

Abstract
The molecular programs involved in regulatory T (Treg) cell activation and homeostasis remain incompletely understood. Here we show that T cell receptor (TCR) signaling in Treg cells induces the nuclear translocation of serine/threonine kinase 4 (Stk4), leading to the formation of a Stk4/NF-κB p65/Foxp3 complex that regulates Foxp3 and p65-dependent transcriptional programs. This complex was stabilized by Stk4-dependent phosphorylation of Foxp3 serine 418. Stk4 deficiency in Treg cells, either alone or in combination with its homologue Stk3, precipitated a fatal autoimmune lymphoproliferative disease in mice characterized by decreased Treg cell p65 expression and nuclear translocation, impaired NF-κB p65/Foxp3 complex formation, and defective Treg cell activation. In an adoptive immunotherapy model, over-expression of p65 or the phosphomimetic Foxp3S418E in Stk3/4-deficient Treg cells ameliorated their immune regulatory defects. Our studies identify Stk4 as an essential TCR-responsive regulator of p65-Foxp3-dependent transcription that promotes Treg cell-mediated immune tolerance.
One Sentence Summary:
Stk4 combines with NF-κB p65 and Foxp3 in a TCR-regulated trimolecular complex to mediate Treg cell activation and homeostasis.
Introduction
Regulatory T cells (Treg) enforce peripheral tolerance and prevent untoward autoimmunity and immune dysregulation (1). They also promote tissue homeostasis and repair (2, 3). A number of monogenic defects that affect Treg cell development and function, such as mutations in the Treg cell transcription factor Foxp3, result in human disorders of autoimmunity and immune dysregulation (4–7). In that regard, a disorder of immune deficiency and immune dysregulation has been described in humans related to mutations in the STK4 (8), encoding the serine/threonine kinase 4. Stk4 and its homologue Stk3 have been defined as regulators of the canonical Hippo signaling pathway, which controls organ size and cell number through modulation of cell proliferation and apoptosis (9, 10). Stk3/4 phosphorylates the Hippo pathway kinase intermediates large tumor suppressor kinase 1/2 (LATS1/2)(11, 12), which in turn control access of the Hippo pathway regulators Yes-associated protein (YAP) and transcriptional coactivator with PDZ binding motif (TAZ) to the nucleus (13). In the nucleus, YAP/TAZ cooperate with the TEAD transcription factors to regulate gene transcription and cellular proliferation (14, 15).
Patients with global STK4 deficiency suffer from recurrent infections, immune dysregulation, and autoimmunity (8, 16–18). Studies in mice have revealed different roles for Stk3/4 in T cell and dendritic cell responses (19, 20). Stk3/4 deletion in mouse Treg cells has been shown to induce defective Treg cell function resulting in a multi-organ autoimmune inflammatory syndrome, which was related to suboptimal IL-2-STAT5 activation (21). However, the molecular mechanisms by which Stk3/4 regulate the different functions of Treg cells, including IL-2-STAT5 signaling, remain obscure.
In this study, we demonstrate that Stk4 is coupled to the T cell receptor and translocates to the nucleus upon T cell receptor activation. Stk4 associates in a tri-molecular complex with Foxp3 and the NF-kB subunit p65 to regulate the respective Treg cell transcriptional programs in a phosphorylation-dependent manner. This complex was stabilized by Stk4-mediated phosphorylation of Foxp3 on Serine 418. Stk3/4 deletion impaired Treg cell transcriptional programs by destabilizing NF-kB complexes in the cytosol and limited their availability in the nucleus, leading to defective Treg cell activation and tissue surveillance. Over-expression of either p65 or phosphomimetic Foxp3E418 ameliorated the immune regulatory deficit of Stk3/4-deficient Treg cells. Treg cells of patients with STK4 deficiency manifested many of the same defects observed in the mouse system, thus confirming the cardinal role of STK4 in human and mouse Treg cell biology. Our results establish Stk3/4 as a TCR-regulated molecular sensor that couples cellular activation with gene transcription in Treg cells to promote their stability and function.
Results
Essential role of Stk4 in Treg cell homeostasis and function.
To elucidate the role of the Stk3/4 in peripheral tolerance, we crossed mice expressing a Cre recombinase fused with the enhanced yellow fluorescent protein under the control of the Foxp3 locus together with loxP-flanked Stk3 and Stk4 either alone or in combination (Foxp3YFPCreStk3∆/∆, Foxp3YFPCreStk4∆/∆ and Foxp3YFPCreStk3∆/∆Stk4∆/∆). Consistent with a previous report, Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice exhibited a rapidly fatal autoimmune lymphoproliferative disease with stunted growth, similar to the phenotype of Foxp3-deficient mice (Fig. S1A and B) (22). Foxp3YFPCreStk3∆/∆ were unaffected by disease, reflected by their normal survival. Foxp3YFPCreStk4∆/∆ mice developed a progressive autoimmune lymphoproliferative disease that was more indolent than that observed with Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice. The difference between Foxp3YFPCreStk3∆/∆ versus Foxp3YFPCreStk4∆/∆ in terms of susceptibility to disease might have reflected the relative abundance of transcripts for the respective kinase in Treg cells. Thus, Stk4 transcripts were expressed up to 13-fold higher in Treg cells compared to those of Stk3 (Fig. S1C). Treg cells of Foxp3YFPCreStk3∆/∆ mice had further upregulation of Stk4 transcripts, which could potentially compensate for the loss of Stk3 transcripts (Fig. S1D). Analysis of Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice at 4 weeks of age showed severe inflammation with decreased Treg cell frequencies in the face of an expanded T cell compartment, increased CD4+CD62LloCD44hi T effector memory cell frequencies and numbers, dysregulated IFNγ expression and hyper-immunoglobulinemia compared to Foxp3YFPCre, Foxp3YFPCreStk3∆/∆ and Foxp3YFPCreStk4∆/∆ mice (Fig. S1E to I). While Foxp3YFPCreStk4∆/∆ mice appeared normal at 4 weeks of age, they evolved similar disease characteristics as they grew older by 3 months of age (Fig. S1J and K).
Analysis of Foxp3YFPCreStk3∆/∆Stk4∆/∆ male mice revealed decreased Treg cells frequencies in the spleen and organ tissues, including the lung and liver. The Treg cells exhibited decreased expression of some markers, including CD25 and CD73, and evidence of increased turnover of Treg cells with heightened apoptosis and proliferation, as measured by staining with AnnexinV and Ki-67, respectively (Fig. S2A to C).
To segregate the Treg cell-intrinsic defects associated with Stk3/4 deficiency from those secondary to the intense autoimmune inflammation of Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice, we analyzed the Treg cells of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ females at 4 weeks of age. These mice are otherwise healthy due to a portion of their Treg cells failing to undergo Stk3 and Stk4 deletion by virtue of random X-chromosome inactivation that silences the heterozygous Foxp3YFPCre allele. Analysis of YFP+CD4+ Treg cells from the spleens, livers and lungs of Foxp3YFPCre/+ and Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ heterozygous females revealed profound depletion of Stk3/4 deficient Treg cells in the latter mice, indicating an obligate role for Stk3/Stk4 in Treg cell fitness under non-inflammatory conditions (Fig. 1A and B). These changes took place in the absence of an overt immune dysregulation in the mice, as reflected by the normal expression of IFNγ and IL-17 in their Teff cells (Fig. 1C and D). Importantly, the Stk3/4-deficient Treg cells of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ females were mostly naïve (CD62L+CD44−) with low expression of the early T cell activation marker CD69 (Fig. 1E and F). Stk3/4-deficient Treg cells of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ were more apoptotic and less proliferative compared to Stk3/4-sufficient Treg cells of Foxp3YFPCre/+ mice (Fig. 1G and H). They also had decreased expression of several Treg cell markers including Foxp3, CD25 and CD73 (Fig. 1I). Overall, these results indicated that Stk3/4 deficiency resulted in a Treg cell-intrinsic activation defect that limited their expansion and promoted their apoptosis.
Figure 1. STK3/4 deficiency leads to activation defects of Treg cells.
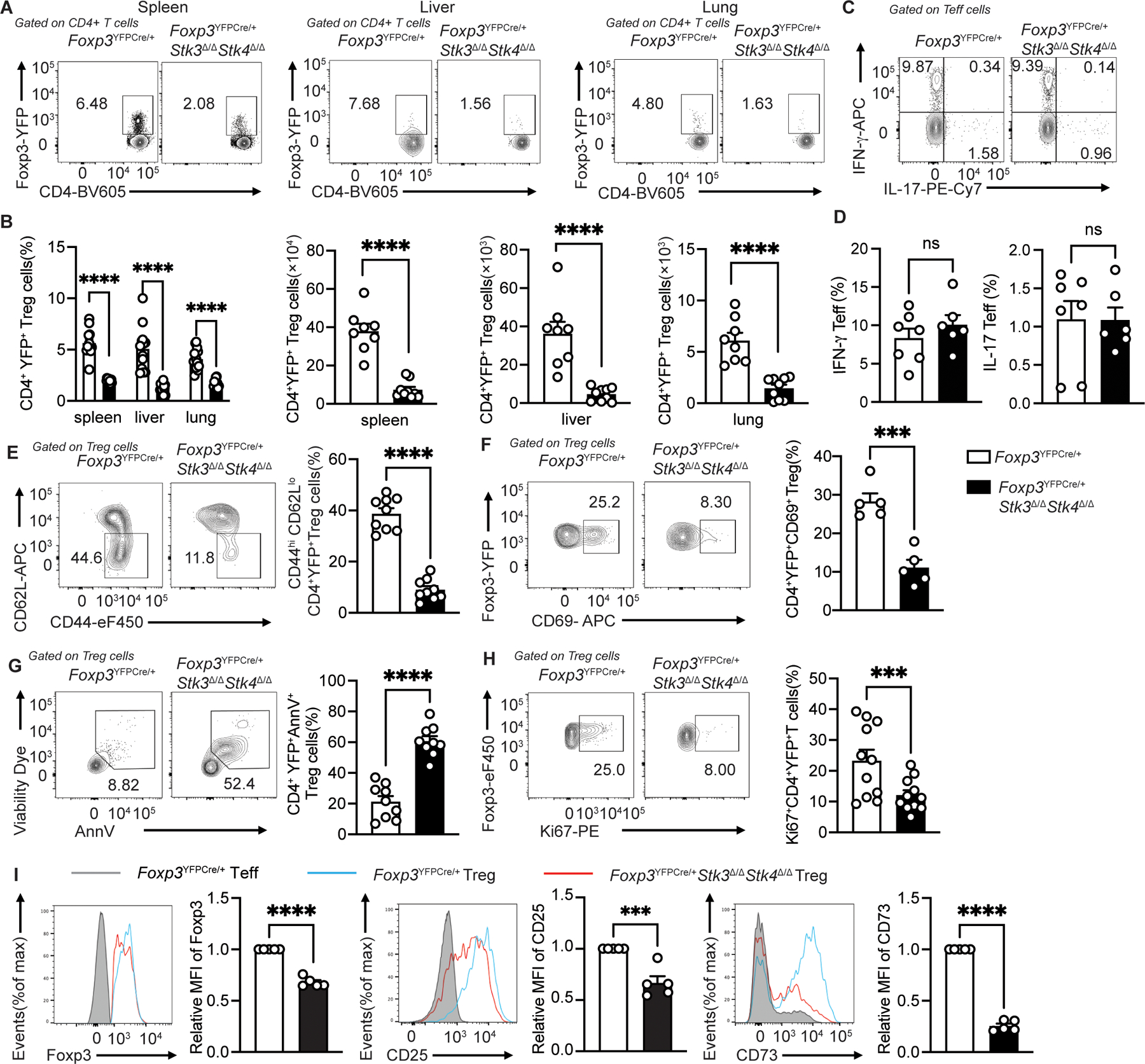
(A) Flow cytometric analysis of CD4+ Foxp3+Treg cells from spleen, liver and lung of Foxp3YFPCre/+ and Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ mice and (B) quantification of frequencies (n=10–13 for each group) and numbers (n=8 for Foxp3YFPCre/+ group; n=9 for Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ group) (scatter plots with means ± S.E.M.) of each contour plot in (A). (C and D) Representative flow cytometric analysis (C) and frequencies (D, scatter plots with means ± S.E.M.) of IFNγ+ and IL-17+ CD4+Foxp3−Teff cells (n=6–7 per group). The results represent one of three independent experiments. (E) Representative flow cytometric analysis and frequencies (scatter plots with means ± S.E.M.) of CD62LloCD44hi CD4+YFP+Treg cells (n=9 per group). The results represent pool of three independent experiments. (F) Representative flow cytometric analysis and frequencies (scatter plots with means ± S.E.M.) of CD4+YFP+CD69+ Treg cells (n=5 per group). (G and H) AnnexinV+ Viability dye+ splenic Treg cells (G) (n=9 for each group) and Ki67+ Treg cells (G) (n=11 for each group). (I) Flow cytometric analysis and scatter plot representation of relative Foxp3 mean fluorescence intensity (MFI), relative CD25 MFI and relative CD73 MFI of CD4+YFP+ Treg cells from spleen of Foxp3YFPCre/+ and Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ mice (n=5 per group). Each point represents one mouse. Error bars indicate the standard error of the means (s.e.m). The results are representative of three independent experiments. Statistical tests: ***, P<0.005, ****, P<0.0001 by two-way ANOVA with Sidak’s multiple comparisons test (B) or Student’s unpaired two tailed t test (B to I).
While Stk3/4 have been implicated in negatively regulating the Hippo pathway, Treg cell-specific deletion of Yap1 and Wwtr1, encoding the hippo pathway effectors Yap and Taz, failed to prevent the development of lethal autoimmunity in Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice (Fig. S3A to E). Reciprocally, Treg cell-specific deletion of Lats1 and Lats2 (Foxp3YFPCreLats1∆/∆Lats2∆/∆), encoding kinases that phosphorylate YAP/TAZ to mediate their sequestration and degradation, failed to induce disease (Fig. S4A to D). The Treg cells of those mice were otherwise phenotypically similar to those of Foxp3YFPCre mice.
To further elucidate the mechanisms involved in TCR-induced Stk3/4 activation, we examined the capacity of inhibitors of signaling intermediates downstream of the TCR to suppress activation-induced Stk3/4 phosphorylation. Inhibitors of the TCR/CD3-associated Zeta-chain-associated protein kinase 70 (Zap70), phosphatidylinositol 3-kinase (PI3-K) and the mucosa-associated lymphoid tissue lymphoma translocation protein 1 (Malt1) all abolished the increase in Stk3/4 phosphorylation post anti-CD3/CD28 mAb stimulation of Treg cells (Figure S5A and B). Furthermore, Stk3/4 deficiency severely impaired anti-CD3 activated Treg cell suppressive function (Fig. S5C). Thus, Stk3/4 are activated in a TCR signaling-dependent to promote Treg cell homeostasis and regulatory function independent of the classical Hippo pathway.
Stk4 regulates Treg cell TCR, Foxp3 and NF-κB p65-dependent transcriptional programs.
To determine the transcriptional regulatory functions of Stk3/4, we analyzed the transcriptomes of splenic YFP+ Treg cells of Foxp3YFPCre versus Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ female mice. The resulting lack of autoimmune inflammation in these mice allows the distinction of cell intrinsic transcriptomic changes induced by Stk3/4 deficiency in YFP+ Treg cells versus extrinsic changes induced by the inflammatory response. Results showed that there was a broad impact of Stk3/4 deficiency on the Treg cell transcriptome that differentially affected key transcriptional modules regulated by TCR, Foxp3 and NF-κB p65 (Fig. 2A to F)(23–25). The Stk3/4-deficient Treg cells exhibited decreased expression of several genes associated with TCR signaling which included Cd83, Nkg7, Penk, Icos, Pdcd1, Ctla4, Ebi3 and Rrm2. There was decreased expression of a large number of genes regulated by NF-κB p65, either alone or in combination with Foxp3, including Icam1, Junb, Batf, Ccl5, Gm2a, Ctsz, Rpl6 as well as several Bcl2 family genes (Bcl2a1a, Bcl2a1b, Bcl2a1d and Bcl2l1), which may contribute to increased apoptosis observed in these cells. We detected decreased expression of several other gene transcripts specifically regulated by Foxp3, including Il2rb, Nrp1, Lta, Nt5e, Id2, Ccr8 and TNF receptor superfamily members Tnfrsf4 and Tnfrsf18). RNA-seq analysis confirmed defects of the Treg cell signature (p<6.4e-10) and IL2 signature (p<0.0014) in Stk3/4-deficient Treg cells without affecting the Teff signature. In addition, Stk3/4 deficiency downregulated Treg cell expression of genes associated with oxidative phosphorylation (Atp5d, Atp5e, Atp5g2, Atp6v0e, Cox5b, Cox6a1, Cox6b1, Cox6c, Cox7b, Cox8a), a pathway critical for Treg cell homeostasis and function(26, 27).
Fig 2. Stk3/4-deficient Treg showed transcriptome defects on TCR/p65/Foxp3 regulated genes.
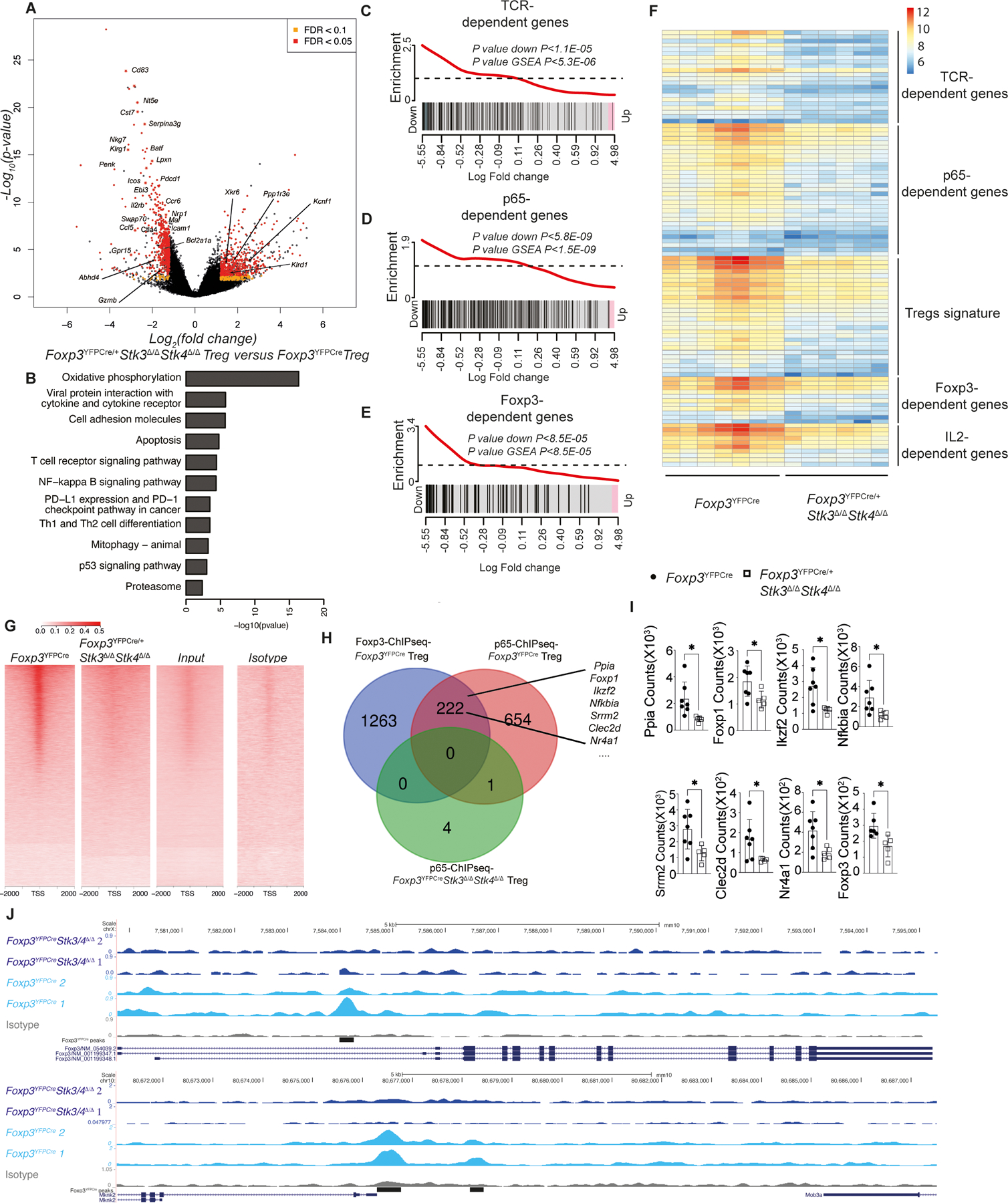
(A) Volcano plot of gene expression and (B) pathway analysis of gene transcripts using the KEGG database in YFP+ CD4+ Treg cells from Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ versus Foxp3YFPCre mice. (C to E) Gene set enrichment of TCR/p65/Foxp3 dependent genes. (F) Heatmap of gene transcripts of YFP+ CD4+ Treg cells isolated from Foxp3YFPCre (n=7) and Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ (n=6) mice. Counts were normalized using DESeq2-VST for heatmap visualization. (G) Heatmap showing the binding profile of p65 ChIP-seq signal for Foxp3YFPCre, Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg, pooled input and IgG isotype control samples at TSS and flanking +/− 2kb region of the expressed genes. The Y-axis indicates the number of counts per million mapped to denoted regions. (H) Venn diagram on the number of p65 pulldown ChIP-Seq enriched genes overlapping between WT Treg cells, Stk3/4-deficient Treg cells and Foxp3 binding genes (I) Arrows indicate the scatter plots of related gene transcripts expression in RNA-seq (n=7 for Foxp3YFPCre group; n=5 for Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ group). (J) Example genes Foxp3 and Mknk2 from the set of genes enriched by p65 Treg ChIP-Seq analysis. Coverage tracks were visualized in UCSC genome browser and display normalized p65 sequencing signal. The black bars indicate the p65 ChIP-seq peaks enriched in Foxp3YFPCre Treg cells group. Each point represents one mouse. Error bars indicate the standard error of the means (s.e.m). Statistical tests: for transcript analysis (A to F) was called at adjusted p values <0.05, fold-changes over ±1.5 and false discovery rate (FDR) were below 0.1. *, P<0.05 by Mann-Whitney test (panel I).
To further decipher the transcriptomic role of Stk3/4 in Treg cells, we performed a chromatin immunoprecipitation (ChIP) study with p65 antibody in Stk3/4-sufficient and deficient Treg cells. NF-κB p65 collaborates with Foxp3 to regulate a large network of downstream genes in Treg cells (28–30). Accordingly, we compared the peaks enriched by p65-ChIP in Stk3/4 sufficient and deficient Treg cells with the Foxp3-ChIP results in WT Treg cells (31). Deletion of Stk3/4 in Treg cells resulted in a profound decrease in p65 binding to both p65 and Foxp3 target gene (Fig. 2G and H). The transcripts identified in those of p65-bound Foxp3 target loci were dramatically decreased in Stk3/4 deficient Treg cells compared to WT Treg cells, which confirmed that Stk3/4-deficiency affects the transcriptomic collaboration between p65 and Foxp3 in Treg cells (Fig. 2H and I). This was illustrated by increased signals in p65-ChIPseq in WT Treg cells at p65-associated loci, such as Foxp3 and Mknk2 (Fig. 2J). Overall, our results revealed a broad dysregulation of gene expression controlled by TCR, Foxp3 and p65 in Stk3/4-deficient Treg cells.
TCR-regulated nuclear translocation of Stk4 and its association with Foxp3 and p65.
In view of the critical role played by Stk3/4 in regulating Treg cell transcriptional pathways, we analyzed the mechanisms by which this regulation took place both spatially and functionally. A previous study showed co-immunoprecipitation of Stk4 with Foxp3 upon transgenic expression in HEK293T cells(32). Confocal microscopic analysis revealed that Stk4 was predominantly localized to the cytosol in quiescent Treg cells, and stimulation with anti-CD3/CD28 mAbs induced time-dependent translocation of Stk4 into the nucleus as early as 0.5 h post-stimulation, where it co-localized with Foxp3 (Fig. 3A and B). Importantly, TCR stimulation of Treg cells induced the association of Foxp3 with p65 as previously reported (23), and also induced the formation of a larger trimolecular complex that includes Stk4, Foxp3 and p65, as revealed by co-immunoprecipitation studies (Fig. 3C). Trimolecular Stk4-Foxp3-p65 complex formation was also directly demonstrated by co-immunoprecipitation studies following the overexpression of the respective proteins in HEK293T cells (Fig. 3D to F). The TCR signaling-induced translocation of Stk4 into the nuclei of Treg cells and its co-localization with Foxp3 was inhibited by treatment with XMU-MP-1, a specific Stk4 kinase inhibitor, indicating that this process proceeded by a Stk4 kinase activity-dependent mechanism (Fig. 3G to H). Similar results were obtained in HEK293T cells using a Stk4 mutant with a lysine to arginine substitution at position 59 (Stk4K59R) that lacked kinase catalytic activity (Fig. 3I)(33, 34). These results established the formation of a Stk4-Foxp3-NF-κB p65 complex in response to TCR signaling relevant to the Treg cell transcriptional regulation.
Figure 3. TCR activation stimulates Stk4 nucleus translocation and colocalization with Foxp3.
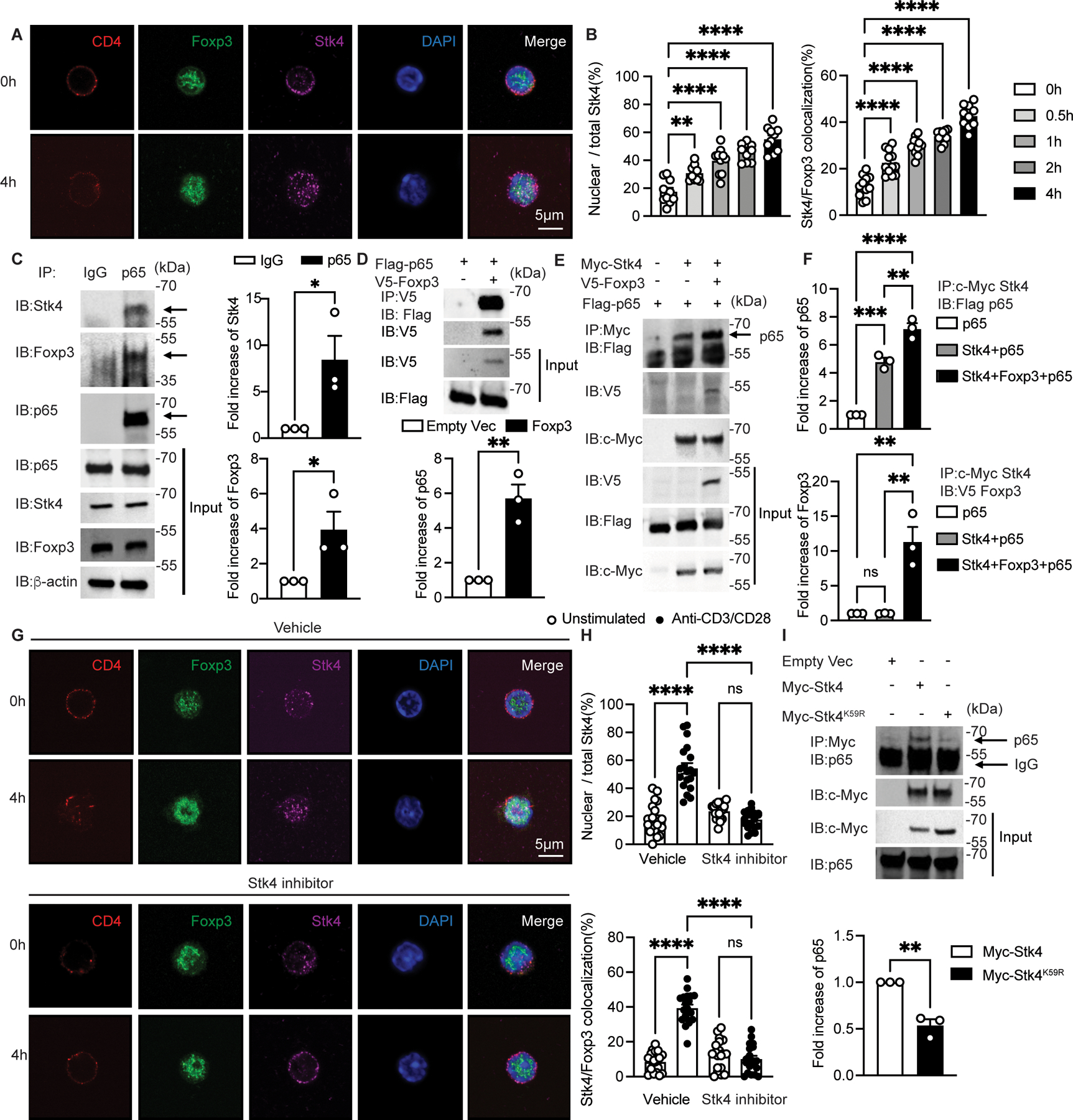
(A and B) Confocal microscopic analysis of CD4, Foxp3, STK4, DAPI and Merge (A), and ratios of nuclear/total Stk4 and frequencies of Stk4 and Foxp3 co-localization (B) in Foxp3YFPCre Treg cell cultures either unstimulated or stimulated with anti-CD3+anti-CD28 mAbs as indicated (n=10 per group). (C) Immunoblot analysis of Stk4 and Foxp3 association with p65 in Foxp3YFPCre Treg cells that were stimulated overnight with anti-CD3/CD28. Cell lysates were immunoprecipitated and immunoblotted with the indicated antibodies. (D and E) Immunoblot analysis of Stk4 and Foxp3 association with p65 in HEK293T cells transfected with the respective plasmids. Cell lysates were immunoprecipitated with anti-V5 mAb (specific for V5-tagged Foxp3) (D) or anti-c-Myc mAb (E), then immunoblotted with the indicated antibodies. (F) densitometric analysis of immunoblots of p65 (upper panel) and Foxp3 (lower panel) shown in (E). (G and H) Confocal microscopic analysis (G) and frequencies (H) of CD4, Foxp3, STK4, DAPI and Merge, ratios of nuclear/total Stk4 and frequencies of Stk4 and Foxp3 co-localization in Foxp3YFPCre Treg cells either unstimulated or stimulated with anti-CD3+anti-CD28 mAbs without or with the Stk4 kinase inhibitor XMU-MP-1(n=19 per group). (I) Stk4-p65 association is Stk4 kinase activity-dependent. Immunoblot analysis of Stk4-p65 association in HEK293T cells transfected with plasmids encoding either Stk4 kinase-competent (Stk4) or deficient (Stk4K59R) proteins. Each point represents one cell for confocal studies and one blot for immunoblot analyses. Error bars indicate the standard error of the means (s.e.m). Statistical tests: one-way ANOVA with post-test analysis (B and F), two-way ANOVA with post- test analysis (H) and Student’s unpaired two tailed t test (C,D,I). *, P<0.05,**, P<0.01, ***, P<0.005, ****, P<0.0001.
To discriminate between the requirement for cytosolic versus nuclear Stk4 kinase activity in the formation of STk4-Foxp3-NF-κB p65 complex, we employed an Stk4 mutant in which canonical lysine/arginine residues in the kinase bipartite nuclear localization sequence (NLS) were mutated to alanine (35). Transfection studies into Jurkat T cells using constructs encoding WT Stk4 (Stk4WT) versus the NLS mutant (Stk4NLS) revealed that Stk4WT translocated from the cytosol to the nucleus following activation with anti-CD3/CD28 mAbs, but the Stk4NLS mutant failed to do so (Fig. S6A). Expression of the respective kinase species in Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells revealed that Stk4WT but not the Stk4NLS mutant corrected the decreased CD25 expression and normalized the otherwise decreased p65 expression in these cells (Fig. S6B). Co-transfection studies revealed that Stk4Wt but not Stk4NLS co-precipitated with p65 even while both kinase species mediated a similar increase in the phosphorylation of the Stk4 substrate mob1 compared to the catalytically inactive Stk4K59R mutant (Fig. S6C and D)(36). These studies indicated that a catalytically competent but cytosolically restricted Stk4 is incapable of associating with NF-κB p65 or correcting NF-κB-associated defects in Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells.
Stk4-mediated Foxp3Ser418 phosphorylation stabilizes Stk4-Foxp3-p65 complex formation.
We next examined the capacity of Stk4 to regulate Foxp3 activity by phosphorylation. Immunoblot analysis with a pan anti-phospho-(p) serine and p-threonine (anti-pS/T) antibody revealed that following TCR signaling, phosphorylation of Foxp3 in Stk3/4-deficient Treg cells was severely decreased compared to that of WT Treg cells (Fig. 4A). Analysis of Foxp3 amino acid sequence for candidate phosphorylation sites based on consensus kinase target sequence homology identified S418 (S418) as a prime target (34, 37). The phosphorylation of this residue has previously been demonstrated to positively regulates Foxp3 function in response to extracellular signals (38). Immunoblot analysis with an anti-pS418Foxp3 antibody confirmed this residue as a key phosphorylation target downstream of Stk3/4 in Treg cells (Fig. 4B). By reconstituting Jurkat leukemic T cells with ectopic Foxp3 either alone or together with either kinase active (WT) or inactive (K59R mutant) Stk4, it could be demonstrated that TCR-mediated phosphorylation of S418Foxp3 was dependent on Stk4 kinase activity (Fig. 4C). We further confirmed the targeting of S418Foxp3 by Stk4 by transfecting Jurkat cells with constructs encoding Stk4 together with ones encoding either WT Foxp3 or with a mutant Foxp3 in which the serine418 residue was mutated into alanine (Foxp3S418A). Results showed that TCR signaling-induced Foxp3 phosphorylation, detected by immunoblotting with anti-pS/T, was decreased in the Foxp3S418A transfected cells as compared to those expressing WT Foxp3 (Fig. 4D).
Figure 4. Foxp3 S418 phosphorylation is Stk4-dependent and stabilizes the Stk4-Foxp3-p65 complex.
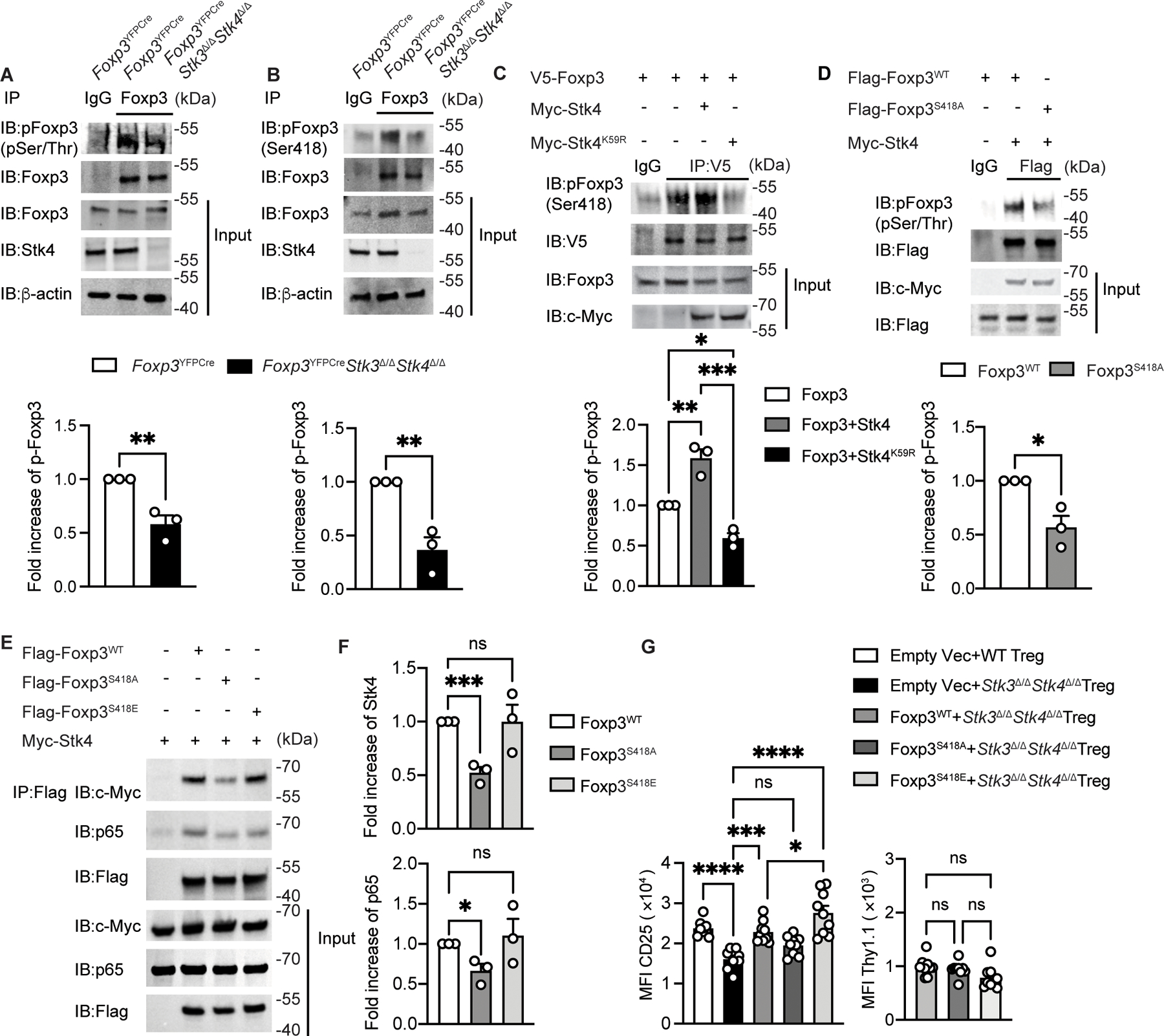
(A,B) Immunoblot analysis (upper panel) and densitometry (lower panel) of phospho-serine/threonine Foxp3 (A) and Foxp3 phospho-S418 Foxp3 (B) in Foxp3YFPCre and Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells stimulated with anti-CD3/ CD28 beads. Cell lysates were immunoprecipitated with anti-Foxp3 antibody and then immunoblotted with the indicated antibody. (C,D) Immunoblot analysis (upper panel) and densitometry (lower panel) of phospho-S418 Foxp3 in Jurkat cells transfected with the indicated plasmids then stimulated with CD3/CD28 beads. Cell lysates were immunoprecipitated with an anti-V5 mAb (C) or with anti-Flag mAb (D) and then immunoblotted with the indicated antibodies. (E,F) immunoblot analysis (E) and densitometry (F) of p65 and Foxp3 association in HEK293T cells transfected with the indicated plasmids. Cell lysates were immunoprecipitated with anti-Flag mAb, then immunoblotted with the indicated antibodies. (G) Scatter plot representation of CD25 and Thy1.1 MFI in Foxp3YFPCre or in transfected Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg with the indicated retrovirus (n=9 for each group). The results represent a pool of three independent experiments. Each point represents one blot for immunoblot analysis and one mouse. Error bars indicate S.E.M. Statistical tests: Student’s unpaired two tailed t test (A,B,D), one-way ANOVA with post-test analysis (C,F,G), ns: Not significant. *, P<0.05, **, P<0.01, ***, P<0.005, ****, P<0.0001.
To determine the impact of Foxp3 phosphorylation at S418 on Stk4-Foxp3-p65 complex formation, we generated a Foxp3 mutant in which the S418 residue was changed to the phosphomemetic glutamic residue (Foxp3S418E). Transfection of the respective proteins into HEK293T cells followed co-immunoprecipitation studies revealed that the Foxp3S418E mutant was more abundantly associated with Stk4/p65 than the phosphorylation-resistant Foxp3S418A mutant (Fig. 4E and F). To further dissect the role of Foxp3 S418 phosphorylation in Treg cells, we examined the capacity of WT Foxp3 and the Foxp3S418A and Foxp3S418E mutants to rescue the decreased CD25 expression when introduced by retroviral transduction in Stk3/4-deficient Treg cells. Whereas enforced expression of the retrovirally encoded WT Foxp3 upregulated CD25 expression to levels comparable to those found in Stk3/4-sufficient Treg cells, that of the Foxp3S418A mutant failed to do so. In contrast, expression of the Foxp3S418E mutant increased CD25 expression to even higher levels than those achieved with WT Foxp3 (Fig. 4G). Overall, these results indicated that Stk4-mediated phosphorylation of Foxp3 on serine 418 promotes Stk4-Foxp3-p65 trimolecular complex assembly and enhances Foxp3 and NF-kB p65-dependent expression in Stk3/4-deficient Treg cells.
Stk4 regulates p65 activation and promotes its nuclear translocation.
Given the contribution of Stk4 to Foxp3-p65 complex formation and impaired NF-κB-dependent gene expression in Stk3/4-deficient Treg cells, we investigated the impact of Stk3/4-deficiency on p65 activation and nuclear translocation. Immunofluorescence analysis revealed that TCR-induced p65 nuclear translocation and p65-Foxp3 co-localization were decreased in Stk3/4-deficient versus sufficient Treg cells (Fig. 5A to B). Decreased TCR-induced nuclear translocation of NF-κB p65 was also observed when Stk3/4 were acutely inhibited by treatment of Treg cells with the kinase inhibitor XMU-MP-1 (Fig. S7A). Stk3/4-deficient Treg cells isolated from Foxp3YFPCreStk3∆/∆Stk4∆/∆ males exhibited increased TCR-mediated p65 phosphorylation but decreased p65 protein content compared to WT Treg cells (Fig. 5C and D). Similar results were found in Treg cells of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ females, indicating these abnormalities were cell intrinsic and a reflection of generalized systemic inflammation in the mutant males (Fig. 5E). Further analysis of Stk3/4-deficient Treg cells of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ revealed that expression of the NF-κB inhibitor IκB in Stk3/4-deficient Treg cells was decreased, whereas phospho-IKKα/β (IκB kinase) was increased compared with WT Treg cells, consistent with the increased p65 phosphorylation (Fig. S7B to C). The decreased p65 expression was normalized upon treatment of the Stk3/4-deficient Treg cells with the proteasome inhibitor MG132, indicating that the decreased p65 expression was due to proteasomal degradation (Fig. 5F).
Figure 5. Impaired p65 nuclear translocation and enhanced degradation in Stk3/4 deficient Treg cells.
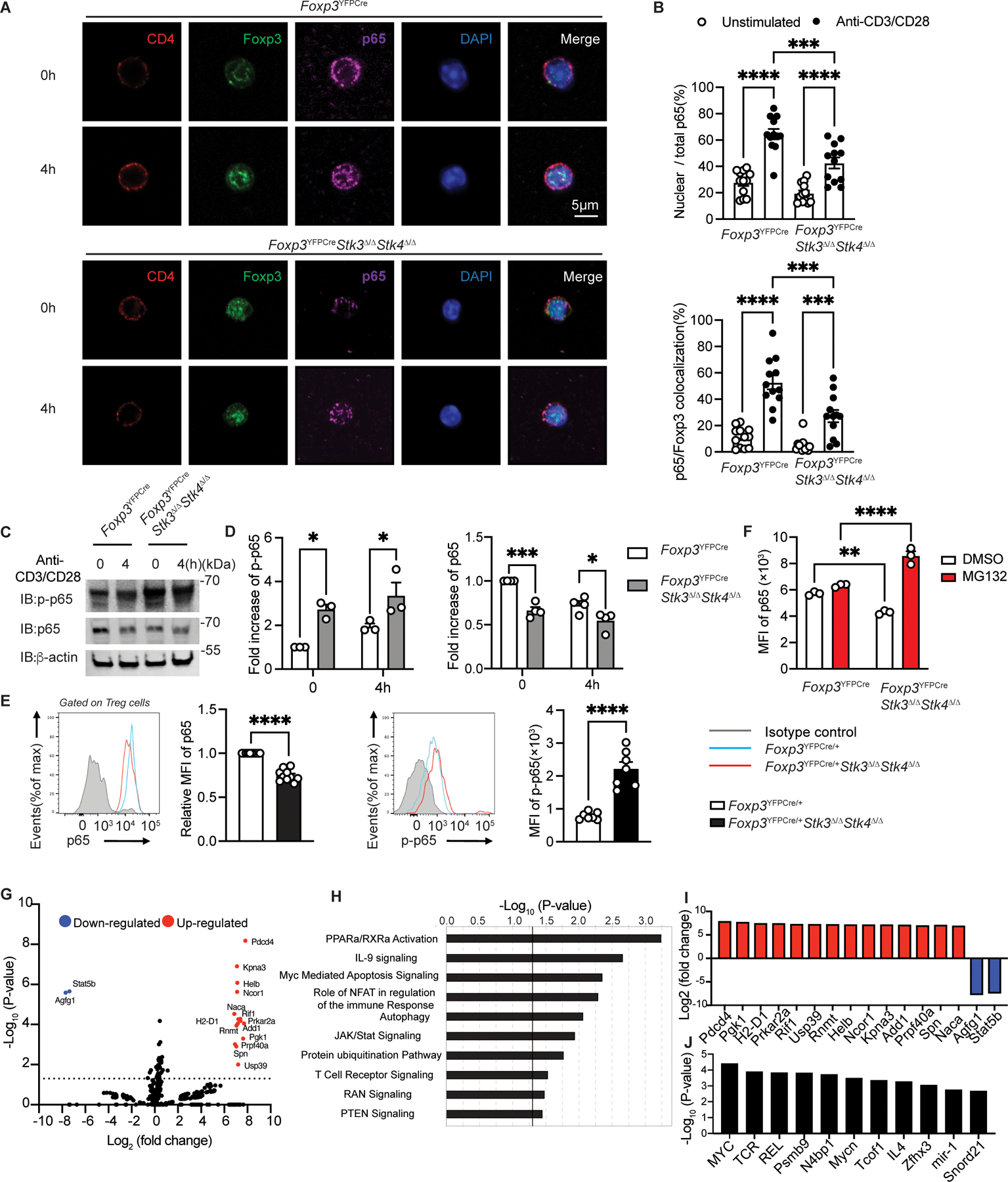
(A) Confocal microscopic analysis of CD4, Foxp3, p65, DAPI and Merge (B) ratios of nuclear/total Stk4 and p65/Foxp3 colocalization in Foxp3YFPCre Treg cells either unstimulated or stimulated with anti-CD3+anti-CD28 mAbs at the indicated time points (n=12 per group). (C to D) Immunoblot analysis (C) and densitometry (D) of p-p65, p65 and β-actin in Foxp3YFPCre and Foxp3YFPcreStk3∆/∆Stk4∆/∆ Treg cells. (E) Representative flow cytometric analysis and scatter plot representation of p65 (n=10 per group) and p-p65 (n=7 per group) MFI in Treg cells from Foxp3YFPCre/+ or Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆. (F) Scatter plot representation of p65 MFI in Foxp3YFPCre or in Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells either untreated or treated with MG132. (G) Volcano plot of phosphorylated proteins identified by phosphoproteomics in Foxp3YFPCreStk3∆/∆Stk4∆/∆ versus Foxp3YFPCre Treg cells. (H) Pathway analysis of phosphorylated protein in Foxp3YFPCreStk3∆/∆Stk4∆/∆ versus Foxp3YFPCre Treg cells. (I) Increased or decreased phosphorylated proteins identified in Foxp3YFPCreStk3∆/∆Stk4∆/∆ versus Foxp3YFPCre Treg. (J) Predicted upstream regulators deduced from phosphoproteomic analysis of Stk3/4 deficient versus WT Treg cells. Each point represents one cell in the confocal analysis, one blot for immunoblot analysis and one mouse for the flow analysis. Error bars indicate S.E.M. Statistical tests: Student’s unpaired two tailed t test (E), two-way ANOVA with post-test analysis (B,D,F), ns: Not significant. P<0.05, **, P<0.01, ***, P<0.005, ****, P<0.0001.
Further insights into mechanisms of action of Stk3/4 in Treg cells was revealed by phospho-proteomics analysis of Stk3/4-sufficient versus -deficient Treg cells (Fig. 5G and Data Set 1). Pathway analysis confirmed that Stk3/4 deletion in Treg cells induced the hyper-phosphorylation of NF-κB (p65 and p100), found in several pathways such as PPARα/RXRα (Fig. 5H). Moreover, this analysis also revealed that Stk3/4 deficiency impacted several other pathways, including Myc mediated apoptosis signaling, autophagy, protein ubiquitination, TCR signaling and Ras related nuclear protein (RAN) signaling (Fig. 5G to I). Activation of the protein ubiquitination pathway is in agreement with the enhanced proteasomal degradation of p65, while decreased RAN signaling, a pathway implicated in p65 transport to the nucleus, is consistent with decreased p65 nuclear translocation Stk3/4 deficiency. Upstream regulator prediction analysis for those pathways impacted by Stk3/4 revealed that Myc, TCR and Rel (p65 NF-κB) were the main regulators (Fig. 5J). Collectively, these results indicated that Stk3/4 deficiency impaired NF-κB p65 nuclear translocation despite hyperactivated NF-κB phosphorylation, reflecting a requirement for Stk3/4-p65 complex formation for effective p65 nuclear translocation.
Overexpression of p65 and Foxp3S418E rescues Stk3/4- Treg cell deficiency.
To elucidate the contribution of impaired Stk3/4-dependent p65-Foxp3 complex formation to the defective function of Stk3/4-deficient Treg cells, we first examined the capacity of p65 over-expression in Stk3/4-deficient Treg cells, effected by transduction of isolated Foxp3YFPCreStk3∆/∆Stk4∆/∆Treg cells with NF-κB p65-encoding retroviral construct, to ameliorate the defective CD25 expression in these cells. Transduction of the Stk3/4-deficient Treg cells with the p65-expressing retroviral construct, but not control retroviral vector, normalized the expression p65 in the recipient cells and upregulated CD25 expression, while Foxp3 expression was unaffected (Fig. 6A).
Figure 6. Overexpression of p65 and Foxp3S418E in Stk3/4-deficient Treg cells restores their regulatory functions.
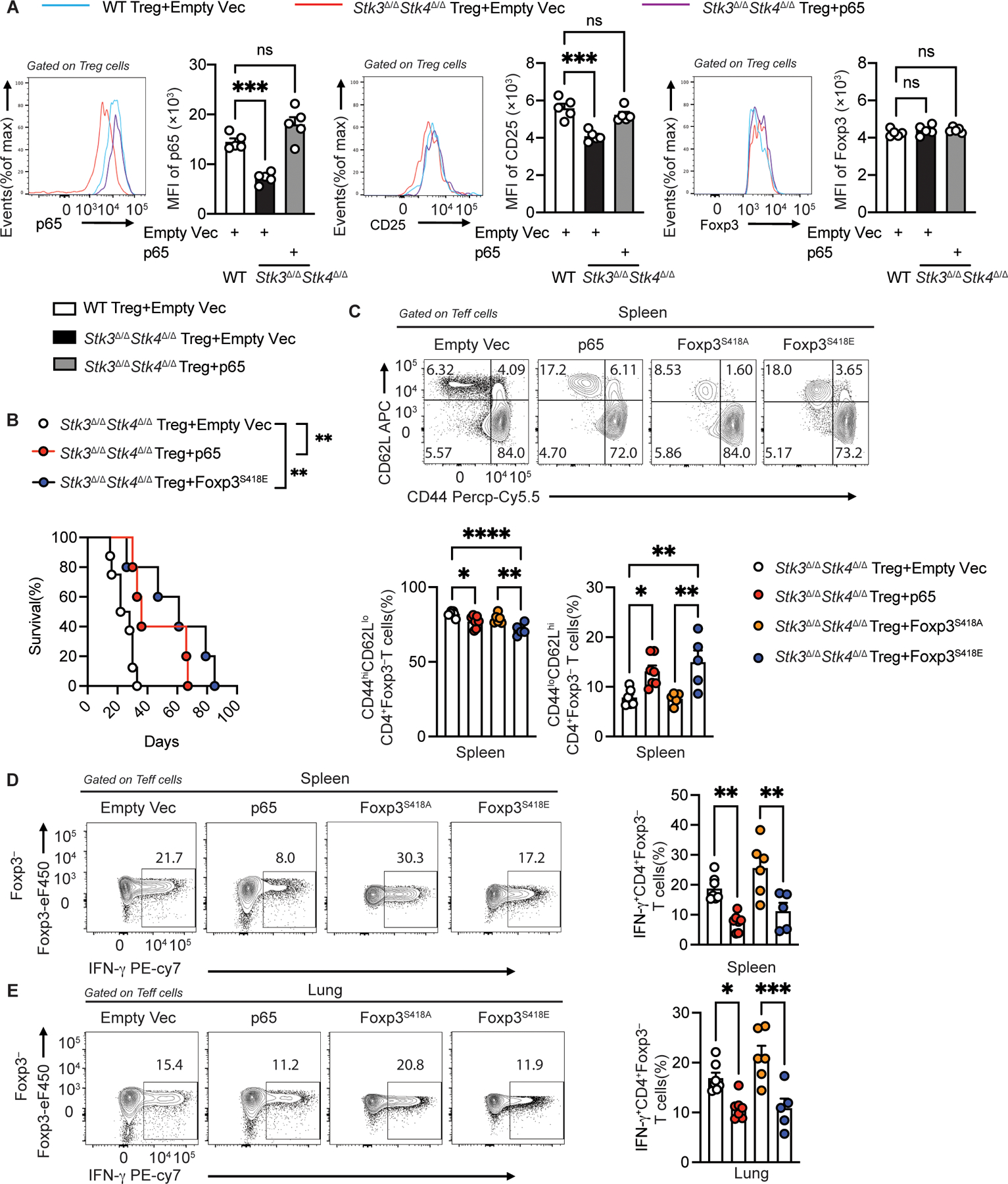
(A) Flow cytometric analysis and scatter plot representation of MFI of p65, CD25 and Foxp3 in Foxp3YFPCre and Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells transfected with either empty vector or with p65-encoding retrovirus, as indicated (n=5 per group). (B) Survival of Foxp3∆EGFPiCre mice injected with empty vector-transfected (n=8), p65-transfected (n=5) or Foxp3S418E transfected Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells (n=5). (C) Flow cytometric analysis and frequencies of CD62LloCD44hi CD4+Foxp3− and CD62LhiCD44lo CD4+Foxp3− Teff cells from spleens of Foxp3∆EGFPiCre mice injected with Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells that had been transfected with either empty vector (n=7), p65 (n=7), Foxp3S418A (n=6) or Foxp3S418E encoding vectors (n=5). (D, E) Flow cytometric analysis and frequencies of IFNγ+ CD4+Foxp3− T cells from spleens (D) and lungs (E) of the respective groups (n=7 for empty vector; n=7 for p65; n=6 for Foxp3S418A; n=5 for Foxp3S418E). The results represent three pooled independent experiments. Each point represents one mouse. Error bars indicate S.E.M. Statistical tests: log-rank-test (B), one-way ANOVA with post-test analysis (A,C,D,E), ns: Not significant, *P<0.05, **P<0.01, ***,P<0.005, ****P<0.0001.
We then further tested the capacity of p65 transduction to ameliorate the defective in vivo regulatory function of Foxp3YFPCreStk3∆/∆Stk4∆/∆Treg cells by employing an adoptive transfer model in which new-born Foxp3ΔEGFPiCre pups, which lacked Foxp3 expression(22), received WT Teff cells mixed with Stk3/4-deficient Treg cells that have been transduced with either an empty vector or with one expressing p65 (39). The Foxp3ΔEGFPiCre mice injected with the p65-transduced Stk3/4-deficient Treg cells survived longer compared to those receiving empty vector-transduced cells (Fig. 6B). Similarly, pups that received Stk3/4-deficient Treg cells transduced with a Foxp3S418E expressing vector survived longer than those receiving empty vector-transduced Treg cells. The Foxp3ΔEGFPiCre mice that received p65 or Foxp3S418E transduced Treg cells had decreased CD4+CD62LloCD44hi T effector memory cell frequencies and increased naïve (CD62L+CD44−) T cell frequencies in the spleen, liver and lung compared to those that received Treg cells transduced with either an empty vector or with one encoding the non-phosphomimetic Foxp3 mutant Foxp3S418A (Fig. 6C and Fig. S8A to B).
We further checked the capacity of the respectively transduced Stk3/4-deficient Treg cells to suppress IFN-γ expression by CD4+Foxp3− T cells in the spleens, lung and liver tissues of the recipient mice. The p65 and Foxp3418E-transduced Stk3/4-deficient Treg cell recipients showed decreased IFN-γ production by their CD4+Foxp3− T cells in the respective tissues compared to those mice receiving empty vector or Foxp3S418A-transduced Stk3/4-deficient Treg cells (Fig. 6D to E and Fig. S8C). Similarly, CD4 Foxp3 T cells transduced with either p65- or Foxp3S418E produced IFN-γ when transferred into Foxp3ΔEGFPiCre mice compared to empty vector or Foxp3S418A-transduced Treg cells (Fig. S8C to E). These results established that both of p65 and Foxp3S418E transduction greatly improved the function of Stk3/4-deficient Treg cells.
Finally, to determine the relevance of the findings in mouse Treg cells to the role of STK4 in human Treg cells, we analyzed patients with loss of function mutations in STK4 for evidence of immune dysregulation and Treg cell abnormalities. These included two patients with previously unidentified STK4 frameshift mutations (c.1103delT and c.1245delA), two other siblings with a previously reported deep intronic mutation (c.1362+28,055A>G) and one patient with a premature stop codon (c.1399c>T) (Fig. 7A) (18). Results revealed evidence of immune dysregulation in the patients with decreased frequencies of naïve (CD4+CD45RA+CCR7+) and central memory (CD4+CD45RA−CCR7+) Treg and Teff cells, and increased frequencies of effector memory (CD4+CD45RA−CCR7−) Treg and Teff cells, while those of CD4+CD45RA+CCR7− terminally differentiated effector memory Treg and Teff cells were unchanged (Fig. 7B and C). Furthermore, there was increased apoptosis and decreased proliferation of Treg cells, as measured by AnnexinV and Ki67 staining, respectively, while changes in those markers were not significant in Teff cells (Fig. 7D). Additional analysis of Treg cell markers revealed decreased CD69 expression, consistent with defective TCR-dependent Treg cell activation, while CD25 expression was unaffected (Fig. 7E). Analysis also revealed decreased NF-kB p65 subunit in Treg cells of STK4 deficient subjects, in agreement with the mouse data (Fig. 7F). Activation of STK4-deficient Treg cells with anti-CD3 mAb+IL-2 resulted in decreased induction of Foxp3 S418 phosphorylation as compared to control Treg cells (Fig. 7G). Overall, these results indicated that the function of STK4 in Treg cells is conserved in mice and humans.
Fig. 7. Immune regulatory abnormalities in STK4-deficient patients.
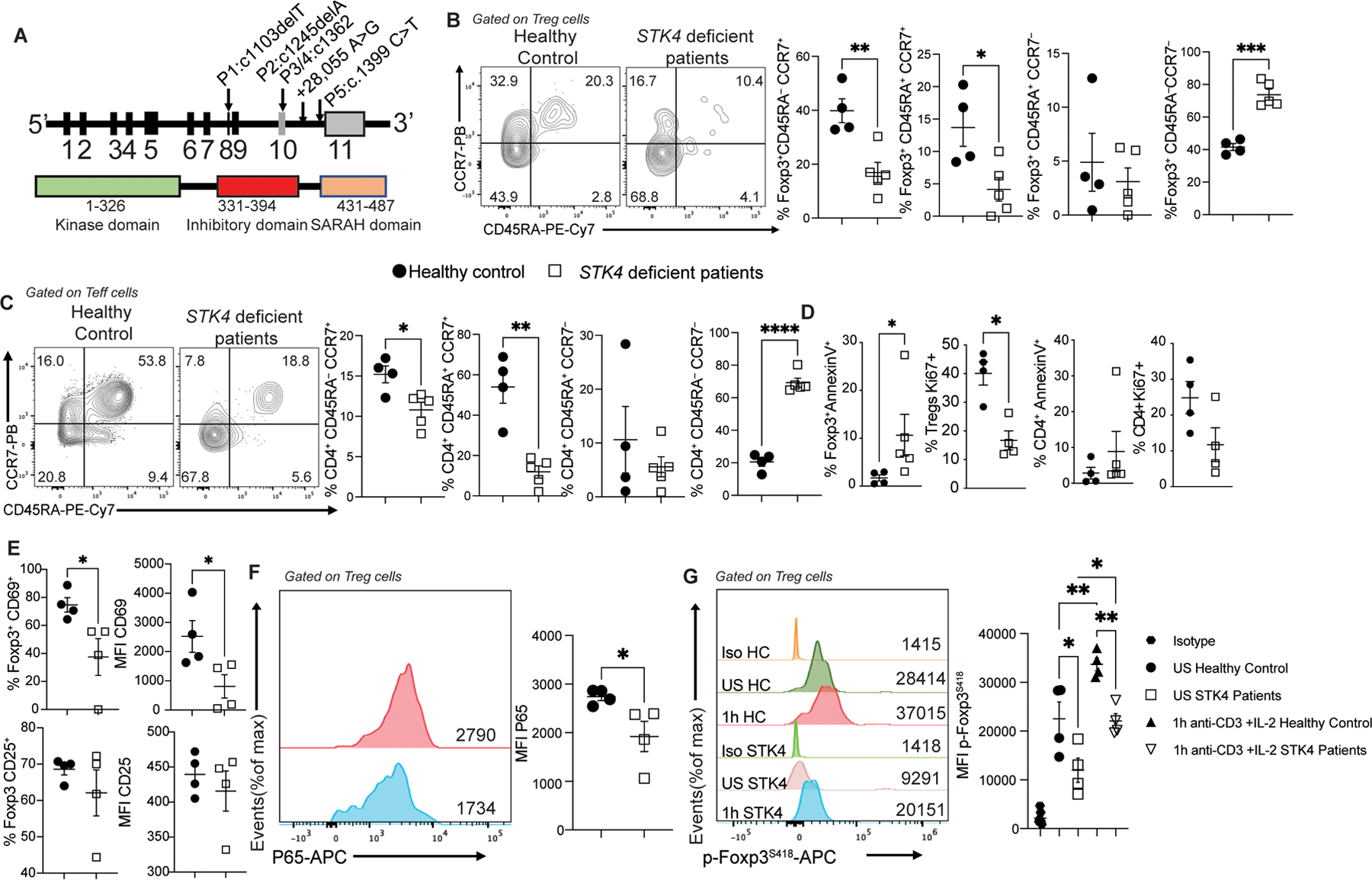
(A) Schematic representation of STK4 illustrating its encoding exons, the protein domains, and mapped mutations of four patients. The kinase domain, the inhibitory domain and the SARAH (Sav/Rassf/Hpo) domain are indicated. (B and C) Flow cytometric analysis and cell frequencies of CD45RA and CCR7 expression on circulating Treg cells (B) and Teff cells (C) of control subjects (n=4) and STK4 deficient patients (n=5). (D) Cell frequencies of AnnexinV and Ki67 expression on circulating Treg and Teff cells of control subjects (n=4) and STK4 deficient patients (n=5 for AnnexinV and n=4 for Ki67). (E) Cell frequencies and MFI of CD69 and CD25 expression on circulating Treg cells of control and STK4 deficient patients (n=4/group). (F) Flow cytometric analysis and MFI of P65 expression on circulating Treg cells of control and STK4 deficient patients (n=4/group). (G) Representative histograms of MFI of phospho-S418 Foxp3 in circulating Treg cells of healthy control (HC) and STK4 deficient patients that were either unstimulated (US) or stimulated with anti-CD3 +IL-2 (n=4/group). Each dot represents one patient. Error bars indicate S.E.M. Statistical tests: Student’s two tailed t test (B to F) or one-way ANOVA with post-test analysis (G). *p<0.05, **,P<0.01, ***,P<0.001, ****,P<0.0001.
Discussion
We demonstrate a TCR-regulated trimolecular complex between Stk4, Foxp3 and NF-κB p65 that plays an essential role in Treg cell homeostasis and function. TCR signaling induced the translocation of Stk4 to the nucleus by a kinase activity-dependent mechanism, where it associated with and stabilized the Foxp3- NF-κB p65 complex and enhanced its transcriptional activity. Combined deficiency of Stk4 and its lesser expressed homologue Stk3 in Treg cells led to a profound activation defect characterized by failure to generate experienced tissue Treg cells, enhanced apoptosis and rapidly lethal autoimmunity. Thus, Stk4 provides a critical link between TCR sensing by Treg cells of their environment with their immunoregulatory functions (24, 40).
Formation of nuclear Stk4-Foxp3-P65 complex involved dyad molecular interactions between Stk4-p65 and Stk4-Foxp3. The impact of Stk4 on p65 involved both enhancement of its translocation to the nucleus and its association with Foxp3. Paradoxically, Stk4 deficiency was associated with enhanced IKK activation and p65 phosphorylation in the face of decreased p65 nuclear translocation, which instead underwent proteasomal degradation(41). The failure of p65 nuclear translocation may reflect defective activation of the RanGTP-Importin pathway implicated in this process, as revealed in phosphoproteomic analysis of Stk3/4-deficient Treg cells (42). Formation of the trimolecular complex was strengthened by Stk4 kinase activity-dependent phosphorylation of Foxp3 at serine 418 (38). Impairment of this complex formation upon Stk3/4 deficiency adversely impacted several Treg cell transcriptional modules including TCR, Foxp3 and p65-dependent transcripts and linked downstream pathways including oxidative phosphorylation. Transgenic expression in Stk3/4-deficient Treg cell of either p65 or Foxp3418E but not Foxp3418A ameliorated the regulatory deficit both in vitro and in vivo, consistent with role of Stk4 in promoting p65 and Foxp3-dependent transcriptional programs.
A critical feature of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ mice is the profound depletion of Treg cells, related to defective Treg cell activation and differentiation into experienced Treg cells. This deficit was also reflected in the RNA-seq analysis on Stk3/4-deficient Treg cells which revealed decreased expression of RNA transcripts encoding tissue homing related molecules including Ccr4, Ccr6, Maf and Itgae (encoding for CD103). Depletion of Treg cells including tissue Treg cells is likely to play a key role in the end organ autoimmunity in these mice.
A previous study on Stk3/4 deficiency in Treg cells incriminated the IL-2 pathway in the immune dysregulation observed in these mice. We also observed decreased expression of Il2ra and its encoded protein CD25, the high affinity IL-2Rα chain subunit, in Treg cells of Foxp3YFPCre/+Stk3∆/∆Stk4∆/∆ mice in association with decreased activation-induced phosphorylation of the downstream transcription factor Stat5. Overexpression of p65 and Foxp3418E specifically in Stk3/4-deficient Treg cells restored the CD25 expression, indicating that the defect in IL-2 signaling in Stk3/4-deficient Treg cells maybe at least in part reflective of defective Stk4-p65-Foxp3 complex formation.
A number of genetic defects involving NF-kB and its upstream Carma (Card11), Bcl10 and Malt1 (CBM complex) have been described to result in immune dysregulatory features both in mice and in human subjects (43). Our results suggest the phenotypic overlap between these defects and those of Treg cell-specific Stk3/Stk4 deficiency and Stk4 deficiency in human subjects can be explained by the underlying impact of these mutations on the NF-κB pathway and its interaction in Treg cells with Foxp3.
There are limitations to our study that need to be mentioned. The rescue of Foxp3ΔEGFPiCre mice with p65 or Foxp3418E-transduced Stk3/4-deficient Treg cells was limited by the number of cells that could be harvested following retroviral transfection and as such may have given rise to partial rescue phenotypes. Another limitation is the small number of human subjects with STK4 deficiency that were employed in our studies, reflective of the rarity of this disease.
Finally, our studies highlight the indispensable function of Stk4, alone or in combination with Stk3, in TCR-mediated sensing by Treg cells of target cells and tissues. Modulating the signal strength of this module by targeting Stk4 maybe exploited therapeutically to alter Treg cell function and tissue homing relevant to cancer immunotherapy and autoimmunity.
Materials and Methods
Study Design.
To address the role of Stk4 and its related kinase Stk3 in Treg cell homeostasis and function, mice with Treg cell-specific deletion of Stk4, Stk3 or both genes, as well as genes encoding various components of the hippo pathway, were analyzed for their state of T cell activation and immune dysregulation. The transcriptional landscape of Treg cells was analyzed by RNA-seq and ChIP-seq. The nuclear translocation of Stk4, the interaction of Stk4 with p65 and Foxp3 and the phosphorylation of Foxp3 on serine 418 were analyzed by co-immune precipitation and/or imaging studies in primary Treg cells and in cell lines transfected with vectors expressing the respective components. The rescue of Stk3/4-deficientTreg cell function by transduced p65 and Foxp3418E was evaluated in vitro and in vivo, the latter by cell transfer into Foxp3-deficient mice. For mouse studies, mice bearing the respective genotypes were identified by PCR analysis, and the genotypes were further confirmed by flow cytometry as appropriate. Human subjects with STK4 deficiency were identified by genetic screening studies. Numbers of mice and of experimental replicates employed in these studies were determined based on the results of pilot studies and on results from previous studies. The studies were not blinded.
Human subjects.
Subjects with loss of function Stk4 mutations and healthy controls were recruited under protocols approved by the local institutional review boards at the respective referring centers. Patients P1 and P2 harbored novel homozygous Stk4 frameshift mutations (c.1103delT and c.1245delA). Patients P3 and P4 were siblings with a previously reported homozygous deep intronic mutation (c.1362+28,055A>G) (reference sequence: NM_006282.5), while patient P5 had a premature stop codon (c.1399C>T). All studies were approved by the Institutional Review Boards (IRBs) at the respective author-affiliated institutes, including Boston Children’s Hospital (IRB-04-09-113R), Marmara University School of Medicine (IRB-09.2018.624), Istanbul University Cerrahpasa Faculty of Medicine (IRB-16/06/2020-73272) and Necmettin Erbakan University Meram Medical Faculty (IRB-2021/3417).
Mice.
Foxp3YFPCre (B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J)(44), Stk3fl/flStk4fl/fl (Stk4tm1.1Rjo Stk3tm1.1Rjo/J)(45) and Wwtr1fl/flYap1fl/fl (Wwtr1tm1Hmc Yap1tm1Hmc/WranJ) (46), Lats1fl/fl (Lats1tm1.1Jfm/RjoJ) and lats2fl/fl (Lats2tm1.1Jfm/RjoJ) (47) mice were obtained from the Jackson Laboratory. Foxp3∆EGFPiCre mice were generated as described (22). Except when it was specified, 25–28 days old mice were used in this study. The mice were housed under specific pathogen-free conditions and used according to the guidelines of the institutional Animal Research Committees at the Boston Children’s Hospital (protocol 00001278).
Cell culture and transfection.
HEK293T and Jurkat cells were obtained from the American Tissue Culture Collection (ATCC). HEK293T were cultured using DMEM (Invitrogen) plus 10% fetal bovine serum (FBS; Gibco), supplemented with 1% penicillin-streptomycin (Invitrogen). Jurkat cells were cultured in RPMI 1640 medium with 10% FBS, 1% penicillin-streptomycin, non-essential amino acids sodium pyruvate. Treg cells were isolated from mouse spleens and cultured in RPMI1640 complete medium with anti-CD3/CD28 beads (from Invitrogen). Lipofectamine 2000 (Invitrogen) was used for transient transfection of HEK293T Cells according to the manufacturer’s instructions. Jurkat cells were transfected with Neon transfection system with program Pulse voltage 1600v, pulse width 10ms and pulse number 3 in pen-strep free complete medium. Transfected Jurkat cells were cultured in complete medium for 24 hours and process further for immunoprecipitation assay.
Real-time PCR.
Total RNA was isolated from sorted cells with RNeasy kit (Qiagen). Reverse transcription was performed with the SuperScript II RT-PCR system (Invitrogen) and quantitative real-time reverse transcription (RT)-PCR with Taqman® Fast Universal PCR master mix, internal house-keeping gene mouse (Gapdh: Mm99999915_g1) and specific target gene primers (Stk3: Mm00490480_m1; Stk4:Mm00451755_m1) (Applied Biosystems) on Step-One-Plus machine. Relative expression was normalized to Gapdh and calculated as fold change compared to Stk3 expression.
Flow cytometry.
Viability dye and antibodies against mouse CD4, CD8, CD90.2, CTLA4, CD39, CD73 (biolegend), CD25, CD44, CD62L, Foxp3, T-Bet, Gata-3, Helios, IFN-γ, IL-4, IL-17A, IL-2, IL-10, OX40, Nrp1 (eBioscience), p65, pS536 p65 (Cell signaling Technology) were used. Cell suspensions were stained for surface markers and viability dye for 30 min on PBS/0.5%FCS. Foxp3, Helios, and CTLA4 staining was performed overnight using the ebioscience Cytofix/CytopermTM kit. For cytokine detection, cell suspensions were pre-incubated with 50 ng/mL PMA, 500 ng/mL ionomycin and 10 µg/mL brefeldin A for 4h in complete medium. Following CD16/32 blocking with specific mAbs, the cells were surface stained for the indicated markers then permeabilized and stained intracellularly overnight with mAbs against IL-4, IL-10, IFN- γ or IL-17 using the ebioscience Cytofix/CytopermTM kit. For pS536p65, and p65 staining, spleen cells were fixed with PBS/2% paraformaldehyde for 20min, permeabilized in 90% methanol for 30 min on ice and stained for CD4, pS536p65 and p65 in PBS. All flow cytometry acquisitions were performed on a BD Fortessa cytometer using DIVA software (BD Biosystems) and analyzed using FlowJo Version 10 (Tree Star). All mouse antibodies used are listed in Supplementary Table 1. The gating strategies for discriminating human and mouse Treg and Teff cells are shown in Fig. S9.
Immunoprecipitation assay and immunoblot analysis.
For immunoprecipitation assay, cells extracts were prepared by using RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with a complete protease inhibitor cocktail (Roche), a PhosSTOP phosphatase inhibitor cocktail (Roche). Lysates were incubated with the appropriate antibody for four hours to overnight at 4°C before adding Protein G Dynabeads™ (ThermoFisher,10004D) for 2 hr. The immunoprecipitates were washed three times with the same buffer and eluted with SDS loading buffer by boiling for 5 min.
For immunoblot analysis, the samples were subjected to SDS-PAGE. The resolved proteins were then electrically transferred to a PVDF membrane (Millipore). Immunoblotting was probed with indicated antibodies. The protein bands were visualized by using a SuperSignal West Pico chemiluminescence ECL kit (Pierce). Signal intensities of immunoblot bands were quantified by Image J software. A list of all antibodies used for immunoblotting is reported in Supplementary Table 2.
ELISA.
Total IgE and IgG1 concentrations were measured in the sera of indicated mice by sandwich ELISAs. For total IgE, the capture and biotinylated detection antibodies (rat anti-mouse IgE clones R35-72 and R35-118, respectively) and purified mouse IgE isotype standard antibody (clone C38-2) were from BD Biosciences (San Jose, Calif). For total IgG1, the capture and biotinylated detection antibodies (Goat anti-mouse IgG1 clones A90-105A-21 and A90-105P-34, respectively) and mouse reference serum (RS10-101-5, 3.0mg/ml IgG1) were from Bethyl Laboratories, Inc.
Transcriptome profiling.
Splenic Treg (CD4+YFP+) cells were double sorted from 4 weeks old female Foxp3YFPCre (n=7) and Foxp3YFPCre/+ Stk3Δ/ΔStk4Δ/Δ mice (n=6). Total RNA was extracted using Qiagen RNeasy mini kit (Qiagen).and converted into double-stranded DNA (dsDNA), using SMART-Seq v4 Ultra Low Input RNA kit (Clontech). dsDNA was then fragmented to 200- to 300-bp-sized fragments, using M220 Focused-ultrasonicator (Covaris), and these were used for the construction of libraries for Illumina sequencing, using the KAPA Hyper Prep Kit (Kapa Biosystems). Libraries were then quantified using Qubit dsDNA HS (high sensitivity) Assay Kit on Agilent High Sensitivity DNA Bioanalyzer.
RNA-seq raw data were aligned using “align” (Rsubread, V2.0.1) and the latest UCSC mouse annotation (GRCm38/mm10). The raw data were trimmed before aligned using Trimmomatic (V0.39) for QC. Gene-level read counts were quantified using “feature Counts” (Rsubread, V2.0.1). To identify differentially expressed genes, we used “edgeR” (version 3.28), “DESeq2” (version 1.26.0) and “Lima” (V3.42.2) Bioconductor packages. We use “barcodeplot” for the visualization of the gene enrichment of the data (Lima, V3.42.2). All software/packages were run using their default parameters. Count tables were normalized to TPM (Transcripts per Million) for visualizations and QC. Sample clustering and standard path analyses (GO and KEGG) were performed using a custom-made pipeline. Transcripts were called as differentially expressed when the adjusted p values were below 0.05, fold-changes over ±1.5 and false discovery rate (FDR) were below 0.1.
Histology.
Large intestine, lung, liver and ear sections were stained with hematoxylin and eosin. Histopathological scoring of tissue was done by a blinded observer, and the final scores reflected averages of scores from 5 different ×200 fields per tissue per mouse. Large intestinal sections were scored as follows (48): 0, no inflammation; 1, mild, scattered infiltrates; 2, moderate infiltrates without loss of epithelium integrity; 3, moderate and diffuse or severe inflammation; 4, Severe inflammation associated with loss of the epithelial barrier integrity. Lung inflammation was scored separately for cellular infiltration around blood vessels and airways, as follows: 0, no infiltrates; 1, few inflammatory cells; 2, a ring of inflammatory cells 1 cell layer deep; 3, a ring of inflammatory cells 2–4 cells deep; 4, a ring of inflammatory cells >4 cells deep. A composite score was determined by adding the inflammatory scores for both vessels and airways. Liver inflammation was scored at portal areas, as follows: 0, no inflammatory cells; 1, mild, scattered infiltrates; 2, moderate infiltrates occupying less than 50% of the portal areas; 3, extensive infiltrates in the portal areas; 4, severe, with infiltrates completely packing the portal area and spilling over into the parenchyma. Ear inflammation was scored as followed: 0, no inflammation, no infiltration; Mild inflammation associated with the infiltration of few cells; 2, moderately inflammation associated with mild to moderate cell infiltration; 3, severe inflammation associated with large infiltration of cells and mild skin dryness; 4, very severe inflammation associated with skin dryness and cartilage erosion.
Cloning and mutagenesis.
Expression plasmid for V5-Foxp3 was from Harvard PlasmID Database (HsCD00416899) (49). Myc-MST1 (#8847) (50), Flag-p65 (#20012) (51), pCL-Eco(#12371) (52), MSCV-IRES-Thy1.1 DEST (#17442) and MSCV-FOXP3-IRES-Thy1.1 retroviral vector (#17443) (53) were from Addgene. Stk4K59R, StkNLS and Foxp3 mutations were generated by directed mutagenesis using the Q5® Site-Directed Mutagenesis Kit (New England Biolabs). Stk4NLS mutant was generated by mutating the two pairs of canonical KR residues in the bipartite NLS (K469/R470/K481/R482) of the Myc-Stk4 into Alanine. All coding sequences in the mutant plasmids were verified by Sanger sequencing.
Retroviral transfections.
Retrovirus production was performed according to the manufacturer’s instructions. Briefly, HEK293T cells plated on 100-mm dishes were transfected with the indicated retroviral expression plasmid (6ug) together with the pCL-Eco (4ug). After 48h, the viral particles were collected, filtered by 0.45um membrane filter and used to infect the indicated cells in the presence of polybrene (10 ug/ml). After transfection for 48h, cells were cultured in complete RPMI 1640 medium with 50U/ml IL-2. After 48h culture, cells are harvest for either analysis or sorting for adoptive transfer model.
Adoptive Transfer Model.
Transduced Treg (Thy1.1+CD4+YFP+) cells and untransfected (Thy1.1−CD4+YFP−) Teff cells were isolated by cell sorting (Sony Sorter Ma900). 2 days old-Foxp3ΔEGFPiCre pups were injected with 350,000 transduced Treg cells and 3 million untransfected Teff cells through intraperitoneal injection. Functional analyses were performed with spleens, livers and lungs from mice 19-days after the intraperitoneal injection.
Confocal microscopy.
Confocal microscopic analysis of Stk4/p65 nuclear and cytoplasmic distribution was carried out as described (54). Treg cells were purified and pretreated with/ without XMU-MP-1 (5 μg /ml) for 4 hours, and then incubated on pre-coated coverslips (poly-L-lysin (100 μg /ml) ± anti-CD3/CD28 (1 μg /ml) at 37 °C for indicated time in RPMI medium/10% FCS. After fixation with PBS/4% paraformaldehyde, cells were permeabilized with PBS/0.25% Triton X-100 for 15 minutes and blocked on PBS/5% bovine serum albumin. Cells were incubated with 1:300 diluted rabbit anti-Stk4 (D8B9Q; Cell Signaling) and FITC-anti mouse Foxp3 (FJK-16s, ebioscience) followed by 1:500 diluted Alexa Fluor 647-anti-rabbit secondary antibody (Life Technologies) in PBS/5% bovine serum albumin. Slides were mounted with gold anti-fade reagent with DAPI (Invitrogen). Images were acquired with a Zeiss LSM880 confocal microscope and ZEN imaging software. Five to ten fields were selected randomly and total cells in the field were analyzed for the percentage of Stk4 nuclear localization using ImageJ software. The percentage of nuclear Stk4 localization was obtained using the following formula: 100 × corrected nuclear fluorescence/corrected total cell fluorescence. Corrected fluorescence was obtained using the formula: integrated density − (area of selected cell or nucleus × mean fluorescence of background).
Chromatin Immunoprecipitation and sequencing
Approximately 3–6 × 106 splenic Treg cells were sorted and cross-linked with fresh 1% formaldehyde (Fisher Scientific) for 8 min at room temperature (RT) and quenched with 125 mM glycine for 5 min at RT. Cells were then washed three times in cold phosphate-buffered saline (PBS) containing protease inhibitors and cell pellets flash-frozen in liquid nitrogen (LN2). Nuclei were isolated using the truChIP kit (Covaris Inc.), and chromatin was sheared at 4°C, 140 W, and 200 cycles per burst with 10% duty factor for 8 min (Covaris). Input fraction was saved (1%), and the remaining sheared chromatin was used for ChIP with a mouse anti-NF-κB p65 antibody (F-6) (sc-8008 X, Santa Cruz) or an immunoglobulin G isotype control (Millipore) in immunoprecipitation buffer [0.1% Triton X-100, 0.1 M tris-HCl (pH 8), 0.5 mM EDTA, and 0.15 M NaCl in 1 × Covaris D3 buffer] at 4 °C, rotating overnight, followed by incubation with Dynabeads Protein G (10003D, Life Technologies) for 4 hours. The chromatin-bead-antibody complexes were then washed sequentially with three wash buffers of increasing salt concentrations and Tris-EDTA (TE) buffer. Chromatin was then eluted using 1% SDS in Tris-EDTA. Cross-linking was reversed by incubation with ribonuclease A (Roche) for 1 hour at 37 °C, and with Proteinase K (Roche) at 65 °C overnight. DNA was purified with the QIAquick PCR Purification Kit (28104, Qiagen). ChIP-Seq libraries were prepared using Swift 2S Accel reagents on a Beckman Coulter Biomek i7 liquid handling platform from approximately 1 ng of DNA according to the manufacturer’s protocol and 14 cycles of PCR amplification. Finished sequencing libraries were quantified by Qubit fluorometer and Agilent TapeStation 2200. Library pooling and indexing were evaluated with shallow sequencing on an Illumina MiSeq. Subsequently, libraries were sequenced on an Illumina NovaSeq 6000 targeting 40 million 150 bp read pairs by the Molecular Biology Core facilities at Dana-Farber Cancer Institute.
ChIP-seq analysis.
Paired-end reads (PE) were obtained for the two replicates per experimental condition, one pooled ChIP input and one IgG isotype control sample. The quality of the reads was assessed using FastQC v0.11.9 (55). The low-quality reads were filtered, and adaptor sequences were trimmed using Cutadapt v2.9 with the following parameters: -q 30 -m 30 -a AGATCGGAAGAG -A AGATCGGAAGAG (56). Post-trimming, the quality was re-assessed across all samples using multiQC tool v1.9 (57). The reads were then aligned to the reference genome (GRCm38/mm10 primary assembly) using Bowtie2 v2.3.4.3 with default settings (58). Aligned read pairs with MAPQ < 20 were discarded with samtools view -f 2 -q 20 options (59, 60). and duplicates were removed using the MarkDuplicates (v2.25.4) command with setting REMOVE_DUPLICATES = true from the Picard tools. Furthermore, reads overlapping the ENCODE mm10 blacklist regions (61) were removed using the intersectBed function in the BEDtools suite v2.27.1 (62). Peak-calling for each ChIP replicate to the Input DNA was performed using MACS2 v2.2.7.1 with the options -f BAMPE -g mm -q 0.05 (63). The peaks were annotated to mm10 genomic features using annotatePeak function from ChIPseeker R package (64). setting TSS regions= c(−2000,1000), txdb= “TxDb.Mmusculus.UCSC.mm10.ensGene”, annoDB= “org.Mm.eg.db’’ and overlap=”all”. Peaks distinctly enriched in wild-type or mutant (absent in IgG isotype) were identified using intersectBed function. The ChIP-seq enrichment profile across the list of expressed genes ranked by the RPKM level in wild-type was represented as heatmaps using ngs.plot program with the following settings: -G mm10 -R TSS -E expressed_gene_list -YAS 0,0.5, -L 2000 (65). The library size normalised BigWig coverage tracks were generated and visualized on UCSC genome browser along with the BED tracks of the peak coordinates. A list of all software, package and tools used for ChIP-seq analysis is reported in Supplementary Table 3.
Mass spectrometry (MS).
Gel bands were excised and washed three times with acetonitrile; the final wash contained ammonium bicarbonate. For the purified samples, the solutions were directly subjected to proteolysis. Trypsin digestion was then carried out (1: 10 molar ratio of trypsin to protein) by incubation at 37°C for 16 h. The non-alkylated cysteine digest samples were then analyzed by LC–MS/MS using either a Q-Exactive Plus or an Orbitrap Fusion mass spectrometer equipped with a Waters nanoACQUITY ultra-performance liquid chromatography (UPLC) system using a Waters Symmetry C18 180 mm by 20 mm trap column and a 1.7 mm (75 mm inner diameter by 250 mm) nanoACQUITY UPLC column (35°C) for peptide separation. Trapping was done at 15 ml/min with 99% buffer A (100% water, 0.1% formic acid) for 1 min. Peptide separation was performed at 300 nL/min with buffer A and buffer B (100% acetonitrile, 0.1% formic acid) over a linear gradient. High-Energy collisional dissociation was utilized to fragment peptide ions via data-dependent acquisition. Mass spectral data were processed with Mascot Distiller, utilizing the high-resolution profile peak-picking algorithm. Protein searches were conducted against the mus musculus SwissProt protein database using Mascot Search Engine (Matrix Science, LLC, Boston, MA; v. 2.6.0). Mascot search parameters included: parent peptide ion tolerance of 10.0 ppm, peptide fragment ion mass tolerance of 0.020 Da, strict trypsin fragments (enzyme cleavage after the C terminus of K or R, but not if it is followed by P), variable modification of phosphorylation (S, T, Y, and H), oxidation (M), and propioamidation(C). Manual examination of the MS/MS spectra and the corresponding assigned fragment ions were conducted to verify the identified phosphopeptide.
Phosphoproteomic analysis.
Phosphorylated peptides were enriched using a TiO2 (titanium dioxide) TopTips column (TiO2; GlySCi, Columbia, MD) with a slightly modified manufacturer protocol. Briefly, the manufacturer protocol was utilized with the addition of 70 mM L-glutamic acid in the loading buffer (65% acetonitrile, 2% trifluoroacetic acid). Bound phosphopeptides on the TiO2 resin were washed with 65% acetonitrile, 2% trifluoroacetic acid, and eluted with 2% ammonium hydroxide solution in water at pH 12. Both enriched phosphopeptides and flowthrough (i.e. un-enriched) peptides fractions were analyzed by LC–MS/MS as described above.
Protein abundances were first log2 transformed before data analysis using Perseus (66) and PRISM software. Proteins were considered to be significantly differentially regulated at 5% FDR using the Benjamini-Hochberg correction. Proteins and phosphoproteins were separately analyzed in the IPA software (QIAGEN Inc.). The gene symbols of the differentially expressed proteins, their Log2 (fold change) values and Log10(p-value) were uploaded in IPA. The Core Analysis was performed under the following parameters: the expression fold change was set as the type of Core Analysis; direct and indirect relationships were considered to generate the networks; the prediction of these networks included the endogenous chemicals, 35 molecules per network and a total of 25 networks enabled per analysis. The cutoff values applied to all datasets included fold change ≥1.5 for up-regulated and ≤−1.5 for down-regulated proteins. Adjusted p values (Benjamini-Hochberg, FDR) of <0.05 were considered significant. Based on the IPA’s analysis, significant canonical pathways, biological functions and diseases, and interaction networks were algorithmically generated.
Analysis of phosphoprotein by phosphoflow.
Total spleen from either Foxp3YFPCre or Foxp3YFPCreStk3/4∆/∆ mice were stimulated at 37°C in non-supplemented RPMI 1640 using pre- formed complexes of biotinylated anti-CD3 mAb (clone 145-2C11, BD, 30 μg/ml) with anti-CD28 mAb (37.51, BD, 30 μg/ml) and streptavidin (60 μg/ml) during 15 min. Cells were incubated with or without different pharmacological inhibitors: Zap70 inhibitor (ZAP #180013, 1mg/ml), PI3-K inhibitor (LY294002 #9901, 50μM), and Malt1 inhibitor (Z-VRPR-FMK trifluoroacetate salt, 75 μM) during the stimulation. The reaction was stopped and cells were permeabilized using a Foxp3/transcription factor staining buffer (eBiosciences) and Perm buffer III (BD Biosciences). Cells were stained using rabbit anti-human Phospho-FOXP3(Ser418), Phospho-MST1 (Thr183)/MST2 (Thr180) (CST), P-p65 S536, and Alexa Fluor 647–conjugated goat anti-rabbit IgG Ab (Thermo Fisher). Samples were acquired on a Fortessa cytometer (BD) and data were analyzed using the FlowJo software.
Treg cell suppression assays.
Mouse spleens and lymph nodes were harvested and lysed in ACK buffer and washed twice. CD4+ T cells were enriched with the Dynabeads CD4 negative selection kit (Invitrogen) according to the manufacturer’s instructions. CD4+ T cells were stained with antibodies directed against CD3, CD4, and CD62L. CD3+CD4+CD62Lhigh (effector T cells) and CD3+CD4+ YFP+(Treg cells) were sorted with a Sony MA900 cell sorter (Sony). The purity of these populations was >98%. Effector T (Teff) cells and Treg cells were co-cultured at different ratios (Treg:Teff cells at 1:1, 1:2, 1:4, 1:8, and 1:16) in the presence of splenic antigen-presenting cells (APCs) and soluble anti-CD3 mAb (1 μg/ml). Teff cells were stained with Cell Trace Violet (2 mM, Life Technologies) to monitor their proliferation. Proliferation was analyzed by flow cytometry after 3 days of culture.
Statistical analysis.
Data was analyzed by paired and unpaired two-tailed Student’s t-test, one- and two-way ANOVA with post-test analyses and log-rank test, as indicated. Differences in mean values were considered significant at a P < 0.05.
Supplementary Material
Fig. S1. Stk3/4 deficiency in Treg cells causes fatal autoimmune lymphoproliferative disease.
Fig. S2. Defective activation and survival of Stk3/4-deficient Treg cells.
Fig. S3. Treg cell-specific inactivation of the Hippo pathway does not rescue the phenotype of Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice.
Fig. S4. Mice with Treg cell specific activation of the Hippo pathway do not phenocopy Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice.
Fig. S5. TCR/CD28 signaling activates Stk4 by a Zap70, PI3K and Malt1-dependent mechanism to promote Treg suppressive function.
Fig. S6. The Stk4 NLS sequence is critical for Stk4-p65 interaction and CD25 upregulation in Treg cells.
Fig. S7. Inhibition of Stk4 kinase activity attenuates the nuclear translocation of p65.
Fig. S8. p65 and Foxp3S418E -transduced Treg cells ameliorate the disease of Foxp3ΔEGFPiCre mice.
Fig. S9. Gating strategies for human and mouse Treg and Teff cell discrimination.
Table S1. Listing of flow cytometry antibodies used.
Table S2. Listing of immunoblotting antibodies used.
Table S3. Listing of software, package and tools used.
Data Set 1. Phospho-proteomic analysis of Foxp3YFPCre and Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells.
Blank Material Transfer Agreement.
Acknowledgements
This work was supported by a National Institutes of Health grants R01AI085090 and R01AI128976 (to T.A.C.) and R01AI153174 (to L.M.C.).
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Data Availability
Data presented in the manuscript will be made available to investigators following request to the corresponding author. Any data and materials to be shared will be released via a Material Transfer Agreement. RNAseq datasets have been deposited in the GEO with the accession code GSE178992. CHIP-Seq datasets have been deposited in the Bioproject database with accession code PRJNA84959.
References and Notes
- 1.Josefowicz SZ, Lu LF, Rudensky AY, Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30, 531–564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell C, Rudensky A, Roles of Regulatory T Cells in Tissue Pathophysiology and Metabolism. Cell Metab 31, 18–25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panduro M, Benoist C, Mathis D, Tissue Tregs. Annu Rev Immunol 34, 609–633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM, JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest 106, R75–81 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27, 20–21 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME, X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27, 18–20 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Georgiev P, Charbonnier LM, Chatila TA, Regulatory T Cells: the Many Faces of Foxp3. J Clin Immunol 39, 623–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, Gertz EM, Schambach A, Kreipe HH, Pfeifer D, Engelhardt KR, Rezaei N, Grimbacher B, Lohrmann S, Sherkat R, Klein C, The phenotype of human STK4 deficiency. Blood 119, 3450–3457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia J, Zhang W, Wang B, Trinko R, Jiang J, The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev 17, 2514–2519 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D, The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev Cell 25, 388–401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hergovich A, Schmitz D, Hemmings BA, The human tumour suppressor LATS1 is activated by human MOB1 at the membrane. Biochem Biophys Res Commun 345, 50–58 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D, Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 154, 1342–1355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL, Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21, 2747–2761 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL, TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF, The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. Int J Mol Sci 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nehme NT, Schmid JP, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P, Rieux-Laucat F, Lutz P, Picard C, Mahlaoui N, Fischer A, de Saint Basile G, MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119, 3458–3468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crequer A, Picard C, Patin E, D’Amico A, Abhyankar A, Munzer M, Debre M, Zhang SY, de Saint-Basile G, Fischer A, Abel L, Orth G, Casanova JL, Jouanguy E, Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS One 7, e44010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cagdas D, Halacli SO, Tan C, Esenboga S, Karaatmaca B, Cetinkaya PG, Balci-Hayta B, Ayhan A, Uner A, Orhan D, Boztug K, Ozen S, Topaloglu R, Sanal O, Tezcan I, Diversity in Serine/Threonine Protein Kinase-4 Deficiency and Review of the Literature. J Allergy Clin Immunol Pract 9, 3752–3766 e3754 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Ueda Y, Katagiri K, Tomiyama T, Yasuda K, Habiro K, Katakai T, Ikehara S, Matsumoto M, Kinashi T, Mst1 regulates integrin-dependent thymocyte trafficking and antigen recognition in the thymus. Nat Commun 3, 1098 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, Li Y, Guy C, Nguyen TM, Dhungana Y, Neale G, Peng J, Yu J, Chi H, Hippo/Mst signalling couples metabolic state and immune function of CD8alpha(+) dendritic cells. Nature 558, 141–145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi H, Liu C, Tan H, Li Y, Nguyen TM, Dhungana Y, Guy C, Vogel P, Neale G, Rankin S, Feng Y, Peng J, Tao W, Chi H, Hippo Kinases Mst1 and Mst2 Sense and Amplify IL-2R-STAT5 Signaling in Regulatory T Cells to Establish Stable Regulatory Activity. Immunity 49, 899–914 e896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, Garcia-Lloret MI, Chen K, Ozen A, Carmeliet P, Li MO, Pellegrini M, Chatila TA, Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol 20, 1208–1219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon HK, Chen HM, Mathis D, Benoist C, Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat Immunol 18, 1238–1248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine AG, Arvey A, Jin W, Rudensky AY, Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 15, 1070–1078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C, Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, Jiao J, Veasey SC, Sims CA, Baur JA, Wallace DC, Hancock WW, Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J 29, 2315–2326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC, Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronin E, Lubrano di Ricco M, Vallion R, Divoux J, Kwon HK, Gregoire S, Collares D, Rouers A, Baud V, Benoist C, Salomon BL, The NF-kappaB RelA Transcription Factor Is Critical for Regulatory T Cell Activation and Stability. Front Immunol 10, 2487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, Wang J, Bhatt DM, Heise N, Schmid RM, Hayden MS, Klein U, Rabadan R, Ghosh S, An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 47, 450–465 e455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long M, Park SG, Strickland I, Hayden MS, Ghosh S, Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 31, 921–931 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, Le DT, Liu W, Brusko TM, Anderson MS, Rudensky AY, Marson A, Chang HY, Bluestone JA, A Mutation in the Transcription Factor Foxp3 Drives T Helper 2 Effector Function in Regulatory T Cells. Immunity 50, 362–377 e366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Du X, Shi H, Deng K, Chi H, Tao W, Mammalian Sterile 20-like Kinase 1 (Mst1) Enhances the Stability of Forkhead Box P3 (Foxp3) and the Function of Regulatory T Cells by Modulating Foxp3 Acetylation. J Biol Chem 290, 30762–30770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Xiao J, Zhou X, Xu M, Hu C, Xu X, Lu Y, Liu C, Xue S, Nie L, Zhang H, Li Z, Zhang Y, Ji F, Hui L, Tao W, Wei B, Wang H, STK4 regulates TLR pathways and protects against chronic inflammation-related hepatocellular carcinoma. J Clin Invest 125, 4239–4254 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glantschnig H, Rodan GA, Reszka AA, Mapping of MST1 kinase sites of phosphorylation. Activation and autophosphorylation. J Biol Chem 277, 42987–42996 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Lee KK, Yonehara S, Phosphorylation and dimerization regulate nucleocytoplasmic shuttling of mammalian STE20-like kinase (MST). J Biol Chem 277, 12351–12358 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Praskova M, Xia F, Avruch J, MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr Biol 18, 311–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller CJ, Lou HJ, Simpson C, van de Kooij B, Ha BH, Fisher OS, Pirman NL, Boggon TJ, Rinehart J, Yaffe MB, Linding R, Turk BE, Comprehensive profiling of the STE20 kinase family defines features essential for selective substrate targeting and signaling output. PLoS Biol 17, e2006540 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nie H, Zheng Y, Li R, Guo TB, He D, Fang L, Liu X, Xiao L, Chen X, Wan B, Chin YE, Zhang JZ, Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med 19, 322–328 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA, Williams CB, A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 35, 109–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li MO, Rudensky AY, T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 16, 220–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saccani S, Marazzi I, Beg AA, Natoli G, Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J Exp Med 200, 107–113 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fagerlund R, Kinnunen L, Kohler M, Julkunen I, Melen K, NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J Biol Chem 280, 15942–15951 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Lu HY, Biggs CM, Blanchard-Rohner G, Fung SY, Sharma M, Turvey SE, Germline CBM-opathies: From immunodeficiency to atopy. J Allergy Clin Immunol 143, 1661–1673 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., Muller W, Rudensky AY, Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, Johnson RL, Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 107, 1437–1442 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim DS, Pawson T, Wrana J, McNeill H, Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet 9, e1003380 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF, Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rivas MN, Koh YT, Chen A, Nguyen A, Lee YH, Lawson G, Chatila TA, MyD88 is critically involved in immune tolerance breakdown at environmental interfaces of Foxp3-deficient mice. J Clin Invest 122, 1933–1947 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, Root DE, A public genome-scale lentiviral expression library of human ORFs. Nature methods 8, 659–661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin Y, Khokhlatchev A, Figeys D, Avruch J, Death-associated protein 4 binds MST1 and augments MST1-induced apoptosis. J Biol Chem 277, 47991–48001 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Sanjabi S, Williams KJ, Saccani S, Zhou L, Hoffmann A, Ghosh G, Gerondakis S, Natoli G, Smale ST, A c-Rel subdomain responsible for enhanced DNA-binding affinity and selective gene activation. Genes Dev 19, 2138–2151 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naviaux RK, Costanzi E, Haas M, Verma IM, The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol 70, 5701–5705 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A, FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Charbonnier LM, Wang S, Georgiev P, Sefik E, Chatila TA, Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. Nat Immunol 16, 1162–1173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrews S, FastQC: A Quality Control Tool for High Throughput Sequence Data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- 56.Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10.14806/ej.17.1.200 (2011). [DOI] [Google Scholar]
- 57.Ewels P, Magnusson M, Lundin S, Käller M, MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP, The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Picard toolkit http://broadinstitute.github.io/picard/ (2019).
- 61.Amemiya HM, Kundaje A, Boyle AP, The ENCODE blacklist: identification of problematic regions of the genome. Scientific reports 9, 1–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quinlan AR, BEDTools: the Swiss‐army tool for genome feature analysis. Current protocols in bioinformatics 47, 11.12. 11–11.12. 34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng J, Liu T, Qin B, Zhang Y, Liu XS, Identifying ChIP-seq enrichment using MACS. Nature protocols 7, 1728–1740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu G, Wang L-G, He Q-Y, ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Shen L, Shao N, Liu X, Nestler E, ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC genomics 15, 284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Stk3/4 deficiency in Treg cells causes fatal autoimmune lymphoproliferative disease.
Fig. S2. Defective activation and survival of Stk3/4-deficient Treg cells.
Fig. S3. Treg cell-specific inactivation of the Hippo pathway does not rescue the phenotype of Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice.
Fig. S4. Mice with Treg cell specific activation of the Hippo pathway do not phenocopy Foxp3YFPCreStk3∆/∆Stk4∆/∆ mice.
Fig. S5. TCR/CD28 signaling activates Stk4 by a Zap70, PI3K and Malt1-dependent mechanism to promote Treg suppressive function.
Fig. S6. The Stk4 NLS sequence is critical for Stk4-p65 interaction and CD25 upregulation in Treg cells.
Fig. S7. Inhibition of Stk4 kinase activity attenuates the nuclear translocation of p65.
Fig. S8. p65 and Foxp3S418E -transduced Treg cells ameliorate the disease of Foxp3ΔEGFPiCre mice.
Fig. S9. Gating strategies for human and mouse Treg and Teff cell discrimination.
Table S1. Listing of flow cytometry antibodies used.
Table S2. Listing of immunoblotting antibodies used.
Table S3. Listing of software, package and tools used.
Data Set 1. Phospho-proteomic analysis of Foxp3YFPCre and Foxp3YFPCreStk3∆/∆Stk4∆/∆ Treg cells.
Blank Material Transfer Agreement.
Data Availability Statement
Data presented in the manuscript will be made available to investigators following request to the corresponding author. Any data and materials to be shared will be released via a Material Transfer Agreement. RNAseq datasets have been deposited in the GEO with the accession code GSE178992. CHIP-Seq datasets have been deposited in the Bioproject database with accession code PRJNA84959.