SAEs), NCT03171220 (AEs), NCT05020119 (AEs and DLTs) |
| TCR-T cell + chemical drug | Endocrine/neuroendocrine,non-small cell lung cancer, breast cancer, gastrointestinal/genitourinary cancers, ovarian cancer, melanoma, head and neck squamous cell carcinoma, urothelial carcinoma, renal cell carcinoma, small-cell lung cancer, cutaneous squamous cell carcinoma, anal squamous cell carcinoma, merkel cell carcinoma, vaginal cancer, cervical cancer, anal cancer, penile cancer, oropharyngeal cancer | NCT04102436 (Response rate), NCT04596033 (AEs), NCT02280811 (DLTs and ORR), NCT02858310 (Overall response rate and AEs) | |
| TCR-T cell + ICBs | Malignant epithelial neoplasms, solid tumor | NCT05349890 (AEs and SAEs), NCT04520711 (Safety, tolerability and DLTs), NCT03970382 (DLTs and AEs) | |
| TCR-T cell + ICB + chemical drug | Endocrine tumors, non-small cell lung cancer, ovarian cancer, breast cancer, gastrointestinal /genitourinary cancers, neuroendocrine tumors, multiple myeloma, Hpv-16 positive squamous cell anal cancer | NCT03412877 (Response rate), NCT04536922 (Treatment effect) | |
| TCR-T cell + radiotherapy | Hepatocellular carcinoma | NCT03199807 (AEs) | |
| TILs | TILs | Malignant epithelial tumors, malignant solid tumor | NCT05141474 (AEs, SAEs and treatment-limiting toxicity (TLT)) |
| TILs + chemical drug | Gastric cancer, colorectal cancer, pancreatic cancer, gall bladder cancer, esophageal cancer, recurrence tumor, metastatic cancer, solid tumor | NCT04426669 (Maximum tolerated dose (MTD)), NCT03658785 (ORR), NCT02959905 (AEs) | |
| TILs + ICBs | Melanoma, advanced non-small cell lung cancer | NCT03997474 (AEs), NCT04032847 (AEs) | |
| TILs + ICBs + chemical drug | Non-small cell lung cancer, squamous cell carcinoma, adenosquamous carcinoma | NCT03215810 (DLTs) | |
| CAR-T | CAR-T therapy + Chemical drug | Glioblastoma multiforme | NCT02844062 (Safety) |
| Immune checkpoint blockade (ICB) | |||
| ICBs | ICBs | Acute myeloid leukemia, myelodysplastic syndrome, bladder cancer, melanoma, colon cancer, glioma, glioblastoma, head and neck squamous cell carcinoma, HPV16-positive cancer, locally recurrent cancer, nasopharyngeal carcinoma, non-small cell lung cancer, oesophageal cancer, prostate cancer, rectal cancer, uterine cancer | NCT03600155 (Optimal dose), NCT02553642 (ORR), NCT03827044 (3-years DFS), NCT03718767 (6-months PFS), NCT03925246 (24-weeks PFS), NCT03082534 (ORR), NCT03357757 (Treatment effect), NCT03813394 (2-years ORR and PFS), NCT02437279 (Safety), NCT04825990 (ORR), NCT03130764 (Clinical response), NCT03653052 (Clinical response), NCT02113657 (Clinical response), NCT03040791 (Clinical response), NCT04019964 (Clinical response), NCT04293419 (Clinical response), NCT04262089 (Clinical response) |
| ICB + chemical drug | Acute myeloid leukemia, bladder cancer, breast cancer, colorectal cancer, hormone receptor positive tumor, endometrium cancer, gastric cancer, hepatocellular carcinoma, ovarian cancer, triple-negative breast cancer, peritoneal cancer, fallopian tube cancer, prostate cancer, head and neck squamous cell carcinoma | NCT04214249 (Clinical response), NCT03978624 (Clinical response), NCT02990845 (8-months PFS rate), NCT02453620 (AEs), NCT03409198 (3-years toxicity and PFS), NCT05456165 (AEs), NCT05201612 (10-months ORR), NCT03832621 (8-month PFS), NCT03186326 (12-months PFS), NCT04659382 (9-months PFS), NCT04262687 (10-months PFS), NCT05141721 (5-years PFS), NCT04014530 (AEs and ORR), NCT03918499 (Safety), NCT03655002 (DLTs and AEs), NCT03126812 (Clinical response), NCT03554317 (Clinical response), NCT04336943 (Clinical response), NCT04068194 (MTD and ORR), NCT05317000 (Clinical response), NCT02883062 (tumor infiltrating lymphocyte (TIL) percentage) | |
| ICB + radiotherapy | Cutaneous T cell lymphoma | NCT03385226 (Overall response rate) | |
| ICB + chemical drug + radiotherapy | Colorectal cancer, meningioma, rectal cancer | NCT03854799 (Rate of complete pathologic response), NCT03604978 (MTD, AEs and ORR), NCT04340401 (Rate of complete pathologic response) | |
Neoantigen peptide vaccines elicit and amplify anti-tumor immune responses in cancers with either high or low mutational burden. A vaccine formulated with the adjuvant poly-ICLC and a synthetic neoantigen long peptide efficiently activates CD8+ T and CD4+ lymphocytes in patients with advanced melanoma, NSCLC, or bladder cancer, all of which have high levels of mutations (NCT02897765). This neoantigen vaccine prevents recurrence for 25 months after treatment in four out of six high-risk melanoma patients.15 In NSCLC patients who have failed in multiple conventional therapies, personalized neoantigen peptide vaccination triggers specific T cell responses targeting EGFR mutations, including the relatively prevalent mutations L858R and T790M. Accordingly, a large subset of NSCLC patients responding relatively poorly to ICB approaches may benefit from the neoantigen vaccines based on shared immunogenic EGFR mutations.308 In addition, the peptide-based neoantigen vaccination can potentially modify the immune milieu of immunologically cold tumors with a relatively low mutational burden, inducing neoantigen-specific T cells to infiltrate and destroy tumor cells. For example, administration of neoantigen vaccines induces T cell immune responses in HLA-A*24:02 or HLA-A*02:01-positive glioblastoma patients. These neoantigen-specific T cells are able to cross the blood-brain barrier (BBB) and infiltrate the tumor, thereby altering the immune milieu of glioblastoma and extending the median overall survival of patients to 29.0 months.309–314
Personalized neoantigen peptide vaccines can expand the durability and repertoire of tumor-specific T cells.30 According to a retrospective analysis of the circulating immune responses in melanoma patients after vaccination, neoantigen-specific T lymphocytes exhibit a memory phenotype that lasts for an average of ~4 years following vaccination (NCT01970358). The neoantigen-specific T cells have evolved overtime into a variety of clones with different functional avidities. Meanwhile, non-vaccine antigen-directed T cell responses are also detected, suggesting epitope spreading after vaccination. The epitope spreading is associated with prolonged progression-free survival.15,315,316 The long-term persistence and diversification of functional neoantigen-specific T cell clones support the neoantigen peptide vaccines as a potent strategy for controlling the continuously evolving metastatic tumors.317
The immunogenicity of peptide vaccines can be further enhanced through improving the neoantigen presentation and using immunostimulatory adjuvants.318–322 For example, KRAS-G12D mutant peptides are fused to the C-terminal of diphtheria toxin to produce a more immunogenic peptide vaccine. This vaccine boosts CD8+ T cells while decreases T regulatory cells in mice with CT26 tumor.323 Heat shock proteins (HSPs), like HSP70, have also been complexed with synthetic peptides derived from tumor-specific neoantigens to enhance the presentation and recognition of antigens, which are widely used for treating advanced tumors resistant to conventional therapies (NCT02992977, NCT03673020).324,325 Nanoparticle formation is another technique for improving the immunogenicity of peptide vaccines. B16.F10 and CT26 neoantigens formulated with polyethyleneimine (PEI)-adsorbed mesoporous silicas micro-rod (MSR) can completely eradicate existing lung metastases in tumor-bearing mice.325,326 Another advantage of nanoparticle platform is capable of co-delivering peptides and adjuvants. Self-assembled intertwining DNA-RNA nanocapsules have been used to efficiently deliver tumor-specific neoantigen peptide and synergistic adjuvants, DNA CpG and shRNA to APCs in lymph nodes. These neoantigen vaccines induce peripheral memory neoantigen-specific CD8+ T lymphocyte, suppressing the progression of neoantigen-associated colorectal cancers.327–329 High density lipoprotein-mimicking nanodiscs elevate the efficient co-delivery of peptides and adjuvants to lymphoid organs and maintain the neoantigen presentation on DCs. In clinical trials, neoantigen-specific CTLs activated by nanodisc vaccines are 31 times more frequencies than the strongest adjuvant and up to 47 times more than soluble vaccines.330 The formulation of SNP-7/8a derived from charge-modified peptide-TLR-7/8a can effectively activate specific CD8+ T lymphocytes against 50% of neoantigens with high predicted MHC-I affinity binding, thereby enhancing anti-tumor efficacy.331 Collectively, a generic approach can be utilized to improve the anti-tumor immune response of personalized peptide vaccines.
Nucleic acid vaccines
Like peptide vaccines, nucleic acid vaccines, such as RNA and DNA vaccines, also have the advantage of being low-cost and non-HLA-specific (Table 3). Nucleic acid vaccines can deliver multiple tumor neoantigens in a single vaccination, triggering both cellular and humoral anti-tumor immune responses.245,262,325
Currently, mRNA technology has been widely used in the clinical treatment of tumors, the prevention of infectious diseases and protein-encoding therapies. The recent success of the COVID-19 mRNA vaccine has revealed the therapeutic potential of mRNA technology.332 mRNA vaccines offer considerable anti-tumor potential due to their advantages in safety, high potency, rapid and low-cost industrial production, and ability to encode entire antigens.333 Currently, in vitro transcription (IVT) is the major method used to create mRNA that contains the sequence for neoantigens. A cap structure is added to mRNAs post-IVT to increase their stability and decrease their immunogenicity. After purification through SEC or tangential flow filtration (TFF), appropriate delivery systems, such as liposomes and polymers, are selected to introduce mRNA into cells and tissues to translate the target neoantigens, thereby activating the immune response.334,335 Personalized mRNA vaccines based on tumor-specific neoantigens induce a more potent immune response than shared tumor-associated self-antigens due to the absence of central immune tolerance. For example, neoantigen-specific mRNA vaccines in 13 evaluable melanoma patients activated several neoepitope-specific CD4+ and CD8+ T cells, greatly reducing the cumulative incidence of recurrences and leading to persistent progression-free survival.302,335,336 The mRNA-4650 vaccine, which contains defined neoantigens, novel neoantigens derived from driver gene mutations and predicted HLA-I epitopes, elicits both CD8+ and CD4+ T cell response, with a preference for CD4+ T cell responses with no severe side effects.337 Clinical studies for the personalized mRNA-4157 and BNT122 vaccines are currently underway. mRNA-4157 monotherapy or in combination with the PD-1 inhibitor is well tolerated and induces a neoantigen-specific T cell response in clinical trials (NCT03313778; NCT03897881).338 A phase I trial of RNA vaccine (NCT02316457) in triple negative breast cancer (TNBC) patients demonstrate a highly effective at eliciting robust poly-epitopic T cell responses, increasing the clinical benefit for TNBC patients following surgery and (neo-)adjuvant chemotherapy.339 Moreover, the RO7198457 vaccines have been explored by BioNTech to treat various solid tumors, including melanoma, NSCLC and colorectal cancer, in combination with PD-L1 antibody.340
mRNA-encoded neoantigen vaccines may offer a proper but more potent immunogenic response and therapeutic efficacy when compared with peptide vaccines. This superiority may arise from the biological function of mRNA as a template for protein synthesis. The mRNA vaccine enables post-translational modification of protein products in human, which has the potential to present various epitopes without being constrained to a specific HLA type. In addition, numerous neoantigen epitopes can be incorporated into the same backbone, producing myriad neoantigens that can exist either as independent molecules or as a series of multiple coding sequences.302,337 One such example is the RNA-based poly-neoepitope approach developed by Sahin and colleagues. Ten selected mutations per patient are engineered into two synthetic pharmacologically optimized RNA molecules, each of which encodes five linker-connected 27mer peptides (NCT02035956).302 Another example is the personalized cancer vaccines in clinical trials, including mRNA-4157 and mRNA-4650, containing an mRNA backbone that can encode up to 30 different neoantigens.337 As a result, mRNA vaccine can express a variety of neoantigens originating from patient’s own tumor, resulting in a stronger immune response.177
Effective application of mRNA vaccines in vivo requires maintaining mRNA stability and effective intracellular distribution of the mRNA moiety to target cells. Since RNA is intrinsically unstable, early attempts focused mostly on its stabilization. The 5′ cap structure, the length of 3′ poly(A) tail and regulatory elements in the untranslated regions have all been optimized for this purpose.177,341 Efficient intracellular delivery is also required for effective mRNA therapies in vivo. Nanoformulations, such as lipid, calcium, and phosphate nanoparticles, are one method for shielding RNA from extracellular ribonucleases, resulting in improved delivery efficiency and immunogenicity.342–344 Clinical studies have been initiated for several personalized cancer vaccines based on lipid nanoparticle-mRNA formulations.177 The lipid nanoparticle-formulated mRNA-4157 and mRNA-4650 vaccines are used alone in individuals with primary solid tumors or in combination with PD-1 inhibitor (NCT03313778, NCT03897881, NCT03480152).338 Advanced RNA-lipoplex formulations have been developed and explored as therapeutic cancer vaccines in several clinical studies owing to their advantage in systemic DC targeting and synchronized induction of highly potent adaptive and innate immune responses (NCT02410733, NCT02316457).345,346 Another point worth noting in mRNA vaccine delivery is the various oncology-related administration routes.177 Intravenous administration is preferable over intradermal or subcutaneous injection for mRNA-lipoplex vaccination, which induces a higher level of T cell responses in syngeneic tumor models.345 The route of administration mechanically determines the antagonistic effects of IFN on mRNA-lipoplex vaccines-induced T cell response. When mRNA-lipoplex vaccine is delivered subcutaneously, IFN signaling inhibits the antigen-specific T cell response; conversely, IFN increases T cell responses when administered intravenously.345,347,348 Intravenous injection has been widely used for the clinical administration of mRNA vaccines, which can deliver mRNA vaccine into direct intratumoral injection-inaccessible malignancies or those without reachable lymph nodes (NCT03897881, NCT03480152, NCT03908671, and NCT03948763).177 Altogether, neoantigen-based mRNA vaccines benefit from approaches that preserve their stability and improve the delivery efficiency.
In contrast to RNA and peptide vaccines, DNA vaccines are a multifunctional platform with numerous benefits, such as the ability to accommodate any sequence without affecting its stability or solubility, rapid industrial manufacturing at low cost, and easy storage without complicated cold-chain procedures. The DNA sequence encoding the predicted neoantigens is constructed into a suitable expression vector, which is amplified and purified in prokaryotic cells like Escherichia coli. Plasmid DNA is then introduced into cells or tissues via intramuscular or subcutaneous injection in combination with electroporation, where neoantigen is expressed to induce immune responses.349 DNA vaccines also offer a significant advantage in boosting immunity, including activation of humoral immunity via antigen-induced CD4+ and CD8+ T cell responses and stimulation of innate immune response by recognition of the double-stranded DNA structure.350–353 Rational selection of tumor-specific neoantigens can improve the immunogenicity of DNA vaccines by broadening immune responses and overcoming concerns, like antigen loss, modification and tolerance. A DNA vaccine based on polyepitopic neoantigens induces similar therapeutic anti-tumor responses achieved by peptide vaccines in mice bearing mammary tumors E0771 or 4T1.308,350,354 Combining a therapeutic DNA vaccine and anti-PD-1 therapy synergistically controls tumor growth in mice.336,355 An optimized polyepitope neoantigen DNA vaccine that encodes long epitopes linked with mutant ubiquitin also elicits strong neoantigen-specific immune responses in patients with pancreatic neuroendocrine tumors when paired with ICB therapy.308 There are numerous neoantigen-based DNA vaccine clinical trials being conducted for solid tumors, including TNBC, advanced small cell lung cancer, glioblastoma, pancreatic cancer, and pediatric recurrent brain tumor (Table 4).
Even though the personalized mRNA and DNA vaccines show less efficacy and success than ICBs and T cell therapies, tremendous improvements are still being made in the formulations and preparations of nucleic acid cancer vaccines, which will further accelerate the clinical application of neoantigen-based personalized nucleic acid vaccines in cancer patients.156,334
Dendritic cell vaccines
APCs like DCs continuously present antigens to the immune system, making them an effective platform for delivering neoantigens. Autologous DCs can be isolated from patients and exposed to neoantigens, which are then injected back into the patient to elicit neoantigen-specific immune responses. Ex vivo loading of blood-isolated monocytes or hematopoietic progenitor cells with tumor neoantigens effectively improves the anti-tumor effects of neoantigen-based vaccines. Neoantigen-loaded DC vaccines can expand the antigenic breadth and clonal diversity of anti-tumor immunity.9,112,356–361 Several clinical trials are investigating the efficacy and safety of personalized neoantigen DC vaccines in solid tumors, such as melanoma, bladder cancer, colorectal cancer, esophageal cancer, breast cancer, ovarian cancer, pancreatic cancer, hepatocellular carcinoma, lung cancer, and gastric cancer (Table 4).
Neoantigens can be loaded to DCs by a variety of techniques, including pulses with the whole mRNA derived from autologous tumors, pulses with synthetic peptides and pulses with autologous whole tumor lysate (WTL), and fusion with tumor cells. The mRNA transfection is the simplest method for intracellular neoantigen production in DCs. Beyond introducing neoantigens, mRNA electroporation can also deliver functional proteins into the DCs, providing additional activation and maturation signals.362 The whole tumor mRNA-transfected DC vaccines induce T cell responses in vitro and improve the survival of immune responders with advanced melanoma (NCT01278940).363 The whole tumor mRNA-loaded DC vaccines also elicit neoantigen-specific T cell responses and exhibit safety in patients with various tumors, including melanoma, renal cancer, prostate cancer, uterine and ovarian cancer, colorectal cancer, pancreatic cancer, multiple myeloma and AML.335
Direct pulsing with synthetic peptides is another easy technique to load DCs with neoantigen-derived epitopes, which induces the necessary immune responses. This method requires the accurate identification and prediction of existing suitable epitopes in individuals, which are then synthesized into peptides or even full-length proteins to properly trigger an antigen presentation by the patient’s HLA repertoire on DCs.362,364 In several clinical trials, personalized neoantigen peptide-pulsed DCs have been tested against cancers, including melanoma, ovarian cancer, NSCLC and pancreatic cancers. DCs pulsed with synthetic long peptides and adjuvant Poly(I:C) broaden the breadth and diversity of neoantigen-specific T lymphocytes in melanoma. The t(2;13) translocation in 80% of alveolar rhabdomyosarcomas results in a PAX-FKHR fusion protein that is endogenously processed to generate a breakpoint epitope presented by HLA-B7. Stimulation of DCs with the SPQNSIRHNL fusion peptide derived from the PAX-FKHR neoantigen produced a specific CTL effect, resulting in lysis of rhabdomyosarcoma tumor cells.365 DCs pulsed with AR and ESFT fused neoantigen-specific breakpoint peptides, including EWS/FLI-1, EWS/FLI-2, PAX3/FKHR, and rhIL-2-treated autologous lymphocytes were reinfused to patients, and this regimen produced an immune response rate of 39% against the fusion breakpoint peptide.100,366 A personalized neoantigen peptide-pulsed autologous DC vaccine is also combined with chemotherapy or ICBs to treat patients with advanced lung cancer and pancreatic cancer (NCT05195619, NCT04627246, NCT02956551).
DCs pulsed with autologous WTL are safe and effective at inducing a broad anti-tumor immunity which have been extensively studied in various malignancies. In recurrent ovarian cancer patients, autologous DCs pulsed with oxidized WTL are well tolerated and elicit potent anti-tumor T cell responses. The vaccination amplifies T cell responses against neoepitopes originated from somatic mutations, including T cell clones against novel neoepitopes and clones with significantly higher avidity against known neoepitopes.367–369 Furthermore, neoantigens can be loaded into DCs by electrofusion technology, which fuses only the cytoplasm of two cell types without damaging the nucleus, thus maintaining the cellular function of these cells. In addition to expressing the tumor antigens, the fusion cells also enhance the co-stimulation ability of DCs.315 DC-tumor cell fusion vaccines have been tested in renal cancers, breast cancers, multiple myeloma and melanoma. In a subset of patients with renal cancer, the fusion cells induce tumor-specific immune responses and disease regression.325,370–372 Collectively, these preclinical and clinical studies have proven that neoantigen-based DC vaccines can elicit tumor-specific T cell responses, suggesting a feasible, safe, and effective immunotherapy for solid tumors.373
Neoantigen-based adoptive cell therapies
Neoantigens with high immunogenicity, as described above, provide excellent targets for the ACT, which employs patients’ own naturally existing or genetically engineered anti-tumor lymphocytes. Neoantigen-based adoptive cell therapies, including TILs and genetically engineered immune cells with novel TCRs or CARs, are currently successfully used to treat multiple malignancies.374
Adoptive transfer of TILs
CD8+ T lymphocytes have the capacity to identify and eradicate cancer cells, as discovered over 50 years ago.375 It has been demonstrated that adoptive transfer of in vitro expanded autologous TILs without genetic modifications can induce a full remission of certain human cancers. These TILs are taken from the patient, expanded under particular circumstances, and primed to increase their anti-cancer activity. Then, this cell product is reinfused back into the same patient, who have previous non-myeloablative lymphodepleting chemotherapy and subsequent cytokine therapy, like IL-2, thereby stimulating a potent anti-tumor immune response (Fig. 5).376,377 TILs enriched in specificity for neoantigens are preferable to unselected TILs at achieving complete and durable tumor regression. Compared to the low avidities of tumor antigen-specific TCRs, the majority of neoantigen-specific TCRs display significantly higher avidities, even towards cognate antigens expressed at relatively lower levels.378 Even a modest number of T lymphocytes with an affinity for scarcely tumor-specific neoantigens can be expanded for therapeutic application with the proper manufacturing process. Adoptive transfer of TILs enriched in neoantigen-targeted T cells is a promising treatment strategy, even for tumors with a low mutational burden.379
Neoantigen-reactive TILs mediate a remarkable regression of epithelial cancers, including advanced breast cancer, metastatic cholangiocarcinoma, colorectal cancer, melanoma, and cervical cancers.28,380–385 In the earliest prospective study of neoantigen-reactive T cells in epithelial cancers, metastatic cholangiocarcinoma patients with low TMB showed effective tumor regression lasting up to 35 months, offering the first concrete proof that neoantigen-targeted TILs can induce regression of metastatic epithelial cancer. Retrospective analysis of the infusion product has shown that the CD4+ T-helper 1 cells were reactive to an ERBB2IP mutation, suggesting a potential function of neoantigen-specific CD4+ T cells in the control of a metastatic epithelial cancer.386 TILs from individuals with metastatic gastrointestinal cancers have CD4+ and/or CD8+ T cells that recognize neoantigens resulting from somatic tumor mutations. Even though no common immunogenic epitopes are shared in these patients, a prevalent hotspot driver mutation KRAS-G12D in numerous patients can be targeted by CD8+ TILs.271 Similarly, in patients with metastatic colorectal cancer, KRAS-G12D mutant-targeted CD8+ TILs induce an efficient anti-tumor immune response against lung metastases that expressed HLA-C*08:02.387 The potential anti-tumor effect of neoantigen-reactive T cells has also been supported by retrospective investigations on the infusion of TIL products in patients with solid tumors. Patients with HPV16+ metastatic cervical squamous cell carcinoma have a full response to TILs that were initially selected based on their sensitivity to HPV antigens together with a high-dose of IL-2.388 Follow-up studies found that nearly 35% of the TILs could recognize the antigens resulting from tumor mutations compared to the 14% of the viral antigen-reactive TILs, indicating that the personalized neoantigen-reactive CD8+ T cells were responsible for tumor regression.219
TILs have been utilized to treat patients with metastatic malignancies who are refractory to current therapies, including chemotherapies, radiotherapies and anti–PD-1 therapies.389–394 Adoptive transfer of TILs targeting specific mutations in four genes, CTSB, CADPS2, KIAA0368, and SLC3A2, along with IL-2 and pembrolizumab results in a full durable regression of chemo-refractory HR+ metastatic breast cancer, which is still active at the last follow-up, 5.5 years after therapy.377 Patients with metastatic melanoma who are resistant to current therapies might achieve objective response rates of 50% to 70% with autologous TIL transfer and IL-2 after host lymphodepletion by total-body irradiation or chemotherapy.395 Patients who have metastatic NSCLC and are refractory to anti–PD-1 therapies showed a clinical response to immunotherapy combining the TILs, IL-2, and anti–PD-1 (NCT03215810, NCT04032847).389 Altogether, these studies have provided strong evidence that neoantigen-reactive T cells can improve the clinical outcome of epithelial cancers resistant to current therapies.
The frequency and breadth of TILs are key determinants of their therapeutic efficacy. The quantity and quality of tumor-reactive TILs are unambiguous variable across cancers with complex correlations with anti-tumor immune responses. For example, tumor-reactive TILs are limited to a small number of cells, as only about 10% of intratumoral CD8+ T cells can recognize autologous TSAs in ovarian and colorectal cancers, even no tumor-reactive TCRs have been found in some patients with the presence TILs.396 In contrast, neoantigen-reactive TILs have been detected in infusion products derived from nonresponding patients with metastatic breast cancer, gastrointestinal cancer, and NSCLC.389,397,398 Therefore, assessing the proportion of intratumoral T cell repertoires and their ability to recognize autologous tumors is critical for predicting the clinical activity of human cancer immunotherapies. Human CD8+ TILs can recognize a wide range of epitopes other than tumor antigens, such as antigens derived from viruses, forming bystander T cells that may infiltrate the tissue as effector cells. Neoantigen-specific TILs frequently exhibit stronger anti-tumor activity and tumor-specific expansion as compared to blood-emigrant bystander and regulatory TILs at various signatures and phenotypes.399–403 CD39, a marker of T cell reactivity to tumors and T cell exhaustion, can be used to identify the tumor-reactive T cells in a variety of malignancies. The bystander CD8+ TILs have overlapping characteristics with tumor-specific cells but lack CD39 expression and signs of persistent antigen stimulation at the tumor site.396 Furthermore, the frequency of CD39 expression in CD8+ TILs correlates with several important clinical parameters, such as the mutation burden and survival rate.396,404 Therefore, CD39 may be a promising indicator for evaluating the prognosis of cancer immunotherapy.405 The expression of CD39 could also be a viable biomarker for identification, isolation and expansion of tumor-reactive T cell populations in cancers.404 Using Cellular Indexing of Transcriptome and Epitopes by sequencing (CITE-seq) and TCR sequencing based on the signatures, such as CD39 and CXCL13 expression, neoantigen-reactive TCRs in NSCLC TILs can be identified with a success rate of 45% for CD8+ and 66% for CD4+ T cells.406 The immunomagnetic cell sorting of stem-like, self-renewable and tumor-specific TILs based on CD39 expression increases the median survival of mice by 60%.407 Collectively, optimizing the quality of the intratumoral TCR repertoire that is tumor-specific will improve the therapeutic potency of ACT.396
The intrinsic properties of the transferred T cells, including phenotype, avidity and persistent time, also influence the efficiency of neoantigen-directed ACT.397 High-dimensional analysis of TIL products identified two CD8+ T cell populations: one has a memory-progenitor CD39-negative stem-like phenotype (CD39-CD69-) and the other has a highly differentiated exhausted CD39-positive state (CD39+CD69+) TILs.375,408 The persistent exposure of TILs to antigens within the intratumoral microenvironment markedly shifted their phenotype towards an exhausted cell state (PD1+CD39+), accompanying by a progressive loss of CD8+ T cell activities and overexpression of inhibitory receptors like PD-1.375,378 It has been recently discovered that PD-1+CD8+ T cells retain a less differentiated subpopulation of stem-like TILs with ability of self-renewal, expansion, persistence, terminally differentiation and superior anti-tumor activity in vivo. These memory-like or progenitor-exhausted PD-1+CD8+ T cells serve as a source of terminally exhausted T cells that are capable of killing target cells.375,378,409 In contrast to the ACT non-responders, ACT responders have a reservoir of stem-like neoantigen-reactive TILs that expand prolifically and supply differentiated subsets, promoting T cell persistence and long-term tumor control.375,408 Consistent with their exhausted status, progenitor exhausted cells displayed inadequate enrichment for a central memory signature as opposed to an effector memory signature, relative to that of true central memory cells.409 When compared to T cells produced from an effector memory source, those from a central memory population show a stronger replicative potential in response to antigen and a longer in vivo persistence.410 The disentanglement of TIL exhaustion through isolating and expanding a desirable neoantigen-specific T cells with memory phenotype, engineering T cells to have stem-like properties, boosting the memory specificities outside the tumor with cancer vaccines, could pave the way for the creation of more effective T cell-based immunotherapies.
Genetically engineered anti-tumor immune cells
Immune cells, including T cells, natural killer (NK) cells and macrophages, can be genetically modified in vitro to generate TCRs and CARs that redirect their specificity to neoantigens. These engineered immune cells circumvent the issues such as limited proportion of tumor antigen-reactive TILs.411–415 Since tumor neoantigens encoded by tumor-specific somatic mutations have emerged as primary antigenic targets of CD8+ and CD4+ T cells in ACT therapy without the toxicity of targeting normal tissues, rapid development of neoantigen-based immune cells holds promising effects for the treatment of solid tumors.416 A number of neoantigen-targeted TCR-T and CAR-T therapies are being actively investigated in early phase clinical studies, which show an intriguing therapeutic prospect (Table 4).417
TCR-T cells
TCR-transduced T cells can target any surface or intracellular antigens. Several groups have proved the viability of an efficient approach from neoantigen identification to the engineering of the neoantigen-targeting cytotoxic TCR-T cells.416,418–420 When neoantigens are identified and predicted, neoepitope-specific T cells are isolated and their TCRs are sequenced. Candidate TCR sequences with known neoantigen reactivity can be introduced into T cells by transposon or CRISPR/Cas9 systems. These engineered cells expressing TCRs that are specific to neoantigens were infused into the patient after being verified for their tumor reactivity.416
The engineered high-avidity TCRs render CD8+ T cells specifically cytotoxic to neoantigen-containing tumors. The TCRs specifically targeting recurrent fusion genes CBFB-MYH11 confer CD8+ T cells antileukemic activity in vitro and in patient-derived murine xenograft (PDX) models with fusion gene-driven AML.88,419,421 Similarly, peripheral blood lymphocytes transduced with TCRs highly reactive to the mutated KRAS variants G12V and G12D could recognize multiple HLA-A*11:01+ pancreatic cell lines bearing the appropriate KRAS mutations in a xenograft model.419,420 The safety and efficacy of autologous T cells that have been engineered to express TCRs particularly targeting the HLA-A*11:01-presented public neoantigens, KRAS-G12V or G12D, are investigated in a clinical trial enrolling patients with advanced pancreatic cancer (NCT04146298, NCT05438667). Moreover, autologous T cells engineered with personalized neoantigen-specific TCRs are also being conducted in solid tumors, such as ovarian cancer, lung cancer, colorectal cancer, pancreatic cancer, cholangiocarcinoma and gynecologic cancer (NCT05292859, NCT05194735, NCT04520711).
In TCR-T therapy, replacing the endogenous TCR with a neoantigen-specific TCR (neoTCR) can precisely redirect the T cells to tumor cells with specific neoantigens presented by HLA. A recently developed non-viral precision genome editing technique can simultaneously knock-out the endogenous TCR or CAR genes and introduce a neoTCR or CAR, allowing a faster production of clinical-grade T cells.422,423 Based on this non-viral precision TCR replacement technology, a variety of T cell products with distinctly personalized neoTCRs for one patient are available to improve the anti-tumor effect. Three TCR-T cell products with unique personalized neoTCRs were administered to each of sixteen patients with refractory solid cancers, five of which had stable disease and the other 11 had disease progression as best response on therapy.422 Therefore, it is feasible and safe to create a broadly applicable, tumor-specific, and tailored T cell treatment for patients with solid malignancies based on this non-viral precision TCR replacement approach.
CAR-T cells
CAR-T cell approaches have a substantial advantage over TCR-T cells since they do not rely on HLA expression and neoantigen presentation, the loss of which are commonly exploited by cancer cells for immune evasion. The engineered expression of CAR molecules, which contain an intracellular signaling and co-signaling domain and an extracellular antigen-binding domain, enable CAR-T cells to bind any cell surface protein once for which there is an antibody and then activate CAR-T cells independent of MHC.424,425 Early clinical trials using CD19-targeted CAR-T cells for the treatment of B-cell malignancies patients showed outstanding results, while CAR-T cells for the treatment of patients with solid cancer showed poor outcome because of the limited antigens.424 Tumor neoantigens have inspired creative solutions and given solid tumor patients hope for CAR-T therapy. The limited number of tumor-specific surface neoantigens that are suited for CAR-T can be overcome by integrating a single-chain variable fragment (scFv) that recognizes a neoantigenic pMHC complex on the tumor surface. CAR-T cells with an scFv that recognizes the oncogene nucleophosmin (NPM1c) epitope-HLA-A2 complex demonstrated strong cytotoxicity against NPM1c+HLA-A2+ leukemia cells and AML blasts with no or minimal on-target/off-tumor toxicity.336,376,426
CAR-T cells redirected at novel neoantigens are being tested in ongoing clinical trials in hematological and solid tumors.424,427 The most well-known example of neoantigen-based CAR-T therapy in solid tumors is neoantigens from EGFRvIII mutation, which are caused by in-frame deletion of a piece of the extracellular domain spontaneously in 30% of glioblastoma patients,428 making it a desirable target for CAR-T therapy. A CAR that can recognize the EGFRvIII neoantigen has been created as a part of a lentiviral vector and a truncated EGFR that lacks the ligand binding domain and cytoplasmic kinase domain is incorporated for in vivo tracking and ablation of CAR-T cells in necessary. Human EGFRvIII+ xenogeneic subcutaneous and orthotopic models showed that EGFRvIII-directed CAR-T cells could control tumor growth.429 The safety and effectiveness of autologous anti-EGFRvIII CAR-T cells are also tested in a pilot project in patients with recurrent glioblastoma (NCT02844062). However, only a small portion of tumor cells would be killed by targeting EGFRvIII due to the highly heterogeneous of glioblastoma.
Even if the antigens are imperfectly specific individually, a Boolean logic gate can be used in CAR-T cells to improve the specificity of tumor recognition by priming with tumor-specific neoantigens and boost the eradication efficiency of tumor cells by targeting antigens uniformly expressed by tumors. The T cells can generate CARs that target antigens universally expressed by tumors, like EphA2 and IL13R2, after being primed by a highly tumor-specific neoantigen, like EGFRvIII, and being trained to carry out complete tumor destruction. In addition, a synthetic Notch (synNotch)-regulated CAR activation maintains a significant proportion of T cells in a naive/stem cell memory state, leading to improved anti-tumor immunity. In immunodeficient animals bearing intracerebral PDXs with a heterogeneous expression of EGFRvIII, EGFRvIII synNotch-CAR-T cells outperformed conventional constitutively expressed CAR-T cells in terms of anti-tumor activity and T cell persistence without causing off-tumor damage. T cells engineered with prime-and-kill circuits induce CAR-driven cytotoxicity that is spatially limited only to the proximity of priming cells, preventing off-tumor killing in distant normal tissues that carry the killing antigen but lack the priming antigen.231,430,431
CAR-NK
In addition to T cells, NK cells can also be engineered to express CARs. NK cells have the same capabilities as CD8+ cytotoxic T cells but they are not dependent on MHC-I -mediated tumor neoantigen presentation. As a result, the CAR-NK cells have the potential for immunotherapy against tumors with an extremely low mutational load and deficient neoantigen presentation. Arming NK cells with neoepitope-specific CARs remarkably improve their anti-tumor responses to NPM1-mutated AML without causing off-target toxicity.432 Moreover, NK cells further prime the DC maturation and neoantigen presentation via releasing GM-CSF, and recruit neoantigen-specific CCR5+CD8+ T cells by producing CCL5.433 Thus, the variety of cancer types amenable to immunotherapy increases as a result of modified NK cells.7
Antibody-based therapy against neoantigens
Antibody therapies have been successfully used to treat cancers, such as the anti-PD1/PD-L1/CTLA4 antibody for ICBs. Compared to the conventional antibodies that are incapable of targeting intracellular proteins, TCR-mimic (TCRm) antibodies or mutation-associated neoantigens (MANA)-specific antibodies can recognize the intracellular neoantigens by focusing on pMHC complexes. TCRm antibodies have a greater affinity than TCRs, which has been shown to be essential for minimizing the on-target, off-tumor effects.434–439 These neoantigen-targeted antibodies are simple to transform into a variety of therapeutic formats, including full-length antibodies, antibody-drug conjugates (ADCs) and BsAbs. As mentioned above, TCRm antibody moieties can also be employed to drive specific activity for neoantigens by CAR-T therapy, which has proved remarkably effective in treating certain cancers.440 Moreover, these antibody-based immunological strategies have the potential to develop off-the-shelf products for any patient whose tumors exhibit the targeted public neoantigens.300
Phage display, yeast display and genetic platform are some of the technologies used to determine human TCRm antibodies with exquisite specificity for the neoantigen as presented on HLA. In order to identify scFvs specific for mutant pMHC complex, a phage or yeast display library encoding a vast number of scFv sequences was initially created. Using a competitive selection technique, clones specific for mutant peptides bound to predetermined HLA types were subsequently identified.441,442 A high-throughput genetic platform, PresentER, is comprised of minigenes that encode MHC-I peptide libraries. By assessing the reactivities of TCR-like therapeutic agents against vast libraries of MHC-I ligands, PresentER could be utilized to determine the on-and-off targets of T cells and TCRm antibodies.443 Combining structural analysis of a reagent with its corresponding pMHC complexes with library screening helps improve TCRm antibody specificity evaluations.300 According to a crystal structure, a human TCRm antibody called ESK1 attaches to Wilms tumor (WT1)-derived peptide/HLA-A*02:01 in a manner distinct from TCRs. The possible patient pool for ESK1 therapy can be expanded by using the structure to anticipate high-affinity binding of ESK1 with several different HLA-A*02 subtypes and potential off-target binding.444
Public neoantigens originating from recurrent driver mutations, including oncogenes and TSGs, provide shared targets that could benefit a substantial proportion of patients. The scFvs that target the public neoantigens coming from oncogene mutations, such as EGFR, KRAS, PIK3CA, and CTNNB1, have been identified and transformed into therapeutic formats.300,441,445–447 For example, one scFv specific for KRAS mutant-derived peptide and one for EGFR mutant-derived peptide has been identified by phage display. These scFvs recognize the peptides only in complexes with HLA, such as KRAS peptide/HLA-A2 or EGFR peptide/HLA-A3 complexes. The scFv specific for KRAS(G12V)-HLA-A2 is converted to a full-length antibody, which responds with mutant peptide-HLA complexes even when the peptide differs from the normal wild-type form by just one amino acid.441
In contrast to oncogenes, public neoantigens coming from recurrent mutations in TSGs are unable to trigger an immune response because they are either rendered inactive by non-recurrent mutations or produced at low levels due to nonsense-mediated RNA decay. The well-characterized TSG p53 is a special case due to the identification of TCRm antibodies that target the p53 pMHC complex. Due to MHC-binding restrictions, peptides containing mutant p53 sequences are uncommon; however, tumors expressing mutant p53 may have increased expression and the MHC molecule-mediated presentation of wild-type p53 peptide, which distinguishes the tumors with mutant p53 from healthy cells expressing wild-type p53.448–451 Therefore, a TCR-like antibody P1C1TM that is specific for the wild-type p53125-134 peptide in complex with the HLA-A24:02 (HLA-24) MHC allele can target tumors harboring mutant p53 and HLA-A24. This specificity for the p53 peptide/HLA-A24 complex enables P1C1TM as an antibody-drug conjugate to effectively deliver a cytotoxic payload to tumors with mutant p53, as demonstrated by the lethal effects of PNU-159682-P1C1TM restricted to mutant p53-expressing colorectal cancer cells in in vivo models.451
BsAbs can be employed to address the issue that the density of the mutant p53 pMHC complex on the cell surface was insufficient to recruit T lymphocytes to the tumor site. Bispecific T cell engager (BiTE) is a bsAb construct that provides an efficient and potent signal for T cell activation through simultaneously binding a neoantigen on tumor cells and a CD3 complex on T cells. Even when the neoantigen-MHC complex is expressed at low levels, the highly powerful bsAb is able to decisively reverse the undruggable reputation of p53.317,440 A peptide produced from the p53 missense mutant (R175H) can be presented by HLA-A*02:01 to form a mutant p53 pMHC complex at the cell surface, which serves as a natural TCR ligand to activate T cells. An H2 antibody fragment with enhanced affinity for the HLA-A*02:01-restricted p53 R175H neoantigen has been discovered by screening utilizing a large phage library. This TCRm antibody fragment was fused with a CD3-specific antibody fragment to create a bsAb that could improve the activation of T cells to recognize and destroy cancer cells and grafts in animal models expressing the p53 R175H pMHC complex.440
Dimeric T cell engaging bsAbs are also created based on human TCRm antibodies with exquisite specificity for the mutant LMP2A peptide-HLA-A*02:01 and mutant RAS peptide-HLA complexes. These bsAbs were effective in precisely activating T cells and killing target cancer cells that expressed endogenous, incredibly low quantities of the mutant neoantigens and cognate HLA alleles.452,453 In addition, bsAbs were also employed to target public neoantigens originating from dysregulated PTM in malignancies. BsAbs engaging CD3 with TCRm specific for a pIRS2-derived phosphopeptide in complex with HLA-A*02:01 were capable of killing tumor cells in a pIRS2- and HLA-A*02:01-restricted manner.189 Alternatively, soluble structures guided by monoclonal TCR moieties specific for tumor neoantigens can also be coupled to an anti-CD3 antibody component to generate a group of bispecific molecules, known as immune-mobilizing monoclonal TCRs against cancer (ImmTACs). ImmTACs get over the biophysical constraints that prevent TCR-based immunotherapeutic methods in the past and might make it possible to target any cell based on its proteomic traits. Cancer cells with extraordinarily low surface epitope concentrations were successfully killed by T lymphocytes that had been guided by ImmTACs.300,454,455
As a result, TCRm antibodies-based strategy could be used to target neoantigens originating from mutations in both oncogenes and TSGs that are challenging to eradicate using traditional methods, enabling the development of more targeted anti-cancer therapies.452,456–459 Given TCR mimic antibodies have a much better affinity to peptide-HLA molecules than natural TCRs. To prevent cross-reactivity or binding of the HLA component unrelated to the given peptide, TCR mimic antibodies must be properly screened. Similar to designed TCRs, cross-reactivity can be prevented by using negative selection against off-target peptides.426,441,447 At least one instance of synthetic reagents has been developed that exhibits lower cross-reactivity than equivalent natural receptors.460
Combinational therapies
The therapeutic efficacy of a single immunotherapy for patients with advanced cancer is inadequate due to the heterogeneity of the neoantigen landscape and the continually evolving cancer immune evasion mechanisms. Combining several immunotherapies can improve the efficacy against cancers by simultaneously targeting various stages of the cancer-immunity cycle, including antigen release and presentation, immune cell priming and activation, immune cell trafficking and infiltration into tumors, and recognition and killing of cancer cells.7,461 Another strategy is combining therapies with different mechanisms of action to overcome the resistance induced by tumor heterogeneity. All targeted cancer cells must have the same pattern of neoantigen expression and presentation, otherwise, a resistance clone without the predicted neoantigens can survival and confer a clonal growth advantage. Therefore, precision immunotherapy can be combined with conventional treatments like radiotherapy and chemotherapy that kills cancer cells independent of the neoantigens, achieving a more prominent and durable therapeutic effect (Fig. 6).
Fig. 6.

Combinational neoantigens-based anti-tumor strategies. The “Cancer-Immunity Cycle” refers to the sequential events that must be initiated, proceeded, and expanded to achieve an anti-cancer immune response, resulting in the efficient eradication of cancer cells. Briefly, neoantigens generated by oncogenesis are released and captured by DCs (step 1). DCs convey the collected neoantigens on MHC-I and MHC-II molecules to T cells (step 2), resulting in priming and activation of effector T cell responses against cancer-specific neoantigens (step 3). Subsequently, activated effector T cells migrate to (step 4) and infiltrate into (step 5) the tumor bed, where they recognize and finally destroy their target cancer cells (step 6). The death of cancer cells produces additional tumor-associated neoantigens (step 1 once more), which broadens and intensifies the immune response in subsequent cycles. Therefore, cancer immunotherapies have been designed to reinitiate or amplify a self-sustaining cycle of cancer immunity. Multiple immunotherapies have been developed to target the rate-limiting steps in “Cancer-Immunity Cycle”, including enhancing the neoantigen release by chemotherapy, radiation therapy and oncolytic virus, increasing the quantity and quality of tumor-reactive T cells through cancer vaccine and ACTs, and boosting the infiltration and cytotoxicity efficacy of immune cells via checkpoints inhibitors
Neoantigen-based immunotherapies and ICBs
Checkpoint inhibitor-based immunotherapy has achieved prolonged anti-tumor effects in several malignancies, including renal cell carcinoma, NSCLC and melanoma. Patients, however, do not react to ICB therapy in the absence of tumor-specific effector T cells.245 Moreover, ICB therapy only affects one or two phases of the anti-cancer immunity pathways, such as anti-CTLA4 antibodies regulate the immune cell priming and activation, while anti-PD-1/PD-L1 antibodies focus on the final negative regulation of T effector cells. Therefore, only a small percentage of patients have anti-tumor response with a single agent. The neoantigen load and intratumor heterogeneity can be predictive biomarkers for the ICB response.38 It is reasonable to suspect that more effective anti-tumor response would be achieved by combining ICBs with neoantigen-based immunotherapy approaches that boost the tumor-reactive T cells.8 ICBs enhance specific T cell responses by targeting neoantigens, including PRKDC, EVI2B and S100A9, in a relapsed multiple myeloma patient.41 Compared to monotherapy, the neoantigen vaccine (PancVAX) in combination with two checkpoint modulators, such as anti-PD-1 and agonist OX40 antibodies, causes an improved and more substantial tumor regression.462 For patients with solid tumors who are unresponsive to, or relapsed following anti-PD-1 therapy, mRNA-based neoantigen vaccines, such as mRNA-4157, mRNA-5671, and BNT122, are used together with immune checkpoint inhibitors in multiple clinical trials (Table 4).177 Frequently, immunosuppressant regulators, such as PD-1, PD-L1, CTLA-4, and TIM3, are upregulated by neoantigen vaccines.245,433 ICBs could mitigate this negative effect of neoantigen vaccinations, leading to fast and long-lasting CD8+ T cell control of malignancies.177,433 Therefore, the combination of neoantigen vaccines and ICBs can achieve a better expected effect of anti-tumor immune response.245
The anti-tumor efficacy of CTLs, including those specific for mutation-associated neoantigens, can be further boosted by ICB therapy. TILs often exist in small quantities within a tumor and demonstrate an irreversible hypo-responsiveness as a result of the suppressive microenvironment. Therefore, most cancer patients are not eligible for TIL therapy.389,421 Patients with immunotherapy response to PD-1 inhibitors have a high proportion of TILs, indicating that ICBs can promote the infiltration of neoantigen-reactive lymphocytes into tumors.376,463 Blocking the PD-1 inhibitory signals induce the expansion of PD-1+ CD8+ T cells, resulting in a transient elevated cycling PD-1+ CD8+ T cells and an increasing amount of effector T lymphocytes at the tumor site.376,400,464–467 In addition, ICBs can reinvigorate the exhausted neoantigen-specific T cells via overcoming the suppressive microenvironment. Persistent exposure to TSAs promotes the exhaustion of CD8+ T cells, which characteristically expressed high levels of PD-1 and CD39.465,466 The intratumoral CD8+ T cells with high PD-1 expression show an intrinsically high capacity for tumor recognition.468 Given the potent activation of CD39+CD8+ T cells by high-affinity neoantigens, patients with hepatocellular carcinoma in high-affinity neoantigens-high group benefited more from anti-PD-1 therapy than high-affinity neoantigen-low group.123
Combinations of neoantigen vaccine and ACT
Combinations of neoantigen vaccination and ACT have also been utilized successfully to boost clinical efficacy in tumor treatment.245 Recent exciting findings showed that vaccination can increase the amount of neoantigen-reactive T cells in circulation, possibly by boosting better outgrowth of T lymphocytes. Alternatively, the vaccines can induce de novo T cell responses that overcome the insufficient recognition of neoepitope by T cells due to inadequate cross-presentation of a neoantigen by tumor cells. In addition, vaccines can be made to shield neoantigen-reactive T cells from immune checkpoint signaling or FasL-mediated apoptosis, allowing T cells to infiltrate the immunosuppressive tumor microenvironment (TME) and durably reduce epithelial malignancies. In order to increase the clinical efficacy of subsequent ACT therapy, vaccinations may be utilized to prime the patient’s neoantigen-reactive TILs or PBMCs before in vitro T cell culture. This could result in the induction of a known memory T cell response.374
Vaccine is also used to enhance the efficacy of CAR-T therapy to eliminate solid tumors. A booster vaccine for CAR-T cells has been designed, in which the peptide neoantigens can be trafficked to lymph nodes and subsequently decorated the membrane of resident APCs by their albumin-binding phospholipid-polymers. Vaccine-boosting donor cells enhance CAR-T function in solid tumors through their chimeric receptor directly in vivo. This amph-ligand vaccine can significantly elicit the amplification and intratumoral infiltration of EGFRvIII-specific CAR-T cells compared to CAR-T cell delivery alone. This vaccine strategy safely expands CAR-T cells in vivo and boosts their function and anti-tumor activity in multiple models of solid tumors, showing the significant promise of neoantigen vaccine and CAR-T combinatorial therapy.245,469–471
Neoantigen-based immunotherapies and conventional therapies
The majority of chemotherapeutic agents and radiation therapy were designed based on their direct cytotoxic effects without considering their impact on immune system. The genomic damage and altered gene transcription during these conventional therapies can promote the production of tumor-specific neoantigens, hence exhibiting potential of stimulating the anti-tumor immune response. Therefore, several FDA-approved combination therapies using conventional therapy together with immunotherapy have been developed.7
Chemotherapy and radiotherapy can be used to increase the release of tumor-specific neoantigens, circumventing issues such as an insufficient number of neoantigens to stimulate T cell response. In a patient with metastatic NSCLC who has completed response to the combined CTLA4 blockade and radiotherapy, neoantigenic mutation in KPNA2 is upregulated by radiation. Peptides derived from mutant KPNA2 trigger neoantigen-reactive CD8+ T cells and induce IFNγ production, which may trigger antigen spread.472,473 In addition, radiation can enhance the levels of existing peptide presentation by increasing the surface expression of MHC-I on tumor cells. Though expanding intracellular neoantigen pools and increasing the MHC-I-dependent presentation, radiation would promote cell killing by neoantigen-specific CD8+ T cells.472,474,475 In a poorly immunogenic mouse model of TNBC, radiotherapy increases the expression of genes with immunogenic mutations. The neoantigen vaccines based on the immunogenic mutations induced by radiotherapy elicit CD8+ and CD4+ T cells that improved the therapeutic efficacy of radiotherapy.476 Notably, highly subclonal neoantigens induced by radiation, which might be worsened by DNA-damage response (DDR) inhibitors, would interfere with the production of T lymphocytes against clonal tumor neoantigens. Additional investigations on the formation of subclonal neoantigens, as well as a thorough investigation of combined radiation, DDR inhibitors, and neoantigen-based therapies are needed to address these concerns.472
During the chemotherapy and targeted therapy, the tumor cells often occur new mutations, including reversion mutation, contributing to drug resistance. Many reversions are predicted to encode tumor-specific neoantigens, offering a potential strategy for combating resistance with CAR-T cell therapies, immune checkpoint inhibitors or anti-cancer vaccines. Reversion mutations in breast cancer-related genes are just one example that occurs during clinical platinum and PARP inhibitor resistance.477 The amount and functional activity of neoantigen-specific T lymphocytes can also be increased by administering a tumor vaccination followed by pretreatment with cyclophosphamide (CTX) and other drugs.177 Together, these studies show proof-of-principle that conventional treatments can enhance tumor control when used in conjunction with immune therapies based on neoantigens.
Challenges and opportunities for clinical application
Despite the success in hematological malignancies and solid tumors mentioned previously, neoantigen-based immunotherapies have only shown objective efficacy in a small number of well-documented patient responses. Consequently, considerable improvements are required to improve clinical results, including increasing the accuracy of neoantigen prediction, overcoming immune evasion, and optimizing the streamlining of the production process. This section focuses on the barriers that must be surmounted to enable potent immune response specifically based on tumor-specific neoantigens and the possible solutions for offering a safe and effective therapy for solid tumors.
Limited accuracy of neoantigen prediction
The widespread application of personalized immunotherapies has been constrained by the limited discovery of targetable cancer neoantigens due to the heterogeneity of mutational burdens and significantly distinct neoantigen presentation among various tumor types.81 Only 10% of non-synonymous tumor cell mutations can produce mutant peptides with high MHC affinity, and only 1% of the MHC-binding peptides are recognized by patient T cells. Theoretically, the higher the TMB, the greater the number of neoantigen-specific T cells in the tumor can be detected, resulting in a greater immunotherapy response rate. Nevertheless, low TMB can produce neoantigen-reactive lymphocytes in hematological malignancies and certain epithelial cancers, such as gastrointestinal cancers.10,240,478 The insufficient neoantigen density in malignancies with low TMB, such as AML and pediatric brain cancers, requires greater powerful strategies for the accurate identification of immunogenic neoepitopes that can be detected by CD8+ T cells.479,480 High-throughput technologies enable systematic assessment of suitable neoantigens for immunotherapies, overcoming the limited neoantigens caused by low TMB.333 For example, a proteogenomic method that integrates NGS and MS data supports the development of highly target-specific, autologous, personalized neoantigen immunotherapy, especially for tumors with low TMB.479,481,482
The prediction of neoantigens is also constrained by genetic heterogeneity, particularly the diverse somatic mutations in distinct cancer types, in different individuals and even within tumor subclone cells. A major cause of genetic heterogeneity in cancer is genomic instability, which is dynamically altered in distinct tumors and different stages. For example, TNBC patients have a higher load of frameshift and mutation-associated neoantigens (MANA) and a higher response rate to immunotherapy compared with patients with other invasive breast cancer subtypes. Furthermore, BRCA-1-mutated TNBC has an even higher mutational load.53,483–485 Therefore, identification and prediction of neoantigens should be conducted uniquely for individuals with specific cancer.245 Additional problems may develop depending on how the tumor sample from a patient is obtained for neoantigen identification. Recent technologies enable the investigation of the genomes and transcriptomes of single tumor samples taken at specified time points; however, this does not disclose heterogeneous mutations occurring in different lesions across a patient. The diversity of neoantigen-specific T cells present in a patient may not be fully captured by a single excised lesion due to restricted T lymphocyte infiltration, which constrains the TCR repertoire that may be established for therapy.245 Furthermore, mutational heterogeneity within tumors contributes an additional degree of intricacy for neoantigen prediction. The genome of tumor cells undergoes extensive generation, cloning, alteration and loss of mutations. Thus, tumor clone cells that do not respond to the neoantigen-specific T cells may exist, which may outgrow other clones due to a selection advantage, thereby restricting clinical benefit.40
Escape from immunological surveillance
A significant barrier to eliminating cancer is immune evasion, particularly for anti-cancer immunotherapies. Tumors can evade neoantigen-based immunotherapies through a number of mechanisms, including the loss of neoantigens, modification of antigen peptide presentation, and immunosuppressive TME.
Loss of neoantigens
The loss of tumor-specific neoantigens may be a significant immune escape strategy for tumors, especially if many neoantigens are by-products of tumorigenesis and do not have a critical function in tumor cell survival. The depletion of neoantigens can also present as a refractory mechanism to anti-tumor immunity, limiting the application of individualized neoantigen-specific immunotherapy. Neoantigen depletion can be induced by multiple pathways, such as copy number loss, transcriptional repression, epigenetic silencing and post-translational mechanisms. In a cohort of early-stage NSCLC tumors, 48.9% (43/88) showed evidence of neoantigen loss due to subclonal copy number events. The mutant genes encoding non-expressed neoantigens have enriched hypermethylation at their promoter regions compared with the wild-type parental genes in other purity/ploidy matched samples.486 In addition, tumors can alter the presentation of neoantigens via modulating protein turnover. Mutant proteins are more likely to misfold and degrade quickly through the proteasome, resulting in elevated antigen presentation. Molecular chaperone HSP90, however, can be used by tumors to stabilize altered proteins, preventing them from entering the antigen presentation pathway.487,488 Neoantigens that exclusively existed in specific tumor cell subpopulations can also be lost as a result of the CD8+ T cell-mediated eradication of the entire subclonal cell population. Many of the deleted mutations are identified by patients' T cells and neoantigen-encoding genes are unlikely to be produced in tumors with extensive immune cell infiltration, suggesting that neoantigen-expressing tumor subclones may be preferentially removed by the immune system.487,489 Furthermore, neoantigen loss through the deletion of chromosomal regions or the elimination of tumor subclones can lead to acquired resistance to immunotherapies such as ICBs.489 Therefore, to compensate for the loss of targetable neoantigens during immunotherapy, personalized neoantigen-specific immunotherapy should target multiple neoantigens, therefore expanding the scope of neoantigen reactivity.487
Disrupted presentation of neoantigen peptides
Tumors may evolve mutations that change not just neoantigen expression but also HLA heterozygosity and MHC stability in response to anti-tumor immune pressure. These changes impede neoantigen processing and presentation, hence inhibiting T cell recognition and tumor killing.316,490 The tumors may be able to avoid recognition by adoptively transferred T lymphocytes if there are mutations in key antigen presentation genes like β2M or a lack of HLA allele heterozygosity.316,380 For example, all seven lung metastases from a colorectal cancer patient regressed after receiving an infusion of TILs containing four unique T cell clonotypes that target KRAS-G12D. However, an evaluation nine months after treatment found that one of these lesions had advanced progress. Further analysis of this excised lesion discovered that the chromosome 6 haplotype responsible for the HLA-C*08:02 MHC-I molecule had been deleted, contributing to the tumor immune evasion.387 A second verified mechanism for epitope loss has been found as the downregulation of MHC molecules in tumor cells owing to aberrant transcription, translation or protein stability events.491,492 In multiple myeloma cell lines, higher levels of splicing factor expression are associated with lower levels of MHC-II activity, while spliceosome inhibition improved MHC-II activity, suggesting that abnormal alternative splicing is responsible for the loss of MHC-II.123 Moreover, autophagy-dependent degradation causes the lower expression of MHC-I in pancreatic ductal adenocarcinomas that are resistant to ICB treatment. Inhibiting autophagy improves antigen presentation and anti-tumor T cell responses, slowing tumor growth in syngeneic host mice.493 Due to the decreased neoantigen presentation, these mechanisms together may help to partially explain why higher neoantigen loads in some cancers were not linked to better prognostic outcomes. Based on these findings, ICB therapy for cancers may be more effective if MHC-I presentation is activated using splicing inhibitors or autophagy inhibitors.123,493
The immunosuppressive TME
Loss of neoantigens and insufficient presentation are only two of the many immune evasion mechanisms possessed by tumor cells. The recognition of neoantigens and activation of T cells can also be compromised by immunosuppressive TME processes, including the suppression of immunological checkpoints, the immunosuppressive effects of various TME cells, and the release of ions or proteins from within tumor cells following necrosis.245 The immunosuppressive checkpoint ligand molecules like PD-L1 and CTLA-4, which can restrict T cell growth and function biologically, are often upregulated in tumor cells during immunotherapeutic treatment.245,433,491 ICBs are combined with neoantigen vaccines to prevent immune escape.245
Inducible neoantigen expression, combinatorial CARs, and leveraging epitope spreading are compensatory strategies that may be used to address the immune escape of tumor cells.494 The MHC-I immunopeptidomes can be extended by splicing-derived neoepitopes that result from defective interactions of splicing complex with RNA, improper degradation of the accessory splicing factors, or aberrant splicing factor PTMs. The considerable number of highly immunogenic neoantigens created by pharmacologically altered splicing enhances tumor immunogenicity and improves the immune response to ICB treatment in mouse model.495 These findings open the exciting possibility of employing immunotherapy, which was until now only effective in diseases with greater mutation burdens like melanoma, to treat cancers that are resistant to current treatment.
Insufficient production of neoantigen-specific T cells
The immunotherapies, including vaccination, adoptive transfer of tumor-reactive TILs and TCR-T cell therapy, as well as ICBs, all rely on neoantigen-specific T cells. Thus, streamlining the sufficient production of tumor-reactive T cells from cancer patients or healthy donors should accelerate the application of neoantigen-based therapies, including broadening the TCR repertoire, enhancing the neoantigen presentation and the proper expansion of T cells.
Limited neoantigen-reactive T cell repertoire
TILs include a large number of neoantigen-reactive T cells, making them a valuable source of T lymphocytes for ACTs.380 However, the implementation of TILs treatment will be hampered by the scarcity of fresh tumor samples and cold tumors with low TILs.389,421 First, the acquisition of TILs requires invasive surgery to remove a resectable lesion, which enables only some patients to be suitable. Second, in cold tumors, the suppressive TME may reduce the efficacy and quantity of TIL-derived neoantigen-specific T cells.379,389,421 Therefore, the majority of cancer patients are ineligible for TIL therapy due to insufficient TCR repertoire.379 Efforts are currently being made to develop efficient strategies for the isolation and rapid expansion of neoantigen-specific T cells, which could benefit neoantigen-based ACTs. Utilizing particular cell-surface markers such as CD39, immunomagnetic cell sorting can efficiently detect and separate potent tumor-specific TILs with self-renewing ability from solid tumors, enabling the long-term effectiveness of ACTs.407
The easily accessible peripheral blood could be a suitable source for generating large amounts of neoantigen-reactive T cells for ACTs.379 Both circulating neoantigen-specific T cells and peripheral blood lymphocytes modified to express neoantigen-specific TCRs recognize and kill autologous malignancies.467,496,497 There are several clinical trials utilizing neoantigen-reactive T cells generated from peripheral blood to treat patients with epithelial cancer (NCT02959905, NCT05020119, and NCT04596033).498 Nonetheless, these T cells are significantly scarce and need to be precisely isolated by identified neoantigens or stimulated in vitro to proliferate to detectable levels.374 Cancer patient-derived PBMCs can be stimulated in vitro with mutant peptides to enrich neoantigen-reactive T lymphocytes. However, it should be highlighted that the substantial in vitro expansion can both further differentiate T cells and expand falsely positive neoantigen-reactive T cells.499,500 Moreover, the tumor-reactive TCR repertoire of PBMC-derived T cells can be expanded by transducing TCRs specific for tumor neoantigens of each patient.374 In addition, numerous neoantigen-reactive TCRs can be simultaneously transduced into the PBMC of a patient to boost the immune response. Based on these advantages, minimally cultured T cells with individualized TCRs will have better cytolytic capacity than exhausted and senescent TILs. Therefore, future studies improving neoantigen-reactive T lymphocytes generated from peripheral blood will benefit for their clinical applications.
The efficacy of ACTs can be restricted when tumor-reactive T cells are weakly persistent and terminally differentiated effector cells.501–503 Naive and very early memory T cells with stem cell-like properties can be produced using induced pluripotent stem cell (iPSC) technology.504 The functionally regenerated CTLs produced from iPSC that are specific for the EWS/FLI1 fusion gene-derived neoantigen elicit an anti-tumor immune response in both cultured EWS/FLI1+ sarcoma cells and Ewing sarcoma xenograft mouse model.505 iPSC-derived immature T cell lineages can now be further differentiated into neoantigen-specific T cells based on the unique three-dimensional thymic organ culture system that faithfully recapitulates the disease in vitro. The ability of iPSC-derived thymic emigrants to express heterodimeric αβ co-receptor of T cells can be maintained by imitating the thymus in vitro.506 Another benefit is that iPSCs can be established from a single neoantigen-specific CTL clone and re-differentiated into a large number of CTLs.505 In the near future, iPSCs derived from a single CTL clone will be employed to generate a sufficient number of neoantigen-specific TCR-T cells that preserve a naive-like condition and contain the TCRs in its endogenous state, significantly boosting neoantigen-based immunotherapies.
Neoepitope presentation
Presently, mature DCs or EBV-transformed B cell lines that are pulsed with peptide or transfected with TMGs for APCs are used to present antigens to T cells.374 Neoantigen-expressing mRNA-transfected DCs are also employed to prime the autologous naive CD8+ T lymphocytes in healthy donors, who are not exposed to the immunosuppressive milieu of tumor hosts.101 However, TCRs triggered by APCs pulsed with mRNA-encoding or synthetic peptides might not recognize tumor cells that present antigens endogenously.507 Directly detecting the neoantigens presented by tumor cells may be the most efficient method to establish neoantigen-reactive T cells, ensuring they can recognize the neoepitope in vivo. Tumor single-cell suspension, PDXs in highly immunodeficient mice and three-dimensional patient-derived tumor organoid cultures can be used for the recognition. Notably, PDXs and tumor organoids exhibit more typical features in neoantigen processing and presentation in contrast to the synthetic peptides or TMGs presented by DCs or B cells. PDXs maintain the endogenous expression, natural processing, and presentation of neoepitopes.508 When little tumor biopsies are available, PDX tumors can be employed to steadily extend the authentic peptidomes, creating possibilities for identifying neoantigens and sufficiently presenting neoepitopes to T cells.509 The tumor organoid cultures that extend the in vitro capacity of patient tumor cells are another alternative for autologous neoepitope presentation. A unique preclinical therapeutic model was created using tumor organoid-T cell co-culture systems to precisely measure each patient’s sensitivity to various immunotherapies. This approach was able to assess the effectiveness of cellular immunotherapies in vitro while also preserving the heterogeneity and microenvironment of tumors. In addition, co-culturing autologous PBMCs with tumor organoids produced personalized tumor-reactive CD8+ T lymphocytes that significantly reduced the growth of tumors.405,510 Although further improvements are required to conserve TME, including myeloid and stromal components, the organoid model may offer a better in vitro opportunity for neoantigen presentation and recognition by T cells.
Improved expansion approaches for tumor-reactive T cells
A significant mechanism of immunotherapy resistance is the death of tumor-reactive T lymphocytes. Since the finding that the expression of Fas-ligand (FasL) in melanoma cells caused TIL mortality, the role of the Fas/Fas-ligand pathway in establishing tumoral immune resistance has been debated for several decades.511 T cells separated from cancer patients express more Fas than healthy donors. Moreover, most cancer cells and intratumoral vascular endothelial cells express high levels of Fas ligand (FasL), which is related to a lack of CD8+ infiltration.374,512 Fas-FasL interactions in the TME during ACT may lead to T cell death. Therapies to control the Fas-mediated apoptosis and differentiation may be helpful to generate the appropriate cell products for efficient ACT. The mutant Fas that failed to bind FADD are used as a dominant negative receptor (DNR) to prevent FasL-mediated apoptosis in Fas-competent T cells.380 The Fas DNR can be transduced together with a neoantigen-reactive TCR or CAR into T cells, which can be further enriched using a magnetic bead of the introduced TCR or Fas. These Fas DNR-engineered TCR-T or CAR-T cells showed improved persistent anti-tumor activity against established solid and hematologic malignancies.380 Other strategies involve the systemic administration of Fas-Fc or anti-FasL to neutralize FasL. These FasL-neutralizing methods may reduce the death of TILs, enhance the tumoral infiltration of CD8+ T cells, and improve the persistence and activity of T cells at the tumor site.513 Notably, when administered in concert with ACT, these cell-extrinsic reagents may impair the capacity of T cells to use Fas/FasL signaling to cause cytolysis in tumor cells.
Determination and monitoring of neoantigen-specific T cell responses
Reliable immune monitoring will be essential to assess whether neoantigen-based immunotherapy achieves its expected immunologic effects and to expand the immunologically effective candidates to larger and suitable patient subsets.514 The tumor-reactive T cell responses are crucial for anti-tumor efficacy of various therapies, including cancer vaccines, ACTs, bsAbs and ICBs. The quantity and quality of tumor-reactive T cells can be measured and tracked in order to anticipate how well cancer immunotherapy will work.378,515,516 A multiparameter phenotypic and functional readout of T cell reactivity will be likely necessary in the absence of extensive evaluation using several types of T cells and APCs.380 Numerous effective markers, including CD39, PD-1, TIM-3, OX40, 4-1BB, IFNγ, and TNFα, can be used to determine the proportion of neoantigen-reactive T cells in infusion products and their capacity to recognize autologous tumors. CCR5 and CXCL13 can also be used as T cell-intrinsic indicators for CPI sensitivity. In addition, the neoepitope-specific CD8+ T cells in the blood can be used to identify the ongoing anti-tumor immune response at the tumor location. Blood neoepitope-specific T cells analyzed on a single-cell level revealed substantial clonal T-cell expansions with different effector transcription patterns, which are also present in the corresponding malignancies, indicating the recognition and destruction of tumor cells.401 It should be noted that the level of circulating tumor DNA (ctDNA), a proxy for tumor burden, can be utilized to dynamically detect the mutations that generate neoantigens. The ctDNA sequencing-base method can also monitor the neoantigen evolution during ICB treatment, thereby guiding personalized immunotherapy.517
The function and specificity of T cells can also be understood through examination of cellular states, which can also serve as a predictor for how well neoantigen-directed ACT would work. A reservoir of stem-like neoantigen-reactive TILs, such as CD8+ cells expressing activation or exhaustion markers (PD-1, TIM-3, and LAG-3), expand prolifically and supply differentiated subsets promotes T cell persistence and long-term tumor control, thereby strengthening the anti-tumor response.518,519 With high-throughput transcriptomic and TCR sequencing, neoantigen-specific TCR clonotypes dysfunctional characteristics were utilized to detect anti-tumor TCRs with minimal TIL material. Signatures of neoantigen-specific TCR clonotypes characterize the landscape of TILs across tumors, allowing TCR prediction based solely on TIL transcriptomic states for neoantigen-based cancer immunotherapy.520 These signatures may provide a degree of clonality predicting clinical response to various immunotherapies by identifying anti-cancer TCRs in the blood and, more crucially, in the tumor without the need for functional screening of putative neoantigens.378,516
Conclusion and perspective
In summary, neoantigens play a pivotal role in cancer immunotherapies, including cancer vaccines, ACTs, antibody-based therapies, and ICBs. This review summarizes the compelling evidence indicating the therapeutic strategies by targeting these cancer-specific neoantigens without normal tissue destruction and provides a strong rationale that supports the relevance of neoantigens in clinically successful immunotherapies. Numerous initiatives are being made to develop personalized or off-the-shelf anti-cancer medicines based on neoantigens. Nevertheless, experimental and theoretical improvements are required to address the time and economic issues for advanced personalized neoantigen-based immunotherapies, including efficient recruitment of patients, optimizing sequencing technology and neoantigen prediction algorithms, and taking off-the-shelf therapies against public neoantigens.15,245,302,309
Effective patient recruitment is essential for neoantigen-based immunotherapy. First, early resection may improve clinical outcomes by giving clinicians more time to carefully design, produce and test neoantigen-based therapeutic medicines. Second, early resection makes it easier to select patients who might be eligible for the off-the-shelf therapies, including vaccines, TCRs or TCRm antibodies that target well-characterized cancer driver mutations. Third, given cancer treatment, including chemotherapy, radiation therapy and ICBs will stimulate T cells to become excessively differentiated, isolating autologous T cells or TILs as soon as a patient is diagnosed with cancer may enable the collection of the highest-quality and least differentiated T cells from patients.409 In addition, early patient enrolment in neoantigen-based therapies enable effective infusion of superior neoantigen-based cellular products and ameliorate the severity of co-morbidities caused by advanced metastatic cancer clones.
The precise identification of immunogenic neoantigens and their cognate TCRs is the most crucial and rate-limiting step in the creation of personalized cancer immunotherapies.101 Immunogenic neoantigens can be identified by both immunogenomic approach that builds virtual peptidomes using in silico algorithm based on NGS data and immunopeptidomic approach that uses MS to analyze the MHC-loaded peptides. The genomic and transcriptomic sequencing data have also been integrated with MS profiling of HLA-associated peptidomes to improve the sensitivity and specificity of neoantigens identification. Neoantigen-based therapies could, however, be quite affordable due to the less expensive high-throughput sequencing and the application of powerful deep learning algorithms.245,521 A comprehensive and efficient one-stop computational workflow or a categorized benchmark of the available in silico neoantigen detection approaches is still necessary for clinical application.522 The efficient one-stop computational methods might also enable determining the potential of neoantigens as biomarkers for survival prognosis or ICB response prediction using huge data cohorts. Most crucially, the accuracy of these epitope prediction pipelines should also be confirmed by thorough immunomonitoring in early clinical investigations to improve the development of neoantigen-based cancer therapies. In addition to these in silico approaches that predict the immunogenic neoantigens and cognate TCRs based on high-throughput sequencing data, several T cell antigen discovery strategies have recently been developed to unbiased map the immunogenic neoantigens. A variety of pMHCs libraries, including yeast display library, SABRs, BATTLES, have been established, which allow for the flexible and scalable screening of antigenic epitopes. By relying on the physiological activity of T cell killing rather than evaluating TCR-pMHC binding affinity, T-Scan enables the interrogation of a significantly larger antigen space independent of predictive algorithms. Thus, the simplicity and scalability of T cell ligand discovery techniques will be useful for studying the immunogenicity of candidate neoantigens and aiding the development of novel neoantigen-based immunotherapies.
Off-the-shelf precision immunotherapies against public neoantigens is another possible strategy to overcome the time and financial issues in individualized treatment based on personalized neoantigens.300 An excellent public neoantigen shared across patients would be produced by a peptide having a hotspot mutation in a driver gene or a TSG that is presented by a relatively common HLA allele. Public neoantigens are more possibility of being clonally conserved across metastases and systematically reappearing among patients.523 Numerous general therapeutic techniques are readily available to target public neoantigens, including vaccines, bsAbs, adoptive transfer of CTLs and TCR-T cells.300,524 A library of TCRs that specifically target shared neoantigens in an HLA-specific manner has subsequently been developed for patients with advanced cancers.446,525–530 As more public neoantigens and corresponding TCRs are discovered, more patients with frequent genetic alterations that drive cancers will benefit from the public neoantigen-reactive TCR library. In addition, the widespread use of cancer genome sequencing and neoantigen prediction pipelines will facilitate matching patients with therapies that target the public neoantigens of their tumors. Therefore, this off-the-shelf strategy based on public neoantigens is anticipated to shorten the time needed for neoantigen identification and extensive T cell cultures, increasing the application of neoantigen-based therapies in a significant portion of patients.
In addition to the neoantigens generated by spontaneous mutations during carcinogenesis, certain covalent molecules can be employed to induce the production of tumor-specific public neoantigens by modifying hotspot residues in highly recurrent somatic mutations. Covalent KRAS-G12C inhibitors, like ARS1620, irreversibly modify the mutant cysteine. The haptenated peptides that carry a covalently attached small molecule can be presented by MHC-I on the cell surface. The haptenated peptide:MHC complexes can serve as tumor-specific neoantigens, triggering a cytotoxic T cell response.191,192 Based on this principle, the mutant tumor suppressor proteins can be targetable by a new class of molecules, which induce neoantigen generation and trigger a specific immune response by covalently modifying the hotspot residues like TP53Y220C and TP53R273C. Therefore, the range of tumor-specific neoantigens suitable for therapeutic targeting can be significantly expanded by haptens that specifically modify a mutant oncoprotein rather than functioning as pharmacologic inhibitors.
The neoantigens provide powerful targets for cancer vaccines, which can not only precisely eliminate residual tumor lesions, but also effectively target distant metastatic cells due to their systemic characteristics. Personalized neoantigen vaccines are produced in accordance with the individual tumor conditions in the following steps: collection of tumor tissues and normal samples, sequencing, and analysis of unique mutations, prediction, and validation of immunogenic neoantigens as well as design and production of vaccines. A variety of platforms, including peptides, nucleic acids, and DCs, can be used to develop vaccines based on predicted personalized or matched public neoantigens. Peptide, RNA and DNA-based neoantigen vaccines are high feasibility, generally safety and economical manufacture. However, the majority of patients have not been reliably induced by peptide-based neoantigen vaccinations to elicit substantial neoantigen-specific CD8+ T cell responses. The recent success of the COVID-19 mRNA vaccine has accelerated the development of mRNA-based vaccination for cancers. All the active components of tumor vaccines, such as neoantigens, formulations and delivery systems, have undergone ongoing improvement. Synthetic self-amplifying mRNAs (samRNAs), which contain replicase genes that encode RNA-dependent RNA polymerase (RdRp), are gaining interest due to their higher and longer-lasting expression of antigens compared to conventional mRNA.531 The in vivo expression of vaccine neoantigens can also be enhanced by natural carriers of genetic instructions, including adenoviruses (Ads), retroviruses and adeno‐associated viruses (AAV).532 In addition, various nanoparticle formations, such as lipid nanoparticles, exosomes, virus-like particles, caged protein nanoparticles, bacterial membrane materials-based nanocarrier, high density lipoprotein-mimicking nanodiscs, polyplexes and polymeric nanoparticles, are being developed to enhance the capacity of transport and tissue penetration, boosting the immunogenicity of personalized vaccinations.533–539 Compared to the viral vectors, the nanoparticles can also efficiently co-delivery vaccines and immune adjuvants to lymphoid organs, strengthening the neoantigen presentation.
Ex vivo loading of blood-isolated monocytes or hematopoietic progenitor cells with tumor neoantigens effectively improves the anti-cancer effects of neoantigen-based vaccines. Autologous DCs can be loaded the neoantigens in the forms of peptides, RNA and DNA. Compared to the time-and cost-intensive sequencing and computational analysis of patient-specific neoantigens, using autologous WTLs is a more convenient and economical method to induce neoantigen-specific immune responses. Whole tumor cells have both MANA and non-mutated TAAs, which might overcome the potential immune escape and resistance.367–369 However, the higher abundance of nonimmunogenic self-antigens might limit the capacity of neoantigens to elicit immune responses. Various immunosuppressive factors are also present in WTLs, which inhibit DC maturation and T cell activation.30,540 To overcome these issues, extracellular vesicles (EVs) produced by tumor cells have recently been shown to be a vaccination platform that supports DC maturation and neoantigen presentation. Tumor cell-derived EVs can deliver tumor antigen repertoires into DCs and facilitate neoantigen cross-presentation. EVs also have high immunostimulatory factors that trigger DCs to release innate immune signals.541–543 In addition to tumor cell-derived EVs, DC-derived EVs can also serve as a neoantigen-presenting unit to immune cells.544,545 Though modulating the tumor immune microenvironment and systemic immune responses, EV-based vaccination can turn a ‘cold’ tumor into a ‘hot’ one. Therefore, EVs may provide an option format for neoantigen-based cancer vaccines, which may potentially be given orally.
Although neoantigen-based immunotherapies have shown promising outcomes in earlier preclinical and clinical investigations, significant advancements are still required, especially for patients with epithelial malignancies. Cancer cells have evolved inherent defenses to evade immune recognition at every stage of the cancer-immunity cycle.7,461 Given a complicated mechanisms of immune escape in cancers, combination therapies that simultaneously target different stages of the cancer-immunity cycle may be more effective (Fig. 6). The neoantigen generation and release will be boosted by the cell death following chemotherapy, radiation, targeted therapy, photodynamic therapy and oncolytic viral therapies, which further enhance the anti-tumor immunity cycle. Neoantigen presentation can be facilitated by the administration of IFN-α, GM-CSF, anti-CD40, TLR agonist and STING agonists.546–553 To promote the infiltration of immune cells into tumors, the TME modification method and intratumor cytokines can be used.554,555 The ICBs and IDO inhibitor will also alter the immunosuppressive TME to enhance neoantigen-based immunotherapy.556,557 Nano- and EV-based drug delivery systems have been recently employed as an integrated platform for simultaneous administration of numerous drugs or therapeutic medications that work in concert to activate different stages of cancer-immunity cycle, reverse the immunosuppression and create an immunosupportive TME.558–560 These combining strategies using therapeutic agents with different mechanisms of action induce a robust effective, long-lasting and tumor-specific immune response in cancer patients.
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2020YFA0509400), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), the National Natural Science Foundation of China (81821002, 82130082, 81790251, 81972665, 82173003, 82102738 and 82103168), 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD22007 and ZYJC21004), and the Science and Technology Foundation of Shenzhen (JCYJ20200109113810154). BioRender was used to create the figures.
Author contributions
N.X. and G.S. wrote the manuscript and made the figures, W.G. and Z.H. organized the tables, C.H. and L.F. conceived, designed, and edited the manuscript. All authors have read and approved the article.
Competing interests
The authors declare that they have no potential conflicts of interest. C.H. is the editorial board member of Signal Transduction and Targeted Therapy, but he has not been involved in the process of the manuscript handling.
Footnotes
These authors contributed equally: Na Xie, Guobo Shen.
Contributor Information
Canhua Huang, Email: hcanhua@hotmail.com.
Li Fu, Email: gracelfu@szu.edu.cn.
References
- 1.Minati R, Perreault C, Thibault P. A roadmap toward the definition of actionable tumor-specific antigens. Front. Immunol. 2020;11:583287. doi: 10.3389/fimmu.2020.583287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Z, et al. Neoantigen: a new breakthrough in tumor immunotherapy. Front. Immunol. 2021;12:672356. doi: 10.3389/fimmu.2021.672356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jou J, et al. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin. Cancer Res. 2021;27:689–703. doi: 10.1158/1078-0432.CCR-20-0245. [DOI] [PubMed] [Google Scholar]
- 4.Apavaloaei A, Hardy MP, Thibault P, Perreault C. The origin and immune recognition of tumor-specific antigens. Cancers. 2020;12:2607. doi: 10.3390/cancers12092607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, et al. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J. Hematol. Oncol. 2022;15:28. doi: 10.1186/s13045-022-01247-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J. Clin. Invest. 2015;125:3413–3421. doi: 10.1172/JCI80008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu S, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021;14:156. doi: 10.1186/s13045-021-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, et al. Gene fusion neoantigens: Emerging targets for cancer immunotherapy. Cancer Lett. 2021;506:45–54. doi: 10.1016/j.canlet.2021.02.023. [DOI] [PubMed] [Google Scholar]
- 9.Carreno BM, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 11.Cortes-Selva D, Dasgupta B, Singh S, Grewal IS. Innate and innate-like cells: the future of chimeric antigen receptor (CAR) cell therapy. Trends Pharm. Sci. 2021;42:45–59. doi: 10.1016/j.tips.2020.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Ladle BH. Moving toward the ideal autologous adoptive T-cell therapy for cancer. Cancer Res. 2021;81:1940–1941. doi: 10.1158/0008-5472.CAN-21-0302. [DOI] [PubMed] [Google Scholar]
- 13.Li WH, Li YM. Chemical strategies to boost cancer vaccines. Chem. Rev. 2020;120:11420–11478. doi: 10.1021/acs.chemrev.9b00833. [DOI] [PubMed] [Google Scholar]
- 14.Manieri NA, Chiang EY, Grogan JL. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. 2017;38:20–28. doi: 10.1016/j.it.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Ott PA, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rapoport AP, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015;21:914–921. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robbins PF, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somarribas Patterson LF, Vardhana SA. Metabolic regulation of the cancer-immunity cycle. Trends Immunol. 2021;42:975–993. doi: 10.1016/j.it.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, et al. The evolution of alternative splicing in glioblastoma under therapy. Genome Biol. 2021;22:48. doi: 10.1186/s13059-021-02259-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahin U, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–112. doi: 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- 21.Leko V, Rosenberg SA. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell. 2020;38:454–472. doi: 10.1016/j.ccell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang E, Aifantis I. RNA splicing and cancer. Trends Cancer. 2020;6:631–644. doi: 10.1016/j.trecan.2020.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, et al. The roles of alternative splicing in tumor-immune cell interactions. Curr. Cancer Drug Targets. 2020;20:729–740. doi: 10.2174/1568009620666200619123725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front. Immunol. 2017;8:1679. doi: 10.3389/fimmu.2017.01679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018;18:168–182. doi: 10.1038/nri.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buonaguro L, Tagliamonte M. Selecting target antigens for cancer vaccine development. Vaccines. 2020;8:615. doi: 10.3390/vaccines8040615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith CC, et al. Alternative tumour-specific antigens. Nat. Rev. Cancer. 2019;19:465–478. doi: 10.1038/s41568-019-0162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang T, et al. Tumor neoantigens: from basic research to clinical applications. J. Hematol. Oncol. 2019;12:93. doi: 10.1186/s13045-019-0787-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richters MM, et al. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019;11:56. doi: 10.1186/s13073-019-0666-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021;18:215–229. doi: 10.1038/s41571-020-00460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Jia Q, Zhang J, Zhu B. Neoantigens in precision cancer immunotherapy: from identification to clinical applications. Chin. Med. J. 2022;135:1285–1298. doi: 10.1097/CM9.0000000000002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pritchard AL, et al. Exome sequencing to predict neoantigens in Melanoma. Cancer Immunol. Res. 2015;3:992–998. doi: 10.1158/2326-6066.CIR-15-0088. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Cao YJ. Adoptive cell therapy targeting neoantigens: a frontier for cancer research. Front. Immunol. 2020;11:176. doi: 10.3389/fimmu.2020.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mardis ER. Neoantigens and genome instability: impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019;11:71. doi: 10.1186/s13073-019-0684-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capietto AH, Hoshyar R, Delamarre L. Sources of cancer neoantigens beyond single-nucleotide variants. Int. J. Mol. Sci. 2022;23:10131. doi: 10.3390/ijms231710131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brueffer C, et al. The mutational landscape of the SCAN-B real-world primary breast cancer transcriptome. EMBO Mol. Med. 2020;12:e12118. doi: 10.15252/emmm.202012118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey P, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016;6:35848. doi: 10.1038/srep35848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou C, Zhu C, Liu Q. Toward in silico identification of tumor neoantigens in immunotherapy. Trends Mol. Med. 2019;25:980–992. doi: 10.1016/j.molmed.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Schischlik F, et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood. 2019;134:199–210. doi: 10.1182/blood.2019000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charoentong P, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 41.Perumal D, et al. Mutation-derived neoantigen-specific T-cell responses in Multiple Myeloma. Clin. Cancer Res. 2020;26:450–464. doi: 10.1158/1078-0432.CCR-19-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chatterjee A, Dasgupta S, Sidransky D. Mitochondrial subversion in cancer. Cancer Prev. Res. (Philos.) 2011;4:638–654. doi: 10.1158/1940-6207.CAPR-10-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaberi E, et al. Identification of unique and shared mitochondrial DNA mutations in neurodegeneration and cancer by single-cell mitochondrial DNA structural variation sequencing (MitoSV-seq) EBioMedicine. 2020;57:102868. doi: 10.1016/j.ebiom.2020.102868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grasso D, et al. Mitochondria in cancer. Cell Stress. 2020;4:114–146. doi: 10.15698/cst2020.06.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reznik E, et al. Mitochondrial respiratory gene expression is suppressed in many cancers. Elife. 2017;6:e21592. doi: 10.7554/eLife.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 47.Gattermann N. Mitochondrial DNA mutations in the hematopoietic system. Leukemia. 2004;18:18–22. doi: 10.1038/sj.leu.2403209. [DOI] [PubMed] [Google Scholar]
- 48.Parsons TJ, et al. A high observed substitution rate in the human mitochondrial DNA control region. Nat. Genet. 1997;15:363–368. doi: 10.1038/ng0497-363. [DOI] [PubMed] [Google Scholar]
- 49.Beadnell TC, et al. Mitochondrial genetics cooperate with nuclear genetics to selectively alter immune cell development/trafficking. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165648. doi: 10.1016/j.bbadis.2019.165648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deuse T, et al. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat. Biotechnol. 2019;37:1137–1144. doi: 10.1038/s41587-019-0227-7. [DOI] [PubMed] [Google Scholar]
- 51.Seo YH, et al. Bone reconstruction using two-layer porcine-derived bone scaffold composed of cortical and cancellous bones in a rabbit calvarial defect model. Int. J. Mol. Sci. 2022;23:2647. doi: 10.3390/ijms23052647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills RE, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;21:830–839. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turajlic S, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 54.Juhari WKW, et al. Whole-genome profiles of Malay Colorectal Cancer patients with intact MMR proteins. Genes. 2021;12:1448. doi: 10.3390/genes12091448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen UK, et al. Tumor-infiltrating T cells from clear cell renal cell carcinoma patients recognize neoepitopes derived from point and frameshift mutations. Front. Immunol. 2020;11:373. doi: 10.3389/fimmu.2020.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willis JA, et al. Immune activation in mismatch repair-deficient carcinogenesis: more than just mutational rate. Clin. Cancer Res. 2020;26:11–17. doi: 10.1158/1078-0432.CCR-18-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roudko V, et al. Lynch Syndrome and MSI-H Cancers: from mechanisms to "Off-The-Shelf" cancer vaccines. Front. Immunol. 2021;12:757804. doi: 10.3389/fimmu.2021.757804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He Y, et al. The role of DNA mismatch repair in immunotherapy of human cancer. Int. J. Biol. Sci. 2022;18:2821–2832. doi: 10.7150/ijbs.71714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thol K, McGranahan N. Potential use of shared frameshift mutations in’Off-the-Shelf’ neoantigen vaccines. Trends Cancer. 2021;7:175–177. doi: 10.1016/j.trecan.2021.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Kloor M, et al. A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: a phase I/IIa clinical trial. Clin. Cancer Res. 2020;26:4503–4510. doi: 10.1158/1078-0432.CCR-19-3517. [DOI] [PubMed] [Google Scholar]
- 61.Ballhausen A, et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020;11:4740. doi: 10.1038/s41467-020-18514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roudko V, et al. Shared immunogenic poly-epitope frameshift mutations in microsatellite unstable tumors. Cell. 2020;183:1634–1649.e1617. doi: 10.1016/j.cell.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kloor M, von Knebel Doeberitz M. The immune biology of microsatellite-unstable cancer. Trends Cancer. 2016;2:121–133. doi: 10.1016/j.trecan.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 64.Maby P, Galon J, Latouche JB. Frameshift mutations, neoantigens and tumor-specific CD8(+) T cells in microsatellite unstable colorectal cancers. Oncoimmunology. 2016;5:e1115943. doi: 10.1080/2162402X.2015.1115943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sena LA, et al. Tumor frameshift mutation proportion predicts response to immunotherapy in mismatch repair-deficient Prostate Cancer. Oncologist. 2021;26:e270–e278. doi: 10.1002/onco.13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maby P, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res. 2015;75:3446–3455. doi: 10.1158/0008-5472.CAN-14-3051. [DOI] [PubMed] [Google Scholar]
- 67.Spaanderman IT, et al. Framing the potential of public frameshift peptides as immunotherapy targets in colon cancer. PLoS ONE. 2021;16:e0251630. doi: 10.1371/journal.pone.0251630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gebert J, et al. Recurrent frameshift neoantigen vaccine elicits protective immunity with reduced tumor burden and improved overall survival in a lynch syndrome mouse model. Gastroenterology. 2021;161:1288–1302.e1213. doi: 10.1053/j.gastro.2021.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leoni G, et al. A genetic vaccine encoding shared cancer neoantigens to treat tumors with microsatellite instability. Cancer Res. 2020;80:3972–3982. doi: 10.1158/0008-5472.CAN-20-1072. [DOI] [PubMed] [Google Scholar]
- 70.Abbott CW, et al. Prediction of immunotherapy response in melanoma through combined modeling of neoantigen burden and immune-related resistance mechanisms. Clin. Cancer Res. 2021;27:4265–4276. doi: 10.1158/1078-0432.CCR-20-4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Budczies J, et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann. Oncol. 2019;30:1496–1506. doi: 10.1093/annonc/mdz205. [DOI] [PubMed] [Google Scholar]
- 72.Wu HX, et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann. Transl. Med. 2019;7:640. doi: 10.21037/atm.2019.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chae YK, et al. Clinical and immunological implications of frameshift mutations in lung cancer. J. Thorac. Oncol. 2019;14:1807–1817. doi: 10.1016/j.jtho.2019.06.016. [DOI] [PubMed] [Google Scholar]
- 74.Cimen Bozkus C, et al. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. 2019;9:1192–1207. doi: 10.1158/2159-8290.CD-18-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat. Genet. 2004;36:331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- 76.Taniue K, Akimitsu N. Fusion genes and RNAs in cancer development. Noncoding RNA. 2021;7:10. doi: 10.3390/ncrna7010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinez-Lage M, et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat. Commun. 2020;11:5060. doi: 10.1038/s41467-020-18875-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang W, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hindson J. Gene-fusion neoantigens stimulate T cells. Nat. Rev. Cancer. 2019;19:364. doi: 10.1038/s41568-019-0160-6. [DOI] [PubMed] [Google Scholar]
- 80.Wei Z, et al. The landscape of tumor fusion neoantigens: a pan-cancer analysis. iScience. 2019;21:249–260. doi: 10.1016/j.isci.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frankiw L, Baltimore D, Li G. Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 2019;19:675–687. doi: 10.1038/s41577-019-0195-7. [DOI] [PubMed] [Google Scholar]
- 82.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 83.McGranahan N, et al. Allele-specific HLA loss and immune escape in Lung Cancer evolution. Cell. 2017;171:1259–1271.e1211. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Velaga R, Koo KM, Mainwaring PN. Harnessing gene fusion-derived neoantigens for ’cold’ breast and prostate tumor immunotherapy. Immunotherapy. 2022;14:1165–1179. doi: 10.2217/imt-2022-0081. [DOI] [PubMed] [Google Scholar]
- 85.Fotakis G, et al. NeoFuse: predicting fusion neoantigens from RNA sequencing data. Bioinformatics. 2020;36:2260–2261. doi: 10.1093/bioinformatics/btz879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clark RE, et al. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood. 2001;98:2887–2893. doi: 10.1182/blood.V98.10.2887. [DOI] [PubMed] [Google Scholar]
- 87.Yang J, et al. Recurrent LRP1-SNRNP25 and KCNMB4-CCND3 fusion genes promote tumor cell motility in human osteosarcoma. J. Hematol. Oncol. 2014;7:76. doi: 10.1186/s13045-014-0076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Biernacki MA, et al. CBFB-MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia. J. Clin. Invest. 2020;130:5127–5141. doi: 10.1172/JCI137723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bolouri H, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018;24:103–112. doi: 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu P, et al. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 91.Grimwade D, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–365. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 92.Crew AJ, et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995;14:2333–2340. doi: 10.1002/j.1460-2075.1995.tb07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sato Y, et al. Detection and induction of CTLs specific for SYT-SSX-derived peptides in HLA-A24(+) patients with synovial sarcoma. J. Immunol. 2002;169:1611–1618. doi: 10.4049/jimmunol.169.3.1611. [DOI] [PubMed] [Google Scholar]
- 94.Ida K, et al. Crisscross CTL induction by SYT-SSX junction peptide and its HLA-A*2402 anchor substitute. J. Immunol. 2004;173:1436–1443. doi: 10.4049/jimmunol.173.2.1436. [DOI] [PubMed] [Google Scholar]
- 95.Gao Q, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23:227–238 e223. doi: 10.1016/j.celrep.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kalina JL, et al. Mutational analysis of gene fusions predicts novel MHC class I-restricted T-cell epitopes and immune signatures in a subset of prostate cancer. Clin. Cancer Res. 2017;23:7596–7607. doi: 10.1158/1078-0432.CCR-17-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Srivastava RM, Purohit TA, Chan TA. Diverse neoantigens and the development of cancer therapies. Semin Radiat. Oncol. 2020;30:113–128. doi: 10.1016/j.semradonc.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 98.Yoshimura M, et al. Identification of a novel HLA-A 02:01-restricted cytotoxic T lymphocyte epitope derived from the EML4-ALK fusion gene. Oncol. Rep. 2014;32:33–39. doi: 10.3892/or.2014.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cathcart K, et al. A multivalent bcr-abl fusion peptide vaccination trial in patients with chronic myeloid leukemia. Blood. 2004;103:1037–1042. doi: 10.1182/blood-2003-03-0954. [DOI] [PubMed] [Google Scholar]
- 100.Mackall CL, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin. Cancer Res. 2008;14:4850–4858. doi: 10.1158/1078-0432.CCR-07-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ali M, et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019;14:1926–1943. doi: 10.1038/s41596-019-0170-6. [DOI] [PubMed] [Google Scholar]
- 102.van Belzen I, Schonhuth A, Kemmeren P, Hehir-Kwa JY. Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology. NPJ Precis Oncol. 2021;5:15. doi: 10.1038/s41698-021-00155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rheinbay E, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature. 2020;578:102–111. doi: 10.1038/s41586-020-1965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dixon JR, et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018;50:1388–1398. doi: 10.1038/s41588-018-0195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Inaki K, Liu ET. Structural mutations in cancer: mechanistic and functional insights. Trends Genet. 2012;28:550–559. doi: 10.1016/j.tig.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 106.Xia L, et al. Multiplatform discovery and regulatory function analysis of structural variations in non-small cell lung carcinoma. Cell Rep. 2021;36:109660. doi: 10.1016/j.celrep.2021.109660. [DOI] [PubMed] [Google Scholar]
- 107.Du Y, et al. Dynamic interplay between structural variations and 3D genome organization in pancreatic cancer. Adv. Sci. 2022;9:e2200818. doi: 10.1002/advs.202200818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu Z, et al. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol. 2022;23:68. doi: 10.1186/s13059-022-02636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hu T, et al. Detection of structural variations and fusion genes in breast cancer samples using third-generation sequencing. Front. Cell Dev. Biol. 2022;10:854640. doi: 10.3389/fcell.2022.854640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mansfield AS, Peikert T, Vasmatzis G. Chromosomal rearrangements and their neoantigenic potential in mesothelioma. Transl. Lung Cancer Res. 2020;9:S92–S99. doi: 10.21037/tlcr.2019.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blasco RB, et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014;9:1219–1227. doi: 10.1016/j.celrep.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 112.Cosenza MR, Rodriguez-Martin B, Korbel JO. Structural variation in cancer: role, prevalence, and mechanisms. Annu. Rev. Genomics Hum. Genet. 2022;23:123–152. doi: 10.1146/annurev-genom-120121-101149. [DOI] [PubMed] [Google Scholar]
- 113.Fennell DA, Dulloo S, Harber J. Immunotherapy approaches for malignant pleural mesothelioma. Nat. Rev. Clin. Oncol. 2022;19:573–584. doi: 10.1038/s41571-022-00649-7. [DOI] [PubMed] [Google Scholar]
- 114.Hsiao YE, et al. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res. 2018;28:812–823. doi: 10.1101/gr.231209.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sahin I, George A, Seyhan AA. Therapeutic targeting of alternative RNA splicing in gastrointestinal malignancies and other cancers. Int. J. Mol. Sci. 2021;22:11790. doi: 10.3390/ijms222111790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chao Y, et al. Regulatory roles and mechanisms of alternative RNA splicing in adipogenesis and human metabolic health. Cell Biosci. 2021;11:66. doi: 10.1186/s13578-021-00581-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kahles A, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211–224 e216. doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat. Rev. Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shukla GC, Singh J. Mutations of RNA splicing factors in hematological malignancies. Cancer Lett. 2017;409:1–8. doi: 10.1016/j.canlet.2017.08.042. [DOI] [PubMed] [Google Scholar]
- 120.Hoyos LE, Abdel-Wahab O. Cancer-specific splicing changes and the potential for splicing-derived neoantigens. Cancer Cell. 2018;34:181–183. doi: 10.1016/j.ccell.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jayasinghe RG, et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 2018;23:270–281 e273. doi: 10.1016/j.celrep.2018.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong C, et al. Intron retention-induced neoantigen load correlates with unfavorable prognosis in multiple myeloma. Oncogene. 2021;40:6130–6138. doi: 10.1038/s41388-021-02005-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang D, et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat. Commun. 2020;11:2089. doi: 10.1038/s41467-020-15815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Han XJ, et al. Progress in neoantigen targeted cancer immunotherapies. Front. Cell Dev. Biol. 2020;8:728. doi: 10.3389/fcell.2020.00728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.David JK, et al. Putatively cancer-specific exon-exon junctions are shared across patients and present in developmental and other non-cancer cells. NAR Cancer. 2020;2:zcaa001. doi: 10.1093/narcan/zcaa001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Haen SP, Loffler MW, Rammensee HG, Brossart P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020;17:595–610. doi: 10.1038/s41571-020-0387-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang TY, Yang R. Integrated protocol for exitron and exitron-derived neoantigen identification using human RNA-seq data with ScanExitron and ScanNeo. STAR Protoc. 2021;2:100788. doi: 10.1016/j.xpro.2021.100788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang TY, et al. A pan-cancer transcriptome analysis of exitron splicing identifies novel cancer driver genes and neoepitopes. Mol. Cell. 2021;81:2246–2260.e2212. doi: 10.1016/j.molcel.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Venkataramany AS, et al. Alternative RNA splicing defects in pediatric cancers: new insights in tumorigenesis and potential therapeutic vulnerabilities. Ann. Oncol. 2022;33:578–592. doi: 10.1016/j.annonc.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brooks AN, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Graubert TA, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Furney SJ, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122–1129. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Alsafadi S, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016;7:10615. doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu J, Chen Z, Yong L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol. Oncol. 2018;148:368–374. doi: 10.1016/j.ygyno.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 136.Bjorklund SS, et al. Widespread alternative exon usage in clinically distinct subtypes of Invasive Ductal Carcinoma. Sci. Rep. 2017;7:5568. doi: 10.1038/s41598-017-05537-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Robertson AG, et al. Integrative analysis identifies four molecular and clinical subsets in Uveal Melanoma. Cancer Cell. 2018;33:151. doi: 10.1016/j.ccell.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 138.Marcelino Meliso F, Hubert CG, Favoretto Galante PA, Penalva LO. RNA processing as an alternative route to attack glioblastoma. Hum. Genet. 2017;136:1129–1141. doi: 10.1007/s00439-017-1819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.De Kesel J, Fijalkowski I, Taylor J, Ntziachristos P. Splicing dysregulation in human hematologic malignancies: beyond splicing mutations. Trends Immunol. 2022;43:674–686. doi: 10.1016/j.it.2022.06.006. [DOI] [PubMed] [Google Scholar]
- 140.Bigot J, et al. Splicing patterns in SF3B1-mutated uveal melanoma generate shared immunogenic tumor-specific neoepitopes. Cancer Discov. 2021;11:1938–1951. doi: 10.1158/2159-8290.CD-20-0555. [DOI] [PubMed] [Google Scholar]
- 141.Celik A, Kervestin S, Jacobson A. NMD: at the crossroads between translation termination and ribosome recycling. Biochimie. 2015;114:2–9. doi: 10.1016/j.biochi.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Perrin-Vidoz L, et al. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002;11:2805–2814. doi: 10.1093/hmg/11.23.2805. [DOI] [PubMed] [Google Scholar]
- 143.Oka M, et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol. 2021;22:9. doi: 10.1186/s13059-020-02240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu C, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014;20:596–598. doi: 10.1038/nm.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lejeune F. Nonsense-mediated mRNA decay, a finely regulated mechanism. Biomedicines. 2022;10:141. doi: 10.3390/biomedicines10010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tan K, Stupack DG, Wilkinson MF. Nonsense-mediated RNA decay: an emerging modulator of malignancy. Nat. Rev. Cancer. 2022;22:437–451. doi: 10.1038/s41568-022-00481-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Popp MW, Maquat LE. Nonsense-mediated mRNA decay and cancer. Curr. Opin. Genet. Dev. 2018;48:44–50. doi: 10.1016/j.gde.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 149.Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017;18:18–30. doi: 10.1038/nrm.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yeh HS, Yong J. Alternative polyadenylation of mRNAs: 3’-untranslated region matters in gene expression. Mol. Cells. 2016;39:281–285. doi: 10.14348/molcells.2016.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Curinha A, et al. Implications of polyadenylation in health and disease. Nucleus. 2014;5:508–519. doi: 10.4161/nucl.36360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ren F, et al. Alternative polyadenylation: a new frontier in post transcriptional regulation. Biomark. Res. 2020;8:67. doi: 10.1186/s40364-020-00249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li W, et al. Systematic profiling of poly(A)+ transcripts modulated by core 3’ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11:e1005166. doi: 10.1371/journal.pgen.1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rehfeld A, Plass M, Krogh A, Friis-Hansen L. Alterations in polyadenylation and its implications for endocrine disease. Front. Endocrinol. 2013;4:53. doi: 10.3389/fendo.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Singh I, et al. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun. 2018;9:1716. doi: 10.1038/s41467-018-04112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhao X, Pan X, Wang Y, Zhang Y. Targeting neoantigens for cancer immunotherapy. Biomark. Res. 2021;9:61. doi: 10.1186/s40364-021-00315-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee SH, et al. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature. 2018;561:127–131. doi: 10.1038/s41586-018-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Desterro J, Bak-Gordon P, Carmo-Fonseca M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 2020;19:112–129. doi: 10.1038/s41573-019-0042-3. [DOI] [PubMed] [Google Scholar]
- 159.Mackay A, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade Glioma from the HERBY Phase II randomized trial. Cancer Cell. 2018;33:829–842.e825. doi: 10.1016/j.ccell.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Han L, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Paz-Yaacov N, et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;13:267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- 162.Kurkowiak M, et al. The effects of RNA editing in cancer tissue at different stages in carcinogenesis. RNA Biol. 2021;18:1524–1539. doi: 10.1080/15476286.2021.1877024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liang W, et al. mRNA modification orchestrates cancer stem cell fate decisions. Mol. Cancer. 2020;19:38. doi: 10.1186/s12943-020-01166-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Coltri PP, Dos Santos MGP, da Silva GHG. Splicing and cancer: challenges and opportunities. Wiley Interdiscip. Rev. RNA. 2019;10:e1527. doi: 10.1002/wrna.1527. [DOI] [PubMed] [Google Scholar]
- 165.Shiromoto Y, et al. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 2021;12:1654. doi: 10.1038/s41467-021-21921-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang M, et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018;9:3919. doi: 10.1038/s41467-018-06405-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Peng X, et al. A-to-I RNA editing contributes to proteomic diversity in cancer. Cancer Cell. 2018;33:817–828 e817. doi: 10.1016/j.ccell.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bazak L, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li J, et al. Genome-wide expression changes mediated by A-to-I RNA editing correlate with hepatic oncogenesis. Transl. Cancer Res. 2021;10:2725–2737. doi: 10.21037/tcr-21-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Park J, Chung YJ. Identification of neoantigens derived from alternative splicing and RNA modification. Genomics Inf. 2019;17:e23. doi: 10.5808/GI.2019.17.3.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kracht MJ, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat. Med. 2017;23:501–507. doi: 10.1038/nm.4289. [DOI] [PubMed] [Google Scholar]
- 172.Laumont CM, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018;10:eaau5516. doi: 10.1126/scitranslmed.aau5516. [DOI] [PubMed] [Google Scholar]
- 173.Rosenberg SA, et al. Identification of BING-4 cancer antigen translated from an alternative open reading frame of a gene in the extended MHC class II region using lymphocytes from a patient with a durable complete regression following immunotherapy. J. Immunol. 2002;168:2402–2407. doi: 10.4049/jimmunol.168.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Khurana E, et al. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016;17:93–108. doi: 10.1038/nrg.2015.17. [DOI] [PubMed] [Google Scholar]
- 176.Zhao Q, et al. Proteogenomics uncovers a vast repertoire of shared tumor-specific antigens in ovarian cancer. Cancer Immunol. Res. 2020;8:544–555. doi: 10.1158/2326-6066.CIR-19-0541. [DOI] [PubMed] [Google Scholar]
- 177.Barbier AJ, et al. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022;40:840–854. doi: 10.1038/s41587-022-01294-2. [DOI] [PubMed] [Google Scholar]
- 178.Erhard F, Dolken L, Schilling B, Schlosser A. Identification of the cryptic HLA-I immunopeptidome. Cancer Immunol. Res. 2020;8:1018–1026. doi: 10.1158/2326-6066.CIR-19-0886. [DOI] [PubMed] [Google Scholar]
- 179.Ruiz Cuevas MV, et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021;34:108815. doi: 10.1016/j.celrep.2021.108815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dersh D, Hollý J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2021;21:116–128. doi: 10.1038/s41577-020-0390-6. [DOI] [PubMed] [Google Scholar]
- 181.Vlad AM, et al. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J. Exp. Med. 2002;196:1435–1446. doi: 10.1084/jem.20020493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Apostolopoulos V, et al. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc. Natl Acad. Sci. USA. 2003;100:15029–15034. doi: 10.1073/pnas.2432220100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Marijt KA, Doorduijn EM, van Hall T. TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class I(low) cancers. Mol. Immunol. 2019;113:43–49. doi: 10.1016/j.molimm.2018.03.029. [DOI] [PubMed] [Google Scholar]
- 184.van Hall T, et al. Selective cytotoxic T-lymphocyte targeting of tumor immune escape variants. Nat. Med. 2006;12:417–424. doi: 10.1038/nm1381. [DOI] [PubMed] [Google Scholar]
- 185.RodrIguez E, Schetters STT, van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018;18:204–211. doi: 10.1038/nri.2018.3. [DOI] [PubMed] [Google Scholar]
- 186.Vogt G, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat. Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 187.Malaker SA, et al. Identification of glycopeptides as posttranslationally modified neoantigens in Leukemia. Cancer Immunol. Res. 2017;5:376–384. doi: 10.1158/2326-6066.CIR-16-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mohammed F, et al. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat. Immunol. 2008;9:1236–1243. doi: 10.1038/ni.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Dao T, et al. A TCR mimic monoclonal antibody reactive with the "public" phospho-neoantigen pIRS2/HLA-A*02:01 complex. JCI Insight. 2022;7:e151624. doi: 10.1172/jci.insight.151624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Engelhard VH, et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer. 2020;8:e000262. doi: 10.1136/jitc-2019-000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang Z, et al. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. 2022;40:1060–1069.e1067. doi: 10.1016/j.ccell.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Freed-Pastor WA, Aguirre AJ. Getting a handle on KRAS inhibitor resistance with hapten-mediated anti-tumor immunity. Cancer Cell. 2022;40:908–910. doi: 10.1016/j.ccell.2022.08.018. [DOI] [PubMed] [Google Scholar]
- 193.Tcyganov EN, et al. Peroxynitrite in the tumor microenvironment changes the profile of antigens allowing escape from cancer immunotherapy. Cancer Cell. 2022;40:1173–1189.e1176. doi: 10.1016/j.ccell.2022.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Capietto AH, Delamarre L. Peroxynitrite promotes immune evasion by reducing tumor antigenicity. Cell Rep. Med. 2022;3:100787. doi: 10.1016/j.xcrm.2022.100787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Niedermann G, et al. Contribution of proteasome-mediated proteolysis to the hierarchy of epitopes presented by major histocompatibility complex class I molecules. Immunity. 1995;2:289–299. doi: 10.1016/1074-7613(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 196.Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. 2004;427:252–256. doi: 10.1038/nature02240. [DOI] [PubMed] [Google Scholar]
- 197.Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nat. Rev. Immunol. 2010;10:73–78. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- 198.Vigneron N, et al. An antigenic peptide produced by peptide splicing in the proteasome. Science. 2004;304:587–590. doi: 10.1126/science.1095522. [DOI] [PubMed] [Google Scholar]
- 199.Liepe J, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. 2016;354:354–358. doi: 10.1126/science.aaf4384. [DOI] [PubMed] [Google Scholar]
- 200.Tran MT, et al. T cell receptor recognition of hybrid insulin peptides bound to HLA-DQ8. Nat. Commun. 2021;12:5110. doi: 10.1038/s41467-021-25404-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dalet A, et al. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Natl Acad. Sci. USA. 2011;108:E323–E331. doi: 10.1073/pnas.1101892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Warren EH, et al. An antigen produced by splicing of noncontiguous peptides in the reverse order. Science. 2006;313:1444–1447. doi: 10.1126/science.1130660. [DOI] [PubMed] [Google Scholar]
- 203.Robbins PF, et al. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patient responding to immunotherapy. Cancer Res. 1994;54:3124–3126. [PubMed] [Google Scholar]
- 204.Doorduijn EM, et al. TAP-independent self-peptides enhance T cell recognition of immune-escaped tumors. J. Clin. Invest. 2016;126:784–794. doi: 10.1172/JCI83671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Doorduijn EM, et al. T cells specific for a TAP-independent self-peptide remain naive in tumor-bearing mice and are fully exploitable for therapy. Oncoimmunology. 2018;7:e1382793. doi: 10.1080/2162402X.2017.1382793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Marijt KA, et al. Identification of non-mutated neoantigens presented by TAP-deficient tumors. J. Exp. Med. 2018;215:2325–2337. doi: 10.1084/jem.20180577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Durgeau A, et al. Human preprocalcitonin self-antigen generates TAP-dependent and -independent epitopes triggering optimised T-cell responses toward immune-escaped tumours. Nat. Commun. 2018;9:5097. doi: 10.1038/s41467-018-07603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Krump NA, You J. From Merkel cell polyomavirus infection to Merkel cell carcinoma oncogenesis. Front. Microbiol. 2021;12:739695. doi: 10.3389/fmicb.2021.739695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.DeCaprio JA. Merkel cell polyomavirus and Merkel cell carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372:20160276. doi: 10.1098/rstb.2016.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Morales-Sanchez A, Fuentes-Panana EM. Human viruses and cancer. Viruses. 2014;6:4047–4079. doi: 10.3390/v6104047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clin. Cancer Res. 2004;10:803–821. doi: 10.1158/1078-0432.CCR-0670-3. [DOI] [PubMed] [Google Scholar]
- 212.Bauer M, et al. Epstein-Barr Virus-associated malignancies and immune escape: the role of the tumor microenvironment and tumor cell evasion strategies. Cancers. 2021;13:5189. doi: 10.3390/cancers13205189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhang WT, et al. Association of PD-1/PD-L1 expression and Epstein-Barr virus infection in patients with invasive breast cancer. Diagn. Pathol. 2022;17:61. doi: 10.1186/s13000-022-01234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yeo-Teh NSL, Ito Y, Jha S. High-risk human papillomaviral oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int J. Mol. Sci. 2018;19:1706. doi: 10.3390/ijms19061706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pal A, Kundu R. Human papillomavirus E6 and E7: the Cervical Cancer hallmarks and targets for therapy. Front. Microbiol. 2019;10:3116. doi: 10.3389/fmicb.2019.03116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Eckhardt M, et al. Multiple routes to oncogenesis are promoted by the human papillomavirus-host protein network. Cancer Discov. 2018;8:1474–1489. doi: 10.1158/2159-8290.CD-17-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Chan CK, et al. Human papillomavirus infection and cervical cancer: epidemiology, screening, and vaccination-review of current perspectives. J. Oncol. 2019;2019:3257939. doi: 10.1155/2019/3257939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Draper LM, et al. Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin. Cancer Res. 2015;21:4431–4439. doi: 10.1158/1078-0432.CCR-14-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Stevanovic S, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356:200–205. doi: 10.1126/science.aak9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Norberg SM, et al. Regression of epithelial cancers following T cell receptor gene therapy targeting human papillomavirus-16 E7. Blood. 2018;132:1. doi: 10.1182/blood-2018-99-117017. [DOI] [Google Scholar]
- 221.Doran SL, et al. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J. Clin. Oncol. 2019;37:2759–2768. doi: 10.1200/JCO.18.02424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Doran SL, et al. Genetically engineered T-cell therapy for HPV-associated epithelial cancers: A first in human, phase I/II clinical trial. J. Clin. Oncol. 2018;36:1. doi: 10.1200/JCO.2018.36.15_suppl.3019. [DOI] [Google Scholar]
- 223.Khairkhah N, Bolhassani A, Najafipour R. Current and future direction in treatment of HPV-related cervical disease. J. Mol. Med. 2022;100:829–845. doi: 10.1007/s00109-022-02199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Borden ES, Buetow KH, Wilson MA, Hastings KT. Cancer neoantigens: challenges and future directions for prediction, prioritization, and validation. Front. Oncol. 2022;12:836821. doi: 10.3389/fonc.2022.836821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Borden ES, et al. NeoScore integrates characteristics of the neoantigen:MHC class I interaction and expression to accurately prioritize immunogenic neoantigens. J. Immunol. 2022;208:1813–1827. doi: 10.4049/jimmunol.2100700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Garcia-Garijo A, Fajardo CA, Gros A. Determinants for neoantigen identification. Front. Immunol. 2019;10:1392. doi: 10.3389/fimmu.2019.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gopanenko AV, Kosobokova EN, Kosorukov VS. Main strategies for the identification of neoantigens. Cancers. 2020;12:2879. doi: 10.3390/cancers12102879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Thind AS, et al. Demystifying emerging bulk RNA-Seq applications: the application and utility of bioinformatic methodology. Brief. Bioinform. 2021;22:bbab259. doi: 10.1093/bib/bbab259. [DOI] [PubMed] [Google Scholar]
- 229.Liu XS, Mardis ER. Applications of immunogenomics to cancer. Cell. 2017;168:600–612. doi: 10.1016/j.cell.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.De Mattos-Arruda L, et al. Neoantigen prediction and computational perspectives towards clinical benefit: recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020;31:978–990. doi: 10.1016/j.annonc.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Choe JH, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021;13:eabe7378. doi: 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Seki M, et al. Evaluation and application of RNA-Seq by MinION. DNA Res. 2019;26:55–65. doi: 10.1093/dnares/dsy038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wu J, et al. TSNAdb: a database for tumor-specific neoantigens from immunogenomics data analysis. Genomics Proteom. Bioinforma. 2018;16:276–282. doi: 10.1016/j.gpb.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bassani-Sternberg M, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016;7:13404. doi: 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Yi X, et al. caAtlas: an immunopeptidome atlas of human cancer. iScience. 2021;24:103107. doi: 10.1016/j.isci.2021.103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang X, Qi Y, Zhang Q, Liu W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019;120:109542. doi: 10.1016/j.biopha.2019.109542. [DOI] [PubMed] [Google Scholar]
- 237.Purcell AW, Ramarathinam SH, Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019;14:1687–1707. doi: 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- 238.Bulik-Sullivan B, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019;37:55–63. doi: 10.1038/nbt.4313. [DOI] [PubMed] [Google Scholar]
- 239.Solleder M, et al. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. Mol. Cell Proteom. 2020;19:390–404. doi: 10.1074/mcp.TIR119.001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yadav M, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 241.van der Lee DI, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Invest. 2019;129:774–785. doi: 10.1172/JCI97482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Meyer VS, et al. Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. J. Proteome Res. 2009;8:3666–3674. doi: 10.1021/pr800937k. [DOI] [PubMed] [Google Scholar]
- 243.Zarling AL, et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl Acad. Sci. USA. 2006;103:14889–14894. doi: 10.1073/pnas.0604045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Engelhard VH, Altrich-Vanlith M, Ostankovitch M, Zarling AL. Post-translational modifications of naturally processed MHC-binding epitopes. Curr. Opin. Immunol. 2006;18:92–97. doi: 10.1016/j.coi.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 245.Peng M, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol. Cancer. 2019;18:128. doi: 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Finotello F, Rieder D, Hackl H, Trajanoski Z. Next-generation computational tools for interrogating cancer immunity. Nat. Rev. Genet. 2019;20:724–746. doi: 10.1038/s41576-019-0166-7. [DOI] [PubMed] [Google Scholar]
- 247.Fotakis G, Trajanoski Z, Rieder D. Computational cancer neoantigen prediction: current status and recent advances. Immunooncol. Technol. 2021;12:100052. doi: 10.1016/j.iotech.2021.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Calmeiro J, et al. Biomaterial-based platforms for in situ dendritic cell programming and their use in antitumor immunotherapy. J. Immunother. Cancer. 2019;7:238. doi: 10.1186/s40425-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Okada M, Shimizu K, Fujii SI. Identification of neoantigens in cancer cells as targets for immunotherapy. Int. J. Mol. Sci. 2022;23:2594. doi: 10.3390/ijms23052594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Robinson J, et al. IPD-IMGT/HLA database. Nucleic Acids Res. 2020;48:D948–D955. doi: 10.1093/nar/gkz950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Abelin JG, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Montesion M, et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 2021;11:282–292. doi: 10.1158/2159-8290.CD-20-0672. [DOI] [PubMed] [Google Scholar]
- 253.Szolek A, et al. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–3316. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Shukla SA, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kim D, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Liu C, et al. ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 2013;41:e142. doi: 10.1093/nar/gkt481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kawaguchi S, et al. HLA-HD: an accurate HLA typing algorithm for next-generation sequencing data. Hum. Mutat. 2017;38:788–797. doi: 10.1002/humu.23230. [DOI] [PubMed] [Google Scholar]
- 258.Orenbuch R, et al. arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics. 2020;36:33–40. doi: 10.1093/bioinformatics/btz474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Boegel S, et al. HLA typing from RNA-Seq sequence reads. Genome Med. 2012;4:102. doi: 10.1186/gm403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Buchkovich ML, et al. HLAProfiler utilizes k-mer profiles to improve HLA calling accuracy for rare and common alleles in RNA-seq data. Genome Med. 2017;9:86. doi: 10.1186/s13073-017-0473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Mardis ER. Genomic prediction of neoantigens: immunogenomics before NGS. Nat. Rev. Genet. 2021;22:550–551. doi: 10.1038/s41576-021-00374-4. [DOI] [PubMed] [Google Scholar]
- 262.Esprit A, et al. Neo-antigen mRNA vaccines. Vaccines. 2020;8:776. doi: 10.3390/vaccines8040776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mirza N, et al. An integrative in silico system for predicting dysregulated genes in the human epileptic focus: application to SLC transporters. Epilepsia. 2016;57:1467–1474. doi: 10.1111/epi.13473. [DOI] [PubMed] [Google Scholar]
- 264.Nielsen M, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Jurtz V, et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.O’Donnell TJ, et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 2018;7:129–132 e124. doi: 10.1016/j.cels.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 267.Reynisson B, et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Chen B, et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019;37:1332–1343. doi: 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Khodadoust MS, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature. 2017;543:723–727. doi: 10.1038/nature21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Linnemann C, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015;21:81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 271.Tran E, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Jensen KK, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406. doi: 10.1111/imm.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Andreatta M, et al. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics. 2015;67:641–650. doi: 10.1007/s00251-015-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Stronen E, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 275.Kalaora S, et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in Melanoma. Cancer Discov. 2018;8:1366–1375. doi: 10.1158/2159-8290.CD-17-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bagaev DV, et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 2020;48:D1057–d1062. doi: 10.1093/nar/gkz874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Grazioli F, et al. On TCR binding predictors failing to generalize to unseen peptides. Front. Immunol. 2022;13:1014256. doi: 10.3389/fimmu.2022.1014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Goncharov M, et al. VDJdb in the pandemic era: a compendium of T cell receptors specific for SARS-CoV-2. Nat. Methods. 2022;19:1017–1019. doi: 10.1038/s41592-022-01578-0. [DOI] [PubMed] [Google Scholar]
- 279.Shugay M, et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018;46:D419–D427. doi: 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Tickotsky N, et al. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics. 2017;33:2924–2929. doi: 10.1093/bioinformatics/btx286. [DOI] [PubMed] [Google Scholar]
- 281.Huang H, et al. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol. 2020;38:1194–1202. doi: 10.1038/s41587-020-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hayashi F, et al. A new clustering method identifies multiple sclerosis-specific T-cell receptors. Ann. Clin. Transl. Neurol. 2021;8:163–176. doi: 10.1002/acn3.51264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Wang C, Huang H, Davis MM. Grouping T-cell antigen receptors by specificity. Methods Mol. Biol. 2022;2574:291–307. doi: 10.1007/978-1-0716-2712-9_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Glanville J, et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547:94–98. doi: 10.1038/nature22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Lu T, et al. Deep learning-based prediction of the T cell receptor-antigen binding specificity. Nat. Mach. Intell. 2021;3:864–875. doi: 10.1038/s42256-021-00383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lang F, et al. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022;21:261–282. doi: 10.1038/s41573-021-00387-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Nogueira C, Kaufmann JK, Lam H, Flechtner JB. Improving cancer immunotherapies through empirical neoantigen selection. Trends Cancer. 2018;4:97–100. doi: 10.1016/j.trecan.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 288.Mysore V, et al. Protective heterologous T cell immunity in COVID-19 induced by the trivalent MMR and Tdap vaccine antigens. Med. 2021;2:1050–1071.e1057. doi: 10.1016/j.medj.2021.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Cimen Bozkus C, Blazquez AB, Enokida T, Bhardwaj N. A T-cell-based immunogenicity protocol for evaluating human antigen-specific responses. STAR Protoc. 2021;2:100758. doi: 10.1016/j.xpro.2021.100758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lu YC, et al. An efficient single-cell RNA-seq approach to identify neoantigen-specific T cell receptors. Mol. Ther. 2018;26:379–389. doi: 10.1016/j.ymthe.2017.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Lu YC, et al. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing. J. Immunother. Cancer. 2021;9:e002595. doi: 10.1136/jitc-2021-002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Danilova L, et al. The mutation-associated neoantigen functional expansion of specific T Cells (MANAFEST) assay: a sensitive platform for monitoring antitumor immunity. Cancer Immunol. Res. 2018;6:888–899. doi: 10.1158/2326-6066.CIR-18-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Gee MH, et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell. 2018;172:549–563.e516. doi: 10.1016/j.cell.2017.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Joglekar AV, et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. methods. 2019;16:191–198. doi: 10.1038/s41592-018-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Li G, et al. T cell antigen discovery via trogocytosis. Nat. Methods. 2019;16:183–190. doi: 10.1038/s41592-018-0305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Feng Y, et al. A bead-based method for high-throughput mapping of the sequence- and force-dependence of T cell activation. Nat. Methods. 2022;19:1295–1305. doi: 10.1038/s41592-022-01592-2. [DOI] [PubMed] [Google Scholar]
- 297.Kula T, et al. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell. 2019;178:1016–1028.e1013. doi: 10.1016/j.cell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Chen H, et al. Neoantigen-based immunotherapy in pancreatic ductal adenocarcinoma (PDAC) Cancer Lett. 2020;490:12–19. doi: 10.1016/j.canlet.2020.06.011. [DOI] [PubMed] [Google Scholar]
- 299.Liu S, et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer. 2019;7:156. doi: 10.1186/s40425-019-0629-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Pearlman AH, et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer. 2021;2:487–497. doi: 10.1038/s43018-021-00210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Lin MJ, et al. Cancer vaccines: the next immunotherapy frontier. Nat. Cancer. 2022;3:911–926. doi: 10.1038/s43018-022-00418-6. [DOI] [PubMed] [Google Scholar]
- 302.Sahin U, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 303.Chen I, Chen MY, Goedegebuure SP, Gillanders WE. Challenges targeting cancer neoantigens in 2021: a systematic literature review. Expert Rev. Vaccines. 2021;20:827–837. doi: 10.1080/14760584.2021.1935248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Nelde A, Rammensee HG, Walz JS. The peptide vaccine of the future. Mol. Cell Proteom. 2021;20:100022. doi: 10.1074/mcp.R120.002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kawaguchi S, et al. Phase I vaccination trial of SYT-SSX junction peptide in patients with disseminated synovial sarcoma. J. Transl. Med. 2005;3:1. doi: 10.1186/1479-5876-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Pinilla-Ibarz J, et al. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood. 2000;95:7. doi: 10.1182/blood.V95.5.1781.005k46_1781_1787. [DOI] [PubMed] [Google Scholar]
- 307.Stephens AJ, Burgess-Brown NA, Jiang S. Beyond just peptide antigens: the complex world of peptide-based cancer vaccines. Front. Immunol. 2021;12:696791. doi: 10.3389/fimmu.2021.696791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Li L, et al. Optimized polyepitope neoantigen DNA vaccines elicit neoantigen-specific immune responses in preclinical models and in clinical translation. Genome Med. 2021;13:56. doi: 10.1186/s13073-021-00872-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Keskin DB, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Baratta MG. Glioblastoma is 'hot' for personalized vaccines. Nat. Rev. Cancer. 2019;19:129. doi: 10.1038/s41568-019-0118-8. [DOI] [PubMed] [Google Scholar]
- 311.Ridler C. Personalized vaccines use tumour fingerprint to target glioblastoma. Nat. Rev. Neurol. 2019;15:59. doi: 10.1038/s41582-019-0135-y. [DOI] [PubMed] [Google Scholar]
- 312.Editorial. Progress in the fight against brain cancer. Nature565, 134 (2019). [DOI] [PubMed]
- 313.Zaidi N, Jaffee EM. Immune cells track hard-to-target brain tumours. Nature. 2019;565:170–171. doi: 10.1038/d41586-018-07728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Hilf N, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565:240–245. doi: 10.1038/s41586-018-0810-y. [DOI] [PubMed] [Google Scholar]
- 315.Hu Z, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021;27:515–525. doi: 10.1038/s41591-020-01206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kreiter S, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Haas L. Neoantigens take center stage. Nat. Cancer. 2021;2:1288. doi: 10.1038/s43018-021-00310-9. [DOI] [PubMed] [Google Scholar]
- 318.Ni Q, et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci. Adv. 2020;6:eaaw6071. doi: 10.1126/sciadv.aaw6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Backlund CM, et al. Cell-penetrating peptides enhance peptide vaccine accumulation and persistence in lymph nodes to drive immunogenicity. Proc. Natl Acad. Sci. USA. 2022;119:e2204078119. doi: 10.1073/pnas.2204078119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Khong H, Overwijk WW. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer. 2016;4:56. doi: 10.1186/s40425-016-0160-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Schijns V, et al. Modulation of immune responses using adjuvants to facilitate therapeutic vaccination. Immunol. Rev. 2020;296:169–190. doi: 10.1111/imr.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Veatch JR, et al. A therapeutic cancer vaccine delivers antigens and adjuvants to lymphoid tissues using genetically modified T cells. J. Clin. Invest. 2021;131:e144195. doi: 10.1172/JCI144195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Wan Y, et al. Recombinant KRAS G12D protein vaccines elicit significant anti-tumor effects in mouse CT26 tumor models. Front. Oncol. 2020;10:1326. doi: 10.3389/fonc.2020.01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Aldous AR, Dong JZ. Personalized neoantigen vaccines: a new approach to cancer immunotherapy. Bioorg. Med. Chem. 2018;26:2842–2849. doi: 10.1016/j.bmc.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 325.Fritah H, Rovelli R, Chiang CL, Kandalaft LE. The current clinical landscape of personalized cancer vaccines. Cancer Treat. Rev. 2022;106:102383. doi: 10.1016/j.ctrv.2022.102383. [DOI] [PubMed] [Google Scholar]
- 326.Tornesello AL, et al. Nanoparticles to improve the efficacy of peptide-based cancer vaccines. Cancers. 2020;12:1049. doi: 10.3390/cancers12041049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Zhu G, et al. Intertwining DNA-RNA nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nat. Commun. 2017;8:1482. doi: 10.1038/s41467-017-01386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Scheetz L, et al. Engineering patient-specific cancer immunotherapies. Nat. Biomed. Eng. 2019;3:768–782. doi: 10.1038/s41551-019-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Saeed M, et al. Engineering nanoparticles to reprogram the tumor immune microenvironment for improved cancer immunotherapy. Theranostics. 2019;9:7981–8000. doi: 10.7150/thno.37568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kuai R, et al. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017;16:489–496. doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Lynn GM, et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol. 2020;38:320–332. doi: 10.1038/s41587-019-0390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Wei J, Hui AM. The paradigm shift in treatment from Covid-19 to oncology with mRNA vaccines. Cancer Treat. Rev. 2022;107:102405. doi: 10.1016/j.ctrv.2022.102405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Cheng R, et al. Identification of alternative splicing-derived cancer neoantigens for mRNA vaccine development. Brief. Bioinform. 2022;23:bbab553. doi: 10.1093/bib/bbab553. [DOI] [PubMed] [Google Scholar]
- 334.Qin S, et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct. Target Ther. 2022;7:166. doi: 10.1038/s41392-022-01007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Beck JD, et al. mRNA therapeutics in cancer immunotherapy. Mol. Cancer. 2021;20:69. doi: 10.1186/s12943-021-01348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Fan C, et al. Cancer/testis antigens: from serology to mRNA cancer vaccine. Semin Cancer Biol. 2021;76:218–231. doi: 10.1016/j.semcancer.2021.04.016. [DOI] [PubMed] [Google Scholar]
- 337.Cafri G, et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest. 2020;130:5976–5988. doi: 10.1172/JCI134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021;6:1078–1094. doi: 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Schmidt M, et al. T-cell responses induced by an individualized neoantigen specific immune therapy in post (neo)adjuvant patients with triple negative breast cancer. Ann. Oncol. 2020;31:1. doi: 10.1016/j.annonc.2020.08.209. [DOI] [Google Scholar]
- 340.Deng Z, et al. mRNA vaccines: the dawn of a new era of cancer immunotherapy. Front. Immunol. 2022;13:887125. doi: 10.3389/fimmu.2022.887125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Liu L, et al. Combination immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol. Ther. 2018;26:45–55. doi: 10.1016/j.ymthe.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Salomon N, et al. A liposomal RNA vaccine inducing neoantigen-specific CD4(+) T cells augments the antitumor activity of local radiotherapy in mice. Oncoimmunology. 2020;9:1771925. doi: 10.1080/2162402X.2020.1771925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Zhang H, et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc. Natl Acad. Sci. USA. 2021;118:e2005191118. doi: 10.1073/pnas.2005191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Van Hoecke L, et al. The opposing effect of type I IFN on the T cell response by non-modified mRNA-lipoplex vaccines is determined by the route of administration. Mol. Ther. Nucleic Acids. 2020;22:373–381. doi: 10.1016/j.omtn.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Kranz LM, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 347.De Beuckelaer A, et al. Type I interferons interfere with the capacity of mRNA lipoplex vaccines to elicit cytolytic T cell responses. Mol. Ther. 2016;24:2012–2020. doi: 10.1038/mt.2016.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Pollard C, et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013;21:251–259. doi: 10.1038/mt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat. Rev. Cancer. 2008;8:108–120. doi: 10.1038/nrc2326. [DOI] [PubMed] [Google Scholar]
- 350.Lopes A, Vandermeulen G, Preat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019;38:146. doi: 10.1186/s13046-019-1154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Li L, Petrovsky N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev. Vaccines. 2016;15:313–329. doi: 10.1586/14760584.2016.1124762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Ori D, Murase M, Kawai T. Cytosolic nucleic acid sensors and innate immune regulation. Int. Rev. Immunol. 2017;36:74–88. doi: 10.1080/08830185.2017.1298749. [DOI] [PubMed] [Google Scholar]
- 353.Tang CK, Pietersz GA. Intracellular detection and immune signaling pathways of DNA vaccines. Expert Rev. Vaccines. 2009;8:1161–1170. doi: 10.1586/erv.09.79. [DOI] [PubMed] [Google Scholar]
- 354.Duperret EK, et al. A synthetic DNA, multi-neoantigen vaccine drives predominately MHC class I CD8(+) T-cell responses, impacting tumor challenge. Cancer Immunol. Res. 2019;7:174–182. doi: 10.1158/2326-6066.CIR-18-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Tondini E, et al. A poly-neoantigen DNA vaccine synergizes with PD-1 blockade to induce T cell-mediated tumor control. Oncoimmunology. 2019;8:1652539. doi: 10.1080/2162402X.2019.1652539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Delamarre L, Mellman I, Yadav M. Cancer immunotherapy. Neo approaches to cancer vaccines. Science. 2015;348:760–761. doi: 10.1126/science.aab3465. [DOI] [PubMed] [Google Scholar]
- 357.Jones B. Clinical genetics. Sequencing for tailored melanoma immunotherapy. Nat. Rev. Genet. 2015;16:259. doi: 10.1038/nrg3945. [DOI] [PubMed] [Google Scholar]
- 358.Guo Z, et al. Durable complete response to neoantigen-loaded dendritic-cell vaccine following anti-PD-1 therapy in metastatic gastric cancer. NPJ Precis. Oncol. 2022;6:34. doi: 10.1038/s41698-022-00279-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Peng S, et al. Combination neoantigen-based dendritic cell vaccination and adoptive T-cell transfer induces antitumor responses against recurrence of Hepatocellular Carcinoma. Cancer Immunol. Res. 2022;10:728–744. doi: 10.1158/2326-6066.CIR-21-0931. [DOI] [PubMed] [Google Scholar]
- 360.Cannon MJ, Block MS, Morehead LC, Knutson KL. The evolving clinical landscape for dendritic cell vaccines and cancer immunotherapy. Immunotherapy. 2019;11:75–79. doi: 10.2217/imt-2018-0129. [DOI] [PubMed] [Google Scholar]
- 361.Pao SC, Chu MT, Hung SI. Therapeutic vaccines targeting neoantigens to induce T-cell immunity against cancers. Pharmaceutics. 2022;14:867. doi: 10.3390/pharmaceutics14040867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Dorrie J, Schaft N, Schuler G, Schuler-Thurner B. Therapeutic cancer vaccination with ex vivo RNA-transfected dendritic cells-an update. Pharmaceutics. 2020;12:92. doi: 10.3390/pharmaceutics12020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Kyte JA, et al. Immune response and long-term clinical outcome in advanced melanoma patients vaccinated with tumor-mRNA-transfected dendritic cells. Oncoimmunology. 2016;5:e1232237. doi: 10.1080/2162402X.2016.1232237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Reynolds CR, Tran S, Jain M, Narendran A. Neoantigen cancer vaccines: generation, optimization, and therapeutic targeting strategies. Vaccines. 2022;10:196. doi: 10.3390/vaccines10020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.van den Broeke LT, et al. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res. 2006;66:1818–1823. doi: 10.1158/0008-5472.CAN-05-2549. [DOI] [PubMed] [Google Scholar]
- 366.Zhang H, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat. Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 367.Tanyi JL, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018;10:eaao5931. doi: 10.1126/scitranslmed.aao5931. [DOI] [PubMed] [Google Scholar]
- 368.Kandalaft LE, et al. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J. Transl. Med. 2013;11:149. doi: 10.1186/1479-5876-11-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Chiang CL, et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin. Cancer Res. 2013;19:4801–4815. doi: 10.1158/1078-0432.CCR-13-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Liu Y, et al. Engineered fusion hybrid vaccine of IL-4 gene-modified myeloma and relative mature dendritic cells enhances antitumor immunity. Leuk. Res. 2002;26:757–763. doi: 10.1016/S0145-2126(02)00002-4. [DOI] [PubMed] [Google Scholar]
- 371.Avigan D, et al. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin. Cancer Res. 2004;10:4699–4708. doi: 10.1158/1078-0432.CCR-04-0347. [DOI] [PubMed] [Google Scholar]
- 372.Rosenblatt J, et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood. 2011;117:393–402. doi: 10.1182/blood-2010-04-277137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Ding Z, et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct. Target Ther. 2021;6:26. doi: 10.1038/s41392-020-00448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019;25:1488–1499. doi: 10.1038/s41591-019-0596-y. [DOI] [PubMed] [Google Scholar]
- 375.Held W, Speiser DE. Not all tumor-infiltrating CD8(+) T cells are created equal. Cancer Cell. 2021;39:145–147. doi: 10.1016/j.ccell.2021.01.015. [DOI] [PubMed] [Google Scholar]
- 376.Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 2021;18:842–859. doi: 10.1038/s41423-020-00565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Zacharakis N, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018;24:724–730. doi: 10.1038/s41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Oliveira G, et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature. 2021;596:119–125. doi: 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Kumar A, Watkins R, Vilgelm AE. Cell therapy with TILs: training and taming T cells to fight cancer. Front Immunol. 2021;12:690499. doi: 10.3389/fimmu.2021.690499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Yamamoto TN, et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest. 2019;129:1551–1565. doi: 10.1172/JCI121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Zhao Y, et al. Tumor infiltrating lymphocyte (TIL) therapy for solid tumor treatment: progressions and challenges. Cancers. 2022;14:4160. doi: 10.3390/cancers14174160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Lu YC, et al. Single-cell transcriptome analysis reveals gene signatures associated with T-cell persistence following adoptive cell therapy. Cancer Immunol. Res. 2019;7:1824–1836. doi: 10.1158/2326-6066.CIR-19-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Parkhurst MR, et al. Unique neoantigens arise from somatic mutations in patients with Gastrointestinal Cancers. Cancer Discov. 2019;9:1022–1035. doi: 10.1158/2159-8290.CD-18-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Rohaan MW, Wilgenhof S, Haanen J. Adoptive cellular therapies: the current landscape. Virchows Arch. 2019;474:449–461. doi: 10.1007/s00428-018-2484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Kverneland AH, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes supported by checkpoint inhibition across multiple solid cancer types. J. Immunother. Cancer. 2021;9:e003499. doi: 10.1136/jitc-2021-003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Tran E, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Tran E, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Stevanovic S, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015;33:1543–1550. doi: 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Creelan BC, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med. 2021;27:1410–1418. doi: 10.1038/s41591-021-01462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Zhu Y, et al. Adoptive tumor infiltrating lymphocytes cell therapy for cervical cancer. Hum. Vaccin Immunother. 2022;18:2060019. doi: 10.1080/21645515.2022.2060019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Yin H, et al. TILs and anti-PD1 therapy: an alternative combination therapy for PDL1 negative metastatic Cervical Cancer. J. Immunol. Res. 2020;2020:8345235. doi: 10.1155/2020/8345235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Teng F, et al. Tumor infiltrating lymphocytes (TILs) before and after neoadjuvant chemoradiotherapy and its clinical utility for rectal cancer. Am. J. Cancer Res. 2015;5:2064–2074. [PMC free article] [PubMed] [Google Scholar]
- 393.Matsutani S, et al. Significance of tumor-infiltrating lymphocytes before and after neoadjuvant therapy for rectal cancer. Cancer Sci. 2018;109:966–979. doi: 10.1111/cas.13542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Schiza A, et al. Tumour-infiltrating lymphocytes add prognostic information for patients with low-risk DCIS: findings from the SweDCIS randomised radiotherapy trial. Eur. J. Cancer. 2022;168:128–137. doi: 10.1016/j.ejca.2022.01.016. [DOI] [PubMed] [Google Scholar]
- 395.Dudley ME, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Scheper W, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019;25:89–94. doi: 10.1038/s41591-018-0266-5. [DOI] [PubMed] [Google Scholar]
- 397.Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol. Rev. 2019;290:127–147. doi: 10.1111/imr.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Zacharakis N, et al. Breast cancers are immunogenic: immunologic analyses and a Phase II pilot clinical trial using mutation-reactive autologous lymphocytes. J. Clin. Oncol. 2022;40:1741–1754. doi: 10.1200/JCO.21.02170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Feola S, Chiaro J, Martins B, Cerullo V. Uncovering the tumor antigen landscape: what to know about the discovery process. Cancers. 2020;12:1660. doi: 10.3390/cancers12061660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Simoni Y, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 401.Holm JS, et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat. Commun. 2022;13:1935. doi: 10.1038/s41467-022-29342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Liu CJ, et al. Treatment of an aggressive orthotopic murine glioblastoma model with combination checkpoint blockade and a multivalent neoantigen vaccine. Neuro-Oncol. 2020;22:1276–1288. doi: 10.1093/neuonc/noaa050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Tsuji T, et al. Clonality and antigen-specific responses shape the prognostic effects of tumor-infiltrating T cells in ovarian cancer. Oncotarget. 2020;11:2669–2683. doi: 10.18632/oncotarget.27666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Kortekaas KE, et al. CD39 identifies the CD4(+) tumor-specific T-cell population in human cancer. Cancer Immunol. Res. 2020;8:1311–1321. doi: 10.1158/2326-6066.CIR-20-0270. [DOI] [PubMed] [Google Scholar]
- 405.Zou F, et al. The CD39(+) HBV surface protein-targeted CAR-T and personalized tumor-reactive CD8(+) T cells exhibit potent anti-HCC activity. Mol. Ther. 2021;29:1794–1807. doi: 10.1016/j.ymthe.2021.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Hanada KI, et al. A phenotypic signature that identifies neoantigen-reactive T cells in fresh human lung cancers. Cancer Cell. 2022;40:479–493.e476. doi: 10.1016/j.ccell.2022.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Wang Z, et al. Efficient recovery of potent tumour-infiltrating lymphocytes through quantitative immunomagnetic cell sorting. Nat. Biomed. Eng. 2022;6:108–117. doi: 10.1038/s41551-021-00820-y. [DOI] [PubMed] [Google Scholar]
- 408.Krishna S, et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science. 2020;370:1328–1334. doi: 10.1126/science.abb9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Miller BC, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Chapuis AG, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc. Natl Acad. Sci. USA. 2012;109:4592–4597. doi: 10.1073/pnas.1113748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Poorebrahim M, et al. Genetically modified immune cells targeting tumor antigens. Pharm. Ther. 2020;214:107603. doi: 10.1016/j.pharmthera.2020.107603. [DOI] [PubMed] [Google Scholar]
- 412.Arnesen VS, Gras Navarro A, Chekenya M. Challenges and prospects for designer T and NK cells in Glioblastoma immunotherapy. Cancers. 2021;13:4986. doi: 10.3390/cancers13194986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Khawar MB, Sun H. CAR-NK cells: from natural basis to design for kill. Front. Immunol. 2021;12:707542. doi: 10.3389/fimmu.2021.707542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Habif G, et al. Targeting natural killer cells in solid tumors. Cell Mol. Immunol. 2019;16:415–422. doi: 10.1038/s41423-019-0224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Chu J, et al. Natural killer cells: a promising immunotherapy for cancer. J. Transl. Med. 2022;20:240. doi: 10.1186/s12967-022-03437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Arnaud M, Bobisse S, Chiffelle J, Harari A. The promise of personalized TCR-based cellular immunotherapy for cancer patients. Front. Immunol. 2021;12:701636. doi: 10.3389/fimmu.2021.701636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Ye L, Creaney J, Redwood A, Robinson B. The current lung cancer neoantigen landscape and implications for therapy. J. Thorac. Oncol. 2021;16:922–932. doi: 10.1016/j.jtho.2021.01.1624. [DOI] [PubMed] [Google Scholar]
- 418.Poorebrahim M, et al. TCR-like CARs and TCR-CARs targeting neoepitopes: an emerging potential. Cancer Gene Ther. 2021;28:581–589. doi: 10.1038/s41417-021-00307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Matsuda T, et al. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin. Cancer Res. 2018;24:5357–5367. doi: 10.1158/1078-0432.CCR-18-0142. [DOI] [PubMed] [Google Scholar]
- 420.Wang QJ, et al. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 2016;4:204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.He J, et al. Defined tumor antigen-specific T cells potentiate personalized TCR-T cell therapy and prediction of immunotherapy response. Cell Res. 2022;32:530–542. doi: 10.1038/s41422-022-00627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Foy, S. P. et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature10.1038/s41586-022-05531-1 (2022). [DOI] [PMC free article] [PubMed]
- 423.Zhang J, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. 2022;609:369–374. doi: 10.1038/s41586-022-05140-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016;13:370–383. doi: 10.1038/nrclinonc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Schubert ML, et al. Chimeric antigen receptor transduced T cells: tuning up for the next generation. Int. J. Cancer. 2018;142:1738–1747. doi: 10.1002/ijc.31147. [DOI] [PubMed] [Google Scholar]
- 426.Xie G, et al. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat. Biomed. Eng. 2021;5:399–413. doi: 10.1038/s41551-020-00625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Shang S, et al. The role of neoantigens in tumor immunotherapy. Biomed. Pharmacother. 2022;151:113118. doi: 10.1016/j.biopha.2022.113118. [DOI] [PubMed] [Google Scholar]
- 428.Dunn GP, Sherpa N, Manyanga J, Johanns TM. Considerations for personalized neoantigen vaccination in Malignant glioma. Adv. Drug Deliv. Rev. 2022;186:114312. doi: 10.1016/j.addr.2022.114312. [DOI] [PubMed] [Google Scholar]
- 429.Johnson LA, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015;7:275ra222. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Villanueva MT. New CAR“s bells and whistles. Nat. Rev. Drug Discov. 2021;20:425. doi: 10.1038/d41573-021-00084-w. [DOI] [PubMed] [Google Scholar]
- 431.Chen LC, Hou AJ, Chen YY. Getting better mileage with logically primed CARs. Med. 2021;2:785–787. doi: 10.1016/j.medj.2021.06.002. [DOI] [PubMed] [Google Scholar]
- 432.Dong H, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc. Natl Acad. Sci. USA. 2022;119:e2122379119. doi: 10.1073/pnas.2122379119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat. Rev. Cancer. 2020;20:437–454. doi: 10.1038/s41568-020-0272-z. [DOI] [PubMed] [Google Scholar]
- 434.Boutros C, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016;13:473–486. doi: 10.1038/nrclinonc.2016.58. [DOI] [PubMed] [Google Scholar]
- 435.Huang AC, Zappasodi R. A decade of checkpoint blockade immunotherapy in melanoma: understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022;23:660–670. doi: 10.1038/s41590-022-01141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Upadhaya S, Neftelinov ST, Hodge J, Campbell J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat. Rev. Drug Discov. 2022;21:482–483. doi: 10.1038/d41573-022-00030-4. [DOI] [PubMed] [Google Scholar]
- 437.Marable J, et al. Nanobody-based CTLA4 inhibitors for immune checkpoint blockade therapy of canine cancer patients. Sci. Rep. 2021;11:20763. doi: 10.1038/s41598-021-00325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front. Oncol. 2018;8:86. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Qin S, et al. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol. Cancer. 2019;18:155. doi: 10.1186/s12943-019-1091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Hsiue EH, et al. Targeting a neoantigen derived from a common TP53 mutation. Science. 2021;371:eabc8697. doi: 10.1126/science.abc8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Skora AD, et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc. Natl Acad. Sci. USA. 2015;112:9967–9972. doi: 10.1073/pnas.1511996112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Chao G, et al. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 443.Gejman RS, et al. Identification of the targets of T-cell receptor therapeutic agents and cells by use of a high-throughput genetic platform. Cancer Immunol. Res. 2020;8:672–684. doi: 10.1158/2326-6066.CIR-19-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Ataie N, et al. Structure of a TCR-mimic antibody with target predicts pharmacogenetics. J. Mol. Biol. 2016;428:194–205. doi: 10.1016/j.jmb.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Editorial. PIK3CA hotspot mutation generates a shared neoantigen targetable by TCR gene therapy. Nat. Med. 28, 907–908, (2022). [DOI] [PubMed]
- 446.Chandran SS, et al. Immunogenicity and therapeutic targeting of a public neoantigen derived from mutated PIK3CA. Nat. Med. 2022;28:946–957. doi: 10.1038/s41591-022-01786-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Miller MS, et al. An engineered antibody fragment targeting mutant beta-catenin via major histocompatibility complex I neoantigen presentation. J. Biol. Chem. 2019;294:19322–19334. doi: 10.1074/jbc.RA119.010251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Umano Y, et al. Generation of cytotoxic T cell responses to an HLA-A24 restricted epitope peptide derived from wild-type p53. Br. J. Cancer. 2001;84:1052–1057. doi: 10.1054/bjoc.2000.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Barfoed AM, et al. Cytotoxic T-lymphocyte clones, established by stimulation with the HLA-A2 binding p5365-73 wild type peptide loaded on dendritic cells in vitro, specifically recognize and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand. J. Immunol. 2000;51:128–133. doi: 10.1046/j.1365-3083.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- 450.Eura M, et al. A wild-type sequence p53 peptide presented by HLA-A24 induces cytotoxic T lymphocytes that recognize squamous cell carcinomas of the head and neck. Clin. Cancer Res. 2000;6:979–986. [PubMed] [Google Scholar]
- 451.Low L, et al. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat. Commun. 2019;10:5382. doi: 10.1038/s41467-019-13305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Douglass J, et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021;6:eabd5515. doi: 10.1126/sciimmunol.abd5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Ahmed M, et al. TCR-mimic bispecific antibodies targeting LMP2A show potent activity against EBV malignancies. JCI Insight. 2018;3:e97805. doi: 10.1172/jci.insight.97805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Liddy N, et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012;18:980–987. doi: 10.1038/nm.2764. [DOI] [PubMed] [Google Scholar]
- 455.Lowe KL, et al. Novel TCR-based biologics: mobilising T cells to warm 'cold' tumours. Cancer Treat. Rev. 2019;77:35–43. doi: 10.1016/j.ctrv.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 456.Dao T, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015;33:1079–1086. doi: 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Yang C, Lou G, Jin WL. The arsenal of TP53 mutants therapies: neoantigens and bispecific antibodies. Signal Transduct. Target Ther. 2021;6:219. doi: 10.1038/s41392-021-00635-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Lum LG, Tushir-Singh J. Arming "old guards" with "new dual-targeting weapons". Cancer Cell. 2021;39:604–606. doi: 10.1016/j.ccell.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.O'Leary K. Bispecifics target cancers’ most wanted. Nat. Rev. Cancer. 2021;21:279. doi: 10.1038/s41568-021-00354-0. [DOI] [PubMed] [Google Scholar]
- 460.Sharma P, Harris DT, Stone JD, Kranz DM. T-cell receptors engineered de novo for peptide specificity can mediate optimal T-cell activity without self cross-reactivity. Cancer Immunol. Res. 2019;7:2025–2035. doi: 10.1158/2326-6066.CIR-19-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 462.Kinkead HL, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight. 2018;3:e122857. doi: 10.1172/jci.insight.122857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Caushi JX, et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. 2021;596:126–132. doi: 10.1038/s41586-021-03752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Kamphorst AO, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl Acad. Sci. USA. 2017;114:4993–4998. doi: 10.1073/pnas.1705327114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Lopez de Rodas M, Schalper KA. Tumour antigen-induced T cell exhaustion-the archenemy of immune-hot malignancies. Nat. Rev. Clin. Oncol. 2021;18:749–750. doi: 10.1038/s41571-021-00562-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Duhen T, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018;9:2724. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Gros A, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016;22:433–438. doi: 10.1038/nm.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Thommen DS, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018;24:994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Ma L, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–168. doi: 10.1126/science.aav8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Singh N, June CH. Boosting engineered T cells. Science. 2019;365:119–120. doi: 10.1126/science.aax6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Dart A. Steering CARs in the right direction. Nat. Rev. Cancer. 2019;19:487. doi: 10.1038/s41568-019-0189-6. [DOI] [PubMed] [Google Scholar]
- 472.McLaughlin M, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer. 2020;20:203–217. doi: 10.1038/s41568-020-0246-1. [DOI] [PubMed] [Google Scholar]
- 473.Formenti SC, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018;24:1845–1851. doi: 10.1038/s41591-018-0232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Garnett CT, et al. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64:7985–7994. doi: 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 475.Sharma A, et al. gamma-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS ONE. 2011;6:e28217. doi: 10.1371/journal.pone.0028217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Lhuillier C, et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Invest. 2021;131:e138740. doi: 10.1172/JCI138740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Pettitt SJ, et al. Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. 2020;10:1475–1488. doi: 10.1158/2159-8290.CD-19-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Kristensen VN. The antigenicity of the tumor cell - context matters. N. Engl. J. Med. 2017;376:491–493. doi: 10.1056/NEJMcibr1613793. [DOI] [PubMed] [Google Scholar]
- 479.Cieri N, Wu CJ. Splice it up: atypical transcripts to boost leukemia immunotherapy. Immunity. 2021;54:608–610. doi: 10.1016/j.immuni.2021.03.016. [DOI] [PubMed] [Google Scholar]
- 480.Ehx G, et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity. 2021;54:737–752.e710. doi: 10.1016/j.immuni.2021.03.001. [DOI] [PubMed] [Google Scholar]
- 481.Kanaseki T, Torigoe T. Proteogenomics: advances in cancer antigen research. Immunol. Med. 2019;42:65–70. doi: 10.1080/25785826.2019.1640500. [DOI] [PubMed] [Google Scholar]
- 482.Rivero-Hinojosa S, et al. Proteogenomic discovery of neoantigens facilitates personalized multi-antigen targeted T cell immunotherapy for brain tumors. Nat. Commun. 2021;12:6689. doi: 10.1038/s41467-021-26936-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Benvenuto M, et al. Tumor antigens heterogeneity and immune response-targeting neoantigens in breast cancer. Semin Cancer Biol. 2021;72:65–75. doi: 10.1016/j.semcancer.2019.10.023. [DOI] [PubMed] [Google Scholar]
- 484.Nanda R, et al. Pembrolizumab in patients with advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 2016;34:2460–2467. doi: 10.1200/JCO.2015.64.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Nolan E, et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017;9:eaal4922. doi: 10.1126/scitranslmed.aal4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Rosenthal R, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–485. doi: 10.1038/s41586-019-1032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer. 2021;21:298–312. doi: 10.1038/s41568-021-00339-z. [DOI] [PubMed] [Google Scholar]
- 488.Jaeger AM, et al. Rebalancing protein homeostasis enhances tumor antigen presentation. Clin. Cancer Res. 2019;25:6392–6405. doi: 10.1158/1078-0432.CCR-19-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Anagnostou V, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7:264–276. doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Lee MY, Jeon JW, Sievers C, Allen CT. Antigen processing and presentation in cancer immunotherapy. J. Immunother. Cancer. 2020;8:e001111. doi: 10.1136/jitc-2020-001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Zhang H, et al. Progress and challenges of personalized neoantigens in the clinical treatment of tumors. Med. Drug Discov. 2020;6:7. doi: 10.1016/j.medidd.2020.100030. [DOI] [Google Scholar]
- 492.Thibodeau J, Bourgeois-Daigneault MC, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology. 2012;1:908–916. doi: 10.4161/onci.21205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Yamamoto K, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581:100–105. doi: 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Zheng S, Asnani M, Thomas-Tikhonenko A. Escape from ALL-CARTaz: Leukemia immunoediting in the age of chimeric antigen receptors. Cancer J. 2019;25:217–222. doi: 10.1097/PPO.0000000000000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Lu SX, et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell. 2021;184:4032–4047 e4031. doi: 10.1016/j.cell.2021.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Chiozzini C, et al. Tumor cells endowed with professional antigen-presenting cell functions prime PBLs to generate antitumor CTLs. J. Mol. Med. 2019;97:1139–1153. doi: 10.1007/s00109-019-01797-7. [DOI] [PubMed] [Google Scholar]
- 497.Labarriere N, et al. PBMC are as good a source of tumor-reactive T lymphocytes as TIL after selection by Melan-A/A2 multimer immunomagnetic sorting. Cancer Immunol. Immunother. 2008;57:185–195. doi: 10.1007/s00262-007-0361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Li B, et al. Phase Ia clinical trial of adoptively transferring peripheral blood-derived cytotoxic T lymphocytes targeting individual neo-antigens to treat patients with advanced solid tumor. J. Clin. Oncol. 2019;37:1. [Google Scholar]
- 499.Tran E. Neoantigen-specific T cells in adoptive cell therapy. Cancer J. 2022;28:278–284. doi: 10.1097/PPO.0000000000000605. [DOI] [PubMed] [Google Scholar]
- 500.B., L. & S., Q. A phase Ia study of a personalized TSA-CTL (tumor specific antigen-induced cytotoxic T lymphocytes) therapy in metastatic Melanoma. Ann. Oncol. 29, 1, (2018).
- 501.Gattinoni L, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Rosenberg SA, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med. 2016;8:320ra323. doi: 10.1126/scitranslmed.aad5222. [DOI] [PubMed] [Google Scholar]
- 504.Maeda T, et al. Regeneration of CD8alphabeta T cells from T-cell-derived iPSC imparts potent tumor antigen-specific cytotoxicity. Cancer Res. 2016;76:6839–6850. doi: 10.1158/0008-5472.CAN-16-1149. [DOI] [PubMed] [Google Scholar]
- 505.Ishii M, et al. iPSC-derived neoantigen-specific CTL therapy for Ewing Sarcoma. Cancer Immunol. Res. 2021;9:1175–1186. doi: 10.1158/2326-6066.CIR-21-0193. [DOI] [PubMed] [Google Scholar]
- 506.Vizcardo R, et al. Generation of tumor antigen-specific iPSC-derived thymic emigrants using a 3D thymic culture system. Cell Rep. 2018;22:3175–3190. doi: 10.1016/j.celrep.2018.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Jaigirdar A, Rosenberg SA, Parkhurst M. A High-avidity WT1-reactive T-cell receptor mediates recognition of peptide and processed antigen but not naturally occurring WT1-positive tumor cells. J. Immunother. 2016;39:105–116. doi: 10.1097/CJI.0000000000000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Malekzadeh P, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest. 2019;129:1109–1114. doi: 10.1172/JCI123791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Rijensky NM, et al. Identification of tumor antigens in the HLA peptidome of patient-derived xenograft tumors in mouse. Mol. Cell Proteom. 2020;19:1360–1374. doi: 10.1074/mcp.RA119.001876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Dijkstra KK, et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174:1586–1598.e1512. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Hahne M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 512.Motz GT, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014;20:607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Rossin A, Miloro G, Hueber AO. TRAIL and FasL functions in cancer and autoimmune diseases: towards an increasing complexity. Cancers. 2019;11:639. doi: 10.3390/cancers11050639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Kohrt HE, et al. Immunodynamics: a cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials. J. Immunother. Cancer. 2016;4:15. doi: 10.1186/s40425-016-0118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Robins HS, et al. Digital genomic quantification of tumor-infiltrating lymphocytes. Sci. Transl. Med. 2013;5:214ra169. doi: 10.1126/scitranslmed.3007247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Jia Q, et al. Tracking neoantigens by personalized circulating tumor DNA sequencing during checkpoint blockade immunotherapy in Non-Small Cell Lung Cancer. Adv. Sci. 2020;7:1903410. doi: 10.1002/advs.201903410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Gros A, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Di Giacomo AM, et al. Ipilimumab and fotemustine in patients with advanced melanoma (NIBIT-M1): an open-label, single-arm phase 2 trial. Lancet Oncol. 2012;13:8. doi: 10.1016/S1470-2045(12)70324-8. [DOI] [PubMed] [Google Scholar]
- 520.Lowery FJ, et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science. 2022;375:877–884. doi: 10.1126/science.abl5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Sidhom JW, et al. Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy. Sci. Adv. 2022;8:eabq5089. doi: 10.1126/sciadv.abq5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Peng S, et al. Sensitive detection and analysis of neoantigen-specific T cell populations from tumors and blood. Cell Rep. 2019;28:2728–2738.e2727. doi: 10.1016/j.celrep.2019.07.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Klebanoff CA, Wolchok JD. Shared cancer neoantigens: making private matters public. J. Exp. Med. 2018;215:5–7. doi: 10.1084/jem.20172188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Kim SP, et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol. Res. 2022;10:932–946. doi: 10.1158/2326-6066.CIR-22-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Biankin AV, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Poulain S, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121:4504–4511. doi: 10.1182/blood-2012-06-436329. [DOI] [PubMed] [Google Scholar]
- 527.Long GV, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011;29:1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 528.Kim JK, et al. Intratumoral T-cell repertoires in DNA mismatch repair-proficient and -deficient colon tumors containing high or low numbers of tumor-infiltrating lymphocytes. Oncoimmunology. 2022;11:2054757. doi: 10.1080/2162402X.2022.2054757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Klebanoff CA. T-cell receptor gene therapy clinically targeting a TP53 public neoantigen. Cancer Immunol. Res. 2022;10:919. doi: 10.1158/2326-6066.CIR-22-0386. [DOI] [PubMed] [Google Scholar]
- 530.Inderberg EM, et al. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. 2017;6:e1302631. doi: 10.1080/2162402X.2017.1302631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Palmer CD, et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat. Med. 2022;28:1619–1629. doi: 10.1038/s41591-022-01937-6. [DOI] [PubMed] [Google Scholar]
- 532.Gebre MS, et al. Novel approaches for vaccine development. Cell. 2021;184:1589–1603. doi: 10.1016/j.cell.2021.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021;20:817–838. doi: 10.1038/s41573-021-00283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Kon E, Elia U, Peer D. Principles for designing an optimal mRNA lipid nanoparticle vaccine. Curr. Opin. Biotechnol. 2022;73:329–336. doi: 10.1016/j.copbio.2021.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Ward BJ, et al. Phase 1 randomized trial of a plant-derived virus-like particle vaccine for COVID-19. Nat. Med. 2021;27:1071–1078. doi: 10.1038/s41591-021-01370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Wang Y, et al. mRNA vaccine: a potential therapeutic strategy. Mol. Cancer. 2021;20:33. doi: 10.1186/s12943-021-01311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Zhao X, Zhao R, Nie G. Nanocarriers based on bacterial membrane materials for cancer vaccine delivery. Nat. Protoc. 2022;17:2240–2274. doi: 10.1038/s41596-022-00713-7. [DOI] [PubMed] [Google Scholar]
- 538.Wang Z, et al. Exosomes decorated with a recombinant SARS-CoV-2 receptor-binding domain as an inhalable COVID-19 vaccine. Nat. Biomed. Eng. 2022;6:791–805. doi: 10.1038/s41551-022-00902-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Li Y, et al. Rapid surface display of mRNA antigens by bacteria-derived outer membrane vesicles for a personalized tumor vaccine. Adv. Mater. 2022;34:e2109984. doi: 10.1002/adma.202109984. [DOI] [PubMed] [Google Scholar]
- 540.Mestrallet G, Sone K, Bhardwaj N. Strategies to overcome DC dysregulation in the tumor microenvironment. Front. Immunol. 2022;13:980709. doi: 10.3389/fimmu.2022.980709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Ma J, et al. Mechanisms by which dendritic cells present tumor microparticle antigens to CD8(+) T cells. Cancer Immunol. Res. 2018;6:1057–1068. doi: 10.1158/2326-6066.CIR-17-0716. [DOI] [PubMed] [Google Scholar]
- 542.Zhang H, et al. Cell-free tumor microparticle vaccines stimulate dendritic cells via cGAS/STING signaling. Cancer Immunol. Res. 2015;3:196–205. doi: 10.1158/2326-6066.CIR-14-0177. [DOI] [PubMed] [Google Scholar]
- 543.Ma J, Zhang H, Tang K, Huang B. Tumor-derived microparticles in tumor immunology and immunotherapy. Eur. J. Immunol. 2020;50:1653–1662. doi: 10.1002/eji.202048548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Mehanny M, Lehr CM, Fuhrmann G. Extracellular vesicles as antigen carriers for novel vaccination avenues. Adv. Drug Deliv. Rev. 2021;173:164–180. doi: 10.1016/j.addr.2021.03.016. [DOI] [PubMed] [Google Scholar]
- 545.Xiong X, et al. Neoantigen-based cancer vaccination using chimeric RNA-loaded dendritic cell-derived extracellular vesicles. J. Extracell. Vesicles. 2022;11:e12243. doi: 10.1002/jev2.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Spadaro F, et al. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood. 2012;119:1407–1417. doi: 10.1182/blood-2011-06-363564. [DOI] [PubMed] [Google Scholar]
- 547.van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;119:3383–3393. doi: 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- 548.Helft J, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. 2015;42:1197–1211. doi: 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 549.Li A, et al. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 2019;12:35. doi: 10.1186/s13045-019-0721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Ahn J, et al. Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer Cell. 2018;33:862–873.e865. doi: 10.1016/j.ccell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Yi M, et al. Combination of oral STING agonist MSA-2 and anti-TGF-β/PD-L1 bispecific antibody YM101: a novel immune cocktail therapy for non-inflamed tumors. J. Hematol. Oncol. 2022;15:142. doi: 10.1186/s13045-022-01363-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.de Goër de Herve MG, et al. Differential effect of agonistic anti-CD40 on human mature and immature dendritic cells: the Janus face of anti-CD40. Blood. 2005;106:2806–2814. doi: 10.1182/blood-2004-12-4678. [DOI] [PubMed] [Google Scholar]
- 553.Katakam AK, et al. Dendritic cells require NIK for CD40-dependent cross-priming of CD8+ T cells. Proc. Natl Acad. Sci. USA. 2015;112:14664–14669. doi: 10.1073/pnas.1520627112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi: 10.1158/2159-8290.CD-13-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Xu N, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2021;218:e20200844. doi: 10.1084/jem.20200844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Yi M, et al. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol. Cancer. 2022;21:28. doi: 10.1186/s12943-021-01489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Prendergast GC, et al. Inflammatory reprogramming with IDO1 inhibitors: turning immunologically unresponsive 'Cold' tumors 'Hot'. Trends Cancer. 2018;4:38–58. doi: 10.1016/j.trecan.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Martin JD, Cabral H, Stylianopoulos T, Jain RK. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020;17:251–266. doi: 10.1038/s41571-019-0308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Yong, T., Wei, Z., Gan, L. & Yang, X. Extracellular-vesicle-based drug delivery systems for enhanced antitumor therapies through modulating the cancer-immunity cycle. Adv. Mater. e2201054, (2022). [DOI] [PubMed]
- 560.Li Q, et al. Symphony of nanomaterials and immunotherapy based on the cancer-immunity cycle. Acta Pharmaceutica Sin. B. 2022;12:107–134. doi: 10.1016/j.apsb.2021.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Zhang J, Mardis ER, Maher CA. INTEGRATE-neo: a pipeline for personalized gene fusion neoantigen discovery. Bioinformatics. 2017;33:555–557. doi: 10.1093/bioinformatics/btw674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Hundal J, et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 2020;8:409–420. doi: 10.1158/2326-6066.CIR-19-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Rubinsteyn A, et al. Computational pipeline for the PGV-001 neoantigen vaccine trial. Front. Immunol. 2017;8:1807. doi: 10.3389/fimmu.2017.01807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Rech AJ, et al. Tumor immunity and survival as a function of alternative neopeptides in human cancer. Cancer Immunol. Res. 2018;6:276–287. doi: 10.1158/2326-6066.CIR-17-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Richman LP, Vonderheide RH, Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019;9:375–382.e374. doi: 10.1016/j.cels.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Cygan KJ, Sanford CH, Fairbrother WG. Spliceman2: a computational web server that predicts defects in pre-mRNA splicing. Bioinformatics. 2017;33:2943–2945. doi: 10.1093/bioinformatics/btx343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Mort M, et al. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol. 2014;15:R19. doi: 10.1186/gb-2014-15-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Ioannidis NM, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Shen S, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl Acad. Sci. USA. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Kim S, et al. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018;29:1030–1036. doi: 10.1093/annonc/mdy022. [DOI] [PubMed] [Google Scholar]
- 572.Bjerregaard AM, et al. MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017;66:1123–1130. doi: 10.1007/s00262-017-2001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Bais P, et al. CloudNeo: a cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics. 2017;33:3110–3112. doi: 10.1093/bioinformatics/btx375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 575.Zhou Z, et al. TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection. R. Soc. Open Sci. 2017;4:170050. doi: 10.1098/rsos.170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Kim TH, Jeon YJ, Kim WY, Kim HS. HESAS: HERVs expression and structure analysis system. Bioinformatics. 2005;21:1699–1700. doi: 10.1093/bioinformatics/bti194. [DOI] [PubMed] [Google Scholar]
- 577.Tongyoo P, et al. EnHERV: enrichment analysis of specific human endogenous retrovirus patterns and their neighboring genes. PLoS ONE. 2017;12:e0177119. doi: 10.1371/journal.pone.0177119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Smith CC, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Invest. 2018;128:4804–4820. doi: 10.1172/JCI121476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Huang Y, et al. HLAreporter: a tool for HLA typing from next generation sequencing data. Genome Med. 2015;7:25. doi: 10.1186/s13073-015-0145-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Bai Y, Wang D, Fury W. PHLAT: Inference of high-resolution HLA types from RNA and whole exome sequencing. Methods Mol. Biol. 2018;1802:193–201. doi: 10.1007/978-1-4939-8546-3_13. [DOI] [PubMed] [Google Scholar]
- 581.Ka S, et al. HLAscan: genotyping of the HLA region using next-generation sequencing data. BMC Bioinforma. 2017;18:258. doi: 10.1186/s12859-017-1671-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Racle J, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019;37:1283–1286. doi: 10.1038/s41587-019-0289-6. [DOI] [PubMed] [Google Scholar]
- 583.Abelin JG, et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51:766–779.e717. doi: 10.1016/j.immuni.2019.08.012. [DOI] [PubMed] [Google Scholar]